
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the base-mediated deuteration of pyridyl phosphonium and ammonium salts†
Arianna Montoli a,
Alessandro Dimasia,
Miriana Guarnacciaa,
Andrea Citarellaa,
Paolo Ronchib,
Delia Blasia,
Sergio Rossia,
Daniele Passarellaa and
Valerio Fasano*a
a,
Alessandro Dimasia,
Miriana Guarnacciaa,
Andrea Citarellaa,
Paolo Ronchib,
Delia Blasia,
Sergio Rossia,
Daniele Passarellaa and
Valerio Fasano*a
aDepartment of Chemistry, Università degli Studi di Milano, Via Camillo Golgi, 19, 20133 Milano, Italy. E-mail: valerio.fasano@unimi.it; Web: https://www.fasanolab.com
bMedicinal Chemistry and Drug Design Technologies Department, Global Research and Preclinical Development, Chiesi Farmaceutici S.p.A, Largo Francesco Belloli 11/a, 43126 Parma, Italy
First published on 10th January 2025
Abstract
Pyridines can be deuterated at the remote sites by treatment with KOtBu in DMSO-d6, although without discrimination between the meta- and para-position. Herein, base-catalyzed deuterations have been studied, computationally and experimentally, using a series of pyridyl phosphonium salts with a temporary electron-withdrawing group to block the para-position while increasing the acidity in the other positions.
The past decade has witnessed spectacular progress in the field of “deuterium switching”, a strategy based on the replacement of hydrogen with deuterium.1,2 Indeed, while the H-to-D substitution is one of the most conservative examples of bioisosterism, the C–D bond is associated with greater activation energy for cleavage (with a difference of 1.2–1.5 kcal mol−1), thus resulting in a more robust C–D bond.3 As a result, the metabolic stability of a drug can improve as well as its efficacy and safety compared with the non-deuterated counterparts (Scheme 1A). Nevertheless, the deuterium switch in drugs is still in its infancy, with the first deuterated drug Austedo (deutetrabenazine) approved by FDA only in 2017.4 Furthermore, deuterium is mainly installed on sp3-hybridized carbon atoms (e.g. a –CD3 group), whereas incorporation of deuterium on sp2-hybridized carbons of drugs has been less explored.5 This is particularly true for pyridines, for which only a handful of strategies are available for the deuteration of the aromatic ring (Scheme 1B).6,7 For instance, taking 2-phenyl-pyridine 1 as a model substrate, deuteration has been achieved at the ortho-position (of the pyridine or the phenyl ring) using transition metal-catalysis8–12 or at the para-position upon conversion into the corresponding pyridyl phosphonium triflate.13 Alternatively, complete deuteration has been possible with supercritical D2O,14,15 whereas a base-mediated deuteration (KOtBu in DMSO-d6) has allowed for remote labelling at the meta/para-positions.16,17
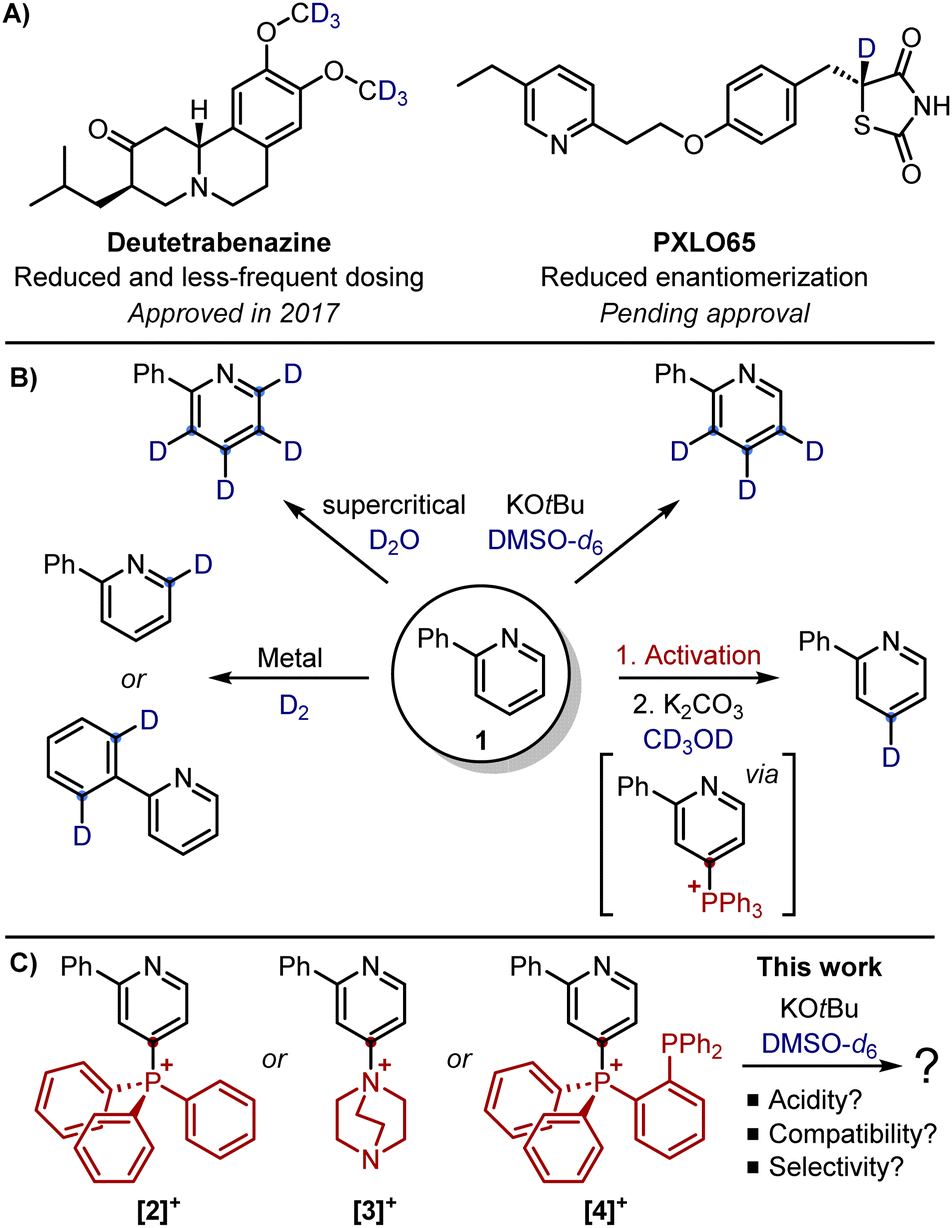 | ||
| Scheme 1 (A) Examples of deuterated drugs. (B) Most relevant modes of deuteration of 2-phenyl-pyridine 1. (C) Mechanistic insights into the base-mediated deuteration of pyridyl phosphonium and ammonium salts. | ||
