
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ generation and conversion of a half-zirconocene catalyst for the synthesis of N-acylpyrazoles†
Juan Wu‡
a,
Mingming Yang‡ *a,
Deying Lengb,
Qiuping Hua,
Yanxiu Yaoc,
Huaming Sunc and
Ziwei Gao*ac
*a,
Deying Lengb,
Qiuping Hua,
Yanxiu Yaoc,
Huaming Sunc and
Ziwei Gao*ac
aSchool of Chemistry and Chemical Engineering, Yan'an University, Yan'an 716000, P.R. China. E-mail: mmyang@yau.edu.cn
bSchool of Physics and Electronic Information, Yan'an University, Yan'an 716000, P.R. China
cKey Laboratory of Applied Surface and Colloid Chemistry, MOE, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P.R. China. E-mail: zwgao@snnu.edu.cn
First published on 20th January 2025
Abstract
Pyrazoles are an important class of five-membered nitrogen heterocyclic compounds that have been widely used in agriculture and medicine. Exploring their synthesis methods under mild conditions has always been a hot research topic. Herein, a new strategy was developed to enhance the activity of a zirconium metal centre for the synthesis of N-acylpyrazole derivatives using Cp2ZrCl2 as a pre-catalyst. Cp2ZrCl2 was activated in situ within the catalytic system through cyclopentadienyl ring dissociation, leading to the formation of an activated species, [CpZrCl(acac)2]. The new approach demonstrates broad substrate scope under mild reaction conditions, leading to formation of (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanones with yields of up to 97%.
Introduction
Pyrazoles, a significant class of five-membered nitrogen heterocyclic compounds, serve as versatile building blocks1–3 and crucial intermediates4,5 in organic synthesis. They exhibit a wide range of pharmaceutical activities and are extensively studied in medical research6–9 for their anti-tubercular, anti-cancer,10 anti-depressant, anti-bacterial,11,12 and anti-parkinsonism activities (Scheme 1a). Several methods, such as cyclocondensation of hydrazine and its derivatives with carbonyl systems,13–18 dipolar cycloadditions,19–23 and multicomponent reactions,24,25 have been employed in the development of pyrazoles (Scheme 1b). However, certain existing synthetic approaches for pyrazole synthesis necessitate expensive metal catalysts, harsh reaction conditions, and inconvenient operations as well as yield low product quantities. These limitations impose constraints on the practicality of these reactions, thereby necessitating the establishment of convenient, mild, high-yielding methods that employ cost-effective and less toxic reagents for N-acylpyrazole synthesis.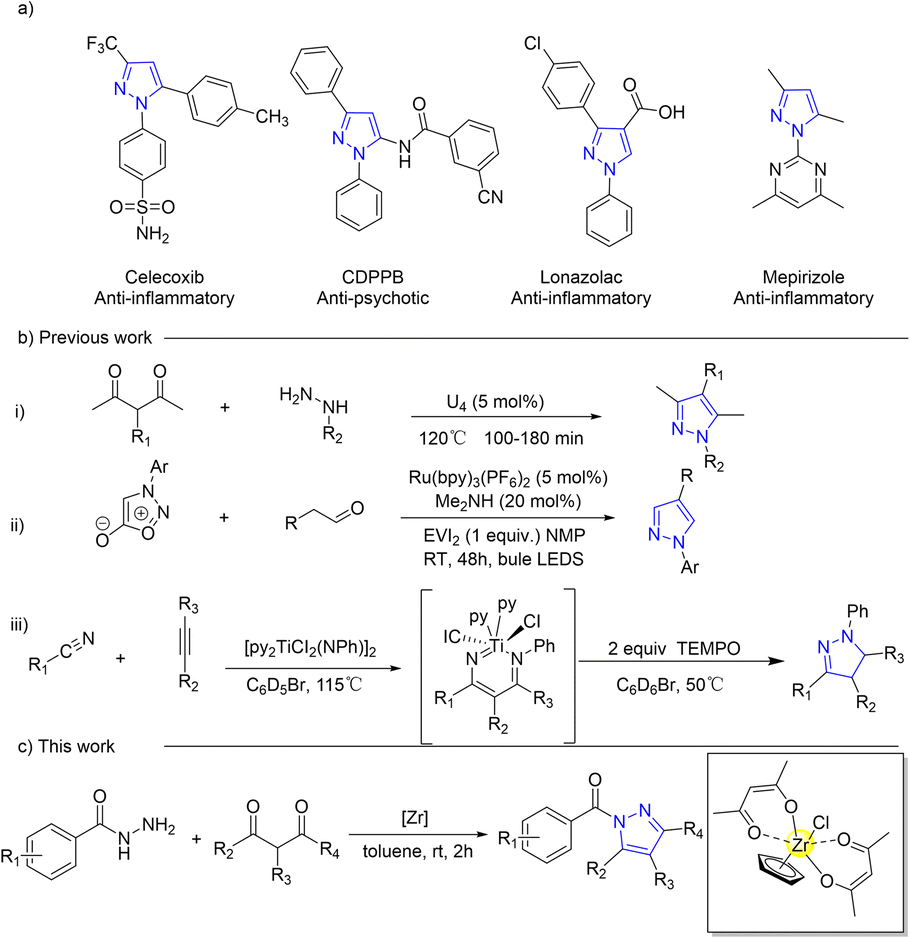 | ||
| Scheme 1 Structures of significant pyrazoles and novel synthetic routes for N-acylpyrazole. | ||
The use of tetravalent IVB Lewis acid catalysts coordinated with cyclopentadienyl (Cp), such as titanocene26–32 and zirconocene,33–37 has been demonstrated to be more diverse, efficient, and practical for C–C38–43 and C–N44–47 formation reactions. This is attributed to their cost-effectiveness,48 low toxicity,49 high stability,50 tunable Lewis acidity,51,52 and high coordination number.53–57 By incorporating the Cp ligand into the catalyst system, the requirement for stringent anhydrous conditions can also be eliminated due to its less electron deficiency and steric hindrance, which provides a hydrophobic domain for protecting the Lewis acid centre against water.58–60 However, increased catalyst stability comes at the expense of catalytic activity. The development of a titanocene or zirconocene catalyst with a single Cp ligand is advisable in order to maintain the original catalytic stability and further enhance its catalytic performance.61,62 Nowadays, the synthesis of these catalysts typically involves the use of precursors such as CpZrCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 63,64 and CpTiCl3,65–68 which require intricate procedures and a stringent anhydrous and oxygen-free environment, in addition to being costly. The establishment of an in situ, convenient, and mild conversion method for a half-zirconocene catalyst is therefore imperative to enable its direct application in catalytic reactions. Additionally, these diverse coordination modes between zirconium and both the ligand69–71 and substrate72–74 facilitate facile capture of adduct products and thereby enable comprehensive investigation into catalyst–substrate interaction.
