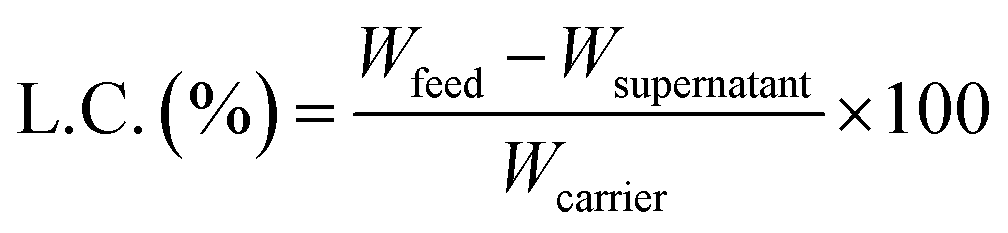DOI:
10.1039/D4RA07142E
(Paper)
RSC Adv., 2025,
15, 1115-1124
Preparation of a CNF porous membrane and in situ synthesis of silver nanoparticles (AgNPs)†
Received
4th October 2024
, Accepted 27th December 2024
First published on 13th January 2025
Abstract
We prepared a cellulose nanofiber (CNF)-based porous membrane with three dimensional cellular structures. CNF was concentrated via a surfactant-induced assembly by mixing CNF with a cationic surfactant, domiphen bromide (DB). Furthermore, they were accumulated by centrifugation to obtain a CNF-DB sol. Next, when the CNF-DB sol was naturally dried, a membrane composed of densely packed CNF was obtained. On the other hand, when the CNF-DB sol was freeze-dried, a porous membrane with the anisotropic cellular structure could be obtained. The interspace between layered CNF sheets was tunable by the DB concentration in the assembly process and the centrifugal force in the accumulation process. FT-IR analysis of the porous membrane showed the formation of hydrogen bonds between the CNF, resulting in facilitation of crosslinking of the CNF and formation of the cellular structures. The obtained CNF-DB membrane exhibited high water resistance. They showed a high ability to absorb hydrophobic dyes such as Nile red and rhodamine B (RhB) due to the presence of the hydrophobic core of DB micelles. Then, the release of RhB could be controlled by the ionic strength in the medium. In addition, they possessed a high ability to adsorb cationic metals such as Ag ions due to the presence of carboxyl moieties of CNF. Next, in situ synthesis of silver nanoparticles (AgNPs) was carried out by employing the CNF-DB membrane as a template for Ag ion adsorption and reduction. Deposition of AgNPs could be observed on the CNF-DB membrane, which suppressed aggregation of AgNPs. Almost all AgNPs were arrayed apart from each other to generate the hotspots, which could enhance surface-enhanced Raman scattering (SERS) of AgNPs. Such an AgNPs-CNF composite membrane could be applied for a label-free analysis of adsorbed RhB.
Introduction
Naturally derived biopolymers including cellulose, lignin and chitin have attracted increasing interest as abundant and non-edible biomass. Recently, a cellulose nanofiber (CNF) has been exploited by mechanical defibration or 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) oxidation of cellulose.1 It possesses nanofibrous structures composed of a bundle of cellulose chains, which make it possible to form a densely packed film with high transparency and good gas barrier properties.2 Also, as the CNF possesses high aspect ratios, it is expected to produce functional materials in terms of not only mechanical properties but electrical and thermal conductivities by tuning directions.3,4 In this manner, CNF has high potential as a source to generate a new class of materials.
Porous materials are promising for various applications including thermal insulating materials, energy storage devices, sensors, bioscaffolds, and supports for pollutant treatment and catalysts owing to their properties such as light weight, high specific surface area and high porosity.5 There have been many approaches to create porous structures, such as phase inversion, porogen incorporation, and templating.6 Biomass-based porous materials have been prepared by using bacteria cellulose, chitin, and pectin.7 CNF has often been used as a raw material to prepare porous structures by using the ice crystals-templated approach.8 However, it is challenging to tune the pore structure and size in the fabrication of the biomass-based porous materials. For instance, anisotropic porous structures with layered structures have attracted much attention due to anisotropic functions in thermal conductivity, compressibility and liquid transport.3,9 Alternatively, Hu et al. have reported a top-down approach to fabricate an anisotropic wood aerogel with a layered structure from natural wood by a chemical treatment and a removal of lignin and hemicellulose.9
Our research groups have reported the preparation of nano-to micron-sized particles from various biopolymers with the aid of ionic surfactants.10–12 In this surfactant-induced assembly, the surfactant acts as a crosslinker to create coacervates with the oppositely charged biopolymer as follows. The surfactants bind with a biopolymer through the ionic complex formation and then their alkyl chains are hydrophobically associated to form coacervates through compaction and assembly of biopolymers. Catchmark et al. have investigated the binding interactions of cellulose nanocrystals (CNC) with oppositely charged surfactants and reported that the structure of the CNC–surfactant complex in the solution was largely changed by modulating the concentration of the surfactant.13
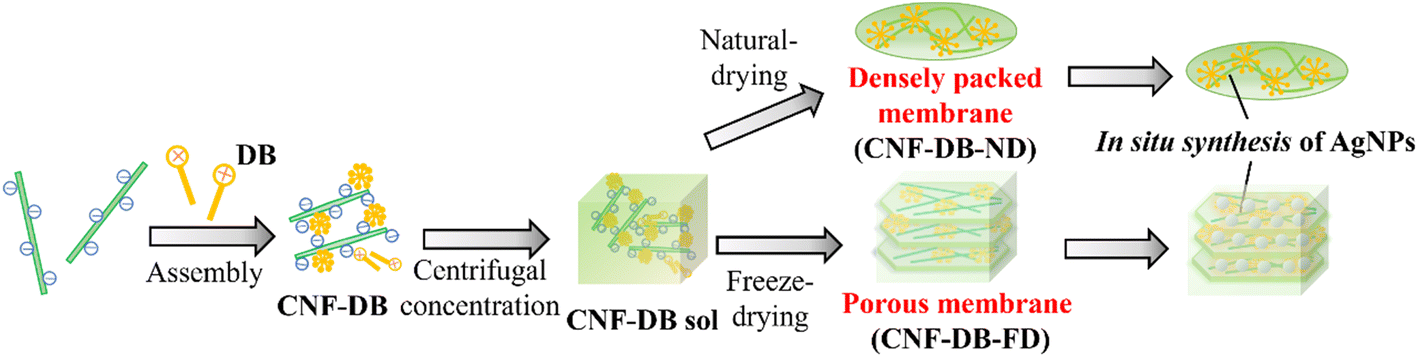
In this study, we intended to fabricate a CNF-based porous material by combining the surfactant-induced assembly and the ice crystals-templated pore formation. As shown in Scheme 1, CNF was assembled with a cationic surfactant, domiphen bromide (DB), to prepare CNF assembly (CNF-DB) through the ionic complex formation between CNF and DB and the hydrophobic association between alkyl chains of DB. Next, CNF-DB was concentrated by centrifugation to produce a CNF sol (CNF-DB-sol), which was expected to be accumulated perpendicular to the centrifugal force. Next, the sol was naturally dried to prepare a dried membrane (CNF-DB-ND). In addition, it was lyophilized to prepare a freeze-dried membrane (CNF-DB-FD).
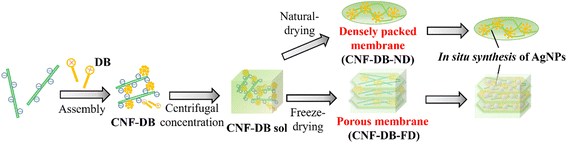 |
| | Scheme 1 Schematic illustration for preparation of a CNF densely packed membrane and a CNF porous membrane via surfactant-induced assembly. In situ reduction of Ag ions was conducted to prepare an AgNPs-CNF composite membrane (in situ synthesis of AgNPs). | |
CNF-DB-FD showed a high specific surface area derived from porous structures. Therefore, we intended to load lipophilic substances to hydrophobic cores derived from associated alkyl chains of DB, which aimed to apply CNF-DB-FD as drug delivery carriers, cosmetic products and human health care products. In addition to its high surface area, CNF-DB-FD possessed negative charges derived from carboxy moieties of CNF. Thus, we intended to load rhodamine B (RhB) as a positive charged substance. Reducing end groups derived from CNF was expected to provide sites for the redox reaction. Therefore, we selected Ag ion and intended to conduct in situ synthesis of silver nanoparticles (AgNPs) by using CNF-DB-FD as a template for Ag ion adsorption and reduction. It is known that AgNPs exhibit fascinating properties originated from localized surface plasmon resonance (LSPR) including coloration of colloidal solutions,14 photothermal effects,15 catalytic activity16 and surface-enhanced Raman scattering (SERS).17,18 We intended to array AgNPs over the surface of CNF porous materials to generate hot spots which lead to enhancement of SERS signals.
Materials and methods/experimental procedure
Materials
Cellulose nanofiber (Rheocrysta I-2SX) was donated by DKS Co. Ltd (Kyoto, Japan). Domiphen bromide was purchased from Sigma-Aldrich Co. (St. Louis, MO, U.S.A.). Rhodamine B (RhB), sulfuric acid, and silver nitrate were purchased from Junsei Chemical Co. (Tokyo, Japan). Phenol, trisodium citrate dihydrate and sodium tetrahydroborate were purchased from FUJIFILM Wako Pure Chemical Co. (Osaka, Japan). All chemicals were used as received. The water used in all experiments was prepared in a water purification system (WT-100, Yamato Scientific Inc., Tokyo, Japan) and had a resistivity higher than 18.2 MΩ cm. Perfluoro alkoxyalkane (PFA) film with a thickness of 0.1 mm was purchased from Junkosha Co. (Tokyo, Japan).
Preparation of a CNF assemblage and a CNF sol
CNF was suspended at the final concentration of 2000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm in Milli-Q water or 50 mM NaCl aq. by ultrasonication for 5 min. After stirring for 24 h at 25 °C, an aqueous solution of DB (1.5 mL, Milli-Q or 50 mM NaCl aq.) was added dropwise to 1.5 mL of the CNFs suspension at the final DB concentration of 0.25–100 mM, and then stirred for 30 min at 25 °C (the surfactant-induced assembly). The obtained CNF assemblage was referred to as CNF-DBx where the subscript x stands for the DB concentration. The photo images of CNF-DBx were taken and their structures were observed with an optical microscope by placing them onto a glass slide.
000 ppm in Milli-Q water or 50 mM NaCl aq. by ultrasonication for 5 min. After stirring for 24 h at 25 °C, an aqueous solution of DB (1.5 mL, Milli-Q or 50 mM NaCl aq.) was added dropwise to 1.5 mL of the CNFs suspension at the final DB concentration of 0.25–100 mM, and then stirred for 30 min at 25 °C (the surfactant-induced assembly). The obtained CNF assemblage was referred to as CNF-DBx where the subscript x stands for the DB concentration. The photo images of CNF-DBx were taken and their structures were observed with an optical microscope by placing them onto a glass slide.
Next, CNF-DBx was accumulated by centrifugation at 10000 rpm for 20 min at 25 °C to obtain a sol-like concentrate (CNF-DBx-sol). The supernatant was collected, and the amount of DB in the supernatant was measured by ion-exchange chromatography (Prominence HIC-SP, Shimadzu, Kyoto, Japan) to estimate the content of DB in CNF-DBx. To estimate the content of CNF in CNF-DBx-sol, the amount of CNF in the supernatant was measured by phenol–sulfuric acid method,19 which is based on a colorimetric analysis to determine the total sugar content of the sample.
Preparation of a CNF-DB membrane and a CNF porous membrane
CNF-DB-sol as prepared was lyophilized to prepare a freeze-dried CNF membrane. On the other hand, 6 mg CNF-DBx-sols (x = 5, 10, 50, and 100) were resuspended in 1 mL Milli-Q water, and then they were drop-casted onto a water-repellent PFA film followed by natural drying. Then, each membrane was peeled off the PFA surface to obtain a free-standing membrane. The freeze-dried CNF membrane and the naturally dried one were referred to as CNF-DBx-FD and CNF-DBx-ND, respectively. Morphologies of these two membranes were observed with a scanning electron microscope (SEM, F-50, Hitachi High-Tech corporation Japan). Prior to SEM observations, all the dried samples were placed on a carbon tape and further sputter-coated with Os with an ion sputter (Neoc-Pro, Meiwafosis Co., Ltd Tokyo, Japan). Also, the average interspaces between layers in CNF-DBx-FD were calculated by measuring the widths of at least 50 interspaces from different regions of SEM images. Fourier transform infrared (FT-IR) spectra were measured using a spectral reflectometer (ALPHA, Bruker Optik FmbH, Germany) to detect the formation of hydrogen bonds in the CNF membranes. To evaluate the water resistance, CNF-DBx-FD or CNF-DBx-ND was immersed in Milli-Q water for 1 day at 25 °C and their morphologies were observed with SEM. They were further incubated in Milli-Q water for 10 days and the amount of dissolved CNF was measured by phenol–sulfuric acid method as described above.
Loading of Nile red and loading and release of rhodamine B
To investigate the ability to absorb (load) hydrophobic dyes to hydrophobic cores in CNF-DBx-FD, Nile red (NR) was selected as a lipophilic dye model. NR was dissolved at 1000 ppm in ethanol and then 1.0–90 ppm aqueous solutions were prepared by dilution with 50 mM NaCl aq. This aqueous solution (1.0 mL) was added to CNF-DB5-FD and the incubation was carried out for 30 min at 25 °C. The supernatant was collected, and the fluorescent intensity (λex = 533 nm, λem = 624 nm) of NR was measured with a spectrofluorometer (FP-6500, JASCO, Tokyo, Japan). The loading capacity (L.C.) of a substance into a carrier is estimated by calculation according to the following eqn (1).| |
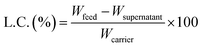 | (1) |
where Wfeed is the weight of each substance in the feed, Wsupernatant is the weight of each substance remaining in the supernatant solution after loading, and Wcarrier is the total weight of the carrier (CNF-DB5-FD).
As well, loading of rhodamine B (RhB) to CNF-DB5-FD was conducted using 10 ppm RhB in 50 mM NaCl aqueous solution. Then, the dialysis against 150 mM NaCl aq. was carried out for 3 h with a dialysis membrane (MWCO: 14000) to remove the unloaded RhB. After RhB-loaded CNF-DB5-FD (10 ppm) was rinsed with 150 mM NaCl aqueous solution, release experiments were carried out using a dialysis cup in each 20 mL of Milli-Q water, 50 mM NaCl aq. and 150 mM NaCl aq. and further dialyzed to investigate the release profile for 2 days. At the predetermined time intervals, an aliquot (1.0 mL) was collected from the dialyzed solution and the release amount of RhB (S) was calculated by measuring the fluorescent intensity of RhB (λex = 555 nm, λem = 573 nm) with the spectrofluorometer (FP-6500, JASCO, Tokyo, Japan). The release rate was calculated using the following eqn (2) where S0 is the amount of RhB loaded.
