
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpiro thiochromene–oxindoles as novel anti-inflammatory agents: design, sustainable synthesis, in vitro and in silico evaluations†
Manju a,
Ashok Kumar Raigara,
Kamlesh Sainia,
Nirmal Jyotia,
Ravi Kumar Kapavarapub and
Anjali Guleria*a
a,
Ashok Kumar Raigara,
Kamlesh Sainia,
Nirmal Jyotia,
Ravi Kumar Kapavarapub and
Anjali Guleria*a
aDepartment of Chemistry, University of Rajasthan, Jaipur-302004, Rajasthan, India. E-mail: dranjaliguleria@uniraj.ac.in
bDepartment of Pharmaceutical Chemistry and Phytochemistry, Nirmala College of Pharmacy, Atmakur, Mangalgiri, Andhra Pradesh, India
First published on 2nd January 2025
Abstract
A one-pot, acid-, base-, and metal-free, multicomponent strategy has been developed to synthesize spiro thiochromene–oxindole derivatives as potential anti-inflammatory agents. The synthesized compounds were screened in vitro for their anti-inflammatory activity by inhibiting heat-induced Bovine Serum Albumin (BSA) denaturation assay, revealing moderate to good efficacy. Compounds 4e, 4k, and 4h exhibited the highest activity, inhibiting BSA denaturation by 90.97–95.45% at 800 μg mL−1 concentration with half maximal inhibitory concentration (IC50) values of 127.477 ± 2.285, 190.738 ± 3.561, and 285.806 ± 8.894 μg mL−1, respectively. For mechanistic insights in silico studies were conducted, revealing binding affinities of the active compounds with cyclooxygenase-2 (COX-2) protein, with binding energies of −8.9 kcal mol−1 (4e), −8.7 kcal mol−1 (4k), and −8.6 kcal mol−1 (4h). Bioactivity and pharmacokinetic parameters were further analyzed, encompassing ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) characteristics. This study highlights the potential of spiro thiochromene–oxindoles as anti-inflammatory agents, warranting further exploration as potential leads. The synthetic strategy for these target compounds utilizes taurine as an eco-friendly bio-organic catalyst, facilitating an acid-, base-, and metal-free intramolecular C–S and C–C bond formation in aqueous media. The reaction involves a one-pot, three-component Knoevenagel–Thia-Michael cascade between substituted isatins, 1,3-dicarbonyls, and 2-naphthalene thiol. Key features of this green protocol include high yields, cost-efficiency, non-toxicity, atom economy, and acid-, base-, and metal-free synthesis in water. Additionally, the catalyst exhibits excellent reusability, maintaining its activity across three cycles with easy recovery, while product isolation is achieved through simple filtration, eliminating the need for chromatographic purification and organic solvents. These attributes underscore this approach's synthetic and environmental advantages, highlighting its potential for broader application in the development of anti-inflammatory agents.
Introduction
Inflammation is a multifaceted physiological and pathological response that mobilizes immune and non-immune cells to safeguard host tissues by eliminating pathogens, promoting tissue repair, and restoring organ function. This essential defense mechanism plays a pivotal role in countering infections, repairing damage, and clearing invasive agents.1–4 In well-regulated inflammation, the initiation phase activates the immune system for host defense, while the resolution phase terminates the response and restores tissue homeostasis. However, dysregulated or chronic inflammation can lead to inflammatory diseases like arthritis, depression, colitis, Alzheimer's disease, and asthma, with significant tissue damage and increased risks of cancer, cardiovascular diseases, and osteoporosis.5–9 Thus, there is an urgent need for potent anti-inflammatory agents, given the prevalence of conditions characterized by inflammation and associated complications.Over the years, extensive research has firmly established that COX-2 is a key mediator in the inflammatory response.10–15 Although COX-2 is typically expressed at low levels in healthy tissues, its expression can increase dramatically during inflammatory episodes, leading to the overproduction of prostaglandins at sites of inflammation. This upregulation contributes to the exacerbation of tissue damage and the development of various inflammatory disorders. Given its important involvement in these pathological processes, COX-2 has emerged as a key therapeutic target.
Recognizing the essential role of COX-2 in inflammation, there is an ongoing effort among researchers to develop drugs that specifically inhibit this enzyme. The need for effective COX-2 inhibitors is underscored by the limitations of conventional nonsteroidal anti-inflammatory drugs (NSAIDs), which broadly inhibit both COX-1 and COX-2 enzymes, often leading to gastrointestinal side effects due to the suppression of protective COX-1 activity.16,17 Over the past decade, COX-2 selective inhibitors have been developed and are widely used in clinical settings to manage conditions such as rheumatoid arthritis, osteoarthritis, postoperative pain, toothache, and certain cancers.18–23 These selective inhibitors specifically target COX-2, thereby reducing inflammation and minimizing the adverse effects of non-selective NSAIDs. However, despite these advancements, significant challenges persist, as some COX-2 inhibitors are still linked to side effects, including cardiovascular risks and other toxicities, which restrict their broader clinical application. This ongoing issue reveals an important research gap and underscores the need for the development of newer and more potent COX-2 inhibitors that can provide effective anti-inflammatory benefits with minimal, if any, side effects. Therefore, continued research efforts are essential to design and synthesize COX-2 inhibitors with improved safety profiles and therapeutic efficacy.
Hence, in our quest to develop more effective COX-2 inhibitors, we have strategically focused on designing novel compounds by fusing two privileged scaffolds known for their established anti-inflammatory properties. This approach is grounded in extensive research highlighting the therapeutic potential of these scaffolds as COX-2 inhibitors. During our search for suitable privileged structures, we identified the work of Kim et al.,24 which demonstrated the potent anti-inflammatory activity of 3-substituted oxindoles. These oxindoles exhibit structural resemblance to aurones, a naturally occurring class of flavones in plants known for their anti-inflammatory effects. This structural similarity has inspired further exploration of 3-substituted oxindoles, ultimately confirming their anti-inflammatory potential and COX-2 inhibitory activity, as reported by Pan et al.25
For the second privileged scaffold, we turned our attention to thiopyrans, based on the findings of Rogier Jr et al.,26 who established that thiopyrans also possess significant anti-inflammatory activity as COX-2 inhibitors. Recognizing the potential of these scaffolds, we hypothesized that their fusion could lead to compounds with enhanced efficacy and safety profiles. By integrating 3-substituted oxindoles and thiopyrans, our design aims to leverage the complementary mechanisms of action inherent in these two scaffolds. This approach seeks to enhance the selective inhibition of COX-2, thereby improving anti-inflammatory efficacy while minimizing off-target effects commonly associated with other COX inhibitors.
The fusion of these two scaffolds is intended to yield novel drug leads with superior anti-inflammatory properties, addressing the critical need for COX-2 inhibitors that offer improved safety profiles and reduced side effects. By selecting and fusing these privileged scaffolds, we aim to synthesize newer COX-2 inhibitors that exhibit potent anti-inflammatory activity and demonstrate a favorable therapeutic index (Fig. 1). This approach represents a significant advancement in the design of COX-2 inhibitors, offering the potential to develop safer and more effective anti-inflammatory agents.
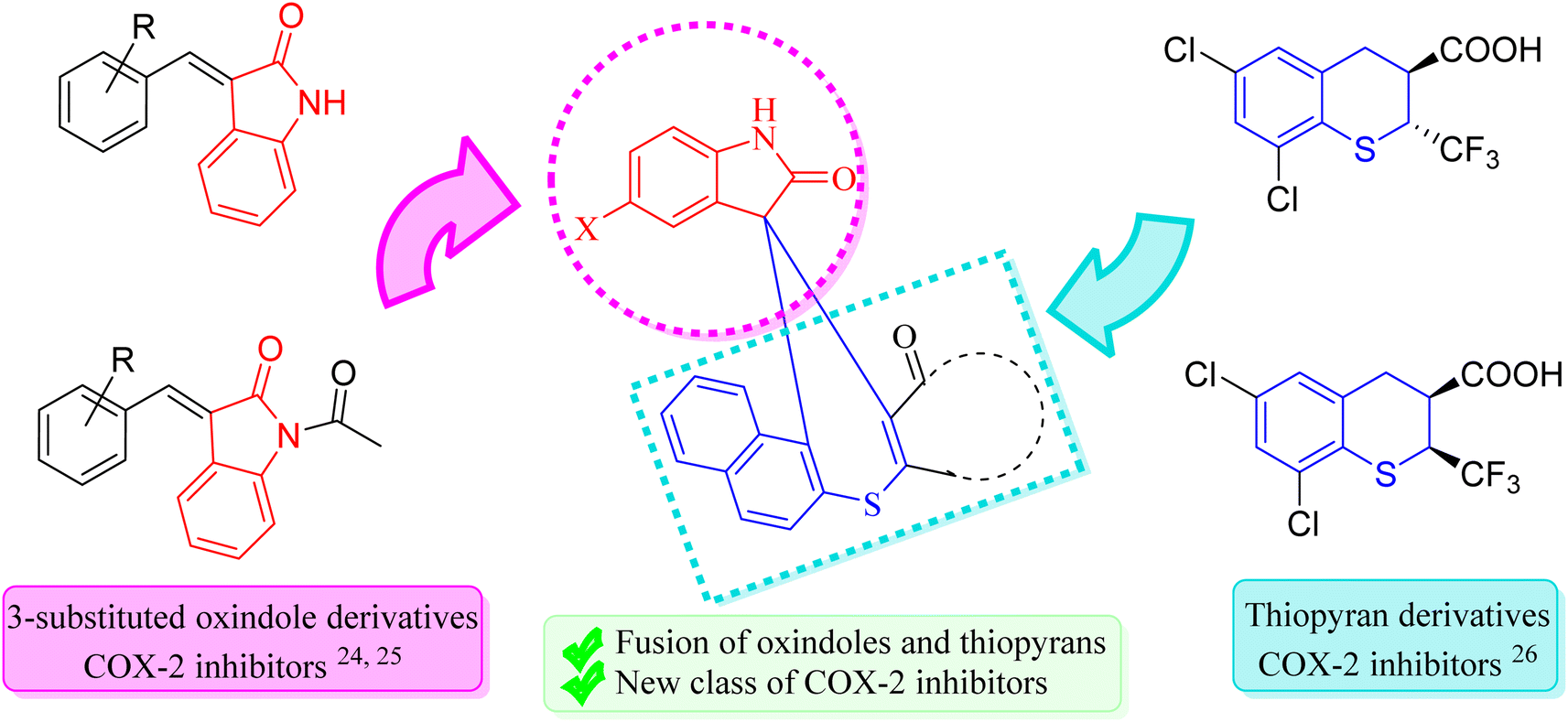 | ||
| Fig. 1 Rationale of spiro thiochromene–oxindole derivatives. | ||
After identifying the potential anti-inflammatory COX-2 inhibiting scaffolds, our subsequent objective was to develop a sustainable synthetic route for their synthesis. The importance of sustainable synthetic methodologies in the pharmaceutical industry cannot be overstated, as they offer several critical advantages, including reduced environmental impact by reducing waste, lowering energy consumption, cost-effectiveness, and enhanced safety. To achieve this, we focused on optimizing reaction conditions that minimize the use of hazardous volatile organic solvents, prioritized milder reaction conditions with simple operations that do not require chromatographic steps, and using non-toxic, reusable, and easily recoverable green bio-organic catalysts. By integrating green chemistry principles, our approach aims to produce the desired COX-2 inhibitors effectively while reducing the environmental impact and enhancing the overall sustainability of the synthesis.
Hence, to identify the most efficient route for synthesizing the target compound, we employed a retrosynthetic analysis that suggested the feasibility of a Knoevenagel–Michael cascade reaction followed by intramolecular ring closure through C–S bond formation (Scheme 1).
