
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic peptide/hemin complex from ester–amide exchange reaction mediated by deep eutectic solvents†
Cheng-Hsi Wanga,
Yao-Yu Jhanga and
Sheng-Sheng Yu *abc
*abc
aDepartment of Chemical Engineering, National Cheng Kung University, Tainan 70101, Taiwan. E-mail: ssyu@mail.ncku.edu.tw
bCore Facility Center, National Cheng Kung University, Tainan, 70101, Taiwan
cProgram on Smart and Sustainable Manufacturing, Academy of Innovative Semiconductor and Sustainable Manufacturing, National Cheng Kung University, Tainan, 70101, Taiwan
First published on 2nd January 2025
Abstract
The functions of peptides often emerge upon their self-assembly or binding with other co-factors. However, the synthetic complexity makes these functional peptides intractable. Here, we utilize the ester–amide exchange reaction in deep eutectic solvents to generate peptide libraries from unactivated amino acids. This strategy leads to peptide mixtures that exhibit hemin-binding capability and peroxidase-like activity.
Peptides are versatile biomolecules for catalysis,1 drug delivery,2 and understanding the origin of life on prebiotic Earth.3 The functions of peptides emerge from their assembly by van der Waals, electrostatic, hydrogen bonding, and π–π stacking interactions.3 In particular, the assembled peptides may exhibit catalytic activities by providing microenvironments to bind substrates and co-factors, such as metal ions and porphyrinic compounds.4 For instance, horseradish peroxidase (HRP) relies on hemin-based co-factors for its oxidase activity.5 Indeed, several recent works use peptides6–9 or modified amino acids10,11 to provide hydrophobic coordinating environments for hemin-binding. The modern route to catalytic peptides employs a rational design that selectively adopts known active sequences from natural proteins.4,12 Some approaches also utilize the high-throughput combinatorial strategy that surveys functional candidates from extensive libraries of peptides.13–15 Nevertheless, complex intermolecular interactions are often difficult to predict. The current synthetic strategy of peptides, especially from unactivated amino acids, is still time-consuming and expensive.16 For example, conventional peptide coupling reactions, such as those used in solid-phase peptide synthesis (SPPS), require stoichiometric amounts of expensive coupling agents and a large amount of toxic solvents.17,18 Despite these challenges, they are widely used to synthesize long peptides with precise sequence control. Ring-opening polymerization of N-carboxy anhydride needs strict reaction environments.19 Genetic engineering for peptide expression is limited by intensive labor and low yield.20
Our previous works on the ester–amide exchange reaction provide a simple method to synthesize a wide range of peptides directly from unactivated amino acids.21–23 Hydroxy acids, such as glycolic acid and lactic acids, form ester linkages upon heating. Subsequently, the ester bonds are exchanged by amine groups of amino acids to form peptide bonds. This process leads to depsipeptides, copolymers containing both ester and amide bonds. This simple reaction can be further modulated by deep eutectic solvents (DES). DES are typically mixtures of hydrogen bond donors and hydrogen bond acceptors.24,25 The intense hydrogen bonding between the two components leads to enthalpic-driven negative deviations from thermodynamic ideality and a significant reduction of melting points of the eutectic mixtures.26 DES has been applied as an alternative media for extraction27 and polymer synthesis.28 We found that hydroxy acids and quaternary ammonium salts form DES that selectively enhance the amide bond formation, leading to peptides with their N-terminus capped by hydroxy acids.29,30 The simplicity of this approach should enable a rapid survey of oligopeptides toward specific functions. Although this method produces mostly short peptides with 2 to 10 residues, several reports have found short or even ultrashort peptides are known to self-assemble and exhibit catalytic activity.9,31,32 These short peptides may provide a cost-effective route to active catalysts.33 Nevertheless, only a few works demonstrate that peptides or depsipeptides from polycondensation and ester–amide exchange exhibit hydrolase-like activity.9,34 More complex functions, such as binding co-factors and oxidase-like activity, have not been achieved.