Given that pyridines are some of the prevailing N-heterocycles in drugs,18 novel methodologies that can give access to diverse patterns of deuteration are still highly desirable. Inspired by the base-mediated meta/para-deuteration,16,17 we wondered how the deuteration of 1 could be modified to allow for a different labelling selectivity. In particular, we envisaged that the temporary installation of an electron-withdrawing group (EWG) on the para-position could block this position and increase the relative acidity of the adjacent positions (Scheme 1C). In this work, we report our investigation of the base-promoted deuteration of pyridyl phosphonium and ammonium salts, hoping to reverse the selectivity reported for these activated pyridines. The use of 1 as a benchmark allowed the direct comparison with the literature as well as avoiding alternative deuterations not induced by the EWG group but rather by the stereoinductive effects of other substituents (as observed for pyridines containing halogens or methyl groups).16
In 2022, the base-promoted deuteration of 1 was reported in two related studies, as shown in Scheme 2A.16,17
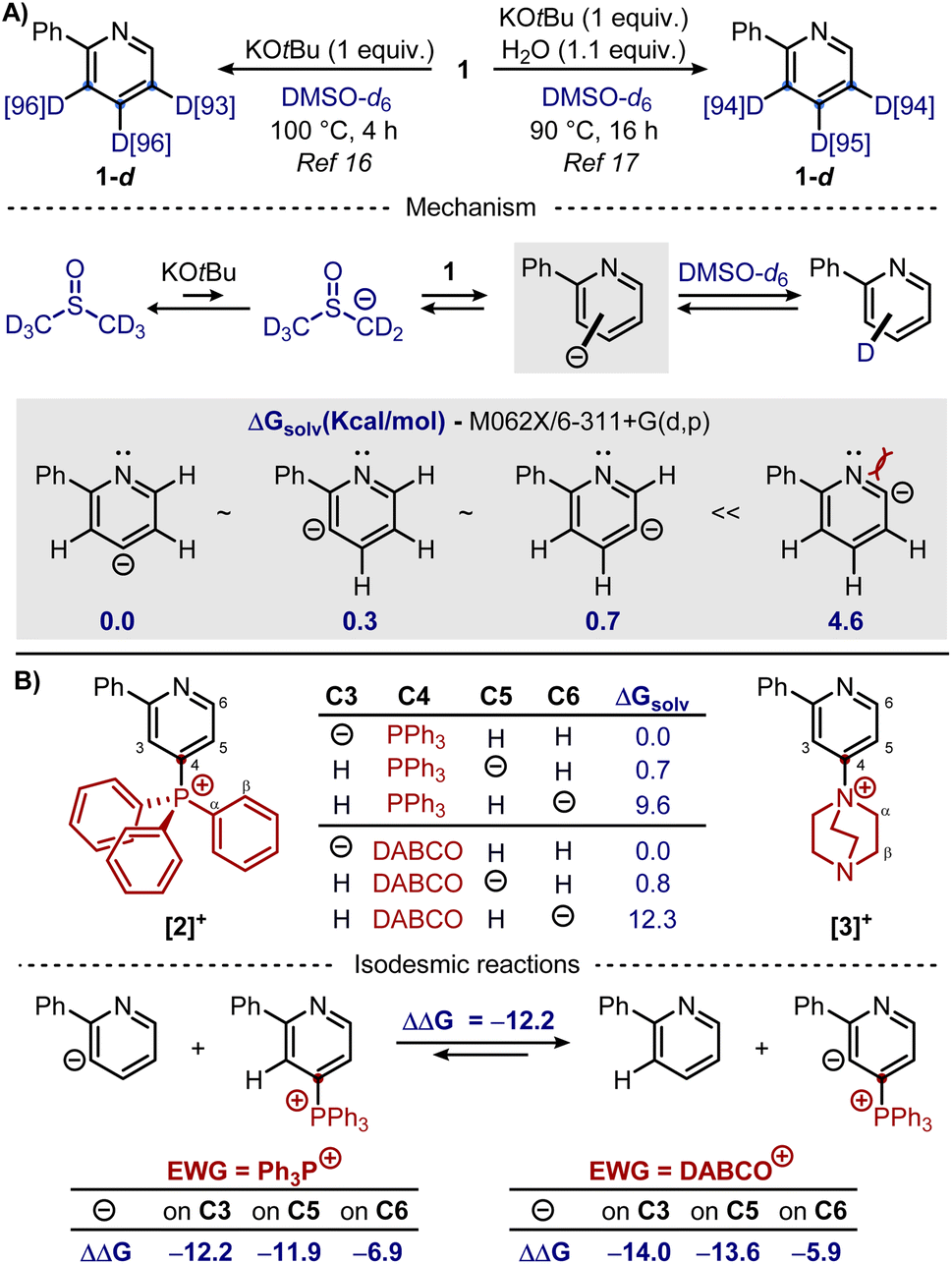 | ||
| Scheme 2 (A) Base-mediated deuteration of 1. (B) DFT calculations of the stability of carbanions deriving from the deprotonation of [2]+ and [3]+. | ||
Gao and co-workers showed that treatment of 1 with 1 equiv. of KOtBu in DMSO-d6 at 100 °C for 4 hours resulted in high deuterium incorporation in distal positions 3, 4 and 5 (>90%), with neglectable deuteration in the ortho-positions of the pyridine or the phenyl ring. Similar results were obtained by Beller and co-workers running the reaction at 90 °C for 16 hours in the presence of 1.1 equiv. of H2O (to slightly decrease the overall basicity of the reaction mixture). While the reaction conditions were marginally different, both studies showed that this reaction proceeded via a pyridyl anion mechanism, with the latter obtained upon the deprotonation of 1 by the in situ generated dimsyl anion. Indeed, the distribution and degree of deuteration could be explained by the relative thermodynamic stability of the pyridyl anions, whereas isotopic studies suggested a reversible reaction, with the rate-determining step not involving C–H cleavage but rather the deuteration of the pyridyl anions or the generation of the dimsyl anion (in DMSO, pKa(tBuOH) = 32 vs. pKa(DMSO) = 35).19 Furthermore, kinetic studies showed that the para-position underwent a slightly faster deuteration compared to the meta-positions, although deuteration of the latter was reached within one hour. Based on these premises, we envisaged that phosphonium [2]+ or ammonium [3]+ could promote ortho/meta-deuterations by increasing the corresponding acidity in these positions while blocking the para-position. Our investigation started evaluating the stability of pyridyl anions deriving from the deprotonation of [2]+ and [3]+ (Scheme 2B). In analogy with previous theoretical studies on 1,17 we chose M062X functional with 6-311+G(d,p) basis set for optimization in DMSO-d6 solvent based on solute electron density (SMD). For compounds [2]+ and [3]+, being the para-position substituted, the most stable anions derive from the deprotonation in meta-positions, as reported for 1. Interestingly, the deprotonation in ortho-position, to give the least stable pyridyl anion, could now compete with the deprotonation of the EWG (in β for [2]+ and in α for [3]+, with ΔG = 4.0 kcal mol−1 and 10.1 kcal mol−1, respectively). To