
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study of supported heteropolyacid catalysts for one-step DME synthesis from CO2 and H2†
Anne Wesner,
Nick Herrmann ,
Lasse Prawitt,
Angela Ortmann,
Jakob Albert and
Maximilian J. Poller*
,
Lasse Prawitt,
Angela Ortmann,
Jakob Albert and
Maximilian J. Poller*
Institute of Technical and Macromolecular Chemistry, University of Hamburg, Bundesstraße 45, Hamburg 20146, Germany. E-mail: maximilian.poller@uni-hamburg.de; Tel: +49 40 42838 3172
First published on 2nd January 2025
Abstract
Dimethyl ether (DME) is a versatile molecule, gaining increasing interest as a viable hydrogen and energy storage solution, pivotal for the transitioning from fossil fuels to environmentally friendly and sustainable energy supply. This research explores a novel approach for the direct conversion of CO2 to DME in a fixed-bed reactor, combining the Cu/ZnO/Al2O3 methanol synthesis catalyst with supported heteropolyacids (HPAs). First, various HPAs, both commercially available and custom-synthesized, were immobilized on Montmorillonite K10. Using a wet impregnation procedure an almost ideal mono-layer of HPA on the support was achieved. The catalysts were further evaluated for their efficiency in direct synthesis of DME from CO2/H2 in combination with the Cu/ZnO/Al2O3 catalyst. Among the catalysts tested, tungstosilicic acid (HSiW) supported on K10 exhibited the most promising performance, achieving a DME yield (YDME) of 7.06% and a molar productivity (Pmol) of 77.84 molDME molHPA−1 h−1. In a subsequent step, further tests using HSiW on various support materials identified ZrO2 as the most effective support, increasing the molar productivity to 125.44 molDME molHPA−1 h−1, while maintaining the DME yield. The results highlight the potential of applying HPA-based catalysts for sustainable DME synthesis directly from CO2, emphasizing the critical role of the catalyst support for optimizing catalytic performance.
Introduction
In view of climate change and geopolitical challenges, Europe is turning to renewable energy sources like the sun and wind to reduce dependence on fossil fuels. However, aligning renewable electricity supply with demand is challenging. A viable solution is converting surplus electricity into so-called ‘green’ hydrogen via electrolysis, which can then be transformed into methanol (MeOH) or dimethyl ether (DME), effectively storing the hydrogen.1,2 DME offers a higher volumetric energy density of 21 MJ L−1 compared to hydrogen with 8.5 MJ L−1,3 is environmentally benign, and easily liquefies under slightly elevated pressure for use with existing liquid gas infrastructure. It already has several applications from propellant to diesel substitute, highlighting its potential as a green energy solution.4–6Typically, DME is produced in a two-step process: first, converting syngas (CO/H2) to methanol using a Cu/ZnO/Al2O3 catalyst, then, in a second step, dehydrating MeOH into DME with a solid acid catalyst.7,8 A more efficient approach is the direct synthesis, converting CO or CO2 with H2 into DME in one step. This method has several advantages, such as simplified operational procedures, increased reaction rates and enhanced equilibrium conversion, achieved through the continuous removal of MeOH as an intermediate from the reaction mixture. Although this process is not yet ready for commercial application, it has gained significant interest from major players in the DME production industry, such as Topsoe, Air Products & Chemicals for its efficiency and potential.9,10
The conversion of CO2 to DME via catalytic hydrogenation is favored from a thermodynamic perspective (eqn (1)). This process requires two different catalytic functionalities: a metallic catalyst for the conversion of CO2 to methanol, and a solid acid catalyst for the subsequent dehydration of methanol to DME.8,11
| 2CO2 + 6H2 ↔ CH3OCH3 + 3H2O ΔH298 K = −123 kJ mol−1 | (1) |
Within the scientific literature, various catalysts with Brønsted or Lewis acidic functionalities have shown to be effective for dehydrating MeOH to DME, with performance depending on the acidic sites' density and strength. Weak and medium acid centers favor DME production, while very strong acid centers may cause formation of other hydrocarbons and coke.12–14 Notable catalysts include γ-Al2O3, H-ZSM-5, mesoporous silicates such as MCM-41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 or aluminophosphates,16 whereby Al2O3 and H-ZSM-5 are most commonly used.8,17 Al2O3 faces challenges due to the adsorption of water produced during the reaction, which inhibits the active sites.18 Conversely, in zeolites like H-ZSM-5, there is a tendency to generate methane or other hydrocarbons as undesirable by-products due to the excessively strong acidic sites.19
15 or aluminophosphates,16 whereby Al2O3 and H-ZSM-5 are most commonly used.8,17 Al2O3 faces challenges due to the adsorption of water produced during the reaction, which inhibits the active sites.18 Conversely, in zeolites like H-ZSM-5, there is a tendency to generate methane or other hydrocarbons as undesirable by-products due to the excessively strong acidic sites.19
To overcome the drawbacks of using alumina or zeolites for methanol dehydration, an alternative emerges in the form of Keggin-type heteropolyacids (HPAs) immobilized on supports with high surface areas.20,21 These anionic metal-oxide clusters, with the general formula [XM12O40]n−, feature a central heteroatom X (typically P or Si) and a metal atom M (usually Mo or W). Their properties can be customized by modifying counterions or metal atoms, tailoring charge, acidity, and pH stability for optimal catalytic performance.22–24 Due to their low surface area (approximately 5–10 m2 g−1), HPAs benefit significantly from being supported on high surface area supports (such as TiO2, SiO2, ZrO2). This approach gains enhanced access to active centers, boosting their activity in methanol dehydration.6,25–27
Attributable to their high Brønsted acidity, lacking the excessively strong acidic sites of zeolites, HPAs exhibit remarkable catalytic activity in the dehydration of methanol and have been subject of various studies.9,12,20,25,28–31 These studies highlight the strong catalytic performance of HPAs, especially supported H3PW12O40 (HPW) and H4SiW12O40 (HSiW) due to their high acidity.30,32 In some instances, these have even outperformed the catalytic activity of H-ZSM-5.33 Notably, HPW supported on MCM-41 exhibited a 100% selectivity towards DME from MeOH at equilibrium conversion.34 The inherent advantages of HPAs, such as operating under mild conditions, minimizing byproduct formation, thermal stability and resisting deactivation by water, make them especially promising for converting methanol to DME.9
To the best of our knowledge, only a limited range of unsubstituted, commercially available HPAs have been utilized in DME synthesis. In this study, the research scope is extended to include transition-metal substituted HPAs to examine the effects of incorporating different heteroatoms such as vanadium and indium. The incorporation of these heteroatoms allow for the modification of the acid sites within the HPAs.35 This study aims to explore how varying the acidity through different heteroatoms influences their performance as catalysts in the conversion of methanol to DME. Additionally, this research marks the first instance where both commercial and specially designed catalysts have been evaluated under uniform experimental conditions, enabling a detailed comparative and comprehensive analysis of their catalytic performance. Moreover, diverse supports were employed to further investigate the HPA–support interactions.