In this work, we utilized the DES-mediated peptide synthesis to survey peptides for hemin-binding capability and peroxidase-like catalytic behavior (Fig. 1). DES of tetraethylammonium chloride (TEACl) and lactic acid (a) were used to promote peptide bond formation, as reported in our early works.29,30 A high reaction temperature (120 °C) was used to ensure most amino acids have been reacted. Because most proteins utilized histidine (H) to coordinate hemin,35 we mainly studied peptides synthesized from mixtures of histidine and other amino acids. The co-assembly of peptides and hemin was achieved by a common solvent-switching method.36 Hemin and peptides were first dissolved in dimethyl sulfoxide (DMSO) and then transferred to aqueous phosphate buffer solution for co-assembly. UV-Vis spectra of assembled complexes were analyzed to evaluate the hemin-binding capability of different peptide mixtures.
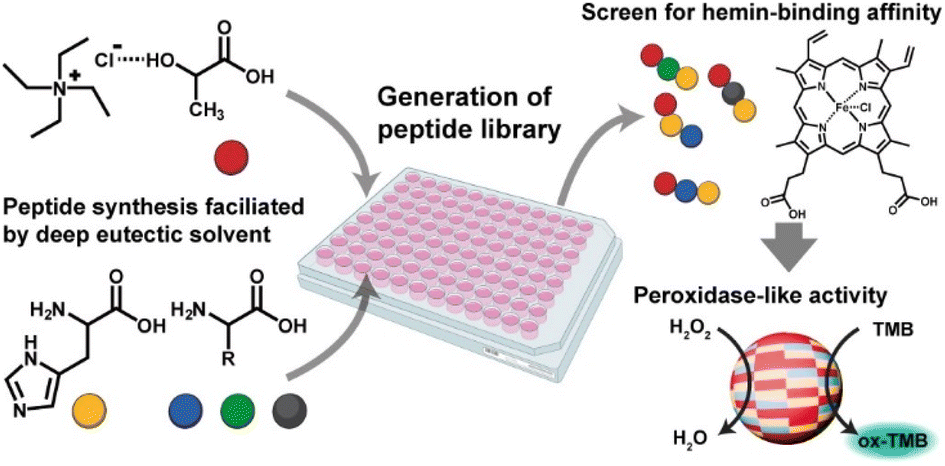 | ||
| Fig. 1 The proposed approach to generate and screen catalytic peptides from DES-mediated ester–amide exchange reaction. | ||
First, we investigated the hemin-binding activity of peptides prepared from histidine and various amino acids. Hydrophobic amino acids like phenylalanine (F), leucine (L), valine (V), and alanine (A) were used. Polar amino acids, such as lysine (K) and aspartic acid (D), were also included. As shown in Fig. 2a, the UV-Vis spectrum of hemin in aqueous solution showed a broad Soret band at 370–390 nm and a weak Q band at 635 nm, corresponding to the aggregated hemin dimers.35 Among these pairs of amino acid mixtures, peptides from histidine and phenylalanine exhibited superior hemin binding capability, as characterized by significant shifts in both the Soret band and the Q band. For completeness, we also analyzed the products from other amino acids (cysteine (C), glycine (G), methionine (M), arginine (R), and serine (S)) that are known for hemin-binding.14 Again, none of them showed significant changes in UV-Vis spectra after mixing with hemin (Fig. S1, ESI†). We further utilized liquid chromatography-mass spectrometry (LC-MS) to identify the oligomers produced by various amino acid combinations (Fig. S2 to S12, ESI†). Similar to previous work,37 products were designated by the number of lactic acids (lowercase letter “a”) and amino acid residues (uppercase letters) without specifying their positions in the sequence. Most products were short oligomers with up to 3 amino acid residues. For the H/F combination, the main products observed were 1a1H, 1a1F, and 1a2F, meaning that the first peptide had one lactic acid and one histidine residue in the structure, the second peptide had one lactic acid and one phenylalanine, and the third peptide had one lactic acid and two phenylalanines. The terminal groups were H– and –OH, indicating uncapped (depsi)peptides. Some mixed oligomers like 1a1H1F were also observed. We hypothesize that the aromatic side chain of phenylalanine not only provides a hydrophobic microenvironment but also forms π–π interactions with the heterocyclic structure of hemin to stabilize hemin more effectively than other amino acids.
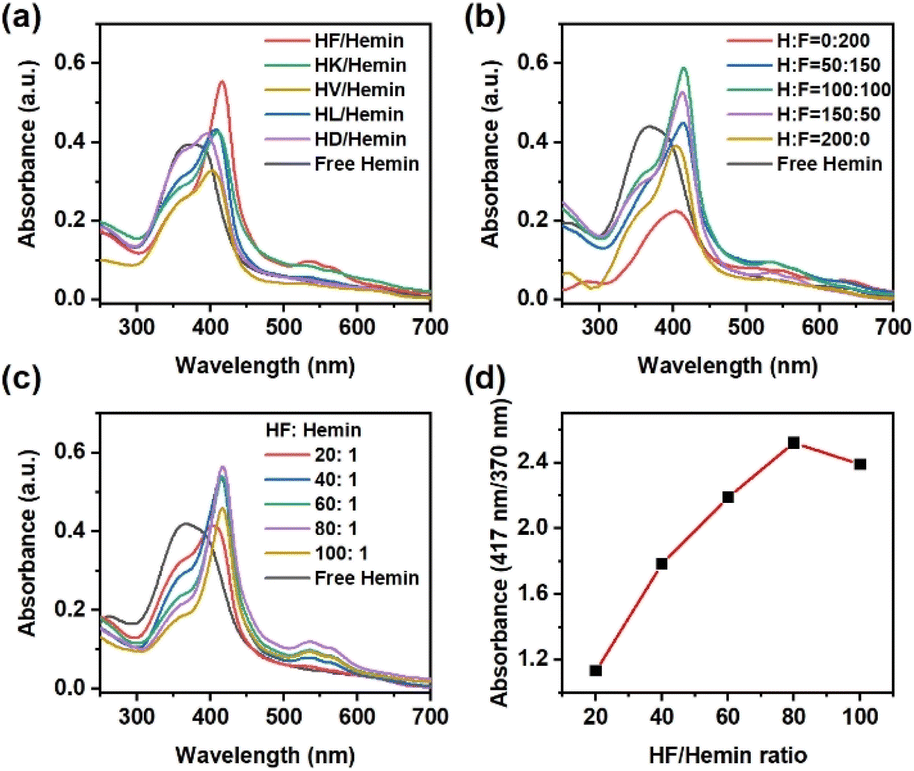 | ||
Fig. 2 (a) UV-Vis spectra of hemin with peptides generated from different amino acids. The ratio of histidine to other amino acids was 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. The molar ratio of amino acids to hemin was 80:1. (b) UV-Vis spectra of hemin with HF peptides using different H/F ratio. The molar ratio of amino acids to hemin was 80:1. (c) UV-vis spectra of hemin with HF peptides using different HF/hemin ratio. (d) Absorbance change vs. HF/hemin ratio. The molar ratio of H to F in the peptide mixture was 100:100. The concentration of hemin was 10 μM. The solution was a 100 mM phosphate buffer solution at pH 7.0. :100. The molar ratio of amino acids to hemin was 80:1. (b) UV-Vis spectra of hemin with HF peptides using different H/F ratio. The molar ratio of amino acids to hemin was 80:1. (c) UV-vis spectra of hemin with HF peptides using different HF/hemin ratio. (d) Absorbance change vs. HF/hemin ratio. The molar ratio of H to F in the peptide mixture was 100:100. The concentration of hemin was 10 μM. The solution was a 100 mM phosphate buffer solution at pH 7.0. | ||
We further changed the molar ratio of histidine to phenylalanine in the starting materials. As shown in Fig. 2b, the ratio of the two monomers significantly changed the hemin-binding affinity of the peptides. Peptides composed solely of phenylalanine showed no hemin-binding capability and tended to precipitate, resulting in a low UV-vis absorbance. Peptides prepared solely from histidine only had minimal hemin-binding capability, consistent with other reports using poly(histidine).38 The product with H:F = 100:100 showed the optimal binding capability to hemin. These peptides were also analyzed by LC-MS and sequenced by tandem MS (Fig. S13 to S21, ESI†). The main product using only phenylalanine was a-F. Peptides composed solely of histidine only contained oligomers like a-H and a-H. The samples with both histidine and phenylalanine showed mixed oligomers like a-H–F and a-F–H. All products contained a lactic acid unit at their N-termini, as expected for the ester–amide exchange reaction.
We also attempted to quantify the amount of HF-derived peptides required to coordinate hemin. Since the ester–amide exchange reaction provides a mixture of peptides, it was difficult to estimate the exact concentration of each peptide in our products. Therefore, we used the total amount of amino acids in the starting materials to represent the quantity of peptide mixture. As shown in Fig. 2c, increasing the concentration of peptides indeed leads to a gradual change in UV-Vis spectra. The changes in UV-Vis spectra were most significant when HF:hemin ratio was 80:1. The ratio of absorbance at 417 nm and 370 nm was used as an indicator for the extent of hemin-binding. The absorbance reached a maximum at 80:1, but decreased slightly at 100:1, potentially because of the large quantity of precipitated peptides (Fig. 2d). We also compared the ability of histidine monomers to bind with hemin, especially since existing literature has found that histidine may directly bind to hemin.38 As shown in Fig. S22a (ESI†), the addition of histidine monomers changed the UV-Vis spectrum of hemin, showing a redshift in the Soret band and the appearance of Q bands at 537 and 565 nm. However, the concentration of histidine needed to be at least 10000 times that of hemin to be effective (Fig. S22b, ESI†). In contrast, the HF peptide requires an amino acid concentration only 80 times that of hemin to achieve significant changes in the UV-Vis spectra, suggesting that the assembly and aggregation of the peptides provide the necessary microenvironment for the hemin complex.
Next, we examined the effect of reaction conditions, such as DES and temperature, on the hemin-binding capability of HF-derived peptides. As shown in Fig. S23a (ESI†), the products synthesized at a lower temperature (95 °C) in DES still changed the UV-Vis absorption of hemin. In the cases without TEACl to form DES, the products from 120 °C also exhibited hemin-binding capability. However, when the reaction temperature was lowered to 95 °C, the product without TEACl could not coordinate hemin (Fig. S23b, ESI†). For the sample prepared at 95 °C with DES, the main products were similar to the one at 120 °C, but at lower abundance (Fig. S24 and S25, ESI†). Some unreactive amino acids could also be found. Similar product distribution were found in the control experiment without TEACl at 120 °C (Fig. S26 and S27, ESI†). Reaction at 95 °C without DES only produced 1a1F and 1a1H with much lower abundance than in previous cases (Fig. S28, ESI†). Based on these results, DES effectively reduces the reaction temperature required to synthesize active peptides that coordinate hemin.
Furthermore, we prepared two batches of peptide products by using only histidine (H:F = 200:0) or phenylalanine (H:F = 0:200). The two batches were then physically mixed for the hemin binding test. The physically mixed peptides exhibited some hemin binding capability, but it was not as effective as the sample synthesized using both histidine and phenylalanine as starting material (Fig. S29a, ESI†). We also varied the molar ratio of physically mixed peptides to hemin and found that the ratio of 80:1 still exhibited the most effective hemin binding (Fig. S29b, ESI†). We then conclude that the combination of short oligomers with only histidine and phenylalanine (a-H and a-F) was sufficient to bind hemin, but peptides with mixed sequences of both amino acids could promote the hemin binding process.
We then examined the assembly and aggregation morphology of HF/hemin complex. As shown in Fig. 3a, both HF peptides and HF/hemin aggregated in water, but their colloidal stability was significantly different. HF peptides exhibited noticeable precipitation after 3 hours, whereas the HF/hemin complex did not precipitate. Fig. 3b shows the particle size distribution measured by dynamic light scattering (DLS). HF peptides tended to form large aggregates, with some beyond the detection limit of the DLS measurement. In contrast, the particle size of the HF/hemin complex was generally small and exhibited two distinct size distributions, approximately 100 nm and 900 nm in diameter. We also used atomic force microscopy (AFM) to observe the morphology of the samples (Fig. 3c and d). The HF peptides formed larger and irregular aggregates, while the HF/hemin complex predominantly formed nearly spherical particles with some aggregation. This result indicates some synergistic interactions between hemin and the HF-derived peptides exist during the assembly process to form colloidally stable particles.
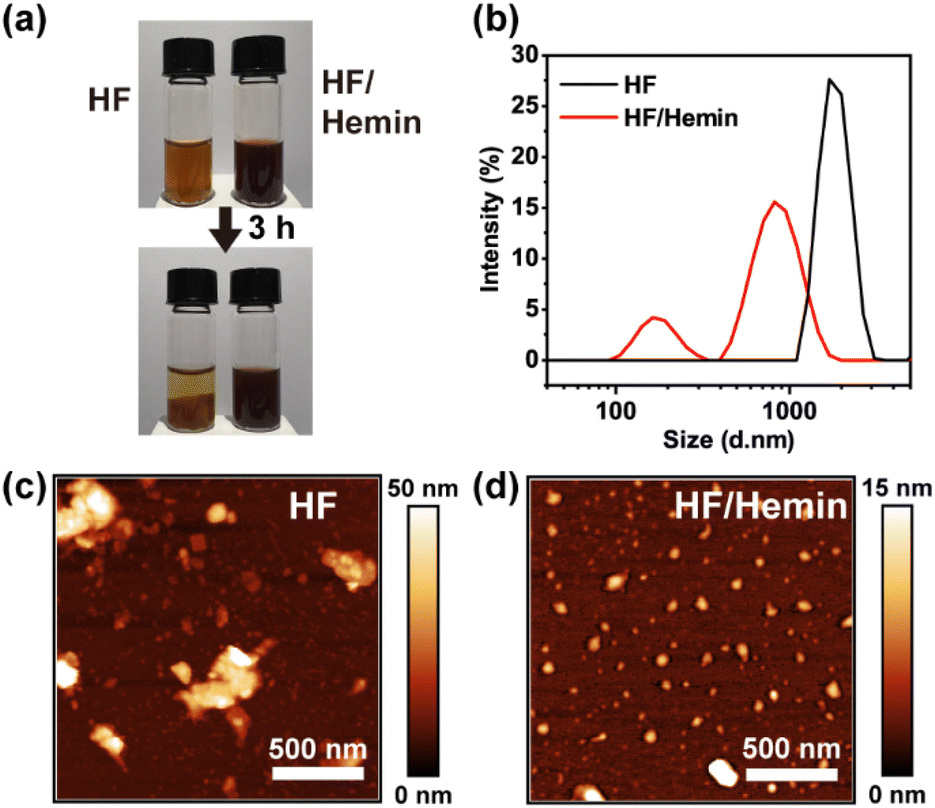 | ||
| Fig. 3 (a) The colloidal stability of HF and HF/hemin complex. (b) Particle size distribution of HF peptides and HF/hemin, determined by DLS. (c) AFM image of HF. (d) AFM image of HF/hemin. The molar ratio of amino acids to hemin was 80:1. HF peptides were prepared from a mixture of TEACl/a/H/F (100:100:100:100) at 120 °C for 3 days. | ||
We further examined the enzymatic activity of the HF/hemin complex by using the H2O2-mediated oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB).5,35 The substrate TMB is a colorless compound, and its oxidized product has a blue-green color with a high absorbance at a wavelength of 652 nm. Thus, the change in color and UV-Vis spectra can be used to verify the activity of the catalysts. As shown in Fig. 4a, samples without any catalyst or with only HF peptides showed no significant color change after 500 seconds. The sample with hemin alone exhibited a weak blue-green color. On the other hand, the sample containing the HF/hemin complex showed a significant blue-green color. Fig. 4b further confirms that under these conditions, the sample with the HF/hemin complex had the highest absorbance at 652 nm. Then, we tracked the reaction kinetics of each sample from 0 to 500 seconds and found that the sample containing the HF/hemin complex had the fastest oxidation rate (Fig. 4c). From these results, HF/hemin complex indeed exhibits the typical activity common enzymes, indirectly verifying that hemin was in an appropriate microenvironment. We also compared the catalytic activities of the products generated by using other amino acids (Fig. S30, ESI†). HF/hemin and HG/hemin had a similar catalytic activity that was higher than peptides prepared by other amino acids. The result of HG/hemin suggests that hemin-binding efficiency may not be the sole factor that governs the peroxidase activity.
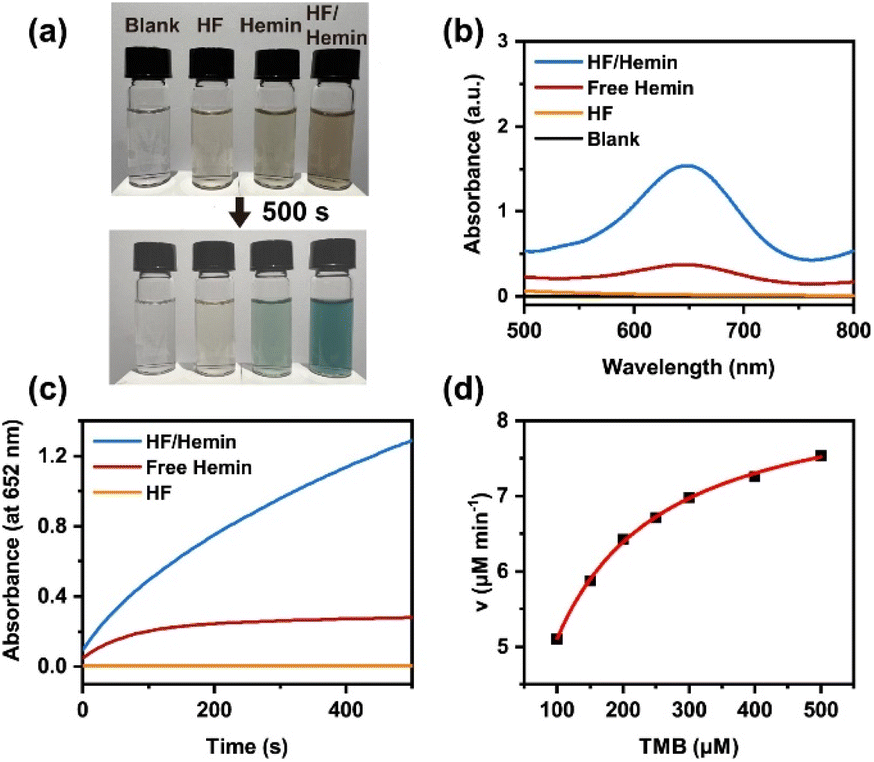 | ||
| Fig. 4 (a) Color changes of TMB oxidation test. (b) UV-Vis spectra of different samples after 500 s of TMB oxidation. (c) Kinetics of TMB oxidation in different samples. (d) Michaelis–Menten kinetic fitting results of HF/hemin at different TMB concentrations. 500 μM TMB, 500 μM H2O2, and 16.67 μM hemin were used. The molar ratio of amino acids to hemin was 80:1. HF peptides were prepared from a mixture of TEACl/a/H/F (100:100:100:100) at 120 °C for 3 days. | ||
Finally, we measured the initial reaction rates at different TMB concentrations to analyze the reaction kinetics. Our results indicate that the kinetics of this reaction followed the Michaelis–Menten model of common enzymes (Fig. 4d). The apparent kinetic parameters, kcat and KM could be estimated from its Lineweaver–Burk plot (Fig. S31a, ESI†). We also analyzed the TMB oxidation kinetics by common HRP enzymes (Fig. S31b, ESI†). As shown in Table S1,† HF/hemin had a lower Km value than that of HRP, which indicated a high affinity of HF/hemin to the TMB substrate. However, the kcat value of HRP was significantly higher than that of HF/hemin, suggesting that the natural enzyme still had a higher catalytic efficiency. In addition, the kcat/Km value was also higher for HRP, indicating that HRP has better overall catalytic efficiency. Based on the above results, the enzymatic catalysis of our HF/hemin peptides is mainly limited by the step that converts the enzyme/substrate to products. In addition, this reaction may be limited by the surface area of HF/hemin nanoparticles. The size of HF/hemin nanoparticles was larger than that of HRP (3.6 nm in diameter).39 As a result, most hemin active sites embedded inside the HF/hemin particles could not participate in the TMB oxidation.
Further engineering on the assembling process is required to enhance the catalytic performance of our materials. The intriguing roles of HF-derived peptides and hemin during assembly also require further study, but common characterizations, like infrared spectroscopy and circular dichroism, are currently limited by the complexity of the peptide products. Further work is ongoing to synthesize different sequences of short HF peptides by SPPS and probe their hemin-binding behavior. Although we only used UV-Vis spectroscopy to probe the hemin-binding behavior of our products, electron paramagnetic resonance spectroscopy may also provide additional information on the coordination state of hemin.8,40
In conclusion, we demonstrated the ester–amide exchange reaction with DES is effective in screening peptides capable of forming complexes with hemin for a typical peroxidase-like activity. In particular, DES enabled the formation of functional peptides at a low reaction temperature. Short peptides composed of histidine and phenylalanine were sufficient to form a catalytic complex with hemin. Although we used intensive LC-MS analysis to characterize the peptide libraries, these peptides could be directly used for hemin-binding without additional treatment. Our findings provide proof-of-principle of a simple strategy to synthesize artificial enzymes from unactivated amino acids. The peptide/hemin assembling process may be further fine-tuned to optimize the size distribution, morphologies, and catalytic activity of the complex. Although we mainly focused on short peptides for simplicity, our library may be expanded by feeding additional amino acids to extend the peptide chain length,41 potentially leading to other functionalities.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science and Technology Council in Taiwan for financial support (111-2314-B-006-046, 112-2314-B-006-010, 113-2314-B-006-006 and 110-2222-E-006-009-MY3). We also thank the support by Higher Education Sprout Project, Ministry of Education to the Headquarters of University Advancement at National Cheng Kung University (NCKU). The authors gratefully acknowledge the use of high-resolution Orbitrap mass spectrometry (MS004000) of NSTC 112-2740-M-006-001 belonging to the Core Facility Center of National Cheng Kung University.Notes and references
- S. Liu, P. Du, H. Sun, H.-Y. Yu and Z.-G. Wang, ACS Catal., 2020, 10, 14937–14958 CrossRef CAS.
- J. Yang, H.-W. An and H. Wang, ACS Appl. Bio Mater., 2021, 4, 24–46 CrossRef CAS.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS.
- L. R. Marshall, O. Zozulia, Z. Lengyel-Zhand and I. V. Korendovych, ACS Catal., 2019, 9, 9265–9275 CrossRef CAS PubMed.
- Y. Liu and Z.-G. Wang, ACS Nano, 2023, 17, 13000–13016 CrossRef CAS.
- H. Christopher Fry, R. Divan and Y. Liu, Nanoscale, 2022, 14, 10082–10090 RSC.
- Y. Zhang and X. Li, J. Mater. Chem. B, 2023, 11, 3898–3906 RSC.
- L. A. Solomon, J. B. Kronenberg and H. C. Fry, J. Am. Chem. Soc., 2017, 139, 8497–8507 CrossRef CAS PubMed.
- O. Zozulia and I. V. Korendovych, Angew. Chem., Int. Ed., 2020, 59, 8108–8112 CrossRef CAS PubMed.
- Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS PubMed.
- R. Geng, R. Chang, Q. Zou, G. Shen, T. Jiao and X. Yan, Small, 2021, 17, 2008114 CrossRef CAS.
- Y. Maeda, O. V. Makhlynets, H. Matsui and I. V. Korendovych, Annu. Rev. Biomed. Eng., 2016, 18, 311–328 CrossRef CAS.
- M. P. Friedmann, V. Torbeev, V. Zelenay, A. Sobol, J. Greenwald and R. Riek, PLoS One, 2015, 10, e0143948 CrossRef PubMed.
- T. Kühl, N. Sahoo, M. Nikolajski, B. Schlott, S. H. Heinemann and D. Imhof, ChemBioChem, 2011, 12, 2846–2855 CrossRef PubMed.
- K. Hilpert, R. Volkmer-Engert, T. Walter and R. E. W. Hancock, Nat. Biotechnol., 2005, 23, 1008–1012 CrossRef CAS PubMed.
- X. Wang, Nat. Catal., 2019, 2, 98–102 CrossRef.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- W. Wang, H. Fu, Y. Lin, J. Cheng and Z. Song, Acc. Mater. Res., 2023, 4, 604–615 CrossRef CAS.
- B. E. I. Ramakers, J. C. M. van Hest and D. W. P. M. Löwik, Chem. Soc. Rev., 2014, 43, 2743–2756 RSC.
- J. G. Forsythe, S.-S. Yu, I. Mamajanov, M. A. Grover, R. Krishnamurthy, F. M. Fernández and N. V. Hud, Angew. Chem., Int. Ed., 2015, 54, 9871–9875 CrossRef CAS.
- M. Frenkel-Pinter, J. W. Haynes, M. C. Solano, A. S. Petrov, B. T. Burcar, R. Krishnamurthy, N. V. Hud, L. J. Leman and L. D. Williams, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16338–16346 CrossRef CAS.
- M. Frenkel-Pinter, A. B. Sargon, J. B. Glass, N. V. Hud and L. D. Williams, RSC Adv., 2021, 11, 3534–3538 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- B. Gurkan, H. Squire and E. Pentzer, J. Phys. Chem. Lett., 2019, 10, 7956–7964 CrossRef CAS PubMed.
- D. O. Abranches and J. A. P. Coutinho, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 141–163 CrossRef CAS PubMed.
- D. J. G. P. van Osch, L. F. Zubeir, A. van den Bruinhorst, M. A. A. Rocha and M. C. Kroon, Green Chem., 2015, 17, 4518–4521 RSC.
- C.-Y. Li and S.-S. Yu, Macromolecules, 2021, 54, 9825–9836 CrossRef CAS.
- C.-Y. Chien and S.-S. Yu, Chem. Commun., 2020, 56, 11949–11952 RSC.
- Y.-T. Tsai, C.-W. Huang and S.-S. Yu, Phys. Chem. Chem. Phys., 2021, 23, 27498–27507 RSC.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- A. M. Garcia, M. Kurbasic, S. Kralj, M. Melchionna and S. Marchesan, Chem. Commun., 2017, 53, 8110–8113 RSC.
- R. Das, B. Gayakvad, S. D. Shinde, J. Rani, A. Jain and B. Sahu, ACS Appl. Bio Mater., 2020, 3, 5474–5499 CrossRef CAS.
- A. J. Surman, M. Rodriguez-Garcia, Y. M. Abul-Haija, G. J. T. Cooper, P. S. Gromski, R. Turk-MacLeod, M. Mullin, C. Mathis, S. I. Walker and L. Cronin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5387–5392 CrossRef CAS.
- N. Cvjetan and P. Walde, Q. Rev. Biophys., 2023, 56, 1–43 CrossRef CAS PubMed.
- I. W. Hamley, ACS Appl. Bio Mater., 2023, 6, 384–409 CrossRef CAS.
- J. G. Forsythe, A. S. Petrov, W. C. Millar, S.-S. Yu, R. Krishnamurthy, M. A. Grover, N. V. Hud and F. M. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7652–E7659 CrossRef CAS PubMed.
- E. Karnaukhova, S. Rutardottir, M. Rajabi, L. Wester Rosenlöf, A. I. Alayash and B. Åkerström, Front. Physiol., 2014, 5, 465 Search PubMed.
- S. Tan, D. Gu, H. Liu and Q. Liu, Nanotechnology, 2016, 27, 155502 CrossRef PubMed.
- M. Ju, J. Kim and J. Shin, Bull. Korean Chem. Soc., 2024, 45, 835–862 CrossRef CAS.
- S.-S. Yu, M. D. Solano, M. K. Blanchard, M. T. Soper-Hopper, R. Krishnamurthy, F. M. Fernández, N. V. Hud, F. J. Schork and M. A. Grover, Macromolecules, 2017, 50, 9286–9294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08607d |
| This journal is © The Royal Society of Chemistry 2025 |