assess the variation in acidity going from 1 to [2]+ and [3]+, isodesmic reactions were calculated too. These hypothetical reactions, in which the number of bonds of each type remains the same on each side of the equation, are useful to compensate for potential systematic errors in the modelling of each species.20 It was found that introducing both electron-withdrawing groups in para-position should increase the acidity in meta-positions, and, to a lesser extent, in ortho-position too, thus supporting our starting hypothesis. From the computational analysis, we can therefore conclude that going from 1 to [2]+ and [3]+ deprotonation at any position of the pyridine ring should become easier, but there should be more discrimination between the favored meta-positions and the unfavored ortho-position. Nevertheless, the modelling was purely based on thermodynamics (i.e. the anion stability), therefore the effect of sterics could dramatically affect the predictions. With this in mind, base-mediated deuterations were performed in the laboratory. Initially, the deuteration of 1 with KOtBu in DMSO-d6 (as reported by Gao and co-workers) was repeated, confirming indeed, upon an aqueous work-up, isolation of 1-d in 88% yield with deuteration occurring at the distal positions. Interestingly enough, conducting the same reaction in a J-Young tube to monitor its progression by in situ 1H NMR (not done in previous studies), showed the expected disappearance of the signals of distal protons, along with a partial loss (up to 50%) of the diagnostic signal at 8.68 ppm of the ortho-proton. Yet no deuteration at this position was observed in 1-d upon an aqueous work-up, even repeating the reaction and quenching it with D2O, followed by extraction with CDCl3. While this could be attributed to a severe broadening of the signal of the ortho-proton (confirmed by NMR analysis with longer acquisition times),21 this effect, caused by the coupling with the quadrupolar 14N and 2D nuclei, should be considered when following the reaction by in situ NMR analysis. The deuteration was then explored on pyridyl phosphonium salt [2][OTf], obtained in one-pot by the sequential addition of Tf2O, Ph3P, and DBU to 1 (Scheme 3A).22
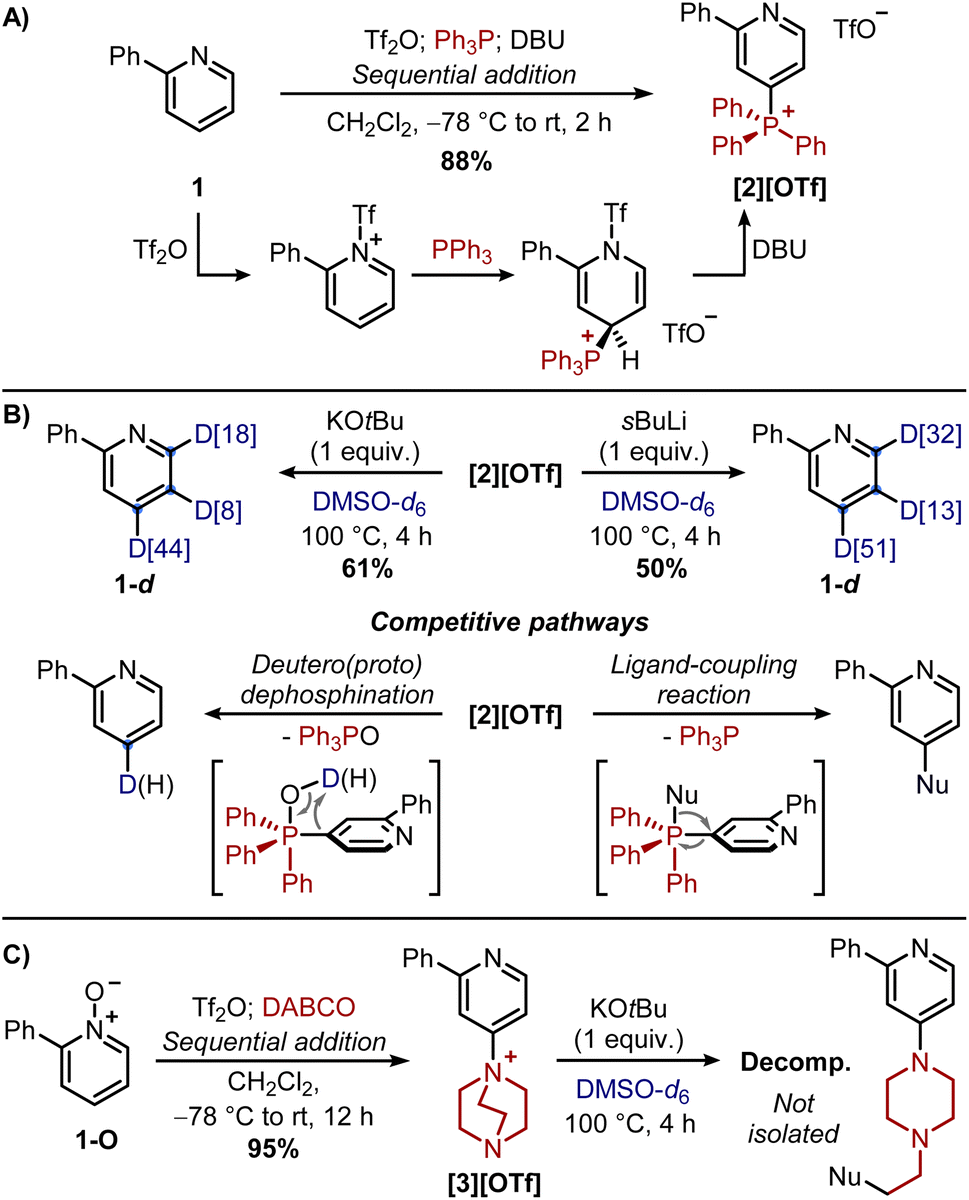 | ||
| Scheme 3 (A and B) Synthesis and reactivity of [2][OTf]. (C) Synthesis and reactivity of [3][OTf]. | ||
McNally and co-workers have shown that the treatment of [2][OTf] with K2CO3 in CD3OD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O allows the replacement of the phosphine with a deuterium atom upon elimination of Ph3PO (a two-step deuterium switch from 1).13 In our case, [2][OTf] was dissolved in DMSO-d6 and treated with 1 equiv. of KOtBu (Scheme 3B). The in situ monitoring of the reaction by 1H NMR and 31P NMR revealed, a few minutes after the mixing, a small amount of Ph3PO, suggesting initial dephosphination of [2]+ probably caused by water traces deriving from hygroscopic KOtBu. This process became prevalent upon heating the reaction mixture at 100 °C, with now significant Ph3PO observed after 4 hours. Notably, partial loss of the signal of the ortho-proton was observed too. Upon an aqueous workup of the reaction mixture, isolation of 1-d revealed 44% D-incorporation in para-position thus confirming