Experimental methods
The following HPAs were supported on Montmorillonite K10 (K10) via wet impregnation: H4SiW12O40 (HSiW), H3PMo12O40 (HPMo), H3PW12O40 (HPW), H8PV5Mo7O40 (HPVMo), H6PInMo11O40 (HPInMo), and H4SiMo12O40 (HSiMo). Furthermore, HSiW was supported on different carriers (Al2O3, ZrO2 TiO2, Celite® 545), using the same method. The supports and catalysts were characterized via inductively coupled plasma optical emission spectroscopy (ICP-OES), N2-physisorption, X-ray diffraction (XRD), NH3-temperature programmed desorption (NH3-TPD), scanning electron microscopy (SEM) and infrared spectroscopy (IR). All catalysts were tested in combination with the commercially available Cu/ZnO/Al2O3 methanol synthesis catalyst in a fixed-bed reactor (Fig. S1†), whereby the two catalyst materials were arranged in two layers separated by a layer of glass wool (Fig. S2†). The reaction conditions were set at 250 °C and 50 bar, with a gas hourly space velocity (GHSV) of 10000 h−1, and a feed gas composition of H2/CO2 at a ratio of 3:1. The gas-phase was analyzed using online gas chromatography (Fig. S3†). An in-depth description of the catalyst synthesis and characterization35–38 including all used chemicals (Table S1†), the catalytic experiments39 and the catalytic evaluation, can be found in the ESI.†
Results and discussion
Initially, monolayers of various HPAs, including both commercially available and custom-synthesized variants, were deposited on K10 and their performance was evaluated as part of a bifunctional catalyst system together with commercial Cu/ZnO/Al2O3 catalyst for DME synthesis. Subsequently, the most promising HPA from the initial screening was combined with different support materials, and their catalytic performance in DME synthesis was systematically evaluated.HPA catalyst selection for DME synthesis – supporting of various HPAs on K10
N2 physisorption data reveal that K10, as expected, is a mesoporous layered silicate with an average pore radius just below 2 nm (Table 1). A single Keggin molecule possesses a diameter of approximately 1 nm, indicating that HPA molecules can infiltrate the pores and potentially cover the entire surface area.35 The application of HPAs on K10 results in a reduction of the BET surface area by about half in all samples, additionally, a significant decrease in pore volume is also observed. This finding aligns with previous studies, which additionally demonstrated an increase in micropore volume upon impregnation of K10 using HPMo and HPW.12
| Catalyst | HSiW | HPMo | HPW | HPVMo | HPInMo | HSiMo | Pure K10 |
|---|---|---|---|---|---|---|---|
| Textural properties | |||||||
| SBET (m2 g−1) | 97 | 100 | 102 | 112 | 106 | 108 | 215 |
| Ø pore diameter (nm) | 1.96 | 1.97 | 1.97 | 1.97 | 1.96 | 1.96 | 1.97 |
| Pore volume (mL g−1) | 0.05 | 0.09 | 0.05 | 0.06 | 0.10 | 0.06 | 0.28 |
|
|||||||
| Elemental analysis | |||||||
| W or Mo (wt%) | 29.45 | 20.27 | 33.53 | 12.55 | 23.51 | 21.50 | — |
| HPA (wt%) | 38.42 | 32.14 | 43.77 | 41.15 | 34.05 | 30.00 | — |
| Loadingeff (μmolHPA gcat−1) | 130 | 180 | 150 | 190 | 220 | 190 | — |
| Loadingtheor (μmolHPA gcat−1) | 160 | 190 | 160 | 200 | 190 | 190 | — |
| NH3-TPD-normalized adsorption capacity | 1.00 | 1.91 | 1.02 | 1.44 | 2.48 | 1.36 | 0.48 |
| Per mass catalyst | 1.00 | 1.91 | 1.02 | 1.44 | 2.48 | 1.36 | 0.48 |
| Per molar mass HPA | 1.00 | 1.38 | 0.88 | 0.98 | 1.46 | 0.93 | — |
The impregnation of K10 with HPAs aimed at achieving a monolayer of HPA on the entire surface of the support material. The results of elemental analysis (Table 1) were used for the calculation of effective loading (Loadingeff), which is compared to the maximum theoretical loading (Loadingtheor) to evaluate the impregnation efficiency. Elemental analysis indicates that the impregnation of all HPAs was successful, achieving the target Loadingtheor. For HPMo, HPInMo, and HSiMo, a higher Loadingeff is observed, which may be attributed to measurement inaccuracies in the elemental analysis.
SEM-EDX mapping indicates macroscopic homogeneous distribution of the HPA on the support (Fig. 1 and S4†). Combined with the Loadingeff values, which align with the predicted Loadingtheor, this supports the assumption that monolayer coverage has been achieved.
 | ||
| Fig. 1 Exemplary SEM EDX-mapping of HSiW supported on K10. | ||
SEM indicates no change in morphology of the catalyst due to the synthesis procedure (Fig. S5†). The preservation of the HPA structure upon supporting on K10 is evident in the IR spectra (Fig. 2 and S6†), apparent by the characteristic Keggin vibration bands: 1049–1060 cm−1 for P–O vibration, 945–962 cm−1 for M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oterminal, 866–877 cm−1 for M–O–Mvertex, and 643–767 cm−1 for M–O–Medge.35 K10 itself displays a very broad vibration band at 1027 cm−1 from the stretching vibration of Si–O groups,42 which overlaps with the PO vibration of the HPAs.
Oterminal, 866–877 cm−1 for M–O–Mvertex, and 643–767 cm−1 for M–O–Medge.35 K10 itself displays a very broad vibration band at 1027 cm−1 from the stretching vibration of Si–O groups,42 which overlaps with the PO vibration of the HPAs.
 | ||
| Fig. 2 Exemplary IR spectra (left) and XRD (right) of pure HSiW (red line), HSiW supported on K10 (black line) and pure K10 (blue line). | ||
Additionally, the samples were characterized by X-ray diffraction (Fig. S7†). It is evident that the characteristic peaks of the support material were preserved after the synthesis, indicating the structure remained intact. However, a reduction in the intensity of the diffraction peaks of pure K10 is observed following impregnation, indicative of a partial loss of crystallinity due to the impregnation process.41,43 Furthermore, no peaks corresponding to the HPAs are detected, this is attributed to the insufficient quantity of HPA on the support, resulting in background noise predominance.
