
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanosized MCM-41 silica from rice husk and its application for the removal of organic dyes from water†
Giorgio Celoriaa,
Federico Begnia,
Geo Paul a,
Stefano Marchesia,
Enrico Boccalerib,
Chiara Bisio*ac and
Leonardo Marchesea
a,
Stefano Marchesia,
Enrico Boccalerib,
Chiara Bisio*ac and
Leonardo Marchesea
aDipartimento di Scienze e Innovazione Tecnologica, Università Del Piemonte Orientale A. Avogadro, Viale T. Michel 11, 15121 Alessandria, Italy. E-mail: giorgio.celoria@uniupo.it; federico.begni@uniupo.it; geo.paul@uniupo.it; stefano.marchesi@uniupo.it; chiara.bisio@uniupo.it; leonardo.marchese@uniupo.it
bDipartimento per Lo Sviluppo Sostenibile e La Transizione Ecologica, Università Del Piemonte Orientale A. Avogadro, Piazza S. Eusebio 5, 13100 Vercelli, Italy. E-mail: enrico.boccaleri@uniupo.it
cCNR-SCITEC Istituto di Scienze e Tecnologie Chimiche “Giulio Natta”, Via G. Venezian 21, 20133 Milano, Italy
First published on 24th January 2025
Abstract
A novel synthesis of a nanometric MCM-41 from biogenic silica obtained from rice husk is here presented. CTABr and Pluronic F127 surfactants were employed as templating agents to promote the formation of a long-range ordered 2D-hexagonal structure with cylindrical pores and to limit the particle growth at the nanoscale level thus resulting in a material with uniform particle size of 20–30 nm. The physico-chemical properties of this sample (RH-nanoMCM) were investigated through a multi-technique approach, including PXRD, 29Si MAS NMR, TEM, Z-potential and N2 physisorption analysis at 77 K. The results were compared to those of a nanometric MCM-41 synthesized from a silicon alkoxide precursor. The adsorption capacity of RH-nanoMCM towards the cationic dye rhodamine B from aqueous phase was investigated at different initial dye concentrations by means of UV-vis spectroscopy. Insight into the non-covalent interactions between the dye molecules and the adsorbent surface was gained by means of 1H and 13C MAS NMR spectroscopy and FT-IR spectroscopy.
1 Introduction
In the last two decades, the research into new nanomaterials for environmental, catalytic and biomedical applications has undergone rapid growth.1–5 Thanks to their excellent thermal and chemical stability, surface areas around 1000 m2 g−1, channels with a diameter of ca. 2–10 nm, pore volume higher than 0.90 cm3 g−1 and tuneable surface and morphological properties, mesoporous silicas are still considered as some of the most porous materials to be exploited for the aforementioned applications.3,5–8 MCM-41 silica belongs to the M41S family,3 and it is characterized by a long-range ordered 2D-hexagonal structure with cylindrical pores and pore walls of amorphous silica.1,9,10 This material is usually prepared through an aqueous sol–gel synthesis from an alkoxysilane, that hydrolyses and condenses in the presence of a cationic surfactant.2,9,11In 2004, K. Suzuki et al.10 demonstrated that, by simultaneously using a cationic surfactant as a templating agent and a non-ionic surfactant to limit the particle growth, an MCM-41 with particle size dimensions below 50 nm could be successfully synthesized. The nanosized silica particles thus obtained can provide several advantages in many application fields.4,12 In fact, such silicas are characterized by pores with short length, and this allows easier diffusion of guest molecules as well as active sites accessibility.12 R. Mokaya and coauthors13 demonstrated that the catalytic activity of Al-MCM-41 increases as the particle size decreases, in that the diffusion of the reactant and product molecules was improved. Nanometric mesoporous silicas could be efficiently employed in biomedical applications,10 as the drug loading was more easily controlled. Gatti et al.14 demonstrated that the CO2 adsorption capacity at 35 °C of MCM-41 functionalized with amino groups increases as the particle size decreases.
Regardless of the multiple advantages related to nanometric mesoporous silicas, it has to be taken into account that the large-scale production of these materials could be limited due to the costs of chemicals, such as the alkoxysilane precursors.15,16 For this reason, research efforts have been recently focused on more sustainable and cost-effective processes. The use of biogenic silica extracted from rice husk instead of alkoxysilanes for the preparation of silicon-based materials goes in this direction. For instance, non-porous silica nanoparticles, silicon nitrides and carbides and porous silicas with tailored textural and morphological properties, such as MCM-41, MCM-48, SBA-15 and SBA-16, have been prepared.16–20
Rice husk is the external hull that covers the rice grain and it is made up of lignin, cellulose, hemicellulose and silica ash, but it can also contain metal oxides impurities, whose presence is related to the soil properties.16–19 Silica ash represents 10–20 wt% of rice husk and is made of amorphous silica.16
The aim of this work is to propose a novel cost-effective and environmentally friendly synthesis to prepare a nanosized MCM-41 from biogenic silica derived from rice husk. To the best of our knowledge, the synthesis of mesoporous MCM-41 silica with particle dimensions below 50 nm from rice husk is not reported in the literature. Indeed, recent studies regarding the synthesis of mesoporous silicas from rice husk are related to the preparation of silicas with particle dimensions not below 200 nm.16–19 In this work, the synthesis method proposed by K. Suzuki,10 related to the use double-surfactant approach to obtain nanosized MCM-41 from tetraethyl orthosilicate (TEOS), was properly modified to allow the use of biogenic silica from rice husk as silicon source. From our literature survey, this double-surfactant method is here applied for the first time to the preparation of nanometric MCM-41 from wastes.
The quality of waterbodies has suffered a sharp decline due to the continuous spreading of water-soluble pollutants, such as toxic dyes.21–25 These molecules, in particular organic dyes, represent an emerging class of water contaminants, whose presence in water basins is of growing concern due to their adverse effect on the local environment and mankind.26 Among the different adsorbent materials, mesoporous silicas (e.g. MCM-41) have demonstrated to be excellent candidates for the adsorption of cationic molecules from polluted water.21,22,27
On the light of these considerations, we have proposed RH-nanoMCM as a cost-effective and more environmentally friendly mesoporous sorbent for the removal of pollutants. In this study, rhodamine B was used as a model molecule of a water-soluble cationic dye.27 Moreover, MAS-NMR and FT-IR spectroscopy were employed to gain additional information about the noncovalent interactions at play between the adsorbate molecules and the adsorbent surface.28 To the best of our knowledge, this aspect is not deeply investigated in the literature though of relevance for improving the adsorption performances.