deuterodephoshination (the other 56% H-incorporation was attributed to water traces or during the workup). Notably, 1-d showed 18% deuterium switch in ortho-position, therefore highlighting a certain level of C2-deprotonation occurring directly on [2]+ since deuteration of 1 occurs only at distal positions. In contrast with the computational predictions, minimal deuteration was observed in meta-positions, probably due to the steric shielding provided by the phosphonium moiety that favoured the ortho-position. These results suggest that slowing down the dephosphination reaction or accelerating the deprotonation of the ortho/meta-positions of [2]+ may favour novel deuteration patterns. Therefore, the first strategy looked at the replacement of the KOtBu with sBuLi with the hope of reducing adventitious protic sources (vide supra). As a control experiment, this change was also tested on 1 still confirming deuteration only at distal positions (1-d isolated in 86% yield). However, upon addition of [2][OTf] to a solution of sBuLi (1 equiv.) in DMSO-d6, in situ monitoring by 31P NMR of the reaction mixture revealed rapid (within 5 min) disappearing of [2]+ with the formation of Ph3PO and Ph3P. While the former could be attributed to dephosphination, the release of the latter suggested a ligand-coupling reaction, a mechanism by which Ph3P is replaced by a nucleophile (tentatively attributed to the dimsyl anion, formed quantitively with sBuLi, although we have not been able to characterize the corresponding product).23–26 Upon workup, isolation of 1-d revealed a similar level of deuteration in para-position (due to deuterodephosphination), but an increased deuterium switch in ortho/meta-position (45% in total). While modest, these increments highlight how the phosphonium moiety enhances the acidity of the adjacent positions (if sterically accessible), given the deprotonation of [2]+ occurs before dephosphination. In this regard, ammonium salts have emerged as a valid alternative to phosphonium salts for selective functionalizations of pyridines with nucleophiles (e.g. alkoxides or halogens).27 However, protodeamination is not predominant since not driven by the formation of Ph3PO as in the case of phosphonium salts. A second approach we investigated was then based on the use of pyridyl ammonium salt [3][OTf], a DABCO-derivative synthesized similarly to phosphonium salts but starting from N-oxide 1-O (Scheme 3C).28 However, the base-mediated deuteration of [3][OTf] resulted in an unproductive consumption of the starting material, with rapid ring-opening of the bicycle as judged by the desymmetrization of the signals associated with DABCO.29 A third strategy we explored looked instead at the installation of a directing group on the Ph3P+ moiety to facilitate the approach of the base by pre-coordination of its cation. We identified phosphines as ideal directing groups since (i) phosphines are excellent ligands for cations, and (ii) symmetrical diphosphines (Ph2P-linker-PPh2), needed for the synthesis, are commercially available. The first diphosphine we targeted was 1,2-bis(diphenylphosphino)-benzene (dppbe) since its electronic properties should not dramatically differ from those of PPh3. In the laboratory, pyridyl phosphine–phosphonium salt [4][OTf] was synthesized in 84% yield by treatment of 1 with Tf2O, dppbe, and DBU (Scheme 4A). This salt, obtained in high purity by simple precipitation in cold ether, showed two diagnostic peaks in the 31P{1H} NMR, confirming the desymmetrisation of the starting dppbe. The peak at 22.32 ppm (d, 3JP–P = 30.3 Hz) and the other at −14.43 ppm (d, 3JP–P = 30.3 Hz) were assigned to the P(V) and P(III) atoms, respectively.
:D2O allows the replacement of the phosphine with a deuterium atom upon elimination of Ph3PO (a two-step deuterium switch from 1).13 In our case, [2][OTf] was dissolved in DMSO-d6 and treated with 1 equiv. of KOtBu (Scheme 3B). The in situ monitoring of the reaction by 1H NMR and 31P NMR revealed, a few minutes after the mixing, a small amount of Ph3PO, suggesting initial dephosphination of [2]+ probably caused by water traces deriving from hygroscopic KOtBu. This process became prevalent upon heating the reaction mixture at 100 °C, with now significant Ph3PO observed after 4 hours. Notably, partial loss of the signal of the ortho-proton was observed too. Upon an aqueous workup of the reaction mixture, isolation of 1-d revealed 44% D-incorporation in para-position thus confirming deuterodephoshination (the other 56% H-incorporation was attributed to water traces or during the workup). Notably, 1-d showed 18% deuterium switch in ortho-position, therefore highlighting a certain level of C2-deprotonation occurring directly on [2]+ since deuteration of 1 occurs only at distal positions. In contrast with the computational predictions, minimal deuteration was observed in meta-positions, probably due to the steric shielding provided by the phosphonium moiety that favoured the ortho-position. These results suggest that slowing down the dephosphination reaction or accelerating the deprotonation of the ortho/meta-positions of [2]+ may favour novel deuteration patterns. Therefore, the first strategy looked at the replacement of the KOtBu with sBuLi with the hope of reducing adventitious protic sources (vide supra). As a control experiment, this change was also tested on 1 still confirming deuteration only at distal positions (1-d isolated in 86% yield). However, upon addition of [2][OTf] to a solution of sBuLi (1 equiv.) in DMSO-d6, in situ monitoring by 31P NMR of the reaction mixture revealed rapid (within 5 min) disappearing of [2]+ with the formation of Ph3PO and Ph3P. While the former could be attributed to dephosphination, the release of the latter suggested a ligand-coupling reaction, a mechanism by which Ph3P is replaced by a nucleophile (tentatively attributed to the dimsyl anion, formed quantitively with sBuLi, although we have not been able to characterize the corresponding product).23–26 Upon workup, isolation of 1-d revealed a similar level of deuteration in para-position (due to deuterodephosphination), but an increased deuterium switch in ortho/meta-position (45% in total). While modest, these increments highlight how the phosphonium moiety enhances the acidity of the adjacent positions (if sterically accessible), given the deprotonation of [2]+ occurs before dephosphination. In this regard, ammonium salts have emerged as a valid alternative to phosphonium salts for selective functionalizations of pyridines with nucleophiles (e.g. alkoxides