NH3-TPD data (Table 1 and Fig. 3) indicate varying acidities among the different supported HPAs. It is evident that supporting the HPAs on K10 results in increased acidity compared to pure K10 for all HPAs. The supported catalysts themselves exhibit distinct acidity strengths (Table 1). For instance, HPInMo demonstrates a five-fold higher normalized adsorption capacity of 2.48, related to mass of the catalyst, compared to commercially available HSiW (1.00) and HPW (1.02). The supported, unsubstituted HPMo exhibits a relatively high adsorption capacity of 1.91. In contrast, the incorporation of vanadium (HPVMo) reduces this capacity to 1.44, while HSiMo exhibits an even lower adsorption capacity of 1.36. Thus, incorporation of different heteroatoms allows for targeted adjustment of the acidity of supported HPAs, allowing specific investigation in this study into the impact of acidity on catalytic activity in DME synthesis.
 | ||
| Fig. 3 NH3-TPD analysis of HPAs supported on K10, normalized to mass of catalyst (left) and normalized to molar mass of supported HPA (right). | ||
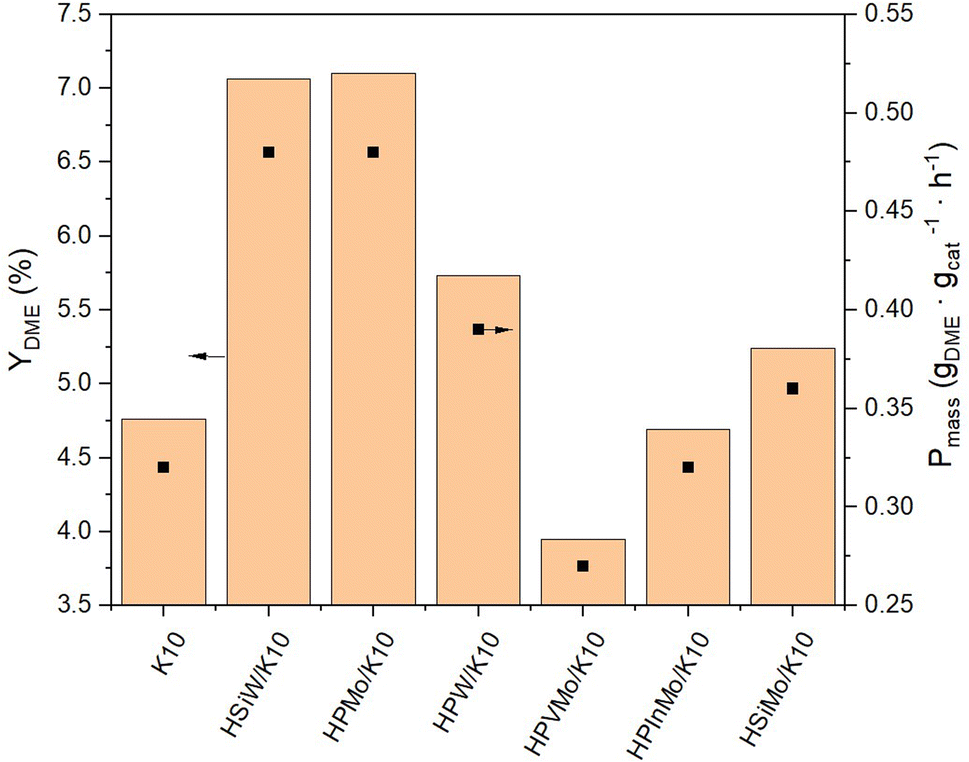 | ||
| Fig. 4 Yield of DME YDME and productivity Pmass of HPAs supported on K10. Reaction conditions: T = 250 °C, p = 50 bar, H2/CO2 3/1, GHSV = 10000 h−1. | ||
NH3-TPD data (Table 1) reveal no direct correlation between the measured acidity and catalytic activity. For instance, impregnation of K10 with HPInMo increases the acidity fivefold, yet the DME yield decreases post-impregnation compared to pure K10. Conversely, K10 impregnated with HSiW and HPMo, which exhibit the highest catalytic activity, show an acidity increase by just two and four times, respectively, compared to pure K10. This discrepancy can be attributed to the reactions being conducted under optimal conditions for methanol synthesis,44 where especially the Brønsted acidic sites of the heteropoly acids have a negligible impact on DME formation.41 These conditions were chosen to maximize methanol yield for its subsequent conversion to DME, but leading to no acidity–activity correlation.
The DME selectivities SDME for each supported HPA catalyst follow the same trend as for YDME (Fig. S8†). The combined selectivities of DME and MeOH make up approximately 50%, with the remaining 50% attributed to the by-product CO (Table S2†) resulting from the competing reverse water–gas-shift (RWGS) reaction. This indicates that in each experiment conducted, the Cu/ZnO/Al2O3 catalyst produced almost an equal amount of MeOH and CO, as no further reaction of CO occurs on the DME catalyst.45 Consequently, the comparison of DME synthesis activities of the catalysts for the second reaction step is based on consistent conditions.
The productivity Pmass follows the same trend as the DME yield (YDME), as a consistent mass of catalyst was used across all synthesis experiments (Fig. 4). However, due to the varying molar masses of the individual HPAs, the molar-based productivity Pmol shows significant differences (Fig. 5). Here too, HSiW and HPMo on K10 exhibit the highest productivities with 77.84 and 59.40 molDME molHPA−1 h−1, respectively, with HSiW/K10 having a higher productivity than HPMo/K10 due to its lower molar mass. HPVMo/K10 and HPInMo/K10 continue to show the lowest Pmol (both around 30 molDME molHPA−1 h−1). The comparison of data between HSiW, HPW, HSiMo, and HPMo on K10 is interesting. Among the tungstates, the Si-containing HPA achieves better results, while HPMo catalyzes the reaction more efficiently than both HSiMo and HPW. Thus, it cannot be stated that either of the metals (W or Mo) offers an advantage, nor is there a trend favoring a central hetero atom (Si or P).