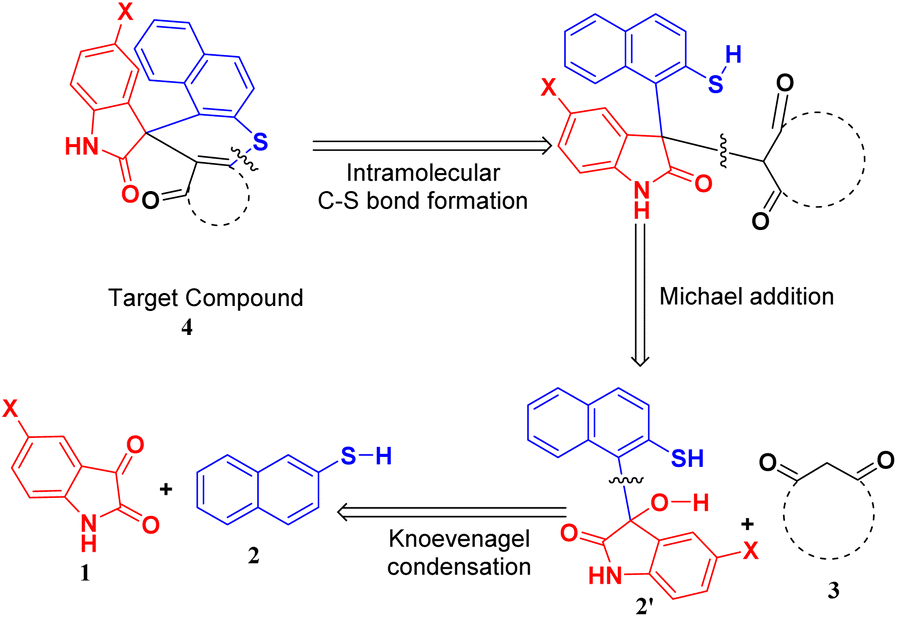 | ||
| Scheme 1 Retrosynthetic approach for the target compound. | ||
This approach leverages the potential of a stepwise reaction sequence, which is advantageous for synthesizing complex molecules with greater efficiency. Recent studies have reported the use of various green catalysts for Knoevenagel–Michael cascade reactions in aqueous media,27–31 aligning with our sustainability objectives. However, the subsequent step of intramolecular ring closure via C–S bond formation using green catalysts and aqueous solvents remains underexplored. The literature indicates that Thia-Michael intramolecular C–S bond formation typically relies on acid-, base-, and metal-based catalysts or reactions conducted in volatile organic solvents under harsh conditions, which are often associated with significant environmental drawbacks, such as the use of toxic and expensive catalysts, high reaction temperatures, and complex operational procedures32–46 (Fig. 2). To address these challenges, in one of our prior studies, we explored a greener approach and successfully achieved C–S bond formation and ring closure under catalyst-free conditions using an azeotropic mixture of water and ethanol in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as solvent under reflux conditions.47 Although this method demonstrated the feasibility of a greener approach, it was limited by a narrow substrate scope, which restricted its broader applicability. Therefore, there is a clear need for an aqueous-mediated green catalytic system capable of facilitating Knoevenagel–Thia-Michael cascade reaction for C–S bond formation in a one-pot process under mild reaction conditions with an expanded substrate scope.
:1 ratio as solvent under reflux conditions.47 Although this method demonstrated the feasibility of a greener approach, it was limited by a narrow substrate scope, which restricted its broader applicability. Therefore, there is a clear need for an aqueous-mediated green catalytic system capable of facilitating Knoevenagel–Thia-Michael cascade reaction for C–S bond formation in a one-pot process under mild reaction conditions with an expanded substrate scope.
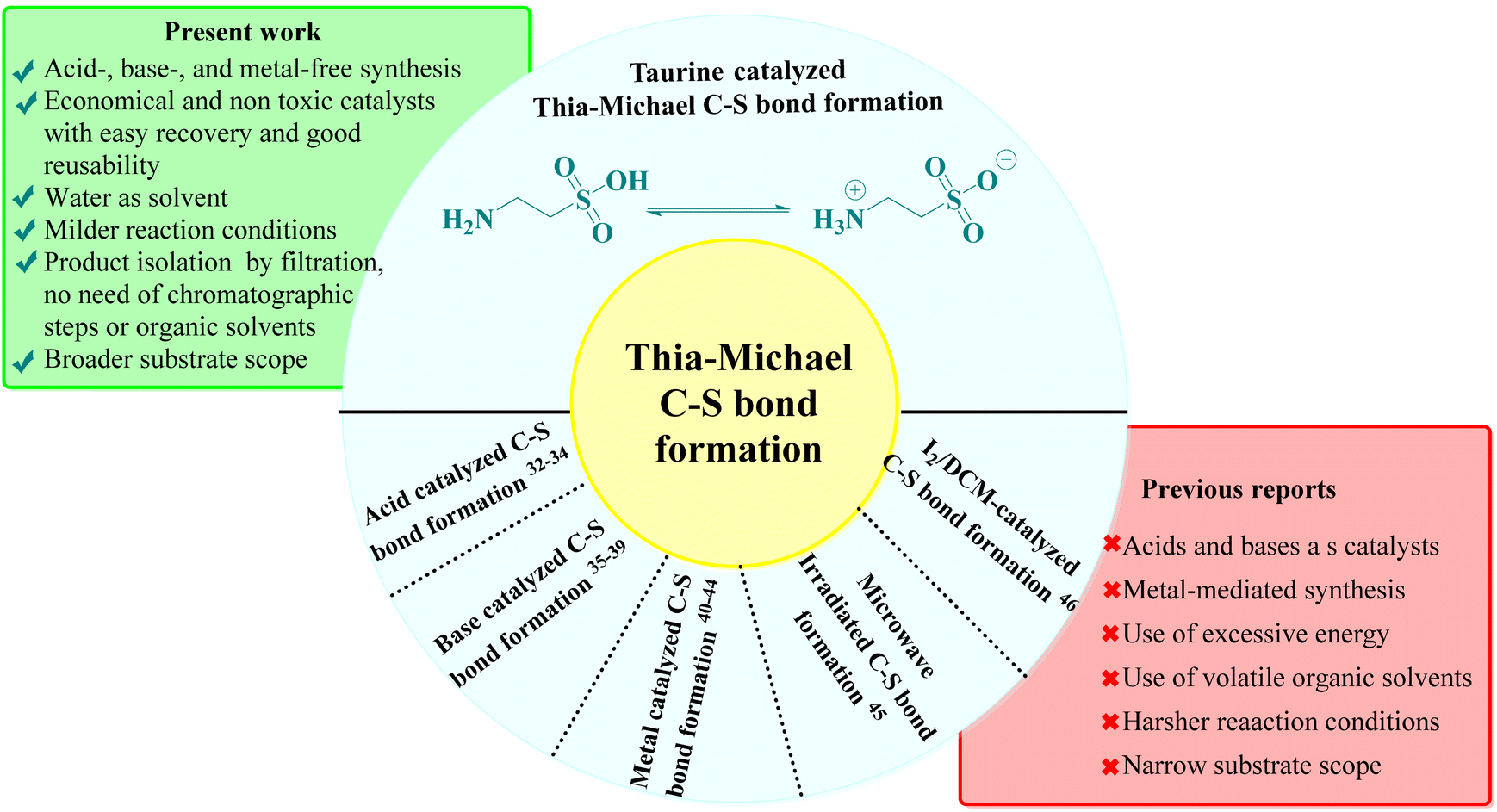 | ||
| Fig. 2 Comparison of previous literature and present work. | ||
To address this need, we identified taurine, a β-amino acid abundant in human and animal tissues, as a promising candidate due to its established efficacy as a green bio-organic bifunctional hydrogen bond donor–acceptor catalyst, known for effectively catalyzing Knoevenagel–Michael cascade reactions.48–54 Taurine is economically viable, water-soluble, and non-toxic, meeting key criteria for sustainable pharmaceutical synthesis. Its catalytic activity is attributed to its ability to adopt a zwitterionic form in aqueous solutions, enhancing its hydrogen-bonding capabilities, as supported by computational modeling and NMR studies.55–57 This characteristic not only facilitates catalytic processes in water, aligning with our environmental goals but could also provide a versatile catalytic environment for both the Knoevenagel–Thia-Michael cascade for subsequent C–S bond formation under mild conditions. By integrating taurine as the catalyst, we envisioned developing a one-pot, aqueous-mediated synthesis that overcomes the limitations of traditional methods for intramolecular C–S bond formation, ultimately broadening the substrate scope and enhancing the sustainability of the synthesis of target compounds.
Finally, the synthesized analogs were evaluated for their anti-inflammatory potential using a heat-induced BSA denaturation assay. Among the tested compounds, 4e, 4k, and 4h exhibited the most significant activity, achieving BSA denaturation inhibition rates between 90.97% and 95.45% at a concentration of 800 μg mL−1 with IC50 values of 127.477 ± 2.285, 190.738 ± 3.561, and 285.806 ± 8.894 μg mL−1, respectively. To further elucidate the in vitro anti-inflammatory effects, in silico studies were performed to assess the binding affinities, molecular interactions, and structural mechanisms of the compounds. Notably, compounds 4e, 4k, and 4h showed binding energies of −8.9 kcal mol−1, −8.7 kcal mol−1, and −8.6 kcal mol−1, respectively, with the COX-2 protein. Additionally, bioactivity assessments and pharmacokinetic profiling, including ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) parameters, were conducted to further characterize the therapeutic potential of these compounds.
In summary, the current study addresses the critical need for novel and effective COX-2 inhibitors by designing and synthesizing spiro thiochromene–oxindole derivatives, leveraging the anti-inflammatory potential of privileged scaffolds like 3-substituted oxindoles and thiopyrans. Given the limitations of conventional COX-2 inhibitors, our approach aims to enhance therapeutic efficacy while minimizing adverse effects through selective inhibition of COX-2. To support this goal, we employed a sustainable synthetic strategy grounded in green chemistry principles, utilizing taurine as a bio-organic bifunctional catalyst that catalyzes the Knoevenagel–Michael cascade reaction and intramolecular C–S bond formation in aqueous media, thus, addressing the challenges associated with traditional metal-based or harsh catalytic conditions. The synthesized compounds were evaluated through in vitro and in silico studies, demonstrating promising anti-inflammatory activity and potential as COX-2 inhibitors. This work underscores the importance of combining sustainable synthesis with strategic scaffold selection to advance the development of next-generation anti-inflammatory agents. Further clinical trials are necessary to fully validate the therapeutic efficacy and safety profiles of these compounds for potential clinical application.
Results and discussion
Chemistry
Preliminary optimization studies were conducted using a model three-component reaction involving 5-bromo isatin (1a), 2-naphthalene thiol (2), and dimedone (3a) to develop an efficient, green catalytic system. Reaction progress was monitored via TLC (ethyl acetate/hexane, 2:8) under various reaction conditions to determine the most favorable setup.
Initially, the reaction was attempted under solvent- and catalyst-free conditions at temperatures ranging from room temperature to 130 °C. However, no product formation was observed, even after 12 hours. This can be attributed to insufficient nucleophilic and electrophilic activation of the starting materials, particularly the carbonyl functionalities, which require external catalysis to promote nucleophilic attack. Recognizing these limitations, the reaction was tested under solvent-free conditions with taurine (28 mol%) as a potential catalyst at various temperatures (RT − 130 °C). In all instances, only trace amounts of the product were detected, confirming the necessity of a catalytic and solvent system to enhance reactivity as higher temperatures alone were insufficient to drive the reaction forward.
Realizing the limitations of solvent-free conditions, we explored solvent-based approaches with a focus on green and sustainable systems. A range of organocatalysts, including taurine, thiourea dioxide, proline, and thiourea, were tested using aqueous media under reflux conditions. Among these, taurine demonstrated the highest catalytic efficiency, yielding 95% of the desired product, compared to thiourea dioxide (15%) and trace amounts produced by proline and thiourea (Fig. 3a). Taurine's superior catalytic performance can be attributed to its zwitterionic nature in aqueous media, which enhances both nucleophilic and electrophilic activation by stabilizing transition states and promoting proton transfer.
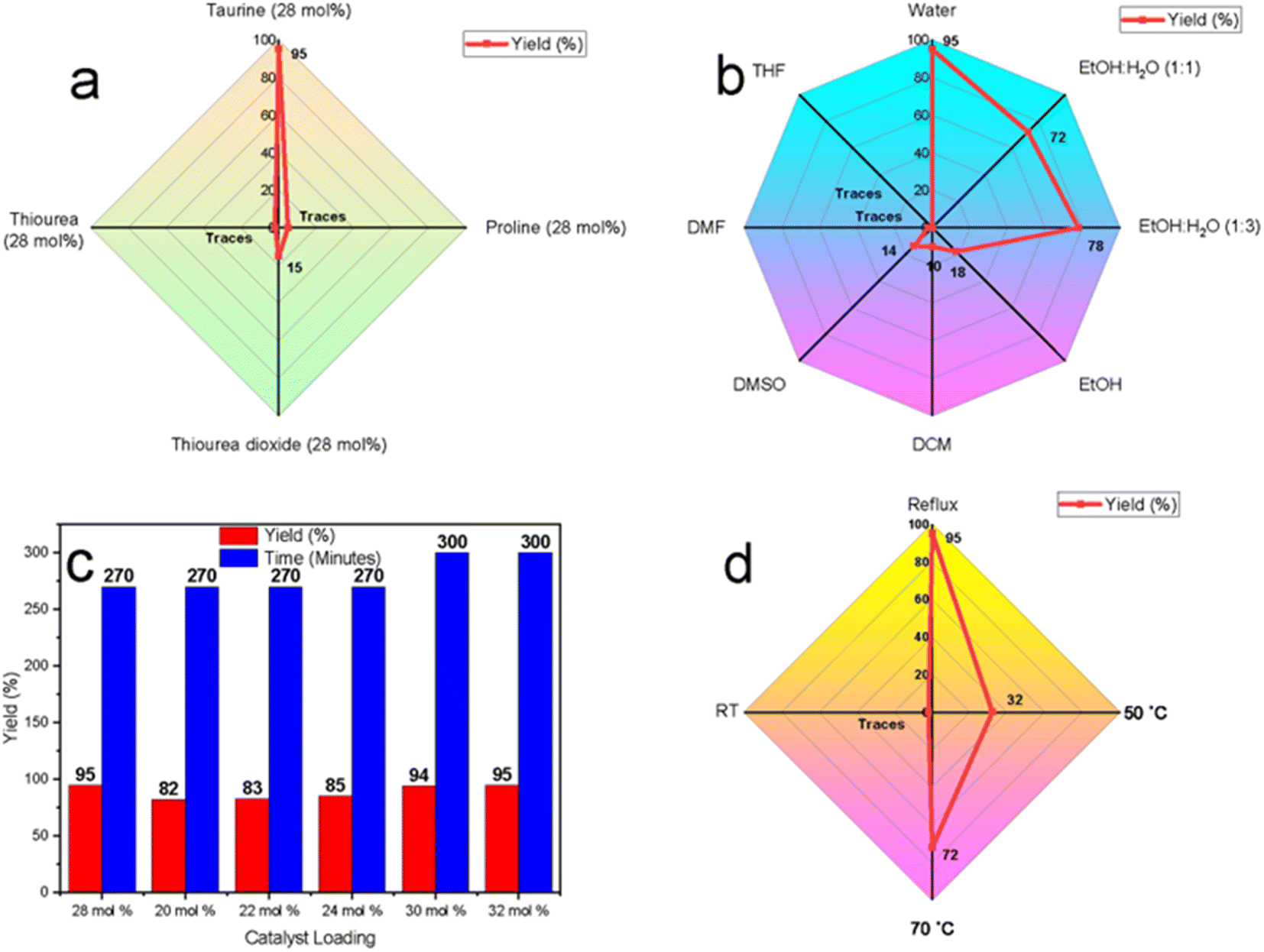 | ||
| Fig. 3 Optimization studies (a) catalyst optimization using water as solvent under reflux conditions, (b) solvent optimization using taurine as the catalyst under reflux conditions, (c) catalyst amount optimization, (d) temperature optimization using taurine (28 mol%) as the catalyst in water. | ||
To further validate water as the optimal solvent, taurine-catalyzed reactions were conducted in a variety of solvents, including ethanol, ethanol–water mixtures (1:1 and 1:3), dichloromethane (DCM), dimethyl sulfoxide (DMSO), dimethylformamide (DMF), and tetrahydrofuran (THF) under reflux (Fig. 3b). The yields were significantly lower in organic solvents, such as ethanol (18%), DCM (10%), and DMSO (14%), with only trace amounts observed in DMF and THF, likely due to taurine's poor solubility. Ethanol–water mixtures improved yields to 72% and 78%, while water alone provided the highest yield of 95%. These results suggest that water enhances the catalytic activity of taurine by enhancing its solubility and promoting its zwitterionic form, leading to improved hydrogen-bonding interactions that stabilize intermediates and accelerate the reaction.
Next, the effect of catalyst loading was evaluated by varying the mol% of taurine in water under reflux (Fig. 3c). The highest yield (95%) was achieved with 28 mol% taurine, within 270 minutes. Finally, to affirm that reflux conditions were the best conditions temperature optimization studies were conducted in the taurine-catalyzed aqueous medium, revealing that no reaction occurred at room temperature. Yield improvements were observed as the temperature increased, with the highest yield (95%) obtained under reflux conditions. This is likely due to enhanced molecular motion and increased energy input, which are necessary to overcome activation barriers and facilitate catalytic turnover.
In conclusion, the optimal reaction conditions were determined to be 28 mol% taurine in water under reflux, yielding the highest efficiency. These findings not only establish taurine as an effective green organocatalyst but also highlight water as the preferred solvent, reinforcing the environmental advantages of this sustainable catalytic system.
After successfully optimizing the reaction conditions, the scope of the reaction was evaluated by synthesizing a series of derivatives of the target compounds (4a–l) using substituted isatins (1a and 1b), 2-aminothiophenol (2), and various cyclic 1,3-dicarbonyl compounds (3a–f), as outlined in Scheme 2. All reactions proceeded efficiently under the established optimal conditions, yielding the desired products in excellent yields within reasonable reaction times across the substrate range. No significant electronic or steric influence of the substituents on the 1,3-dicarbonyl compounds was observed, as indicated by the results summarized in ESI Table 1.† However, the reaction failed with acyclic 1,3-dicarbonyl compounds, which can likely be attributed to increased steric hindrance, preventing Michael's addition, a key step for the synthesis of target compounds.
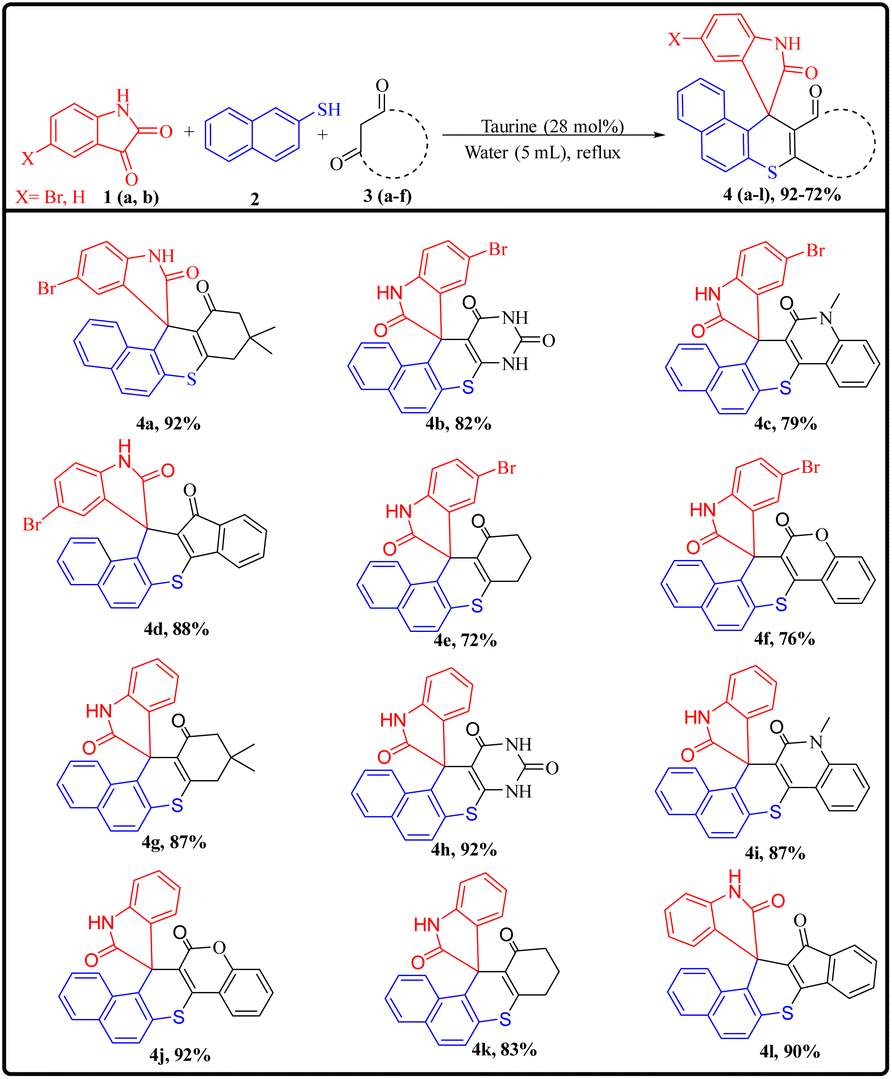 | ||
| Scheme 2 Synthesis and substrate scope of spiro thiochromene–oxindole derivatives. | ||
A plausible mechanism for the synthesis of spiro thiochromene–oxindole derivatives (4a–l) is illustrated in Scheme 4. To determine the reactivity order between the two reactants, substituted 1,3-dicarbonyls (3a–f) or 2-naphthalene thiol (2) with substituted isatin (1a and 1b), we carried out a control experiment, as depicted in Scheme 3. Two separate two-component reactions were performed, one between 1a and 3a, and another between 1a and 2, under the optimal conditions. The results indicated that no reaction occurred between 1a and 3a, even after 24 hours. However, for the reaction between 1a and 2, the intermediate 2′ formed within 2 hours under the same optimal conditions.
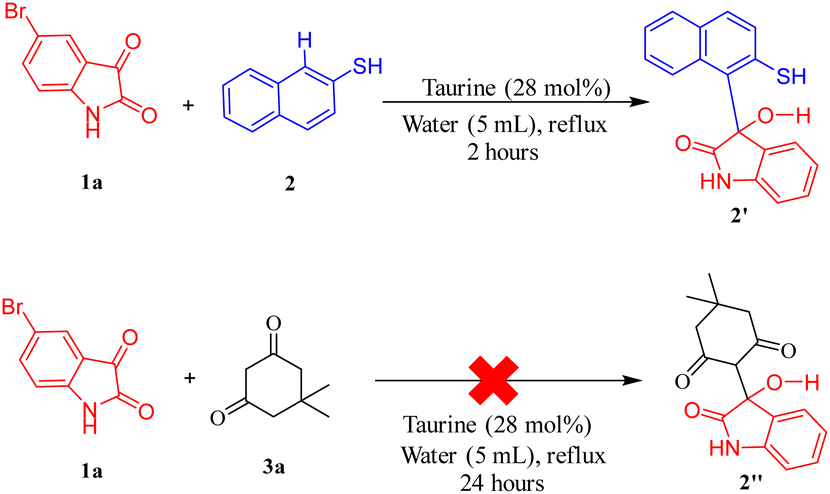 | ||
| Scheme 3 Control experiment to determine reactivity order for Knoevenagel condensation between substituted 1,3-dicarbonyls and 2-naphthalene thiol. | ||
From this, we concluded that the reaction initiates with taurine-facilitated Knoevenagel condensation between 1 and 2, yielding the intermediate 2′.