2 Experimental section
2.1 Materials
HCl 37% w/w (Sigma Aldrich®, Steinheim, Germany), sodium hydroxide (NaOH, CAS: 1310-73-2, M.W. 39.99 g mol−1, Merck KGsA, Darmstadt, Germany), cetyltrimethylammonium bromide (CTABr, C19H42BrN, CAS: 57-09-0, M.W. 364.45 g mol−1, Merck KGsA, Darmstadt, Germany), Pluronic F127 (C3H6O·C2H4OX, CAS: 9003-11-6, Merck KGsA, Darmstadt, Germany) and tetraethyl orthosilicate (TEOS, Si(OC2H5)4, CAS: 78-10-4, M.W. 208.33 g mol−1, Merck KGsA, Darmstadt, Germany) were the chemicals used for the synthesis.2.2 Synthesis of RH-nanoMCM
Rice husk (RH) was supplied by a local farm located in Novara (Piedmont, Italy). Before calcination, in order to remove dust, about 20 g of RH were washed in 500 mL of deionized water for 15 min in 1 L Becher under vigorous stirring. The wet RH was dried overnight at 70 °C. Next, the dried RH was calcined in air at 600 °C for 2 h in a muffle furnace to obtain a white powder named rice husk ashes (RHA). For each gram of RH, ca. 120 mg of RHA were obtained. Next, to remove traces of metal impurities and to obtain a high-purity siliceous precursor, 2 g of RHA were transferred inside a round-bottom flask and were stirred with 200 mL of a 2 mol L−1 HCl solution for 5 h at RT. Finally, the powder was recovered through Buchner filtration and copiously washed with deionized water until pH neutralization and dried overnight.To prepare the sodium silicates solution, 1 g of acid washed RHA were stirred with 1.70 g of NaOH and 30 mL of deionized H2O inside a round-bottom flask for 24 h at 70 °C. At the end of the procedure, a dark suspension was obtained, whose colour was mainly due to traces of suspended carbonaceous residues derived from the calcination of RH. The carbonaceous residues were separated from the sodium silicate solution in this way: the suspension was equally divided into two conical-bottom polypropylene centrifugation tubes, that were centrifugated at 7000 rpm for 4 min. The supernatant (i.e., the silicates solution) was withdrawn with a disposable Pasteur pipette and transferred inside a 100 mL Becher. At this point, 2.6 g of CTABr and 2 g of Pluronic F127 were added to the sodium silicate solution that was carefully stirred until the complete dissolution of the surfactants. Next, the pH was brought to 10 by means of multiple additions of glacial acetic acid and a white suspension was obtained. Subsequently, the gel was carefully stirred for 24 h at RT. Finally, the gel was transferred in a 100 mL sealed PTFE bottle and incubated for 96 h at 60 °C. After the incubation period, the solid was recovered by means of Buchner filtration and copiously washed with deionized water and ethanol to remove the surfactant residues. The sample was then dried for 6 h at 70 °C and calcined at 600 °C for 6 h to remove the surfactants (heating ramp of 2 °C min−1 and a cooling ramp of 20 °C min−1). At the end of the procedure, 0.98 g of material were obtained. The sample was coded as RH-nanoMCM. A scheme of the synthetic procedure of RH-nanoMCM is shown in Fig. 1.
 | ||
| Fig. 1 Graphical representation of the main steps of the synthesis of RH-nanoMCM from rice husk. | ||
2.3 Synthesis of MCM-41 nano
According to the procedure reported by Suzuki et al.,10 a nanometric-sized MCM-41 to be used as reference material was synthesized from an alkoxysilane precursor. In this procedure, 2.6 g of CTAC and 2 g of Pluronic F127 were completely dissolved in 30 g of an HCl 0.3 N solution in a 250 mL round-bottom flask. The solution was carefully stirred at RT until the complete dissolution of the surfactants. Next, 3.75 mL of TEOS were added dropwise and the solution was stirred for 3 h. Subsequently, 3 g of ammonium hydroxide were added to the previously prepared solution, that was stirred at RT for 24 h. Finally, the gel was transferred in a 250 mL sealed PTFE bottle, which was kept in an oven at 60 °C for 24 h. After the incubation time, the gel was recovered through Buchner filtration and copiously washed with deionized water and ethanol until the removal of surfactants residues. The resulting solid was dried at 80 °C overnight. Finally, the material was calcined at 600 °C for 6 h, with a heating ramp of 2 °C min−1 and a cooling ramp of 20 °C min−1. At the end of the procedure, 0.88 g of material were obtained. The sample was named MCM-41 nano.2.4 Characterization methods
The PXRD patterns of the different materials were obtained with a Bruker D8 advance diffractometer (Karlsruhe, Germany) with Bragg–Brentano geometry, with Cu anode (λ = 1.5418 Å) equipped with a Ni filter and operating at 40 kV and 40 mA. The 2θ explored was of 10–50°, with 2θ steps of 0.02°, 0.1 s per step and automatic synchronization of the anti-scatter knife for the rice husk ashes samples and of 0.8–10°, with 2θ steps of 0.02°, 0.3 s per step and the anti-scatter knife fixed at 0.8 mm for the MCM-41 and rice husk ashes samples.The different TEM micrographs were obtained with a JEOL JEM-3010 UHR TEM microscope (JEOL Ltd, Tokyo, Japan) operating at an acceleration voltage of 300 kV. Prior to the measurements, the samples were prepared by depositing the powder over a lacey-carbon copper grid.
Solid-state NMR spectra were acquired on a Bruker Avance III 500 spectrometer and a wide bore 11.74 Tesla magnet with operational frequencies for 29Si and 1H of 99.35 and 500.13 MHz, respectively. A 4 mm triple resonance probe with MAS capabilities and zirconia rotors were employed in all the experiments. The magnitude of radio frequency (RF) fields was 100 and 50 kHz for 1H and 29Si, respectively. For the 13C CPMAS experiments, the magnetic fields υHrf of 55 and 28 kHz were used for initial excitation and decoupling, respectively. During the CP period the 1H RF field υHrf was ramped using 100 increments, whereas the 13C RF field υCrf was maintained at a constant level. During the acquisition, the protons are decoupled from the carbons by using a TPPM decoupling scheme. A moderate ramped RF field υHrf of 62 kHz was used for spin locking, while the carbon RF field υCrf was matched to obtain optimal signal and the CP contact time of 5 ms was used. The proton spin-lattice relaxation time, T1, has been determined using saturation recovery method. The relaxation delay between accumulations was 60 and 5 s for 29Si and 1H, respectively. All chemical shifts are reported using δ scale and are externally referenced to tetramethylsilane (TMS) at 0 ppm.