63,64 and CpTiCl3,65–68 which require intricate procedures and a stringent anhydrous and oxygen-free environment, in addition to being costly. The establishment of an in situ, convenient, and mild conversion method for a half-zirconocene catalyst is therefore imperative to enable its direct application in catalytic reactions. Additionally, these diverse coordination modes between zirconium and both the ligand69–71 and substrate72–74 facilitate facile capture of adduct products and thereby enable comprehensive investigation into catalyst–substrate interaction.
Herein, we present an in situ acetylacetone-coordinated half-zirconocene complex and its catalytic role in the synthesis of N-acylpyrazoles, as depicted in Scheme 1c. The evidence from in situ ESI(+)-MS and 1H NMR experiments elucidated the interaction between the zirconium centre and substrates. The catalyst effectively facilitated the synthesis of pyrazoles, achieving a remarkable yield of 97% under mild, low catalyst loading, non-anhydrous and non-anaerobic conditions.
Results and discussion
Our experimental optimization for N-acylpyrazole synthesis commenced with benzoyl hydrazine (1a) and acetylacetone (acac) (2a), resulting in the formation of two products, namely, N-acylpyrazole (3a) and 5-hydroxy-dihydropyrazole (5a). The reaction was conducted at room temperature for 2 hours in toluene solvent using 5 mol% of Cp2ZrCl2 as a pre-catalyst, leading to the desired N-acylpyrazole 3a ((3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone) with a high yield of 97% (Table 1, entry 1). The control experiment confirmed the essential role of zirconium catalyst in preventing the undesirable generation of compound 5a (entry 2). Initially, various solvents were evaluated employing Cp2ZrCl2 as a pre-catalyst; their results are presented in entries 3–8 of Table 1. DMSO provided a yield of only 47% while THF, DMF, acetonitrile, EtOH and CH2Cl2 exhibited similar yields, ranging from 80% to 90%. Consequently, toluene was identified as the optimal choice due to its superior yield up to 97%. Subsequently, several catalysts were examined (entries 9–18). Considering that oxygen within acetylacetone readily interacts with the zirconium metal centre, ZrCl4 was initially assessed due to its easily breakable Zr–Cl bonds, resulting in an 88% yield. However, the presence of Zr(OH)4 failed to produce desired product 3a while yielding only 22% of compound 5a, possibly due to the inability of stable Zr–OH ligand to be replaced by acetylacetone. Zr(OAc)2 yielded only 83% of compound 5a without any desired product formation. Acetylacetonate zirconium was introduced as a contrasting catalyst to the in situ coordinating complex, resulting in only 22% yield of 5a. CpZrCl3 was employed as a formed half-zirconocene catalyst, giving 90% yield, consistent with Cp2ZrCl2. When FeCl3·6H2O and CuCl2·2H2O were used as Lewis acid catalysts, the desired products were obtained with yields of 74% and 36%, respectively. Several mild acids, including PTSA, sulfamic acid and camphor sulfonic acid, were introduced as alternatives to the Zr catalyst. Consequently, the yields of the target product were 0%, 32% and 79%, respectively.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) | |
| Product 3a | Product 5a | |||
| a 0.5 mmol of benzoyl hydrazine, 0.5 mmol of acetylacetone and 5% of the catalyst in 0.5 mL solvent at room temperature for 2 h.b Yield of 1H NMR. | ||||
| 1 | Cp2ZrCl2 | Toluene | 97 | — |
| 2 | — | Toluene | — | 76 |
| 3 | Cp2ZrCl2 | DMSO | 47 | — |
| 4 | Cp2ZrCl2 | THF | 82 | — |
| 5 | Cp2ZrCl2 | DMF | 80 | — |
| 6 | Cp2ZrCl2 | CH3CN | 81 | — |
| 7 | Cp2ZrCl2 | CH2Cl2 | 87 | — |
| 8 | Cp2ZrCl2 | EtOH | 90 | — |
| 9 | ZrCl4 | Toluene | 88 | — |
| 10 | Zr(OH)4 | Toluene | — | 22 |
| 11 | Zr(OAc)2 | Toluene | — | 83 |
| 12 | Zr(O2C5H7)4 | Toluene | — | 22 |
| 13 | CpZrCl3 | Toluene | 90 | — |