Subsequently, taurine promotes a Michael addition between 2′ and 3, followed by a taurine-catalyzed ring closure via intramolecular C–S bond formation, accompanied by the elimination of water, ultimately yielding the desired spiro thiochromene–oxindole derivatives (4a–l), as depicted in Scheme 4.
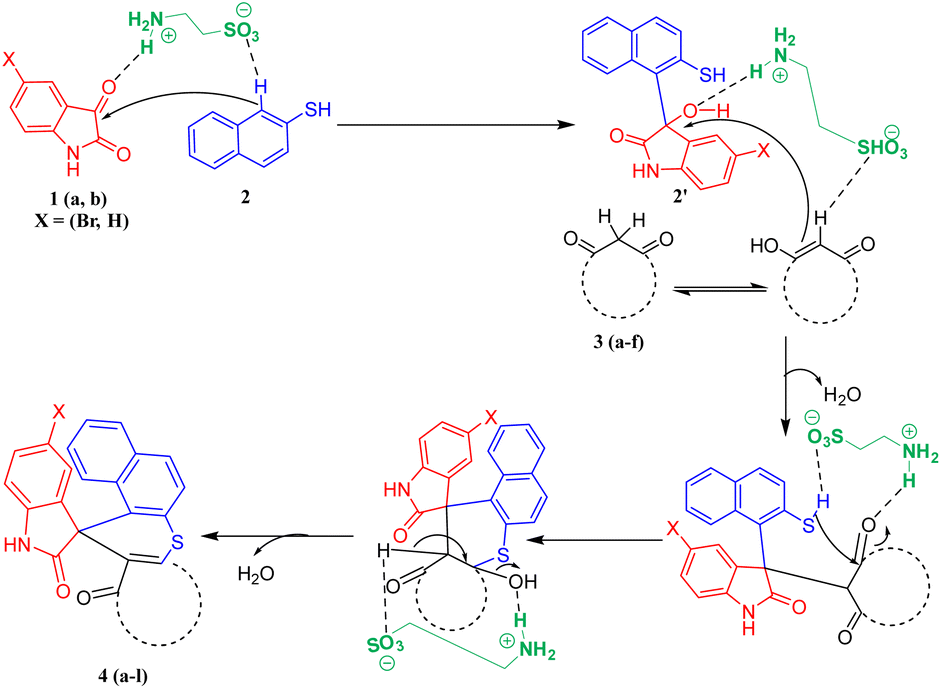 | ||
| Scheme 4 Plausible mechanism for the synthesis of spiro thiochromene–oxindole derivatives (4a–l). | ||
Catalyst reusability is a key consideration in sustainable synthesis. To evaluate the reusability of taurine as a catalyst, we selected 5-bromo isatin (1a), 2-naphthalene thiol (2), and dimedone (3a) as model substrates. The reactions were performed consecutively three times using the same aqueous solution from the previous run, without adding fresh catalyst for subsequent reactions. The results demonstrated excellent reusability of taurine, as it retained its catalytic activity over three cycles, with no significant decrease in product yields or any notable increase in reaction times (Fig. 4a).
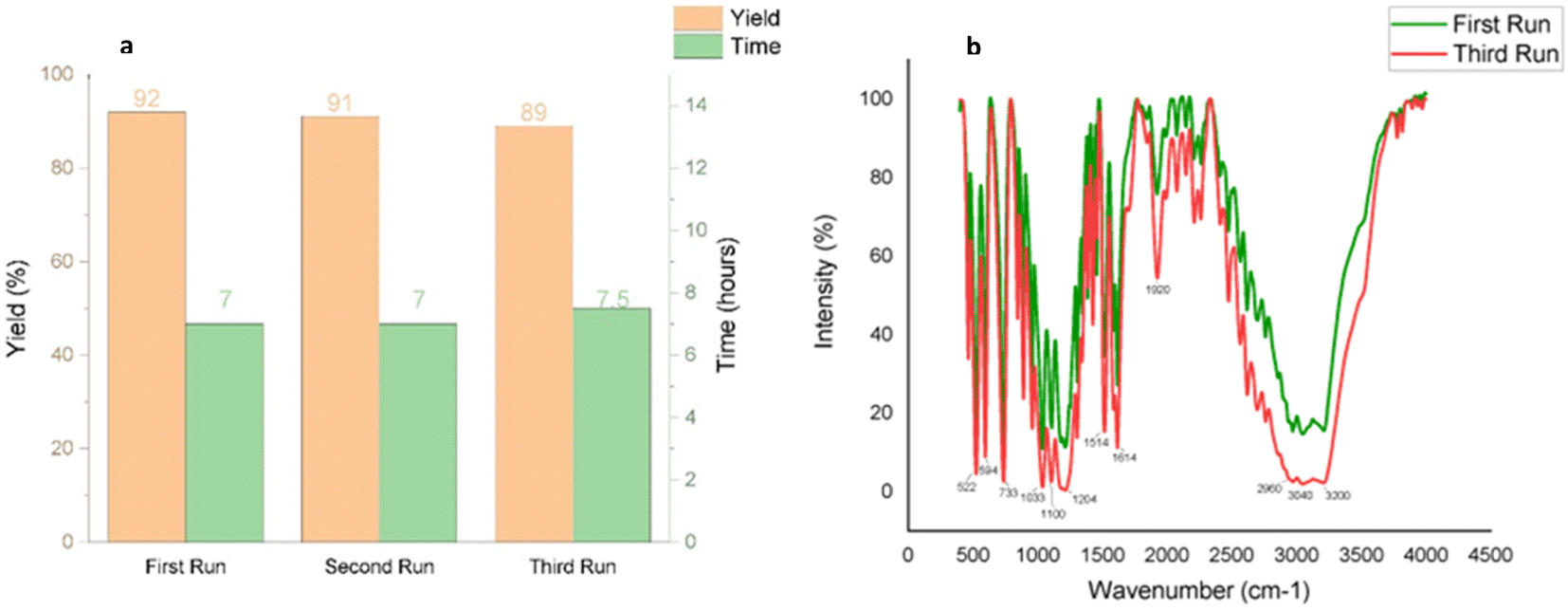 | ||
| Fig. 4 (a) Reusability of the catalyst for synthesizing 4a (b) comparative IR spectra of fresh and recycled catalyst to validate its purity after three runs. | ||
In the final cycle, the catalyst was recovered by evaporating the water and recrystallizing the residue from ethanol, yielding an 89% recovery of taurine after the third run. Comparative IR spectra of the fresh and recycled taurine (post-third run) confirmed the purity of the recycled catalyst, further supporting its efficient reusability (Fig. 4b).
In vitro anti-inflammatory activity screening of synthesized compounds (4a–l)
Upon exposure to heat, BSA undergoes denaturation, leading to the expression of antigens associated with Type III hypersensitivity reactions, which are implicated in various inflammatory disorders, including systemic lupus erythematosus, glomerulonephritis, and rheumatoid arthritis. Inhibiting BSA denaturation serves as an in vitro model to assess the anti-inflammatory properties of compounds by preventing protein unfolding and subsequent antigen presentation.
This assay, therefore, provides a reliable method for preliminary screening of anti-inflammatory activity. The synthesized compounds (4a–l) were tested for their ability to inhibit BSA denaturation, quantitatively measuring their anti-inflammatory potential. The results indicated moderate to high efficacy across the series. Fig. 5a illustrates the percentage inhibition of BSA denaturation by all compounds at 800 μg L−1 concentration. Among these, compounds 4e, 4k, and 4h exhibited the highest activity, with inhibition rates ranging from 90.97% to 95.45% at 800 μg mL−1. Their respective IC50 values were 127.477 ± 2.285, 190.738 ± 3.561, and 285.806 ± 8.894 μg mL−1. These IC50 values were comparable or superior to the reference drug, diclofenac sodium, which had an IC50 value of 243.641 ± 2.686 μg mL−1 (Fig. 5b).
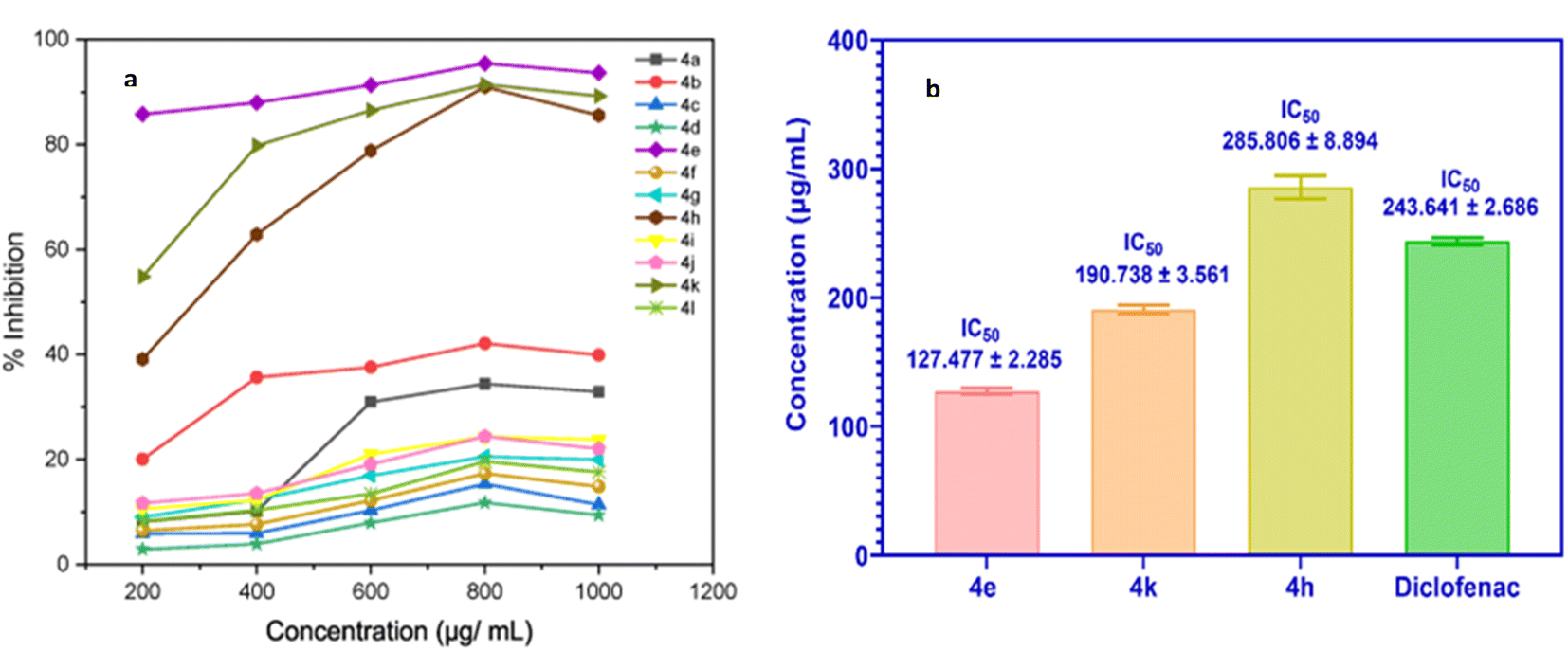 | ||
| Fig. 5 (a) Percent inhibition activity graph of compounds (4a–l); (b) IC50 values of 4e, 4k, 4h, and diclofenac (control). | ||
In silico studies for the anti-inflammatory activity of synthesized compounds (4a–l)
Docking simulations revealed that the synthesized compounds consistently exhibited stronger binding affinities towards COX-2 compared to COX-1. Celecoxib used as a reference, displayed binding energies of −11.6 kcal mol−1 with COX-2 and −10.5 kcal mol−1 with COX-1. Among the evaluated compounds, compound 4e demonstrated the highest binding affinity with COX-2 (−8.9 kcal mol−1). The top three compounds, based on their COX-2 binding affinities, were 4e (−8.9 kcal mol−1), 4k (−8.7 kcal mol−1), and 4h (−8.6 kcal mol−1). Compounds 4a (−8.4 kcal mol−1) and 4b (−7.9 kcal mol−1) also showed promising binding energies, reinforcing their potential as COX-2 selective inhibitors. In contrast, these compounds demonstrated lower affinities towards COX-1: 4e (−6.1 kcal mol−1), 4k (−6.8 kcal mol−1), 4h (−6.5 kcal mol−1), 4a (−6.4 kcal mol−1), and 4b (−6.8 kcal mol−1), highlighting their selectivity for COX-2. However, compound 4j displayed relatively less selectivity, with a binding affinity of −7.9 kcal mol−1 for COX-2 and −7.2 kcal mol−1 for COX-1. These findings suggest that the compounds with better selectivity towards COX-2 warrant further exploration as potential anti-inflammatory agents. Binding energy outcomes for all compounds are presented in Fig. 6.
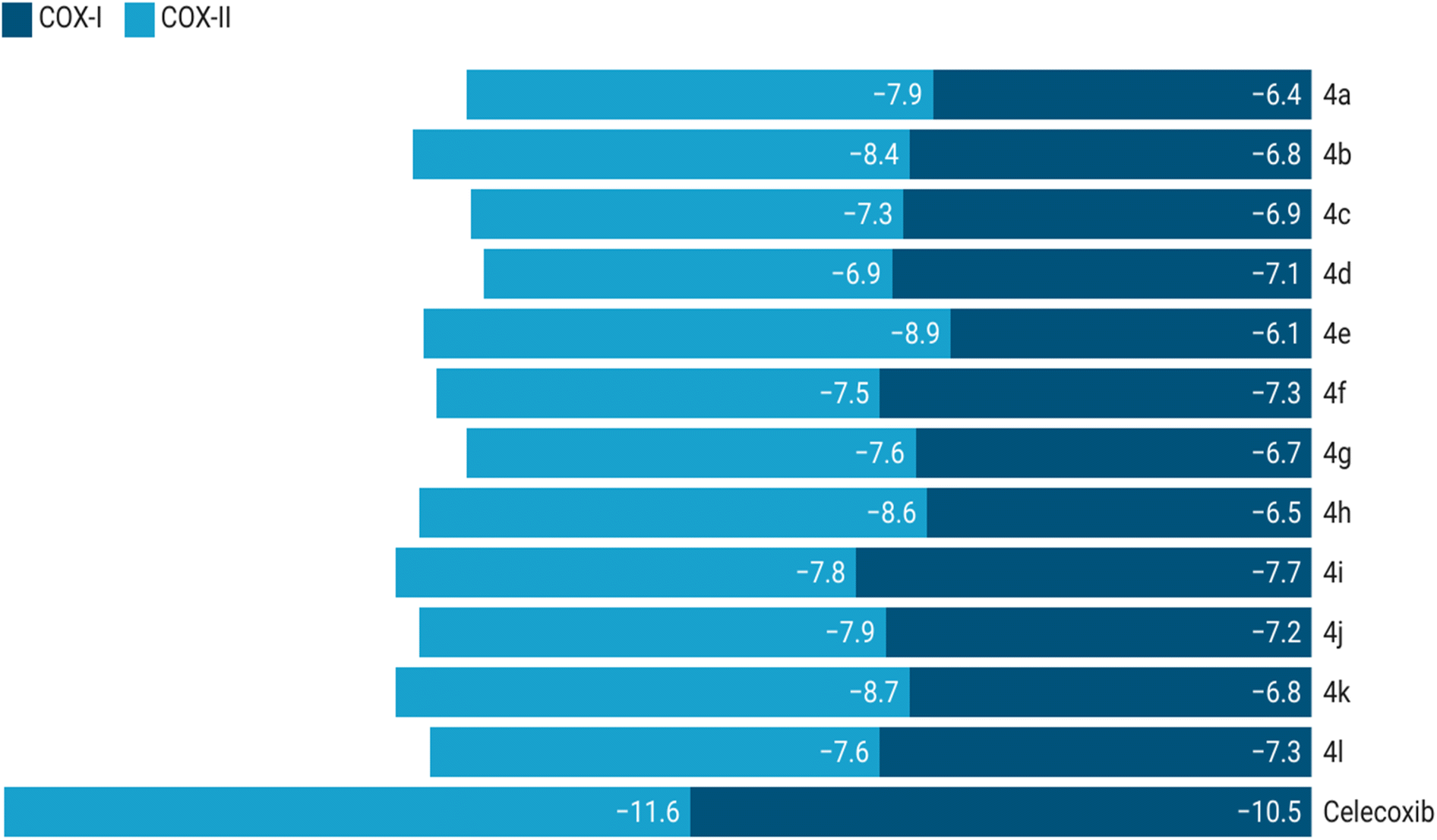 | ||
| Fig. 6 Binding energy plot of compounds with COX-1 and COX-2. | ||
Fig. 6 provides a comparative plot of the binding energies of the synthesized compounds with COX-1 and COX-2. The interaction analysis of selected high-affinity compounds is further depicted through 2D interaction diagrams in Fig. 7, illustrating their molecular interactions with the active site residues of the target proteins. A detailed molecular interaction summary of all synthesized compounds with COX-1 and COX-2 has been displayed in ESI Tables 2 and 3.†
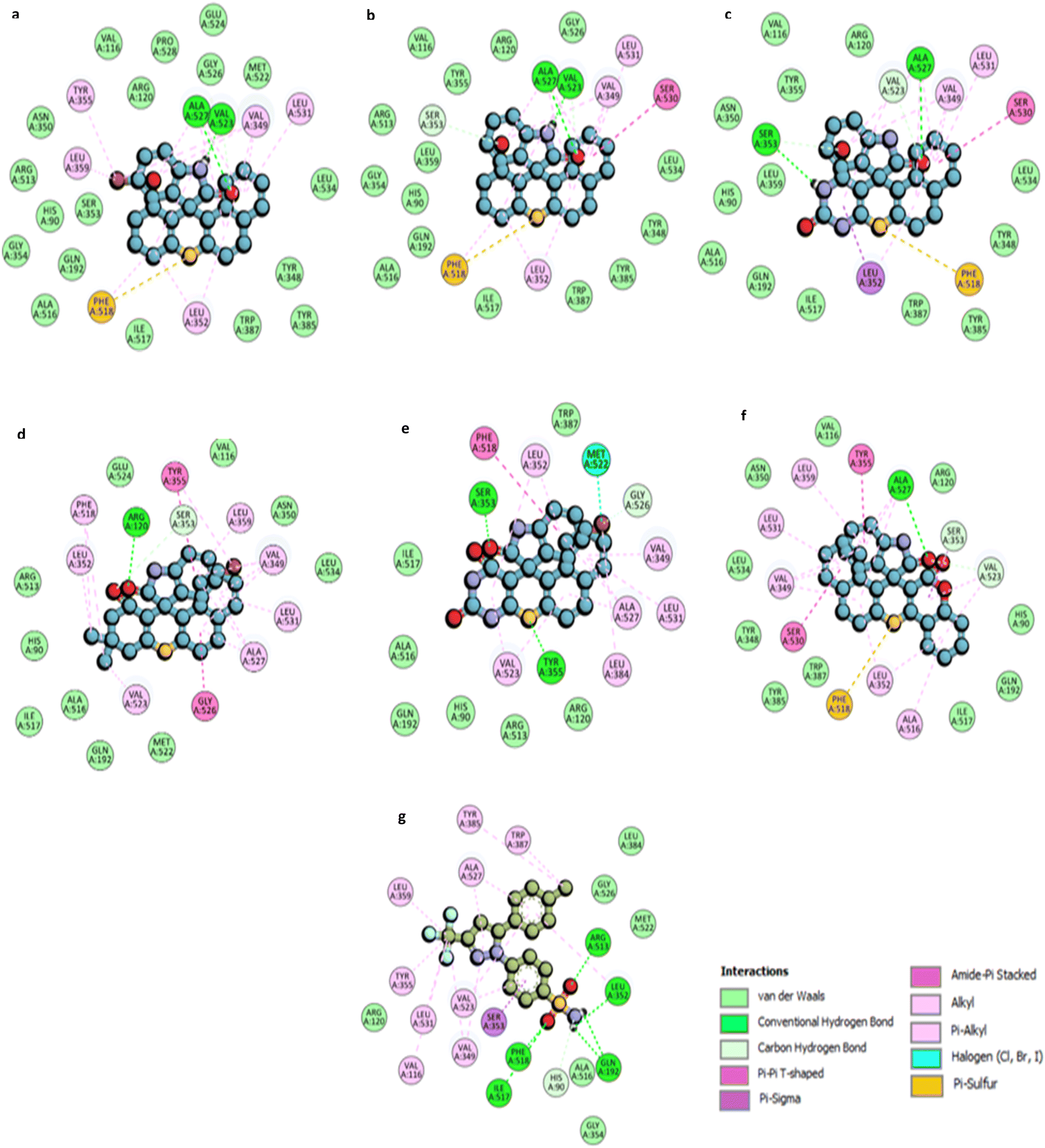 | ||
| Fig. 7 2D molecular representation of interactions of compounds (a) 4e; (b) 4k; (c) 4h; (d) 4a; (e) 4b; (f) 4j, and (g) Celecoxib with the active site residues of the human COX-2 protein. Interactions were displayed as color-coded dashed lines; green lines indicated the H–H-bonds. | ||
For compound 4e, a 5-bromo spiro thioxanthene derivative, two hydrogen bond interactions are observed between the –NH and carbonyl groups of the indoline ring with the ALA527 and VAL523 residues. The sulfur atom of the thioxanthene ring participates in a π–sulfur interaction with PHE518, which also forms π–alkyl interactions. Additional π–alkyl interactions occur between the aromatic rings of the scaffold and residues such as LEU352, LEU359, TYR355, VAL349, and LEU531 (Fig. 7a).
Compound 4k displays similar interaction patterns, with hydrogen bonds forming between the –NH and carbonyl groups of the indoline ring and ALA527 and VAL523. The sulfur of thioxanthene engages in π–sulfur interactions, while SER530 forms an amide–π-stacked interaction and SER353 contributes through carbon-hydrogen bond interactions. Π–alkyl interactions are observed with VAL349, LEU531, and LEU352 (Fig. 7b).
For compound 4h, hydrogen bonding is observed between the carbonyl group of the indoline ring and ALA527, and between the –NH group of the thioxanthene and SER353. Additionally, PHE518 participates in π–sulfur interactions with the sulfur of the thioxanthene ring. The compound also engages in π–alkyl interactions with residues such as VAL523, VAL349, LEU352, and LEU531 (Fig. 7c).
Compound 4a forms a hydrogen bond with ARG120 via the carbonyl moiety on the indoline ring, while TYR355 engages in π–π T-shaped interactions. Π–alkyl interactions are observed with residues such as ALA527, LEU531, VAL349, and LEU359. GLY526 forms an amide–π-stacked interaction, and additional interactions occur between PHE518 and the methyl groups of the thioxanthene scaffold (Fig. 7d).
Compound 4b exhibits two hydrogen bonds: one with TYR355 through the sulfur of the thioxanthene ring and another with SER353 through the carbonyl group of the indoline ring. PHE518 is involved in π–π T-shaped interactions, while MET522 forms a halogen interaction with the bromine group. Several residues, including LEU352, ALA527, and VAL523, contribute to π–alkyl and alkyl interactions (Fig. 7e).
Finally, compound 4j displays hydrogen bond interactions like compound 4h, with ALA527 and PHE518 participating in π–sulfur interactions with the thioxanthene sulfur. Additional π–σ and amide–π-stacked interactions are observed with SER353 and SER530 (Fig. 7f).
Celecoxib, the standard COX-2 inhibitor, forms five hydrogen bond interactions through its sulfonamide group with residues such as ARG513, PHE518, and ILE517. The –CF3 group engages in alkyl interactions with VAL116, LEU531, and TYR355, while π–alkyl interactions are seen with ALA527 and VAL349 (Fig. 7g).
Fig. 8 and 9 further illustrate the binding modes and pharmacophore features of the top compounds and celecoxib, respectively. Fig. 8 depict the binding modes of the top compounds and celecoxib within the active pocket, highlighting their respective interactions and Fig. 9 provides insights into the molecular alignment of binding poses, comparing them with celecoxib, and examines the pharmacophore features within the binding site. Moreover, the molecular alignment of the binding poses reveals notable overlaps in pharmacophoric features among compounds 4a, 4b, 4e, 4h, 4j, 4k, and celecoxib, as depicted in Fig. 9b.
 | ||
| Fig. 8 COX-2 surface image of protein displaying the (a) binding modes and (b) interactions of 4a, 4b, 4e, 4h, 4j, 4k, and Celecoxib with the active site residues of the human COX-2 protein. | ||
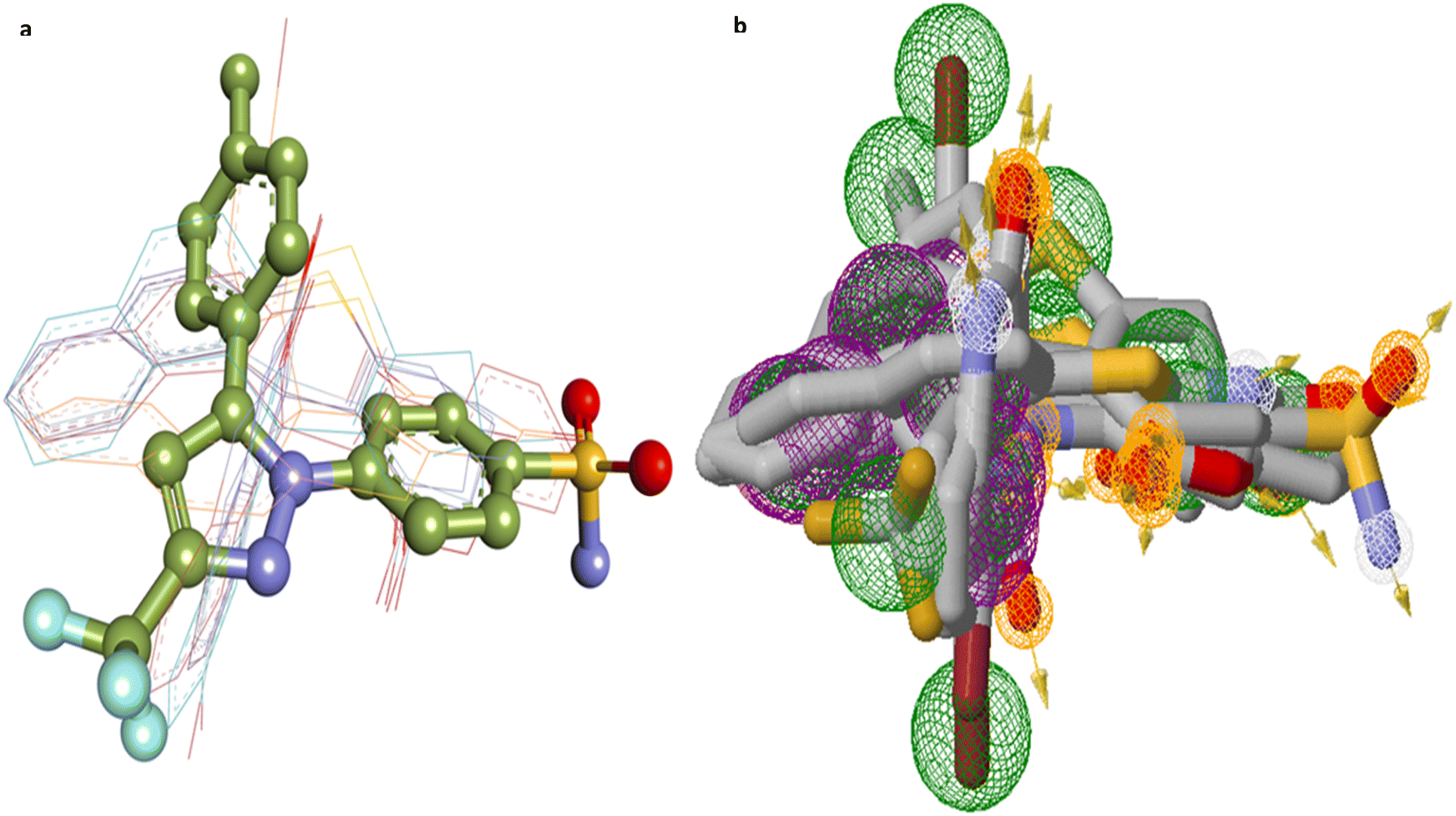 | ||
| Fig. 9 (a) Molecular alignment of binding poses of 4a, 4b, 4e, 4h, 4j, 4k and Celecoxib in the active site of COX2 proteins; (b) pharmacophore features of top compounds along with the Celecoxib (pharmacophore features color coding include: purple rings-aromatic, green rings-hydrophobic, orange rings-hydrogen bond donor, white rings-hydrogen bond acceptor). | ||
Overall, docking simulations suggest that the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –NH moieties adjacent to the indoline ring play key roles in hydrogen bonding across the top compounds, closely mimicking the sulfonamide –SO and –NH2 groups of celecoxib. While the specific active site residues involved in hydrogen bonding vary among the synthesized compounds, their interaction profiles highlight promising avenues for further investigation as selective COX-2 inhibitors with potential anti-inflammatory activity.
O and –NH moieties adjacent to the indoline ring play key roles in hydrogen bonding across the top compounds, closely mimicking the sulfonamide –SO and –NH2 groups of celecoxib. While the specific active site residues involved in hydrogen bonding vary among the synthesized compounds, their interaction profiles highlight promising avenues for further investigation as selective COX-2 inhibitors with potential anti-inflammatory activity.
| Parameters | 4a | 4b | 4e | 4h | 4j | 4k |
|---|---|---|---|---|---|---|
| Molecular weight (g mol−1) | 490.41 | 478.32 | 462.36 | 399.42 | 433.48 | 383.46 |
| logP o/w |
5.35 | 3.60 | 4.80 | 2.96 | 4.81 | 4.17 |
| No. of H-bond donors | 1 | 3 | 1 | 3 | 1 | 1 |
| No. of H-bond acceptors | 2 | 3 | 2 | 3 | 3 | 2 |
| Solubility | Poor | Moderate | Poor | Moderate | Poor | Moderate |
| TPSA (Å2) | 71.47 | 120.12 | 71.47 | 120.12 | 84.61 | 71.47 |
| GI absorption | High | High | High | High | High | High |
| BBB permeation | No | No | No | No | No | Yes |
| P-gp substrate | No | No | No | No | No | No |
| Drug likeness (Lipinski) | Yes with 1 violation MLOGP > 4.5 | Yes | Yes with 1 violation MLOGP > 4.5 | Yes | Yes with 1 violation MLOGP > 4.5 | Yes |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
Notably, all compounds show high gastrointestinal (GI) absorption. However, only compound 4k is predicted to cross the blood–brain barrier (BBB), while none of the other compounds do so, despite being classified as non-substrates for P-glycoprotein (P-gp). These predictions, particularly regarding BBB permeability and P-gp interaction, are represented in the BOILED-Egg model (Fig. 10). In this model, red rings indicate non-substrate status for P-gp. The appearance of a red ring within the “Egg Yolk” signifies compound 4k's potential to cross the BBB.
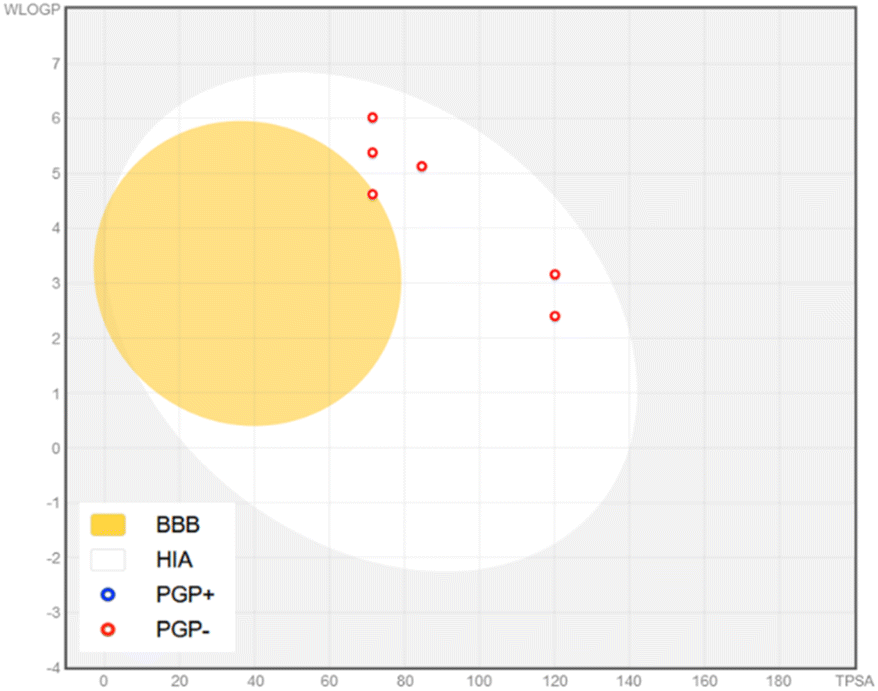 | ||
| Fig. 10 The brain or intestinal estimated permeation (BOILED-Egg) method illustrates the absorption, blood–brain barrier (BBB) permeation, and substrate selectivity for P-glycoprotein (PGP) of compounds 4a, 4b, 4e, 4h, 4j and 4k. Blue indicates the substrate of P-glycoprotein (PGP), while red represents the non-substrate of PGP. | ||
Despite their potential as non-substrates for P-gp, implying minimal interaction with this efflux transporter, it is critical to consider the broader implications for bioavailability and distribution. P-gp plays a vital role in actively transporting xenobiotics and drug molecules out of cells, thereby influencing intracellular concentrations and potentially reducing therapeutic efficacy. Thus, while reduced interaction with P-gp may favor bioavailability, it necessitates further evaluation of intracellular pharmacokinetics.
A key challenge observed is moderate solubility, which could limit their therapeutic effectiveness. Enhanced solubility would significantly improve the overall pharmaceutical properties of these compounds, making it a priority for future optimization efforts. Specifically, compounds 4a, 4e, and 4j exhibit minor deviations from Lipinski's rule of five, particularly concerning MLOGP values (>4.5), which could indicate potential challenges in oral bioavailability. On the other hand, compounds 4b, 4h, and 4k fully conform to Lipinski's criteria for drug-likeness. These findings underscore the importance of optimizing the pharmacokinetic profiles through careful molecular modifications.
To further assess the ADMET characteristics, the pkCSM webserver was employed. The analysis covered key parameters such as water solubility, human intestinal absorption, Caco-2 permeability (absorption), BBB permeability, volume of distribution (VDss) for distribution, interactions with CYP450 isoforms for metabolism, total clearance for excretion, and toxicity assessments, including AMES toxicity, hepatotoxicity, and the maximum tolerated dose. Such parameters are crucial for determining how compounds are absorbed, distributed, metabolized, and excreted, as well as understanding potential toxicity risks, thereby guiding the optimization of their pharmaceutical profiles. Table 2 presents the ADMET data for the top compounds.
| Compounds | Absorption | Distribution | Metabolism | Excretion | Toxicity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Water solubility log mol L−1 | Human intestinal absorption (%) | Caco2 permeability log papp in 10−6 cm s−1 | VDss (log L kg−1) | BBB permeability (log BB) | CYP450 isoform inhibitors | CYP450 isoform substrates | Total clearance (ml min−1 kg−1) | Renal OCT2 substrate | Max. Tolerated dose (log mg per kg per day) | Skin sensitization | Hepatotoxicity | AMES toxicity | |
| 4a | −5.307 | 95.66 | 1.513 | −0.221 | −0.038 | 2C9, 2C19 3A4 | 3A4 | −0.43 | No | 0.118 | No | Yes | Yes |
| 4b | −3.251 | 98.187 | 0.15 | −1.355 | −0.941 | 2C9, 2C19 1A2 | 3A4 | −0.388 | No | 0.38 | No | Yes | Yes |
| 4e | −4.79 | 95.163 | 1.004 | −0.229 | 0.01 | 2C19, 2C9 3A4 | 3A4 | −0.421 | No | 0.193 | No | Yes | Yes |
| 4h | −3.066 | 95.046 | 0.155 | −0.779 | −0.992 | 2C9, 2C19 1A2 | 3A4 | −0.234 | No | 0.541 | No | Yes | Yes |
| 4j | −2.892 | 77.56 | −0.877 | 0.011 | 0.213 | — | — | −1.121 | No | 0.438 | No | Yes | Yes |
| 4k | −3.765 | 96.015 | 0.949 | 0.451 | −0.09 | 2C9, 2C19 1A2 | 3A4 | −0.267 | No | 0.453 | No | Yes | Yes |
In terms of absorption, the predictions indicate that compound 4j exhibits lower human intestinal absorption compared to the other candidates. Conversely, compounds 4a, 4b, 4e, 4h, and 4k demonstrate high absorption rates ranging from 95% to 98%. Despite these high absorption rates, their solubility under physiological conditions remains a concern, which could ultimately limit their bioavailability and therapeutic efficacy. The low solubility, despite favorable intestinal absorption, suggests a significant hurdle that must be addressed to enhance the clinical utility of these compounds.
For distribution, parameters such as VDss and BBB permeability were evaluated. A lower VDss indicates a higher likelihood of non-uniform distribution between the blood and tissues. The VDss (human) values for the compounds range from 0.451 to −1.355, with compounds 4j and 4k exhibiting the most favorable distribution profiles. Regarding BBB permeability, all compounds demonstrated poor BBB permeability, suggesting limited ability to penetrate the central nervous system, which may be advantageous for avoiding central side effects in non-CNS-targeted therapies.
Metabolism-related predictions suggest that all compounds, except for 4j, are likely substrates for CYP3A4, one of the major enzymes responsible for drug metabolism. Additionally, compounds 4a, 4b, 4e, 4h, and 4k may inhibit CYP450 2C9 and 2C19 isoforms, while compounds 4a and 4e have the potential to inhibit CYP3A4, which could lead to drug–drug interactions. These findings emphasize the need to monitor metabolic stability and potential interactions with other drugs metabolized by the CYP450 system.
Excretion profiles indicate poor overall clearance rates for the compounds, with both renal and hepatic pathways predicted to contribute minimally to total drug clearance. Additionally, all compounds were identified as non-substrates for the organic cation transporter 2 (OCT2), which is involved in renal excretion, further corroborating the low clearance profiles.
In terms of toxicity, the compounds do not pose a risk of skin sensitization. However, hepatotoxicity and mutagenic effects (as assessed by the AMES test) remain potential concerns, especially for long-term administration. These toxicity risks underscore the importance of conducting more detailed in vitro and in vivo evaluations to mitigate potential safety issues before clinical consideration.
To sum up, while the ADMET profile of these compounds suggests favorable pharmacokinetic properties such as high intestinal absorption and limited P-gp interactions, several challenges remain, particularly regarding solubility, metabolic stability, and toxicity risks. Addressing these challenges, especially by enhancing solubility, optimizing distribution, and improving metabolic profiles, will be critical in advancing these compounds as viable anti-inflammatory drug candidates. Moreover, refining the toxicity profile to mitigate hepatotoxicity and mutagenic effects will be paramount in ensuring the safety of these potential therapeutic agents.
Experimental
Chemistry
:8). After completion of the reaction, an additional 10 mL of water was added, and the solid product was isolated by filtration. Subsequently, the product was washed with water many times. The pure product (racemic mixture) was thus, obtained after drying and did not require further purification steps or the addition of any organic solvent.:8) M. P.: 160–162 °C IR (ν cm−1 KBr): 1716 (CO group), 1582 (–CONH group), 1242 (C–S–C cyclic thioether) 1H-NMR (400 MHz, CDCl3/TMS, δ PPM): 6.73–7.99 (m, –NH, 9H aromatic hydrogens), 2.46–2.57 (s, 2H), 2.15–2.27 (s, 2H), 1.06–1.12 (s, 6H); 13C {1H} NMR (400 MHz, CDCl3): δ 195.6, 178.5, 164.0, 141.6, 135.7, 134.4, 133.6, 132.6, 129.1, 127.9, 127.6, 126.8, 126.7, 125.8, 125.6, 114.5, 113.3, 110.9, 51.0, 45.9, 41.3, 32.1, 28.8, 27.6, 29.8; HRMS (ESI) m/z:[M + H]+ calcd for C26H20BrNO2S + H+ 490.0476 found 490.0484; Found 97.5% purity by HPLC. HPLC condition: 1260 Infinity II with VWD, Eclipse C18 column, 4.6 × 250 mm, 5 microns, CH3CN/H2O 70:30, λ = 254 nm.where A1 = absorbance of the control, and A2 = absorbance of the test sample.

The IC50 values were determined using the lowess curves and cubic spline curves to fit the data and were generated with GraphPad Prism software (version 10.3.1).
Molecular docking studies. Docking simulations were performed by using AutoDock VINA integrated into the PyRx 0.8 (ref. 59) virtual screening tool to identify compounds with high binding affinity. Insilco docking simulation studies to evaluate the molecular interactions of 4a–l compounds were done with COX1 (PDB ID: 3KK6, co-crystallized ligand inhibitor: Celecoxib),60 COX-2 (PDB ID:5KIR, co-crystallized with Rofecoxib).61 Protein structure was processed to ensure an optimized structure for docking studies and it was executed with the UCSF Chimera Dock Prep module that includes the following steps: elimination of water molecules and other ligands, addition of missing atoms and residues, energy minimization and assigning charges and polar hydrogens and then converted to the pdbqt format. The 2D structure of the ligands was drawn with ChemDraw software and the structures were optimized through energy minimization with MMFF94 force field parameters and conjugate gradient algorithm using the Open Babel module of PyRx and eventually converted the ligands to the AutoDock compatible pdbqt format to carry out docking exploration. Post-docking analysis and visualization of binding poses and molecular interactions were done with BIOVIA Discovery Studio 2021 and Chimera X tools.62 Autodock Vina grid box was created around the antagonist active site with the following details of the vina search space with the following coordinates (Å), X: −33.259, Y: 43.443, Z: −5.595 for COX-1, X: 23.606, Y: 1.782, Z: 34.5785 for COX-2. The validation process of the docking program involved the meticulous redocking of Celecoxib into its co-crystallized site. The remarkably low Root Mean Square Deviation (RMSD) value of 0.64 Å, observed between the docked and native poses, serves as a robust indicator of the program's efficacy in accurately predicting the binding pose of Celecoxib. This outcome underscores the program's proficiency in faithfully reproducing the spatial arrangement of Celecoxib within its intended binding site, affirming the reliability and precision of the docking algorithm.
ADMET studies. To evaluate the pharmacokinetic properties of our compounds, we performed an in silico assessment using online servers such as pkCSM (https://biosig.lab.uq.edu.au/pkcsm/prediction)63 and SwissADME (http://www.swissadme.ch/).64 These platforms analyze the pharmacokinetic profiles of top compounds, focusing on aspects such as water solubility, toxicity, metabolism, distribution, and absorption. They predict pharmacokinetic parameters based on the structural similarity of compounds with known ADMET features. These machine learning-based tools are highly regarded for their reliability and accuracy in ADMET prediction. SWISS ADME webserver had been used for the evaluation of physico-chemical and drug-likeliness properties of compounds which are found to be promising in the in vitro and in silico assessments.
Conclusions
This study advances the development of novel anti-inflammatory agents as COX-2 inhibitors by designing and synthesizing spiro thiochromene–oxindole derivatives, integrating two privileged scaffolds known for their anti-inflammatory properties. Our approach leverages the anti-inflammatory potential of 3-substituted oxindoles and thiopyrans to enhance COX-2 inhibition while minimizing off-target effects. The synthesized compounds demonstrated notable anti-inflammatory activity, with compounds 4e, 4k, and 4h achieving BSA denaturation inhibition rates of 90.97%, 95.45%, and 91.67%, respectively, at 800 μg mL−1. Corresponding IC50 values were 127.477 ± 2.285, 190.738 ± 3.561, and 285.806 ± 8.894 μg mL−1. These IC50 values were comparable or superior to the IC50 value of the reference drug, diclofenac sodium (243.641 ± 2.686 μg mL−1). In silico analysis revealed robust binding affinities with COX-2, with binding energies of −8.9 kcal mol−1, −8.7 kcal mol−1, and −8.6 kcal mol−1 for these compounds.Furthermore, the ADMET profile of these compounds indicates favorable pharmacokinetics, including high intestinal absorption and limited P-gp interactions. However, challenges such as solubility, metabolic stability, and potential toxicity remain to be addressed. Particularly, enhancing solubility and optimizing metabolic profiles will be pivotal in progressing these candidates for possible therapeutic applications.
A core feature of this work is the sustainable synthesis of the target compounds. We employed taurine as a green bio-organic bifunctional catalyst, enabling a one-pot, aqueous-mediated process that significantly reduced environmental impact. This strategy facilitated efficient Knoevenagel–Thia-Michael cascade reactions under mild, acid-, base-, and metal-free conditions. By avoiding toxic solvents and cumbersome purification steps, we aligned our approach with contemporary green chemistry principles, contributing to environmentally responsible pharmaceutical synthesis.
