
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGSAG:Ce scintillator: insights from yttrium admixture†
Ondřej Zapadlík *abc,
Jan Pejchalb,
Vladimir Babinb,
Vítězslav Jarýb,
Vojtěch Vaněčekb,
Romana Kučerkováb,
Karel Jurekb,
Alena Beitlerováb and
Martin Niklb
*abc,
Jan Pejchalb,
Vladimir Babinb,
Vítězslav Jarýb,
Vojtěch Vaněčekb,
Romana Kučerkováb,
Karel Jurekb,
Alena Beitlerováb and
Martin Niklb
aCRYTUR, Ltd, Na Lukách 2283, Turnov, 511 01, Czech Republic
bInstitute of Physics of the Czech Academy of Sciences, Cukrovarnická 10/112, 160 00 Prague, Czech Republic
cFaculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague, Břehová 78/7, 115 19 Prague, Czech Republic
First published on 22nd January 2025
Abstract
The GSAG:Ce scintillator represents a promising and cost-effective alternative to the expensive GGAG:Ce. Recent studies have attributed its low light yield to the thermal quenching effect. In this study, we employed the strategy of adding an yttrium (Y) admixture to the GSAG matrix to increase the thermal activation energy of thermal quenching. The scintillation, optical, and luminescence properties of Gd(3−x)YxSc2Al3O12:Ce (x = 0, 0.2, 0.5, 0.8) grown using the micro-pulling-down method are thoroughly studied and reported. We further investigated the correlation between the Y content and other characteristics (scintillation rise time, light yield, decay, etc.). The magnitude of thermal quenching was evidenced by combination of the temperature-dependent photoluminescence decay kinetics and thermally stimulated luminescence. Excitation spectra were measured down to 30 nm in the VUV range under the synchrotron radiation at DESY to monitor the energy transfer efficiency from the host under excitation above the band gap. Results suggested that thermal ionization was not the primary reason for the low performance of the GSAG:Ce composition, and other defects related to the presence of scandium (Sc) must play a role. Furthermore, it was found that the stoichiometric GSAG host provided a noticeably higher light yield than the previously studied congruent one. This research provides valuable insights into the characteristics of GSAG:Ce scintillators.
1 Introduction
Gd3Al2Ga3O12:Ce (GAGG:Ce), which is an already commercially available1,2 and state-of-the-art scintillator developed by a combination of band-gap and defect engineering, exhibits unmatched scintillation performance (light yield up to 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ph per MeV, energy resolution of 4.2%@662 keV)3–8 or the ability to strongly accelerate the scintillation decay.9 Further dilution of the Gd sublattice by Y ions showed a positive impact on the scintillation characteristics.5 The main disadvantage of the GAGG:Ce composition is that owing to the presence of Ga, the crystal must be grown in an oxygen/inert atmosphere in an iridium crucible.10 The recent surge in the price of iridium due to global events made this already expensive material more expensive. Recently, several studies aimed at addressing the challenges of using iridium crucibles have been published, but they are yet to produce crystals with sufficient performance.11–13
000 ph per MeV, energy resolution of 4.2%@662 keV)3–8 or the ability to strongly accelerate the scintillation decay.9 Further dilution of the Gd sublattice by Y ions showed a positive impact on the scintillation characteristics.5 The main disadvantage of the GAGG:Ce composition is that owing to the presence of Ga, the crystal must be grown in an oxygen/inert atmosphere in an iridium crucible.10 The recent surge in the price of iridium due to global events made this already expensive material more expensive. Recently, several studies aimed at addressing the challenges of using iridium crucibles have been published, but they are yet to produce crystals with sufficient performance.11–13
Cerium-doped gadolinium–scandium–aluminium garnet (GSAG:Ce) has recently attracted much interest due to its distinct similarities to GGAG:Ce. Its main benefit compared to GGAG:Ce lies in the possibility of crystal growth from the non-precious Mo crucible.14 In both compositions, an energy transfer path of the exciton through the Gd3+ sublattice to the Ce3+ centre exists,15,16 competing with Y/Al anti-site defects.17–19 Moreover, both Ga3+ and Sc3+ participate in the formation of the bottom of the conduction band (CB) of the Gd-based garnet crystals.20 The electronic structure of Gd3Sc2Ga3O12, Gd3Sc2Al3O12, and Gd3Ga5O12 have been previously studied by means of first-principles local-density calculation.21 They have shown that the 3d electronic orbital states of Sc3+ strongly participate in the formation of the bottom of the conduction band (CB), while in the case of Ga3+, the CB is dominated by contribution from its 4s orbital states. For Ga3+, this has been experimentally validated.22 However, Sc3+ apparently shifts the bottom of the conduction band even more than Ga3+ into the close vicinity of the 5d1 state level of Ce3+, which results in noticeable thermal quenching at room temperature (RT) of Ce3+ luminescence.20,23–26 Furthermore, scandium substitution results in a larger unit cell (12.39 Å (ref. 27)) and thus in better solubility of Ce3+.28
Recently, several works have been reported focusing on the Ga-admixed GSAG:Ce grown by various crystal growth methods.20,23,24,26 The simultaneous contribution of Ga and Sc to the formation of the bottom of the CB results in significant modification of the band gap structure. Compared to GGAG, the overall band gap width of Ga-admixed GSAG:Ce is reduced by approximately 0.2 eV, and there is a shift of the conduction band's bottom and the 5d1 state into the proximity of each other. The consequence is that the scintillation LY increases (up to 23000 ph per MeV (ref. 24)), but it will also give rise to an intense delayed luminescence (afterglow) due to ionization of the Ce3+ 5d1 state.
As of today, the highest reported scintillation light yield value for GSAG:Ce is approximately 10000 ph per MeV.14 In this study, crystals with the congruent host composition Gd2.88Sc1.89Al3.23O12 were grown by the micro-pulling-down (μ-PD) method. Very recently, the Brigman-grown GSAG:Ce, Mg(Li) high quality crystals were characterized for their optical, luminescence and scintillation performance.29 Although the melt composition was the stoichiometric Gd3Sc2Al3O12 one, the crystal composition was Gd2.91Sc2.08Al3.01O12, i.e., a Gd-deficient composition similar to the congruent composition (Gd2.88Sc1.89Al3.23O12) is obtained. The light yield of GSAG:Ce0.6 at% was 10240 ph per MeV, which is the same value as in ref. 14. These values are considerably lower than the light yield of YAG:Ce.30,31 The thermal quenching at RT caused by partial ionization of the 5d1 state of Ce3+ is believed to be one of the main reasons for the low performance of GSAG:Ce. In this work, we aimed to shift the onset of thermal quenching to prove this hypothesis and further enhance the scintillation LY. This can be achieved by substitution of the Gd with Y in accordance with the research on Gd3−xYxAG:Ce.32 We have grown a series of Gd(3−x)YxSc2Al3O12:Ce (x = 0, 0.2, 0.5, 0.8) crystals by μ-PD method. The Ce concentration was constant for all samples at 0.3 atomic percent with respect to the dodecahedral site and replacing Gd; therefore, we can write the full formula as Gd2.991−xYxCe0.009Sc2Al3O12. For simplicity, the sample composition will be denoted as an undoped matrix composition with the Ce concentration in atomic percentages. We report the optical, luminescence, and scintillation characteristics.
2 Experimental details
The experimental methods employed in this study are summarized in Table 1 below.| Method | Purpose | Conditions/Parameters |
|---|---|---|
| μ-PD crystal growth | Synthesis of the GSAG:Ce crystals | 1820 °C, Ar + 5% H2 reducing atmosphere |
| EPMA analysis | Elemental composition and homogeneity | Beam energy: 20 kV |
| UV/VIS spectroscopy | Absorption spectra | 200–800 nm range |
| PL/PLE spectroscopy | Photoluminescence characterization | Excitation = 445 nm |
| Emission = 530 nm | ||
| PL decay | Decay time and its temperature dependence | Excitation: 452 nm |
| 77–500 K range | ||
| Thermoluminescence glow curves | Analyze trap depths and energy barriers | X-ray irr. 40 kV at 77 K, TSL spectrally unresolved, 77–500 K range |
| RL spectroscopy | Scintillation efficiency | X-ray, 40 kV, 15 mA |
| Pulse-height spectra | Scintillation light yield | Excitation: Cs-137, 662 keV, shaping time 1 μs |
| Scintillation decay | Rise and decay time under pulse X-ray excitation | 40 kV, 75 ps pulse FWHM |
| SUPERLUMI station at DESY | Excitation spectra in the VUV range | 30–330 nm |
Measurement of the absorption spectra was performed using the UV/VIS/NIR spectrophotometer Shimadzu 3101 PC in the range of 200–600 nm. PLE and PL spectra and PL decay curves were measured using a custom-made spectrofluorometer 5000 M (Horiba Jobin Yvon), with a steady state laser-driven xenon lamp (PL and PLE spectra, Energetiq EQ-99X LDLS – A Hamamatsu company), microsecond xenon pulsed flash lamp (delayed recombination decays, IBH Scotland) or nanosecond nanoLED pulsed light sources (IBH Scotland) as the excitation sources (PL decay kinetics/time resolved PL). The detection part of the setup consists of a single-grating monochromator and a photon counting detector TBX04 (IBH Scotland). Measured spectra with the range of 200–800 nm were corrected for the spectral dependence of the excitation energy (PLE) and spectral dependence of the detection sensitivity (PL). A convolution procedure was applied to the decay curves to determine the true decay times using the SpectraSolve software package (Ames Photonics).
RL spectra were measured on the same apparatus as photoluminescence characteristics with the Horiba Jobin Yvon X-ray tube with Mo anode (RL spectra, 40 kV, 15 mA, Seifert GmbH) as the excitation source. The scintillation light yield measurements were performed on the sample wrapped in a reflector (PTFE tape), and optically coupled by silicon grease to a hybrid photomultiplier (HPMT) Photonis PP0475B with a built-in preamplifier. The gamma source, radioisotope Cs-137 (662 keV), was used for the excitation and the amplifier shaping time was set to 1 μs. The details on the HPMT photodetector and the measurement method can be found in ref. 33 and 34. Scintillation decay curves and the rise time of the X-ray excited scintillation response was measured by time-correlated single photon counting technique using the hybrid picosecond photon detector HPPD-860 and Fluorohub unit (Horiba Scientific). Samples were excited by the picosecond (ps) X-ray tube N5084 from Hamamatsu, operating at 40 kV, driven by the ps light laser pulser (Hamamatsu) providing 75 ps FWHM of the instrumental response function. The SpectraSolve software package was applied to determine the rise times by deconvolution.