| 14 | FeCl3·6H2O | Toluene | 74 | — |
| 15 | CuCl2·2H2O | Toluene | 36 | — |
| 16 | PTSA | Toluene | — | — |
| 17 | Sulfamic acid | Toluene | 32 | — |
| 18 | Camphor sulfonic acid | Toluene | 79 | — |
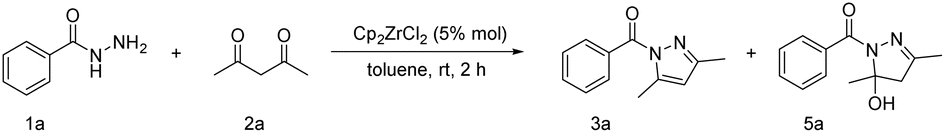
With the optimized conditions established, the substrate scope was explored by reacting various benzoyl hydrazines with acetylacetone. The reactivity of various substituted benzoyl hydrazines was examined and is summarized in Table 2. Electron-donating groups such as –Me and –OMe gave N-acylpyrazole yields in the range of 90–98% (3b–3g), while electron-withdrawing halogen groups (–F, –Cl, –Br) produced the desired products with 87–93% yields (3h–3l). 66% yield (3m) for the benzoyl hydrazine with –OH substitution was obtained, which can be attributed to the facile coordination of the oxygen atom in the hydroxyl group with the zirconium metal centre, thereby causing catalyst poisoning and rendering it inactive. The amino-substituted benzoyl hydrazine underwent a reaction with acetylacetone, leading to the formation of two distinct products. One of these products was the desired compound 3o, while the other product 3n exhibited an imine structure resulting from subsequent interaction between compound 3o and another molecule of acetylacetone. The reaction between p-tert-butyl and p-phenyl-substituted benzoyl hydrazine resulted in a product yield of 93% for compound 3p and 95% for compound 3q, respectively. Heterocyclic substituted substrates including furyl-, thienyl-, 1-naphthyl-, and 2-naphthyl- exhibited favorable yields of 83%, 95%, 78%, and 66%, respectively (3r–3u). Upon the introduction of terephthalic dihydrazide, each of the two hydrazide groups underwent a reaction with one molecule of acetylacetone. Consequently, we increased the amount of acetylacetone to 2 equivalents in order to obtain a product featuring two pyrazole rings, yielding 70% (3v). In the absence of aromatic ring influence (3w–3y), the corresponding N-acylpyrazoles were obtained with moderate yields ranging from 58% to 82%. Simple hydrazine examples, including phenylhydrazine and benzylhydrazine, were also evaluated under the optimized conditions. The reactions gave 89% yield for compound 6a and 78% for compound 6b. The impact of changes in β-diketone derivatives on the reaction was subsequently investigated, resulting in excellent yields ranging from 85% to 91% (4a, 4d, 4e). The introduction of 2,4-hexanedione led to the formation of two distinct structures, namely, 4b and 4c, due to variations in the order of reaction between the 2-carbonyl and 4-carbonyl groups. These findings suggest that the generation of structure 4c is more favorable. Similarly, 6-methylheptane-2,4-dione resulted in the formation of compounds 4i and 4j with yields of 30% and 59%, respectively. Additionally, 1-cyclopropyl-1,3-butandione produced the corresponding N-acylpyrazoles 4k and 4l, yielding 35% and 40%, respectively. When the substituent of β-diketone is changed to tert-butyl or phenyl, the conversion of enone may be hindered by site-blocking effects, necessitating an increase in reaction temperature and time to achieve moderate yields of the target products (4f–4h). Dipivaloylmethane affords the product with a yield of 62% (4f). Benzoylacetone only yields 23% of the desired product (4g). Dibenzoylmethane provides the corresponding N-acylpyrazole with a yield of 62% (4h). The reaction of 5,5-dimethylhexane-2,4-dione results in the formation of compound 4m with a yield of 29%.
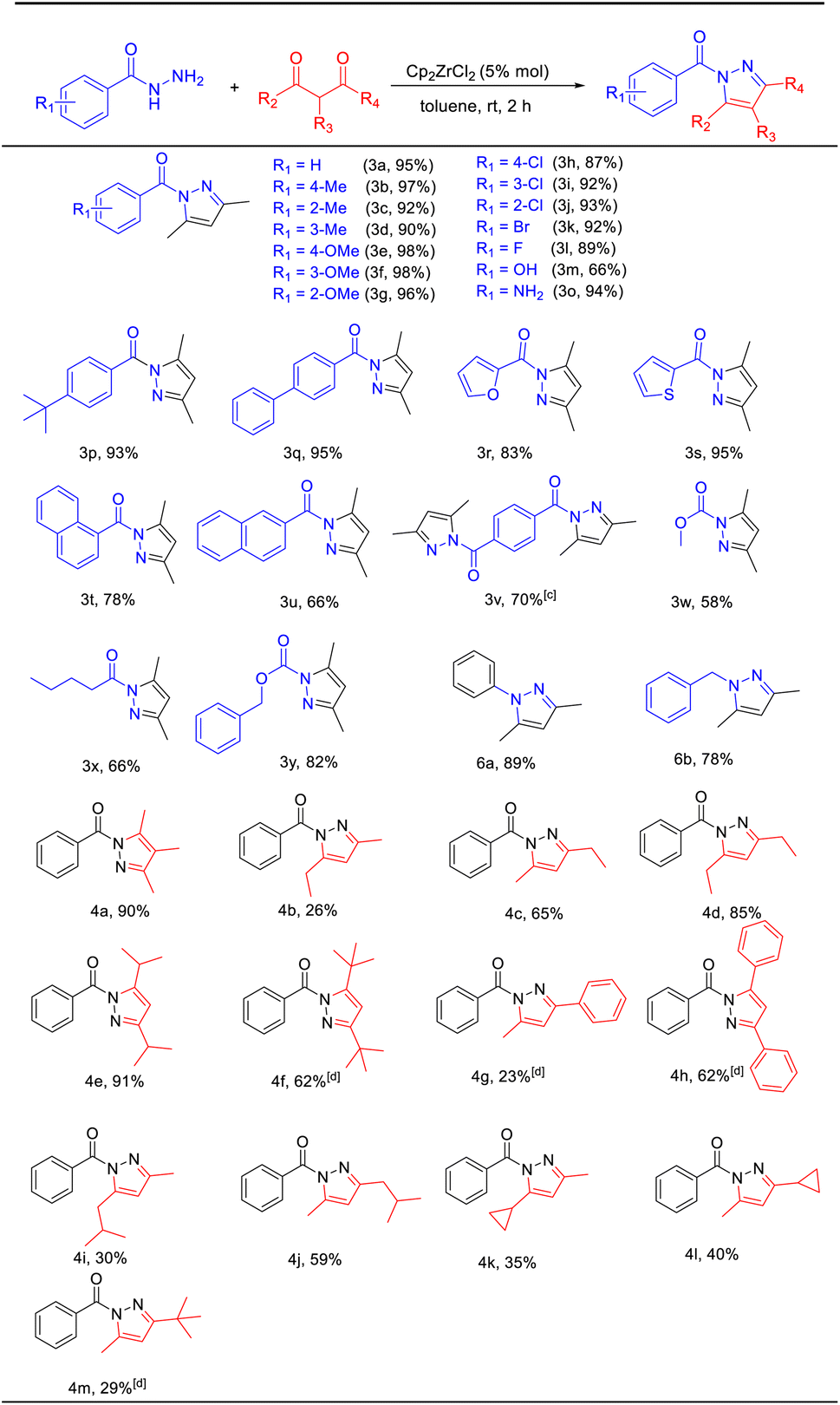
The scalability of this half-zirconocene catalytic system was assessed by conducting a 20 mmol scale reaction in a reaction tube under standard conditions (Scheme 2). The reaction between 1a and 2a yielded the desired product (3,5-dimethyl-1H-pyrazol-1-yl)-(phenyl)methanone with a yield of 74%.
