
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable ultrasound-assisted synthesis of hydroxy imidazole N-oxides and evaluation of their anti-proliferative activities; mechanistic insights into the deoximation of dioximes†
Munna Mukhia a,
Sagar Raia,
Yachna Raia,
Koushik Chakrabortya,
Sangita Deyb,
Bipransh Kumar Tiwaryd,
Dhiraj Brahmanc,
Anoop Kumarb and
Kiran Pradhan*a
a,
Sagar Raia,
Yachna Raia,
Koushik Chakrabortya,
Sangita Deyb,
Bipransh Kumar Tiwaryd,
Dhiraj Brahmanc,
Anoop Kumarb and
Kiran Pradhan*a
aDepartment of Chemistry, University of North Bengal, Raja Rammohunpur, Dist. Darjeeling, 734013, India. E-mail: kpradhan@nbu.ac.in
bDepartment of Chemistry, St. Joseph's College, P.O. North Point, Dist. Darjeeling, 734104, India
cDepartment of Biotechnology, University of North Bengal, Raja Rammohunpur, Dist. Darjeeling, 734013, India
dDepartment of Microbiology, St. Xavier's College, Rajganj, Dist. Jalpaiguri, 734013, India
First published on 15th January 2025
Abstract
The development of synthetic methodologies that promote greener reactions have become so essential that it has slowly shaped the way chemists think about the construction of physiologically and chemically active compounds. The acid-catalyzed iminoketone – aldehyde condensations leading to Hydroxy imidazole N-oxides serve as robust strategies for forming C–N bonds. Considering all the existing challenges that come with the use of solvent and energy-intensive methodologies, herein a green synthetic strategy using ultrasound with optimization of reaction conditions and thorough investigation into the mechanism for obtaining the best yields are reported. Additionally, the importance of the Hydroxy Imidazole N-oxides synthesis is highlighted by their in vitro antiproliferative activity study. Though only satisfactory, the results are promising. It gives us scope for building upon this simple scaffold with appropriate functionalization, which may lead to furhter enhancement in their anti-proliferative activity.
Introduction
The improvement of synthetic methodologies using energy-efficient methods lies at the heart of upholding the principles of Green Chemistry. Organic synthesis using ultrasonics, akin to mechanochemical synthesis is a well-known method used for carrying out synthetic transformations since the early 1980s as they are energy efficient and often give better yields.1,2 The presence of a liquid medium in a system undergoing ultrasonic irradiation is imperative to facilitate the cavitation process, however, it is also found that the coupling of ultrasound into chemically useful cavitation events is a low-yield event even though the production of ultrasound from electrical power is extremely efficient.3 Nevertheless, the plausible explanation for the enhancement of reaction rates with the help of the cavitation principle carries a lot of weight. However, sonochemical reactions carried out either in the absence of solvent or in slurries (liquidus state) are less studied compared to reactions in solvents.The solvent-free syntheses of imidazoles, privileged heterocycles with diverse applications, have been extensively carried out while the neat syntheses of its N-oxide derivatives still remain unambiguously absent, except for a few. The syntheses of these compounds are equally important as their applications are unequivocally diverse (Fig. 1); from being potent HIV-1 integrase4 and HPPD inhibitors5 and showing potent antitrypanosomal activity,6 they have also been used as precatalysts in aqueous Suzuki coupling reactions7 and as co-formers in the formation of pharmaceutical co-crystals.8 They also serve as synthetic intermediates in fields as diverse as drug development (in the synthesis of anti-inflammatory agent9), supramolecular chemistry (anion recognition element7), and catalysis (additive for Grubbs II catalyst10). They also act as synthons for a variety of significant transformations e.g. in the halogenation of imidazole at the 2-position,11,12 in the hydroheteroarylation of unactivated alkenes,13 and many others.14–19
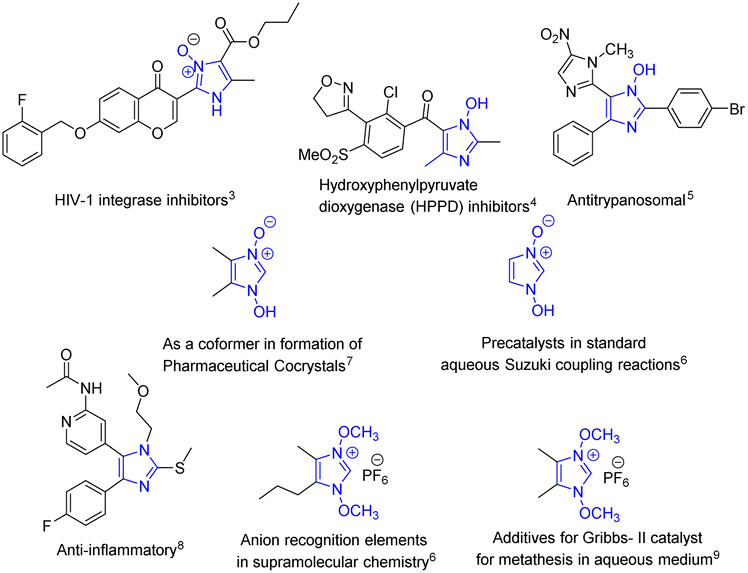 | ||
| Fig. 1 Imidazole N-oxide derivatives in diverse applications. | ||
As such, there is continued interest in the development of procedures for the efficient synthesis of these compounds, especially under mild conditions.
Further, the N-oxide motif has been successfully employed in several recent drug development projects, however not many heterocyclic N-oxides have been found to show anti-proliferative activity.20 Available studies indicate that the presence of the N-oxide group in a compound significantly increases its inhibitory potency. This is highly probable because of the high electron density of the heterocyclic N-oxide oxygen making them strong hydrogen bond acceptors.21 In light of the recent developments; we envisaged that the 1-hydroxyimidazole-3-oxides may as well exhibit some anti-proliferative activities. Hence, the effects of some of the compounds were investigated for their efficacy as anti-proliferative agents. The promising results bode well for the design and development of these scaffolds as efficient anticancer agents and their potential application in the pharmaceutical industry. Due to our continued interest in the design and development of these scaffolds for improving their potency and candidacy as effective anticancer agents, efficient and greener synthetic methodology for their syntheses became equally imperative.
Since the first reported synthesis of 1-hydroxy- imidazole-3-oxide derivatives over a century ago,22 various methods have been developed for the syntheses of these compounds but the most general methods still continue to be from α-dicarbonyl compounds, aldehydes and hydroxylamine (Pathway A in Scheme 1)7 and their variations, involving monoximes of α-dicarbonyl compounds, aldehydes and hydroxylamine (Pathway B in Scheme 1),23 by reaction of glyoximes with aldehydes (LaParola's method,24 Pathway C in Scheme 1) and by the reaction of monoximes with aldehyde oximes (Diel's method,25 Pathway D in Scheme 1). All these traditional methods of syntheses involve either fusion of the reactants at high temperatures or carrying out the reaction in alcohol in the presence of acetic acid or other Brønsted acids. However, the yields of the reactions via the traditional methods are found to be satisfactory and studies on developing energy efficient high yielding methodologies were surprisingly found to be absent. Herein, we report the syntheses of the compounds via a solvent-free ultrasonic-assisted protocol with very high yields achieved in very less time. The rate of formation of the products from all four pathways was thoroughly investigated and compared and plausible insights into the mechanism derived, which has hitherto not been done. Most significantly, this study investigates the deoximation of hydroxyiminoketones and dioximes (in the presence of aldehyde) that have been found to play a crucial role in the formation of the products.