The measurements of the excitation spectra down to the VUV spectral region were performed at the SUPERLUMI station of the P66 beamline at DESY under pulsed excitation by synchrotron radiation in the range of 30–330 nm from the PETRA III storage ring. The excitation monochromator was 2 m normal incidence McPherson equipped with a holder for two Al and Pt interchangeable gratings. An Al grating with 1200 lines per mm (blazed at 180 nm) was optimized for the spectral range of 100–330 nm; Pt grating with 1200 lines per mm (blazed at 55 nm), and intended for the spectral range of 30–100 nm. Excitation spectra were measured with an instrumental resolution of about 0.3 nm. Luminescence spectra in the UV and visible range (200–1100 nm) were recorded with a Czerny–Turner type Kymera 328i spectrograph (Andor) equipped with a cooled CCD detector Newton 920. The analyzing monochromator was equipped with three interchangeable 300 lines per mm gratings blazed at 300, 500 and 1200 nm. For measurements of the excitation spectra, as well as for time-resolved measurements, the luminescence was detected by a photomultiplier (Hamamatsu R6358) working in the 185–830 nm range with a time resolution of about 1 ns or by a photomultiplier microchannel plate (Hamamatsu R3809U) working in the 160–850 nm range with a time resolution of about 55 ps. The detectors were attached to the second exit port of the same spectrograph. The set of the long-pass sharp cut-off filters was also available for the detection line. The samples were located on the cold finger of the He flow cryostat, enabling temperature regulation in the range of 15–300 K. All luminescence spectra were corrected for the quantum efficiencies of the CCD camera and gratings. The excitation spectra were corrected using sodium salicylate, assuming constant quantum efficiency in the measured range.
2.1 Crystal growth
The GSAG crystals were grown by μ-PD method35,36 with radiofrequency inductive heating (see Table 2 for a list of prepared compositions and their designations). The growth was performed using a Mo crucible with a die of 3 mm in diameter and nozzle diameter of 2 mm under Ar + 5% H2 reducing atmosphere, using the 〈111〉 oriented YAG single crystal as a seed. The crucible was placed on a Mo after heater with two windows and an alumina pedestal. The hot-zone around the crucible consisted of 3 layers of alumina shielding for thermal insulation. The Ar + 5% H2 reducing atmosphere was used to protect the Mo crucible, as it shows a tendency for oxidization. The gas flow was always kept at 0.5 l min−1. The crucible with the starting material was heated up to the melting temperature of around 1820 °C. Then, the YAG single-crystal seed was brought into contact with the melt coming through the nozzle due to gravity and wetting forces. The pulling speed was 0.03 mm min−1. In all the growth runs, it was possible to pull out all the melt. The crystal rods had a diameter of 3 mm, and were around 25 mm in length. One-millimeter-thick circular samples were cut approximately from the center of each rod and mirror-polished. Due to blackening of all the samples after the growth, the samples were annealed in air for 12 h at a temperature of 1200 °C and further investigated. Fig. 1 shows the photograph of Gd2.2Y0.8 (see Fig. S1† for pictures of other crystals) as a representative example. | ||
| Fig. 1 Photograph of the Gd2.2Y0.8 crystal annealed at 1200 °C for 12 hours (See F in the ESI data† for photographs of other crystals). | ||
| Chemical formula | Designation |
|---|---|
| Gd2.2Y0.8Sc2Al3O12:0.3% Ce | Gd2.2Y0.8 |
| Gd2.5Y0.5Sc2Al3O12:0.3% Ce | Gd2.5Y0.5 |
| Gd2.8Y0.2Sc2Al3O12:0.3% Ce | Gd2.8Y0.2 |
| Gd3Sc2Al3O12:0.3% Ce | Gd3Y0 |
In our previous work,14 we were unsuccessful in growing anything other than crystals from a congruent composition of the melt. In this work, we succeeded in growing crystals from the melt with the stoichiometric nominal composition Gd3Sc2Al3O12. This is allowed by the large nozzle of the crucible used. The growing crystal is hardly separated from the melt in the crucible, and the equilibrium between the two phases can be established at the solid–liquid interface. If the nozzle has a small diameter, establishment of the equilibrium at the solid–liquid interface is hampered and the garnet structure cannot be created (see discussion in ref. 14). The EPMA analysis of the stoichiometric crystal has shown that such a stoichiometric crystal has a core, whose composition is not far from the congruent one reported for example in ref. 14 and 37 and a mantle, whose composition is slightly deviated from the nominal stoichiometric one. Moreover, in the Y-containing samples, the smaller radius of Y noticeably stabilizes the garnet phase especially at higher concentrations due to a decrease of the dodecahedral site size. This in turn allows for a higher incorporation of larger atoms at the octahedral sites. Thus, the Sc-concentration can be increased, while keeping the stability of the garnet phase.
2.2 Chemical analysis
Chemical analysis was performed using the electron probe microanalysis (EPMA) method. It seems that the final crystal composition in all cases except the Y-absent one is slightly Sc-poor compared to the nominal composition. In Fig. 2, the EPMA analysis of Gd2.2Y0.8 is shown (see Fig. S2† for the analysis of the rest of the crystals). The sample composition is practically homogeneous all over the diameter. The only elements showing a departure from the nominal composition are Sc and Y. The matrix composition corresponds to Gd2.2Y0.9Sc1.9Al3O12 (see Table 3 for the real composition of the other samples). This would suggest an admixture of Y to the octahedral sites replacing Sc. No other parasitic phases were found.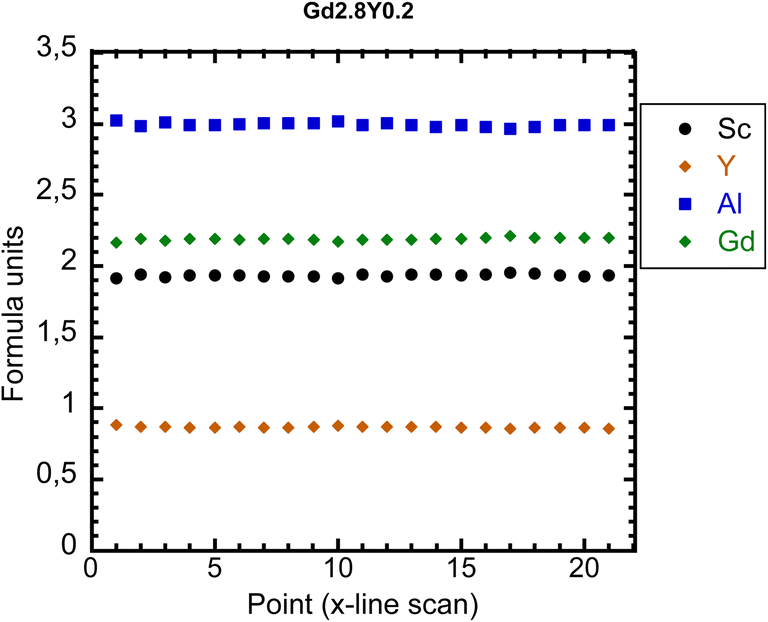 | ||
| Fig. 2 EPMA analysis of Gd2.2Y0.8. | ||
| Nominal composition | EPMA-real composition |
|---|---|
| Gd2.2Y0.8Sc2Al3O12 | Gd2.2Y0.9Sc1.9Al3O12 |
| Gd2.5Y0.5Sc2Al3O12 | Gd2.55Y0.5Sc1.96Al2.99O12 |
| Gd2.8Y0.2Sc2Al3O12 | Gd2.87Y0.2Sc1.94Al2.99O12 |
| Gd3Sc2Al2.97O12 | Gd3Sc2Al3O12 |
3 Results and discussion
3.1 Absorption spectra
The absorption spectra of Gd(3−x)Sc2YxAl3O12:Ce (Gd2.2Y0.8, Gd2.5Y0.5, Gd2.8Y0.2, Gd3Y0) at RT are displayed in Fig. 3. The 4f–5d1,2 transitions of Ce3+ at 450 nm and 340 nm, respectively, are well resolved. It should be noted that there is a small shift of transitions 4f–5d1,2 of Ce3+ by a few nm as the quantity of Gd increases. This shift effect correlates with the increasing crystal field strength at the dodecahedral site with increasing Gd content.38,39 A broad shoulder in the 200–250 nm region can be ascribed to transitions towards higher excited states of Ce3+ (5d3,4,5). The spectra below also display evident absorption lines, corresponding to the Gd3+ 4f–4f transitions situated around 245–255 nm (8S7/2 → 6Dx), 275 nm (8S7/2 → 6I7/2,9/2) and 305–310 nm (8S7/2 → 6P3/2,5/2,7/2). The host band edge is situated at around 220 nm.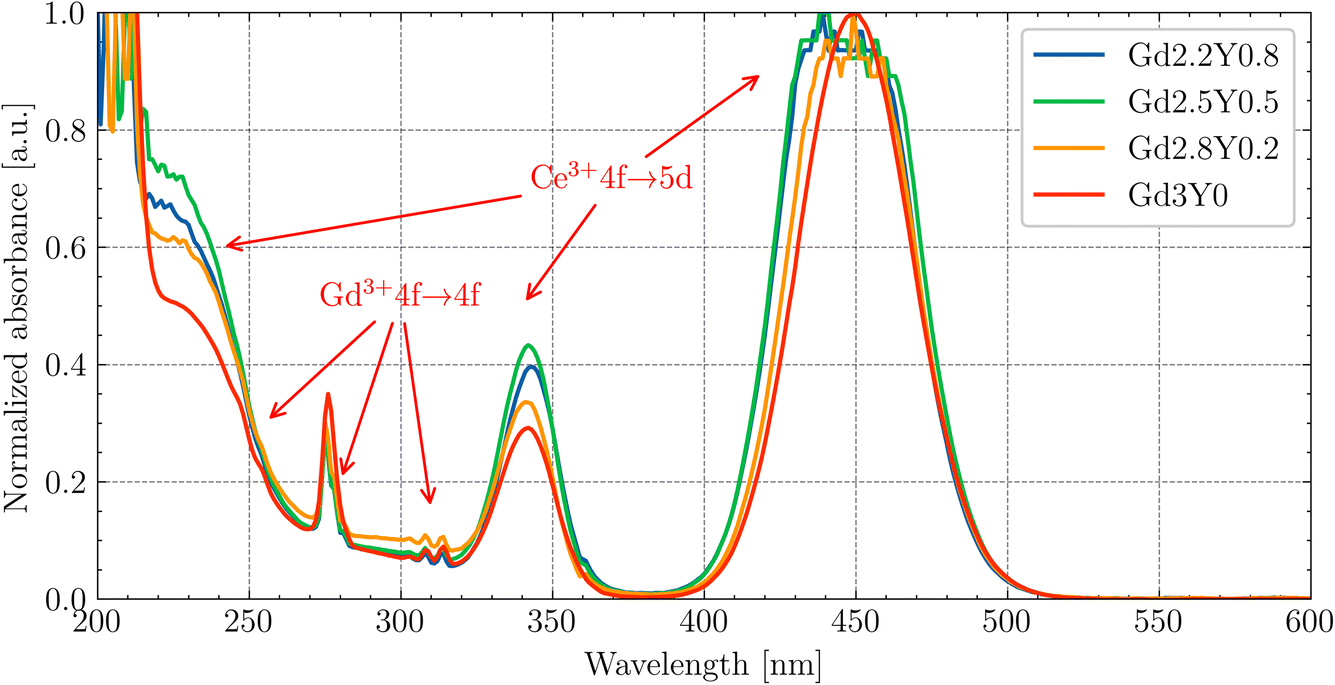 | ||
| Fig. 3 Absorption spectra of the Gd2.2Y0.8, Gd2.5Y0.5, Gd2.8Y0.2, Gd3Y0 samples at RT. | ||
The Y-rich samples Gd2.2Y0.8 and Gd2.5Y0.5 have a slightly higher concentration of Ce3+. This deviation in the Ce3+ concentration is too small to have a meaningful impact on the luminescence/scintillation properties. The EPMA analysis showed that the Y, while predominantly occupying at the Gd site, can also enter the octahedral Sc site. This leads to an increase in the garnet lattice parameter, which in turn increases the Ce segregation coefficient.