 | ||
| Scheme 2 Reaction scale-up. | ||
To elucidate the activation mechanism of the reaction between acetylacetone and benzoyl hydrazine in the presence of Cp2ZrCl2, 1H NMR titration experiments were conducted (Fig. 1a). The gradual addition of acetylacetone and benzoyl hydrazine to a solution of Cp2ZrCl2 resulted in changes in the Cp signal, as detected in the 1H NMR spectra, which can be attributed to the transformation of zirconocene species. The addition of acetylacetone alone induced a minor new signal at δ 6.29 ppm ( ), indicating coordination between Cp2Zr and acetylacetone. Upon further addition of benzoyl hydrazine, the peak corresponding to Cp2ZrCl2 rapidly vanished, while the immediate appearance of a new signal at δ 6.44 ppm (
), indicating coordination between Cp2Zr and acetylacetone. Upon further addition of benzoyl hydrazine, the peak corresponding to Cp2ZrCl2 rapidly vanished, while the immediate appearance of a new signal at δ 6.44 ppm ( ) suggested that conversion had occurred from Cp2ZrCl2 to a novel zirconium species inconsistent with direct coordination between Cp2ZrCl2 and acetylacetone. The chemical conversion of Cp2ZrCl2 might be attributed to the induced effect of benzoyl hydrazine, which not only served as the reaction substrate but also played a crucial role in removing one Cp ligand. In addition to the change in the characteristic hydrogen of Cp ligand, the newly generated peak at δ 6.13 ppm (
) suggested that conversion had occurred from Cp2ZrCl2 to a novel zirconium species inconsistent with direct coordination between Cp2ZrCl2 and acetylacetone. The chemical conversion of Cp2ZrCl2 might be attributed to the induced effect of benzoyl hydrazine, which not only served as the reaction substrate but also played a crucial role in removing one Cp ligand. In addition to the change in the characteristic hydrogen of Cp ligand, the newly generated peak at δ 6.13 ppm ( ) gradually increased during the progression of the reaction. This peak is assigned to the alkene hydrogen in the enol structure after acetylacetone coordinates with zirconium. As shown in Fig. 1b, there is a proton number ratio of 2:5 for acetylacetone (Hb) and Cp ligand (Ha), indicating that the ligand ratio for acetylacetone and Cp is 2:1 in zirconocene catalytic species (more proton number ratio shown in Fig. S8†). The experimental results indicate that the structure of the catalyst possesses a zirconium metal centre coordinated with two acetylacetonate molecules, as depicted in Fig. 1b.
) gradually increased during the progression of the reaction. This peak is assigned to the alkene hydrogen in the enol structure after acetylacetone coordinates with zirconium. As shown in Fig. 1b, there is a proton number ratio of 2:5 for acetylacetone (Hb) and Cp ligand (Ha), indicating that the ligand ratio for acetylacetone and Cp is 2:1 in zirconocene catalytic species (more proton number ratio shown in Fig. S8†). The experimental results indicate that the structure of the catalyst possesses a zirconium metal centre coordinated with two acetylacetonate molecules, as depicted in Fig. 1b.
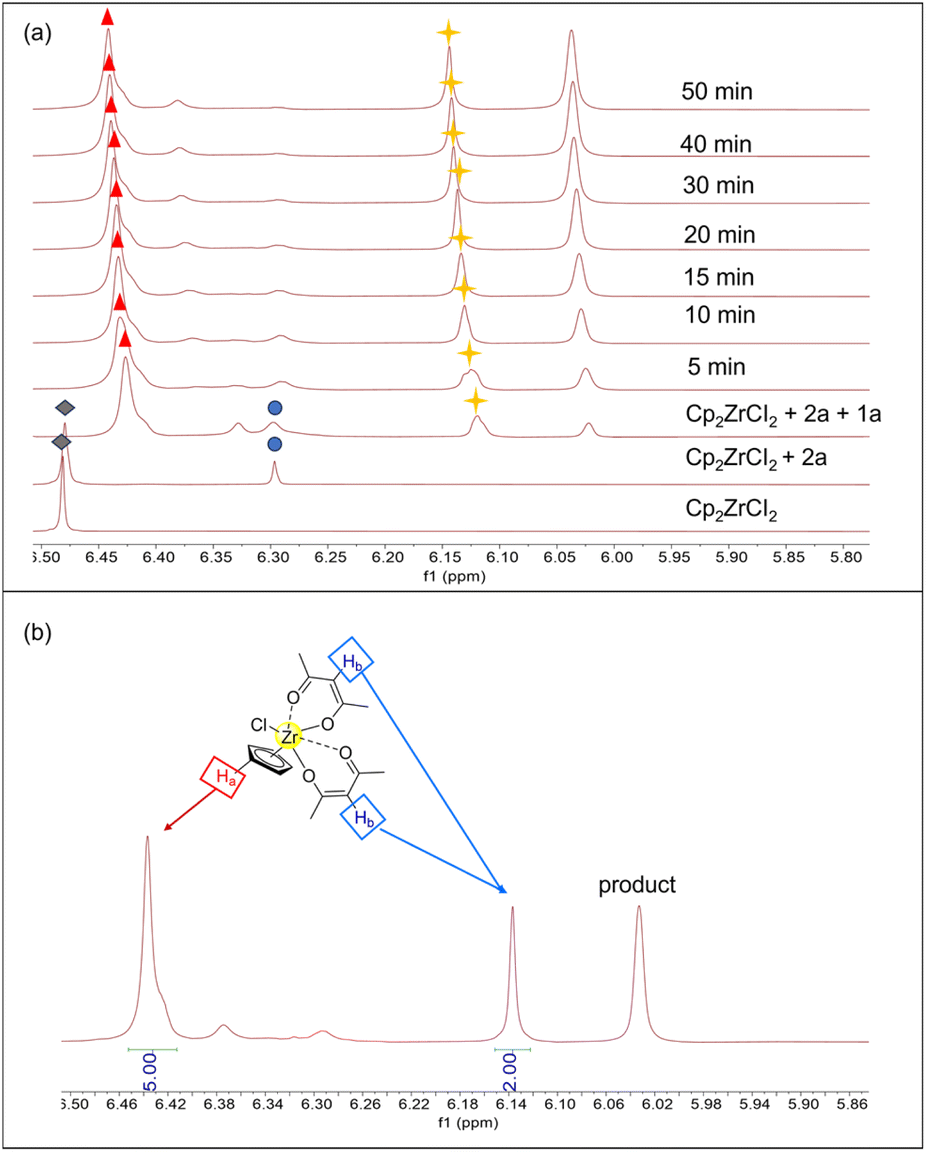 | ||
| Fig. 1 (a) 400 MHz 1H NMR titration experiments. 6.48 ppm [Cp2ZrCl2]; 6.29 ppm [Cp2ZrCl(acac)]; 6.44 ppm and 6.13 ppm [CpZrCl(acac)2]. (b) Proton number ratio for acetylacetone (Hb) and the Cp ligand (Ha). | ||