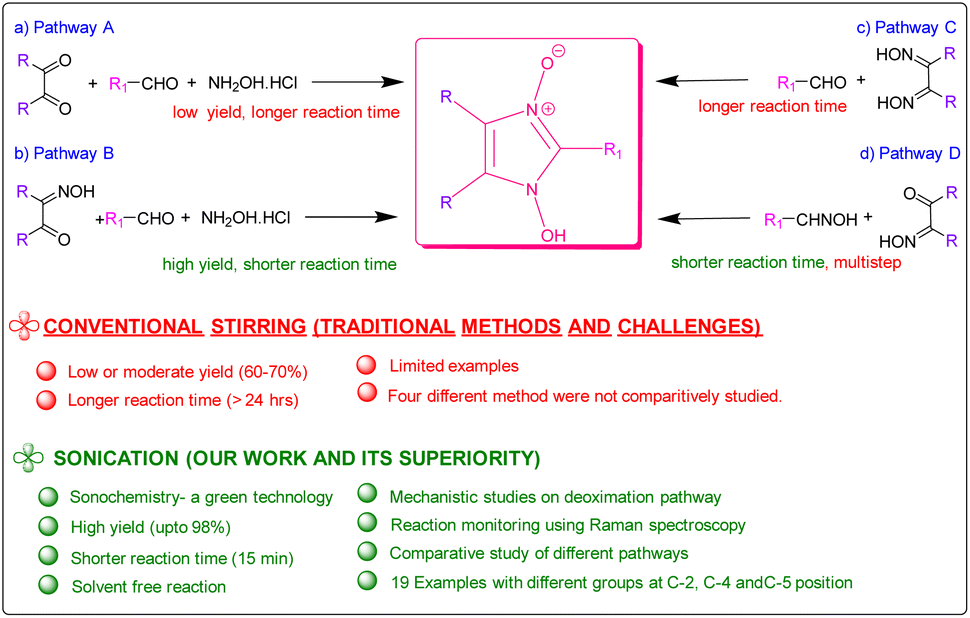 | ||
| Scheme 1 Summary of methods for the synthesis of hydroxy imidazole N-oxides. | ||
Thus, focusing on the Hydroxy Imidazole N-oxide synthesis here had two attractive features: first, we aimed to demonstrate the applicability of Sonochemistry under neat conditions to access these versatile heterocycles. Second, and more generally, it was hoped that a concise analysis of the reaction path by employing spectroscopic methods would allow the identification of intermediates and by-products known to reduce the yield of the final product.
Results and discussion
The viability of two mechanochemical synthetic procedures, mechanical stirring and ultrasound irradiation as better alternatives to the traditional synthetic methods for the synthesis of hydroxy Imidazole N-oxides was investigated under similar conditions. Out of the four pathways, Pathway B was arbitrarily chosen, and equimolar amounts of the reactants diacetyl monoxime, para-hydroxy benzaldehyde, and conc. HCl and an excess of hydroxylammonium hydrochloride were taken together for the synthesis of 1-hydroxy, 2-(p-hydroxyphenyl) 4,5-dimethyl imidazole 3-oxide (1c) as shown in Scheme 2.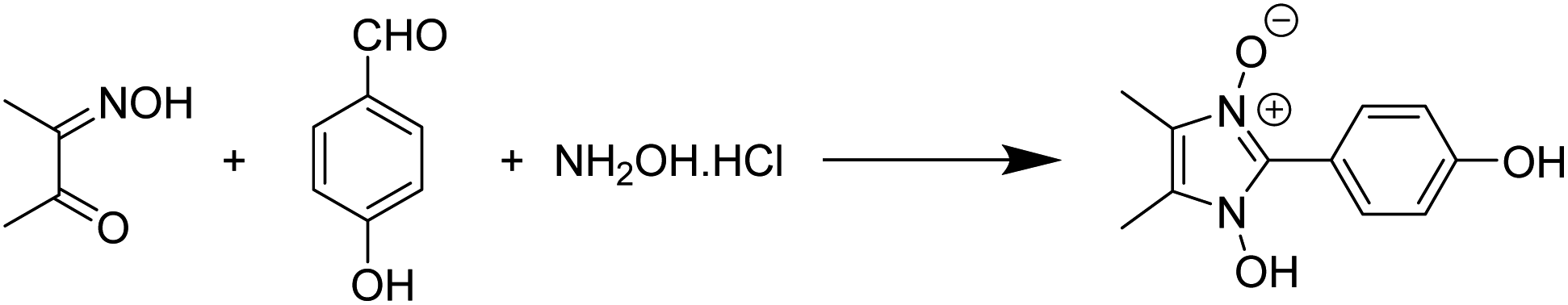 | ||
| Scheme 2 Synthesis of 1-hydroxy, 2-(p-hydroxyphenyl) 4,5-dimethyl imidazole 3-oxide (1c) via Pathway B. | ||
The reaction performed under mechanical stirring in a magnetic stirrer at room temperature gave a conversion of only 60% into the product (1c) after 24 h in methanol as the reaction medium (entry 1, Table 1). Traditional synthesis under refluxing conditions was also reported to give similar yields.15 Very interestingly, a simple mechanical stirring of the neat reaction mixture also yielded 60% of the product in just 1 hour of reaction (entry 3, Table 1). However, during mechanical stirring, it was found that in the absence of any solvent, certain parts of the reaction vessel, such as the sides, were prone to the caking of the solid reactants and thus “dead space” where the reactants were not well-mixed was formed. Despite the enhanced reaction rates initially, an intermediate (discussed in detail in the following sections) remained detectable for a long period of time and the reaction did not progress further. Multiple repetitions and scaling up of the reaction did not give better results. To reduce the compaction and stickiness of the reactants during mechanical stirring, the reaction was performed in silica as a solid support. It was found that mechanical stirring of the reaction mixture at room temperature in silica gave the product in 60% yield after 6 hours (entry 2, Table 1), which was much better than the reaction performed in a solvent but only satisfactory in comparison to the reaction performed under neat conditions. Increasing the reaction time did not result in an improved yield.
| Entry | Medium | Method | Reaction time | Yield (%) |
|---|---|---|---|---|
| a Equivalent amount of HCl was used in all cases. All reactions were performed keeping the ultrasonic bath temperature at 30 °C. | ||||
| 1 | MeOH | Stirring | 24 h | 60 |
| 2 | Silica | Stirring | 6 h | 60 |
| 3 | Solvent-free | Stirring | 1 h | 60 |
| 4 | Silica | Sonication | 5 h | 75 |
| 5 | Solvent-free | Sonication | 50 min | 85 |
| 6 | MeOH | Sonication | 5 h | 65 |
| 7 | H2O | Sonication | 5 h | 60 |
In contrast, by replacing mechanical stirring as well as conventional heating, with ultrasound irradiation at room temperature, better conversion into (1c) was obtained in all cases where reactions were conducted under solvent-free conditions. Although, the reaction in silica gave 75% yield in 5 hours (entry 4, Table 1), reaction in the absence of silica significantly gave higher yields (85% yield) in much lesser time, ca 50 minutes under ultrasound irradiation (entry 5, Table 1), while the reactions in methanol and water as solvents gave yields of 65% and 60% respectively (entries 6 & 7, Table 1). Undoubtedly, ultrasound irradiation of the reaction mixture under neat conditions was found to give better results. Though the conditions were solvent-free and the initial mixing of the reactants resulted in a solid mix, sonication led to a gradual change of the solid mix into a liquidus state, contrary to the caking observed during mechanically stirred reactions. The mechanically stirred neat reactions are also equally efficient under room temperature initially, but they fail to go to completion because of compaction and caking. Sonication on the other hand, by initiating the formation of a liquidus state, provides effective mass transfer and avoids compaction of the reaction mixture thereby increasing reaction rates and yields.