3.2 PL and PLE spectra
In Fig. 4, the normalized PL emission spectra (exc = 445 nm) are given for Gd2.2Y0.8, Gd2.5Y0.5, Gd2.8Y0.2 and Gd3Y0 in the range of 490–750 nm at RT. There is a small blue shift for samples Gd2.2Y0.8 and Gd2.5Y0.5; for the rest of the samples, there is PL emission from the 5d1–4f transition peaks at 545 nm. This difference is due to the lower content of Gd at the dodecahedral site in the mentioned samples, resulting in a reduced crystal field strength.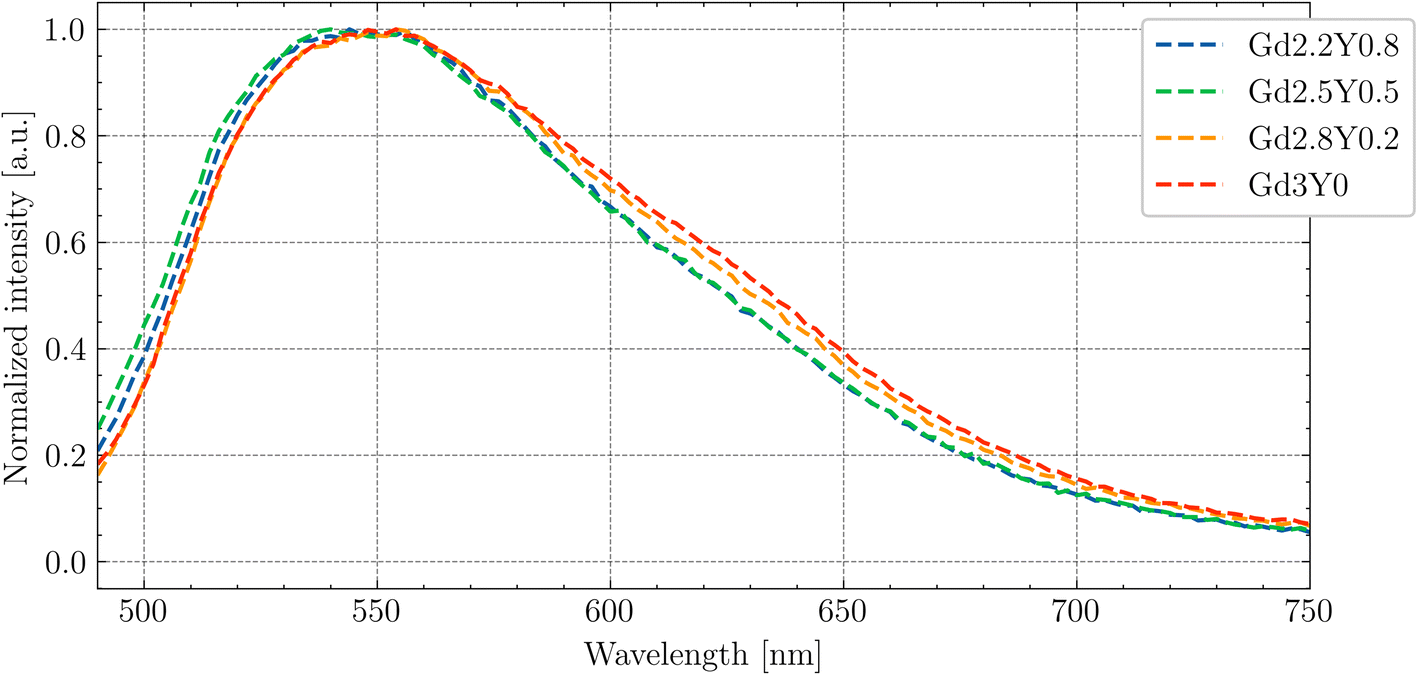 | ||
| Fig. 4 PL (exc = 445 nm) spectra of Gd2.2Y0.8, Gd2.5Y0.5, Gd2.8Y0.2 and Gd3Y0 at RT. | ||
In Fig. 5, the PLE spectrum (em = 530 nm) at RT for Gd3Y0 is displayed. The peaks at 335 and 460 nm in the excitation spectra match well with the position of the 4f–5d2 and 4f–5d1 absorption peaks, respectively (compared with Fig. 3).
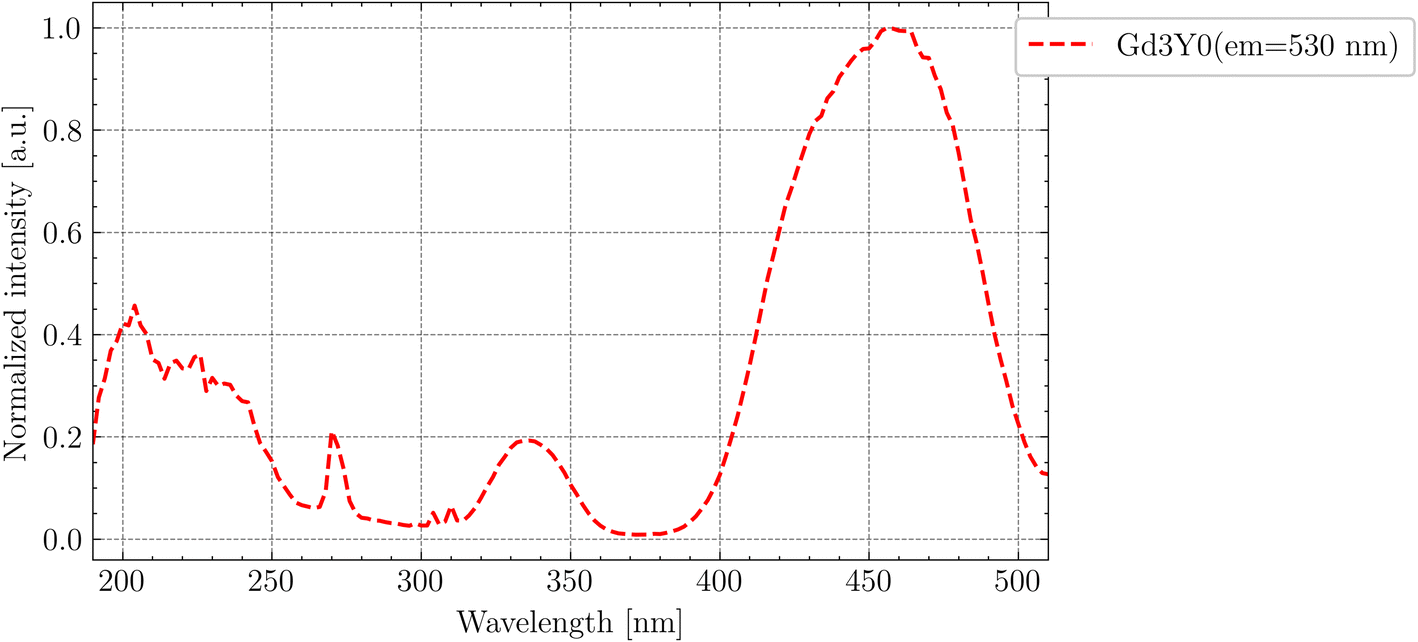 | ||
| Fig. 5 PLE (em = 530 nm) spectrum of Gd3Y0 at RT. | ||
Fig. 6 shows the PLE spectrum (em = 530 nm) at RT for the Gd3Y0 sample measured at the SUPERLUMI station of P66 beamline up to 8 eV. The excitation to the higher 5d3,4,5 states of Ce3+ forming the broad band above 4 eV can be influenced due to their position in the conduction band, and thus enables excited state ionization, followed by electron capture at defects. In the 3.9 eV to 4.4 eV region, there are clear sharp lines (compared with Fig. 3 and 5), corresponding to Gd3+ 4f–4f transitions. The appearance of these Gd3+ lines in the PLE spectra prove the transport of the excitation energy through the Gd3+ sublattice to the Ce3+ luminescent centres. The observed spectra exhibit a similar pattern for all studied samples, except for Gd2.2Y0.8 at the region above the exciton creation energy (>6.7 eV).
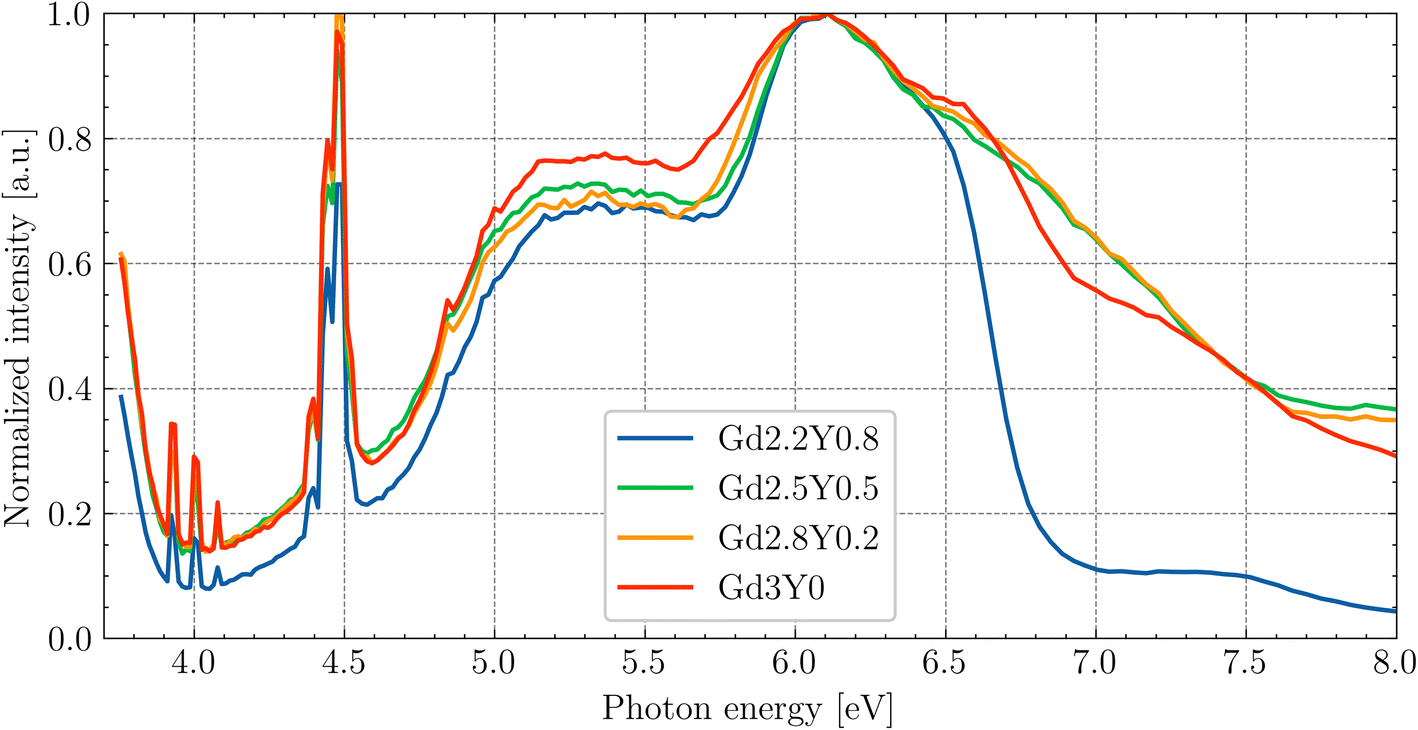 | ||
| Fig. 6 PLE of Ce3+ (em = 530) under synchrotron radiation at RT. The data are normalized to the excitonic band (6.7 eV). | ||
Since the absorption coefficient for the wavelength above the band edge (see Fig. 3) drastically increases, the actual thickness of the layer (which is excited) is only on the order of tens-hundreds of nanometres. Near the surface, there is often a higher density of defects and impurities that can act as non-radiative recombination centres. These defects can capture either the electron or the hole, leading to non-radiative recombination. This explains the drop above 6.7 eV. In the case of Gd2.2Y0.8, the drop is the steepest. So, in this composition, it appears more difficult for the exciton to recombine at the Ce centres. Since the drop is most pronounced in the composition with the least amount of Gd, it may indicate a problem with the Gd3+ → Ce3+ energy transport. The quality of the surface layer also plays a role, so the exact nature of the pronounced drop cannot be definitively assigned.
In Fig. 7, the emission spectra for different excitation wavelengths are shown. The shift of the emission peak under the wavelength close to the band–band transition is associated with the nature of the Ce3+ centres. As was mentioned above, the layer which is excited at this wavelength is very thin, and the lattice structure near the surface is disrupted. The 5d–4f transition of Ce3+ is sensitive to the local crystal field environment, leading to a change in the emission wavelength.
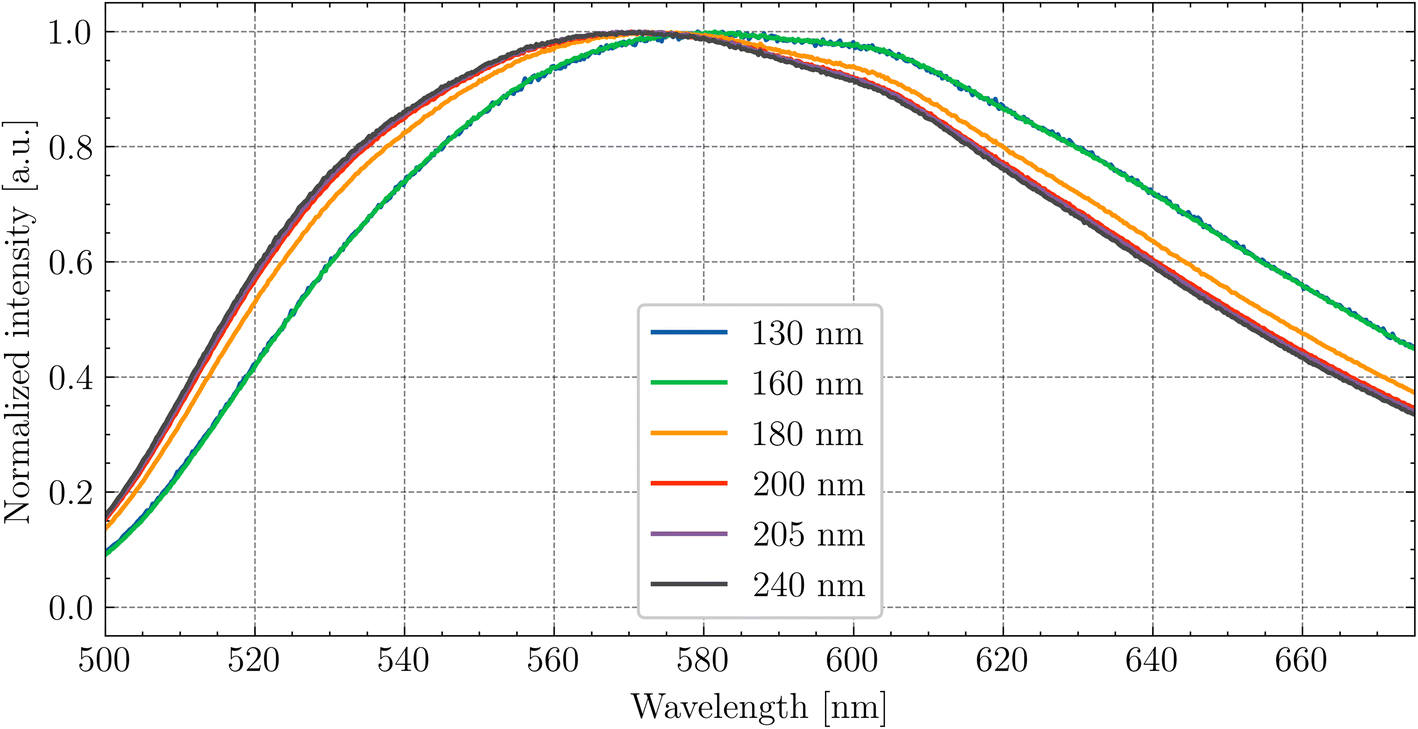 | ||
| Fig. 7 Dependence of the peak position of Ce3+ emission on the excitation wavelength at RT for Gd2.8Y0.2. | ||
The PLE spectra for energies above 12 eV to 45 eV of the studied sample set are presented in Fig. S3† of the ESI.† The increase in intensity above approximately twice the band gap is associated with the process of multiplication of the electronic excitation (MEE). The highest excitation intensity was observed for the Gd3Y0, and the lowest for Gd2.2Y0.8.
3.3 PL decay and its temperature dependence
The room temperature PL decay curves at the emission of 550 nm under the nanoLED-pulse excitation of 452 nm are shown in the ESI in Fig. S4.† The PL decays were fit by the following single exponential function:
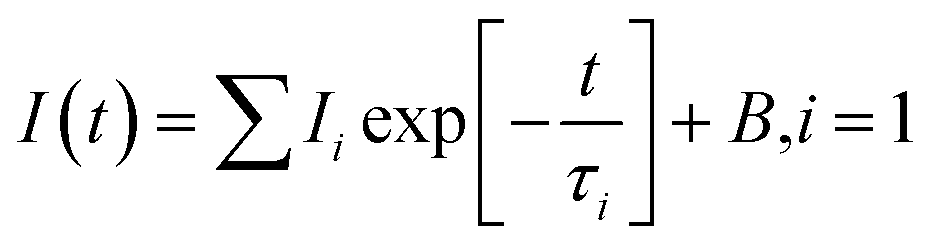 | (1) |
The decay component of 45–50 ns at RT for all samples indicates the presence of a quenching effect, specifically thermal ionization (refer to the temperature-dependent measurement below). The PL decay of 5d1–4f of Ce3+ slows down with Y content, which is attributed to there being a less intense thermal quenching effect at RT (see comparison in Fig. 8).
 | ||
| Fig. 8 Comparison of the PL decay fits of the GYSAG:Ce set under nanoLED-pulse excitation at RT (exc = 452 nm, em = 550 nm). | ||
The temperature dependence of the Ce3+ decay times of Gd2.2Y0.8, Gd2.8Y0.2, and Gd3Y0 for the temperature range of 77–500 K are displayed in Fig. 9. The PL decay curves at a temperature range of 77–200 K can be well fitted with a single exponential function with a decay time of 64 ns for both compositions. This value is very close to the lifetime of Ce3+ in other garnets (60 ns). Above 200 K, the decay curve becomes non-exponential due to the thermal ionization effect. Thus, fitting with a two-exponential function is applied. Using the Mott formula40 single-barrier quenching model:
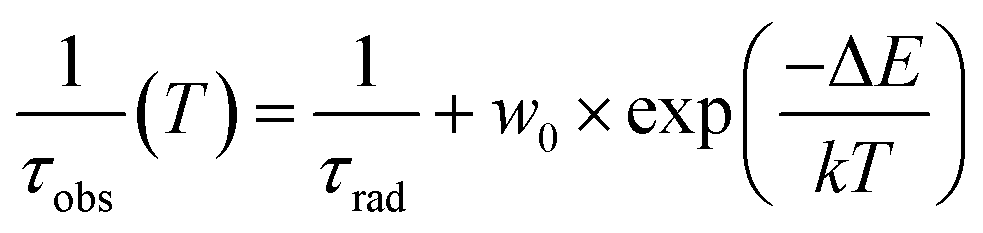 | (2) |
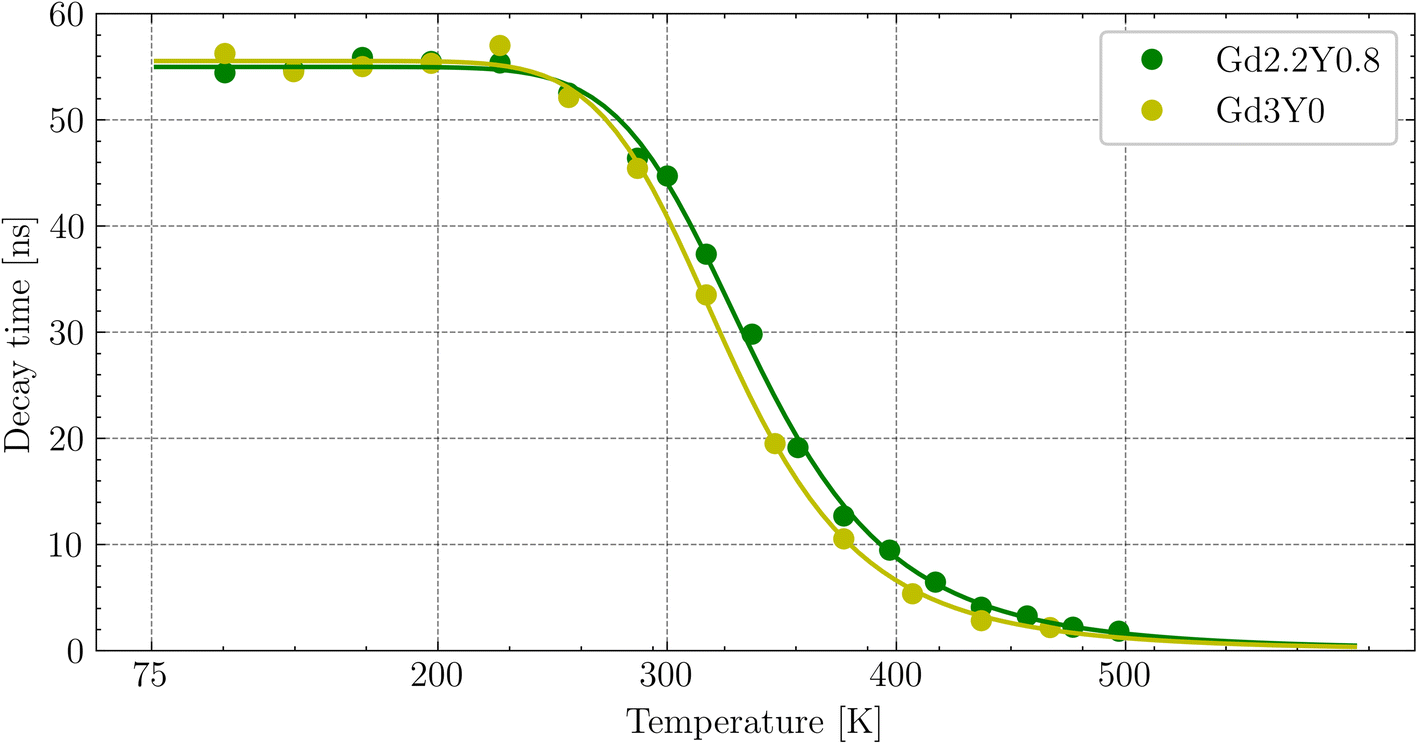 | ||
| Fig. 9 Temperature dependence of the photoluminescence decay time of Ce3+ (first component of the two-exponential fit) fitted by the curve (green and yellow lines) calculated using the single-barrier quenching model. | ||
3.4 Thermoluminescence glow curves
The thermoluminescence (TSL) glow curves after irradiation with the X-ray source at 77 K are shown in Fig. 10. The TSL glow curves were corrected for thermal quenching to account for the enhanced ionization of the 5d1 state of Ce3+ at higher temperatures. This correction was based on the temperature dependence of the PL decay times (excitation at 450 nm and emission at 550 nm) and PL emission integrals measured over the range of 77–500 K. The correction curve was constructed by interpolating the data points of the PL emission integrals, which closely matched the decay time measurements, allowing for an accurate adjustment of the TL intensity at elevated temperatures. The Gd3Y0 composition features a total of seven distinguishable glow peaks. For the Gd2.2Y0.8, except for the absent last peak around 480 K, all peaks are linearly shifted towards higher temperatures by 10–30 K. This could be due to an expected upshift of the bottom of the CB with decreasing Gd content. The TSL triple peak structure at lower temperatures (90–200 K) corresponding to the shallow electron traps is generally attributed to the anti-site defects-based traps.18,41 The glow peaks above RT (deep traps) are of different origin, most probably containing oxygen vacancies,42 and typically exhibit variability among samples of the same composition.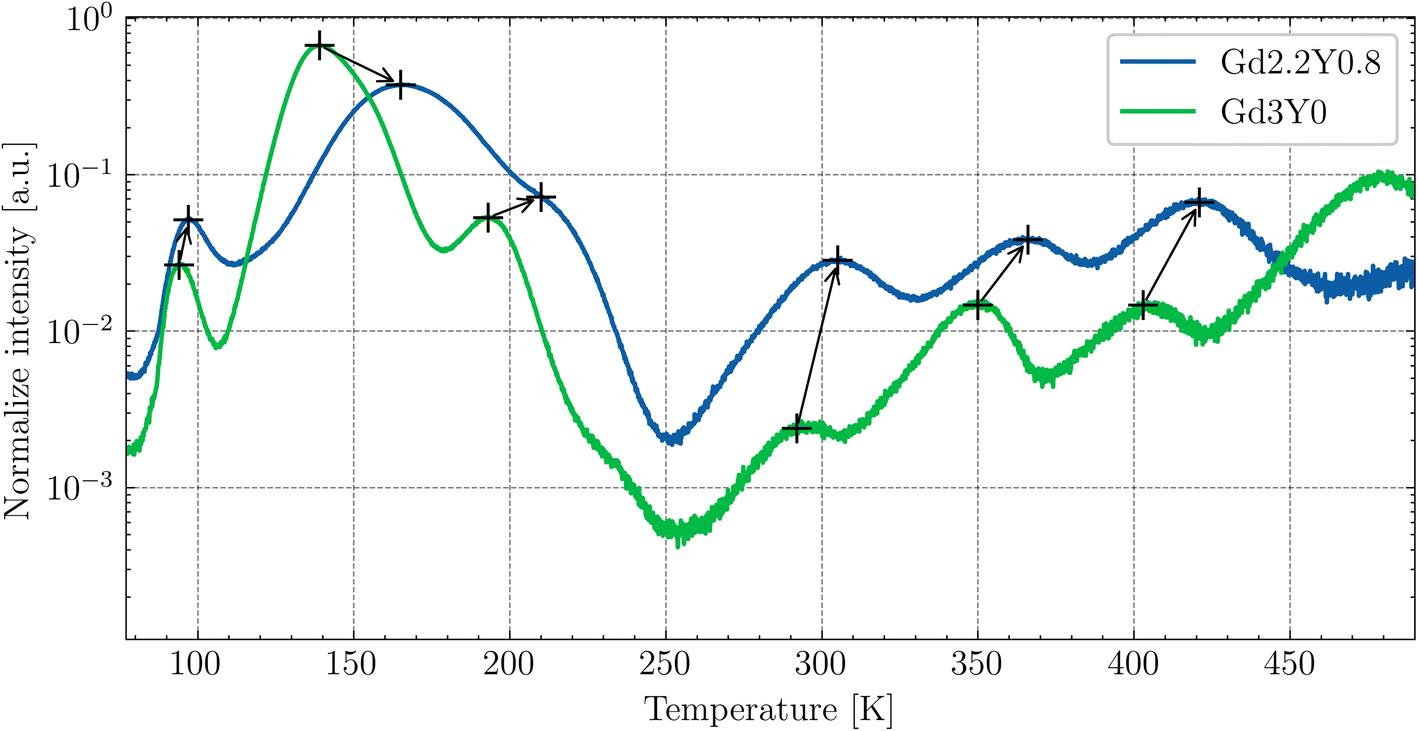 | ||
| Fig. 10 TSL glow curves for the temperature range of 77–500 K for Gd2.2Y0.8 and Gd3Y0 after irradiation with an X-ray source of 40 kV at 77 K. The peaks are shifted to the higher temperatures with Y-substitution. | ||
3.5 RL spectra
Fig. 11 displays the RL spectra of the GYSAG set, ranging from 350 to 750 nm. The intense peak of Ce3+ 5d1–4f is dominant, peaking at 546–551 nm depending on the Gd content (see Fig. S5†). The highest RL intensity value is observed for Gd3Y0 and the lowest value for Gd2.2Y0.8. The RL intensity increases with increasing Gd content from Gd2.2 to Gd2.8. However, further increase in Gd does not lead to further enhancement. It should be noted that the RL light output can be strongly affected by the crystal quality, and the surface quality can also play a role.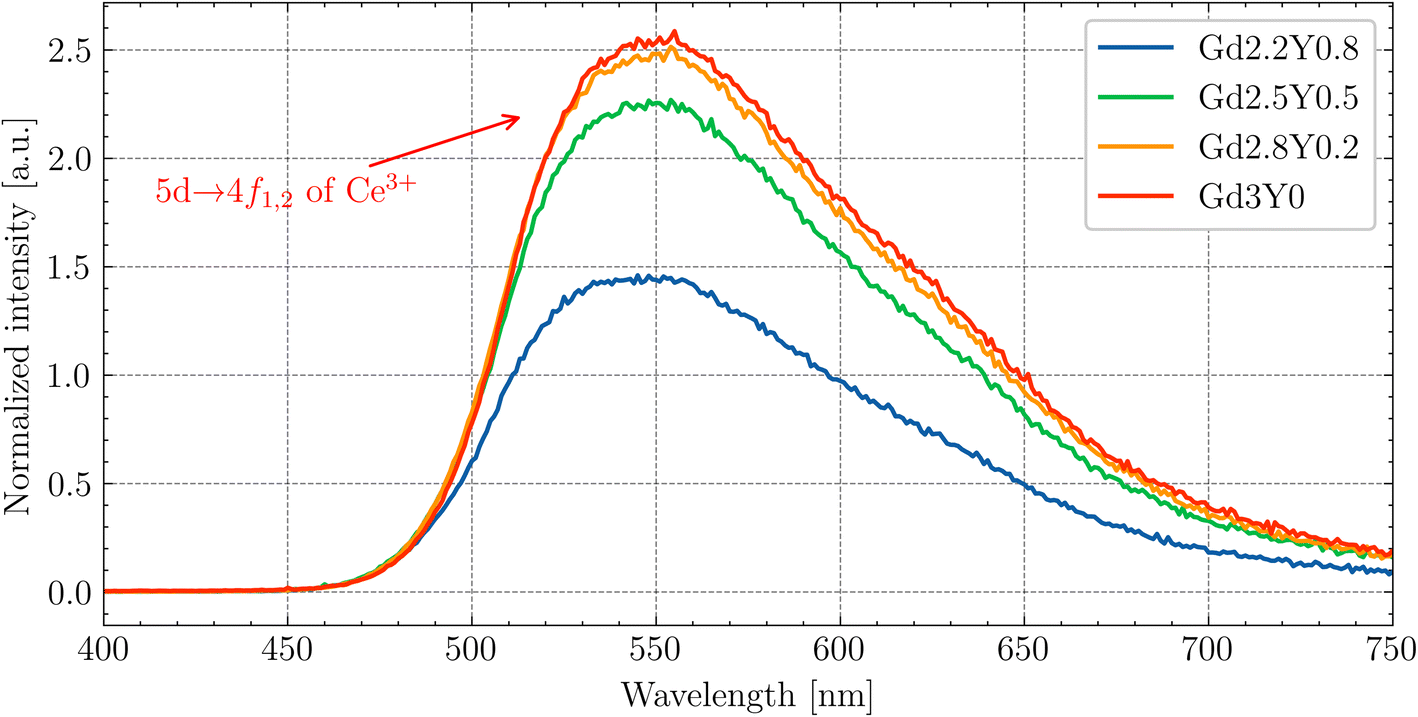 | ||
| Fig. 11 Radioluminescence spectra of the GYSAG:Ce set. The dominant peak corresponds to the Ce3+ 5d1–4f1,2 transition. | ||
Fig. 12 shows the enlarged RL spectrum ranging from 250 to 430 nm six distinguishable peaks. Peaks at 275 nm and 310 nm can be attributed to the Gd3+ transitions. The peaks at 335 nm and 355 nm have an unknown origin. The peaks at 380 nm and 415 nm can be assigned to the impurity Tb3+ (5D3 → 7F6,5) transitions. The peaks are present in all of the samples.
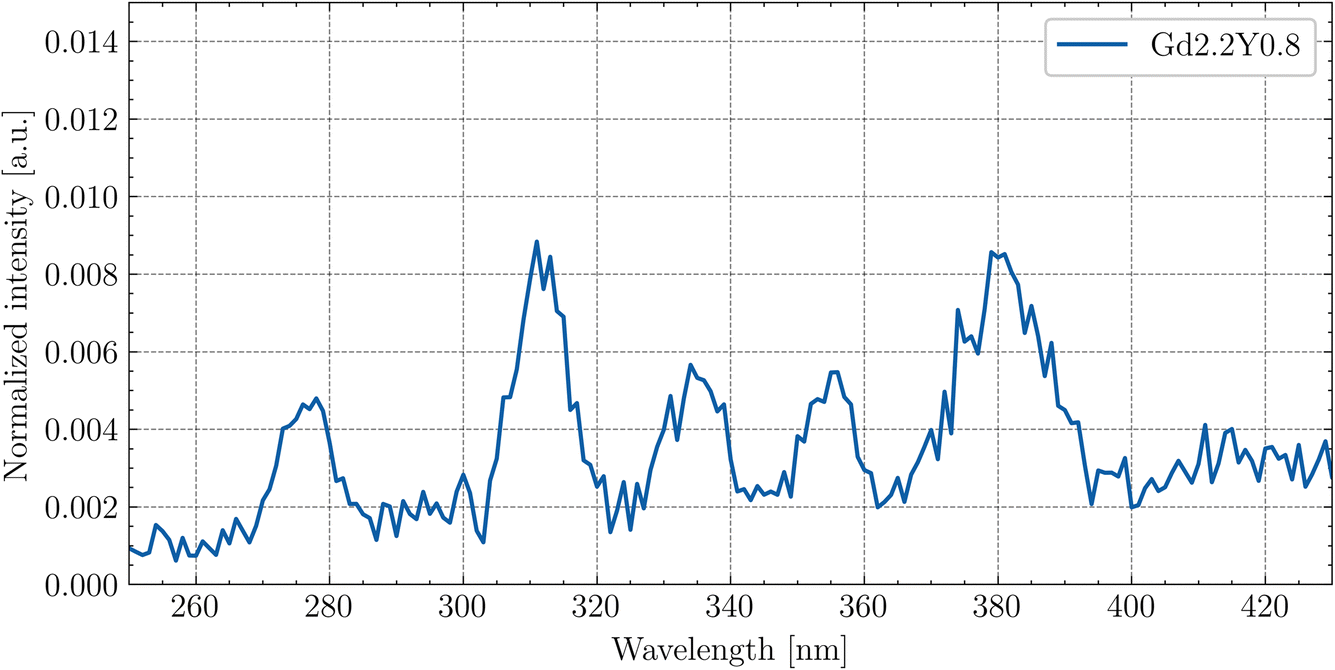 | ||
| Fig. 12 Enlarged RL spectrum of Gd2.2Y0.8 at RT in the 250–430 nm range. | ||
3.6 Scintillation properties
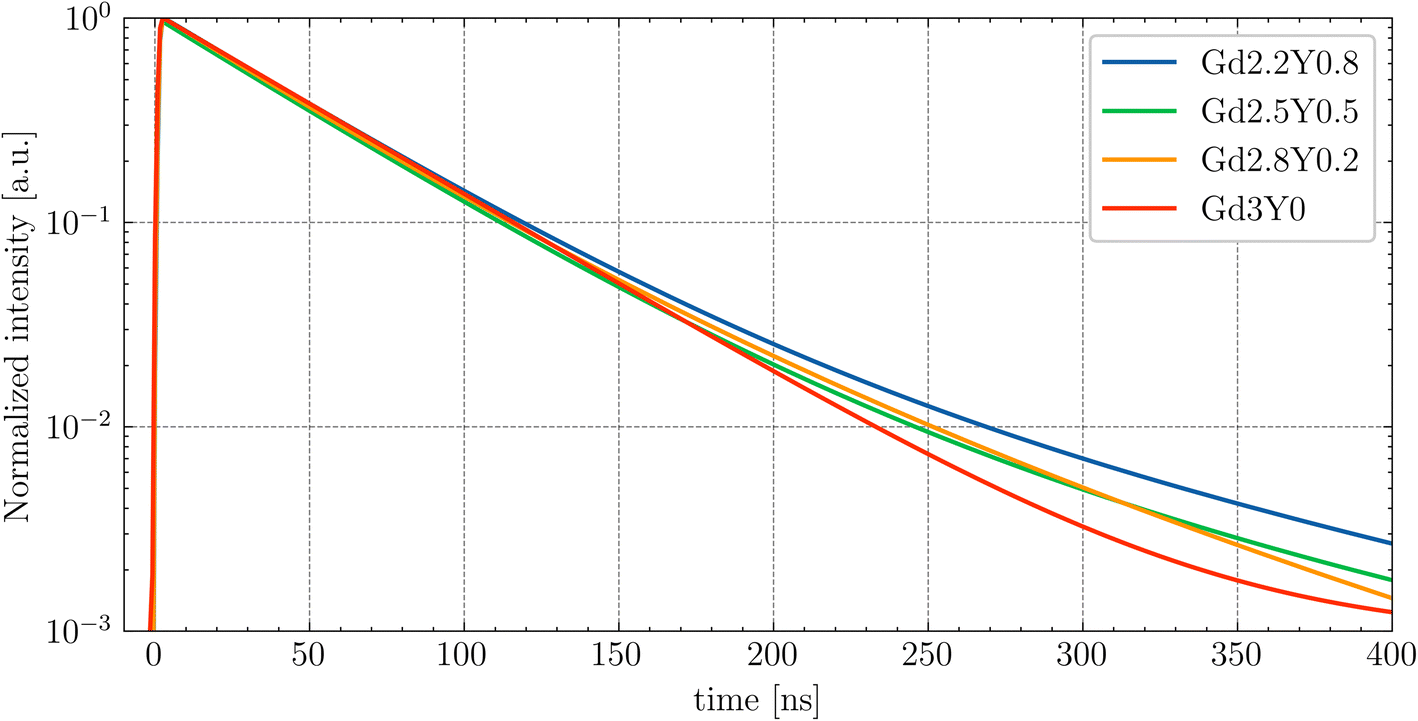
The picosecond laser-driven X-ray excitation source was used for the measurement of the scintillation rise times and scintillation decay curves (Fig. 13). The curves were fitted to multiple exponential functions, as the method used for the PL decay curves (see eqn (2)). | ||
| Fig. 13 Scintillation rise time and decay measurement of Gd2.2Y0.8, Gd2.8Y0.2 and Gd3Y0 with their fits. Excitation by a picosecond laser-driven X-ray source (40 keV, 75 ps pulse FWHM). | ||
From the fitting analysis, two key observations emerge:
(1) The sample dependence of the rise times.
(2) The scintillation decay time accelerates with decreasing Y content.
(1) There is no noticeable trend of rise time values among the sample set. The fastest rise time was observed for the Gd-poor sample, Gd2.2Y0.8, and the slowest rise time was observed for the sample Gd2.8Y0.2. The values of the scintillation rise time of other Ce-doped garnet scintillators, such as YAG:Ce, are typically on the order of a few nanoseconds.31 Since the 3d states of Sc3+ form the bottom of the CB, it seems that the Sc ion, either on its own or within more complex structures, can act as an electron trap.
(2) The observed acceleration of the scintillation decay with decreasing Y content can be attributed to the decrease of the CB bottom (see TSL in Fig. 10), which makes all of the electron traps shallower and is consistent with the observation in ref. 43. The longer decay components are caused by delayed radiative recombination processes, resulting from a combination of re-trapping and energy migration through the Gd3+ sublattice. The best value of scintillation decay was obtained for the Y-free sample Gd3Y0, with a first component of scintillation decay equal to 116 ns, consistent with values reported elsewhere.14,44 The scintillation decay and rise time can be potentially improved by Mg2+ codoping,9,24 which would result in the formation of Ce4+ as an effective electron trap as competing with the crystal lattice defects.45
The normalized afterglow curves of the sample set (Gd2.5Y0.5, Gd3Y0) are shown in Fig. 14. All the samples have very low afterglow, with the intensity drop by 3–4 orders of magnitude in a few tens of ms after X-ray cut-off. Fig. 14B compares the samples based on the average value of the intensity of afterglow in a 20 ms time window after the X-ray cut-off. Without a distinct trend among the sample set, Gd3Y0 exhibits the best afterglow, while Gd2Y0.5 displays the worst one.
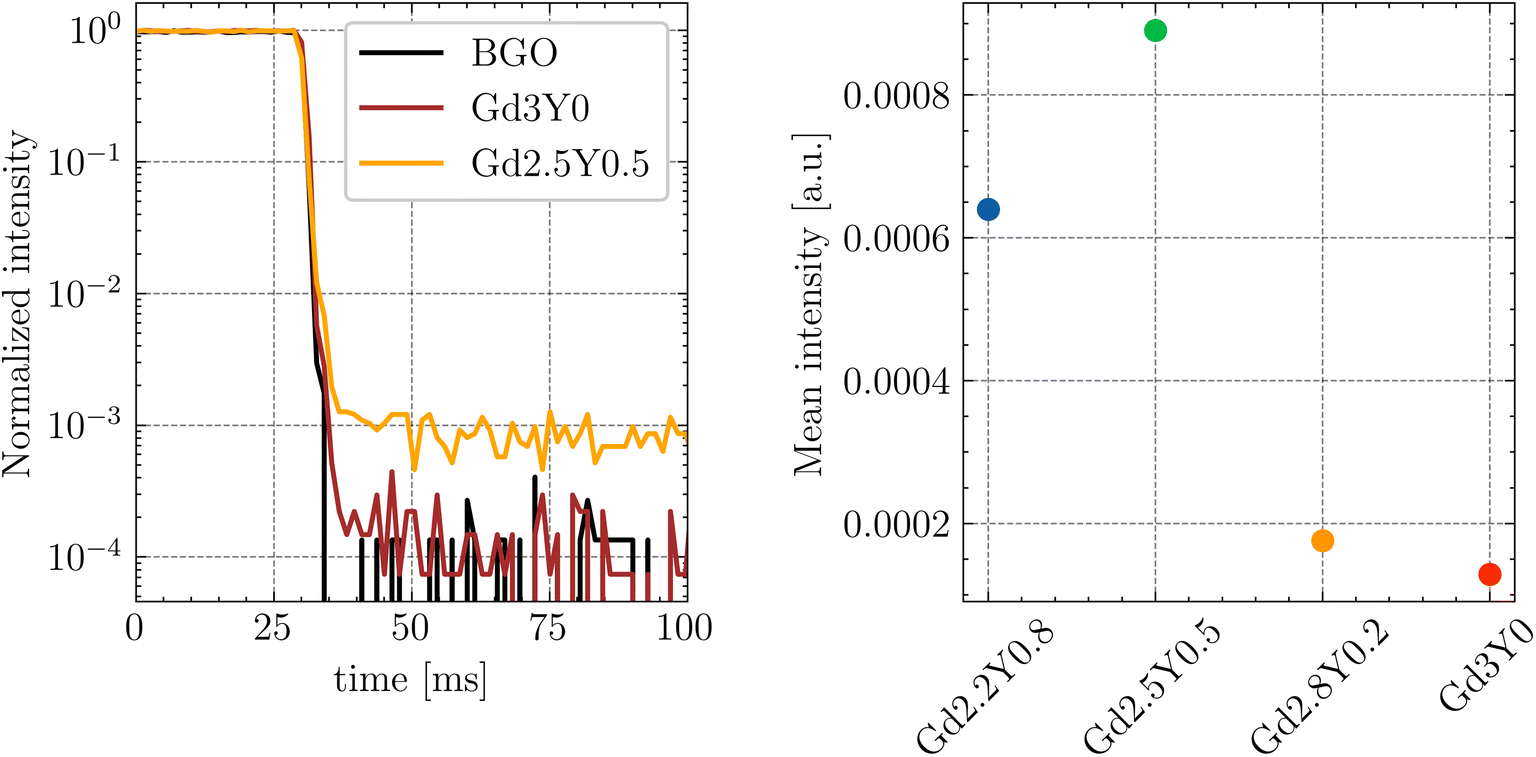 | ||
| Fig. 14 (A) Normalized afterglow curves of Gd3Y0 and Gd2.5Y0.5 samples. (B) Mean afterglow value calculated as a mean of intensities in the range of 20 ms after X-ray cut-off. | ||
The LY was measured at RT under the same conditions with 1 μs shaping time, under the excitation of the Cs-137 radioisotope (662 keV). The LY value gradually increases with the increasing content of Gd from Gd2.2Y0.8 to Gd3Y0 (see Table 4). Interestingly, the sample without yttrium (Gd3Y0), which has a stoichiometric Gd3Sc2Al3O12 host, shows 50% higher LY compared to the congruent Gd2.88Sc1.89Al3.23O12 host composition reported in ref. 14. However, it is worth noting that the samples prepared by the same technology and with the composition where the Ga instead of Sc is in the formula (namely, Gd2YGa2Al3O12:Ce and Gd2YGa3Al2O12:Ce) show much higher LY of 25700 and 44000 ph per MeV, respectively.46
| Sample | LY (ph per MeV) |
|---|---|
| Gd2.2Y0.8 | 8500 |
| Gd2.5Y0.5 | 12000 |
| Gd2.8Y0.2 | 14000 |
| Gd3Y0 | 15100 |
4 Conclusion
In this study, we have successfully grown Gd(3−x)Sc2YxAl3O12:Ce (x = 0, 0.2, 0.5, 0.8) single crystals by μ-PD method. Our goal was to increase the light yield by reducing the thermal quenching of Ce3+ at room temperature, i.e., increasing the activation energy for the quenching process at the 5d1 state of Ce3+. This was achieved through partial substitution of Gd by Y. Indeed, the temperature-dependent measurement of the photoluminescence decay and TSL glow curves indicate an increase of the ionization barrier by approximately 5% for Gd2.2Sc2Y0.8Al3O12:Ce (Gd2.2Y0.8) compared to Gd3Sc2Al3O12:Ce (Gd3Y0). However, the scintillation light yield and integral efficiency decrease with increasing Y amount. The highest LY value of 15100 ph per MeV was measured for Gd3Y0. The scintillation decay accelerates towards Gd-rich composition with a first component equal to 116 ns for Gd3Y0. A very low afterglow (drop by 3–4 orders of magnitude after X-ray cut-off) was observed for all samples. The excitation spectra in the VUV region suggest a rather poor efficiency of the energy transfer through the host in the Y-admixed GSAG host. These results show that the thermal quenching of Ce3+ around room temperature is not the primary cause of the low light yield in GSAG host. Furthermore, we found that the stoichiometric composition of the host enables much higher scintillation performance compared to the congruent one, although it is still behind that of similar compositions where the Ga cation is present in the formula instead of the Sc one.
Data availability
The data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Czech Science Foundation project no. 21-17731S.References
- GAGG+|Crytur.cz [Internet], [cited 2024 Jan 10], available from: http://www.crytur.com/materials/gagg/.
- GAGG(Ce): gamma-ray detector|C&A Corporation [Internet], [cited 2022 May 13], available from: https://c-and-a.jp/products_details/products_detail_GAGG.html.
- C. Wang, Y. Wu, D. Ding, H. Li, X. Chen and J. Shi, et al., Optical and scintillation properties of Ce-doped (Gd2Y1)Ga2.7Al2.3O12 single crystal grown by Czochralski method, Nucl. Instrum. Methods Phys. Res., Sect. A, 2016, 820, 8–13 CrossRef CAS.
- Y. Wu, F. Meng, Q. Li, M. Koschan and C. L. Melcher, Role of Ce4+ in the Scintillation Mechanism of Codoped Gd3Ga3Al2O12:Ce, Phys. Rev. Appl., 2014, 2(4), 044009 CrossRef CAS.
- M. Korzhik, V. Alenkov, O. Buzanov, G. Dosovitskiy, A. Fedorov and D. Kozlov, et al., Engineering of a new single-crystal multi-ionic fast and high-light-yield scintillation material (Gd 0.5 –Y 0.5)3 Al 2 Ga 3 O 12 :Ce,Mg, CrystEngComm, 2020, 22(14), 2502–2506 RSC.
- J. Iwanowska-Hanke, K. Brylew, M. E. Witkowski, P. Sibczynski, T. Szczesniak and M. Moszynski, et al., Cerium-doped gadolinium fine aluminum gallate in scintillation spectrometry, Nucl. Instrum. Methods Phys. Res., Sect. A, 2020, 979, 164464 CrossRef CAS.
- M. Nikl, K. Kamada, V. Babin, J. Pejchal, K. Pilarova and E. Mihokova, et al., Defect Engineering in Ce-Doped Aluminum Garnet Single Crystal Scintillators, Cryst. Growth Des., 2014, 14(9), 4827–4833 CrossRef CAS.
- K. Kamada, S. Kurosawa, P. Prusa, M. Nikl, V. V. Kochurikhin and T. Endo, et al., Cz grown 2-in. size Ce:Gd3(Al,Ga)5O12 single crystal; relationship between Al, Ga site occupancy and scintillation properties, Opt. Mater., 2014, 36(12), 1942–1945 CrossRef CAS.
- L. Martinazzoli, S. Nargelas, P. Boháček, R. Calá, M. Dušek and J. Rohlíček, et al., Compositional engineering of multicomponent garnet scintillators: towards an ultra-accelerated scintillation response, Mater. Adv., 2022, 3(17), 6842–6852 RSC.
- K. Kamada, M. Nikl, T. Kotaki, H. Saito, F. Horikoshi and M. Miyazaki, et al., Multiple shaped-crystal growth of oxide scintillators using Mo crucible and die by the edge defined film fed growth method, J. Cryst. Growth, 2020, 535, 125510 CrossRef CAS.
- T. Wu, L. Wang, Y. Shi, T. Xu, H. Wang and J. Fang, et al., Fast (Ce,Gd)3Ga2Al3O12 Scintillators Grown by the Optical Floating Zone Method, Cryst. Growth Des., 2022, 22(1), 180–190 CrossRef CAS.
- F. Zajíc, J. Pospíšil, R. Kučerková, P. Boháček and M. Nikl, Growth of GSAG:Ce scintillator by floating zone method under pressurized oxygen atmosphere, J. Cryst. Growth, 2024, 627, 127479 CrossRef.
- Y. Akira, K. Vladimir, Y. Masao, M. Rikito, T. Taketoshi and T. Isao, et al., Bulk Single-Crystal Growth of Ce/Gd3 (Al,Ga)5 O12 from Melt without a Precious Metal Crucible by Pulling from a Cold Container, Cryst. Growth Des., 2023, 23(4), 2048–2054 CrossRef CAS.
- O. Zapadlík, J. Pejchal, R. Kučerková, A. Beitlerová and M. Nikl, Composition-Engineered GSAG Garnet: Single-Crystal Host for Fast Scintillators, Cryst. Growth Des., 2021, 21(12), 7139–7149 CrossRef.
- U. Happek, J. Choi and A. M. Srivastava, Observation of cross-ionization is Gd3Sc2Al3O12:Ce3+, J. Lumin., 2001, 94–95, 7–9 CrossRef.
- Y. Wu, M. Nikl, V. Jary and G. Ren, Thermally induced ionization of 5d1 state of Ce3+ ion in Gd3Ga3Al2O12 host, Chem. Phys. Lett., 2013, 574, 56–60 CrossRef CAS.
- V. Babin, K. Blazek, A. Krasnikov, K. Nejezchleb, M. Nikl and T. Savikhina, et al., Luminescence of undoped LuAG and YAG crystals, Phys. Status Solidi C, 2005, 2(1), 97–100 CrossRef CAS.
- M. Nikl, E. Mihokova, J. Pejchal, A. Vedda, Y. Zorenko and K. Nejezchleb, The antisite LuAl defect-related trap in Lu3Al5O12:Ce single crystal, Phys. Status Solidi B, 2005, 242(14), R119–R121 CrossRef CAS.
- Y. Zorenko, V. Gorbenko, I. Konstankevych, A. Voloshinovskii, G. Stryganyuk and V. Mikhailin, et al., Single-crystalline films of Ce-doped YAG and LuAG phosphors: advantages over bulk crystals analogues, J. Lumin., 2005, 114(2), 85–94 CrossRef CAS.
- D. Spassky, N. Kozlova, E. Zabelina, V. Kasimova, N. Krutyak and A. Ukhanova, et al., Influence of the Sc cation substituent on the structural properties and energy transfer processes in GAGG:Ce crystals, CrystEngComm, 2020, 22(15), 2621–2631 RSC.
- Y. N. Xu, W. Y. Ching and B. K. Brickeen, Electronic structure and bonding in garnet crystals Gd 3 Sc 2 Ga 3 O 12 , Gd 3 Sc 2 Al 3 O 12 , and Gd 3 Ga 3 O 12 compared to Y 3 Al 3 O 12, Phys. Rev. B:Condens. Matter Mater. Phys., 2000, 61(3), 1817–1824 CrossRef CAS.
- M. Fasoli, A. Vedda, M. Nikl, C. Jiang, B. P. Uberuaga and D. A. Andersson, et al., Band-gap engineering for removing shallow traps in rare-earth Lu 3 Al 5 O 12 garnet scintillators using Ga 3 + doping, Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 84(8), 081102 CrossRef.
- W. Chewpraditkul, N. Pattanaboonmee, W. Chewpraditkul, O. Sakthong, K. J. Kim and M. Yoshino, et al., Luminescence and scintillation properties of Gd3Sc2(Al3-xGax)O12:Ce (x = 1, 2, 3) garnet crystals, Radiat. Phys. Chem., 2021, 187, 109559 CrossRef CAS.
- O. Zapadlík, J. Pejchal, F. Levchenko, R. Kučerková, A. Beitlerová and V. Vaněček, et al., The Ga-admixed GSAG:Ce single crystal scintillator: Composition tuning, J. Lumin., 2023, 263, 119984 CrossRef.
- W. Chewpraditkul, K. Wantong, W. Chewpraditkul, N. Pattanaboonmee, R. Kucerkova and A. Beitlerova, et al., Effects of Sc3+ admixture on luminescence and scintillation properties of Ce3+-doped Lu2Y(Al5-xScx)O12 (x = 1, 1.5, 2) garnet single-crystalline films, Opt. Mater., 2023, 145, 114417 CrossRef CAS.
- K. Wantong, W. Chewpraditkul, W. Chewpraditkul, M. Rathaiah, M. Kucera and S. Danis, et al., Optical, luminescence and scintillation characteristics of Gd3Sc2(Al3-xGax)O12: Ce,Mg (x = 0, 1, 2) single crystalline films, Opt. Mater., 2022, 134, 113240 CrossRef CAS.
- S. Yamazaki, F. Marumo, K. Tanaka, H. Morikawa, N. Kodama and K. Kitamura, et al., A Structural Study of Facet and Off-Facet Parts of Rare-Earth Garnets, Gd3Sc2Al3O12, Gd3Sc2Ga3O12, and La3Lu2Ga3O12, J. Solid State Chem., 1994, 108(1), 94–98 CrossRef CAS.
- G. B. Lutts, A. L. Denisov, E. V. Zharikov, A. I. Zagumennyi, S. N. Kozlikin and S. V. Lavrishchev, et al., GSAG and YSAG: a study on isomorphism and crystal growth, Opt. Quantum Electron., 1990, 22(S1), S269–S281 CrossRef CAS.
- K. L. Hovhannesyan, M. V. Derdzyan, G. Badalyan, G. Kharatyan, J. Pejchal and M. Nikl, et al., Growth of GSAG:Ce scintillation crystals by the Bridgman method: influence of Ce concentration and codoping, CrystEngComm, 2024, 26(35), 4812–4819 RSC.
- M. Moszyński, T. Ludziejewski, D. Wolski, W. Klamra and L. O. Norlin, Properties of the YAG:Ce scintillator, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 345(3), 461–467 CrossRef.
- O. Zapadlík, M. Nikl, J. Polák, P. Průša and V. Linhart, Engineering of YAG:Ce to improve its scintillation properties, Opt. Mater.:X, 2022, 15, 100165 Search PubMed.
- K. Bartosiewicz, V. Babin, K. Kamada, A. Yoshikawa, J. A. Mares and A. Beitlerova, et al., Luminescence quenching and scintillation response in the Ce3+ doped GdxY3−xAl5O12 (x = 0.75, 1, 1.25, 1.5, 1.75, 2) single crystals, Opt. Mater., 2017, 63, 134–142 CrossRef CAS.
- C. D'Ambrosio, H. Leutz, D. Piedigrossi, E. Rosso, V. Cencelli and F. De Notaristefani, et al., Gamma spectroscopy and optoelectronic imaging with hybrid photon detector, Nucl. Instrum. Methods Phys. Res., Sect. A, 2003, 497(1), 186–197 CrossRef.
- J. A. Mares, M. Nikl, A. Beitlerova, C. D'Ambrosio, F. De Notaristefani and K. Blazek, et al., Scintillation photoelectron Nphels(E) and light LY(E) yields of YAP:Ce and YAG:Ce crystals, Opt. Mater., 2003, 24(1–2), 281–284 CrossRef CAS.
- A. Yoshikawa, M. Nikl, G. Boulon and T. Fukuda, Challenge and study for developing of novel single crystalline optical materials using micro-pulling-down method, Opt. Mater., 2007, 30(1), 6–10 CrossRef CAS.
- A. Yoshikawa and V. Chani, Growth of Optical Crystals by the Micro-Pulling-Down Method, MRS Bull., 2009, 34(4), 266–270 CrossRef CAS.
- I. A. Kaurova, E. N. Domoroshchina, G. M. Kuz’micheva and V. B. Rybakov, Evaluation of stability region for scandium-containing rare-earth garnet single crystals and their congruent-melting compositions, J. Cryst. Growth, 2017, 468, 452–456 CrossRef CAS.
- J. L. Wu, G. Gundiah and A. K. Cheetham, Structure–property correlations in Ce-doped garnet phosphors for use in solid state lighting, Chem. Phys. Lett., 2007, 441(4–6), 250–254 CrossRef CAS.
- Z. Song, Z. Xia and Q. Liu, Insight into the Relationship between Crystal Structure and Crystal-Field Splitting of Ce 3+ Doped Garnet Compounds, J. Phys. Chem. C, 2018, 122(6), 3567–3574 CrossRef CAS.
- N. F. Mott and R. W. Gurney, Electronic Processes in Ionic Crystals, Clarendon Press, 1948 Search PubMed.
- E. Mihóková, M. Nikl, J. A. Mareš, A. Beitlerová, A. Vedda and K. Nejezchleb, et al., Luminescence and scintillation properties of YAG:Ce single crystal and optical ceramics, J. Lumin., 2007, 126(1), 77–80 CrossRef.
- A. Vedda, D. Di Martino, M. Martini, V. V. Laguta, M. Nikl and E. Mihokova, et al., Thermoluminescence of Zr-codoped Lu3Al5O12:Ce crystals, Phys. Status Solidi A, 2003, 195(3), R1–R3 CrossRef CAS.
- J. A. Mares, M. Nikl, A. Beitlerova, P. Horodysky, K. Blazek and K. Bartos, et al., Scintillation Properties of ${\rm Ce}^{3+}$- and ${\rm Pr}^{3+}$-Doped LuAG, YAG and Mixed ${\rm Lu}_{\rm x}{\rm Y}_{1-{\rm x}}{\rm AG}$ Garnet Crystals, IEEE Trans. Nucl. Sci., 2012, 59(5), 2120–2125 CAS.
- A. Kling, D. Kollewe and D. Mateika, Scintillation properties of cerium-doped gadolinium-scandium-aluminum garnets, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 346(1), 205–212 CrossRef CAS.
- V. Babin, P. Herman, M. Kucera, M. Nikl and S. Zazubovich, Effect of Mg2+ co-doping on the photo- and thermally stimulated luminescence of the (Lu,Gd)3(Ga,Al)5O12:Ce epitaxial films, J. Lumin., 2019, 215, 116608 CrossRef CAS.
- K. Kamada, T. Yanagida, J. Pejchal, M. Nikl, T. Endo and K. Tsutumi, et al., Scintillator-oriented combinatorial search in Ce-doped (Y,Gd)3 (Ga,Al)5 O12 multicomponent garnet compounds, J. Phys. D: Appl. Phys., 2011, 44(50), 505104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07622b |
| This journal is © The Royal Society of Chemistry 2025 |