| | |
Release (%) = S/S0 × 100
| (2) |
In situ reduction of Ag ions to form AgNPs-composite membranes
To investigate the ability to adsorb (load) cationic substances to carboxy moieties in CNF membranes, Ag ion was selected as a cationic substance for CNF-DB100-ND and CNF-DB5-FD. A cellulose filter paper was used as a control. Silver nitrate (AgNO3) was dissolved at 100 mM in Milli-Q water. Each sample was immersed in 1.0 mL of a AgNO3 aqueous solution and then the incubation was carried out for 1 or 24 h at 25 °C in the dark. The amount of Ag ions remaining in the solution was measured by ion-exchange chromatography (Prominence HIC-SP, Shimadzu, Kyoto, Japan). After the adsorption of Ag ions, each sample was rinsed with Milli-Q water and in situ synthesis of silver nanoparticles (AgNPs) was conducted according to the following methods. One mL of NaBH4 (10 mM) was added to the sample and the incubation was carried out for 1 h at 25 °C. Alternatively, two mL of trisodium citrate (100 mM) was added to the sample and simultaneously microwave irradiation at 700 W was conducted for 10 min using a microwave oven (NE-KA1, Panasonic, Osaka, Japan). The resultant AgNPs composite CNF porous membrane was referred to as CNF-DBx-FD-AgNPsy where y stands for the Ag ion concentration. Morphologies of AgNPs composite membranes were observed with SEM (SEM F-50, Hitachi High-Tech corporation, Tokyo, Japan) and elemental analysis in the Ag composite membrane was conducted using energy dispersive X-ray analysis (EDX). Also, X-ray diffraction (XRD D8 advance, Bruker, USA) was carried out to detect the crystal structure of AgNPs.
SERS activity of AgNPs-composite membranes
RhB was selected as a SERS probe, and CNF-DB100-ND-AgNPs100 and CNF-DB5-FD-AgNPs100 were used as SERS substrates. A cellulose filter paper-AgNPs100 and CNF-DB5-FD were selected as control. RhB aqueous solutions were prepared at 0.005, 0.01, 0.05, 0.1, 0.5, and 1.0 mM and each 50 μL was dropped onto the SERS substrate.20 SERS spectra were obtained in the range of 900–1700 cm−1 with 1 s exposure time using Raman imaging microscope (inVia, Renishaw, UK).
Results and discussion
Preparation of CNF assembly via the surfactant-induced assembly
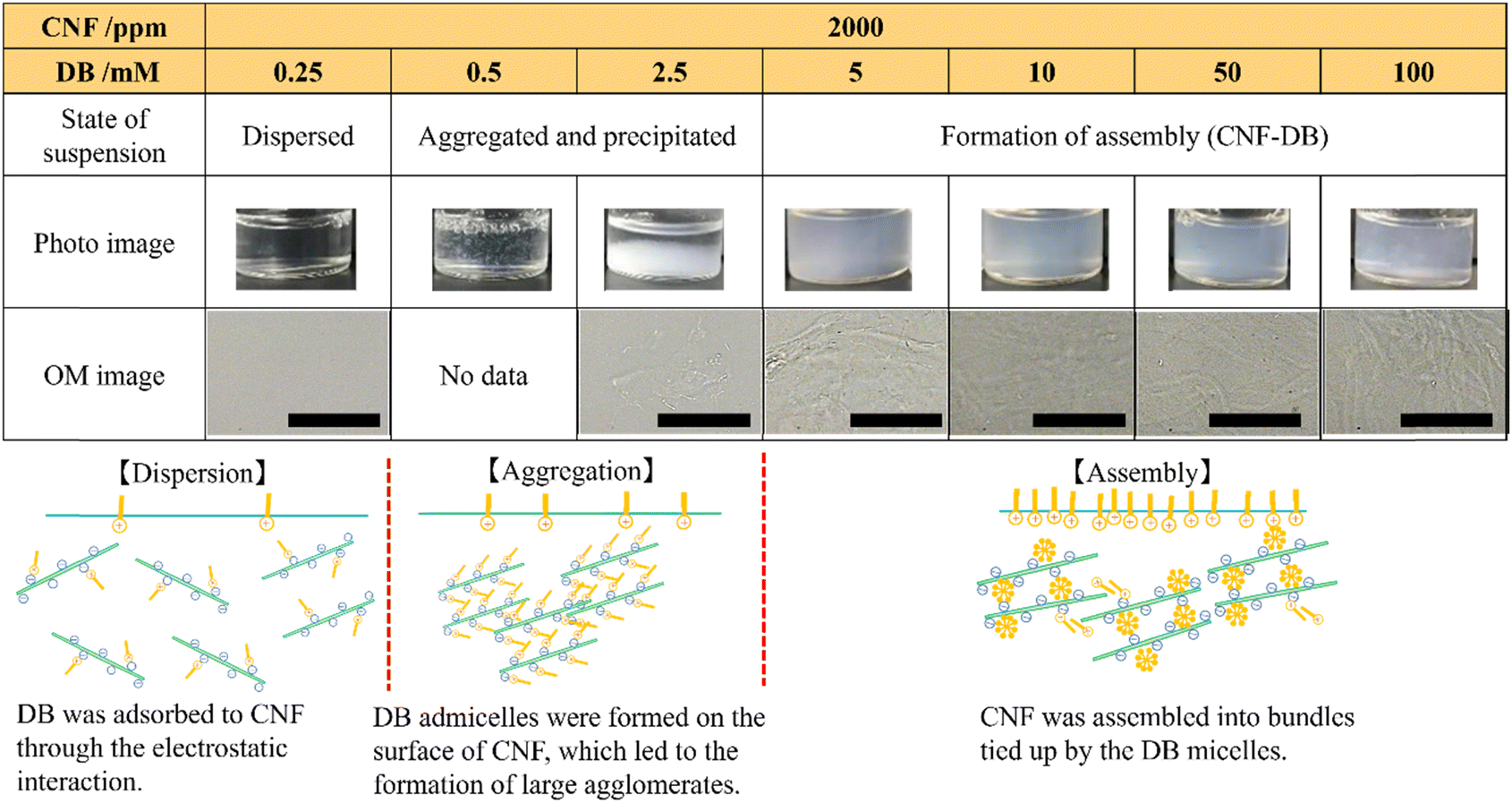
An aqueous solution of CNF isolated by TEMPO oxidation was diluted and then their entanglement was decreased by ultrasonication to obtain a well-suspended CNFs in water. They showed a high aspect ratio with the widths of approximately 5 nm and the length of 2–3 μm (Fig. S1†). We have previously reported that nanoparticles could be prepared from biopolymers with large persistence length such as DNA and mucin by crosslinking them with surfactants through electrostatic attraction.10–12 Catchmark et al. have investigated the binding interactions of cellulose nanocrystals (CNC) with lauric arginate with an opposite charge.13 They have reported that the surfactant aggregates bound to the CNC surface in the form of bilayers to form large and crosslinked aggregates by increasing the surfactant concentration. Tardy et al. have reported that the surfactant adsorption could induce a lowering of the percolation threshold, which results in gelling of the CNF and surfactant complex.21 Here, we employed DB as a cationic surfactant to assemble CNFs. DB is known to be a biosafe surfactant that makes it an essential component in many topical formulations, mouthwashes and throat lozenges. It possesses low critical micellar concentration (c.m.c.) of 0.35 mM (ref. 22) and it can lower the concentration needed for gelling of CNFs. Also, its c.m.c. can be modulated with NaCl concentration. DB was added to the aqueous suspension of CNF (2000 ppm). We investigated the influence of CNF and DB concentrations and the CNF:DB mass ratio on the formation of coacervates and the structure of their assembly (CNF-DB). When increasing the DB concentration, no change was observed at 0.5–1.4 mM DB whereas it was partially aggregated and precipitated at 2.5–5.0 mM DB. Above 10 mM DB, the re-suspension was observed appeared probably due to the coverage of the CNF surface with DB micelles (Table S1†). Next, the same experiment was performed in the presence of 50 mM NaCl. The photo and OM images showed that the mixture remained dispersed when the DB concentration was below 0.5 mM whereas aggregation took place at 0.5 mM DB (Table 1). This is because the presence of NaCl induces great lowering of c.m.c. of DB.22 At 0.5 and 2.5 mM, large aggregates and precipitates were observed probably due to the formation of admicelles over the CNF surface. With increasing the DB concentration to 5 mM, which is much higher than c.m.c. of DB, the CNF suspension turned translucent thanks to the formation of large agglomerates. This is probably because CNF assembly were formed by crosslinking CNF with DB micelles. OM images revealed that further addition of DB (10, 50 and 100 mM) caused the formation of micron-sized assembly. It is thought that they could be assembled from CNF bundles tied up by the DB micelles. These are similar results to those reported by Catchmark et al.13 When the CNF concentration was 5000 ppm, the CNF suspension showed opaque and large assembly was observed with OM (Table S2†). With increasing the CNF concentration to 10000 ppm, large assembly was observed at 10 mM DB. On the other hand, they were separated to form precipitates at 100 mM DB (Table S2†). This is because the electrostatic repulsion between CNFs covered with DB micelles may induce the formation of assembly. In this way, we could obtain such assembly via the surfactant-induced association from CNF and DB.
Table 1 Photo and OM images of CNF assembly (CNF-DB) prepared by mixing CNF (2000 ppm) and DB of different concentrations in 50 mM NaCl aqueous solution. All scale bars are 50 μm
Preparation of a CNF membrane
Next, we intended to build up a CNF-based membrane from CNF-DB assembly. First, CNF-DB assembly was prepared at different DB concentrations and then centrifugation was carried out to concentrate them. We could not obtain a condensed CNF-DB complex at 0.25 mM and 2.5 mM DB (Table 2). On the other hand, we could obtain a sol-like CNF-DB complex (CNF-DB-sol) above 5.0 mM DB (Table 2). The incorporation efficiencies of CNF and DB into the CNF-DB-sol were 95.1 ± 6.7% and 83.0 ± 4.9%, respectively. This suggests that free DB micelles in the solution should act as crosslinkers to induce accumulation of CNF-DB assembly. Next, the obtained CNF-DB-sol was naturally dried on the PFA film. We obtained opaque CNF membranes with a rough and uneven surface irrespective of the DB concentration (Table S3†). SEM observations indicated that CNF-DB assembly were tightly packed to form non-porous membranes. When DB concentration increased to 100 mM, the membrane became thicker probably due to the disassociation of large aggregates via strong electrostatic repulsion in the membrane through the polymer-induced micellization and the formation of free micelles in solution. At 5.0 mM and 10 mM DB, the membrane ruptured during the peeling process probably due to weak crosslinking of CNF with DB. Above 50 mM, the free-standing membrane could be obtained without rupturing but swelled in water whereas the PFA remained the same due to its water-repellent property.
Table 2 Photo images of CNF-DB-sol prepared by centrifugal concentration of CNF-DB assembly formed at different DB concentrations
At 100 mM DB, a free-standing membrane (CNF-DB100-ND) could be obtained. This membrane remained intact after the immersion in Milli-Q water for 24 h (Fig. S2†) and only 0.4% of CNF was eluted for 11 days. On the other hand, a dried CNF sample (CNF-ND) was prepared as a control by natural drying of CNF without DB. It easily dissolved and 84 ± 12% of CNF was eluted for 24 h. It has been reported that dried CNF samples usually possess poor water resistance due to their hydrophilicity and they can easily dissolve in water.23 We suppose that addition of DB facilitated the formation of molecular interactions between CNFs, leading to the improvement in water resistance.
Preparation of a CNF porous membrane via the ice crystals-templated pore formation
Instead of natural drying, we conducted freeze-drying of the CNF-DB-sol to fabricate a porous membrane (the ice crystals-templated pore formation). Freeze-drying can allow polymers to condense and cover the surface of ice crystals. This process could promote molecular interactions such as the hydrogen binding between polymers, resulting in the formation of a porous structure.24–27 From SEM observations (Table 3), the uneven coverage of CNF fibrils was observed over the surface of a freeze-dried CNF sample (CNF-FD) and its cross section showed uneven accumulation of CNF fibrils.
Table 3 SEM images of freeze-dried CNF-DBx-FD. The samples were prepared by centrifugal concentration of CNF-DB assembly at 10000 rpm. All scale bars are 50 μm
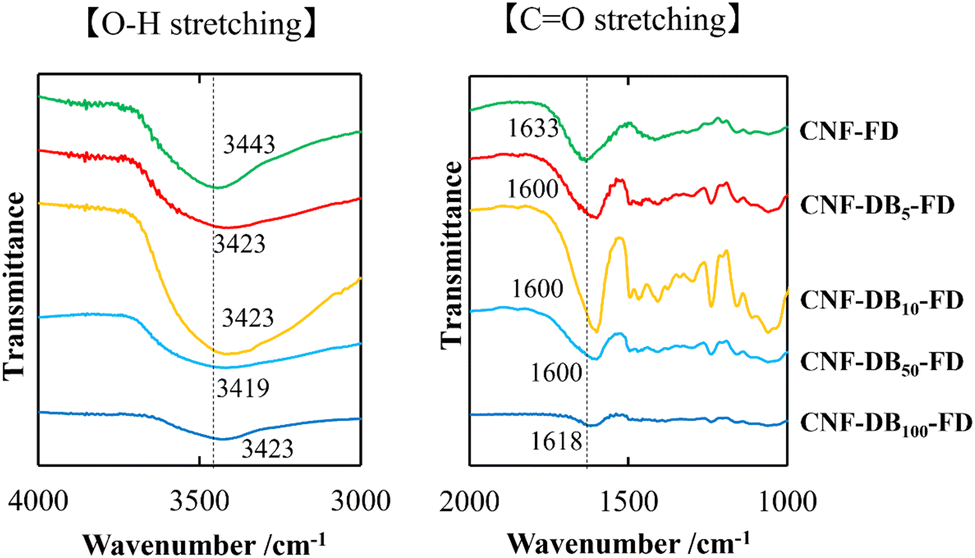
On the other hand, we could observe cellular porous structures on the surface of freeze-dried CNF-DB sol (CNF-DBx-FD), which reflected the ice crystals-templated pore formation (CNF-DBx-FD) (Table 3) and the thickness of CNF-DB5-FD with a porous structure was estimated to be approximately 200 μm (Fig. S3†). The cross section of a freeze-dried CNF-DB sol showed uniform layering of thin CNF sheets with a regular interspace between layers (Table 3). We hypothesized that the CNF sheets could be accumulated perpendicular to the centrifugal force to form the layered structure and then the ice crystals would be formed at the interspace between the layers to create the anisotropic pore structure. It is noteworthy that the interspace and the pore size became smaller with increasing the DB concentration (Table 3). From the FT-IR analysis, no shift in the peak position of the carboxylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and the O–H stretching vibration could be observed at different DB concentrations (Fig. 1). Therefore, this is probably because the surfactant-induced association from CNF and DB was facilitated by the DB concentration, leading to variations of the interspace and the pore size. Next, the effect of centrifugal force in the accumulation process was investigated with CNF-DB5 (Table S4†). CNF-DB sol could not be formed at the rotational speed of 2500 rpm. Above 5000 rpm, the interspace of CNF-DB5-FD decreased with increasing the centrifugal force. In other words, these indicate that the anisotropy in the pore structure can be tuned by the DB concentration and the centrifugal force.
O stretching vibration and the O–H stretching vibration could be observed at different DB concentrations (Fig. 1). Therefore, this is probably because the surfactant-induced association from CNF and DB was facilitated by the DB concentration, leading to variations of the interspace and the pore size. Next, the effect of centrifugal force in the accumulation process was investigated with CNF-DB5 (Table S4†). CNF-DB sol could not be formed at the rotational speed of 2500 rpm. Above 5000 rpm, the interspace of CNF-DB5-FD decreased with increasing the centrifugal force. In other words, these indicate that the anisotropy in the pore structure can be tuned by the DB concentration and the centrifugal force.
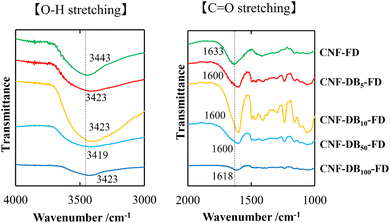 |
| | Fig. 1 FT-IR spectra of CO stretching band (right) and O–H stretching band (left) for lyophilized CNF (CNF-FD) and CNF-DBx-FD. The numbers indicate the wavenumber corresponding to each vibration mode. | |
Next, we analyzed the chemical structure of CNF-DBx-FD with FT-IR. IR spectra of the CO stretching vibrations and the O–H stretching vibrations derived from CNF-FD, CNF-DBx-FD and DB are shown in Fig. 1. The band at 1633 cm−1 observed for CNF-FD corresponds to the carboxylic CO stretching vibration of CNF. On the other hand, DB showed sharp peaks at 1589 cm−1, 1493 cm−1 and 1230 cm−1, which are associated with CC ring stretching vibration, methylene in-plane bending vibration and C–N stretching vibration, respectively (Fig. S4†). The powder mixture of CNF and DB showed peaks at 1635 and 1589 cm−1 (Fig. S4†), corresponding to the carboxylic CO stretching vibration of CNF and CC ring stretching vibration of DB, respectively, whereas CNF-DBx-FD showed each single peak at 1600 cm−1 for CNF-DB5-FD, CNF-D10-FD and CNF-DB50-FD and at 1618 cm−1 for CNF-DB100-FD (Fig. 1). According to the literature, the peak position of carboxylic CO stretching vibration shifts to the lower wavenumber mainly due to the formation of stronger intra or intermolecular hydrogen bonds.28–30 On the other hand, the broad peak in the range of 3000 cm−1–3700 cm−1 corresponds to the O–H stretching vibration in CNF.28,31 The peak position shifted slightly to the lower wavenumber irrespective of the DB concentration, probably because of the formation of hydrogen bonding. From the results described above, we thought that CNF-DBx-FD could be built and maintained by the formation of hydrogen bonding between CNFs, which would give rise to its anisotropic porous structure and water-resistance.
Next, we investigated water resistance of CNF-DB5-FD. After the immersion in Milli-Q water for 24 h, CNF-DB5-FD retained the porous structure (Fig. 2). Furthermore, only 5.3 ± 0.4% of CNF was eluted after 11 days and the porous structure was still maintained with high robustness due to the physical crosslinking via hydrogen bonding. On the other hand, CNF-FD dissolved instantly in Milli-Q water and 66 ± 7.0% of CNF was eluted for 24 h. From these results, we can say that we could fabricate a CNF porous membrane by combining the surfactant-induced assembly and the ice crystals-templated pore formation.
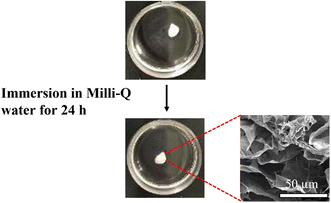 |
| | Fig. 2 Photo images showing the structural stability of CNF-DB5-FD in Milli-Q water. The inset shows its cross-sectional SEM image 24 h after immersion in Milli-Q water. | |
Loading of chemical substances in CNF porous membranes
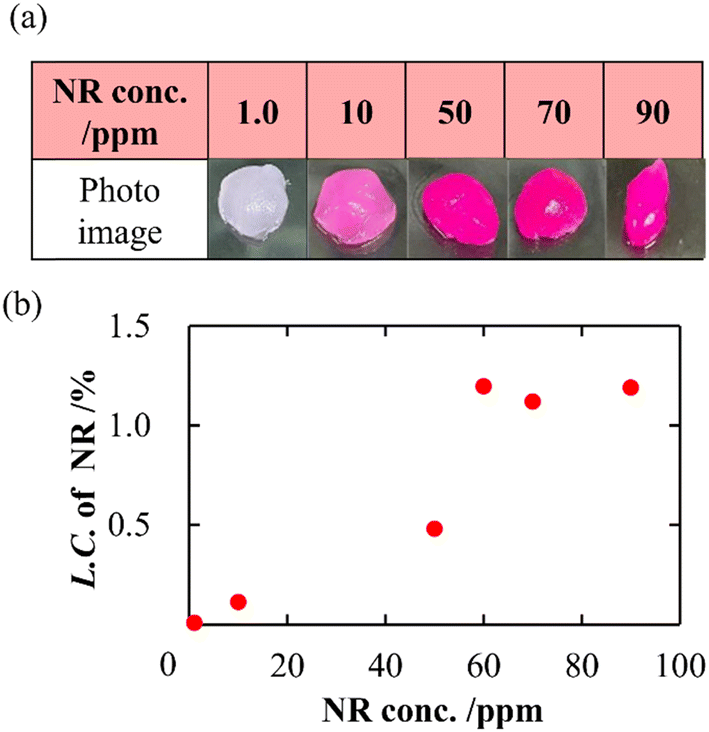
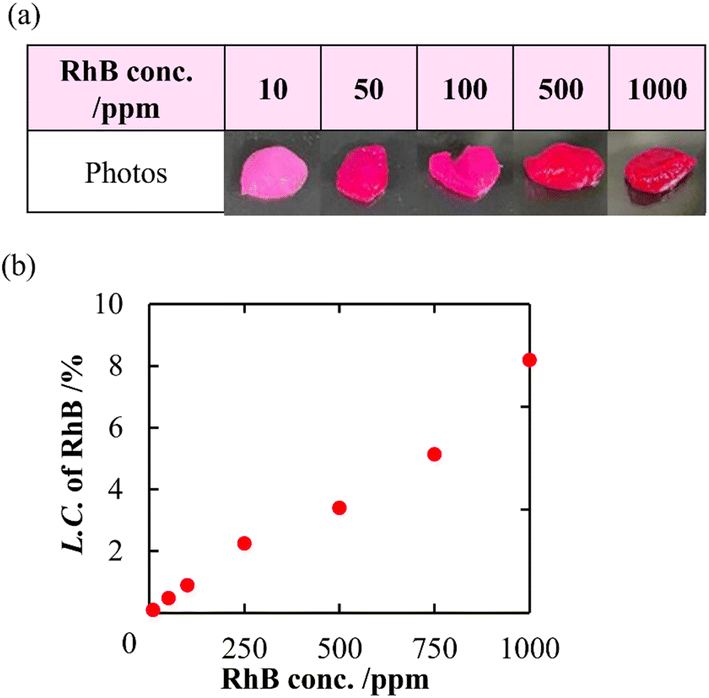
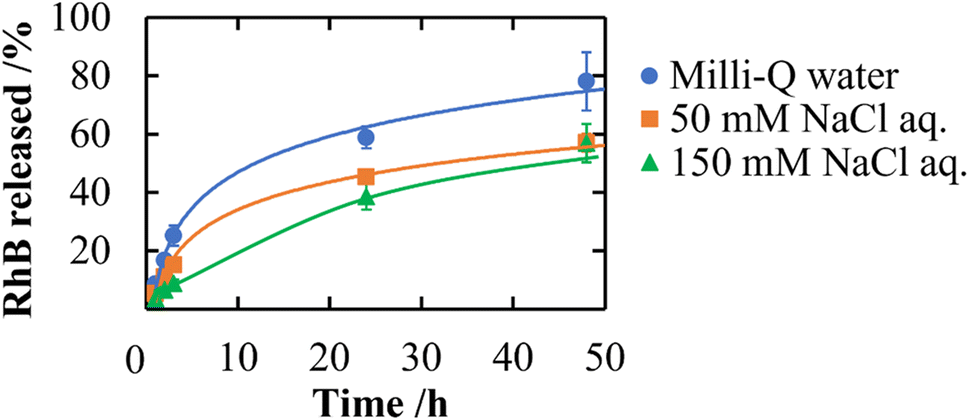
In addition to a high specific surface area derived from porous structures, CNF-DB5-FD possesses hydrophobic cores derived from associated alkyl chains of DB. We have previously reported that nanoparticles prepared with surfactant-induced assembly possessed hydrophobic cores derived from associated surfactants, enabling hydrophobic compounds to be loaded and retained within the cores.10 Therefore, we intended to load NR (miLogP: 4.58) as a lipophilic substance. NR-loaded CNF-DB5-FD turned red indicative of incorporation of NR (Fig. 3a) and the L.C. increased with the NR concentration and leveled off at 60 ppm (Fig. 3b). Next, we intended to load rhodamine B (RhB) as a positively charged and hydrophobic substance (miLogP: 1.91) through its adsorption onto the surface of CNF-DB-FD. Then, we investigated the interactions between RhB and CNF-DB-FD by the addition of NaCl to control the RhB release. Loading of RhB (miLogP: 1.91) gradually increased with the RhB concentration due to both the electrostatic attraction between RhB and CNF in addition to the hydrophobic interaction (Fig. 4). It did not reach a peak even at 1000 ppm RhB. Release experiments were carried out in Milli-Q water (Fig. 5). Burst release was observed and approximately 80% of loaded RhB was released within 48 h. To control the RhB release, we investigated the interactions between RhB and CNF-DB-FD by the addition of NaCl.22 The RhB release was suppressed in 50 and 150 mM NaCl aqueous solutions. This is because the c.m.c. of DB was shifted to the lower concentration by the salt effect, resulting in suppression of the release of RhB.