 | ||
| Fig. 5 Productivity Pmol of HPAs supported on K10. Reaction conditions: T = 250 °C, p = 50 bar, H2/CO2 3/1, GHSV = 10000 h−1. | ||
The IR spectra indicate that the Keggin structure is preserved after the reaction across all catalysts (Fig. S9†). The Keggin bands are most distinct for the HSiW/K10 and HPW/K10 catalysts. For all molybdenum-containing HPAs, the vibrational bands are identifiable but exhibit weaker intensity. Additionally, all of the molybdates show a dark blue coloration after the reaction (Fig. S10†), suggesting a reduction process has occurred during the reaction to form molybdenum blue (eqn (2)).46,47 The darker coloration and weakening of IR bands indicate that this reduction is incomplete, suggesting the presence of the reduced species of the catalyst as well as poorer catalyst stability.
| [PMoVI12O40]3− + 4e− ⇌ [PMoV4MoVI8O40]7− | (2) |
As an interim conclusion, it is notable that the impregnation of K10 with HSiW and HPMo particularly lead to increased DME yields compared to pure K10. By considering molar-based productivity Pmol, HSiW/K10 is identified as the most efficient catalyst. To validate these findings, the reproducibility of the experimental procedure was investigated using HSiW/K10 in multiple repetitions. These experiments resulted in consistent yields and selectivities for the by-products, MeOH and CO, as well as stable catalyst productivity across the experiments (Fig. S11 and Table S3†), and thereby confirmed the initial results.
Support selection for DME synthesis – supporting HSiW on different supports
Following the identification of HSiW as the optimal HPA for DME synthesis, its performance was further evaluated on various support materials. To this end, HSiW was immobilized on ZrO2, Al2O3, TiO2, and Celite® 545 (hereafter simply referred to as Celite). Celite, primarily composed of SiO2, possesses a unique internal structure with vacuoles surrounded by interconnected pores within its silica walls, providing an ideal surface for physical adsorption. Due to its adsorptive and insulating properties, Celite is widely used in applications such as filtration, chromatography, and mild abrasives.48 ZrO2, Al2O3, and TiO2, on the other hand, are established support materials for supported catalysts, valued for their stability and compatibility with a variety of catalytic processes.49–52 The influence of support materials in enhancing the catalytic activity of HPAs for DME synthesis is pivotal, as demonstrated in previous studies, which have highlighted the beneficial effects of utilizing various supports such as SiO2 or TiO2 for HPAs.25,53 However, detailed analyses of the support's influence for HPAs remain insufficiently explored in existing research.The amount of HSiW used for synthesis was adjusted to the surface area of each support to create a monolayer. The impregnation was carried out as described above. In Table 2 the elemental analysis as well as the effective loading Loadingeff and the maximum theoretical loading Loadingtheor and the point of zero charge of the supports are listed. For all supports, the actual and theoretical loadings closely match, indicating complete impregnation of HSiW on each support. IR spectra confirm the preservation of the Keggin structure of all supported catalysts (Fig. S12†).
| HSiW/ZrO2 | ZrO2 | HSiW/Al2O3 | Al2O3 | HSiW/TiO2 | TiO2 | HSiW/Celite | Celite | |
|---|---|---|---|---|---|---|---|---|
| Textural properties | ||||||||
| SBET (m2 g−1) | 81 | 91 | 161 | 277 | 106 | 163 | 4 | 1 |
| Ø pore diameter (nm) | 3.40 | 4.07 | 1.97 | 4.48 | 1.86 | 2.37 | 1.57 | 1.85 |
| Pore volume (mL g−1) | 0.18 | 0.28 | 0.23 | 0.75 | 0.13 | 0.33 | 0.01 | 0.00 |
| Point of zero charge | 6.52 | 7.6 | 5.9 | 7.08 | ||||
|
||||||||
| Elemental analysis | ||||||||
| W (wt%) | 18.32 | — | 33.24 | — | 28.91 | — | 45.02 | — |
| HPA (wt%) | 27.19 | — | 49.34 | — | 42.91 | — | 68.81 | — |
| Loadingeff (μmolHPA gcat−1) | 80 | — | 150 | — | 130 | — | 210 | — |
| Loadingtheor (μmolHPA gcat−1) | 90 | — | 150 | — | 120 | — | 210 | — |
Celite, like K10, represents another silicate used for supporting HSiW. It exhibits a notably low surface area of just 1 m2 g−1 and no measurable pore volume (Table 2). The minimal surface area measured can be attributed to Celite's very large pores of ≥200 nm, visible in SEM (Fig. S13†). These pores are too large to be quantified using the available BET measurement equipment. Post-impregnation, SEM images indicate pore blockage (Fig. S13†), and the clustering effect increases the measured surface area to 4.35 m2 g−1.
For the three oxide materials (ZrO2, Al2O3, and TiO2), SEM images (Fig. S13†), combined with SEM-EDX images (Fig. S14†), indicate that the particles remain approximately the same size, thus undamaged post-synthesis, and reveal a homogeneous distribution of the HPA across the entire surface. Among these materials, ZrO2 has the smallest surface area at 91 m2 g−1, while Al2O3 possesses the largest of 277 m2 g−1. Post-impregnation, the surface areas of Al2O3 and TiO2 decrease by approximately 40%, with a significant reduction in pore volumes as well. Conversely, ZrO2 shows only an 11% reduction of surface area, with smaller decreases in pore radius and volume, suggesting a particularly uniform distribution of HPA molecules across the entire surface of the support (Table 2).
The supported catalysts as well as the supports themselves were employed in the synthesis of DME (Fig. 6). Among the tested supports, pure K10 demonstrates significant inherent catalytic activity. The incorporation of HPAs onto the supports invariably lead to an enhanced catalytic performance compared to the unmodified supports. The DME yield across all HPA-modified catalysts is observed to be around 7%, with a Pmass of 0.5 gDME gcat−1 h−1. Due to the limited precision of the measurements, the productivity data do not decisively distinguish the most effective HPA-support combination. Remarkably, the mass-normalized productivity of unsupported HSiW, matches that of the supported catalyst materials.