The structure of the real catalytic species was further determined through in situ ESI(+)-MS experiments, providing insights into potential reaction mechanisms by capturing several reaction intermediates in the ESI-MS spectra (Fig. 2). The dominant signal at an m/z value of 353.0321 is attributed to Int-II, where two acetylacetone ligands and one Cp ligand are coordinated to the zirconium metal centre. The other major peak, with an m/z value of 471.0852, corresponds to the compound formed by the coordination of a zirconium centre with an acetylacetone, a Cp ligand, and an imide structure, resulting from the reaction of acetylacetone and imine (Int-IV). This peak also arises from a product where the zirconium centre is coordinated with an acetylacetone, a Cp ligand, and an undehydrated intermediate (Int-V). The signal at an m/z value of 489.0962 corresponds to the intermediate structure Int-III, which is formed through the coordination of a half-zirconocene with an acetylacetone and the nucleophilic addition of acetylacetone to benzoyl hydrazine. The analysis of these three main peaks reveals that the initial step involves the reaction between acetylacetone and the catalyst, followed by the subsequent reaction between benzoyl hydrazine and acetylacetone. The overall reaction occurs with the participation of the catalytic species. Fig. 2 also illustrates additional peaks that can be identified, facilitating speculation on the catalytic process. The m/z value of 437.0799 corresponds to Int-VII, which consists of two Cp ligands and an imide intermediate structure connected to the zirconium centre. The presence of Int-VII indicates that the process of Cp ligand removal has not been completed, which corresponds to the gradual disappearance of the signal at δ 6.29 ppm ( ) in the 1H NMR titration experiments. Int-VIII, with a mass–charge ratio of m/z 589.1395, comprises one Cp ligand and two undehydrated intermediates connected to zirconium. The peak at m/z 689.1917 corresponds to the intermediate structure Int-IX, which bears two undehydrated hydroxy-dihydropyrazole and an acetylacetone. The structures Int-VIII and Int-IX illustrate that the catalyst possesses the ability to incorporate multiple substrates at a single half-zirconocene catalytic centre, thereby enabling simultaneous catalysis of two or even three molecular reactions during the catalytic process. ESI-MS analysis reveals that Cp2ZrCl2 undergoes Cp ligand dissociation during catalysis, while the presence of acetylacetone in a coordinated form69,70 significantly enhances the reaction rate.
) in the 1H NMR titration experiments. Int-VIII, with a mass–charge ratio of m/z 589.1395, comprises one Cp ligand and two undehydrated intermediates connected to zirconium. The peak at m/z 689.1917 corresponds to the intermediate structure Int-IX, which bears two undehydrated hydroxy-dihydropyrazole and an acetylacetone. The structures Int-VIII and Int-IX illustrate that the catalyst possesses the ability to incorporate multiple substrates at a single half-zirconocene catalytic centre, thereby enabling simultaneous catalysis of two or even three molecular reactions during the catalytic process. ESI-MS analysis reveals that Cp2ZrCl2 undergoes Cp ligand dissociation during catalysis, while the presence of acetylacetone in a coordinated form69,70 significantly enhances the reaction rate.