The FT-IR spectra of the different samples were obtained from a Thermo Electron Corporation (Whaltham, MA, USA) FT Nicolet 5700 spectrometer with 4 cm−1 resolution. The self-supported pellets were prepared with a mechanical press at ca. 5 tons per g and were placed into an IR cell with a KBr window, which was permanently attached to a vacuum line (residual pressure <10−3 mbar). Prior to the measurements, the samples were treated at beam temperature (35 °C) under vacuum (residual pressure <10−3 mbar) for 3 hours to remove residual traces of water.
Z-potential experiments were carried out on the different materials by using a Malvern Zetasizer NanoZS (Malvern Panalytical, Malvern, UK). Typically, 10 mg of powder were dispersed in 10 mL of deionized water and a 0.01 mol L−1 HCl (37% w/w (Sigma Aldrich®)) solution and a 0.01 mol L−1 NaOH solution (Sigma Aldrich®) were used to vary the pH between 2 to 11. Each measurement was repeated three times in the same conditions. A capillary cell (supplied with the Malvern instrument) was used to carry out the Z potential measurements, and a disposable Pasteur pipette was employed to fill up the cell.
The N2 physisorption measurements between 10−4 and 1 P/P0 at 77 K were performed using a Quantachrome Autosorb iQ2. Prior to adsorption, the samples were outgassed and treated for 10 h at 150 °C, under vacuum. The specific surface area (SSA) of the samples was determined by the Brunauer–Emmett–Teller (BET) multipoint method while the pore size distributions was obtained by applying the NLDFT method on the adsorption branch of the isotherms (silicas and cylinder pore model).
The rhodamine B adsorption was investigated through UV-vis spectroscopy using a Lambda 900 UV-visible spectrometer (PerkinElmer, Waltham, MA, USA). A calibration line was obtained using five different rhodamine B standard solutions: 2.0 × 10−2, 1.0 × 10−2, 6.6 × 10−3, 5.0 × 10−3 and 3.3 × 10−3 mmol L−1. The obtained equation of the calibration line was: A = 101.6 × C with an R2 of 0.9999. The different adsorption measurements were performed at room temperature (ca. 25 °C) placing 25 mg of adsorbent material in 20 mL of rhodamine B solutions at different concentrations (2.0 × 10−2, 1.0 × 10−2, 5.0 × 10−3, 2.5 × 10−3 and 1.2 × 10−3 mmol L−1) inside 50 mL conical-bottom polypropylene centrifugation tubes (Corning®, New York, USA), that were kept under stirring at 300 rpm using a magnetic stirrer. After 20 min, the centrifugation tubes were spun at 8000 rpm for 5 min and aliquots of about 3 mL of clear rhodamine B solution were and introduced inside a quartz cuvette to record the UV-vis spectrum. The same procedures were performed three times for each mesoporous silicas after a contact time of 20 min with the different rhodamine B solutions and the standard deviations over three measurements were obtained.
3 Results and discussion
A comparison of the properties of mesoporous silica samples prepared from rice husk ashes (RH-nanoMCM) and TEOS (MCM-41 nano), respectively, is done in the following starting with the PXRD analysis (Fig. 2). Moreover, an extended physico-chemical characterization of rice husk and rice husk ashes before and after the acid treatment, along with the 29Si NMR spectrum of the sodium silicate solution is reported in the ESI (see Fig. S1–S4†).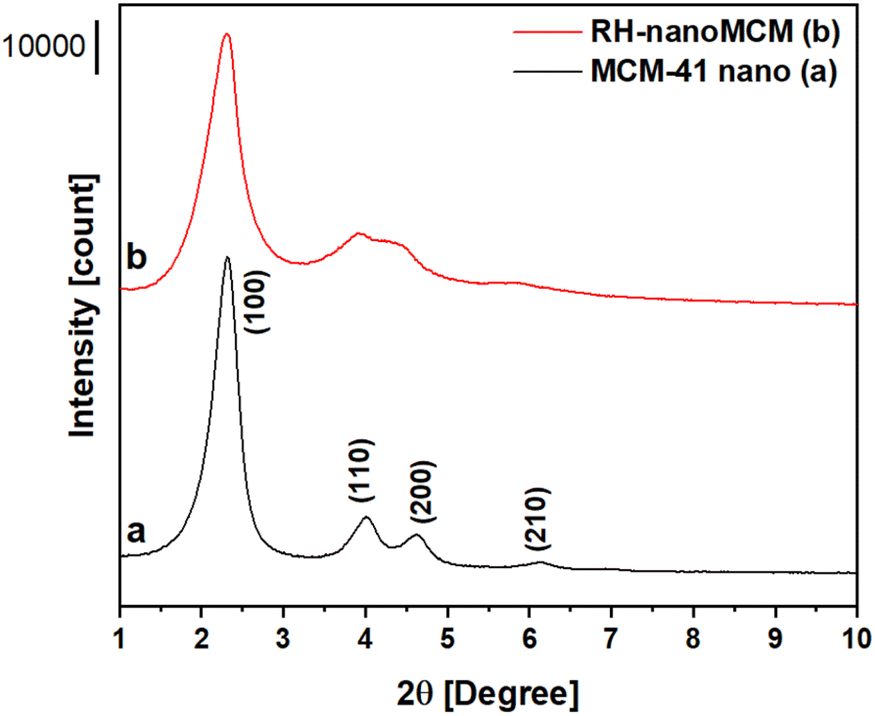 | ||
| Fig. 2 Low-angle PXRD patterns of MCM-41 nanometric (a) and RH-nanoMCM (b). | ||
The RH-nanoMCM shows the presence of four different reflections at ca. 2.3°, 3.7°, 4.2° and 5.8° 2θ, assigned to the (100), (110), (200) and (210) planes, respectively. These reflections are due to a long-range ordered 2D hexagonal structure with cylindrical pores, typical of MCM-41 materials.10,14,29 Though the peaks are slightly larger, the PXRD pattern of RH-nanoMCM is similar to that of the reference sample.