or halogens).27 However, protodeamination is not predominant since not driven by the formation of Ph3PO as in the case of phosphonium salts. A second approach we investigated was then based on the use of pyridyl ammonium salt [3][OTf], a DABCO-derivative synthesized similarly to phosphonium salts but starting from N-oxide 1-O (Scheme 3C).28 However, the base-mediated deuteration of [3][OTf] resulted in an unproductive consumption of the starting material, with rapid ring-opening of the bicycle as judged by the desymmetrization of the signals associated with DABCO.29 A third strategy we explored looked instead at the installation of a directing group on the Ph3P+ moiety to facilitate the approach of the base by pre-coordination of its cation. We identified phosphines as ideal directing groups since (i) phosphines are excellent ligands for cations, and (ii) symmetrical diphosphines (Ph2P-linker-PPh2), needed for the synthesis, are commercially available. The first diphosphine we targeted was 1,2-bis(diphenylphosphino)-benzene (dppbe) since its electronic properties should not dramatically differ from those of PPh3. In the laboratory, pyridyl phosphine–phosphonium salt [4][OTf] was synthesized in 84% yield by treatment of 1 with Tf2O, dppbe, and DBU (Scheme 4A). This salt, obtained in high purity by simple precipitation in cold ether, showed two diagnostic peaks in the 31P{1H} NMR, confirming the desymmetrisation of the starting dppbe. The peak at 22.32 ppm (d, 3JP–P = 30.3 Hz) and the other at −14.43 ppm (d, 3JP–P = 30.3 Hz) were assigned to the P(V) and P(III) atoms, respectively.
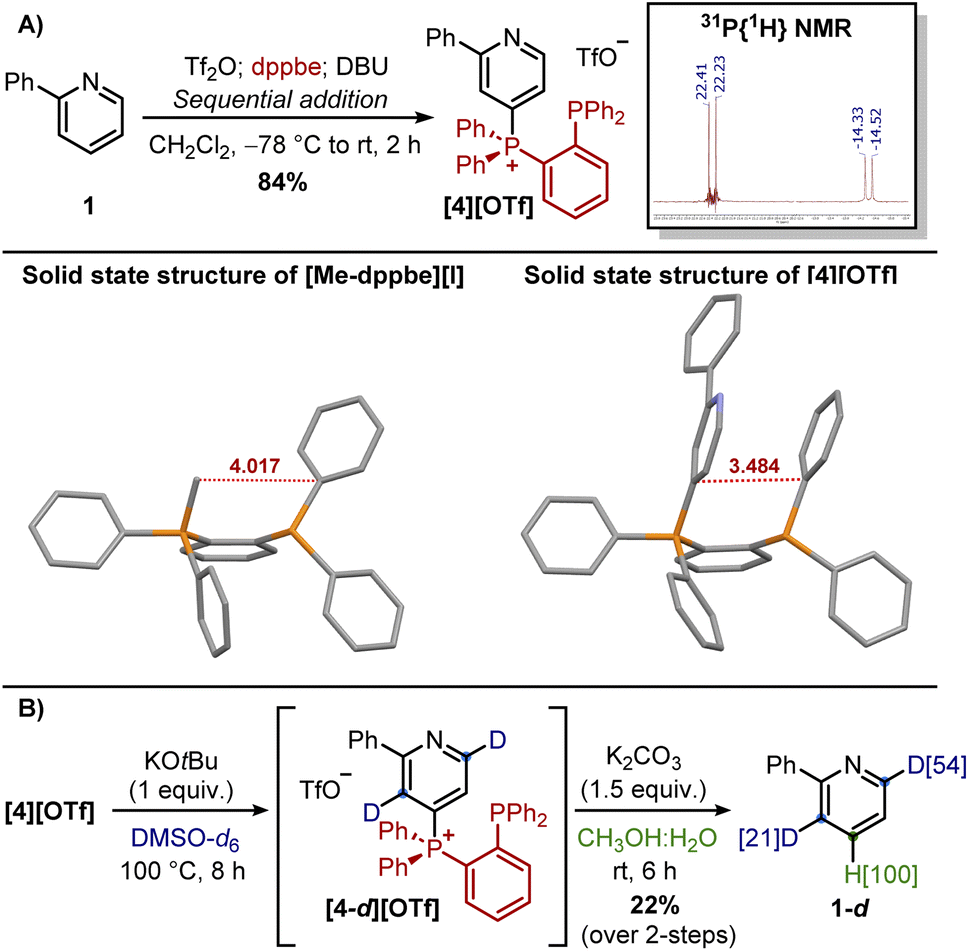 | ||
| Scheme 4 (A) Synthesis of [4][OTf] and its solid-state structure compared with [Me-dppbe][I]30 (anions omitted for clarity). (B) Reactivity of [4][OTf]. | ||
Crystals of [4][OTf] were obtained by slow evaporation from a CHCl3 solution: the solid-state structure showed a π–π interaction between the pyridine and one of the phenyl rings on the P(III) center, with the latter oriented toward the C5-proton. We believe this arrangement is specific to [4]+ since in the solid-state of [Me-dppbe]+, reported by Webster and co-workers and where the π–π interaction is missing,30 the ligands attached to the two phosphorus centers are further away, probably to release steric destabilization. Attempts to replace dppbe with other diphosphines (e.g. linker = –CH2–, –CH2CH2–, –CH2CH2CH2–) was not successful, with the corresponding phosphonium salts obtained as impure products, often with the pendant phosphine easily oxidized in air to its oxide. With compound [4][OTf] in hand, the base-mediated deuteration was explored on this novel pyridyl phosphonium salt (Scheme 4B). While the use of sBuLi as a base gave complete dephosphination within a few minutes, performing the reaction with KOtBu gave interesting results. Specifically, a significantly slower dephosphination was observed, with the pyridyl phosphonium salt still present after prolonged heating (8 hours at 100 °C). Upon workup, a mixture of deuterated pyridines was obtained, but their isolation was complicated by similar polarities. The corresponding [4-d][OTf] could instead be isolated in 22%: this was then subjected to protodephosphination to reveal its level of deuteration. To our delight, deuteration at C6- and C3-position occurred in 54% and 21%, respectively, with the latter preferred over the other meta-position probably for steric reasons. While modest, this result suggests that the use of a temporary EWG group promotes ortho/meta-deuterations while blocking the para-position.
Conclusions
Pyridyl phosphonium and ammonium salts [2][OTf]–[4][OTf] were synthesized aiming for a diverse deuteration of model-pyridine 1. Initial computational analysis validated the working hypothesis since, going from 1 to [2]+ and [3]+, deprotonation at the meta-position became thermodynamically favourable (due to electron-withdrawing effects), with the ortho-position being less convenient than the meta and with the para-position blocked. Nevertheless, sterics played an important role since the deuteration of [2][OTf] took place mainly at the ortho-position before the deutero(proto)dephosphination event occurred at the para-position. Deuteration of [3][OTf] was instead unsuccessful due to incompatibility with the use of bases. A final strategy (based on directing the base by coordination to its cation) brought to [4][OTf], a novel phosphonium salt fully characterized by heteronuclear NMR and XRD analyses. Deuteration of [4][OTf] afforded indeed the highest level of ortho/meta-deuteration with phosphonium salts, although these temporary directing groups are not ideal when it comes to compatibility with bases, thus more robust directing groups are currently being developed in our laboratory.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Prof. Fabrizio Ortu (University of Leicester) for support in X-ray diffraction analysis and Giacomo W. Lombardo (University of Milan) for preliminary results. Dr Daniele Fiorito (Politecnico di Milano) is acknowledged for useful discussions. Computational resources provided by INDACO Core facility, which is a project of High Performance Computing at the University of MILAN https://www.unimi.it. We acknowledge Chiesi Farmaceutici for funding.Notes and references
- R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Deuterium in drug discovery: progress, opportunities and challenges, Nat. Rev. Drug Discovery, 2023, 22, 562 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, C–H Functionalisation for Hydrogen Isotope Exchange, Angew. Chem., Int. Ed., 2018, 57, 3022 CrossRef CAS PubMed.
- S. Scheiner and M. Čuma, Relative Stability of Hydrogen and Deuterium Bonds, J. Am. Chem. Soc., 1996, 118, 1511 CrossRef CAS.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, Applications of Deuterium in Medicinal Chemistry, J. Med. Chem., 2019, 62, 5276 CrossRef CAS PubMed.
- D. Hesk, Highlights of C (sp2)–H hydrogen isotope exchange reactions, J. Labelled Compd. Radiopharm., 2020, 63, 247 CrossRef CAS PubMed.
- A. Tortajada and E. Hevia, Alkali-metal bases in catalytic hydrogen isotope exchange processes, Catal. Sci. Technol., 2023, 13, 4919 RSC.
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Recent Developments for the Deuterium and Tritium Labeling of Organic Molecules, Chem. Rev., 2022, 122, 6634 CrossRef CAS PubMed.
- M. Parmentier, T. Hartung, A. Pfaltz and D. Muri, Iridium-Catalyzed H/D Exchange: Ligand Complexes with Improved Efficiency and Scope, Chem.–Eur. J., 2014, 20, 11496 CrossRef CAS PubMed.
- K. A. Guy and J. R. Shapley, H–D Exchange between N-Heterocyclic Compounds and D2O with a Pd/PVP Colloid Catalyst, Organometallics, 2009, 28, 4020 CrossRef CAS.
- S. C. Schou, The effect of adding Crabtree's catalyst to rhodium black in direct hydrogen isotope exchange reactions, J. Labelled Compd. Radiopharm., 2009, 52, 376 CrossRef CAS.
- G. Pieters, C. Taglang, E. Bonnefille, T. Gutmann, C. Puente, J.-C. Berthet, C. Dugave, B. Chaudret and B. Rousseau, Regioselective and Stereospecific Deuteration of Bioactive Aza Compounds by the Use of Ruthenium Nanoparticles, Angew. Chem., Int. Ed., 2014, 53, 230 CrossRef CAS PubMed.
- H. Yang, C. Zarate, W. N. Palmer, N. Rivera, D. Hesk and P. J. Chirik, Site-Selective Nickel-Catalyzed Hydrogen Isotope Exchange in N-Heterocycles and Its Application to the Tritiation of Pharmaceuticals, ACS Catal., 2018, 8, 10210 CrossRef CAS.
- J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, A General Strategy for Site-Selective Incorporation of Deuterium and Tritium into Pyridines, Diazines, and Pharmaceuticals, J. Am. Chem. Soc., 2018, 140, 1990 CrossRef CAS PubMed.
- N. H. Werstiuk and C. Ju, Protium-deuterium exchange of substituted pyridines in neutral D2O at elevated temperatures, Can. J. Chem., 1989, 67, 5 CrossRef CAS.
- J. Yao and R. F. Evilia, Deuteration of extremely weak organic acids by enhanced acid-base reactivity in supercritical deuteroxide solution, J. Am. Chem. Soc., 1994, 116, 11229 CrossRef CAS.
- Y. Li, C. Zheng, Z.-J. Jiang, J. Tang, B. Tang and Z. Gao, Potassium tert-butoxide promoted regioselective deuteration of pyridines, Chem. Commun., 2022, 58, 3497 RSC.
- S. Kopf, J. Liu, R. Franke, H. Jiao, H. Neumann and M. Beller, Base-Mediated Remote Deuteration of N-Heteroarenes – Broad Scope and Mechanism, Eur. J. Org Chem., 2022, e202200204 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- F. G. Bordwell, Equilibrium acidities in dimethyl sulfoxide solution, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- D. A. Ponomarev and V. V. Takhistov, What are isodesmic reactions?, J. Chem. Educ., 1997, 74, 201 CrossRef CAS.
- S. Castellano, C. Sun and R. Kostelnik, Analysis of the NMR Spectrum of Pyridine, J. Chem. Phys., 1967, 46, 327 CrossRef CAS.
- R. G. Anderson, B. M. Jett and A. McNally, A Unified Approach to Couple Aromatic Heteronucleophiles to Azines and Pharmaceuticals, Angew. Chem., Int. Ed., 2018, 57, 12514 CrossRef CAS PubMed.
- M. C. Hilton, R. D. Dolewski and A. McNally, Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts, J. Am. Chem. Soc., 2016, 138, 13806 CrossRef CAS PubMed.
- R. D. Dolewski, M. C. Hilton and A. McNally, 4-Selective Pyridine Functionalization Reactions via Heterocyclic Phosphonium Salts, Synlett, 2018, 29, 8 CrossRef CAS.
- X. Zhang and A. McNally, Phosphonium Salts as Pseudohalides: Regioselective Nickel-Catalyzed Cross-Coupling of Complex Pyridines and Diazines, Angew. Chem., Int. Ed., 2017, 56, 9833 CrossRef CAS PubMed.
- M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Heterobiaryl synthesis by contractive C-C coupling via P(V) intermediates, Science, 2018, 362, 799 CrossRef CAS PubMed.
- C. Li, Z. Yan, B. Wang, J. Li, W. Lyu, Z. Wang, N. Jiao and S. Song, Regioselective synthesis of 4-functionalized pyridines, Chem, 2024, 10, 628 CAS.
- H. Xiong, A. T. Hoye, K.-H. Fan, X. Li, J. Clemens, C. L. Horchler, N. C. Lim and G. Attardo, Facile Route to 2-Fluoropyridines via 2-Pyridyltrialkylammonium Salts Prepared from Pyridine N-Oxides and Application to 18F-Labeling, Org. Lett., 2015, 17, 3726 CrossRef CAS PubMed.
- D. I. Bugaenko, M. A. Yurovskaya and A. V. Karchava, Quaternary N-(2-Pyridyl)-DABCO Salts: One-Pot In Situ Formation from Pyridine-N-oxides and Reactions with Nucleophiles: A Mild and Selective Route to Substituted N-(2-Pyridyl)-N′-ethylpiperazines, J. Org. Chem., 2017, 82, 2136 CrossRef CAS PubMed.
- W. Levason, G. Reid and M. Webster, 1,2-Bis(diphenylphosphino)benzene and two related mono-methiodides, [o-C6H4(PR2)(PR2Me)]I (R = Ph or Me), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o438 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2352491. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra07557a |
| This journal is © The Royal Society of Chemistry 2025 |