This novel combination of strategic scaffold design and sustainable synthesis introduces a significant advancement in the search for COX-2 inhibitors with improved efficacy and reduced adverse effects. Further clinical validation will be essential to confirm the therapeutic potential and safety profiles of these promising new compounds.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
We strongly encourage authors to include author contributions and recommend using CRediT for standardised contribution descriptions. Please refer to our general author guidelines for more information about authorship. M. performed the experiments and collected the needed literature; A. K. R., K. S., and N. J. performed a supporting role in the collection of the needed literature; R. K. K. carried out in silico studies and co-wrote the part involving these studies, A. G. conceptualized and worked on the methodology, wrote the complete manuscript, characterized the data, and supervised the experimental work. Manju: validation, resources, investigation. Ashok K. Raigar: resources. Kamlesh Saini: resources. Nirmal Jyoti: resources. Ravi Kumar Kapavarapu: writing – original draft, validation, formal analysis. Anjali Guleria: writing – review & editing, writing – original draft, visualization, validation, supervision, project administration, methodology, formal analysis, data curation, conceptualization. Finally, all the authors have approved the final version of the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work has been provided by the HRDG (CSIR), New Delhi, sanction letter no./file no. 09/149(0801)/2020-EMR-I. The authors are grateful to the Department of Chemistry, University of Rajasthan, Jaipur for providing the infrastructure; to Biomitra Life Sciences Pvt. Ltd, Jaipur for carrying out the in vitro heat-induced BSA denaturation assay; to Malviya National Institute of Technology and the Central University of Rajasthan, Kishangarh for providing the facility of IR, 1H NMR, 13C NMR, and Mass spectroscopy.References
- A. Jubert, N. E. Massa, L. L. Te’vez and N. B. Okulik, Vib. Spectrosc., 2005, 37, 161–178 CrossRef CAS.
- M. E. Kotas and R. Medzhitov, Cell, 2015, 160, 816–827 CrossRef CAS PubMed.
- M. G. Netea, F. Balkwill, M. Chonchol, F. Cominelli, M. Y. Donath, E. J. Giamarellos-Bourboulis, D. Golenbock, M. S. Gresnigt, M. T. Heneka and H. M. Hoffman, Nat. Immunol., 2017, 18, 826–831 CrossRef CAS.
- D. Furman, J. Campisi, E. Verdin, P. Carrera-Bastos, S. Targ, C. Franceschi, L. Ferrucci, D. W. Gilroy, A. Fasano and G. W. Miller, Nat. Med., 2019, 25, 1822–1832 CrossRef CAS.
- C. Nathan and A. Ding, Cell, 2010, 140, 871–882 CrossRef CAS.
- O. H. Nielsen and M. A. Ainsworth, N. Engl. J. Med., 2013, 369, 754–762 CrossRef CAS.
- G. Schett, D. Elewaut, I. B. McInnes, J.-M. Dayer and M. F. Neurath, Nat. Med., 2013, 19, 822–824 CrossRef CAS.
- I. B. McInnes and G. Schett, Lancet, 2017, 389, 2328–2337 CrossRef CAS PubMed.
- G. Schett and M. F. Neurath, Nat. Commun., 2018, 9, 3261 CrossRef PubMed.
- A. Zarghi, L. Najafnia, B. Daraee, O. G. Dadrass and M. Hedayati, Bioorg. Med. Chem. Lett., 2007, 17, 5634–5637 CrossRef CAS PubMed.
- T. Zebardast, A. Zarghi, B. Daraie, M. Hedayati and O. G. Dadrass, Bioorg. Med. Chem. Lett., 2009, 19, 3162–3165 CrossRef CAS PubMed.
- Y. Gao and Y. Z. Duan, Anat. Rec., 2010, 293, 485–491 CrossRef CAS.
- E. K. A. Abdelall, P. F. Lamie and W. A. M. Ali, Bioorg. Med. Chem. Lett., 2016, 26, 2893–2899 CrossRef CAS PubMed.
- C. B. Mishra, S. Kumari, A. Prakash, R. Yadav, A. K. Tiwari, P. Pandey and M. Tiwari, Eur. J. Med. Chem., 2018, 151, 520–532 CrossRef CAS PubMed.
- A. Kumar, G. Kour, P. Chibber, D. Saroch, C. Kumar and Z. Ahmed, Cytokine, 2022, 158, 155978 CrossRef CAS.
- P. E. Lipsky, Am. J. Orthod., 1999, 28, 8–12 CAS.
- R. A. Adelizzi, J. Am. Osteopath. Assoc., 1999, 99, 7–12 CrossRef.
- R. A. Dionne and C. W. Berthold, Crit. Rev. Oral Biol. Med., 2001, 12, 315–330 CrossRef CAS.
- M. H. Nekoofar, M. Sadeghipanah and A. R. Dehpour, J. Endod., 2003, 29, 634–637 CrossRef.
- M. A. Huber and G. T. Terezhalmy, J. Am. Dent. Assoc., 2006, 137, 480–487 CrossRef CAS.
- L. Laine, W. B. White, A. Rostom and M. Hochberg, Semin. Arthritis Rheum., 2008, 38, 165–187 CrossRef CAS PubMed.
- B. J. Al-Hourani, S. K. Sharma, J. Y. Mane, J. Tuszynski, V. Baracos, T. Kniess, M. Suresh, J. Pietzsch and F. Wuest, Bioorg. Med. Chem. Lett., 2011, 21, 1823–1826 CrossRef CAS PubMed.
- A. Gupta and M. Bah, Curr. Pain Headache Rep., 2016, 20, 62 CrossRef.
- S. J. Kim, S. H. Lee, H. Lee, M.-S. Shin and J. W. Lee, Int. J. Mol. Sci., 2023, 24, 2066 CrossRef CAS.
- T. Pan, M. He, L. Deng, J. Li, Y. Fan, X. Hao and S. Mu, Molecules, 2023, 28, 4668 CrossRef CAS PubMed.
- D. J. Rogier Jr, J. S. Carter and J. J. Talley, Chem. Abstr., 2001, 135, 107252 Search PubMed.
- S. N. Jadhav, S. P. Patil, D. P. Sahoo, D. Rath, K. Parida and C. V. Rode, Catal. Lett., 2020, 150, 2331–2351 CrossRef CAS.
- Y. Fu, Z. Lu, X. Ma, K. Fang, X. He, H. Xu and Y. Hu, Bioorg. Chem., 2020, 99, 103888 CrossRef CAS.
- P. Choudhary, A. Sen, A. Kumar, S. Dhingra, C. M. Nagaraja and V. Krishnan, Mater. Chem. Front., 2021, 5, 6265–6278 RSC.
- F. Mohamadpour, Sci. Rep., 2023, 13, 13142 CrossRef CAS.
- L. Selvaraj, R. Eswaran, V. K. Natesan and S. P. Muthu, J. Mol. Struct., 2024, 1302, 137373 CrossRef CAS.
- C. F. H. Allen, J. O. Fournier and W. J. Humphlett, Can. J. Chem., 1964, 42, 2616–2620 CrossRef CAS.
- D. M. Pore, M. S. Soudagar, U. V. Desai, T. S. Thopate and P. P. Wadagaonkar, Tetrahedron Lett., 2006, 47, 9325–9328 CrossRef CAS.
- G. L. Khatik, G. Sharma, R. Kumar and A. K. Chakraborti, Tetrahedron, 2007, 63, 1200–1210 CrossRef CAS.
- M. Bandini, P. G. Cozzi, M. Giacomini, P. Melchiorre, S. Selva and A. Umani-Ronchi, J. Org. Chem., 2002, 67, 3700–3704 CrossRef CAS PubMed.
- B. M. Fetterly, N. K. Jana and J. G. Verkade, Tetrahedron, 2006, 62, 440–456 CrossRef CAS.
- P. K. Sasmal, S. Sridhar and J. Iqbal, Tetrahedron Lett., 2006, 47, 8661–8665 CrossRef CAS.
- W. Guo, G. Lv, J. Chen, W. Gao, J. Ding and H. Wu, Tetrahedron, 2010, 66, 2297–2300 CrossRef CAS.
- S. Bruschi, M. Moccia and M. F. A. Adamo, Tetrahedron Lett., 2011, 52, 3602–3604 CrossRef CAS.
- S. K. Garg, R. Kumar and A. K. Chakraborti, Tetrahedron Lett., 2005, 46, 1721–1724 CrossRef CAS.
- C.-M. Chu, W.-J. Huang, C. Lu, P. Wu, J.-T. Liu and C.-F. Yao, Tetrahedron Lett., 2006, 47, 7375–7380 CrossRef CAS.
- C.-T. Chen, Y.-D. Lin and C.-Y. Liu, Tetrahedron, 2009, 65, 10470–10476 CrossRef CAS.
- M. Winkler, Y. S. Raupp, L. A. M. Köhl, H. E. Wagner and M. A. R. Meier, Macromolecules, 2014, 47, 2842–2846 CrossRef CAS.
- L. Hua, Z. Yao, F. Xu and Q. Shen, RSC Adv., 2014, 4, 3113–3120 RSC.
- P. Wadhwa, S. Bagchi and A. Sharma, ChemistrySelect, 2017, 2, 1386–1391 CrossRef CAS.
- A. K. Mallik, T. K. Pal, R. Ghosal and A. Patra, Synlett, 2012, 23, 2459–2462 CrossRef CAS.
- Manju, K. Saini, A. K. Raigar and A. Guleria, Polycycl. Aromat. Comp., 2023, 1–17 Search PubMed.
- S. Agarwal, P. Kalal, A. Sethiya and J. Soni, Mini-Rev. Org. Chem., 2022, 19, 617–628 CrossRef CAS.
- M. Biglari, F. Shirini, N. O. Mahmoodi, M. Zabihzadeh, M. Safarpoor Nikoo Langarudi and M. Alipour Khoshdel, Polycycl. Aromat. Comp., 2022, 42, 1452–1473 CrossRef CAS.
- A. V. Chate, P. P. Rudrawar, G. M. Bondle and J. N. Sangeshetti, Synth. Commun., 2020, 50, 226–242 CrossRef CAS.
- C. K. Jadhav, A. S. Nipate, A. V. Chate and C. H. Gill, Polycycl. Aromat. Comp., 2021, 1–17 Search PubMed.
- M. M. Khan, B. Saigal, S. Shareef, S. Khan and S. C. Sahoo, Synth. Commun., 2018, 48, 2683–2694 CrossRef CAS.
- K. Verma, Y. K. Tailor, S. Khandelwal, E. Rushell, M. Agarwal and M. Kumar, Mol. Divers., 2020, 24, 1355–1365 CrossRef CAS.
- K. Saini, A. K. Raigar, Manju, D. K. Jangid, J. Mathur, S. Dhadda and A. Guleria, J. Org. Chem., 2022, 87, 13734–13743 CrossRef CAS PubMed.
- R. J. Huxtable, Taurine in Nutrition and Neurology, Plenum, New York, 1982, pp. 1–4 Search PubMed.
- R. J. Huxtable and L. A. Sebring, Pharmacol. Sci., 1986, 7, 481–485 CAS.
- I. K. Song and Y. K. Kang, Comput. Theor. Chem., 2013, 1025, 8–15 CrossRef CAS.
- S. Chandra, P. Chatterjee, P. Dey and S. Bhattacharya, Asian Pac. J. Trop. Biomed., 2012, 2, S178–S180 CrossRef.
- D. Sargis and J. O. Arthur, Methods Mol. Biol., 2015, 1263, 243–250 CrossRef.
- G. Rimon, R. S. Sidhu, D. A. Lauver, J. Y. Lee, N. P. Sharma, C. Yuan, R. A. Frieler, R. C. Trievel, B. R. Lucchesi and W. L. Smith, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 28–33 CrossRef CAS.
- B. J. Orlando and M. G. Malkowski, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2016, 72, 772–776 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- E. V. P. Douglas, L. B. Tom and B. A. David, J. Med. Chem., 2015, 58(9), 4066–4072 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07990f |
| This journal is © The Royal Society of Chemistry 2025 |