The TEM micrographs (Fig. 3) reveal that, thanks to the presence of F127 micelles, the particle growth of both RH-nanoMCM and the nanometric MCM-41 samples has been limited at the nanoscale level10 (average diameter of about 20–30 nm).
 | ||
Fig. 3 TEM micrographs of RH-nanoMCM (frames A, A′) and of MCM-41 nano (frames B, B′) at magnifications of 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00× (top images) and 100000× (bottom images). 00× (top images) and 100000× (bottom images). | ||
As already reported, the nanoparticles are still characterized by hexagonally ordered mesopores.
The structure of the silica framework was studied by 29Si MAS-NMR spectroscopy (Fig. 4). The spectra of the RH-nanoMCM and MCM-41 nano samples show three signals at −110, −100 and −90 ppm, which are due to Q4 [Si(OSi)4], Q3 [Si(OH)(OSi)3] and Q2 [Si(OSi)2(OH)2] Si nuclei.30,31 These results suggested that the two types of nano MCM-41 samples show a similar distribution of Si sites within the mesoporous silica framework.
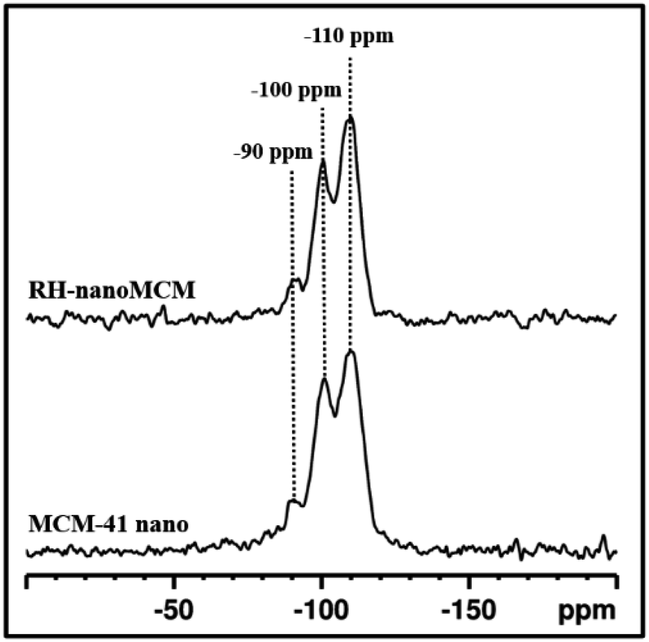 | ||
| Fig. 4 29Si MAS NMR spectra of RH-nanoMCM and MCM-41 nano. | ||
The textural properties of both RH-nanoMCM and nano MCM-41 were studied by N2 physisorption at 77 K. The adsorption/desorption isotherms and the calculated NLDFT pore size distributions of both samples are shown in Fig. 5, whereas the SSABET and the pore volumes of the samples are reported in Table 1.
 | ||
| Fig. 5 N2 adsorption–desorption isotherms at 77 K (frame A) and NLDF pore size (frame B) of RH-nanoMCM (triangles) and MCM-41 nano (circles). | ||
| SSABET [m2 g−1] | Pore volume [cm3 Å−1 g−1] | ||||
|---|---|---|---|---|---|
| Vmicro [<20 Å] | Vmeso [20–500 Å] | Vmacro [>500 Å] | Vtot | ||
| RH-nanoMCM | 1050 | — | 1.57 | 0.09 | 1.66 |
| MCM-41 nano | 900 | — | 1.35 | 0.33 | 1.68 |
Both materials show N2 physisorption isotherms corresponding to a IVa model of the IUPAC classification, typical of mesoporous materials, i.e., when the gas condensation is accompanied by hysteresis, that usually occurs in the presence of pores wider than 40 Å.32 The isotherm of the RH-nanoMCM (Fig. 5A) is characterized by H1 and H3 hysteresis loops at ca. 0.35 and 0.9 P/P0, respectively.32 H1 hysteresis loop, typical of mesoporous materials with cylindrical pores (i.e., MCM-41), is a strong indication of the presence of uniform mesopores, in the case of the RH-nanoMCM sample the pore diameter is 42 Å.32 The H3 hysteresis loop is typically formed in presence of large mesopores and/or macropores, arising from non-rigid aggregates where gas condensation between nanometric-size particles occurs.32 H1 hysteresis loop is missing in the N2 physisorption isotherm of MCM-41 nano because this sample displays mesopores centred at 37 Å. However, H3 hysteresis loop was found at 0.8 P/P0. The thickness of the silicate walls of RH-nanoMCM and MCM-41 nano correspond to 3.3 Å and 8.1 Å, respectively (for more details about the equations, vide ESI, Paragraph S1†). The SSABET of RH-nanoMCM corresponds to 1050 m2 g−1, whereas the reference material shows a SSABET of 900 m2 g−1. RH-nanoMCM and MCM-41 nano are characterized by a total pore volume of 1.66 and 1.68 cm3 Å−1 g−1, respectively (Table 1). The textural properties (i.e., SSABET and pore volume) of RH-nanoMCM are similar to those reported for this type of materials (vide Table S3†).