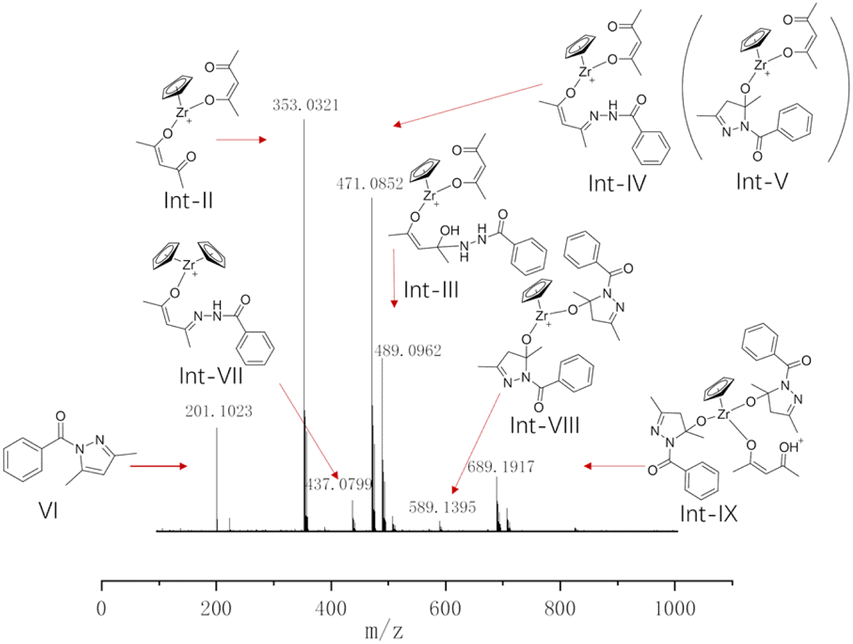 | ||
| Fig. 2 ESI-MS analysis for the synthesis of N-acylpyrazole under standard conditions. | ||
After carefully considering the aforementioned 1H NMR titration experiments, control experiment results, and ESI-MS analysis collectively, a plausible mechanism for the synthesis of (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone derivatives is proposed (Scheme 3). Initially, benzoyl hydrazine promotes the pre-catalyst Cp2ZrCl2 to remove Cp ligand and coordinate with two acetylacetonate ligands, forming the catalytic species [CpZrCl(acac)2]. Then, the primary amine on benzoyl hydrazine undergoes nucleophilic addition to attack acetylacetone and generate intermediate III. This intermediate then proceeds to eliminate a molecule of water and form imine intermediate IV. Subsequently, the enol carbon in the imine intermediate IV undergoes nucleophilic attack by the secondary amine of benzoyl hydrazine, resulting in the formation of N,O-acetal intermediate V. Finally, under the facilitation of the half-zirconocene centre, ligand dissociation occurs to release N,O-acetal and facilitate its dehydration, enabling the formation of target compound (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone while regenerating the catalyst species.
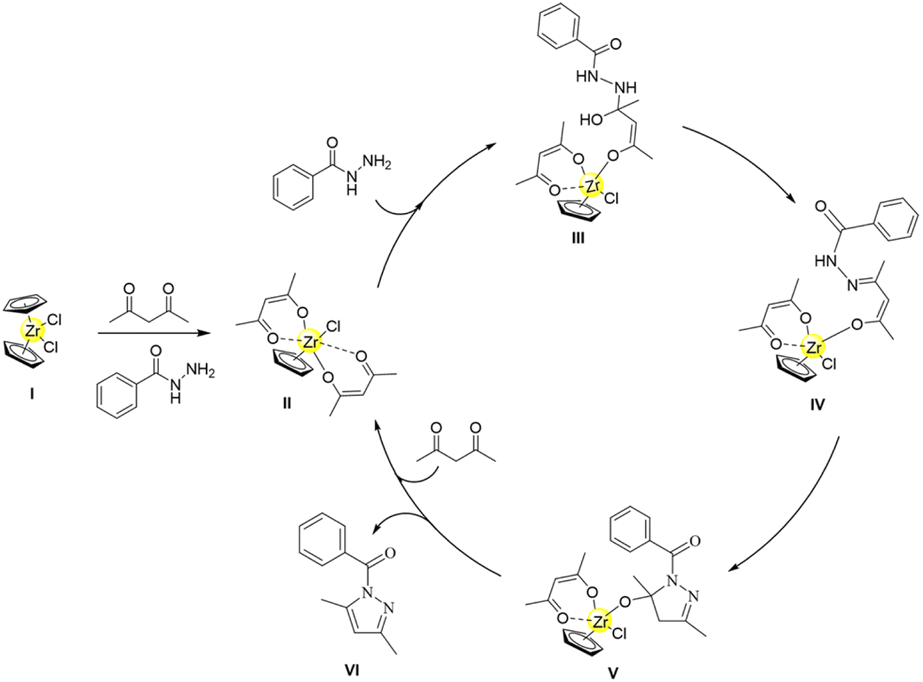 | ||
| Scheme 3 Plausible mechanism for the synthesis of (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone catalyzed by in situ formed half-zirconocene. | ||
Experimental
General procedure for synthesis of compound 3a
A representative example of the preparation of (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone (3a) is described as follows. The reactants, acetylacetone (50.06 mg, 0.5 mmol), benzoyl hydrazine (68.08 mg, 0.5 mmol), Cp2ZrCI2 (7.45 mg, 0.025 mmol), and toluene (0.5 mL), were combined in a reactor to establish the reaction system. The reaction was carried out under magnetic stirring at room temperature for 2 hours. Upon completion of the reaction, as confirmed by TLC analysis, the reaction mixture was subjected to silica gel flash column chromatography using an eluent mixture of petroleum ether and EtOAc in a ratio of 10:1 to yield the desired product 3a (97 mg) as a yellow liquid with a yield of 97%.
Conclusions
In conclusion, the development of an efficient catalytic system that facilitates the condensation and cyclization reaction between benzoyl hydrazine and acetylacetone has been achieved. This system involved the in situ formation of half-zirconocene using Cp2ZrCl2 as a pre-catalyst. Under mild conditions, high yields of N-acylpyrazoles were obtained with as low as 5% Cp2ZrCl2 catalyst loading. Mechanistic studies, including 1H NMR, ESI(+)-MS analyses, and control experiments, revealed that the coordination of Cp2ZrCl2 during the reaction led to the generation of the catalytically active species. The detection of half-zirconocene-based catalyst–substrate adducts by ESI-MS elucidated the process of catalyst–substrate interaction.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (21771122), the Start-up Funds for the PhD of Yan'an University (YAU202407432, YAU202407402), the Fundamental Research Funds for the Central Universities (GK202103027) and the Key Research and Development Project of Shaanxi Province (2021GY-308).References
- F. Franco, S. Meninno, M. Benaglia and A. Lattanzi, Chem. Commun., 2020, 56, 3073–3076 RSC.
- Y. Grell, X. Xie, S. I. Ivlev and E. Meggers, ACS Catal., 2021, 11, 11396–11406 Search PubMed.
- K. Otrubova, S. Chatterjee, S. Ghimire, B. F. Cravatt and D. L. Boger, Bioorg. Med. Chem., 2019, 27, 1693–1703 CrossRef CAS PubMed.