Since 1-hydroxy imidazole-3-oxides have also been synthesized via other pathways as shown in Scheme 1, we also investigated the formation of products via all the pathways in the presence of HCl under neat conditions at room temperature under ultrasound activation. As was expected, through pathway A, with diacetyl, hydroxylamine hydrochloride and para-hydroxy benzaldehyde, the yield of the product was found to be low at 60%, that too at a much longer reaction time because of the possibility of formation of several synthons viz. diacetyl monoxime, dimethyl glyoxime and para-hydroxy benzaldehyde oxime. Thus, with the possibility of formation of only two synthons, Pathway B undoubtedly, gave better results (85% yields) with a much lesser reaction time. With this understanding, we envisaged total conversion to products via Pathways C and D as both routes involved only two components and due to the absence of hydroxylamine hydrochloride the possibility of formation of synthons was not there. However, the yield of the product from Pathway C, as well as D, was still found to be less than expected (80–85%) even though one of the reactants (hydroxyiminoketones/dioxime) was totally consumed in each case.
Close monitoring of the reaction Pathway C with the help of TLC revealed the presence of an extra spot which remained even after the total consumption of the starting dioxime. The spot matched with neither the dioxime nor the aldehyde. Isolation and characterization of the compound pertaining to this spot interestingly revealed it to be the aldehyde oxime (see ESI for NMR data, Pages S13 and S33†). Since pathway C did not involve the addition of hydroxylamine, the possibility of the formation of the aldehyde oxime was only via a deoximation of the glyoxime. Accounts of the deoximation of aldehyde oxime in reactions via Pathway D have been mentioned in earlier reports,26 where there is a mention of the formation of by-products. However, the deoximation of dioximes and subsequent oximation of the aldehyde has not been thoroughly investigated and reported till date but scant mention of it is found in an earlier report.25
The deoximation reactions of oximes have been studied as an important reaction involving the recovery of carbonyl compounds from oximes which also serve as an effective protecting group for them.27 It is also a tried and tested method for the recovery of aldehydes and ketones from the corresponding oximes via hydrolysis under acidic conditions.28 However, a very significant finding of this study is that the hydrolysis of dimethyl glyoxime or any aldehyde oxime in the presence of HCl under sonochemical conditions did not yield any deoximated product. For instance, even after 2 hours of sonication of dimethyl glyoxime with HCl, no traces of diacetyl monoxime or diacetyl were found in the reaction mixture. Thus, the above substrates undoubtedly behaved differently under sonochemical conditions. However, dimethyl glyoxime was easily deoximated via acidic hydrolysis in the presence of para-hydroxy benzaldehyde to yield para-hydroxy benzaldehyde oxime. Thus, the presence of aldehyde was imperative as the aldehyde oxime resulted from an exchange reaction between the aldehyde and the dioxime. A plausible mechanistic interpretation of the exchange reaction is shown in Scheme 3.
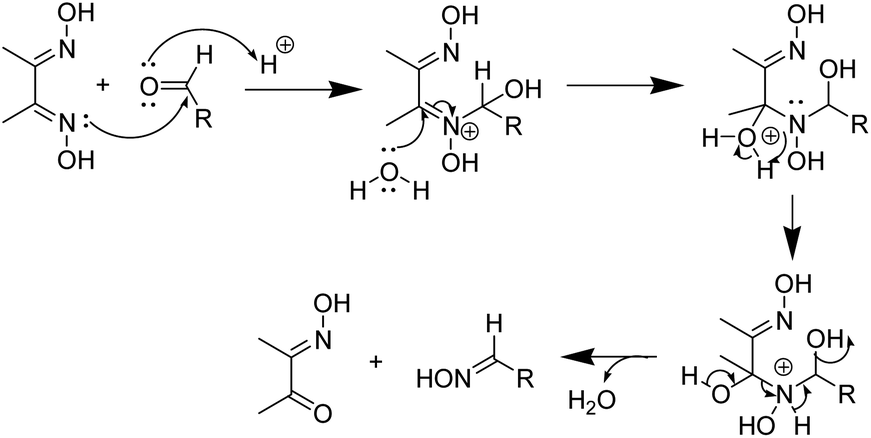 | ||
| Scheme 3 Mechanism of deoximation of dioxime in the presence of an aldehyde. | ||
This was further proved by the deoximation of diacetyl monoxime in the presence of H+. Upon treating diacetyl monoxime with HCl, it yielded traces of dimethyl glyoxime and diacetyl as the deoximated products. Apparently, nucleophilic addition by nitrogen of one molecule to the electrophilic carbon of a second molecule of diacetyl monoxime takes place as shown in Scheme 4.
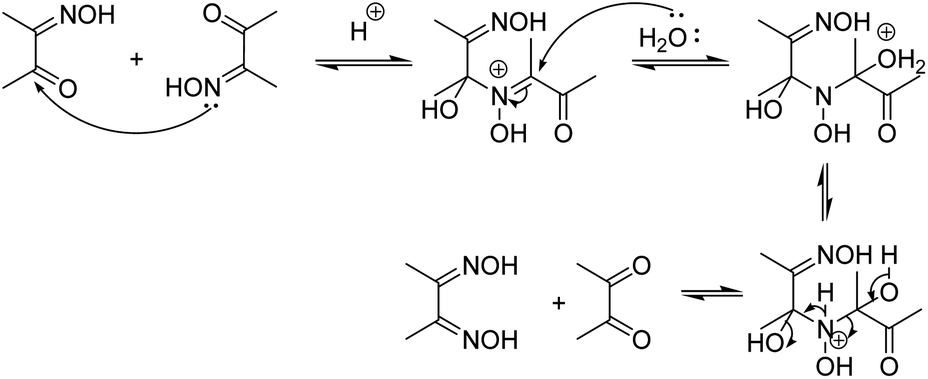 | ||
| Scheme 4 Mechanism of deoximation of diacetyl monoxime. | ||
Moreover, the presence of H+ was also found to be equally important for deoximation since a series of reactions via Pathway C, involving dimethyl glyoxime, carried out in the absence of H+ under ultrasonication did not yield any results. As is evident from Table 2, it is not just the presence of an acidic environment but more specifically Brønsted acidity that was found to play a key role since silica and other Lewis acids did not seem to catalyse the reaction. There was rarely any transformation in the reaction of dimethyl glyoxime to the 1-hydroxy Imidazole-3-oxides in the presence of Lewis acids, even when the reaction was continued for a prolonged period. Graphene oxide, a green carbocatalyst, has been identified to function both as a Brønsted acid29–31 and a Lewis acid.32 However, in our experiment, it failed to yield any product, even after 4 hours of reaction. Extending the reaction time may result in trace amounts of the product. Though Lewis acids did not aid in the deoximation of glyoximes they were, however, found to be effective in catalysing the reaction of diacetyl monoxime with aldehyde oximes (Pathway D). The product started to form after one and half hours of sonication of a stoichiometric mixture of diacetyl monoxime and para-hydroxy benzaldehyde oxime in the presence of anhydrous AlCl3. However, as expected, the same reaction carried out in silica, a weak Lewis acid, did not form any traces of the product even after five hours of sonication, apparently because surface silanol groups of silica, exhibit only weak Lewis acidity and do not show any Brønsted acidity.33 Furthermore, in the absence of any catalyst, sonication of the above reaction mixture did not yield any product even after 8 hours. Longer reaction time might definitely give products as similar reactions via traditional methods carried out in the absence of catalysts have been reported with yields of 68% after prolonged retention times of 48 hours.15 It is thus apparent that strong Lewis acids could be employed for reactions via Pathway D but H+ ions from a Brønsted acid works best not only for the initial deoximation via Pathway C but also for stronger carbonyl activation (in comparison to Lewis acids) in the subsequent steps of reactions via both Pathways C and D. Under sonochemical condition via Pathway D, the product 1c was formed in 85% yields in just 15 minutes in the presence of HCl. Thus, we were able to firmly establish the fact that incomplete conversion to the product was due to the deoximation of the dioxime to the aldehyde oxime.
| Entry | Catalyst/Lewis acid/Bronsted acid | Reaction time | Yield (%) |
|---|---|---|---|
| a Ultrasonic bath temperature maintained at 30 °C throughout. | |||
| 1 | Absence of any | 4 h | No reaction |
| 2 | Absence of any of the above but heated upto (150 °C) | 2 h | No reaction |
| 3 | Silica | 5 h | No reaction |
| 4 | Conc HCl (1 mmol) | 4 h | 60 |
| 5 | Conc HCl (2 mmol) | 4 h | 65 |
| 6 | Conc HCl (2.5 mmol) | 4 h | 70 |
| 7 | Conc HCl (3 mmol) | 1 h 30 min | 86 |
| 8 | Silica + conc HCl | 3.5 h | 80 |
| 9 | Graphene oxide (20 mg) | 4 h | No reaction |
| 10 | Dry AlCl3 | 4 h | No reaction |
| 11 | RuCl3·xH2O | 4 h | No reaction |
Further confirmation of the proposed deoximation mechanism was done with the help of Raman spectroscopy. It was used for checking the progress of the reaction since it is especially suited to in situ reaction monitoring in chemical synthesis where the reactants and products show strong Raman bands. The coupling between the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretchings in glyoximes gives rise to symmetric and antisymmetric NC–CN stretchings.34 However, in the spectra of methylated glyoximes, not only the symmetric NC–CN stretchings, but also the antisymmetric modes exhibit very weak IR intensity.35 On the other hand, the Raman intensity of the antisymmetric and symmetric NC–CN stretching modes, is prominent. Consequently, a very strong Raman band in the region of 1636 cm−1 is characteristic of symmetric NC–CN stretching in glyoximes.
N stretchings in glyoximes gives rise to symmetric and antisymmetric NC–CN stretchings.34 However, in the spectra of methylated glyoximes, not only the symmetric NC–CN stretchings, but also the antisymmetric modes exhibit very weak IR intensity.35 On the other hand, the Raman intensity of the antisymmetric and symmetric NC–CN stretching modes, is prominent. Consequently, a very strong Raman band in the region of 1636 cm−1 is characteristic of symmetric NC–CN stretching in glyoximes.
In an initial monitoring experiment, 2equivalents of dimethyl glyoxime (DMG), 1 equivalent of para-hydroxy benzaldehyde and HCl (3.0 equiv.) were ultrasonicated. The relative intensities of spectra acquired at different time intervals are significantly different. The key feature used for monitoring the reaction kinetics is highlighted at Raman bands around ∼1558, 1612, 1647, 1664 cm−1. In the first 60 min of sonication, we observed a steady reduction in the intensity band of DMG. The aldehyde oxime peak appears in the first 30 minutes while the aldehyde peak intensity diminishes gradually until the sudden loss of its Raman signal after ca. 30 min into sonication and the appearance of the product peak is observed (Fig. 2).
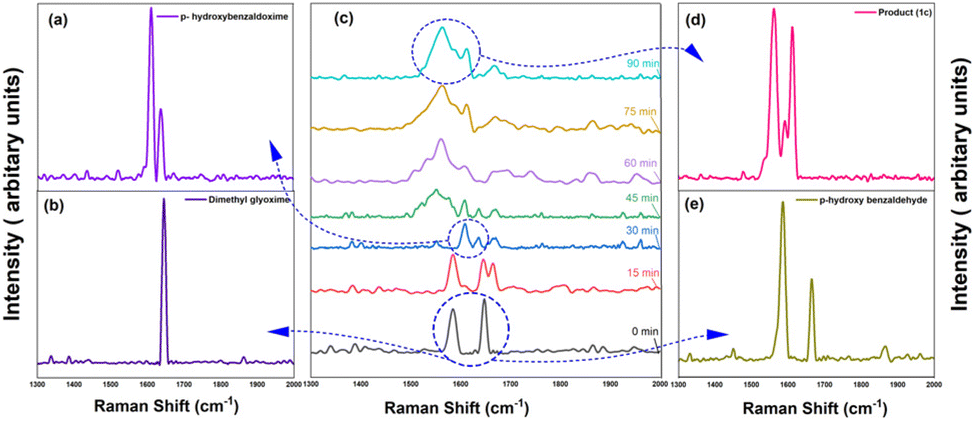 | ||
| Fig. 2 Reaction monitoring of reaction of DMG and p-hydroxy benzaldehyde with the comparison of the Raman ‘fingerprints’ with the product. | ||
Since, HCl was found to work best in the conversions under neat conditions, in the absence of the solvent as well as the support, the reaction condition was thus further optimised with respect to the amount of hydrochloric acid used in the reaction. Upon addition of increasing amounts of HCl (entries 4–7, Table 2), the conversion of dimethyl glyoxime gradually increased and the highest conversion of 92% (entry 8, Table 2) was achieved when 3 equivalents of HCl was used. Increasing the amount of HCl did not increase the yield any further.
Close monitoring of all three pathways A, B and C evidently proved that the aldehyde oxime was present in excess in the reaction mixture at all times. We envisaged that the total consumption of the aldehyde oxime in the reaction would lead to quantitative conversion to products. Therefore, our second task in investigating the mechanochemical route to the target compounds consisted of establishing the optimal stoichiometric ratio for monoxime/dioxime and the aldehyde under solvent-free ultrasonic conditions. For these experiments, the amount of hydroxylamine hydrochloride was set to 1.2 equivalents as the use of lower equivalents of the same gave lesser yields while the use of higher equivalents gave the same yields with similar reaction times. The amount of HCl was also optimised again for all pathways. The results of this screening revealed that high conversion towards 1-hydroxyimidazole-3-oxides could be achieved in less time by reacting 1.5 to 2 equivalents of the starting material diacetyl monoxime/dioxime (Table 3). Thus, a minimum of 1.5 to 2 equivalents of the starting material diacetyl monoxime or dimethyl glyoxime was necessary to consume all of the aldehyde monoxime formed in the reaction mixture and force the reaction to completion.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:Post-optimisation three significant conclusions could be drawn: firstly, the formation of aldehyde oxime was very crucial. Reaction of dimethyl glyoxime with para-hydroxy benzaldehyde does not occur in the presence of any Lewis acid (entries 3, 10 and 11, Table 2). Evidently, the aldehydic carbon does not act as the electrophilic carbon (even when activated by a Lewis acid) for attack by the nucleophilic nitrogen of the dioxime. Secondly, the rate of the reaction depends upon the rate of formation of the aldehyde oxime. Where the aldehyde oxime is formed quite easily from hydroxylamine HCl in Pathway B, the time for completion of the reaction is much less (30 minutes) compared to that from Pathway C (1 h 30 minutes), where the aldehyde oxime is formed only via an exchange reaction with the dioxime. Moreover, as was expected, under the optimised condition the reaction of diacetyl monoxime with p-hydroxy benzaldehyde oxime via Pathway D went to completion in just 15 minutes with almost quantitative yields (Table 3). It is quite apparent that in the absence of the need for an exchange reaction, the product is expeditiously formed. Thirdly and most importantly, the deoximation of aldehyde oximes occurred at a much slower rate than the deoximation of α-hydroxyiminoketones and dioximes since the aldehyde oxime was always found to be present with the product when stoichiometric amounts of dimethyl glyoxime and the aldehyde was taken to form the 1-hydroxyimidazole -3-oxides. Pathway D requires the shortest time for completion since it involves the aldehyde oxime (a nucleophilic form of the aldehyde) which initiates the nucleophilic attack to the carbonyl carbon, the mechanism of which (leading to cyclization and formation of products) we further substantiate (Scheme 5). Hitherto, the mechanism of the cyclization has not been thoroughly investigated though plausible mechanisms have been put forth.36
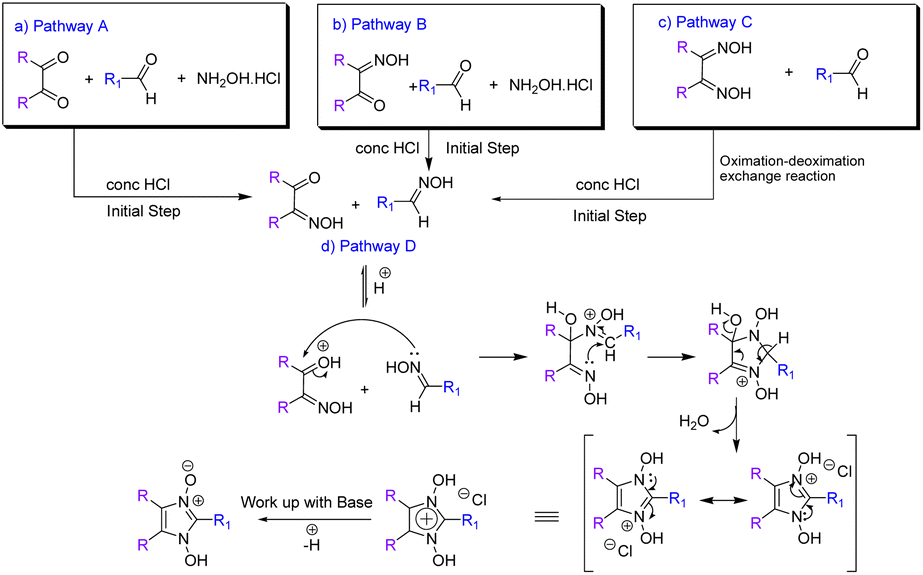 | ||
| Scheme 5 Mechanism of formation of 1-hydroxy imidazole 3-oxides via all pathways. | ||
Although the mechanism apparently is suggestive of being catalysed by H+ the use of the term “acid-catalysed” has been consciously avoided throughout this discussion. Our investigations revealed that the loss of H+ in the final step rarely occurs spontaneously. Base treatment of the reaction mixture during workup resulted in good yields of the hydroxy-imidazole-N-oxide products. Single crystal diffraction (Sc-XRD) data of one of the representative compounds, 1-hydroxy-2-napthylImidazole-3-oxide (2c) obtained via precipitation after base treatment, confirmed the structure. The single crystals of the compound (2c) suitable for Sc-XRD were obtained by its slow evaporation from methanol. The compound crystallizes in a monoclinic crystal system with space group P21/c; a = 8.9333(5) Å, b = 10.9751(6) Å, c = 11.0707(6) Å, β = 90.371(2)° and Z = 4. The ORTEP diagram is presented in Fig. 3.
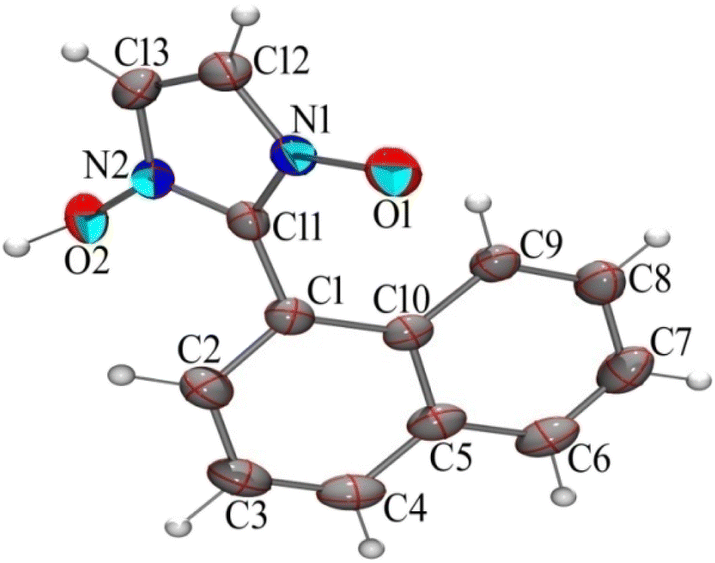 | ||
| Fig. 3 Ortep diagram of compound (2c), ellipsoids drawn in 50% probability. | ||
The presence of a large naphthyl group at C-2 position distorts the usual packing in the crystal structure of the compound as compared to similar compounds reported earlier37 probably due to the C–H⋯Cg and Cg⋯Cg interactions taking place (Figures are included in the ESI†). However,the hydrogen-bonded rings are in a common plane with the OH groups adopting a syn conformation analogous to similar reported compounds as seen in the 3-D packing arrangement of the compound (Fig. 4).
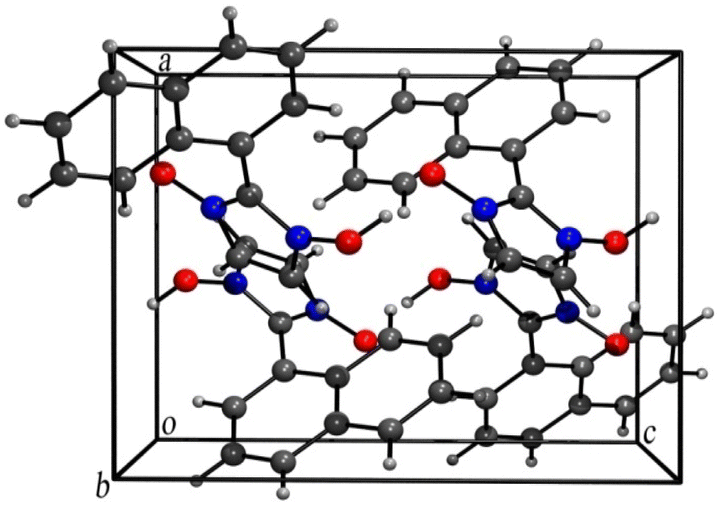 | ||
| Fig. 4 Three-dimensional packing arrangement of compound (2c). | ||
The optimized reaction condition was then extended to the sonochemical synthesis of a variety of 1-hydroxyimidazole-3-oxides (1a–k, 2a–e, and 3a–c) via Pathway B (Scheme 6).
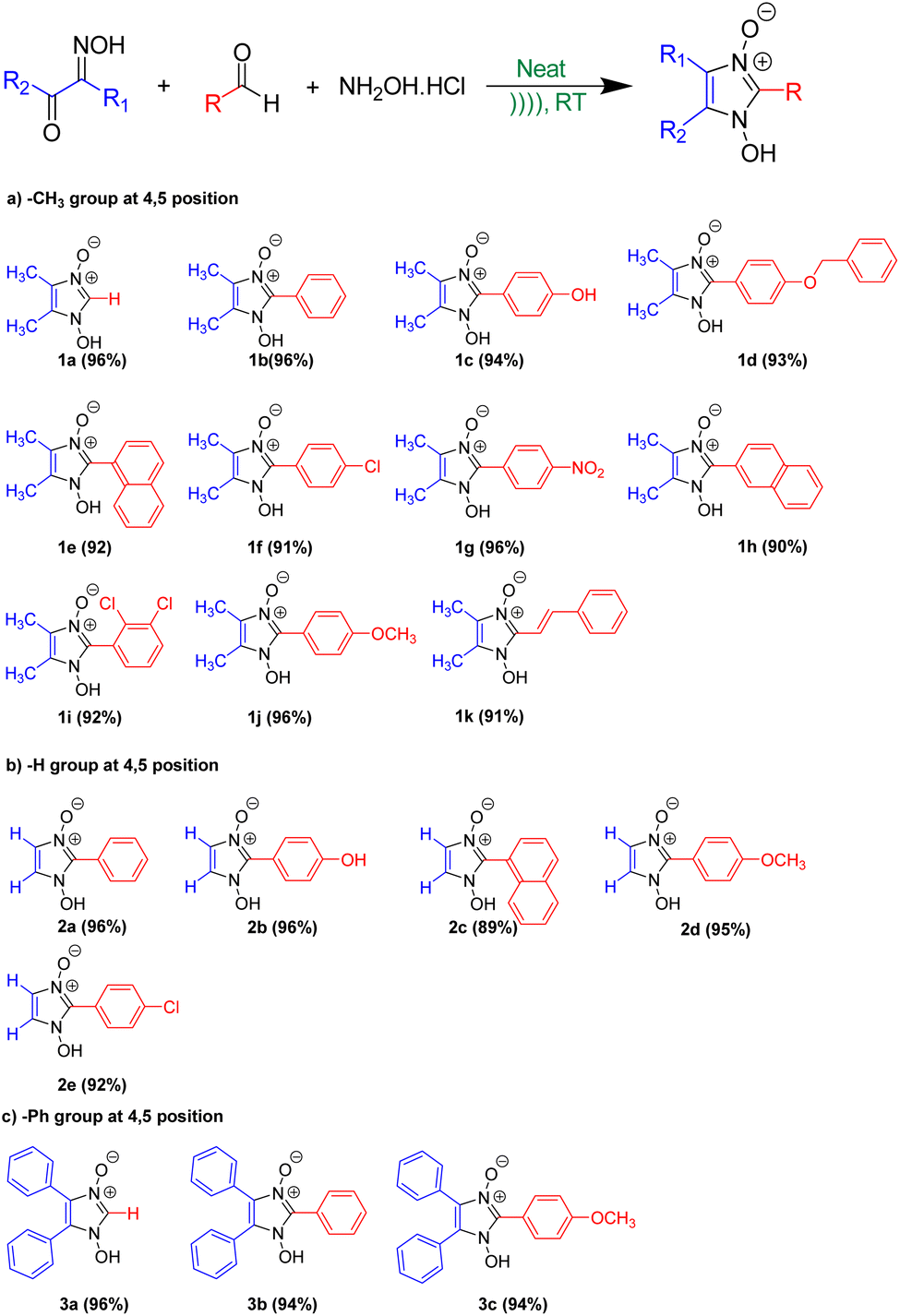 | ||
| Scheme 6 Synthesis of 1-hydroxy, 2-substituted 4, 5-disubstituted Imidazole 3-oxide via Pathway B. | ||
Under the optimised conditions, the sonochemical synthesis of the target compounds was repeated several times. In each case, the reaction was reproducible showing a complete conversion to the product implying that not all sonochemical reactions are irreproducible as is reported.38,39 In addition, a control reaction (for the formation of product 1c) was carried out whereby the reagents (in the same concentrations) were mixed manually and left for several days without being agitated. Although there was some colour change in the mixture, the liquidus phase did not appear and the reaction did not proceed significantly. Only <10% of conversion to the product was observed even after 96 hours. Therefore, it is ultrasonic irradiation that is instigating and accelerating this chemical reaction.40
Anti-cancer studies
To preliminary investigate the potency of compounds containing the 1-hydroxy-imidazole-3-oxide scaffold as anti-proliferative agents; only four of the synthesized compounds were evaluated against non-small cell lung cancer A549 cell line through the MTT assay. The synthesized compounds (1h) and (1c) show potent anticancer properties in comparison to (1i) and (1d). The IC50 values of the drugs (1h) and (1c) were 224.14 ± 12.26 μg ml−1, and 210.03 ± 14.18 μg ml−1 respectively which inhibit 50% proliferation of the A549 cell line as shown in Fig. 5. The non-toxic effect shown by other imidazole derivatives even at higher concentrations indirectly supports our synthetic protocol and the purity of our final products and the absence of toxic chemicals in the end products. However, the IC50 values of (1i) and (1d) were not determined at the highest experimental concentration. It is apparent how slight changes in the structure affect the anticancer property of the different derivatives. The design of lead compounds based on this scaffold, specifically for its anticancer property, is currently being carried out.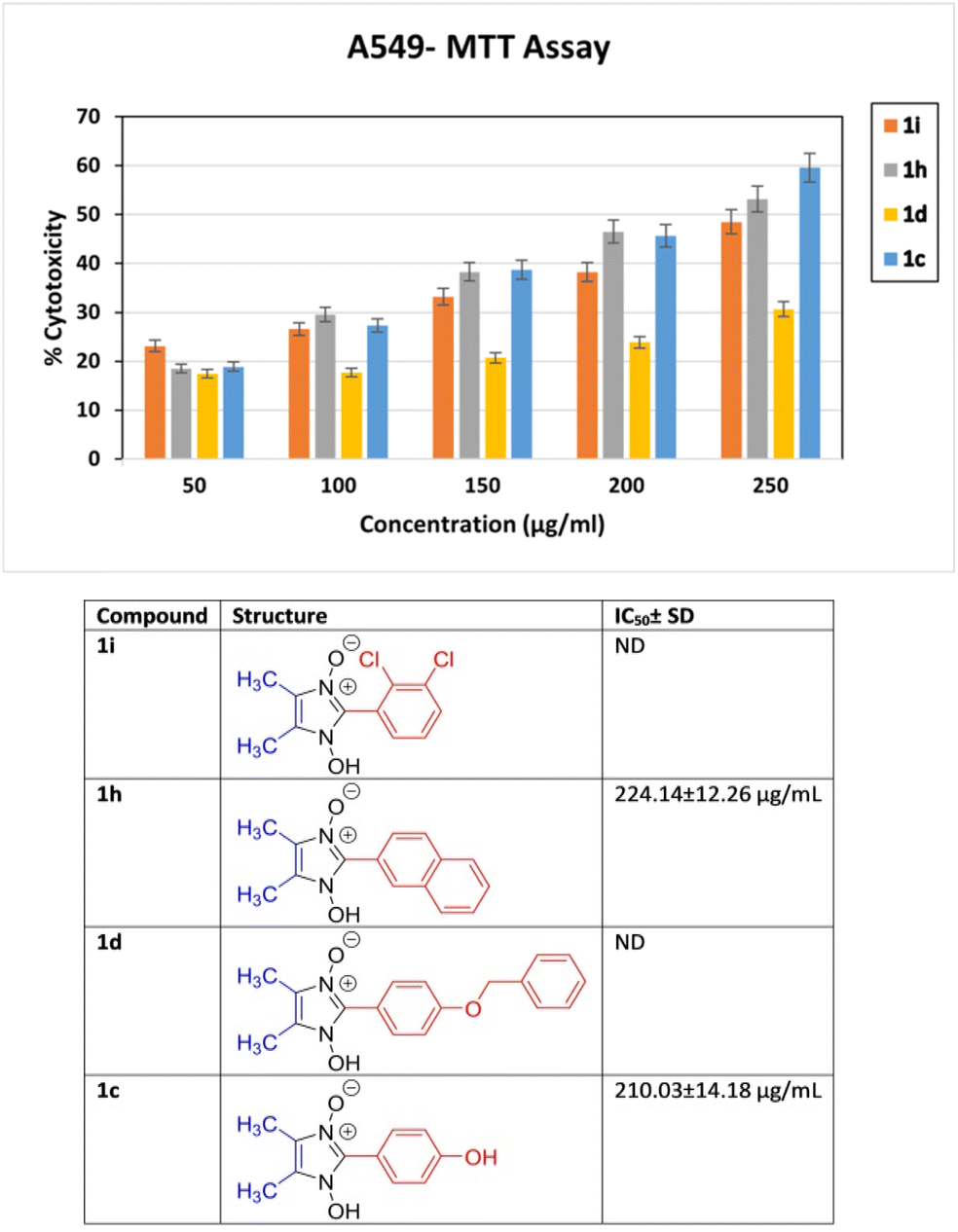 | ||
| Fig. 5 In vitro % cytotoxicity of compound 1i, 1h, 1d and 1c determined by MTT assay at concentrations ranging from 50 to 250 μg ml−1 against non-small cell lung cancer A549 cell line. | ||
Experimental
X-ray single crystal data collection, structural determination and refinement
Single crystal of compound (2c) with the dimension of 0.54 × 0.13 × 0.09 mm, suitable for data collection was mounted on a Bruker APEX-II CCD diffractometer and the Diffraction data was collected using monochromatic Mo Kα radiation (λ = 0.71073 Å) with the ω and φ scan technique. The unit cell was determined using Bruker SMART, the diffraction data were integrated with Bruker SAINT System and the data were corrected for absorption using SADABS; Bruker, 2000.41 The structure was solved by direct method and was refined by full-matrix least squares based on F2 using SHELXT 2018/2.42 All non-hydrogen atoms were refined anisotropically until convergence was reached. H atoms were localized from the difference electron-density map and refined isotropically. ORTEP plot and packing diagram were generated with ORTEP-3 for Windows43 and PLATON.44 WinGX45 was used to prepare the material for publication. The complete crystallographic data for compound (2c) has been deposited as a CIF file in the Cambridge Structural Database (CCDC No. 2388769).Cytotoxicity
Human non-small cell lung cancer cell line (A549) was obtained from National Centre for Cell Science (NCCS), Pune, India. After trypsinization, approximately 100 μL of media-containing cells were seeded at a density of 5 × 103 cells per well into a 96-well microtiter plate and kept for 24 hours of incubation at 37 °C, 5% CO2 incubator. The different concentrations (50 μg ml−1, 100 μg ml−1, 150 μg ml−1, 200 μg ml−1 and 250 μg ml−1) of synthesized samples (dissolved in DMSO) were added in a triplicate manner and kept under the same condition for 24 hours. The next day, the media was replaced with 10 μl of freshly prepared MTT dye solution (5 mg ml−1) and incubated for 3 hours. After 3 hours, 50 μl of isopropanol was added to each well to solubilize the purple formazan crystals. It was gently shaken for 5 minutes until the colour developed. Then, the OD was taken at 620 nm by Spectro Star Nano Spectrophotometer. The Percentage of Cytotoxicity was calculated as Percentage of Cytotoxicity (%) = {(Y − X)/Y × 100}, where Y is the mean OD of cells treated with DMSO and X is the mean OD of cells treated with different concentrations of samples.Conclusions
In conclusion, the first examples of ultrasound induced solvent-free condensation reactions forming 1-hydroxy Imidazole-3-oxides are reported. Hitherto, only studies on the deoximation of aldehyde and ketoximes have been done. Herein we have tried to investigate the deoximation of dioximes and hydroxy-imino ketones and the significance it has on the yields of the products. Raman monitoring of the reaction and phase change to a liquidus state during sonication helped further in visual interpretations. The mechanochemical activation assisted by ultrasound irradiation resulted in a homogeneous mixture leading to quantitative conversion to the products. It also provides a means of applying a gentler form of mechanical energy to a system which may increase the range of organic and inorganic mechanochemical transformations that can be carried out (where grinding results in compaction and possible degradation of the material). Finally, there is potential for the scale-up of sonochemical reactions, therefore aiding in the drive towards sustainable chemistry. In conclusion, we have described a convenient, mild, efficient and scalable method for the synthesis of 1-hydroxy imidazole-3-oxides in presence of HCl at ambient temperature. The compounds are obtained in purified form without strenuous separation and purification techniques. This paper presents, to the best of our knowledge, the first compilation of 19 examples for the synthesis of 2-substituted 1-hydroxy imidazole 3-oxide following a greener procedure and their potency as lead compounds for anti-proliferative activity.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compound 2c has been deposited at the CCDC under CCDC no. 2388769 and can be obtained from https://www.ccdc.cam.ac.ukAuthor contributions
The manuscript was written through the contribution of all the authors. All authors have given approval to the final version of the manuscript. M. M.:investigation, methodology, data analysis, and manuscript preparation – review; S. R.: investigation and data analysis; Y. R.: investigation and data analysis; K. C.:investigation and data analysis; S. D.:investigation and data analysis; B. K. T.:investigation, data analysis and project administration; D. B.: project administration, data analysis and manuscript preparation – review and editing; A. K.: project administration, data analysis and manuscript preparation – review and editing; K. P.: conceptualization, project administration and manuscript preparation – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge DST and SC-XRD LAB, SAIF, IIT Madras for Single Crystal -X-ray diffraction data collection. M. Mukhia acknowledges the financial support of the West Bengal Government’s Swami Vivekananda scholarship.References
- G. Chatel and R. S. Varma, Green Chem., 2019, 21, 6043–6050 RSC.
- P. Cintas and J.-L. Luche, Green Chem., 1999, 1, 115–125 RSC.
- K. Suslick, in Comprehensive Coordination Chemistry II, Elsevier, Oxford, UK, 2004, vol. 1 Search PubMed.
- P. A. Nikitina, I. I. Tkach, E. S. Knyazhanskaya, M. B. Gottikh and V. P. Perevalov, Pharm. Chem. J., 2016, 50, 513–518 CrossRef CAS.
- M. Witschel, Bioorg. Med. Chem., 2009, 17, 4221–4229 CrossRef CAS PubMed.
- R. B. Da Silva, V. B. Loback, K. Salomão, S. L. De Castro, J. L. Wardell, S. M. S. V. Wardell, T. E. M. M. Costa, C. Penido, M. D. G. M. O. De Henriques, S. A. Carvalho, E. F. Da Silva and C. A. M. Fraga, Molecules, 2013, 18, 3445–3457 CrossRef PubMed.
- S. Bartz, B. Blumenröder, A. Kern, J. Fleckenstein, S. Frohnapfel, J. Schatz and A. Wagner, Z. Naturforsch., B, 2009, 64, 629–638 CrossRef CAS.
- A. Wróblewska, J. Śniechowska, S. Kaźmierski, E. Wielgus, G. D. Bujacz, G. Mlostoń, A. Chworos, J. Suwara and M. J. Potrzebowski, Pharmaceutics, 2020, 12, 359 CrossRef PubMed.
- S. A. Laufer, W. Zimmermann and K. J. Ruff, J. Med. Chem., 2004, 47, 6311–6325 CrossRef CAS.
- T. Brendgen, T. Fahlbusch, M. Frank, D. T. Schühle, M. Seßler and J. Schatz, Adv. Synth. Catal., 2009, 351, 303–307 CrossRef CAS.
- E. I. Adiulin, A. V. Kutasevich, V. S. Mityanov, I. I. Tkach and T. Y. Koldaeva, Chem. Heterocycl. Compd., 2015, 51, 500–502 CrossRef CAS.
- M. Hossain, K. Pradhan and A. K. Nanda, Tetrahedron Lett., 2017, 58, 3772–3776 CrossRef CAS.
- X. Ma, H. Dang, J. A. Rose, P. Rablen and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 5998–6007 CrossRef CAS.
- L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291–3306 CrossRef CAS.
- A. V. Kutasevich, A. S. Efimova, M. N. Sizonenko, V. P. Perevalov, L. G. Kuz’mina and V. S. Mityanov, Synlett, 2020, 31, 179–182 CrossRef CAS.
- T. D. Moseev, M. V. Varaksin, D. A. Gorlov, V. N. Charushin and O. N. Chupakhin, J. Org. Chem., 2020, 85, 11124–11133 CrossRef CAS PubMed.
- V. P. Perevalov, V. S. Mityanov, B. V. Lichitsky, A. N. Komogortsev, L. G. Kuz’mina, T. Y. Koldaeva, V. S. Miroshnikov and A. V. Kutasevich, Tetrahedron, 2020, 76, 130947 CrossRef CAS.
- V. S. Mityanov, A. V. Kutasevich, M. M. Krayushkin, B. V. Lichitsky, A. A. Dudinov, A. N. Komogortsev, T. Y. Koldaeva and V. P. Perevalov, Tetrahedron, 2017, 73, 6669–6675 CrossRef CAS.
- G. Mlostoń, M. Celeda, M. Jasiński, K. Urbaniak, P. J. Boratyński, P. R. Schreiner and H. Heimgartner, Molecules, 2019, 24, 4398 CrossRef PubMed.
- A. M. Mfuh and O. V. Larionov, Curr. Med. Chem., 2015, 22, 2819–2857 CrossRef CAS PubMed.
- C. S. Burgey, K. A. Robinson, T. A. Lyle, P. E. Sanderson, S. D. Lewis, B. J. Lucas, J. A. Krueger, R. Singh, C. Miller-Stein, R. B. White, B. Wong, E. A. Lyle, P. D. Williams, C. A. Coburn, B. D. Dorsey, J. C. Barrow, M. T. Stranieri, M. A. Holahan, G. R. Sitko, J. J. Cook, D. R. McMasters, C. M. McDonough, W. M. Sanders, A. A. Wallace, F. C. Clayton, D. Bohn, Y. M. Leonard, T. J. Detwiler, Jr., J. J. Lynch, Jr., Y. Yan, Z. Chen, L. Kuo, S. J. Gardell, J. A. Shafer and J. P. Vacca, J. Med. Chem., 2003, 46, 461–473 CrossRef CAS.
- O. Diels, Ber. Dtsch. Chem. Ges., 1918, 51, 965–976 CrossRef CAS.
- K. Pradhan, B. K. Tiwary, M. Hossain, R. Chakraborty and A. K. Nanda, RSC Adv., 2016, 6, 10743–10749 RSC.
- K. Hayes, J. Heterocycl. Chem., 1974, 11, 615–618 CrossRef CAS.
- J. B. Wright, J. Org. Chem., 1964, 29, 1620–1621 CrossRef CAS.
- K. Bodendorf and H. Towliati, Arch. Pharm. Ber. Dtsch. Pharm. Ges., 1965, 298, 293–297 CrossRef CAS.
- A. Khazaei and A. A. Manesh, Mendeleev Commun., 2006, 16, 109–111 CrossRef.
- D. Li, F. Shi, S. Guo and Y. Deng, Tetrahedron Lett., 2004, 45, 265–268 CrossRef CAS.
- S. Bhattacharya, P. Ghosh and B. Basu, Tetrahedron Lett., 2018, 59, 899–903 CrossRef CAS.
- K. B. Dhopte, D. S. Raut, A. V. Patwardhan and P. R. Nemade, Synth. Commun., 2015, 45, 778–788 CrossRef CAS.
- Z. Chen, Y. Wen, Y. Fu, H. Chen, M. Ye and G. Luo, Synlett, 2017, 28, 981–985 CrossRef CAS.
- V. D. Ebajo, C. R. L. Santos, G. V. Alea, Y. A. Lin and C.-H. Chen, Sci. Rep., 2019, 9, 15579 CrossRef.
- A. A. Tsyganenko, E. N. Storozheva, O. V. Manoilova, T. Lesage, M. Daturi and J. C. Lavalley, Catal. Lett., 2000, 70, 159–163 CrossRef CAS.
- L. Bardet and M. Alain, C. R. Seances Acad. Sci., 1970, 217A, 710 Search PubMed.
- O. R. N. Cherskaya, V. Shlyapochnikov, T. Godovikova and L. Khmel'nitskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 2150To–2152 CrossRef.
- G. V. Nikitina and M. S. Pevzner, Chem. Heterocycl. Compd., 1993, 29, 127–151 CrossRef.
- G. Laus, A. Schwärzler, G. Bentivoglio, M. Hummel, V. Kahlenberg, K. Wurst, E. Kristeva, J. Schütz, H. Kopacka, C. Kreutz, G. Bonn, Y. Andriyko, G. Nauer and H. Schottenberger, Z. Naturforsch., B, 2008, 63, 447–464 CrossRef CAS.
- K. S. Suslick, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1986, vol. 25, pp. 73–119 Search PubMed.
- R. F. Martínez, G. Cravotto and P. Cintas, J. Org. Chem., 2021, 86, 13833–13856 CrossRef.
- D. E. Crawford, Beilstein J. Org. Chem., 2017, 13, 1850–1856 CrossRef CAS PubMed.
- S. S. A. S. B. A. I BRUKER, Madison, Wisconsin, USA, 2000.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2388769. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra07893d |
| This journal is © The Royal Society of Chemistry 2025 |