3.1 Rhodamine B adsorption measurements
The adsorption capacity of RH-nanoMCM towards the cationic dye rhodamine B in aqueous phase has been investigated by means of UV-vis spectroscopy (vide Materials and method section). The rhodamine B removal capacity of the mesoporous silicas, after a contact time of 20 min with dye solutions at different initial concentrations, is reported in Fig. 6.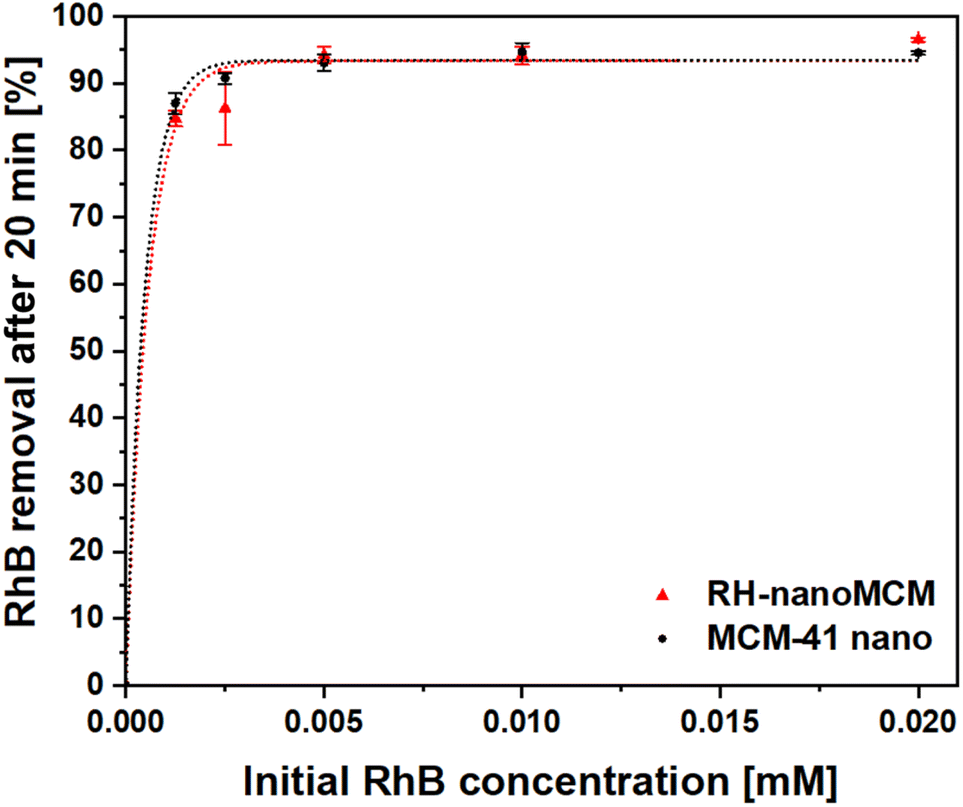 | ||
| Fig. 6 Rhodamine B removal percentages after a contact time of 20 min with RH-nanoMCM (triangles) and MCM-41 nano (circles). The initial RhB concentrations are 2.0 × 10−2, 1.0 × 10−2, 5.0 × 10−3, 2.5 × 10−3 and 1.2 × 10−3 mmol L−1. The error bars represent the standard deviations over three measurements. | ||
RH-nanoMCM showed a removal capacity higher than the 92% for dye solutions of 2.0 × 10−2, 1.0 × 10−2, 5.0 × 10−3 mmol L−1, whereas at lower initial RhB concentrations (i.e., 2.5 × 10−3 and 1.2 × 10−3 mmol L−1), the adsorption capacity of RH-nanoMCM was found above 85%. This result might be related to the higher driving force of the mass transfer process from the aqueous solution to the surface of the adsorbent at higher dye concentration.33–36 No significant differences could be observed between the removal capacity of RH-nanoMCM and MCM-41 nano (Fig. 6). The understanding of the nature of the non-covalent host–guest interactions between the adsorbate molecules and the surface of the adsorbent is relevant for the optimization of the adsorption processes.27,28 Rasalingam et al.27 described the electrostatic interactions between the positively charged rhodamine B molecules and the negatively charged silanoates groups (Si–O−) on a silica surface, along with the possible occurrence of hydrogen bonds between the silanols (Si–OH)/silanoates (Si–O−) and the carboxylate (COO−)/carboxyl (COOH) groups of the dye molecules. However, to the best of our knowledge, an in-depth study on the nature of these interactions is still lacking. Thus, in this work, we tried to fill this gap by carefully investigating the non-covalent interactions at play between the guest organic molecules and host material through 1H and 13C NMR spectroscopy37 and FT-IR spectroscopy.
3.2 Study of the interactions between RH-nanoMCM and rhodamine B molecules
Fig. 7 reports the 1H MAS NMR spectra of the dehydrated RH-nanoMCM, (i.e., the spectrum of the pristine sample treated under vacuum (residual pressure below 10−3 mbar) at room temperature for 3 hours) (Fig. 7A(a)), of the pristine RH-nanoMCM + RhB (i.e., the spectrum of the sample after the rhodamine B adsorption, before the vacuum treatment) (Fig. 7A(b)) and of the dehydrated RH-nanoMCM + RhB (i.e., the spectrum of the sample after the rhodamine B adsorption treated under vacuum (residual pressure below 10−3 mbar) at room temperature for 3 hours) (Fig. 7A(c)). The 13C CPMAS NMR spectrum of RH-nanoMCM + RhB (i.e., the spectrum of the sample after the rhodamine B adsorption treated under vacuum (residual pressure below 10−3 mbar) at room temperature for 3 hours) is reported in Fig. 7B.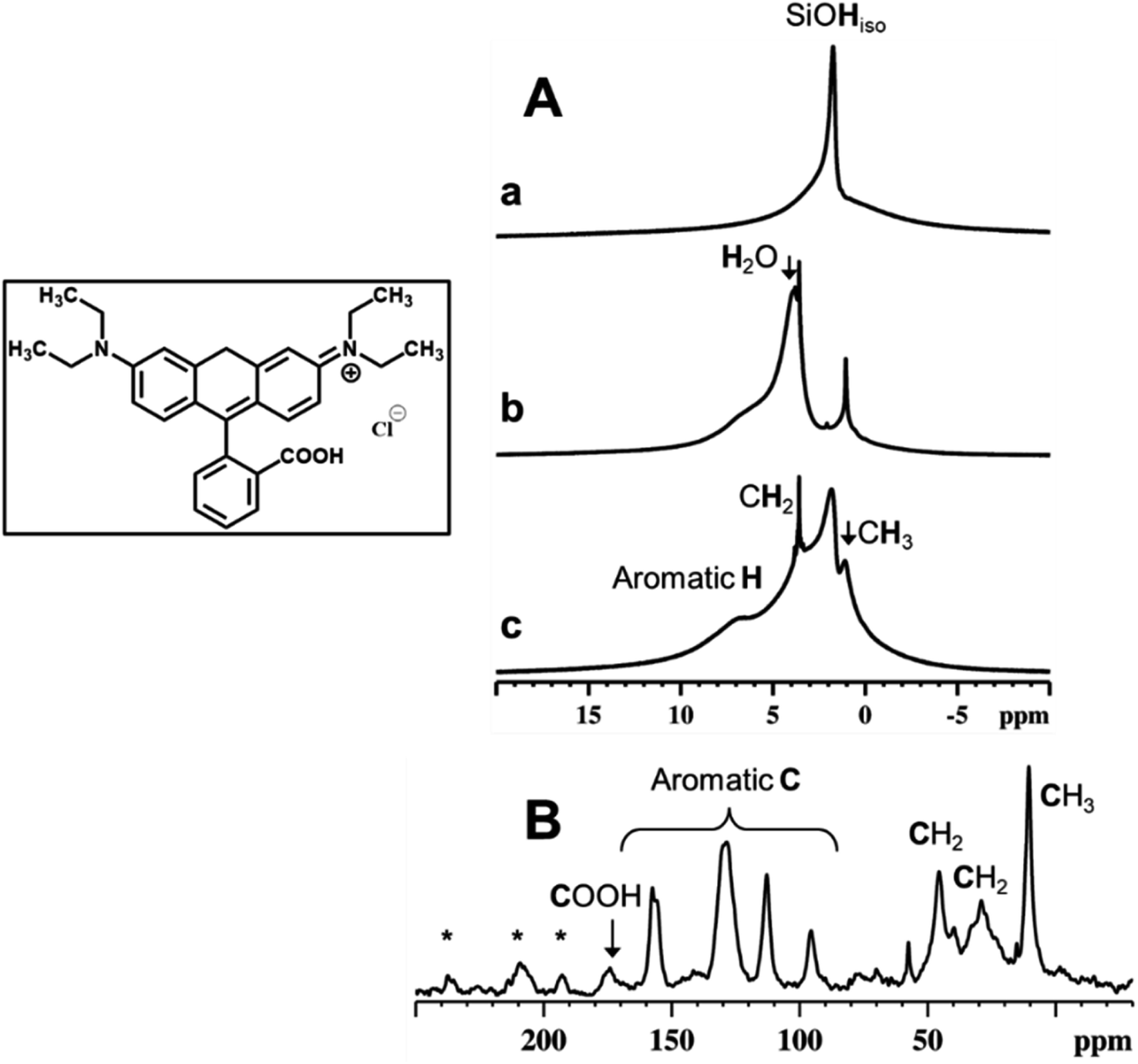 | ||
| Fig. 7 1H MAS NMR (A) spectra of RH-nanoMCM after dehydration (a) and of RH-nanoMCM + RhB before (b) and after (c) dehydration. 13C CPMAS NMR (B) spectrum of RH-nanoMCM + RhB. Inset shows the molecular structure of RhB. * spinning sidebands. | ||
In the 1H MAS NMR spectrum of dehydrated RH-nanoMCM (Fig. 7A(a)) there are two visible contributions: one narrow peak at 1.8 ppm and a broad shoulder at 2–5 ppm. The former is due to isolated silanols while the latter is associated to hydrogen-bonded silanols.38 The 1H MAS NMR spectra of pristine and dehydrated RH-nanoMCM + RhB are shown in Fig. 7A, spectra b and c, respectively. Both the spectra predominantly comprise of contributions from rhodamine B aliphatic protons of CH3 and CH2 moieties at ca. 1 and 3.5 ppm, respectively, and aromatic protons at ca. 7.5 ppm.39 Moreover, the spectrum of the pristine RH-nanoMCM + RhB shows the presence of the proton peak of physisorbed water at 3.9 ppm.
Interestingly, isolated silanol peak is absent in the spectrum of the pristine RH-nanoMCM + RhB (Fig. 7A(b)). Upon dehydration, the peak at 3.9 ppm due to water, disappears while the peak of isolated silanols reappear. Therefore, it is assumed that the protons of silanols are in rapid exchange with water protons in the pristine RH-nanoMCM + RhB sample. On the other hand, silanols peak at 1.8 ppm in dehydrated RH-nanoMCM + RhB is broader than in RH-nanoMCM. This may be due to the existence of host–guest non-covalent interactions associated to RhB and silica.
In a host–guest system, 1H relaxation is governed mainly by the 1H–1H dipolar interactions. On the other hand, molecular motions are responsible for the averaging out of anisotropic homonuclear dipolar interactions. In RH-nanoMCM + RhB system, guest dynamics are associated to molecular motions such as molecular tumbling, methyl group rotations, aromatic ring flips, etc., which will reflect on the NMR spin-lattice relaxation time value, T1 as well as on the spectral line widths. The proton T1 values, determined using saturation recovery method, shows a significant difference between guest-free and host–guest systems. The 1H spin-lattice relaxation time of isolated silanols was determined to be 450 ms for the guest-free system (i.e., without rhodamine B) and 250 ms for the host–guest system (i.e., with rhodamine B). The decrease in the T1 value of the proton of isolated silanols for dehydrated RH-nanoMCM + RhB system is mainly due to the relaxation enhancement generated by the 1H–1H dipolar interactions between rhodamine B and silanols. However, the absence of sharp peaks and the low resolution of the dehydrated RH-nanoMCM + RhB spectrum (Fig. 7A(c)) indicate the noncomplete averaging of the strong homonuclear 1H–1H dipolar interactions. This imply that rhodamine B is experiencing strong interactions inside the mesopores of silica either in the form of guest–guest or host–guest interactions.
The evidence for the rigid nature of rhodamine B molecules within the silica mesopores is also derived from the 13C CPMAS NMR data (Fig. 7B). The effectiveness of the CPMAS technique depends on the successful magnetization transfer between abundant spins (1H) and dilute spins (13C) facilitated by heteronuclear dipolar couplings.40 The relatively higher intensity of the rhodamine B 13C signals with short cross polarization contact time (5 ms) can be attributed to the rigid binding of guest molecules to the silica surface.41 Such bindings can occur either due to electrostatic interactions or non-covalent interactions (both H-bondings and van der Waals dispersion forces).27,41,42 These interactions are feasible in the RH-nanoMCM + RhB systems and can be attributed to the chemical nature of rhodamine B and the morphological landscape of mesoporous silica (i.e., silanols, H-bonded silanols, silanol nests etc.). In fact, the results of Z potential analyses in water (vide Fig. S5†) reveal that the surface of both RH-nanoMCM and MCM-41 nano is negatively charged, allowing electrostatic interactions with the positively charged zwitterionic rhodamine B molecules.27,42
The FT-IR spectra of the RH-nanoMCM and of RH-nanoMCM + RhB are reported in Fig. 8.