- J. Nandi, M. Z. Vaughan, A. L. Sandoval, J. M. Paolillo and N. E. Leadbeater, J. Org. Chem., 2020, 85, 9219–9229 CrossRef CAS PubMed.
- C. Volpe, S. Meninno, G. Mirra, J. Overgaard, A. Capobianco and A. Lattanzi, Org. Lett., 2019, 21, 5305–5309 CrossRef CAS PubMed.
- M. H. Baren, S. A. Ibrahim, M. M. Al-Rooqi, S. A. Ahmed, M. M. El-Gamil and H. A. Hekal, Sci. Rep., 2023, 13, 14680 CrossRef CAS PubMed.
- J. Dwivedi, S. Sharma, S. Jain and A. Singh, Mini-Rev. Med. Chem., 2018, 18, 918–947 CrossRef CAS.
- V. Kumar, K. Kaur, G. K. Gupta, A. K. Gupta and S. Kumar, Recent Pat. Inflammation Allergy Drug Discovery, 2013, 7, 124–134 CrossRef CAS.
- L.-P. Mo and Z.-H. Zhang, Curr. Org. Chem., 2011, 15, 3800–3823 CrossRef CAS.
- Y. Zhang, C. Wu, N. Zhang, R. Fan, Y. Ye and J. Xu, Int. J. Mol. Sci., 2023, 24, 12724 CrossRef CAS.
- R. T. Iminov, A. V. Mashkov, I. I. Vyzir, B. A. Chalyk, A. V. Tverdokhlebov, P. K. Mykhailiuk, L. N. Babichenko, A. A. Tolmachev, Y. M. Volovenko and A. Biitseva, Eur. J. Org Chem., 2015, 2015, 886–891 CrossRef CAS.
- S. Shahbazi, M. A. Ghasemzadeh, P. Shakib, M. R. Zolfaghari and M. Bahmani, Polyhedron, 2019, 170, 172–179 CrossRef CAS.
- Y. R. Girish, K. S. S. Kumar, H. S. Manasa and S. Shashikanth, J. Chin. Chem. Soc., 2014, 61, 1175–1179 Search PubMed.
- Y.-F. Liu, K. Li, H.-Y. Lian, X.-J. Chen, X.-L. Zhang and G.-P. Yang, Inorg. Chem., 2022, 61, 20358–20364 CrossRef CAS.
- J. P. Raj, D. Gangaprasad, K. Karthikeyan, R. Rengasamy, M. Kesavan, M. Venkateswarulu, M. Vajjiravel and J. Elangovan, Tetrahedron Lett., 2018, 59, 4462–4465 Search PubMed.
- M. Stephan, J. Panther, F. Wilbert, P. Ozog and T. J. Müller, Eur. J. Org Chem., 2020, 2020, 2086–2092 Search PubMed.
- M. A. Topchiy, D. A. Zharkova, A. F. Asachenko, V. M. Muzalevskiy, V. A. Chertkov, V. G. Nenajdenko and M. S. Nechaev, Eur. J. Org Chem., 2018, 2018, 3750–3755 Search PubMed.
- G. Yang, Y. Liu, X. Lin, B. Ming, K. Li and C. Hu, Chin. Chem. Lett., 2022, 33, 354–357 CrossRef CAS.
- C. P. Lakeland, D. W. Watson and J. P. Harrity, Chem.–Eur. J., 2020, 26, 155–159 CrossRef CAS PubMed.
- K. Kula, A. Kącka-Zych, A. Łapczuk-Krygier, Z. Wzorek, A. K. Nowak and R. Jasiński, Molecules, 2021, 26, 1364 CrossRef CAS PubMed.
- K. Kula, A. Łapczuk, M. Sadowski, J. Kras, K. Zawadzińska, O. M. Demchuk, G. K. Gaurav, A. Wróblewska and R. Jasiński, Molecules, 2022, 27, 8409 CrossRef CAS.
- M. S. Ledovskaya, V. V. Voronin, M. V. Polynski, A. N. Lebedev and V. P. Ananikov, Eur. J. Org Chem., 2020, 2020, 4571–4580 CrossRef CAS.
- P. Zhao, Z. Zeng, X. Feng and X. Liu, Chin. Chem. Lett., 2021, 32, 132–135 CrossRef CAS.
- G. Mali, B. A. Shaikh, S. Garg, A. Kumar, S. Bhattacharyya, R. D. Erande and A. V. Chate, ACS Omega, 2021, 6, 30734–30742 Search PubMed.
- A. J. Pearce, R. P. Harkins, B. R. Reiner, A. C. Wotal, R. J. Dunscomb and I. A. Tonks, J. Am. Chem. Soc., 2020, 142, 4390–4399 CrossRef CAS.
- M. Heinz, G. Weiss, G. Shizgal, A. Panfilova and A. Gansäuer, Angew. Chem., Int. Ed., 2023, 62, e202308680 CrossRef CAS.
- F. Mühlhaus, H. Weißbarth, T. Dahmen, G. Schnakenburg and A. Gansäuer, Angew. Chem., 2019, 131, 14346–14350 CrossRef.
- T. Oswald, T. Gelert, C. Lasar, M. Schmidtmann, T. Klüner and R. Beckhaus, Angew. Chem., Int. Ed., 2017, 56, 12297–12301 CrossRef CAS.
- Y. Q. Zhang, V. Jakoby, K. Stainer, A. Schmer, S. Klare, M. Bauer, S. Grimme, J. M. Cuerva and A. Gansäuer, Angew. Chem., 2016, 128, 1546–1550 CrossRef.
- Z. Zhang, R. B. Richrath and A. Gansäuer, ACS Catal., 2019, 9, 3208–3212 CrossRef CAS.
- Z. Zhang, D. Slak, T. Krebs, M. Leuschner, N. Schmickler, E. Kuchuk, J. Schmidt, L. I. Domenianni, J. B. Kleine Büning and S. Grimme, J. Am. Chem. Soc., 2023, 145, 26667–26677 CrossRef CAS PubMed.
- E. Le Roux, Coord. Chem. Rev., 2016, 306, 65–85 Search PubMed.
- M. Fischer, M. Jaugstetter, R. Schaper, M. Schmidtmann and R. Beckhaus, Eur. J. Inorg. Chem., 2018, 2018, 5146–5159 Search PubMed.
- R. A. Kehner, M. C. Hewitt and L. Bayeh-Romero, ACS Catal., 2022, 12, 1758–1763 CrossRef CAS.
- R. A. Kehner, G. Zhang and L. Bayeh-Romero, J. Am. Chem. Soc., 2023, 145, 4921–4927 Search PubMed.
- D. J. Mindiola, L. A. Watson, K. Meyer and G. L. Hillhouse, Organometallics, 2014, 33, 2760–2769 Search PubMed.
- N. Romero, Q. Dufrois, L. Vendier, C. Dinoi and M. Etienne, Chem.–Eur. J., 2017, 23, 15766–15774 Search PubMed.
- R. R. Aysin, M. V. Andreev, V. S. Bogdanov, S. S. Bukalov, A. A. Korlyukov, P. V. Dorovatovskii and V. V. Burlakov, Organometallics, 2021, 40, 1344–1350 CrossRef CAS.
- M. Reiß, F. Reiß, A. Spannenberg, P. Arndt and T. Beweries, Organometallics, 2018, 37, 4415–4423 Search PubMed.
- N. Romero, Q. Dufrois, N. Crespo, A. Pujol, L. Vendier and M. Etienne, Organometallics, 2020, 39, 2245–2256 CrossRef CAS.
- Y. Q. Zhang, E. Vogelsang, Z. W. Qu, S. Grimme and A. Gansäuer, Angew. Chem., Int. Ed., 2017, 56, 12654–12657 CrossRef CAS.
- Z. Zhang, J. B. Stückrath, S. Grimme and A. Gansäuer, Angew. Chem., Int. Ed., 2021, 60, 14339–14344 CrossRef CAS.
- S. Xu and E. I. Negishi, Acc. Chem. Res., 2016, 49, 2158–2168 CrossRef CAS PubMed.
- Y. Luo, H. Sun, W. Zhang, X. Wang, S. Xu, G. Zhang, Y. Jian and Z. Gao, RSC Adv., 2017, 7, 28616–28625 RSC.
- Y. Luo, Y. Wu, Y. Wang, H. Sun, Z. Xie, W. Zhang and Z. Gao, RSC Adv., 2016, 6, 66074–66077 RSC.
- M. Yang, Y. Jian, W. Zhang, H. Sun, G. Zhang, Y. Wang and Z. Gao, RSC Adv., 2021, 11, 38889–38893 RSC.
- S. Zheng, Y. Jian, S. Xu, Y. Wu, H. Sun, G. Zhang, W. Zhang and Z. Gao, RSC Adv., 2018, 8, 8657–8661 RSC.
- A. S. Singh, D. R. Naikwadi, K. Ravi and A. V. Biradar, Mol. Catal., 2022, 521, 112189 Search PubMed.
- A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 Search PubMed.
- J. Wang, X. Chen, X. Wang, W.-Q. Zhang, H.-M. Sun, G.-F. Zhang, Y. Wu and Z.-W. Gao, Transit. Met. Chem., 2016, 41, 731–738 Search PubMed.
- X. Wang, Z. H. Wang, G. Zhang, W. Zhang, Y. Wu and Z. Gao, Eur. J. Org Chem., 2015, 2016, 502–507 Search PubMed.
- Y. Wang, Y. Jian, Y. Wu, H. Sun, G. Zhang, W. Zhang and Z. Gao, Appl. Organomet. Chem., 2019, 33, e4925 CrossRef.
- R. J. Burford, A. Yeo and M. D. Fryzuk, Coord. Chem. Rev., 2017, 334, 84–99 Search PubMed.
- S. C. Gagieva, V. A. Tuskaev, D. A. Kurmaev, N. A. Kolosov, A. O. Borissova and B. M. Bulychev, J. Organomet. Chem., 2015, 793, 155–159 CrossRef CAS.
- K. Nikoofar and Z. Khademi, Res. Chem. Intermed., 2016, 42, 3929–3977 Search PubMed.
- A. D. Schwarz, K. R. Herbert, C. Paniagua and P. Mountford, Organometallics, 2010, 29, 4171–4188 CrossRef CAS.
- Z.-H. Zhang and T.-S. Li, Curr. Org. Chem., 2009, 13, 1–30 CrossRef CAS.
- Y. Wu, C. Chen, G. Jia, X. Zhu, H. Sun, G. Zhang, W. Zhang and Z. Gao, Chem.–Eur. J., 2014, 20, 8530–8535 CrossRef CAS.
- Y. Wu, X. Wang, Y. Luo, J. Wang, Y. Jian, H. Sun, G. Zhang, W. Zhang and Z. Gao, RSC Adv., 2016, 6, 15298–15303 RSC.
- M. Yang, Y. Wang, Y. Jian, D. Leng, W. Zhang, G. Zhang, H. Sun and Z. Gao, Mol. Catal., 2020, 498, 111247 CrossRef CAS.
- C. Redshaw and Y. Tang, Chem. Soc. Rev., 2012, 41, 4484–4510 RSC.
- J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004–2016 CrossRef CAS.
- G. Alesso, V. Tabernero and T. Cuenca, J. Organomet. Chem., 2012, 717, 202–210 CrossRef CAS.
- H. Dialer, K. Polborn, W. Ponikwar, K. Sünkel and W. Beck, Chem.–Eur. J., 2002, 8, 691–699 Search PubMed.
- W. Zhao, Q. Yan, K. Tsutsumi and K. Nomura, Organometallics, 2016, 35, 1895–1905 CrossRef CAS.
- J. Yi, N. Nakatani, N. Tomotsu, K. Nomura and M. Hada, Organometallics, 2021, 40, 643–653 CrossRef CAS.
- K. Nomura, I. Izawa, J. Yi, N. Nakatani, H. Aoki, H. