
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, anticancer activity and molecular docking of quinoline-based dihydrazone derivatives†
Jia-Xing Lua,
Hai-Rong Lana,
Dai Zenga,
Jun-Ying Songb,
Ya-Ting Haoa,
Ai-Ping Xing*a,
Ao Shen*a and
Juan Yuan *a
*a
aSchool of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, China. E-mail: hn_xap@163.com; 18515912322@163.com; hnzz_yuan@hactcm.edu.cn
bAcademy of Chinese Medical Sciences, Henan University of Chinese Medicine, Zhengzhou 450046, China
First published on 2nd January 2025
Abstract
Based on the biologically active heterocycle quinoline, we successfully synthesized a series of quinoline-based dihydrazone derivatives (3a–3d). 1H NMR, 13C NMR, ESI-HRMS, IR, element analysis, UV/Vis spectroscopy and fluorescence spectroscopy were performed to comprehensively characterize their chemical structures, spectral properties and stability. Nitrosamine impurities were not detected in 3a–3d, and the systemic toxicological assessment indicated that the toxicity of 3a–3d was lower. Furthermore, their anticancer activity was evaluated by MTT, AO/EB double staining, apoptosis detection and ROS detection. The time-dependent UV/Vis spectra revealed that 3a–3d had good stability in solution. For all the newly synthesized compounds, cytotoxic activities were carried out against human gastric cancer cell line BGC-823, human hepatoma cell line BEL-7402, human breast cancer cell line MCF-7 and human lung adenocarcinoma cell line A549 as well as human normal liver cell line HL-7702. MTT assay indicated that all the tested compounds exhibited important antiproliferative activity against selected cancer cell lines with IC50 values ranging from 7.01 to 34.32 μM, while none of them had obvious cytotoxic activity to human normal liver cell line HL-7702. Further, the most potent compound 3c displayed stronger antiproliferative activity against all the selected cancer cell lines than the clinically used anticancer agent 5-FU. Especially, 3b and 3c displayed cytotoxic activity against MCF-7 cells with IC50 values of 7.016 μM and 7.05 μM, respectively. AO/EB double staining, flow cytometry and ROS detection suggested that 3b and 3c could induce MCF-7 cell apoptosis in a dose-dependent manner. Molecular docking suggests that 3b and 3c could bind with DNA via partial insertion. Additionally, molecular docking also suggests that CDK2 may be one of the targets for 3b and 3c. In a word, 3b and 3c could be suitable candidates for further investigation as chemotherapeutic agents in cancer treatment.
1. Introduction
Cancer is a complicated disease, the rising incidence of which is seriously affecting our health.1 Chemotherapy, as one of the most important treatment pathways for cancer,2 has always been of great concern for researchers and clinical doctors. The development of anticancer agents with better treatment efficiency and fewer clinical side effects has attracted more and more attention of medicinal chemists.3 N-heterocycles play a significant role in the design and synthesis of chemotherapy drugs. Quinoline and its derivatives, as a significant class of pharmaceutically active heterocyclic compounds, demonstrated diverse pharmaceutical activities, such as antiviral,4 anticancer,5,6 inhibitor,7 antihypertensive,8 antibacterial,9,10 antimalarial11 and others. Moreover, quinoline rings are found in various natural products, especially in alkaloids.12,13 So, quinoline derivatives have attracted enormous attention of chemists and biologists. Meanwhile, hydrazones have attracted continuous attention in the medical field because of their good biological activity and reactivity.14–18 The hydrazone moiety cannot only improve the flexibility of a chemical structure to avoid steric hindrance,19 but also be the linker to merge functional groups together. Therefore, hydrazone moieties are often used as an active building block in antiviral and anticancer agents.20,21 Quinoline-based hydrazone derivatives are highly important because of the structural flexibility and pharmacological activities. To our knowledge, a large number of quinoline-based hydrazone derivatives have been studied for their potential anticancer activity (Fig. 1).20,22–26 However, we found that anticancer compounds that integrate two quinoline units into one molecule via hydrazone bonds have not been reported so far. Generally speaking, the number of active functional groups in a compound will be beneficial for its biological activity. Dihydrazone structure makes it possible to increase the number of active functional groups in a molecule, which is expected to improve its biological activity. Based on these facts, supported by our ongoing research for new anticancer agents,27 a series of new quinoline-based dihydrazone derivatives (3a–3d) linked by pyrimidine skeleton were designed, synthesized and investigated for their anticancer properties. Since nitrosamine impurities were detected in pharmaceuticals in 2018,28 people have become aware of these impurities in raw materials. Since 3a–3d are dihydrazone derivatives, nitrosamine impurities were evaluated and detected. Systemic toxicological evaluation of antitumor drug activity is very important because most chemotherapeutic cancer agents are non-specific in action; besides attacking tumors, they rapidly demolish normally dividing cells, and can cause extensive collateral side effects.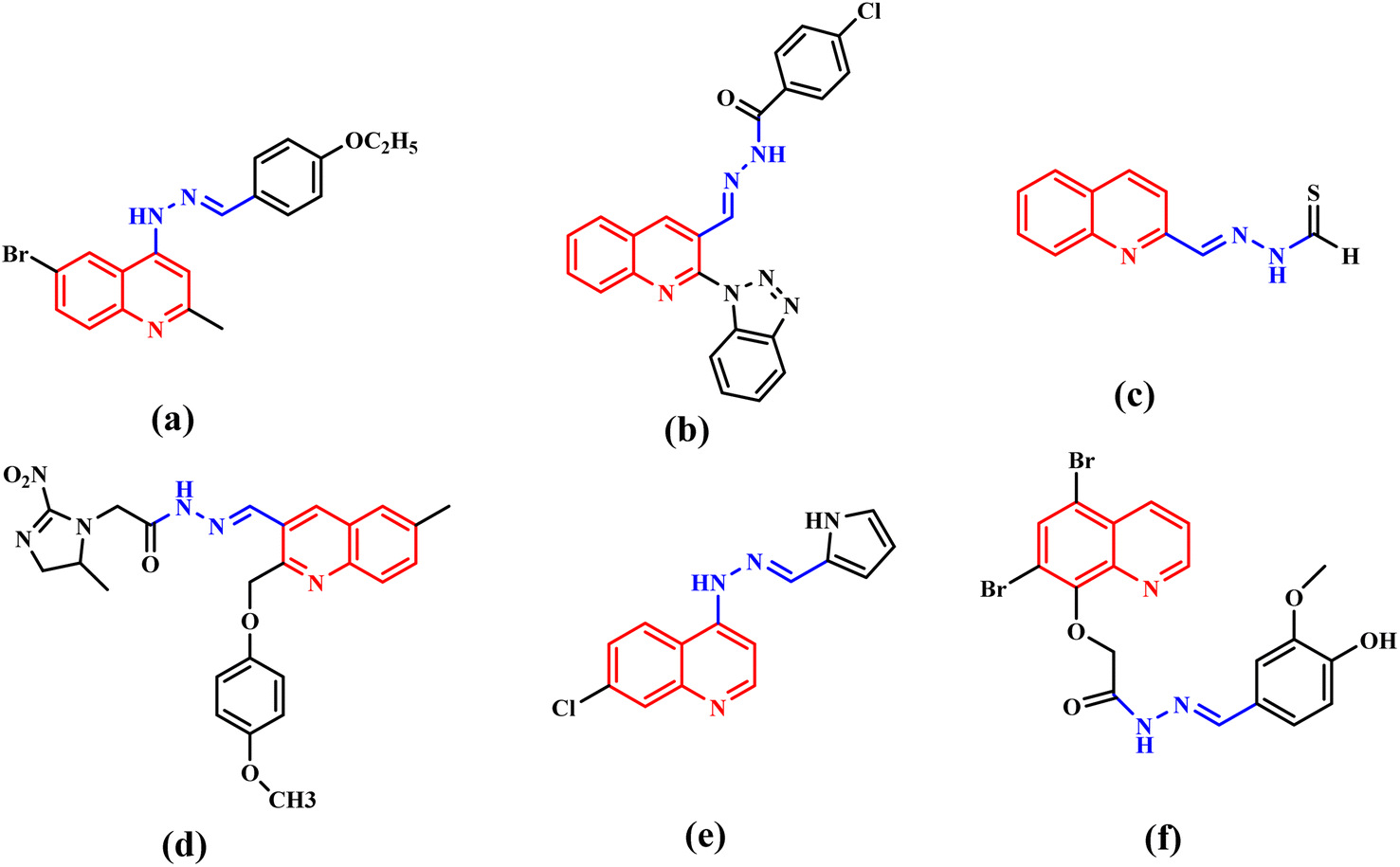 | ||
| Fig. 1 Anticancer agents (a–f) containing quinoline hydrazone moieties. | ||
Quinoline derivatives have been revealed to exert anticancer effects through various mechanisms, which involved disruption of cell migration, growth inhibitors by cell cycle arrest, inhibition of angiogenesis, apoptosis and modulation of nuclear receptor responsiveness.29–33 In addition, it has been reported that many quinoline derivatives exhibit significant anticancer activity through DNA binding.20,34 Deviations within the CDK pathway have been observed in various types of cancer.35,36 CDKs form complexes with cyclins to efficiently regulate tumour growth. CDKs have recently emerged as an extremely promising drug target for treating malignant tumours due to their involvement in various processes such as RNA processing, proliferation, and cell survival.37–39 Therefore, developing anti-cancer drugs with CDK inhibitory activity is increasingly attracting attention. CDK inhibitors have advanced to the third generation, including palbociclib, ribociclib, and abemaciclib. Because they can selectively inhibit CDK4/6, they are also known as CDK4/6 inhibitors. Many non-selective CDK inhibitors, such as milciclib, dinaciclib and NU6300 (Fig. 2), which contain amino-substituted heterocyclic scaffold, have played a crucial role in developing binding affinity on CDK2.40 3a–3d also contain amino-substituted heterocyclic scaffolds, the molecule docking study was performed to explain whether 3a–3d also interact with CDK2. These docking results may indicate that 3a–3d has potential anticancer activity by inhibiting the activity of CDKs. Herein, on the basis of preliminary screening of the cytotoxic activity of 3a–3d by MTT assay, 3b and 3c were further investigated for their pro-apoptotic effect, DNA-binding and CDKs inhibitory activities.
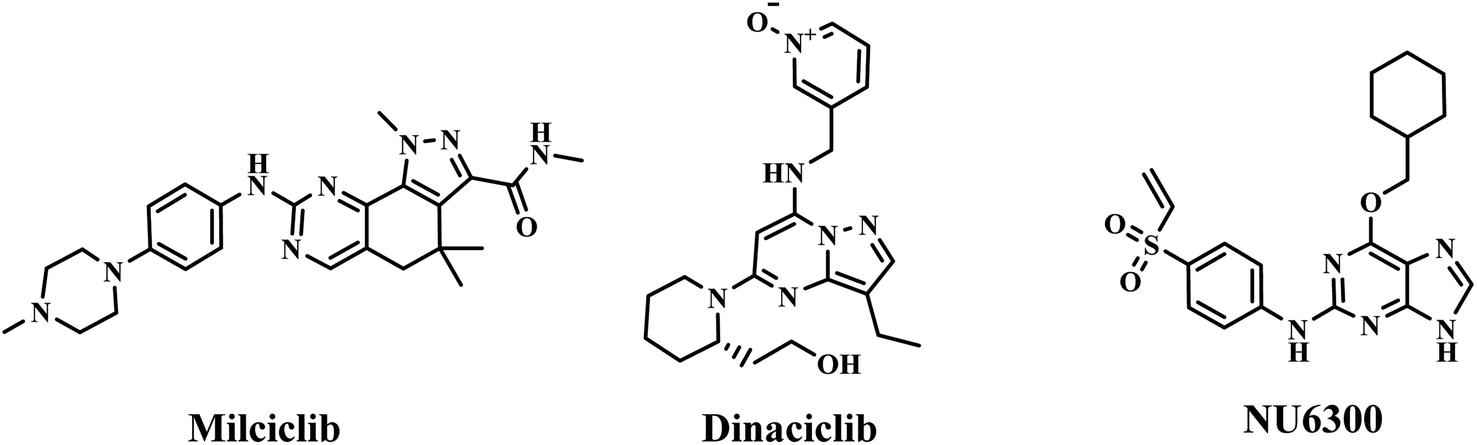 | ||
| Fig. 2 CDK inhibitors containing amino-substituted heterocyclic scaffold. | ||
2. Experimental section
2.1. Materials and measurements
20 Female Blab/c mice (age 6 weeks) of SPF grade were obtained from Changsheng Biotechnology Co., Ltd (SCXK 2020-0001). The animals were housed and kept under a temperature-controlled room (22–25 °C), with 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light–dark cycle and free access to food and water. The experimental protocol was submitted and approved by the Animal Management and Welfare Ethics Committee of Henan University of Chinese Medicine. All chemical raw materials used were obtained from Beijing Inno Chem Science &Technology Co., Ltd (Beijing, China). Biochemical reagents were purchased from Solarbio Science & Technology Co., Ltd. All the above reagents were used directly as purchased. All cell lines (HL-7702, MCF-7, BGC-823, BEL-7402 and A549) were purchased from the Obio Technology (Shanghai) Corp., Ltd (Shanghai, China). Tris-HCl-NaCl buffer solution (0.05 M Tris-HCl/0.1 M NaCl, pH 7.4) was prepared by using ultrapure water. UV/Vis spectra were performed within the range of 200–750 nm on a Shimadzu UV-3600i plus spectrophotometer (Japan). The FLS1000 spectrometer (UK) was used to record fluorescence spectra. 1H NMR and 13C NMR were performed on a Bruker AVANCE III spectrometer (Germany, 500 MHz). ESI-HRMS was carried out on a Bruker Thermo Scientific Q Exactive high resolution mass spectrometry (Germany). Elemental analyses were determined on an Elementar Vario EL elemental analyzer. IR absorption spectra were measured in the range of 4000–400 cm−1 on a WQF-510A FTIR spectrophotometer. MTT assay was collected at 570 nm using a Thermo fisher Multiskan GO (America). AO/EB staining was performed under a Leica DMIL LED inverted fluorescence microscope (Germany). Cell apoptosis and ROS intensity were detected by a flow cytometry (Beckman coulter CytoFLEX, America).
:12 h light–dark cycle and free access to food and water. The experimental protocol was submitted and approved by the Animal Management and Welfare Ethics Committee of Henan University of Chinese Medicine. All chemical raw materials used were obtained from Beijing Inno Chem Science &Technology Co., Ltd (Beijing, China). Biochemical reagents were purchased from Solarbio Science & Technology Co., Ltd. All the above reagents were used directly as purchased. All cell lines (HL-7702, MCF-7, BGC-823, BEL-7402 and A549) were purchased from the Obio Technology (Shanghai) Corp., Ltd (Shanghai, China). Tris-HCl-NaCl buffer solution (0.05 M Tris-HCl/0.1 M NaCl, pH 7.4) was prepared by using ultrapure water. UV/Vis spectra were performed within the range of 200–750 nm on a Shimadzu UV-3600i plus spectrophotometer (Japan). The FLS1000 spectrometer (UK) was used to record fluorescence spectra. 1H NMR and 13C NMR were performed on a Bruker AVANCE III spectrometer (Germany, 500 MHz). ESI-HRMS was carried out on a Bruker Thermo Scientific Q Exactive high resolution mass spectrometry (Germany). Elemental analyses were determined on an Elementar Vario EL elemental analyzer. IR absorption spectra were measured in the range of 4000–400 cm−1 on a WQF-510A FTIR spectrophotometer. MTT assay was collected at 570 nm using a Thermo fisher Multiskan GO (America). AO/EB staining was performed under a Leica DMIL LED inverted fluorescence microscope (Germany). Cell apoptosis and ROS intensity were detected by a flow cytometry (Beckman coulter CytoFLEX, America).
2.2. Chemistry
The intermediates 4,6-dihydrazinylpyrimidine (2a) and 4,6-dihydrazinyl-2-phenylpyrimidine (2b) were similarly prepared by the reaction of 4,6-dichloropyrimidine (1a) or 4,6-dichloro-2-phenylpyrimidine (1b) with an excess of hydrazine hydrate.The detailed synthesis process of 2a was as follows: 1a (10 mmol, 1.49 g) was dispersed in 10 mL hydrazine hydrate. Then, the mixture was slowly heated to 60 °C under stirring. White solid gradually precipitated during the stirring process. The reaction process was monitored by thin-layer chromatography (TLC). After the reaction was completed, the reaction solution was cooled overnight to promote complete precipitation. Finally, the product was filtered, washed with water and then dried, which was named 4,6-dihydrazinyl pyrimidine (2a), yield ca. 72.0%. The intermediate 2b was also obtained by the same method, yield ca. 55.5%.
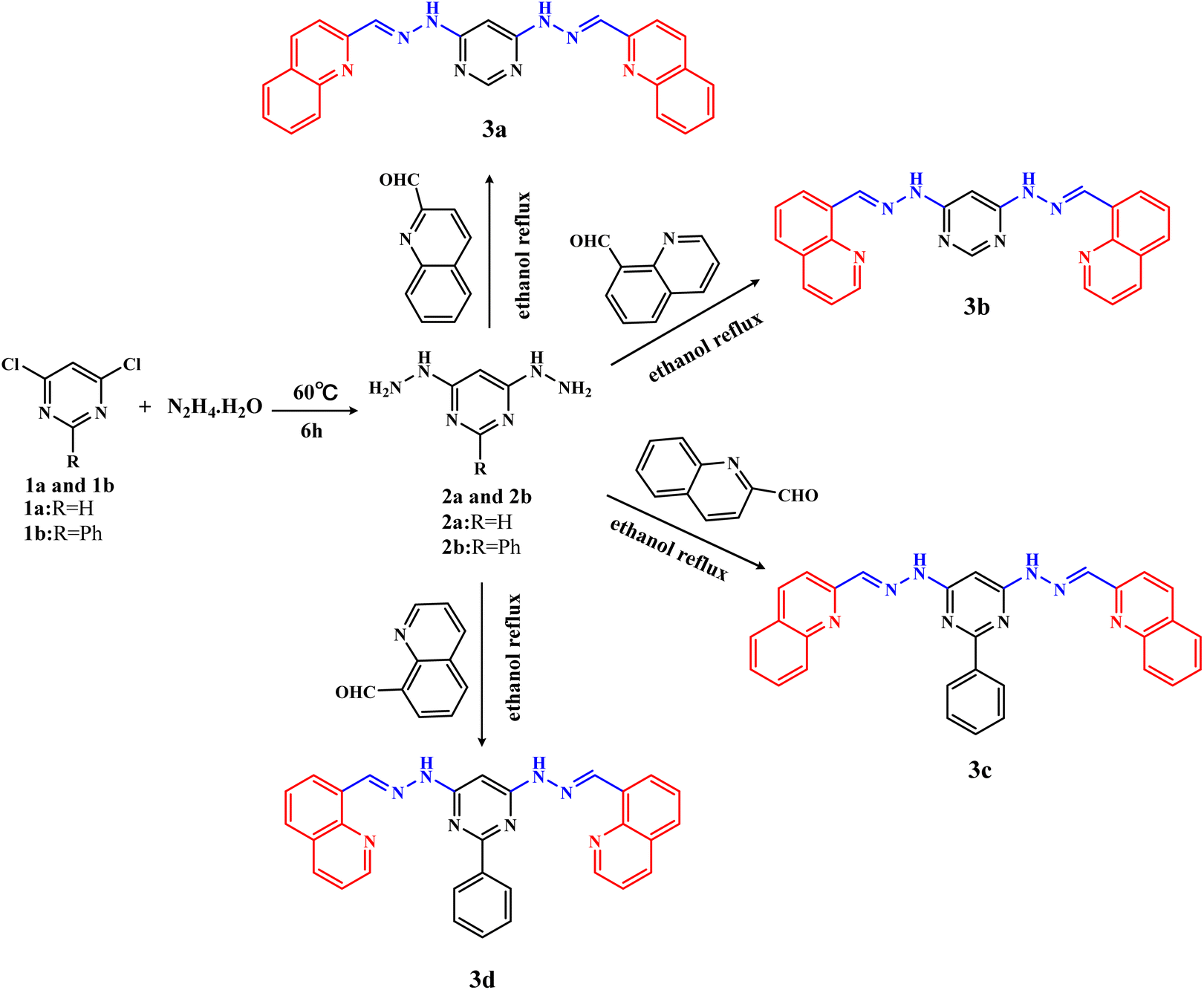 | ||
| Scheme 1 Synthesis of 3a–3d. | ||
4,6-Bis(2-((E)-quinolin-2-ylmethylene)hydrazinyl)pyrimidine (3a).Quinoline-2-formaldehyde (2.1 mmol, 330 mg) and 2a (1 mmol, 140 mg) were added to 20 mL ethanol solvent at room temperature. Then, the reaction mixture was heated and refluxed for 6 h, and yellow precipitation gradually appeared. Finally, the precipitation was filtered, washed thoroughly with ethanol and then purified by column chromatography (petroleum ether:ethyl acetate = 1:1) to give yellow solid, named compound 3a, with a yield of ca. 54.0%. 1H NMR (500 MHz, DMSO-d6) δ 11.64 (s, 2H, NH), 8.47 (d, J = 8.7 Hz, 2H, CHquinoline), 8.31 (s, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 8.28 (s, 1H, N–CHN, CHpyrimidine), 8.21 (d, J = 8.6 Hz, 2H, CHquinoline), 8.02 (d, J = 1.5 Hz, 2H, CHquinoline), 8.00 (d, J = 1.0 Hz, 2H, CHquinoline), 7.77 (t, J = 7.0 Hz, 2H, CHquinoline), 7.61 (t, J = 7.0 Hz, 2H, CHquinoline), 7.08 (s, 1H, CHpyrimidine). 13C NMR (101 MHz, DMSO-d6) δ 161.97, 158.48, 154.63, 148.01, 142.80, 137.20, 130.49, 129.25, 128.45, 128.11, 127.42, 117.78, 82.62. ESI-HRMS: m/z calculated for [C24H18N8+H]+: 419.17327, found: 419.17096. Elemental analysis (%): calcd for C24H18N8 (3a): C 68.9, H 4.3, N 26.8; found: C 68.1, H 4.2, N 27.1. The melting point of 3a is about 315 °C. IR (cm−1, KBr): 3173, 3046, 2964, 2855, 1707, 1667, 1608, 1583, 1558, 1502, 1454, 1415, 1375, 1324, 1247, 1197, 1143, 1112, 985, 827, 746.
N), 8.28 (s, 1H, N–CHN, CHpyrimidine), 8.21 (d, J = 8.6 Hz, 2H, CHquinoline), 8.02 (d, J = 1.5 Hz, 2H, CHquinoline), 8.00 (d, J = 1.0 Hz, 2H, CHquinoline), 7.77 (t, J = 7.0 Hz, 2H, CHquinoline), 7.61 (t, J = 7.0 Hz, 2H, CHquinoline), 7.08 (s, 1H, CHpyrimidine). 13C NMR (101 MHz, DMSO-d6) δ 161.97, 158.48, 154.63, 148.01, 142.80, 137.20, 130.49, 129.25, 128.45, 128.11, 127.42, 117.78, 82.62. ESI-HRMS: m/z calculated for [C24H18N8+H]+: 419.17327, found: 419.17096. Elemental analysis (%): calcd for C24H18N8 (3a): C 68.9, H 4.3, N 26.8; found: C 68.1, H 4.2, N 27.1. The melting point of 3a is about 315 °C. IR (cm−1, KBr): 3173, 3046, 2964, 2855, 1707, 1667, 1608, 1583, 1558, 1502, 1454, 1415, 1375, 1324, 1247, 1197, 1143, 1112, 985, 827, 746.
4,6-Bis(2-((E)-quinolin-8-ylmethylene)hydrazinyl)pyrimidine (3b). Yellow solid, yield: ca. 58.6%. 1H NMR (500 MHz, DMSO-d6) δ 11.35 (s, 2H, NH), 9.38 (s, 2H, CHN), 8.96 (dd, J = 4.1, 1.7 Hz, 2H, CHquinoline), 8.44–8.41 (dd, J = 4.0, 1.7 Hz, 2H, CHquinoline), 8.41 (dd, J = 3.9, 1.5 Hz, 2H, CHquinoline), 8.03 (dd, J = 8.1, 1.2 Hz, 2H, CHquinoline), 7.75 (t, J = 7.7 Hz, 2H, CHquinoline), 7.60 (dd, J = 8.3, 4.1 Hz, 2H, CHquinoline), 7.01 (s, 1H, N–CN, CHpyrimidine), 5.72 (s, 1H, CHpyrimidine). 13C NMR (101 MHz, DMSO-d6) δ 162.31, 158.11, 150.56, 145.72, 139.41, 137.07, 132.31, 129.49, 128.60, 127.20, 125.35, 122.31, 81.88. ESI-HRMS: m/z calculated for [C24H18N8+H]+: 419.17327, found: 419.17102. Elemental analysis (%): calcd for C24H18N8 (3b): C 68.9, H 4.3, N 26.8; found: C 69.1, H 4.3, N 26.9. The melting point of 3b is about 325 °C. IR (cm−1, KBr): 3199, 2960, 2923, 2855, 1587, 1558, 1496, 1461, 1417, 1256, 1203, 1126, 1085, 1035, 989, 792, 652, 547.
2,2′-((1E,1′E)-((2-phenylpyrimidine-4,6-diyl)bis(hydrazin-2-yl-1-ylidene))bis(methanylylidene))diquinoline (3c). Yellow solid, yield: ca. 51.2%. 1H NMR (500 MHz, DMSO-d6) δ 11.79 (s, 2H, NH), 8.61 (d, J = 8.5 Hz, 2H, CHquinoline), 8.43 (d, J = 5.2 Hz, 2H, CHquinoline), 8.37 (s, 2H,CHN), 8.14 (d, J = 8.6 Hz, 2H, CHquinoline), 7.97 (d, J = 8.6 Hz, 2H, CHquinoline), 7.86 (d, J = 8.6 Hz, 2H, CHquinoline), 7.76 (d, J = 7.5 Hz, 2H, CHquinoline), 7.60 (d, J = 6.9 Hz, 2H, CHbenzene), 7.54 (d, J = 7.2 Hz, 3H, CHbenzene), 7.14 (s, 1H, CHpyrimidine). 13C NMR (101 MHz, DMSO-d6) δ 163.98, 163.16, 155.24, 148.40, 143.42, 138.64, 137.64, 131.78, 131.40, 131.07, 129.81, 129.38, 129.03, 128.70, 128.67, 127.99, 118.38, 81.72. ESI-HRMS: m/z calculated for [C30H22N8+H]+: 495.20457, found: 495.20236. Elemental analysis (%): calcd for C30H22N8 (3c): C 72.8, H 4.5, N 22.7; found: C 71.5, H 4.3, N 22.9. The melting point of 3c is about 327 °C. IR (cm−1, KBr): 3203, 3039, 1663, 1604, 1592, 1563, 1533, 1504, 1461, 1411, 1388, 1182, 1147, 1112, 825, 748, 696.
8,8′-((1E,1′E)-((2-Phenylpyrimidine-4,6-diyl)bis(hydrazin-2-yl-1-ylidene))bis(methanylylidene))diquinoline (3d). Yellow solid, yield: ca. 67%. 1H NMR (500 MHz, DMSO-d6) δ 11.47 (s, 2H, NH), 9.43 (s, 2H, CHN), 9.00 (d, J = 8.3 Hz, 2H, CHquinoline), 8.44 (d, J = 7.5 Hz, 2H, CHquinoline), 8.42 (d, J = 7.3 Hz, 2H, CHquinoline), 8.34 (d, J = 7.2 Hz, 2H, CHquinoline), 8.02 (d, J = 6.4 Hz, 2H, CHquinoline), 7.77 (d, J = 7.0 Hz, 2H, CHquinoline), 7.63–7.57 (d, J = 6.9 Hz, 2H, CHbenzene), 7.49 (d, J = 6.4 Hz, 3H, CHbenzene), 7.03 (s, 1H, CHpyrimidine). 13C NMR (101 MHz, DMSO-d6) δ 163.09, 162.88, 158.97, 151.37, 150.68, 145.54, 139.16, 137.24, 132.50, 130.87, 129.45, 128.70, 128.13, 127.60, 127.03, 125.35, 122.57, 81.15. ESI-HRMS: m/z calculated for [C30H22N8+H]+: 495.20457, found: 495.20380. Elemental analysis (%): calcd for C30H22N8 (3d): C 72.8, H 4.5, N 22.7; found: C 72.6, H 4.7, N 23.1. The melting point of 3d is about 271 °C. IR (cm−1, KBr): 3178, 3030, 2998, 1723, 1597, 1589, 1573, 1560, 1496, 1407, 1199, 1126, 825, 788, 755, 694, 567.
2.3. Detection of nitrosamine impurities
The calibration curves for NDMA and NDEA were prepared at concentrations of 0.1, 0.3, 1, 2, 5, 10, 20, 50, 100, 300 and 1000 ng mL−1. The 3a, 3b, 3c and 3d were dissolved in DMSO at a concentration of 5 mg mL−1 at 37 °C and incubated. In each EP tube, 100 μL of the 5 mg mL−1 above compounds was added, followed by 150 μL of DMSO and 250 μL of acetonitrile. The mixture was vortexed and the resulting sample solution was obtained for using. Chromatographic conditions: run time: 6 min; column temperature: 40 °C; injection volume: 5 μL. Chromatographic column: C18, 2.7 μm, 3.0 × 75 mm. Mass spectrometric conditions: ion source: APCI; scan time: 6 min; ion source temperature: 350 °C; CUR: 30 psi; gas 1:60 psi; CAD: MediμM; detection mode: MRM (multiple reaction monitoring); EP: 10.0 V; CXP: 10.0 V.
2.4. Systemic toxicological evaluation
2.5. Biological evaluation
| Inhibition rate (%) = 1−(ODsample − ODblank)/(ODnegative − ODblank) × 100% | (1) |
2.6. Apoptosis detection
2.7. Molecular docking
Compounds were subjected to molecular docking using AutoDock Tools-1.5.6 software. Firstly, the crystal structures of receptors, DNA (PDB ID: 2MG8), CDK1 (PDB ID:6GU7), CDK2 (PDB ID: 4BGH), CDK4 (2W9Z), CDK8 (5i5Z), were downloaded from Protein Data Bank (http://www.rcsb.org./pdb). The molecular structures of ligands were drawn by ChemDraw and converted into MOL2 format by Chem3D. Secondly, the structures of receptors and ligands were all optimized by AutoDock Tools-1.5.6 software and saved as PDBQT format. Then, the appropriate boxes for the interactions of ligand and receptor were set and the set parameters were saved as conf files. Molecular docking was performed, analyzed, and the optimal conformation was selected. The binding modes of tested compounds with DNA or CDKs were conducted to explain the biological results and further understand the binding direction and interaction. The pose with good hydrogen bond geometry and low energy conformation was screened for further analysis. Docking structures and figures were analyzed and generated using the PyMOL molecular graphic system and Discovery Studio 4.5.3. Results and discussion
3.1. Synthesis and spectroscopic characterization
The target compounds 3a–3d were synthesized by a two-step reaction (Scheme 1). In brief, the intermediates 2a and 2b were synthesized by nucleophilic substitution reaction, then 3a–3d were synthesized by condensation reaction of 2a or 2b with quinoline aldehydes, respectively. During the synthesis process, the reaction process was monitored by thin layer chromatography. 3a–3d were all purified by column chromatography using the same eluent, with yields above 50%. It was found that hydrazine hydrate is not only the reactant, but also the solvent of the intermediates (2a, 2b) reaction. If ethanol is used as the reaction solvent, the intermediates 2a and 2b cannot be obtained successfully, only the monosubstituted pyrimidine hydrazine can be obtained.1H NMR, 13C NMR, ESI-HRMS, IR (Fig. S1–S16†), melting, UV/Vis spectra (Fig. S17†) and fluorescence spectra (Fig. S18†) were performed to characterize the chemical structures and spectral properties of 3a–3d. The important spectral data of –NH– and –CHN– and their assignments were listed in Table 1. Characteristic signals for the quinoline ring are multiple peaks at 9.00–7.60 ppm in 1H NMR spectra. The presence of the hydrazone group was confirmed by broad signal of NH protons and CHN protons at 11.79–11.35 ppm, 9.43–8.31 ppm respectively. For 3c and 3d, the presence of the benzene ring was supported by the chemical shift at 7.63–7.49 ppm in 1H NMR spectra. Compounds 3a–3d are symmetric and the results of 1H NMR were consistent with their structures. In the ESI-HRMS analysis, the observed molecular ion peaks of 3a–3d were [M + H]+. The IR absorption bands of the typical functional groups in 3a–3d are very similar. The IR spectra of 3a–3d showed similar absorption bands. The N–H stretching vibrations are in the range of 3173–3203 cm−1, and the CN stretching vibrations were observed in the region of 1589–1608 cm−1. Conventional UV/Vis spectra of 3a–3d were shown in Fig. S17.† The UV/Vis spectra are similar and the structural difference of 3a–3d has little effect on positions of absorption peaks. Because of the presence of CN functional groups and big conjugated system, 3a–3d all demonstrate strong absorption peaks near 205 nm, which are assigned to the transition of lone pair electrons from unbonded heteroatoms to π* antibonding orbitals. Meanwhile, the conjugate system is usually attributed to π → π* transition. As a result, the absorption peaks near 205 nm of 3a–3d are generated by n/π → π* transition. The fluorescence spectra of 3a–3d were recorded in Fig. S18† (EX: 360 nm). As shown in Fig. S18,† the fluorescence spectra of 3a–3d were similar too. The fluorescence emission peaks near 390 nm are attributed to the existence of CN functional groups. The emission peaks near 450 nm are due to quinoline chromophore.
| Compound | Amide –NH– | Imine –CHN– |
|||
|---|---|---|---|---|---|
| 1H NMR δ | FTIR cm−1 | 1H NMR δ | 13C NMR δ | FTIR cm−1 | |
| 3a | 11.64 | 3173 | 8.31 | 142.80 | 1608 |
| 3b | 11.35 | 3199 | 9.38 | 13941 |
1597 |
| 3c | 11.79 | 3203 | 8.37 | 138.64 | 1604 |
| 3d | 11.47 | 3178 | 9.43 | 139.16 | 1589 |
3.2. Risk assessment and detection of nitrosamine contaminants
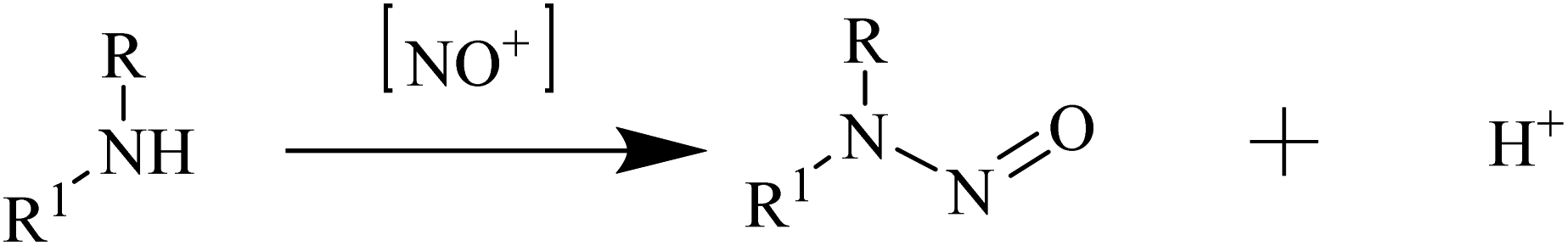 | ||
| Scheme 2 Generalised nitrosation of secondary amines under acidic conditions. | ||
3.3. Systemic toxicological evaluation
| Treatment | Initial weight (g) | Final weight (g) | Liver (mg) | Spleen (mg) | Kidney (mg) |
|---|---|---|---|---|---|
| Control | 18.22 ± 0.53 | 23.10 ± 0.20 | 500 ± 30.1 | 88 ± 3.3 | 200 ± 11.1 |
| 3a | 18.66 ± 0.25 | 23.65 ± 0.33 | 521 ± 19.7 | 78 ± 2.7 | 212 ± 7.7 |
| 3b | 18.31 ± 0.34 | 22.18 ± 0.45 | 517 ± 11.7 | 80 ± 3.6 | 207 ± 6.6 |
| 3c | 18.56 ± 0.33 | 22.35 ± 0.27 | 523 ± 16.1 | 83 ± 4.5 | 218 ± 9.9 |
| 3d | 19.00 ± 0.78 | 24.66 ± 0.95 | 540 ± 15.5 | 89 ± 3.3 | 225 ± 6.3 |
| Treatment | ALT (U L−1) | AST (U L−1) | BUN (mg dL−1) | CREA (μM) |
|---|---|---|---|---|
| Control | 57.23 ± 0.02 | 114.65 ± 0.15 | 15.08 ± 0.32 | 10.85 ± 0.12 |
| 3a | 54.66 ± 0.15 | 115.38 ± 0.23 | 16.08 ± 0.21 | 10.44 ± 0.45 |
| 3b | 53.21 ± 0.38 | 121.60 ± 1.15 | 19.74 ± 0.48 | 12.34 ± 0.78 |
| 3c | 54.35 ± 0.35 | 116.45 ± 2.35 | 21.72 ± 0.36 | 13.42 ± 0.23 |
| 3d | 56.38 ± 0.64 | 119.26 ± 1.24 | 15.96 ± 0.63 | 14.21 ± 0.97 |
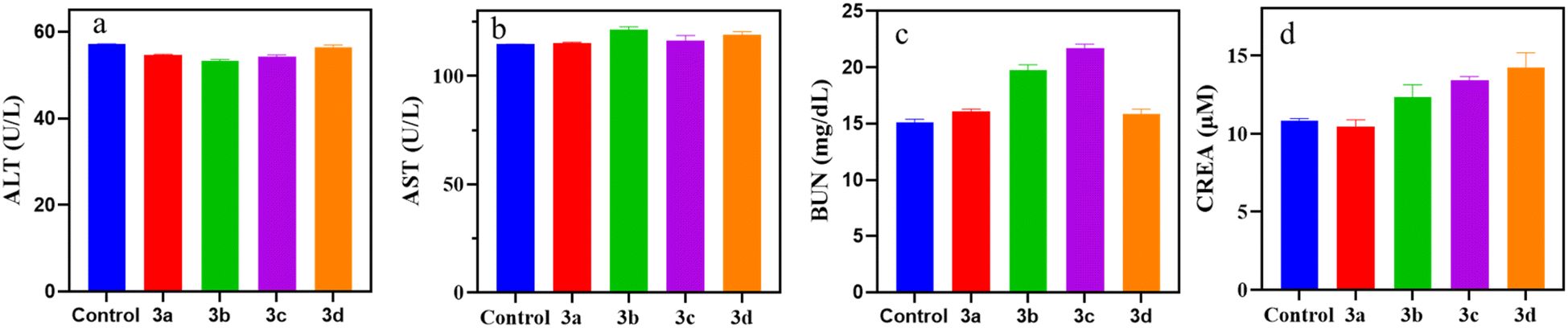 | ||
| Fig. 3 Biochemical indicators of different groups (a: ALT; b: AST; c: BUN; d: CREA). | ||
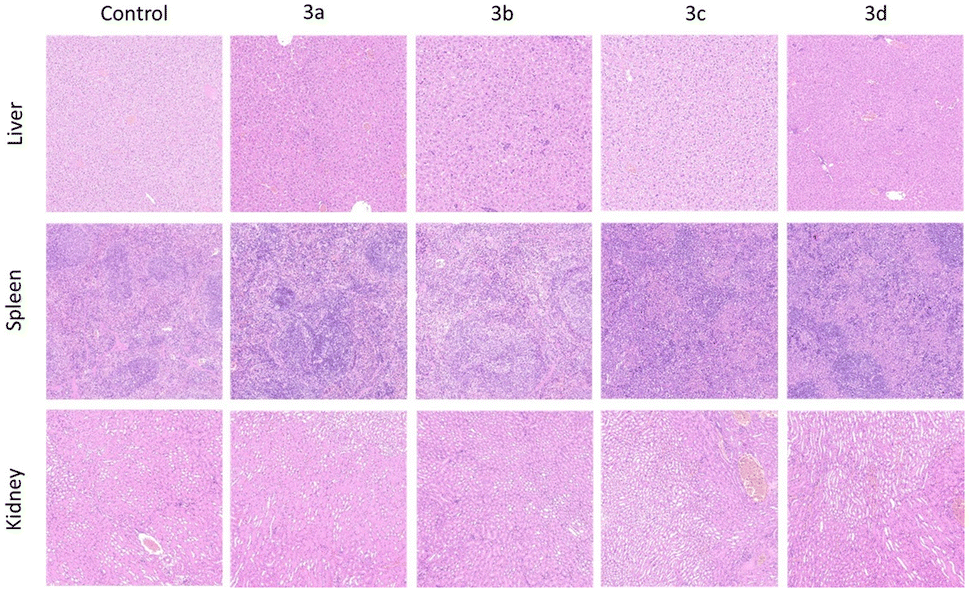 | ||
| Fig. 4 H&E staining of different organ groups (liver, spleen, kidney). | ||
3.4. Biological evaluation
| Compound | BGC-823 | BEL-7402 | MCF-7 | A549 | HL-7702 |
|---|---|---|---|---|---|
| 3a | 12.32 ± 0.37 | 33.97 ± 2.37 | 34.32 ± 0.95 | 16.71 ± 1.50 | >100 |
| 3b | 26.79 ± 0.75 | 17.60 ± 1.11 | 7.01 ± 0.26 | 14.80 ± 0.50 | 31.56 ± 0.71 |
| 3c | 9.12 ± 0.19 | 10.22 ± 0.12 | 7.05 ± 0.05 | 7.32 ± 0.05 | >100 |
| 3d | 16.85 ± 0.66 | 11.14 ± 1.02 | 10.39 ± 0.52 | 12.61 ± 0.09 | >100 |
| 5-FU | 15.18 ± 0.05 | 15.81 ± 0.01 | 11.32 ± 0.78 | 11.77 ± 0.89 | 20.83 ± 0.05 |
| Erlotinib | >20 | >20 | >20 | 17.32 ± 3.11 | — |
| Sorafenib | 6.08 ± 0.06 | 5.06 ± 0.02 | 7.06 ± 0.50 | 12.25 ± 0.42 | — |
In addition, we noticed that 3c showed stronger antiproliferative activity than 3a against all the selected cancer cell lines. Similarly, 3d also displayed better antiproliferative activity than 3b against BGC-823, BEL-7402 and A549. The similar antiproliferative activities in these two different quinoline-based hydrazone derivatives indicated that the introduction of phenyl on pyrimidine skeleton could significantly enhance anticancer activity probably due to the increase of conjugated structure.
3.5. Apoptosis detection analysis
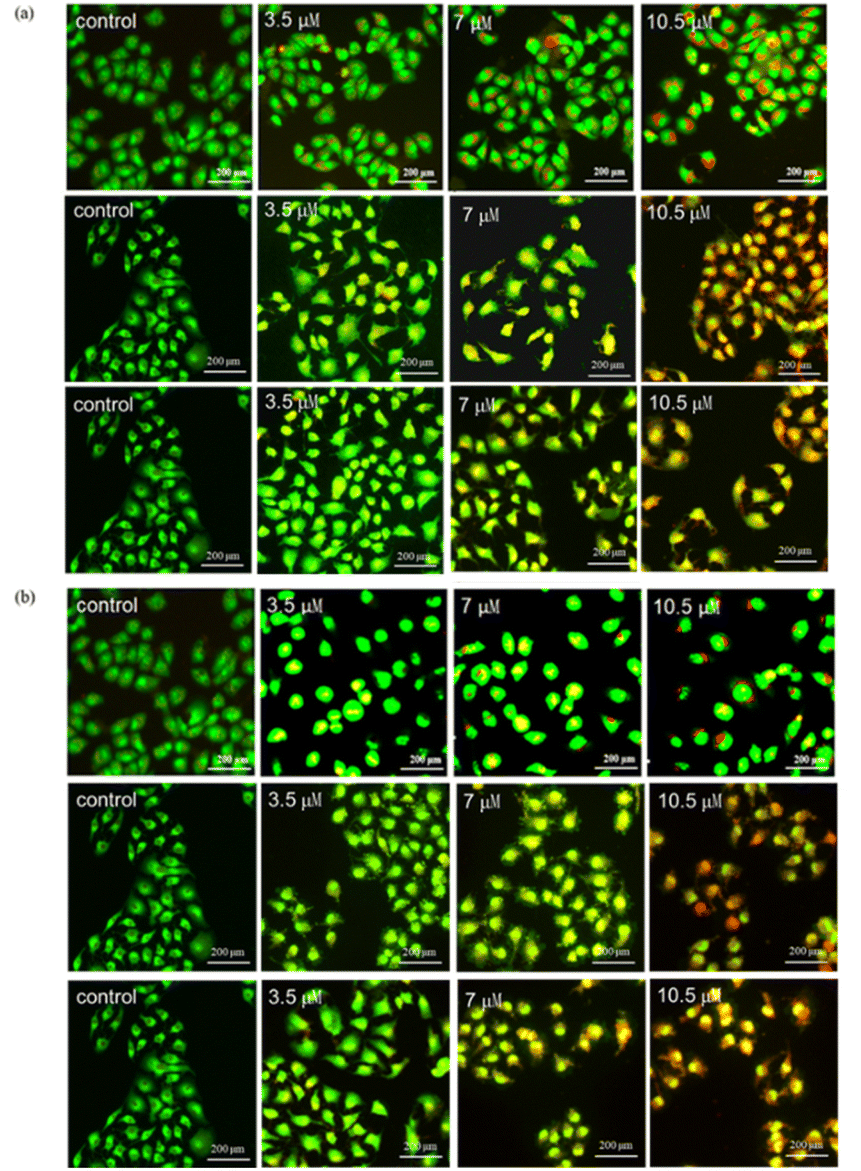 | ||
| Fig. 5 Changes in morphology of AO/EB double stained MCF-7 cells treated with (a) 3b and (b) 3c (3.5 μM, 7 μM and 10.5 μM) for 24 h. (Three independent experiments were performed on each compound.) | ||
 | ||
| Fig. 6 Apoptosis of MCF-7 cells treated with (a) 3b and (b) 3c (0 μM, 3.5 μM, 7 μM and 10.5 μM) for 48 h. (Three independent experiments were performed on each compound.) | ||
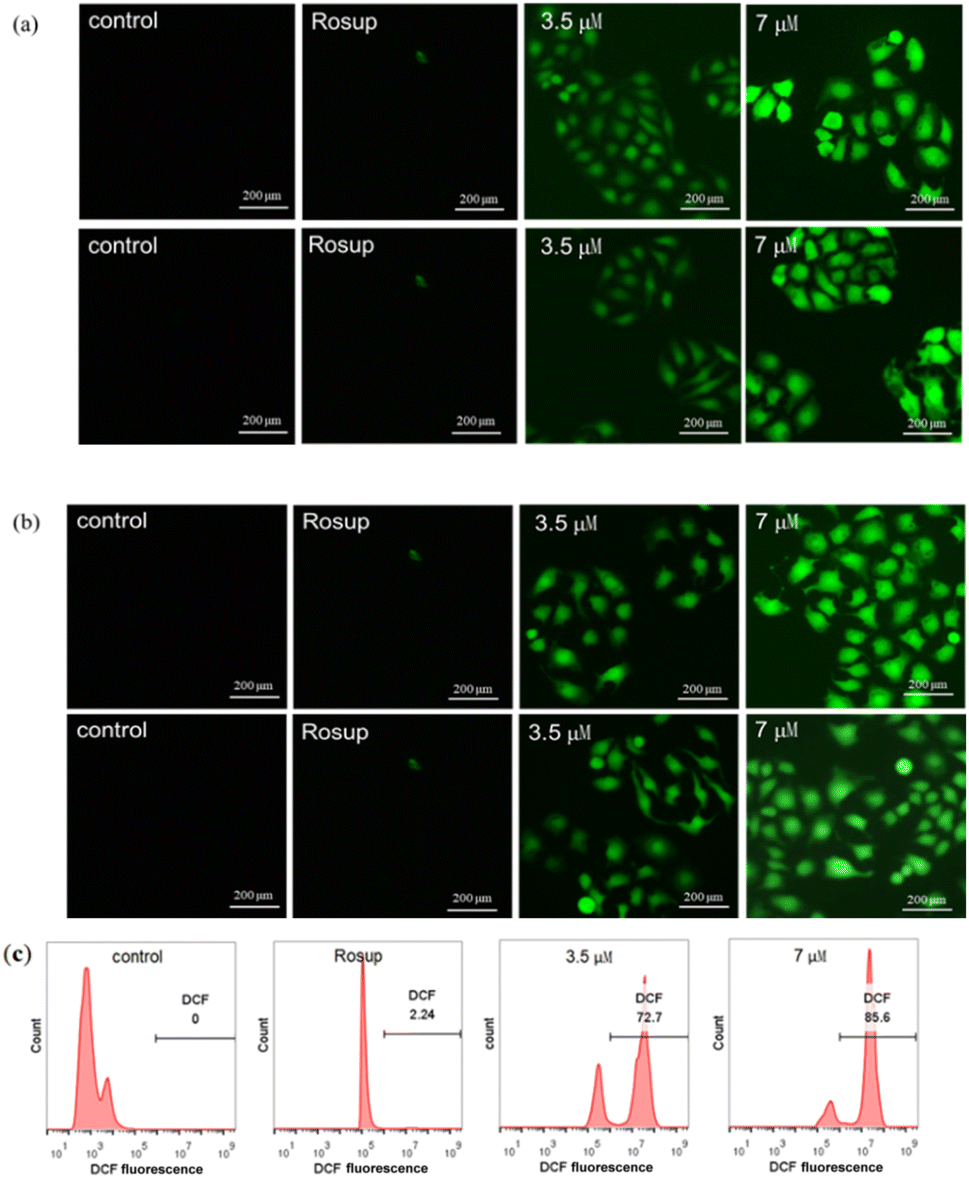 | ||
| Fig. 7 The inverted fluorescence microscope detection for the ROS fluorescence changes of MCF-7 cells treated with (a) 3b and (b) 3c (0 μM, 3.5 μM and 7 μM) for 24 h (independent experiments were performed on each compound). The flow cytometry detection for the ROS fluorescence changes of MCF-7 cells treated with (c) 3b. | ||
3.6. Discussion of molecular docking
Molecular docking is a commonly used computational method aimed at predicting the binding mode and binding affinity of small molecules with DNA or proteins.54 Herein, molecular docking was used to investigate the binding mode of 3b or 3c with DNA (PDB ID: 2MG8) by AutoDock Tools-1.5.6 and PyMOL Molecular Graphics System. In the docking simulations, the structures of all the compounds were kept flexible. Fig. 8a and b depicted the docked conformations, and the visual diagram showed that 3b and 3c binding with DNA adopted a nearly vertical insert mode. The binding energy (ΔGbθ) of 3b and 3c with DNA (PDB ID: 2MG8) was listed in Table 5. The ΔGbθ of 3b and 3c were −7.9 kcal mol−1 and −7.2 kcal mol−1, which indicated that the binding of 3b and 3c with DNA is spontaneous process. In general, the lower the binding energy, the stronger is the binding affinity.55 Thus, the binding affinity of 3b with DNA was slightly larger than that of 3c. The binding constants (Kb) of 3b and 3c calculated according to eqn (1) (ref. 56) are listed in a Table 5. The Kb were 6.53 × 105 M−1 and 2.00 × 105 M−1, respectively, which fall in the range of the insertion mode (1 × 105–1 × 1011 M−1).57,58 According to molecular docking, the interaction mode of 3b and 3c with DNA was insertion. In addition, Fig. 8 and S33† depicted the hydrogen bonding interaction between 3b or 3c with DNA via the N atoms of small molecules (3b or 3c) and the H atoms of DNA base. As shown in Fig. 8, the docking active sites of 3b and 3c with DNA mainly lie on the T and C base pairs of DNA base pairs. The N atoms in 3b interacts with DG-8 and DG-24 bases to form hydrogen bonds (Fig. 8a), while 3c forms hydrogen bonds with DNA only with DG-8 bases (Fig. 8b).|
ΔGbθ = −RTlnKb
| (2) |
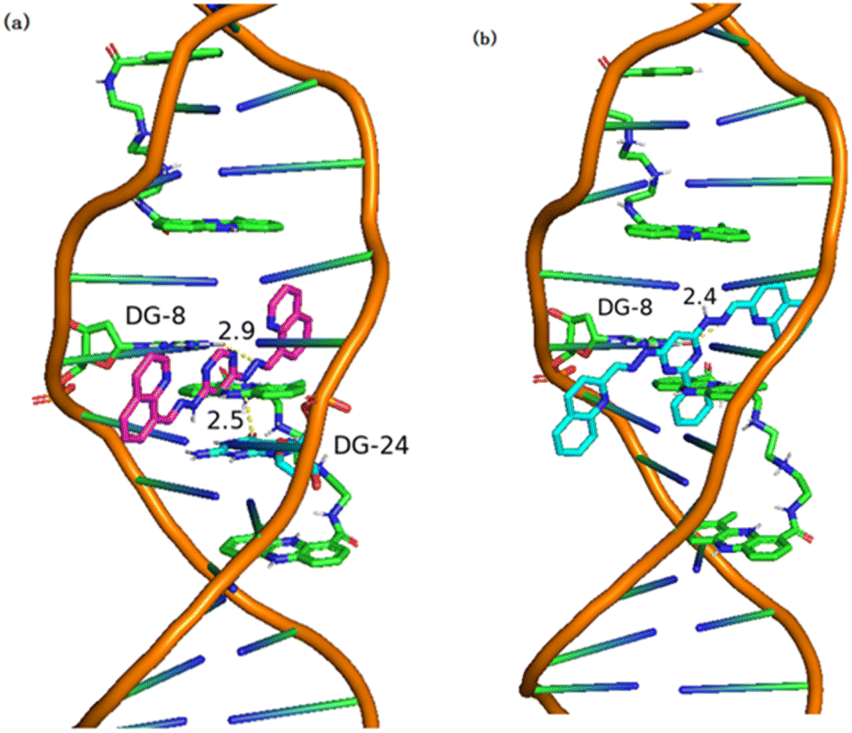 | ||
| Fig. 8 Visualizations of (a) 3b and (b) 3c docking with DNA (PDB ID: 2MG8). | ||
| Compound | ΔGbθ (kcal mol−1) | Kb (M−1) |
|---|---|---|
| 3b | −7.9 | 6.53 × 105 |
| 3c | −7.2 | 2.00 × 105 |
| 5-FU | −4.4 | 1.74 × 103 |
3.7. Architecture of the CDK2 active site
Similarly, molecular docking is performed to predict the active conformation of a compound with target protein active sites and evaluate their binding affinity, molecular docking is typically performed. CDK2 was also regarded as a potentially therapeutic target for compounds containing pyrimidine units.59 Therefore, the interactions of 3a–3d with CDK2 were explored by molecular docking. The ΔGbθ of 3a–3d interacting with CDK2 (PDB ID: 4BGH) were summarized in Table S1.† 3a–3d all showed interaction with CDK2 to some extent. Among them, 3b and 3c showed a slightly stronger interaction with CDK2 than that of 3a and 3d, which was in good agreement with the results of cytotoxicity. The 3D and 2D diagrams of 3b and 3c docking pose in the active site of CDK2 were shown in Fig. 9. The results revealed different molecular interactions, e.g., hydrogen bonding, π–π interactions and electrostatic attraction between hydrazones and CDK2. As shown in Fig. 9, 3b exhibits pronounced hydrogen bonding through the nitrogen atoms of the hydrazone linkage with amino acid residues Lys-88. While for 3c, the hydrogen bonding is formed by hydrogen atoms of hydrazone linkage with amino acid residue Arg-199. In addition, for 3b, the π–π interactions of the quinoline rings or a pyrimidine ring with amino acid residue Ala-201 and Asp-92 are observed. However, for 3c, it can be clearly seen that the π–π interactions exists only in the pyrimidine rings with amino acid residue Asp-92. It is probably due to the introduction of benzene ring, which increased the steric hindrance. The results of docking suggested that CDK2 may be one of the targets for 3b and 3c. To further investigate whether 3b and 3c interact with other CDKs, molecular docking was used to align 3b and 3c with CDK1 (6GU7), CDK4 (2W9Z) and CDK8 (5I5Z). The values of ΔGbθ were summarized in Table S2–S5.† Overall, the interaction between 3c and the three CDKs was stronger than that of 3b, consistent with the previous molecular docking results. CDK1 and CDK8 were selected to dock with 3b and 3c for visualization, and the binding modes were obtained, as shown in Fig. 9e–l. It was observable that 3b forms hydrogen bonds with the amino acid residue Gln-132 of protein CDK1, 3c forms hydrogen bonds with the amino acid residue Asn-133 of protein CDK1, and with the amino acid residue Val-27 of protein CDK8. The distances of the formed hydrogen bonds were significantly shorter than the conventional hydrogen bond distance of 3.5 Å, and the strong binding plays a crucial role in anchoring small molecules within the protein pocket. Besides hydrogen bonding, there were also hydrophobic forces existing with some amino acid residues, such as Lys-133, Phe-80, Leu-83 and Leu-135 of CDK1, and Val-35, Ala-50, Ala-155 and Arg-356 of CDK8. To sum up, these interactions enhanced the stability of 3b and 3c in the binding pockets of CDK1 and CDK8 proteins and formed stable complexes. Consequently, 3b and 3c may possess potential activity against CDK1 and CDK8.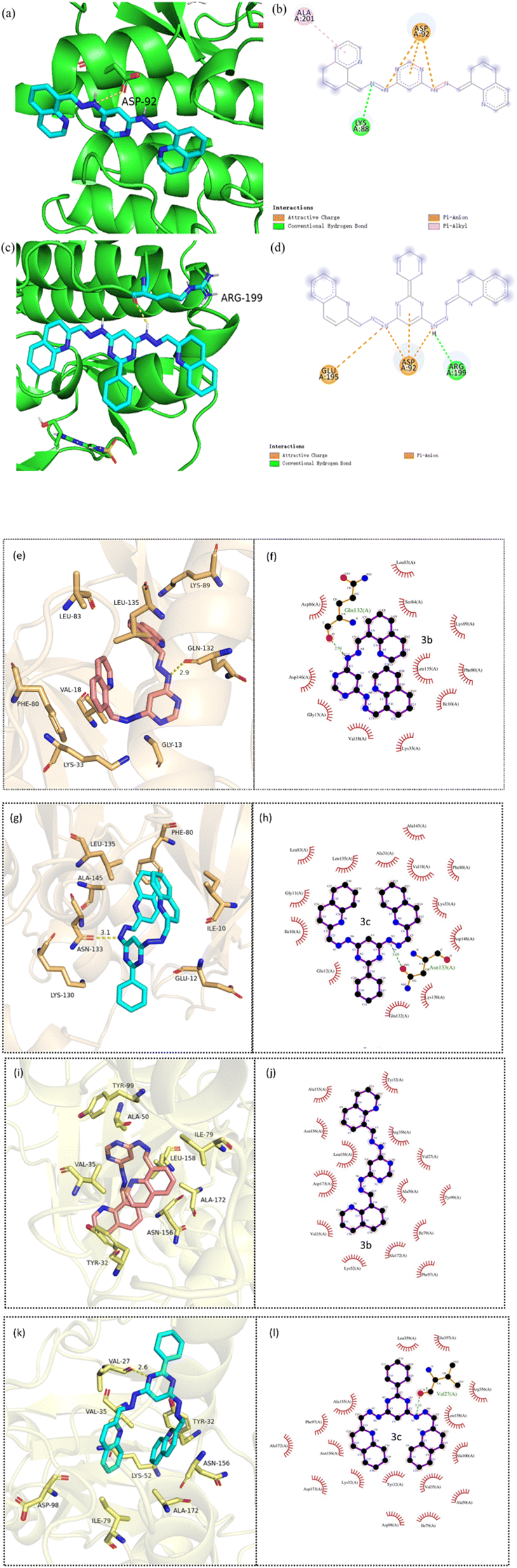 | ||
| Fig. 9 3D diagram of (a, e and i) 3b and (c, g and k) 3c docking pose in the active site of CDK2, CDK1 and CDK8 (PDB: 4BGH; 6GU7; 5I5Z); 2D diagram of (b, f and i) 3b and (d, h and l) 3c docking pose in the active site of CDK2, CDK1 and CDK8. | ||
4. Conclusion
In this work, a series of dihydrazone derivatives (3a–3d) were synthesized and their nitrosamine impurities were evaluated as well as antiproliferative activity and toxicity were evaluated. The assessment results of nitrosamines indicated that the risk of forming nitrosamine impurities in the final drug was relatively low. Moreover, highly carcinogenic NDMA and NDEA were not detected. Toxicological studies examined the safety profile of 3a–3d. MTT results showed that they all displayed broad-spectrum antiproliferative activity against selected cancer cell lines (BGC-823, BEL-7402, MCF-7 and A549). It is worth mentioning that 3b and 3c displayed more potent antiproliferative activity against MCF-7 cells than 5-FU and Erlotinib with IC50 values of 7.016 μM and 7.05 μM, respectively. Results of the apoptosis study showed that compounds 3b and 3c increased the number of late apoptotic and early apoptotic cells. AO/EB staining, elevated intracellular ROS levels demonstrated the induction of apoptosis by 3b and 3c. 3c could induce MCF-7 cell apoptosis in a concentration-dependent manner and bind to DNA via partial insertion. Additionally, the molecular docking simulations performed for the selected active compound – 3b, 3c revealed that they could occupy the active site of CDK1, CDK4 and CDK8 through hydrophobic and H-bonding interactions, which were suggested to be essential for inhibitory activity. This work revealed that quinoline-based dihydrazone derivatives could be used as promising anticancer agents.Data availability
The authors confirm that the data supporting the findings of this study are available within the manuscript and its ESI.†Author contributions
Jia-Xing Lu: conceptualization, validation, formal analysis, writing-original draft, writing-review & editing. Hai-Rong Lan: conceptualization, software, investigation. Dai Zeng: software, data curation, investigation. Jun-Ying Song: visualization, investigation. Ya-Ting Hao: investigation, methodology. Ai-Ping Xing, Ao Shen and juan Yuan: conceptualization, methodology, writing-review & editing. All authors provided their final consent for the publication and accepted responsibility for the work conducted in this study.Conflicts of interest
All authors declare that they have no relevant conflict of interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (Project No. 22101076), the Natural Science Foundation of Henan Province, China (Project No. 202300410261). The authors would like to thank the Shiyanjia lab (https://www.shiyanjia.com/) for the support of 1H NMR, 13C NMR, XRD, TG and ESI-HRMS analysis.Notes and references
- T. I. De Santana, M. De Oliverira Barbosa, P. A. T. De Moraes Gomes, A. C. N. Da Cruz, T. G. Da Silva and A. C. L. Leite, Eur. J. Med. Chem., 2018, 144, 874–886 CrossRef PubMed.
- L. Wayteck, K. Breckpot, J. Demeester, S. C. De Smedt and K. Raemdonck, Cancer Lett., 2014, 352, 113–125 CrossRef CAS PubMed.
- K. Lal and P. Yadav, Med. Chem., 2018, 18, 21–37 CAS.
- R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS.
- S. Kwon, Y. Lee, Y. Jung, J. H. Kim, B. Baek, B. Lim, J. Lee, I. Kim and J. Lee, Eur. J. Med. Chem., 2018, 148, 116–127 CrossRef CAS PubMed.
- A. A. Abu-Hashem, O. Hakami and N. Amri, Heliyon, 2024, 10, e26735 CrossRef CAS.
- Y. Cai, H. Liu and H. F. Chen, Chem. Biol. Drug Des., 2018, 91, 805–816 CrossRef CAS.
- A. Baba, N. Kawamura, H. Makino, Y. Ohta, S. Taketomi and T. Sohda, J. Med. Chem., 1996, 39, 5176–5182 CrossRef CAS.
- R. Chopra, K. Chibale and K. Singh, Eur. J. Med. Chem., 2018, 148, 39–53 CrossRef CAS.
- A. A. Abu-Hashem and N. Amri, Pharmaceuticals, 2022, 15, 1232 CrossRef CAS.
- H. Cheng, W. Q. Wang, L. Huang, P. Cui and Q. Y. Wu, Chin. J. Org. Chem., 2016, 36, 1065–1072 CrossRef CAS.
- V. Srivastava, A. S. Negi, J. K. Kumar, M. M. Gupta and S. P. S. Khanuja, Bioorg. Med. Chem., 2005, 13, 5892–5908 CrossRef CAS.
- K. G. Byler, C. Wang and W. N. Setzer, J. Mol. Model., 2009, 15, 1417–1426 CrossRef CAS PubMed.
- C. H. Xu, W. J. Zhou, G. J. Dong, H. Qiao, J. D. Peng, P. F. Jia, Y. H. Li, H. M. Liu, K. Sun and W. Zhao, Bioorg. Chem., 2020, 105, 104424 CrossRef CAS PubMed.
- P. Kodisundaram, S. Amirthaganesan and T. Balasankar, J. Agric. Food Chem., 2013, 61, 11952–11956 CrossRef CAS.
- J. Amato, R. Morigi, B. Pagano, A. Pagano, A. Ohnmacht, A. De Magis, Y. P. Tiang, G. Capranico, A. Locatelli, A. Graziadio, A. Leoni, M. Rambaldi, E. Novellino, S. Neidle and A. Randazzo, J. Med. Chem., 2016, 59, 5706–5720 CrossRef CAS.
- O. I. El-Sabbagh and H. M. Rady, Eur. J. Med. Chem., 2009, 44, 3680–3686 CrossRef CAS.
- C. M. Moldovan, O. Oniga, A. B. Tiperciuc, P. Verite, A. Pîrnau, O. Crisan, M. Bojita and R. Pop, Eur. J. Med. Chem., 2011, 46, 526–534 CrossRef CAS PubMed.
- M. Qin, X. Zhai, H. B. Xie, J. J. Ma, K. Lu, Y. Wang, L. H. Wang, Y. C. Gu and P. Gong, Eur. J. Med. Chem., 2014, 23, 47–58 CrossRef PubMed.
- K. D. Katariya, S. R. Shah and D. Reddy, Bioorg. Chem., 2020, 94, 103406 CrossRef CAS.
- L. Shi, J. J. Xu, J. J. Bi, Z. G. Zhang, T. X. Liu, X. L. Yang and G. S. Zhang, Chin. J. Org. Chem., 2018, 38, 3016–3025 CrossRef CAS.
- M. Korcz, F. Sączewski, P. J. Bednarski and A. Kornicka, Molecules, 2018, 23, 1497 CrossRef PubMed.
- J. A. Makawana, C. B. Sangani, L. Lin and H. L. Zhu, Bioorg. Med. Chem. Lett., 2014, 24, 734–1736 CrossRef.
- Y. L. Chen, Y. L. Zhao, C. M. Lu, C. C. Tzeng and J. P. Wang, Bioorg. Med. Chem., 2006, 14, 4373–4378 CrossRef CAS PubMed.
- R. C. Montenegro, L. V. Lotufo, M. O. De Moraes, C. Pessoa, F. A. R. Rodrigues, M. De Lima Ferreira Bispo, C. De Alcantara, C. R. Kaiser and M. V. De Souza, Med. Chem. Res., 2012, 21, 3615–3619 CrossRef CAS.
- D. Senthil Raja, N. S. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2012, 47, 73–85 CrossRef CAS PubMed.
- J. Yuan, J. Y. Song, H. H. Yang, H. R. Lan, A. P. Xing, K. H. Li, D. Zeng, Z. Q. Zhang and S. Y. Feng, J. Mol. Struct., 2023, 1276, 134724 CrossRef CAS.
- N. Pawar, A. Bhardwaj, A. Vora and S. Sharma, J. Chromatogr. A, 2024, 1, 1732 Search PubMed.
- O. Afzal, S. Kumar, R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871–910 CrossRef CAS.
- P. Yadav and K. Shah, Bioorg. Chem., 2021, 109, 104639 CrossRef CAS PubMed.
- H. Y. Sun, Z. Nikolovska-Coleska, C. Y. Yang, D. Qian, J. Lu, S. Qiu, L. Bai, Y. Peng, Q. Cai and S. Wang, Acc. Chem. Res., 2008, 41, 1264–1277 CrossRef CAS.
- D. Moreau, C. Jacquot, P. Tsita, I. Chinou, C. Tomasoni, M. Juge, E. Antoniadou-Vyza, L. Martignat, A. Pineau and C. Roussakis, Int. J. Cancer, 2008, 123, 2676–2683 CrossRef CAS.
- J. Králová, T. Bríza, I. Moserová, B. Dolenský, P. Vasek, P. Kejík, Z. Poucková, R. Kaplánek, P. Martásek, M. Dvorák and V. Král, J. Med. Chem., 2008, 51, 5964–5973 CrossRef PubMed.
- S. Okten, O. Cakmak, S. Tekin and T. K. Koprulu, Lett. Drug Des. Discovery, 2017, 14, 1415–1424 CAS.
- M. Peyressatre, C. Prevel, M. Pellerano and M. C. Morris, Cancers, 2015, 7, 179–237 CrossRef CAS PubMed.
- S. R. Whittaker, A. Mallinger, P. Workman and P. A. Clarke, Pharmacol. Ther., 2017, 173, 83–85 CrossRef CAS PubMed.
- A. B. Heptinstall, I. Adiyasa, C. Cano and I. R. Hardcastle, Future Med. Chem., 2018, 10, 1369–1388 CrossRef CAS PubMed.
- C. Thangavel, E. Boopathi, Y. Liu, C. McNair, A. Haber, M. Perepelyuk, A. Bhardwaj, S. Addya, A. Ertel, S. Shoyele, R. Birbe, J. M. Salvino, A. P. Dicker, K. E. Knudsen and R. B. Den, Clin. Cancer Res., 2018, 24, 1402–1414 CrossRef CAS.
- L. M. Spring, S. A. Wander, M. Zangardi and A. Bardia, Curr. Oncol. Rep., 2019, 21, 25 CrossRef PubMed.
- M. M. Al-Sanea, A. J. Obaidullah, M. E. Shaker, G. Chilingaryan, M. M. Alanazi, N. A. Alsaif, H. M. Alkahtani, S. A. Alsubaie and M. A. Abdelgawad, Molecules, 2021, 26, 412 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- T. Efferth, Planta Med., 2010, 76, 1035–1036 CrossRef CAS PubMed.
- D. P. Bezerra, F. O. D. Castro, A. P. N. N. Alves, C. Pessoa, M. O. D. Moraes, E. R. Silveira, M. A. S. Lima, F. J. M. Elmiro, N. M. N. D. Alencar, R. O. Mesquita, M. W. Lima and L. V. Costa-Lotufo, J. Appl. Toxicol., 2007, 28, 156–163 CrossRef.
- E. Ramachandran, V. Gandin, R. Bertani, P. Sgarbossa, K. Natarajan, N. S. P. Bhuvanesh, A. Venzo, A. Zoleo, A. Glisenti, A. Dolmella, A. Albinati and C. Marzano, J. Inorg. Biochem., 2018, 182, 18–28 CrossRef CAS.
- G. B. Jiang, Y. Y. Xie, G. J. Lin, H. L. Huang, Z. H. Liang and Y. J. Liu, J. Photochem. Photobiol., B, 2013, 129, 48–56 CrossRef CAS.
- K. J. Du, J. Q. Wang, J. F. Kou, G. Y. Li, L. L. Wang, H. Chao and L. N. Ji, Eur. J. Med. Chem., 2011, 46, 1056–1065 CrossRef CAS PubMed.
- X. Q. Zhou, Y. Li, D. Y. Zhang, Y. Nie, Z. J. Li, W. Gu, X. Liu, J. L. Tian and S. P. Yan, Eur. J. Med. Chem., 2016, 114, 244–256 CrossRef CAS PubMed.
- Y. Han, Z. Tian, S. Zhang, X. Liu, J. Li, Y. Li, Y. Liu, M. Gao and Z. Liu, J. Inorg. Biochem., 2018, 189, 163–171 CrossRef CAS.
- M. E. Juan, U. Wenzel, H. Daniel and J. M. Planas, J. Agric. Food Chem., 2008, 56, 4813–4818 CrossRef CAS.
- P. Li, Q. L. Zhao, L. H. Wu, P. Jawaid, Y. F. Jiao, M. Kadowaki and T. Kondo, Apoptosis, 2014, 19, 1043–1053 CrossRef CAS.
- L. Diebold and N. S. Chandel, Free Radical Biol. Med., 2016, 100, 86–93 CrossRef CAS PubMed.
- J. Deng, J. Wang, M. Khan, P. Yu, F. Yang and H. Liang, J. Inorg. Biochem., 2018, 185, 10–16 CrossRef CAS PubMed.
- M. H. Khan, M. Cai, J. Deng, P. Yu, H. Liang and F. Yang, Molecules, 2019, 24, 2544 CrossRef CAS.
- M. C. Zhu, X. T. Cui, F. C. Zhao, X. Y. Ma, Z. B. Han and E. J. Gao, RSC Adv., 2015, 5, 47798–47808 RSC.
- M. L. Liu, M. Jiang, K. Zheng, Y. T. Li, Z. Y. Wu and C. W. Yan, J. Coord. Chem., 2014, 67, 630–648 CrossRef CAS.
- M. N. Zafar, A. M. Butt, G. E. Chaudhry, F. Perveen, M. F. Nazar, S. Masood, A. F. Dalebrook, E. U. Mughal, S. U. Sumrra, Y. Y. Sung, T. S. T. Muhammad and L. J. Wright, J. Inorg. Biochem., 2021, 224, 111590 CrossRef CAS PubMed.
- A. Wolfe, H. G. J. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- X. Q. Zhou, Y. Li, D. Y. Zhang, Y. Nie, Z. J. Li, W. Gu, X. Liu, J. L. Tian and S. P. Yan, Eur. J. Med. Chem., 2016, 3, 244–256 CrossRef PubMed.
- I. F. Nassar, M. T. Abdel Aal, W. A. El-Sayed, A. E. Shahin M, E. G. E. Elsakka, M. M. Mokhtar, M. Hegazy, M. Hagras, A. A. Mandour and N. S. M. Ismail, RSC Adv., 2022, 12, 14865–14882 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06954d |
| This journal is © The Royal Society of Chemistry 2025 |