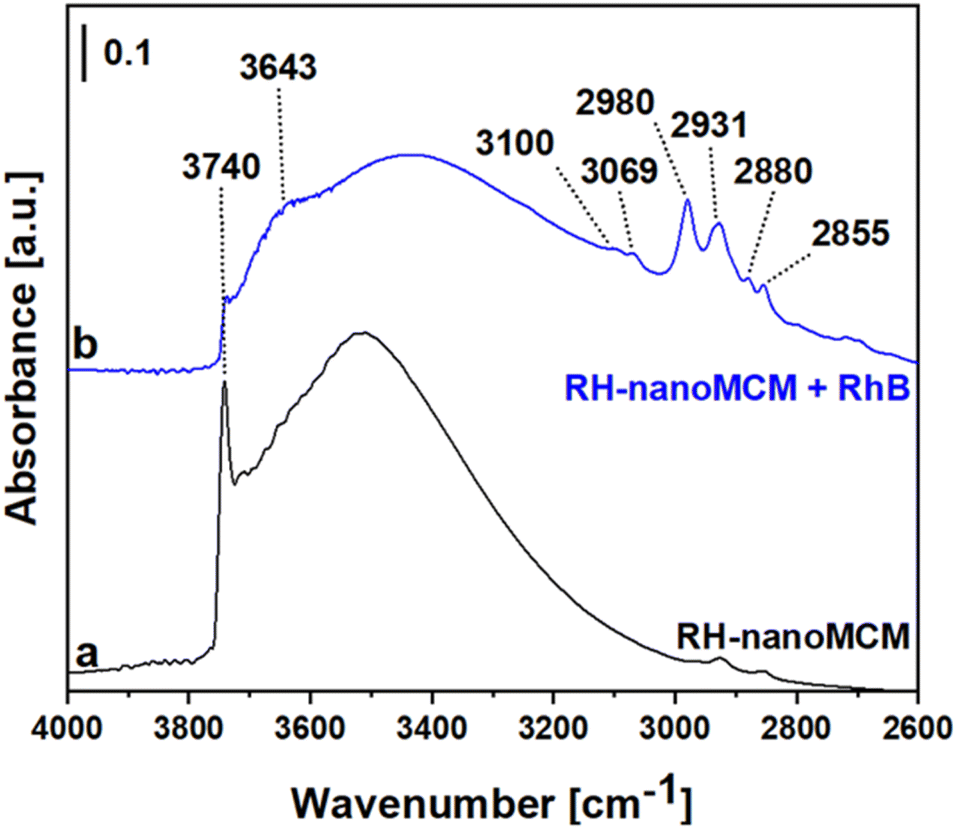 | ||
| Fig. 8 FT-IR spectrum of RH-nanoMCM (a) and of RH-nanoMCM after the adsorption of rhodamine B (b). Prior to the adsorption measurements, the pellets were treated under vacuum (residual pressure below 10−3 mbar) at beam temperature (35 °C) for 3 hours. | ||
The spectrum of RH-nanoMCM (Fig. 8a) is dominated by a sharp signal at 3740 cm−1 due to isolated silanols and the broad band at ca. 3500 cm−1 can be attributed to the mutually H-bonded Si–OH groups.14,43 As already observed from the MAS NMR measurements, after the adsorption of rhodamine B molecules, the signal of silanols is broader due to the occurrence of host–guest non-covalent interactions between rhodamine B and silica surface. In detail, a new band at 3643 cm−1 which could be related to the Si–OH groups in interaction with the dye molecules was formed and, consequently, the peak of isolated silanols at 3740 cm−1 appears less intense in the spectrum of RH-nanoMCM + RhB (Fig. 8b). The signals at 3100 and 3069 cm−1 are related to the stretching modes of aromatic C–H groups, whereas the peaks at 2980, 2931, 2880 and 2855 cm−1 can be attributed to the asymmetric and symmetric stretching modes of C–H bonds of the aliphatic CH3 moieties of rhodamine B.44
4 Conclusions
A synthetic procedure to obtain nanometric MCM-41 mesoporous silicas from alternative silicon sources has been presented. Biogenic silica obtained from rice husk was successfully employed, in addition with two surfactants, CTABr and Pluronic F127, for the synthesis of nanometric MCM-41 (RH-nanoMCM). The particle size was assessed via TEM microscopy confirming the nanometric nature, below 30 nm, of the obtained material. PXRD analysis indicates that the long order structure of the material is the classic hexagonal pore arrangement typical of the MCM-41. The textural properties, assessed via N2 physisorption analysis at 77 K, are also in line with those found for the MCM-41 obtained using the classic nanometric synthetic procedure found in the literature (MCM-41 nano). Both RH-nanoMCM and MCM-41 nano showed similar adsorption capacities of rhodamine B, which was selected as representative of cationic aromatic dyes. The removal of rhodamine B from aqueous solutions was carried out at different concentrations, namely at 2.0 × 10−2, 1.0 × 10−2, 5.0 × 10−3, 2.5 × 10−3 and 1.2 × 10−3 mmol L−1. The adsorption capacity, evaluated after 20 minutes of contact time, was assessed at 85 to 92% of the initial dye concentration. Finally, though the contribution of van der Waals dispersion forces is relevant, the results of the NMR and FT-IR studies reveal that silanols groups on the surface of the adsorbent act as adsorption sites for the RhB molecules. This could be corroborated by the decrease in the T1 value for RH-nanoMCM + RhB due to the relaxation enhancement generated by the 1H–1H dipolar interactions between RhB and silanols.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: G. C., S. M. and C. B.; methodology: G. C., F. B., G. P., C. B. and L. M.; formal analysis: G. C., F. B., S. M. and G. P., data curation: G. C., C. B., F. B. and L. M.; writing, review and editing: G. C., F. B., E. B., C. B. and L. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are fully grateful to Mrs Arianna Fossa for the rhodamine B adsorption measurements (DiSIT, Università del Piemonte Orientale, Alessandria, Italy). This research was funded by the company Syensqo (former Solvay) in the frame of the Joint-lab R&D Center for Environmental Remediation and Protection (Centro RisPA) between the “Università del Piemonte Orientale” - Department of Science and Technological Innovation (DiSIT) - and Solvay Specialty Polymers Italy S.p.A.References
- D. Khan and Shaily, Appl. Organomet. Chem., 2023, 37, e7007 CrossRef CAS.
- A. Mehmood, H. Ghafar, S. Yaqoob, U. F. Gohar and B. Ahmad, J. Dev. Drugs, 2017, 6(2) DOI:10.4172/2329-6631.1000174.
- J. A. S. Costa, R. A. De Jesus, D. O. Santos, J. F. Mano, L. P. C. Romão and C. M. Paranhos, Microporous Mesoporous Mater., 2020, 291, 109698 CrossRef CAS.
- F. Carniato, C. Bisio, G. Paul, G. Gatti, L. Bertinetti, S. Coluccia and L. Marchese, J. Mater. Chem., 2010, 20, 5504 RSC.
- Z. Li, J. C. Barnes, A. Bosoy, J. Fraser Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- G. Martínez-Edo, A. Balmori, I. Pontón, A. Martí Del Rio and D. Sánchez-García, Catalysts, 2018, 8, 617 CrossRef.
- N. Yuan, X. Zhang and L. Wang, Coord. Chem. Rev., 2020, 421, 213442 CrossRef CAS.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139 CrossRef.
- X. S. Zhao, G. Q. Lu and G. J. Millar, Ind. Eng. Chem. Res., 1996, 35, 2075–2090 CrossRef CAS.
- K. Suzuki, K. Ikari and H. Imai, J. Am. Chem. Soc., 2004, 126, 462–463 CrossRef CAS PubMed.
- A. Erigoni and U. Diaz, Catalysts, 2021, 11, 79 CrossRef CAS.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862 RSC.
- R. Mokaya, J. Catal., 1999, 186, 470–477 CrossRef CAS.
- G. Gatti, C. Vittoni, D. Costenaro, G. Paul, E. Mangano, S. Brandani, L. Marchese and C. Bisio, Phys. Chem. Chem. Phys., 2017, 19, 29449–29460 RSC.
- T. Ali-dahmane, M. Adjdir, R. Hamacha, F. Villieras, A. Bengueddach and P. G. Weidler, C. R. Chim., 2014, 17, 1–6 CrossRef CAS.
- A. G. Gebretatios, A. R. Kadiri Kanakka Pillantakath, T. Witoon, J.-W. Lim, F. Banat and C. K. Cheng, Chemosphere, 2023, 310, 136843 CrossRef CAS PubMed.
- N. Soltani, A. Bahrami, M. I. Pech-Canul and L. A. González, Chem. Eng. J., 2015, 264, 899–935 CrossRef CAS.
- J. Chun and J. H. Lee, Sustainability, 2020, 12, 10683 CrossRef CAS.
- M. K. Seliem, S. Komarneni and M. R. Abu Khadra, Microporous Mesoporous Mater., 2016, 224, 51–57 CrossRef CAS.
- M. Shaban, M. R. Abukhadra, A. Hamd, R. R. Amin and A. Abdel Khalek, J. Environ. Manage., 2017, 204, 189–199 CrossRef CAS PubMed.
- W. A. Al-Amrani, M. A. K. M. Hanafiah and A.-H. A. Mohammed, Environ. Sci. Pollut. Res., 2022, 29, 76565–76610 CrossRef CAS PubMed.
- J. A. S. Costa, R. A. de Jesus, D. O. Santos, J. B. Neris, R. T. Figueiredo and C. M. Paranhos, J. Environ. Chem. Eng., 2021, 9, 105259 CrossRef CAS.
- A. A. Adeyemo, I. O. Adeoye and O. S. Bello, Appl. Water Sci., 2017, 7, 543–568 CrossRef CAS.
- S. Bolisetty, M. Peydayesh and R. Mezzenga, Chem. Soc. Rev., 2019, 48, 463–487 RSC.
- P. Samanta, A. V. Desai, S. Let and S. K. Ghosh, ACS Sustainable Chem. Eng., 2019, 7, 7456–7478 CrossRef CAS.
- R. Al-Tohamy, S. S. Ali, F. Li, K. M. Okasha, Y. A.-G. Mahmoud, T. Elsamahy, H. Jiao, Y. Fu and J. Sun, Ecotoxicol. Environ. Saf., 2022, 231, 113160 CrossRef CAS PubMed.
- S. Rasalingam, R. Peng and R. T. Koodali, Appl. Catal., B, 2015, 174–175, 49–59 CrossRef CAS.
- G. Paul, F. Begni, A. Melicchio, G. Golemme, C. Bisio, D. Marchi, M. Cossi, L. Marchese and G. Gatti, ACS Appl. Polym. Mater., 2020, 2, 647–658 CrossRef CAS.
- J. Kong, S.-S. Park and C.-S. Ha, Mater., 2022, 15, 5926 CrossRef CAS PubMed.
- L. F. Chen, L. Noreña, J. Navarrete and J. A. Wang, Mater. Chem. Phys., 2006, 97, 236–242 CrossRef CAS.
- G. Paul, C. Bisio, I. Braschi, M. Cossi, G. Gatti, E. Gianotti and L. Marchese, Chem. Soc. Rev., 2018, 47, 5684–5739 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172–184 CrossRef CAS PubMed.
- Z. Wu, H. Joo and K. Lee, Chem. Eng. J., 2005, 112, 227–236 CrossRef CAS.
- M. S. U. Rehman, M. Munir, M. Ashfaq, N. Rashid, M. F. Nazar, M. Danish and J.-I. Han, Chem. Eng. J., 2013, 228, 54–62 CrossRef CAS.
- S. Karaca, A. Gürses, M. Açıkyıldız and M. Ejder Korucu, Microporous Mesoporous Mater., 2008, 115, 376–382 CrossRef CAS.
- G. Paul, S. Steuernagel and H. Koller, Chem. Commun., 2007, 5194–5196 RSC.
- G. Paul, G. E. Musso, E. Bottinelli, M. Cossi, L. Marchese and G. Berlier, ChemPhysChem, 2017, 18, 839–849 CrossRef CAS PubMed.
- P. Srivastava, P. C. Fürstenwerth, J. F. Witte and U. Resch-Genger, New J. Chem., 2021, 45, 13755–13762 RSC.
- V. Miglio, F. Begni, C. Cassino, G. Paul, A. Bortoli, L. Marchese and C. Bisio, J. Phys. Chem. C, 2024, 128, 2179–2189 CrossRef CAS.
- V. Sacchetto, G. Gatti, G. Paul, I. Braschi, G. Berlier, M. Cossi, L. Marchese, R. Bagatin and C. Bisio, Phys. Chem. Chem. Phys., 2013, 15, 13275 RSC.
- V. Miglio, C. Zaccone, C. Vittoni, I. Braschi, E. Buscaroli, G. Golemme, L. Marchese and C. Bisio, Molecules, 2021, 26, 1316 CrossRef CAS PubMed.
- A. Zecchina, S. Bordiga, G. Spoto, L. Marchese, G. Petrini, G. Leofanti and M. Padovan, J. Phys. Chem., 1992, 96, 4991–4997 CrossRef CAS.
- J. Coates, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, Wiley, 1st edn, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07152b |
| This journal is © The Royal Society of Chemistry 2025 |