
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of pre-treatment conditions on the electrochemical performance of hard carbon derived from bio-waste
Oniye Modupe Mutiat a,
Aibar Alpysbayeva,
Dilshat Abduakhitova,
Kenes Kudaibergenovb,
Zhumabay Bakenova,
Seung-Taek Myungc and
Aishuak Konarov*a
a,
Aibar Alpysbayeva,
Dilshat Abduakhitova,
Kenes Kudaibergenovb,
Zhumabay Bakenova,
Seung-Taek Myungc and
Aishuak Konarov*a
aDepartment of Chemical and Materials Engineering, School of Engineering and Digital Sciences, Nazarbayev University, Astana, Kazakhstan. E-mail: aishuak.konarov@nu.edu.kz
bDepartment of Materials Science, Nanotechnology and Engineering Physics, Satbayev University, 22 Satbaev str., 050013, Almaty, Kazakhstan
cHybrid Materials Research Center, Department of Nanotechnology and Advanced Materials Engineering, Sejong Battery Institute, Sejong University, Seoul 05006, South Korea
First published on 13th January 2025
Abstract
Sodium-ion batteries (SIBs) offer several advantages over traditional lithium-ion batteries, including a more uniform sodium distribution, lower-cost materials, and safer transportation options. A promising development in SIBs is the use of hard carbons as anode materials due to their low insertion voltage and larger interlayer spacing, which improve sodium-ion insertion. Traditionally, hard carbons are made from costly carbon sources, but recent advancements have focussed on using abundant bio-waste, like coffee grounds. This approach reduces costs and helps manage global waste. This research investigates the electrochemical performance of bio-waste-derived hard carbons, which is significantly impacted by various pre-treatment methods. Techniques such as BET, XRD, TEM, and XPS are employed to examine the effects of pre-treatment variables, including washing solvents (organic, acidic, or distilled water), pre-oxidation temperatures, and post-heating processes. These factors influence the structural properties and purity of the hard carbon, impacting its effectiveness as an anode material in SIBs. A significant finding is a mesoporous hard carbon produced from coffee grounds that, after washing with distilled water, pre-oxidation at 150 °C, and thermal treatment at 1300 °C in argon, shows a 23% yield, a reversible capacity of 304 mA h g−1, and Initial coulombic efficiency of 78%. This study underscores the importance of pre-treatments in removing impurities and enhancing the material's sodium storage capabilities.
1 Introduction
Recently, multinational energy companies like CATL Co. Ltd., Faradion UK Ltd. and Natron Energy have announced the commercialisation of Sodium-Ion Batteries (SIBs) for large grid stationary applications and Electric Vehicles (EVs), these new developments are attributed to SIBs prospects as promising materials for the development of sustainable and eco-friendly Energy Storage Systems (ESS).1 SIBs have experienced tremendous growth over the years owing to their intercalation chemistry, physio-electro-chemical characteristics and material synthesis procedures being identical to those of Lithium-Ion Batteries (LIBs), these characteristics allow the implementation of LIBs' battery techniques for the development of SIBs.2,3 Moreover, the availability of sodium metal across the globe provides an excellent price advantage and solves the problem of a monopolised supply of Critical Raw Materials (CRM) that significantly influences LIBs production cost due to the uneven distribution and low relative abundance of about 20 ppm of lithium metal. LIBs Cathode Materials (CM) experienced drastic price turbulence between 2020 and 2023 with an approximately more than 150 percent rise in the cost of LIBs CM while SIBs CM experienced less than 50 percent price turbulence compared with LIBs.4–10Furthermore, SIBs have exceptional safe storage and transportation features, SIBs can be completely discharged to zero voltage at their recorded zero percent State of Charge (SOC) for a long period with no side reactions or effects on the cycling capacity, this further enhances the transportation of SIBs over a long distance unlike the counterpart LIBs. SIBs exhibit excellent cycle life, which makes them withstand several cycles without performance degradation and also makes them a sustainable alternative option for Electronic Vehicles (EVs) compared to LIBs with low cycle life as the Depth of Discharge (DoD) performance of LIBs degrade after approximately 500 cycles.
Owing to sodium's large ionic radius of 1.02 Å, the commonly used anode material (graphite) for LIBs cannot be employed due to its low interlayer spacing (<3.4 Å) which inhibits the sodium ion intercalation and reversibility,4,11 also alloys and NASICON materials are not employed as anode materials for SIBs owing to the existing limitations of significant volume change, high operating voltage and low reversible capacity.9,10 Alternative low-cost material with larger interlayer spacing for Na ion insertion with little to negligible volume change is required to develop good cycling stability and high-performance anode material for SIBs.12,13 Hard-carbon, a carbon-rich amorphous material composed of random turbostratic microstructure with an extended interlayer spacing of over 3.7 Å has demonstrated excellent electrochemical performance with good reversible and cycling capacity within the range of 240–500 mA h g−1,14 due to its distinct disordered structure that provides large interlayer spacing for the de/intercalation of sodium ions without volume change, as sodium intercalation which requires large sufficient interlayer spacing majorly influences the plateau-region capacity of the total voltage-capacity.4,15–19
However, these hard-carbons are synthesised from the thermal treatment of high-composition resin precursors, which are quite expensive, this opposes one of the aims of exploring SIBs, which is to develop low-cost eco-sufficient battery material for large-scale energy storage.20,21 Biomass has been identified as an excellent low-cost, abundant and eco-friendly precursor for hard-carbon synthesis due to its high carbon content and the presence of heteroatoms which aid electronic conductivity.22,23 Readily available biomass like banana peel, rice husk, apple pomace, coconut sheath and other biological waste has demonstrated excellent reversible capacity when explored as anode material for SIBs.24–31 The utilization of this biowaste as anode materials for SIBs not only aids the synthesis of high-capacity anode materials but also provides an efficient waste handling technique by transforming this heavily generated waste to economically important materials.32
Across the globe over seven million tons of waste coffee grounds (WCGs) are generated by beverage industries annually due to the consumption of more than eight million tons of coffee beans with WCGs yield of approximately ninety-one percent, these WCGs have been identified as soil pollutants as they require large oxygen volumes for complete decomposition in the ground.33 WCGs have been identified as an excellent carbon precursor owing to their cellulose and lignocellulose macromolecules composed of aromatic carbons that can be easily carbonized for the synthesis of SIBs negative electrode material.34 Several researches have been carried out to explore the potential of WCG-synthesized hard-carbon as anode material for SIBs, WCGs-synthesized hard-carbon delivered excellent electrochemical performance ranging from 200–500 mA h g−1 attributed to the unique morphology and porosity of the WCG-synthesized hard-carbon which promotes sodium insertion.33–37 Hence utilization of WCGs for the synthesis of anode materials for SIBs not only aids the development of sustainable battery technology but also provides a sustainable solution for the complete disposal of this heavily generated environmental pollutant known as WCGs.