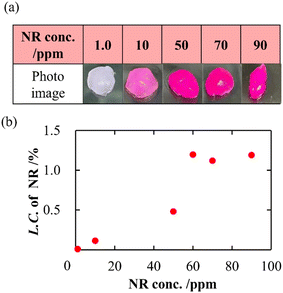 |
| | Fig. 3 Photo images of CNF-DB5-FD after loading of NR (a) and change in the loading capacity of NR into CNF-DB5-FD with NR concentration (b). | |
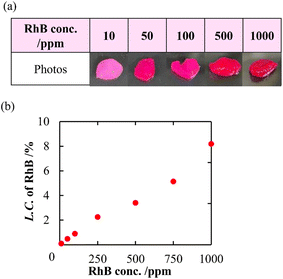 |
| | Fig. 4 Photo images of CNF-DB5-FD after loading of RhB (a) and change in the loading capacity of RhB into CNF-DB5-FD with RhB concentration (b). | |
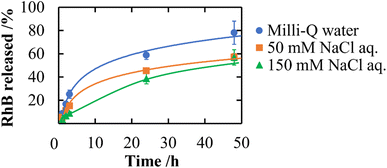 |
| | Fig. 5 Release profiles of RhB from CNF-DB5-FD at different ionic strengths. | |
In situ reduction of Ag ions and SERS of AgNPs-composite membranes
The CNF reducing end groups in addition to its negative moieties was expected to provide sites for the redox reaction. Here, loading of Ag ions to water-resistant CNF porous membranes and in situ reduction of Ag ions were conducted to produce silver nanoparticles (AgNPs). We first evaluated absorption capacity of Ag ions to a cellulose filter paper (Fig. S5†), CNF-DB100-ND, and CNF-DB5-FD. CNF-DB100-ND showed the highest absorption capacity among three kinds of samples. On the other hand, adsorption onto CNF-DB5-FD was significantly suppressed to the same level as the cellulose filter paper (Fig. S6†). This is probably because swelling of CNF-DB100-ND could facilitate absorption of Ag ions whereas CNF-DB5-FD tightly crosslinked via strong intermolecular interactions may induce inhibition of absorption of Ag ions. Contrary to our expectation, in situ reduction of Ag ions did not proceed with no additives. By adding NaBH4 as a reducing agent, CNF-DB5-FD turned brown and it was observed with SEM that large AgNPs were generated at random on the layer surface (Table 4). The percentage of Ag composition in the membrane surface determined by EDX increased with the Ag ion concentration in feed. The treated CNF-DB5-FD exhibited the Ag composition significantly higher than CNF-DB100-ND and the filter paper at 100 mM of the Ag ion concentration. In the XRD analysis for Ag-composite CNF-DB5-FD, especially a peak corresponding to (111) and (200) crystal faces of Ag metal became prominent with increasing the Ag ion concentration20 (Fig. S7†). These results indicate that Ag nanoparticles (AgNPs) were generated on the CNF-DB5-FD by in situ reduction of absorbed Ag ions with NaBH4 (CNF-DB5-FD-AgNPsy).
Table 4 SEM images of AgNPs generated on a filter paper, CNF-DB100-ND and CNF-DB5-FD by reduction with NaBH4. The inset shows the photo images of AgNPs-CNF composite membranes
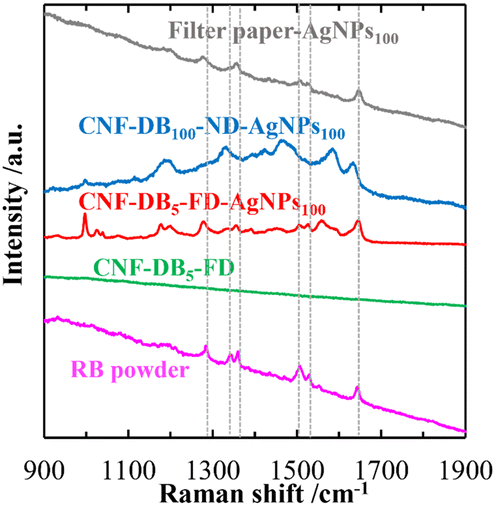
It is known that AgNPs exhibit fascinating properties originated from localized surface plasmon resonance (LSPR) and surface-enhanced Raman scattering (SERS).17,18 We anticipated that a layered structure in CNF porous membranes is advantageous in LSPR enhancement due to its planar surface with the high surface area. If uniform formation of AgNPs is achieved over the layer surface, it is possible to create hotspots, which emerge between the junction of AgNPs where an intense electromagnetic field is generated.32 The Raman spectra of RhB (1.0 mM) adsorbed on CNF-DB5-FD, CNF-DB5-FD-AgNPs100, CNF-DB100-ND-AgNPs100 and filter paper-AgNPs100 were shown in Fig. 6. No peak corresponding to RhB was observed from CNF-DB5-FD and CNF-DB100-ND-Ag100 whereas CNF-DB5-FD-AgNPs100 exhibited a little enhanced Raman peaks of RhB at 1215, 1290, 1346, 1364, 1516, 1537, and 1650 cm−1 resulting from the SERS effect of AgNPs generated on the membrane. This is probably because the size and distribution of generated Ag particles were not sufficient for emerging of the SERS effect that enhances the scattered signal of RhB.
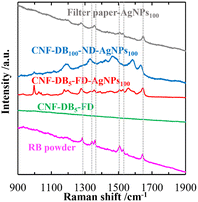 |
| | Fig. 6 Raman spectra of RhB deposited on the filter paper-AgNPs100, CNF-DB100-ND-AgNPs100, CNF-DB5-FD-AgNPs100 and CNF-DB5-FD. The samples were prepared by reduction with NaBH4. The dotted lines indicate the peak positions of RhB. | |
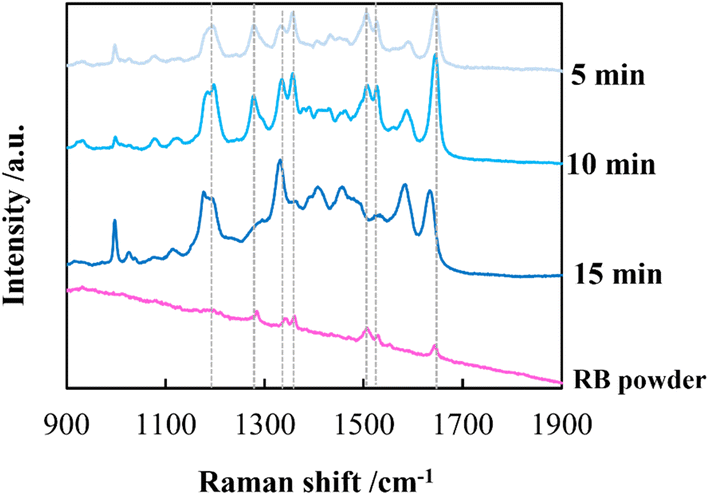
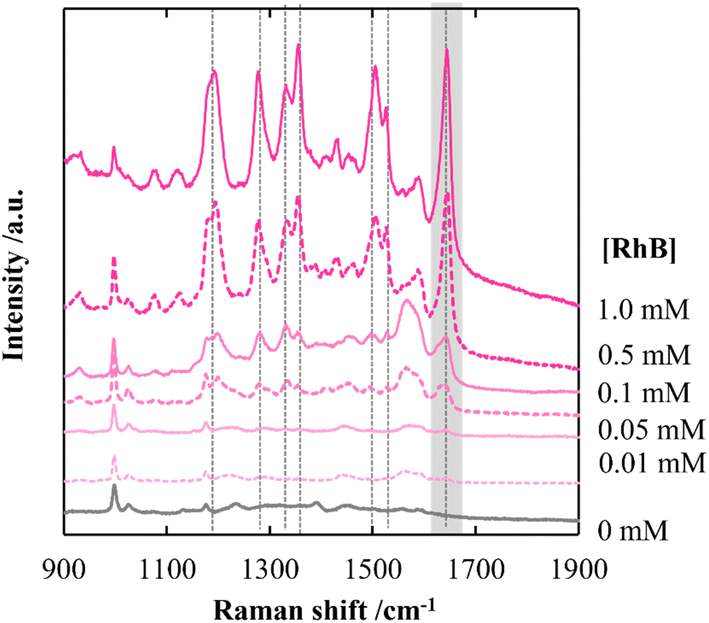
By employing a mild reducing agent such as trisodium citrate, we attempted to achieve the uniform coverage of AgNPs on the layer surface without the formation of AgNPs in the bulk solution.33,34 However, we could obtain few AgNPs due to its weak reduction activity. Then, in situ reduction of Ag ions was carried out by using trisodium citrate and simultaneously microwave (MW) irradiation was carried out. Recently, microwave-assisted methods have been used as green strategies in various chemical syntheses.35 It is expected that MW irradiation enables rapid and homogeneous heating of the entire solution, which would facilitate the formation of AgNPs.36 As expected, AgNPs with almost the same size were instantly generated within 5 min in 100 mM AgNO3 aqueous solution (data not shown). In the same manner, AgNPs were evenly distributed over the layer surface of CNF-DB5-FD by MW irradiation for 5 min and the Ag composition increased with the irradiation time (Table 5). Although a small number of nanoparticles were generated below 50 mM of the Ag ion concentration, it was found that the layer surface was densely covered with nanoparticles at 100 mM (Table S5†). Furthermore, it was confirmed from peaks for Ag (111) and (200) in XRD that AgNPs were generated over the layer surface (Fig. S8†). SEM observation verified that layered structure of CNF-DB-FD was retained even after MW heating process (Fig. S9†). A profound enhancement in the SERS activity was also observed compared with reduction by NaBH4 and SERS peaks were enhanced by increasing the MW irradiation time (Fig. 7). This is because MW irradiation allowed for rapid heating, which could lead to fast reduction reaction of Ag ions and fast nucleation of Ag to generate AgNPs suitable for SERS.37 It might be possible that some portions of Ag ions were desorbed during the reduction process. There is no denying that it had an influence on the formation of AgNPs. Nevertheless, it can be said that each reaction was carried out in the same initial concentration of Ag ions and microwave heating was effective in the formation of AgNPs in terms of SERS activity. Next, we attempted to perform the label-free detection of RhB by using CNF-DB5-FD-AgNPs100 as the SERS substrate. RhB aqueous solutions of different concentrations were dropped onto CNF-DB5-FD-AgNPs100 and then Raman spectra of adsorbed RhB were measured to investigate its detection sensitivity (Fig. 8). It could be observed that the peak intensity increased with increasing the RhB concentration. With decreasing the RhB concentration to 0.005 mM, the Raman band was still detectable. The log–log plot of the peak intensity at 1643 cm−1 against the RhB concentration shows a good linear relationship with the correlation coefficient of 0.97, indicating that the concentration of RhB can be measured by using this plot as a calibration curve (Fig. 9). Our next challenge is to develop such a CNF-based SERS substrate that not only possesses higher sensitivity but also is applicable to a variety of substances.
Table 5 Characterization of CNF-DB5-FD-AgNPs100 prepared by reduction with sodium citrate under microwave irradiation for different periods. All scale bars are 10 μm
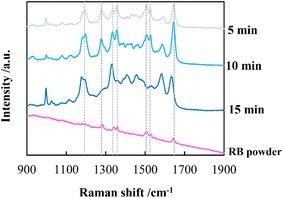 |
| | Fig. 7 Raman spectra of RhB deposited on CNF-DB5-FD-Ag100. The samples were prepared by reduction with sodium citrate under microwave irradiation for different periods. The dotted lines indicate the peak positions of RhB. | |
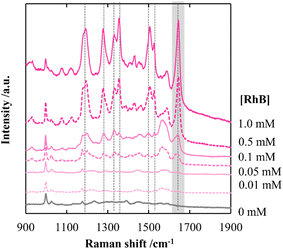 |
| | Fig. 8 Raman spectra of RhB deposited on CNF-DB5-FD-AgNPs100 at different RhB concentrations. CNF-DB5-FD-AgNPs100 was prepared by reduction with sodium citrate under microwave irradiation for 10 min. | |
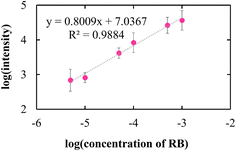 |
| | Fig. 9 The log–log plot of the peak intensity of RhB at 1643 cm−1. | |
Conclusions
CNFs were suspended in water by ultrasonication, and then a cationic surfactant (domiphen bromide, DB) was added to the CNFs suspension. As the DB concentration increased, CNF-DB changed from solubilized, through partially aggregated and precipitated, to disperse assembled state (CNF-DB assembly). CNF-DB assembly were further concentrated by centrifugation, and a sol-like materials (CNF-DB-sol) were obtained. Then, CNF-DB-sol was resuspended in Milli-Q water and subsequently dried naturally to produce a membrane (CNF-DB-ND). Alternatively, CNF-DB-sol was freeze-dried to form a porous structure (CNF-DB-FD). Cross-sectional SEM observations revealed that CNF-DB-FD exhibited a unique layered porous structure and the spacing between the layers was tunable by the ratio of CNF and DB and the centrifugal force during the preparation of CNF-DB-sol. FT-IR peaks derived from the OH and CO groups of CNFs were shifted to a low-wavenumber, indicating that formation of hydrogen bonds between the CNFs was facilitated by DB-induced assembly and further drying process. This resulted in the improvement of the structural stability of CNF-DB-ND and CNF-DB-FD, thus making them water-resistant. Next, we attempted to produce AgNPs-composite CNF porous membrane (CNF-DB-FD-AgNPs) by in situ reduction of Ag ions over the surface of the CNF-DB-FD. When NaBH4 was used for reduction, large AgNPs were non-uniformly produced on the surface. Alternatively, MW heating was carried out in the presence of sodium citrate for the mild reduction of Ag ions. As a result, it was possible to tune the size and Ag content of AgNPs by the concentrations of the Ag ion and sodium citrate. Furthermore, AgNPs were uniformly arrayed to generate the hotspot, which is the advantage for the enhancement of SERS. It was found that the peak for RhB adsorbed on CNF-DB-FD-AgNPs was largely enhanced due to the SERS effect.