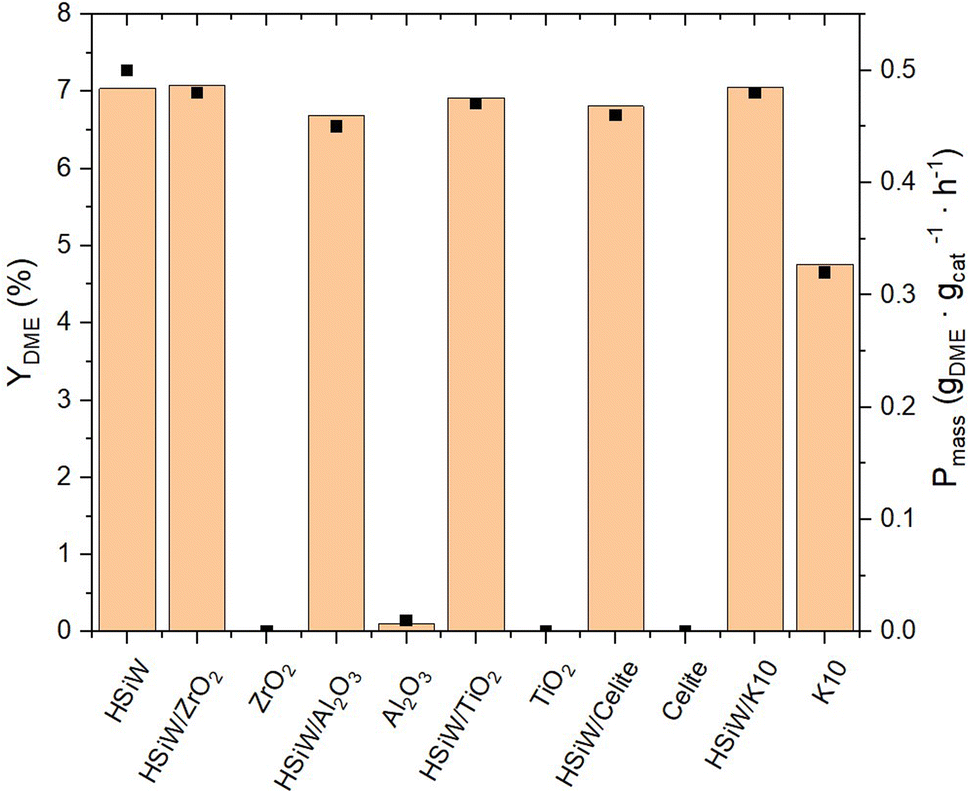 | ||
| Fig. 6 Yield of DME YDME and productivity Pmass of HSiW on different supports. Reaction conditions: T = 250 °C, p = 50 bar, H2/CO2 3/1, GHSV = 10000 h−1. | ||
When normalizing productivity to the molar amount of catalyst (Fig. 7), unsupported HSiW exhibits the lowest productivity of 35.77 molDME molHPA−1 h−1. For each support, it is observed that the catalytic activity is consistently enhanced by the support material. This enhancement is attributed to the generally increased surface area, which improves accessibility to active sites crucial for converting MeOH to DME. Interestingly, catalytic activity does not directly correlate solely with higher surface area and therefore with a higher loading of the HSiW monolayer. Impregnation on Celite slightly increases Pmol to 47.68 molDME molHPA−1 h−1, followed by HSiW on Al2O3, TiO2 and K10, with the HSiW/ZrO2 as combination achieving the highest Pmol of 125.44 molDME molHPA−1 h−1. This suggests a cooperative effect between the support and the HPA, which enhances the catalytic activity.
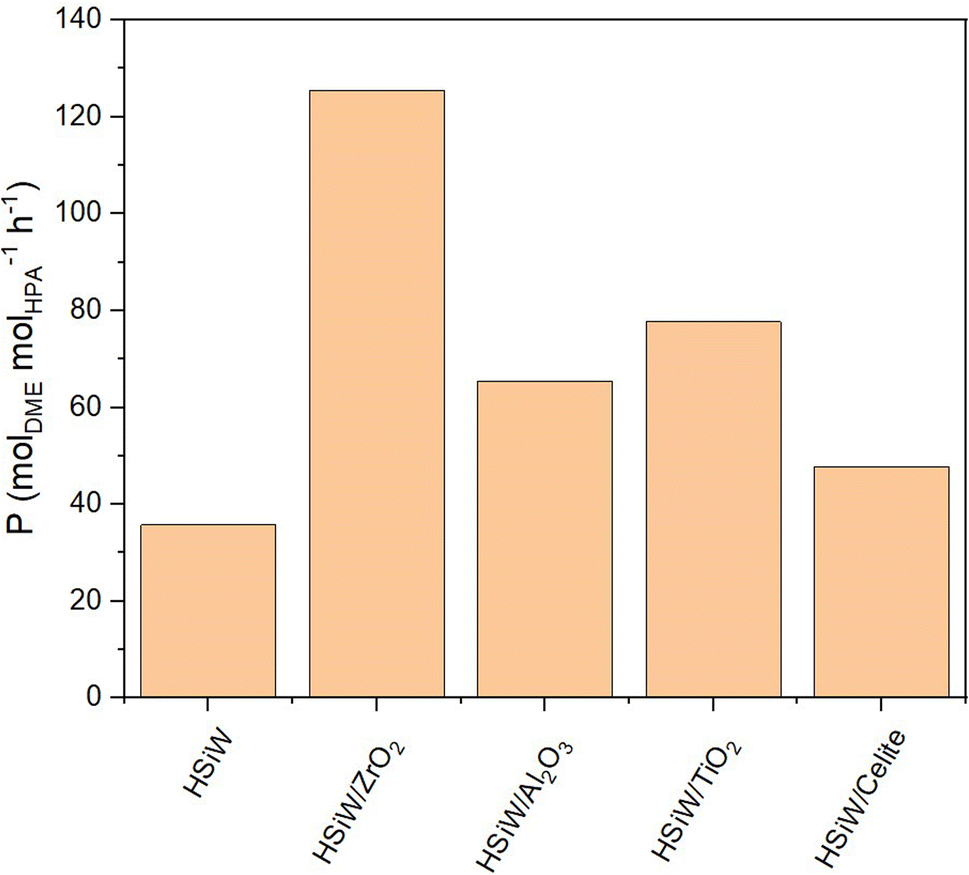 | ||
| Fig. 7 Productivity Pmol of HSiW on different supports. Reaction conditions: T = 250 °C, p = 50 bar, H2/CO2 3/1, GHSV = 10000 h−1. | ||
As previously demonstrated and confirmed in this section, the combined selectivities of DME and MeOH consistently make up about 50%, with the remaining 50% attributed to the by-product CO (Fig. S15 and Table S4†). This steady result indicates that MeOH production by Cu/ZnO/Al2O3 catalyst remains consistent across all experiments, with no further CO conversion by the supported HPA catalyst. This allows for a fair comparison of the DME formation by the supported HPAs in the second reaction step under uniform conditions. The pure supports used for the HPA catalysts showed no catalytic activity for DME synthesis, except for K10, which shows partial conversion of MeOH to DME without any HPA supported.
NH3-TPD analysis (Fig. S16†) indicates that catalytic activity also does not directly correlate with measured Brønsted acidity. Specifically, HSiW/ZrO2 exhibits the second highest acidity after HSiW/Al2O3. These findings suggest additional factors influencing catalytic activity beyond surface area and Brønsted acidity. Previous studies indicate that ZrO2 provides additional sites for methanol adsorption, enhancing methanol conversion and leading to higher DME production.25,54 SEM-EDX analysis and N2-physisorption also confirm that despite ZrO2's smaller surface area, it is fully and uniformly covered by HPA after impregnation, ensuring optimal catalytic activity through enhanced accessibility of acid sites, highlighting ZrO2 as an exceptional support material.
Comparative analysis with previously-reported catalyst
The most effective catalyst identified in this study, hereafter referred to as HSiW/ZrO2W, was compared with the leading literature-reported catalyst for DME synthesis from CO2, HSiW/ZrO2K, as reported by Kubas et al.21 To enable a direct comparison of the catalytic performance, the catalyst was synthesized following the method outlined by Kubas,21 with equivalent HPA-unit loading of 1 HPA unit per nm2 of, and subsequently tested under identical reaction conditions.The catalytic performance (Table 3) of HSiW/ZrO2K shows generally good agreement with HSiW/ZrO2W, with slightly higher values for DME yield (YDME = 7.08%) and selectivity (SDME = 30.91%) for HSiW/ZrO2K, compared to HSiW/ZrO2W with YDME = 6.88% and SDME = 31.09%. The mass-specific productivities for both catalysts are equivalent, with Pmass = 0.48 gDME gcat−1 h−1 (HSiW/ZrO2W) and 0.47 gDME gcat−1 h−1 (HSiW/ZrO2K). However, due to lower HPA loading, the molar productivity of our HSiW/ZrO2W is higher compared to the HSiW/ZrO2K catalyst reported by Kubas et al.,21 indicating a possible improvement in HPA dispersion resulting from the synthesis method we used in this study.
000 h−1
| Catalyst | HSiW/ZrO2W | HSiW/ZrO2K |
|---|---|---|
| XCO2 (%) | 19.36 | 18.70 |
| YMeOH (%) | 3.32 | 3.40 |
| YDME (%) | 7.08 | 6.88 |
| YCO (%) | 12.50 | 11.85 |
| SMeOH (%) | 14.50 | 15.36 |
| SDME (%) | 30.91 | 31.09 |
| SCO (%) | 54.59 | 53.55 |
| Pmass (gDME gcat−1 h−1) | 0.48 | 0.47 |
| Pmol (molDME molHPA−1 h−1) | 125.44 | 108.67 |
Overall, the comparison underscores the enhanced catalytic activity of HSiW supported on ZrO2 as a robust support material, irrespective of specific synthesis or reaction conditions. This study further demonstrates, through the use of tailored heteropoly acid catalysts and a range of supports, that parameters such as support surface area, pore size, and the tuned acidity of heteropoly acids do not have a definitive impact on catalytic activity. Notably, HSiW/ZrO2 consistently outperforms other polyoxometalates, although the exact underlying mechanisms remain unclear and warrant further investigation.
Conclusions
In this study, various HPA catalysts were employed for the single-step synthesis of DME. Therefore, bifunctional catalyst systems, combining commercial Cu/ZnO/Al2O3 catalyst with supported HPAs, have been prepared. Both commercial HPAs (HPW, HPMo, HSiW) and specially synthesized HPAs (HPVMo, HPInMo, HSiMo) were used. The successful impregnation of K10 montmorillonite with monolayers of various HPAs was confirmed by a range of analytical techniques including ICP-OES, SEM-EDX, and N2-physisorption. Subsequently, these catalysts were evaluated, in combination with a methanol synthesis catalyst, for their DME synthesis activity in a fixed-bed reactor. HSiW emerged as the most effective catalyst in this screening, achieving a DME yield of 7.06% (53% of the equilibrium yield) and a molar productivity of 77.84 molDME molHPA−1 h−1. Upon impregnation onto different supports, HSiW supported on ZrO2 proved to be the optimal catalyst, enhancing the molar productivity up to 125.44 molDME molHPA−1 h−1. Overall, we evaluated an unprecedented range of heteropolyacids and support materials for this reaction. The results highlight that, beyond the strengths and numbers of acidic centers, the uniform dispersion of HSiW on ZrO2 enhances accessibility to catalytic active sites.Data availability
The data supporting our article with the title “Study of supported heteropolyacid catalysts for one step DME synthesis from CO2 and H2” have been included as part of the ESI.† Further information is available on request.Author contributions
Anne Wesner was responsible for synthesis and characterization of the catalysts, interpreting data, conceptualizing the experimental workflow, and drafting the manuscript. Nick Herrmann performed supervision and design of catalytic experiments. Lasse Prawitt and Angela Ortmann carried out the catalyst synthesis as well as characterization and conducted all catalytic experiments. Prof. Jakob Albert provided infrastructure and equipment. As principal investigator, Dr Maximilian J. Poller was responsible for conceptualization of this project, acquired financial support, coordinated and supervised the project. All authors contributed to the discussion of the work and the scientific writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the central analytical services of the Chemistry Department at UHH for carrying out ICP-OES (Dr Dirk Eifler and his team), SEM (Dr Charlotte Ruhmlieb and her team), and XRD (Dr Frank Hoffmann and Isabelle Nevoigt) measurements. Furthermore, we thank Philipp Kampe for his advice concerning the operation of the fixed bed reactor setup, and Dr Jan-Christian Raabe for his support concerning the synthesis of HPAs. We also thank Hauke Heller for providing access to the BET apparatus and Sandra König for her valuable guidance in operating the BET device. This project was funded by the “Behörde für Wissenschaft, Forschung, Gleichstellung und Bezirke” of the Free Hanseatic City of Hamburg (BWFGB) via the Hamburg Innovation GmbH through the Call4Transfer funding program (grant number C4T839).References
- P. Schühle, R. Stöber, M. Semmel, A. Schaadt, R. Szolak, S. Thill, M. Alders, C. Hebling, P. Wasserscheid and O. Salem, Dimethyl ether/CO2 – a hitherto underestimated H2 storage cycle, Energy Environ. Sci., 2023, 16, 3002–3013 RSC.
- E. Pawelczyk, N. Łukasik, I. Wysocka, A. Rogala and J. Gębicki, Recent Progress on Hydrogen Storage and Production Using Chemical Hydrogen Carriers, Energies, 2022, 15, 4964 CrossRef.
- M. Hilgers, Alternative Antriebe und Ergänzungen zum konventionellen Antrieb, Springer Fachmedien Wiesbaden, Wiesbaden, 2016 Search PubMed.
- Z. Azizi, M. Rezaeimanesh, T. Tohidian and M. R. Rahimpour, Dimethyl ether: a review of technologies and production challenges, Chem. Eng. Process.: Process Intensif., 2014, 82, 150–172 CrossRef.
- E. Catizzone, G. Bonura, M. Migliori, F. Frusteri and G. Giordano, CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives, Mol., 2018, 23, 31 CrossRef.
- N. Mota, E. M. Ordoñez, B. Pawelec, J. L. G. Fierro and R. M. Navarro, Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems, Catalysts, 2021, 11, 411 CrossRef.
- S. Banivaheb, S. Pitter, K. H. Delgado, M. Rubin, J. Sauer and R. Dittmeyer, Recent Progress in Direct DME Synthesis and Potential of Bifunctional Catalysts, Chem. Ing. Tech., 2022, 94, 240–255 CrossRef.
- K. Krim, A. Sachse, A. Le Valant, Y. Pouilloux and S. Hocine, One Step Dimethyl Ether (DME) Synthesis from CO2 Hydrogenation over Hybrid Catalysts Containing Cu/ZnO/Al2O3 and Nano-Sized Hollow ZSM-5 Zeolites, Catal. Lett., 2023, 153, 83–94 CrossRef.
- C. Peinado, D. Liuzzi, S. N. Sluijter, G. Skorikova, J. Boon, S. Guffanti, G. Groppi and S. Rojas, Review and perspective: Next generation DME synthesis technologies for the energy transition, Chem. Eng. J., 2024, 479, 147494 CrossRef.
- A. Akhoondi, A. I. Osman and A. A. Eslami, Direct catalytic production of dimethyl ether from CO and CO2: a review, Synth. Sinter., 2021, 1, 105–125 CrossRef.
- C. Liu and Z. Liu, Perspective on CO2 Hydrogenation for Dimethyl Ether Economy, Catalysts, 2022, 12, 1375 CrossRef.
- A. Kornas, M. Śliwa, M. Ruggiero-Mikołajczyk, K. Samson, J. Podobiński, R. Karcz, D. Duraczyńska, D. Rutkowska-Zbik and R. Grabowski, Direct hydrogenation of CO2 to dimethyl ether (DME) over hybrid catalysts containing CuO/ZrO2 as a metallic function and heteropolyacids as an acidic function, React. Kinet., Mech. Catal., 2020, 130, 179–194 CrossRef.
- Y. Fu, T. Hong, J. Chen, A. Auroux and J. Shen, Surface acidity and the dehydration of methanol to dimethyl ether, Thermochim. Acta, 2005, 434, 22–26 CrossRef.
- L. Travalloni, A. C. Gomes, A. B. Gaspar and M. A. Da Silva, Methanol conversion over acid solid catalysts, Catal. Today, 2008, 133–135, 406–412 CrossRef CAS.
- D. VARIŞLI, K. C. TOKAY, A. ÇİFTÇİ, T. DOĞU and G. DOĞU, Methanol dehydration reaction to produce clean diesel alternative dimethylether over mesoporous aluminosilicate-based catalysts, Turk. J. Chem., 2009, 33(3), 355–366 Search PubMed.
- W. Dai, W. Kong, G. Wu, N. Li, L. Li and N. Guan, Catalytic dehydration of methanol to dimethyl ether over aluminophosphate and silico-aluminophosphate molecular sieves, Catal. Commun., 2011, 12, 535–538 CrossRef.
- H. Bateni and C. Able, Development of Heterogeneous Catalysts for Dehydration of Methanol to Dimethyl Ether: A Review, Catal. Ind., 2019, 11, 7–33 CrossRef.
- M. Xu, J. H. Lunsford, D. Goodman and A. Bhattacharyya, Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts, Appl. Catal., A, 1997, 149, 289–301 CrossRef.
- N. Khandan, M. Kazemeini and M. Aghaziarati, Determining an optimum catalyst for liquid-phase dehydration of methanol to dimethyl ether, Appl. Catal., A, 2008, 349, 6–12 CrossRef.
- D. Kubas, J. M. Beck, E. Kasisari, T. Schätzler, A. Becherer, A. Fischer and I. Krossing, From CO2 to DME: Enhancement through Heteropoly Acids from a Catalyst Screening and Stability Study, ACS Omega, 2023, 8, 15203–15216 CrossRef PubMed.
- D. Kubas, M. Semmel, O. Salem and I. Krossing, Is Direct DME Synthesis Superior to Methanol Production in Carbon Dioxide Valorization? From Thermodynamic Predictions to Experimental Confirmation, ACS Catal., 2023, 13, 3960–3970 CrossRef.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer Berlin, Berlin, 2013 Search PubMed.
- M. T. Pope and A. Müller, Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- N. I. Gumerova and A. Rompel, Synthesis, structures and applications of electron-rich polyoxometalates, Nat. Rev. Chem, 2018, 2, 0112 CrossRef.
- C. Peinado, D. Liuzzi, R. M. Ladera-Gallardo, M. Retuerto, M. Ojeda, M. A. Peña and S. Rojas, Effects of support and reaction pressure for the synthesis of dimethyl ether over heteropolyacid catalysts, Sci. Rep., 2020, 10, 8551 CrossRef PubMed.
- E. Millán, N. Mota, R. Guil-López, B. Pawelec, J. L. García Fierro and R. M. Navarro, Direct Synthesis of Dimethyl Ether from Syngas on Bifunctional Hybrid Catalysts Based on Supported H3PW12O40 and Cu-ZnO(Al): Effect of Heteropolyacid Loading on Hybrid Structure and Catalytic Activity, Catalysts, 2020, 10, 1071 CrossRef.
- A. Wesner, M. P. Papajewski, L. Schidowski, C. Ruhmlieb, M. J. Poller and J. Albert, Supported H8PV5Mo7O40 on activated carbon: synthesis and investigation of influencing factors for catalytic performance, Dalton Trans., 2024, 53, 14065–14076 RSC.
- H. HAYASHI, The properties of heteropoly acids and the conversion of methanol to hydrocarbons, J. Catal., 1982, 77, 473–484 CrossRef CAS.
- F. M. Ebeid, L. Ali and F. F. Abdalla, Conversion Of Methanol Over Metal-Salts Of 12-Molybdophosphoric Acid, Indian J. Biochem., 1992, 31, 921–928 CrossRef.
- R. M. Ladera, M. Ojeda, J. L. G. Fierro and S. Rojas, TiO2-supported heteropoly acid catalysts for dehydration of methanol to dimethyl ether: relevance of dispersion and support interaction, Catal. Sci. Technol., 2015, 5, 484–491 RSC.
- W. Alharbi, E. F. Kozhevnikova and I. V. Kozhevnikov, Dehydration of Methanol to Dimethyl Ether over Heteropoly Acid Catalysts: The Relationship between Reaction Rate and Catalyst Acid Strength, ACS Catal., 2015, 5, 7186–7193 CrossRef.
- R. M. Ladera, J. L. G. Fierro, M. Ojeda and S. Rojas, TiO2-supported heteropoly acids for low-temperature synthesis of dimethyl ether from methanol, J. Catal., 2014, 312, 195–203 CrossRef.
- B. P. Karaman and N. Oktar, Tungstophosphoric acid incorporated hierarchical HZSM-5 catalysts for direct synthesis of dimethyl ether, Int. J. Hydrogen Energy, 2020, 45, 34793–34804 CrossRef.
- A. Ciftci, D. Varisli, K. C. Tokay, N. A. Sezgi and T. Dogu, Dimethyl ether, diethyl ether & ethylene from alcohols over tungstophosphoric acid based mesoporous catalysts, Chem. Eng. J., 2012, 207–208, 85–93 CrossRef.
- J.-C. Raabe, M. J. Poller, D. Voß and J. Albert, H8 PV5 Mo7 O40 - A Unique Polyoxometalate for Acid and RedOx Catalysis: Synthesis, Characterization, and Modern Applications in Green Chemical Processes, ChemSusChem, 2023, 16, e202300072 CrossRef.
- J.-C. Raabe, J. Aceituno Cruz, J. Albert and M. J. Poller, Comparative Spectroscopic and Electrochemical Study of V(V)-Substituted Keggin-Type Phosphomolybdates and -Tungstates, Inorganics, 2023, 11, 138 CrossRef.
- V. F. Odyakov and E. G. Zhizhina, A novel method of the synthesis of molybdovanadophosphoric heteropoly acid solutions, React. Kinet. Catal. Lett., 2008, 95, 21–28 CrossRef.
- J. D. H. Strickland, The Preparation and Properties of Silicomolybdic Acid. I. The Properties of Alpha Silicomolybdic Acid, J. Am. Chem. Soc., 1952, 74, 862–867 CrossRef.
- A. Wesner, P. Kampe, N. Herrmann, S. Eller, C. Ruhmlieb and J. Albert, Indium-based Catalysts for CO2 Hydrogenation to Methanol: Key Aspects for Catalytic Performance, ChemCatChem, 2023, 15, e202301125 CrossRef.
- J. Zhao, X. Zheng, Q. Liu, M. Xu, S. Yang and M. Zeng, Chitosan supported Pd0 nanoparticles encaged in Al or Al-Fe pillared montmorillonite and their catalytic activities in Sonogashira coupling reactions, Appl. Clay Sci., 2020, 195, 105721 CrossRef.
- M. Śliwa, K. Samson, M. Ruggiero-Mikołajczyk, A. Żelazny and R. Grabowski, Influence of Montmorillonite K10 Modification with Tungstophosphoric Acid on Hybrid Catalyst Activity in Direct Dimethyl Ether Synthesis from Syngas, Catal. Lett., 2014, 144, 1884–1893 CrossRef.
- Z. Danková, A. Mockovčiaková and S. Dolinská, Influence of ultrasound irradiation on cadmium cations adsorption by montmorillonite, Desalin. Water Treat., 2014, 52, 5462–5469 CrossRef.
- G. D. Yadav and N. Kirthivasan, Synthesis of bisphenol-A: comparison of efficacy of ion exchange resin catalysts vis-à-vis heteropolyacid supported on clay and kinetic modelling, Appl. Catal., A, 1997, 154, 29–53 CrossRef.
- I. Kiendl, H. Schmaderer, N. Schödel and H. Klein, Experimentelle und theoretische Betrachtungen der Methanol- und direkten DME-Synthese im Labor- und Technikums-Maßstab, Chem. Ing. Tech., 2020, 92, 736–745 CrossRef.
- P. Kampe, N. Herrmann, C. Ruhmlieb, M. Finsel, O. Korup, R. Horn and J. Albert, Spatially Resolved Reaction Profiles of CO2 Hydrogenation to Methanol Using In-Based Catalysts in a Compact Profile Reactor, ACS Sustainable Chem. Eng., 2024, 12, 9541–9549 CrossRef.
- A. Müller, J. Meyer, E. Krickemeyer and E. Diemann, Molybdenum Blue: A 200 Year Old Mystery Unveiled, Angew. Chem., Int. Ed. Engl., 1996, 35, 1206–1208 CrossRef.
- J. N. Barrows, G. B. Jameson and M. T. Pope, Structure of a heteropoly blue. The four-electron reduced .beta.-12-molybdophosphate anion, J. Am. Chem. Soc., 1985, 107, 1771–1773 CrossRef.
- V. Pace, J. V. Sinisterra and A. R. Alcantara, A. Celite-Supported Reagents in Organic Synthesis: An Overview, COC, 2010, 14, 2384–2408 CrossRef.
- K. Tanabe, Surface and catalytic properties of ZrO2, Mater. Chem. Phys., 1985, 13, 347–364 CrossRef.
- M. Trueba and S. P. Trasatti, γ-Alumina as a Support for Catalysts: A Review of Fundamental Aspects, Eur. J. Inorg. Chem., 2005, 2005, 3393–3403 CrossRef.
- S. Bagheri, N. M. Julkapli and S. B. A. Hamid, Titanium dioxide as a catalyst support in heterogeneous catalysis, Sci. World J., 2014, 2014, 727496 Search PubMed.
- T. W. van Deelen, C. H. Mejía and K. P. Jong, de Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity, Nat. Catal., 2019, 2, 955–970 CrossRef.
- J. Schnee, A. Eggermont and E. M. Gaigneaux, Boron Nitride: A Support for Highly Active Heteropolyacids in the Methanol-to-DME Reaction, ACS Catal., 2017, 7, 4011–4017 CrossRef.
- E. Rafiee and S. Eavani, Heterogenization of heteropoly compounds: a review of their structure and synthesis, RSC Adv., 2016, 6, 46433–46466 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07964g |
| This journal is © The Royal Society of Chemistry 2025 |