Although, SIBs energy density is largely influenced by several factors which include the composition of the electrode materials, electrolyte, separators and operating temperature. The deliverable capacity of biowaste-derived hard-carbon is greatly influenced by precursor composition and synthesis processes.23,38 Biowaste thermal treatment temperature has been identified as one of the main determinants in the synthesis of optimized hard-carbon structure, texture and morphology that enhances the electrochemical performance of the synthesized anode material for SIBs.39 However, the pre-treatment conditions such as sample preparation, pre-pyrolysis, and pre-oxidation before thermal treatment at this temperature directly influence the material structure through factors like the elemental oxygen, carbon and nitrogen compositions, inorganic impurities composition, and Pore Size Distribution (PSD) alongside the graphene and defect bands available in the hard-carbon.32,40 In this study, we investigated the effect of pre-treatment conditions such as just solvent washing (control condition), pre-oxidation and pre-pyrolysis on the electrochemical performance of coffee-waste-derived hard-carbon as anode material for SIBs. The control sample synthesised HC was labelled HC-1300 °C, the pre-oxidized sample synthesised HC was labelled HC-150 °C and the pre-pyrolyzed sample synthesised HC was labelled HC-600 °C.
2 Experimental section
2.1 Material synthesis from waste coffee grounds (WCGs)
The Waste Coffee Grounds (WCGs) were collected from the Nazarbayev University Health Project Coffee House (Astana, Kazakhstan) and dried in a natural convection laboratory oven (Carbolite Gero) at 80 °C for 24 hours for optimal moisture removal and effective yield calculation. These CGs were washed with D.I. water using a laboratory shaker at 100 rpm for 12 hours, followed by vacuum filtration and drying at 80 °C for 48 hours. The dried CGs were stored in air-tight containers.As illustrated in Fig. 1, the coffee-waste-derived hard carbons were synthesised using three different synthesis routes which involved three pre-treatment conditions.
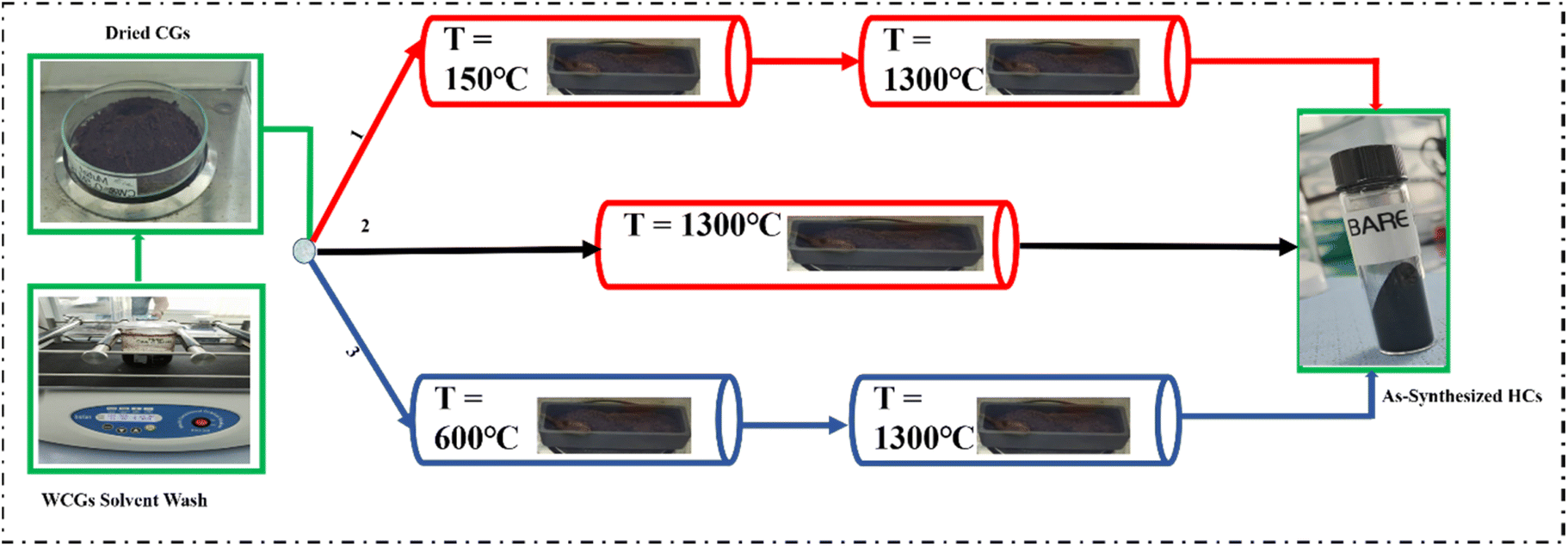 | ||
| Fig. 1 Coffee-waste-derived hard-carbon synthesis routes involving different pre-treatment conditions: (1) pre-oxidation, (2) control, (3) pre-pyrolysis. | ||
2.2 Characterization of the synthesized hard-carbons
The structural properties of the synthesized HCs were analyzed employing electron microscopy techniques, where Scanning Electron Microscopy (SEM) was carried out utilizing the ZEISS Crossbeam 540 to analyze the topography, morphology and surface structure of the as-synthesized HCs and High-Resolution Transmission Electron Microscopy (HR-TEM) was also carried out using JEOL JEM-1400 Plus operated at 120 kV to determine the interparticle structure, orientation and interlayer spacings of the as-synthesized HCs. Raman spectroscopy was used to analyze the graphene and defect bands of the as-synthesized HCs microstructure using the LabRAM Horiba spectrometer with the (532 wavelengths) He–Ne excitation source under ambient temperature and X-ray Diffraction (XRD) analysis with the aid of a Rigaku MiniFlex Benchtop X-ray Diffractometer equipped with Cu-Kα radiation (λ = 1.5405 Å) was used to evaluate the HCs' crystallinity, degree of graphitization and structural phase. The textural properties of the as-synthesized HCs were analyzed utilizing the nitrogen adsorption and desorption analysis, which was carried out using the Micromeritics 3Flex high-performance adsorption analyzer to evaluate the surface area using the Brunauer–Emmett–Teller (BET) theory and the Barrett–Joyner–Halenda (BJH) theory was used to determine the pore volume and pore size distribution.2.3 Electrochemical test of the synthesized anode materials
The negative electrodes of these HCs were obtained via slurries prepared using an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 mixing ratio of active materials (synthesized HCs):conductive material (carbon acetylene black):polymeric binder (polyvinylidene fluoride) all dissolved in NMP (N-methyl-2-pyrrolidone) and coated on Cu current collector and dried overnight at 60 °C in vacuum. The loading of the active materials was approximately 1.5–2 mg cm−2. The standard CR2032 coin cell was employed for the electrochemical analysis utilizing sodium chip, Whatman glass fibre and 1 M NaPF6 salt dissolved in EC:DMC:FEC (49:49:2) as a counter electrode, separator and electrolyte respectively, all these processes were carried out under argon environment in the glove box. The galvanostatic long-term charge–discharge and intermittent titration technique of the assembled coin cells were carried out using a Neware Battery Testing System to investigate the electrochemical performance at a potential window between 0.01 and 2.50 V (vs. Na+/Na) at a current density of 20 mA g−1. Also employing different current densities of 20 mA g−1, 50 mA g−1, 100 mA g−1, and 200 mA g−1 the influence of high-capacity load supply on the stability and deliverable capacity of the assembled coin cells was studied.