We expect that CNF-DB-FD will be used as a porous membrane to retain and release drugs and active substances. Moreover, we believe that a variety of composite membranes in addition to CNF-DB-FD-AgNPs will be created by using CNF-DB-FD as a porous template for many in situ syntheses, which would open new avenues to produce bio-based functional materials. In addition, another challenge is to create bio-based materials with a good mechanical compressibility and thermal insulation property by making use of the anisotropic porous structure.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing interests.
References
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- K. Heise, E. Kontturi, Y. Allahverdiyeva, T. Tammelin, M. B. Linder, O. Ikkala, K. Heise, E. Kontturi, M. B. Linder and O. Ikkala, Adv. Mater., 2020, 33, 2004349–2004379 CrossRef PubMed.
- S. Ahankari, P. Paliwal, A. Subhedar and H. Kargarzadeh, ACS Nano, 2021, 15, 3849–3874 CrossRef CAS PubMed.
- C. Chen and L. Hu, Acc. Chem. Res., 2018, 51, 3154–3165 CrossRef CAS.
- R. Foudazi, R. Zowada, I. Manas-Zloczower and D. L. Feke, Langmuir, 2023, 39, 2092–2111 CrossRef CAS PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS.
- S. Zhao, W. J. Malfait, N. Guerrero-Alburquerque, M. M. Koebel and G. Nyström, Angew. Chem., 2018, 130, 7704–7733 CrossRef.
- M. Zhang, M. Li, Q. Xu, W. Jiang, M. Hou, L. Guo, N. Wang, Y. Zhao and L. Liu, Ind. Crops Prod., 2022, 179, 114701–114714 CrossRef CAS.
- J. Song, C. Chen, Z. Yang, Y. Kuang, T. Li, Y. Li, H. Huang, I. Kierzewski, B. Liu, S. He, T. Gao, S. U. Yuruker, A. Gong, B. Yang and L. Hu, ACS Nano, 2018, 12, 140–147 CrossRef CAS PubMed.
- Y. Fukui, K. Ito and K. Fujimoto, Polym. J., 2022, 54, 1367–1375 CrossRef CAS.
- Y. Fukui, N. Kabayama and K. Fujimoto, Colloids Surf., B, 2015, 136, 168–174 CrossRef CAS.
- Y. Fukui, M. Fukuda and K. Fujimoto, J. Mater. Chem. B, 2018, 6, 781–788 RSC.
- K. Chi and J. M. Catchmark, Carbohydr. Polym., 2017, 175, 320–329 CrossRef CAS PubMed.
- S. Verma, B. T. Rao, A. P. Srivastava, D. Srivastava, R. Kaul and B. Singh, Colloids Surf., A, 2017, 527, 23–33 CrossRef CAS.
- Y. Zhao, J. Zhao, G. Shan, D. Yan, Y. Chen and Y. Liu, Colloids Surf., B, 2017, 154, 150–159 CrossRef CAS.
- M. Kaushik and A. Moores, Green Chem., 2016, 18, 622–637 RSC.
- D. Zhu, Z. Wang, S. Zong, H. Chen, X. Wu, Y. Pei, P. Chen, X. Ma and Y. Cui, Nanoscale, 2014, 6, 8155–8161 RSC.
- P. Joshi and V. Santhanam, RSC Adv., 2016, 6, 68545–68552 RSC.
- M. Dubois, K. Gilles, J. K. Hamilton, P. A. Rebers, F. Smith and R. Dreywood, Indust. Enq. Chem., Anal. Ed, 1951, 476 Search PubMed.
- S. W. Chook, C. H. Chia, C. H. Chan, S. X. Chin, S. Zakaria, M. S. Sajab and N. M. Huang, RSC Adv., 2015, 5, 88915–88920 RSC.
- B. L. Tardy, S. Yokota, M. Ago, W. Xiang, T. Kondo, R. Bordes and O. J. Rojas, Curr. Opin. Colloid Interface Sci., 2017, 29, 57–67 CrossRef CAS.
- P. Held, 2014, preprint, https://www.biotek.com.
- T. Noguchi, Y. Bamba, T. Isogai, M. Endo and A. Isogai, Polym. Degrad. Stab., 2024, 228, 110919–110927 CrossRef CAS.
- S.-H. Hyon, W.-I. Cha and Y. Ikada, Polym. Bull., 1989, 22, 119–122 CrossRef CAS.
- Z. Z. Pan, W. Lv, Q. H. Yang and H. Nishihara, Bull. Chem. Soc. Jpn., 2022, 95, 611–620 CrossRef CAS.
- Y. Sekine, T. Nankawa, S. Yunoki, T. Sugita, H. Nakagawa and T. Yamada, ACS Appl. Polym. Mater., 2020, 2, 5482–5491 CrossRef CAS.
- S. R. Stauffer and N. A. Peppast, Polymer, 1992, 33, 3932–3936 CrossRef CAS.
- P. Verma, A. Srivastava, A. Shukla, P. Tandon and M. R. Shimpi, New J. Chem., 2019, 43, 15956–15967 RSC.
- L. Mo, S. Zhang, F. Qi and A. Huang, Int. J. Biol. Macromol., 2022, 209, 1922–1932 CrossRef CAS PubMed.
- H. Zhao, K. C. Li, W. Wu, Q. Li, Y. Jiang, B. X. Cheng, C. X. Huang and H. N. Li, J. Ind. Eng. Chem., 2022, 110, 150–157 CrossRef CAS.
- L. F. Scatena, M. G. Brown and G. L. Richmond, Science, 2001, 292, 908–912 CrossRef CAS PubMed.
- H. Chen, T. You, L. Jiang, Y. Gao and P. Yin, RSC Adv., 2017, 7, 32743–32748 RSC.
- Y. Murali Mohan, K. Vimala, V. Thomas, K. Varaprasad, B. Sreedhar, S. K. Bajpai and K. Mohana Raju, J. Colloid Interface Sci., 2010, 342, 73–82 CrossRef CAS.
- K. J. Lee, J. T. Park, J. H. Goh and J. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3911–3918 CrossRef CAS.
- A. Kumar, Y. Kuang, Z. Liang and X. Sun, Mater. Today Nano, 2020, 11, 100076–100096 CrossRef.
- F. K. Liu, C. J. Ker, Y. C. Chang, F. H. Ko, T. C. Chu and B. T. Dai, Jpn. J. Appl. Phys., Part 1, 2003, 42, 4152–4158 CrossRef CAS.
- M. Baghbanzadeh, L. Carbone, P. D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 11312–11359 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: TEM image of TEMPO-oxidized CNF, photo images of CNF-DB, photo and SEM images of CNF-DBx-ND, photo images showing the structural stability of CNF-DB100-ND in Milli-Q water, SEM image showing the edge of CNF-DB5-FD, SEM images of CNF-DB5-FD prepared with different rotational speeds of centrifugation, FT-IR spectra of CO stretching band and O–H stretching band for DB and a powder mixture of CNF and DB, SEM images of the surface and the cross section of a filter paper, Ag+ absorption capacity of a filter paper, CNF-DB100-ND and CNF-DB5-FD, XRD profiles for AgNPs generated on CNF-DB5-FD at different AgNO3 concentrations using NaBH4 as a reducing agent, characterization of CNF-DB5-FD-AgNPsy prepared by reduction with sodium citrate under microwave irradiation. SEM image of CNF-DB5-FD-AgNPs25 prepared by reduction with sodium citrate under microwave irradiation for 5 min. See DOI: https://doi.org/10.1039/d4ra07142e. |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Eriko Baba and
Keiji Fujimoto‡
*,
Eriko Baba and
Keiji Fujimoto‡