:10:10 mixing ratio of active materials (synthesized HCs):conductive material (carbon acetylene black):polymeric binder (polyvinylidene fluoride) all dissolved in NMP (N-methyl-2-pyrrolidone) and coated on Cu current collector and dried overnight at 60 °C in vacuum. The loading of the active materials was approximately 1.5–2 mg cm−2. The standard CR2032 coin cell was employed for the electrochemical analysis utilizing sodium chip, Whatman glass fibre and 1 M NaPF6 salt dissolved in EC:DMC:FEC (49:49:2) as a counter electrode, separator and electrolyte respectively, all these processes were carried out under argon environment in the glove box. The galvanostatic long-term charge–discharge and intermittent titration technique of the assembled coin cells were carried out using a Neware Battery Testing System to investigate the electrochemical performance at a potential window between 0.01 and 2.50 V (vs. Na+/Na) at a current density of 20 mA g−1. Also employing different current densities of 20 mA g−1, 50 mA g−1, 100 mA g−1, and 200 mA g−1 the influence of high-capacity load supply on the stability and deliverable capacity of the assembled coin cells was studied.
3 Results and discussion
3.1 Effects of pre-treatment conditions on the yield of the synthesized hard-carbons
The washing solvent (D.I water) along with the bio-waste composition remarkably influenced the yield of the as-synthesized HCs, similar to the reported values between 10 and 30% carbon yield for banana peel, apple peel, and rice husk-derived hard carbon,24,28,29 the WCG-derived hard carbon demonstrated an average yield between 21.5 and 24.5%, this is attributed to the WCG composition with about 25% lignin. Lignin composition along with the thermal treatment temperature significantly influenced the carbon yield, where an increase in lignin composition or the introduction of the lignin isolation process will directly increase the carbon yield to ca. 50% while an increase in the thermal treatment temperature significantly inhibits the carbon yield.29,41 The pre-treatment methods employed for each sample also play a notable role in the yield of the HC, where the control sample (HC-1300 °C) resulted in the highest yield of 24.5%. In comparison, the pre-oxidized sample (HC-150-1300 °C) demonstrated a 23% yield, and the pre-pyrolyzed sample (HC-600-1300 °C) experienced the lowest yield of 21.5%. The pre-treatment options and temperature directly influence the biomass composition especially that of the high carbon yield content (lignin), which is the most suitable precursor for HC.38 Pyrolysis on its own is one of the efficient ways for fast lignin degradation and washing post-pyrolysis removes more impurities, therefore this pre-treatment option demonstrates the lowest HC yield due to lignin degradation and other factors.42Furthermore, modification of the chemical properties, structure and lignin distribution in the pre-oxidized biomass directly influence the quantity of oxygen content formed during the oxidative process; at low pre-oxidation temperature, low oxygen and carbon functional groups are formed which in turn inhibits the quantity of HC obtained from high-temperature carbonization;43,44 High carbon yield is obtained when there are high oxygen and carbon functional groups and vice versa. This shows that pre-oxidation and pre-pyrolysis significantly influence the HC yield owing to lignin re-distribution and degradation during pre-oxidation and pre-pyrolysis respectively.
3.2 Synthesized hard-carbons characterization
 | ||
| Fig. 2 (a–c) SEM and (d–i) TEM images showing the effect of different pre-treatment conditions on the morphology of the synthesized HCs. | ||
TEM analysis was employed to further characterize the synthesized HCs microstructure, TEM images of the synthesized HCs show the typical hard carbon turbostratic structure with the graphite-like carbon domain revealing the degree of their amorphization.23,29,44 As shown in Fig. 2d–f, the pre-treatment condition also plays a major role in the degree of the synthesized HCs disorderliness, stacking and range of the carbon structures where the TEM analysis confirms the presence of long-range disordered carbon structure in HC-150-1300 °C and HC-1300 °C. Although, all the synthesized HCs possessed sufficient interlayer distance (d002 > 0.35 nm) for Na+ into the hard carbon structure, the pre-oxidized sample (HC-150-1300 °C) possessed the highest average interlayer spacing of 0.383 nm while the pre-pyrolyzed sample possessed the interlayer spacing of 0.380 nm with the control sample having an interlayer spacing of 0.378 nm. Furthermore, as seen in Fig. 2d–f, increase in interlayer spacing provides more spaces between carbon layers, which favours the movement and re-ordering of adjacent carbon atoms, which in turn enhance the degree of graphitization by favouring the formation of graphitic structure. Thus, pre-oxidation in air hinders the degree of graphitization due to the formation of flexible long-ranged microstructure with large interlayer distance leading to the formation of more pores and reduction of defects.4,23 Increase in the interlayer spacings are also attributable to the crystallite size and stacking orientation, which is observed in HC-600-1300 °C with low range crystallite size and stacking orientation, thereby increasing microstructure interlayer distance.32,40
Raman spectroscopy was employed to further analyse effects of different pre-treatment conditions on the microstructure and degree of orderliness of the synthesized HCs. The Raman Spectrum of all the synthesized HCs as shown in Fig. 3a confirmed the formation of hardcarbon structures, which were confirmed by the distinct and broad peaks around 1350 cm−1 and 1560 cm−1 assigned to the D-band and G-band of the carbon structure respectively.41,49 Although, similar Raman spectra are observed for all the synthesized HCs, the intensity ratio (ID/IG) of the defect induced and pseudographite bands varied across different pre-treatment conditions which directly influence carbon crystallite length (La). As seen in Fig. 3b the intensity ratios notably increase from a value of 0.79 to 0.92 and highest value of 0.94 for HC-150-1300 °C, HC-1300 °C and HC-600-1300 °C respectively. While the intensity ratios increase, the crystallite length significantly reduce with the pre-oxidized (HC-150-1300 °C) sample showing the highest crystallite length of 24.49 nm followed by control sample (HC-1300 °C) with La of 20.70 nm and pre-pyrolysed (HC-600-1300 °C) sample with the lowest value of 20.5 nm. All the synthesized HCs possessed low intensity ratio, with ID/IG value less than unity which suggest high degree of orderliness and graphitization (ID/IG > 1 indicates high intensity ratio which suggests high degree of defects and disorderliness) this further confirms the degree of the HCs' low defectiveness and their suitability as negative electrode materials due to their high degree of orderliness and porosity,50 HC-150-1300 °C shows higher variation in ID/IG and La, whereas close ID/IG and La values are observed in HC-1300 °C & HC-600-1300 °C, these show that high thermal treatment at a temperature of 1300 °C directly influence the low intensity ratio of approximately 0.92 while pre-oxidation in air at 150 °C further decreases the intensity ratio to about 0.79 owing to the increased graphitization of the HC-150-1300 °C while pre-pyrolysis at 600 °C slightly increase the low ID/IG value to about 0.94, this is due to the increased interlayer distance leading to formation turbostratic carbon.29,40
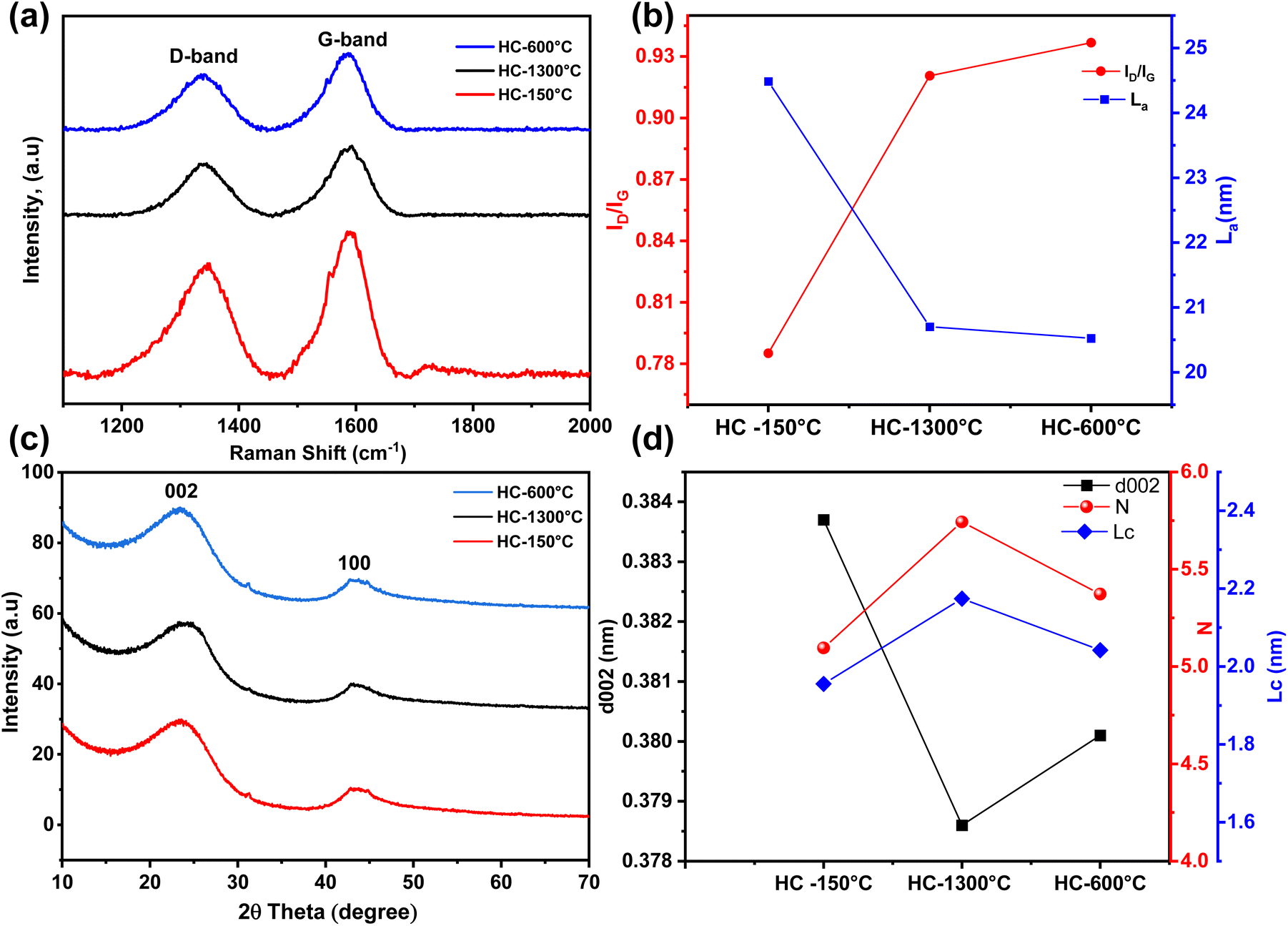 | ||
| Fig. 3 (a and b) Raman spectrum data; (c and d) XRD data and d002 spacing and stacking thickness. | ||
In addition, XRD analysis was carried out to further investigate the effect of these pre-treatment conditions on the microstructure of the synthesized HCs. As shown in Fig. 3c, the XRD diffractograms of the synthesized HCs revealed the typical disordered carbon peaks around 23° and 44° confirming the 002 and 100 planes of the graphene sheets respectively. Comparing the broad 002 peaks of the synthesized HCs, fitted 002 peaks of HC-1300 °C & HC-600-1300 °C occurred at higher angle of 23.47° & 23.38° compared to HC-150-1300 °C at a fitted angle of 23.15° which produces an interlayer distance (d002) of 0.384 nm, 0.379 nm and 0.380 nm as illustrated in Fig. 3d for HC-150-1300 °C, HC-1300 °C & HC-600-1300 °C respectively. Thus demonstrating adequate interlayer spacing for sodium ion insertion as drawn from the Raman & TEM analysis.51 Utilizing the Scherrer equation and ratio of crystallite size to interlayer distance, the synthesized HCs'crystallite sizes (Lc) and carbon sheet stacking order (N) are calculated from the estimated full width half maximum value of the fitted 002 peaks. As depicted in Fig. 3d, Lc and N increased across the samples with HC-1300 °C (control sample) demonstrating highest Lc & N of 2.2 nm and 5.7 respectively. Despite the varying interlayer distance and stacking orientation, all the synthesized HCs demonstrated low graphitization degree with a d002 spacing above 0.335 nm reported for graphite.23,40,50
Finally, nitrogen adsorption–desorption characterization revealed the effects of each pre-treatment condition on the textural property, interaction type, available surface for reaction and porosity of the synthesized HCs. As shown in Fig. 4a, all the synthesized HCs exhibit similar isotherm curves where low gas adsorption occurs at extremely low relative pressure and gradually increases to high adsorption at high relative pressure until it reaches it saturation point, this curve is quite identical to type II isotherm thus revealing the presence of micropores and mesopores owing to adsorption of nitrogen gas at low and high relative pressures respectively.52 As illustrated in Fig. 4b, the surface area of the synthesized HCs slightly varied across the samples, despite different treatment temperature, these pre-treatment conditions also influence the surface area to certain extent, where the control sample (HC-1300 °C) exhibited highest surface area of 17.59 m2 g−1 followed by the pre-oxidized sample (HC-150-1300 °C) with slightly lower surface area of 17.34 m2 g−1 and lastly the pre-pyrolyzed sample (HC-600-1300 °C) with the lowest surface area of 14.38 m2 g−1, thus confirming that pre-oxidation and pre-pyrolysis just like thermal treatment temperature also influence the BET surface area. The PSD as shown in Fig. 4c shows the porosity distribution while delving into the pore volume of these pore sizes, HC-150-1300 °C exhibited basically micro and mesopores majorly distributed within 1.5 nm to 35.1 nm, where over 17% of the PSD are micropores as suggested by the isotherm which reinstate the effect of pre-treatments on the PSD of the synthesized HCs. These pre-treatment conditions shrink the pore volume resulting in declining surface area for the pore sizes.53
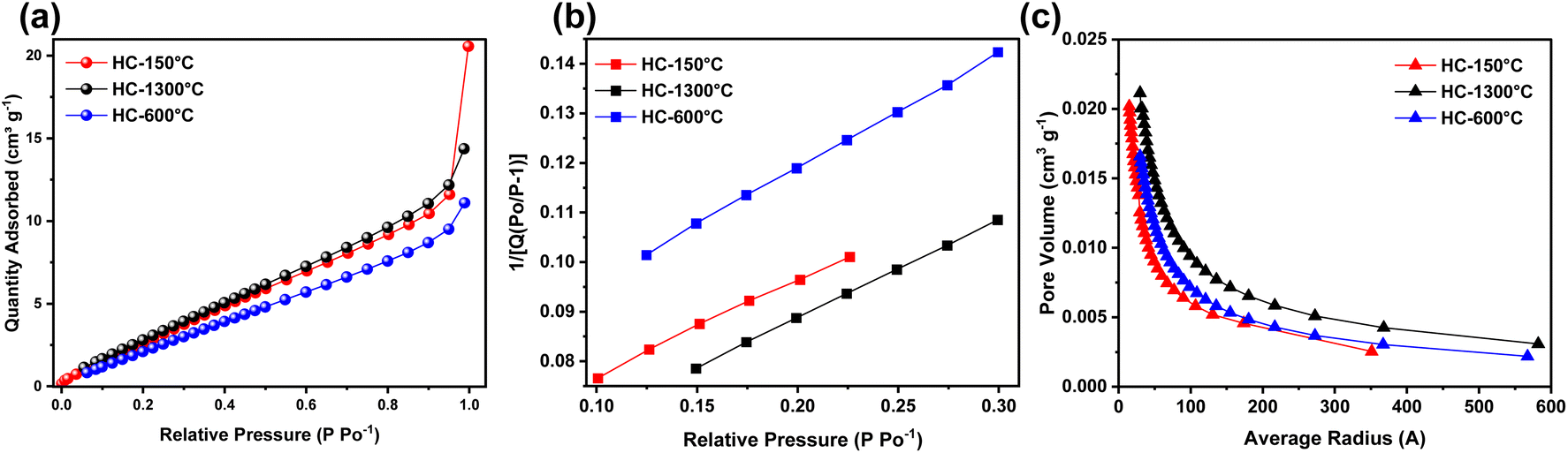 | ||
| Fig. 4 (a) Nitrogen adsorption isotherms, (b) BET isotherms and (c) BJH pore distribution of the synthesized HCs. | ||
3.3 Electrochemical performance of the synthesized hard-carbons
Employing the galvanostatic charge–discharge test, the electrochemical performance of the synthesized HCs as negative electrode material in half-cells sodium-ion batteries was investigated at a current density of 20 mA g−1 as shown in Fig. 5. An irreversible discharge capacity of 372, 339 and 299 mA h g−1 with ICE of 78, 74, and 75% was obtained from HC-150-1300 °C, HC-1300 °C and HC-600-1300 °C respectively, as the electrochemical test progresses, the 2nd cycle gave rise to a reversible capacity of 304, 261 and 250 mA h g−1 for the pre-oxidized, control and pre-pyrolysis HCs as depicted in Fig. 5a–c respectively. As illustrated in Fig. 5d, the control sample (HC-1300 °C) experienced the highest irreversible capacity loss of 78 mA h g−1 accompanied by the pre-oxidized sample (HC-150-1300 °C) with irreversible loss of 67 mA h g−1 and pre-pyrolyzed sample (HC-600-1300 °C) demonstrated the lowest irreversible capacity loss of about 49 mA h g−1, these losses are due to inability to recover all sodium ion stored in the electrode material owing the development of solid electrolyte interface (SEI) due to side reactions and sodium ion trapping owing to the presence of defect sites and degree of modified functional groups of the precursor.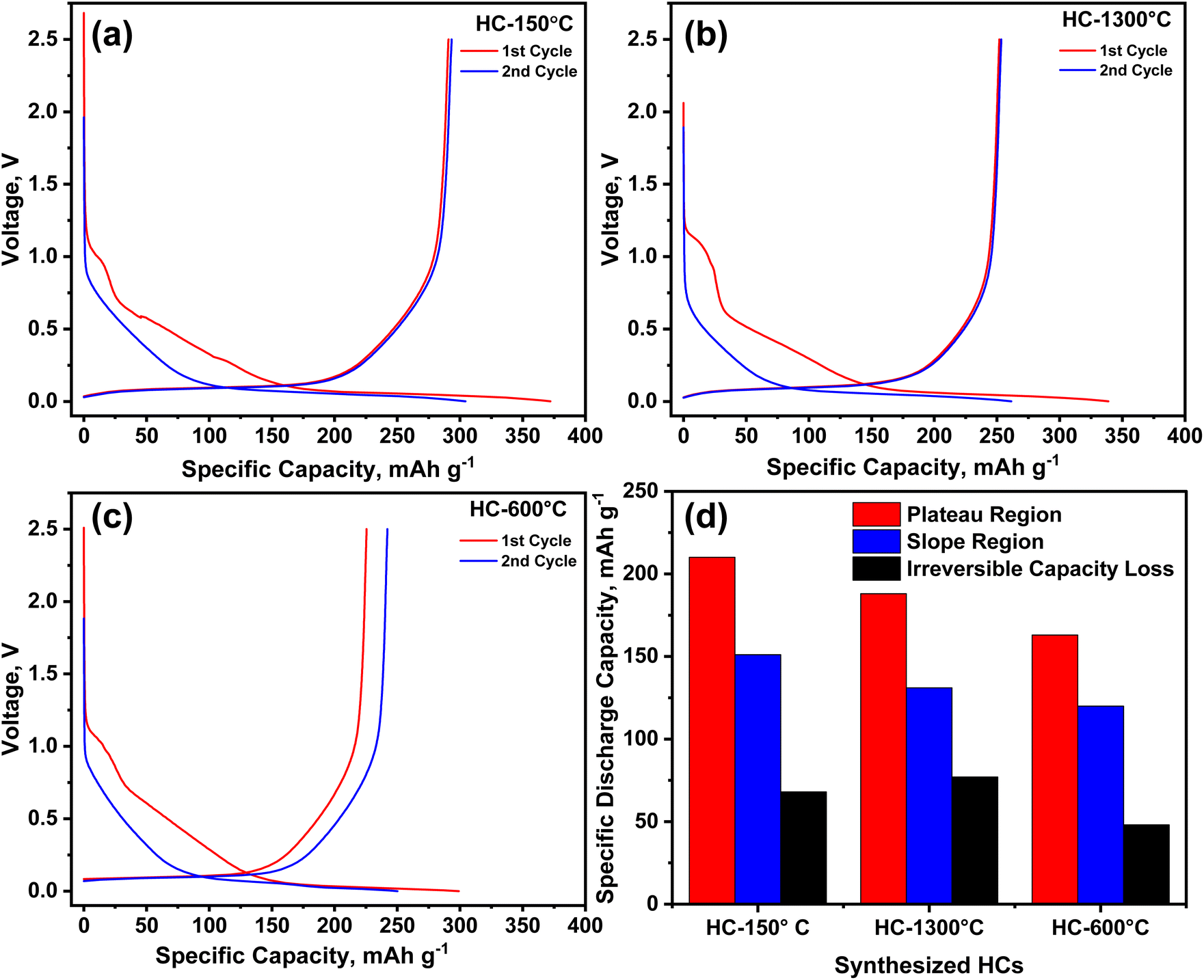 | ||
| Fig. 5 (a–c) 1st and 2nd galvanostatic charge–discharge curves and (d) capacity profile of the synthesized HCs from different pre-treatment conditions. | ||
Identical to the trend of the specific surface area as discussed earlier, since irreversible capacity are majorly influenced by surface adsorption by pores with surface area greater than 10 m2 g−1, it is worthy to note that pre-pyrolysis significantly reduce the irreversible capacity loss owing to it reduced surface area due to pore shrinkage.35,40 Pre-oxidation and pre-pyrolysis collapsed the defect sites through pore shrinking and inhibited irreversible capacity loss but in contrast promote capacity gain at the slope region as seen in Fig. 5d. HC-150-1300 °C & 600–1300 °C exhibit more capacity gain through sodiation process as a result of the large volume of micropores present in these samples.
HC-150-1300 °C demonstrated an excellent cycling stability and capacity retention 5 cycles amongst the synthesized HCs, as depicted in Fig. 6a, HC-600-1300 °C and 1300 °C experienced drastic capacity variation as the cycling progresses which led to the termination of these coin cells cycling at the end of the 15th cycle while HC-150-1300 °C cycling progresses till the 30th cycle with a capacity retention of over 95% and capacity of 291 mA h g−1. HC-150-1300 °C capacity degrades after 30th cycle as a result of the major bottlenecks in the application of hard carbons as anode material for SIBs which is the long-term cyclic stability. This is quite difficult to achieve when utilizing sodium chips as reference electrode owing to the development of SEI which influences the material volume and promotes ageing of the material and capacity degradation.40 C-rate analysis was carried out on HC-150-1300 °C to investigate the effect of current density/load on the stability and capacity degradation the electrode material. As illustrated in Fig. 6c, the cycling stability under different current densities between 20 mA g−1 to 200 mA g−1 was investigated. The pre-oxidized electrode delivered a reversible specific capacity of 279, 256, 217, 169 & 273 mA h g−1 with capacity retentions of 98, 97, 96, 95 & 95% at current densities of 20, 50, 100, 200 and 20 mA g−1 respectively. The capability of the coin cell to retain close to its initial capacity of 278 mA g−1 after cycling under different current rate as illustrated in Fig. 6c, shows the excellent cycling performance and suitability of the synthesized material as negative electrode material for SIBs. When it comes to cycling at higher current density of 50 mA g−1, the cell exhibited excellent performance retaining its 95% of capacity over 150 cycles, as depicted in Fig. 6d.
 | ||
| Fig. 6 (a) Cycling performance of synthesized HCs; (b) cycling at 20 mA g−1; (c) C-rate performance of HC-150 °C and (d) cycling at 50 mA g−1. | ||
These electrochemical performances show that the pre-treatment conditions majorly influence the capacity gain through the microstructure. Where the pre-oxidized sample demonstrated remarkable cycling performance compared to other samples. The pre-oxidation in air at 150 °C not only tune the hard carbon structure but also favors amorphization which enhances sodium ion intercalation into the graphene planes causing improvement in the capacity gain at the plateau region which is responsible for over 58% of its exceptional specific capacity of ca. 304 mA h g−1 but also reduces rate of capacity degradation owing to the formation of SEI by controlling the side reaction which in turn inhibits volume variation and material degradation.
4 Conclusions
Conclusively, this research focused on the effects of pre-oxidation in the air at a temperature of 150 °C and pre-pyrolysis at 600 °C on the microstructure, porosity and electrochemical performance of the anode materials synthesized at a thermal treatment temperature of 1300 °C from coffee-waste for sodium-ion batteries. These pre-treatment conditions significantly tuned the hard carbon microstructure and porosity to various extents. These pre-treatment temperatures were selected after carrying out trial experiments, where the synthesized HCs from pre-oxidation treatment showed maximum yield, reversible capacity and cycling stability at a pre-oxidation temperature of 150 °C. Compared to others obtained at 200 °C, 250 °C and 300 °C.Pre-pyrolysis was carried out at 600 °C to obtain the optimal removal of volatile organic elements, as this temperature is identified as the highest pyrolysis temperature for charring coffee waste in controlled environment. Past research as cited earlier, highlighted the effectiveness of moderate thermal treatment temperature of 1300 °C which produces quality hard-carbon with stable cycling performance as electrode material for SIBs, Hence the selection of 1300 °C as the carbonization temperature.
Although the synthesized materials demonstrated quite similar honeycomb structure for efficient electrolyte transport and ion diffusion, the porosity varies with the pre-treatment type.
Pre-pyrolysis played huge role in increasing the surface area and porosity, but the synthesized electrode experienced a short fall with the pre-oxidation in air at 150 °C demonstrated a remarkable specific capacity of 304 mA h g−1 with an ICE of 78% and capacity retention of over 95% after 30 cycles at a current load of 20 mA g−1. Where pre-oxidation in air played a significant role in promoting amorphization which enhances interlayer spacing, thereby promoting plateau capacity (responsible for a significant portion of the reversible capacity) and inhibiting irreversible capacity loss through a reduced surface area while providing defect sites and pore shrinkage for sodiation reaction. Improving the pre-oxidation temperature and time could provide room for electrochemical improvements and facilitate the development of reliable and high-capacity electrodes for SIBs from coffee grounds.
Data availability
All data supporting findings of this study are available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Ministry of Science and Higher Education of the Republic of Kazakhstan Grant (AP22783602) and by Nazarbayev University under faculty-development competitive research grants program for 2024–2026 Grant No. 201223FD8802, AK.References
- H. Moon, A. Innocenti, H. Liu, H. Zhang, M. Weil, M. Zarrabeitia and S. Passerini, ChemSusChem, 2023, 16, e202201713 CrossRef CAS.
- C. Chen, D. Yan, Y. Von Lim, L. Liu, X. L. Li, J. Chen, T. C. Li, Y. Zhu, J. Cai, Y. Huang, Y. Zhang and H. Y. Yang, Carbon Energy, 2024, 6, e482 CrossRef CAS.
- Y. Yang, C. Wu, X.-X. He, J. Zhao, Z. Yang, L. Li, X. Wu, L. Li and S.-L. Chou, Adv. Funct. Mater., 2024, 34, 2302277 CrossRef CAS.
- P. Liu, Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2016, 4, 13046–13052 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- A. Rudola, C. J. Wright and J. Barker, Energy Mater. Adv., 2021, 2021 DOI:10.34133/2021/9798460.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- Y. Fang, L. Xiao, J. Qian, Y. Cao, X. Ai, Y. Huang and H. Yang, Adv. Energy Mater., 2016, 6, 1502197 CrossRef.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- H. Hou, X. Qiu, W. Wei, Y. Zhang and X. Ji, Adv. Energy Mater., 2017, 7, 1602898 CrossRef.
- Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2015, 4, 96–104 RSC.
- R. Dong, F. Wu, Y. Bai, Q. Li, X. Yu, Y. Li, Q. Ni and C. Wu, Energy Mater. Adv., 2022, 2022 DOI:10.34133/2022/9896218.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- J. C. Hyun, H. M. Jin, J. H. Kwak, S. Ha, D. H. Kang, H. S. Kim, S. Kim, M. Park, C. Y. Kim, J. Yoon, J. S. Park, J.-Y. Kim, H.-D. Lim, S. Y. Cho, H.-J. Jin and Y. S. Yun, Energy Environ. Sci., 2024, 17, 2856–2863 RSC.
- Y. Aniskevich, J. H. Yu, J.-Y. Kim, S. Komaba and S.-T. Myung, Adv. Energy Mater., 2024, 14, 2304300 CrossRef CAS.
- A. Kamiyama, K. Kubota, D. Igarashi, Y. Youn, Y. Tateyama, H. Ando, K. Gotoh and S. Komaba, Angew. Chem., Int. Ed., 2021, 60, 5114–5120 CrossRef CAS.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007 RSC.
- X. Chen, J. Tian, P. Li, Y. Fang, Y. Fang, X. Liang, J. Feng, J. Dong, X. Ai, H. Yang and Y. Cao, Adv. Energy Mater., 2022, 12, 2200886 CrossRef CAS.
- A. Kamiyama, K. Kubota, T. Nakano, S. Fujimura, S. Shiraishi, H. Tsukada and S. Komaba, ACS Appl. Energy Mater., 2020, 3, 135–140 CrossRef CAS.
- A. Baldinelli, X. Dou, D. Buchholz, M. Marinaro, S. Passerini and L. Barelli, Green Chem., 2018, 20, 1527–1537 RSC.
- K. Kubota, S. Shimadzu, N. Yabuuchi, S. Tominaka, S. Shiraishi, M. Abreu-Sepulveda, A. Manivannan, K. Gotoh, M. Fukunishi, M. Dahbi and S. Komaba, Chem. Mater., 2020, 32, 2961–2977 CrossRef CAS.
- L. Wu, D. Buchholz, C. Vaalma, G. A. Giffin and S. Passerini, ChemElectroChem, 2016, 3, 292–298 CrossRef CAS.
- K. Hong, L. Qie, R. Zeng, Z. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q. Fan, W. Zhang and Y. Huang, J. Mater. Chem. A, 2014, 2, 12733–12738 RSC.
- K. Wang, Y. Jin, S. Sun, Y. Huang, J. Peng, J. Luo, Q. Zhang, Y. Qiu, C. Fang and J. Han, ACS Omega, 2017, 2, 1687–1695 CrossRef CAS PubMed.
- Y. Cheng, J. Zhao, L. Zhang, J. Wan, J. Yang and H. Wang, Solid State Ionics, 2023, 402, 116374 CrossRef CAS.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- M. K. Rybarczyk, Y. Li, M. Qiao, Y.-S. Hu, M.-M. Titirici and M. Lieder, J. Energy Chem., 2019, 29, 17–22 CrossRef.
- M. Dahbi, M. Kiso, K. Kubota, T. Horiba, T. Chafik, K. Hida, T. Matsuyama and S. Komaba, J. Mater. Chem. A, 2017, 5, 9917–9928 RSC.
- I. Izanzar, M. Dahbi, M. Kiso, S. Doubaji, S. Komaba and I. Saadoune, Carbon, 2018, 137, 165–173 CrossRef CAS.
- A. Beda, J.-M. Le Meins, P.-L. Taberna, P. Simon and C. Matei Ghimbeu, Sustain. Mater. Technol., 2020, 26, e00227 CAS.
- Y. Liu, D. J. Lee, H.-J. Ahn, S. Y. Nam, K.-K. Cho and J.-H. Ahn, Renew. Energy, 2023, 212, 865–874 CrossRef CAS.
- P.-H. Chiang, S.-F. Liu, Y.-H. Hung, H. Tseng, C.-H. Guo and H.-Y. Chen, Energy Fuels, 2020, 34, 7666–7675 CrossRef CAS.
- M. Kalibek, L. Rakhymbay, Z. Zhakiyeva, Z. Bakenov, S.-T. Myung and A. Konarov, Carbon Resour. Convers., 2024, 7, 100225 CrossRef CAS.
- S. J. Hong, S. S. Kim and S. Nam, Corrosion Sci. Technol., 2021, 20, 15–21 CAS.
- G. Gao, L.-Z. Cheong, D. Wang and C. Shen, Carbon Resour. Convers., 2018, 1, 104–108 CrossRef.
- X. Lin, Y. Liu, H. Tan and B. Zhang, Carbon, 2020, 157, 316–323 CrossRef CAS.
- Y. Jin, S. Sun, M. Ou, Y. Liu, C. Fan, X. Sun, J. Peng, Y. Li, Y. Qiu, P. Wei, Z. Deng, Y. Xu, J. Han and Y. Huang, ACS Appl. Energy Mater., 2018, 1, 2295–2305 CrossRef CAS.
- N. Daher, D. Huo, C. Davoisne, P. Meunier and R. Janot, ACS Appl. Energy Mater., 2020, 3, 6501–6510 CrossRef CAS.
- L. Rakhymbay, N. Bazybek, K. Kudaibergenov, S.-T. Myung, Z. Bakenov and A. Konarov, J. Power Sources, 2024, 602, 234347 CrossRef CAS.
- T. Han, N. Sophonrat, A. Tagami, O. Sevastyanova, P. Mellin and W. Yang, Fuel, 2019, 235, 1061–1069 CrossRef CAS.
- P. Kaparaju and C. Felby, Bioresour. Technol., 2010, 101, 3175–3181 CrossRef CAS PubMed.
- G. Jia, Z. Zhou, Q. Wang, M. T. Innocent, S. Wang, Z. Hu, X. Wang, H. Xiang and M. Zhu, Int. J. Biol. Macromol., 2022, 216, 388–396 CrossRef CAS PubMed.
- E. W. Gabisa and C. Ratanatamskul, Sci. Rep., 2024, 14, 10811 CrossRef CAS PubMed.
- J. H. Um, Y. Kim, C.-Y. Ahn, J. Kim, Y.-E. Sung, Y.-H. Cho, S.-S. Kim and W.-S. Yoon, J. Electrochem. Sci. Technol., 2018, 9, 163–168 CrossRef CAS.
- X.-S. Wu, X.-L. Dong, B.-Y. Wang, J.-L. Xia and W.-C. Li, Renew. Energy, 2022, 189, 630–638 CrossRef CAS.
- S. Alvin, D. Yoon, C. Chandra, H. S. Cahyadi, J.-H. Park, W. Chang, K. Y. Chung and J. Kim, Carbon, 2019, 145, 67–81 CrossRef CAS.
- Z. Zheng, S. Hu, W. Yin, J. Peng, R. Wang, J. Jin, B. He, Y. Gong, H. Wang and H. J. Fan, Adv. Energy Mater., 2024, 14, 2303064 CrossRef CAS.
- T. Meenatchi, V. Priyanka, R. Subadevi, W.-R. Liu, C.-H. Huang and M. Sivakumar, Carbon Lett., 2021, 31, 1033–1039 CrossRef.
- Y. Du, Y. Qiu, R. Zhuang, X. Jing, D. Liu, X. Peng, L. Yan and F. Xu, Carbon, 2024, 221, 118929 CrossRef CAS.
- F. Wu, M. Zhang, Y. Bai, X. Wang, R. Dong and C. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 12554–12561 CrossRef CAS.
- Y.-F. Du, G.-H. Sun, Y. Li, J.-Y. Cheng, J.-P. Chen, G. Song, Q.-Q. Kong, L.-J. Xie and C.-M. Chen, Carbon, 2021, 178, 243–255 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |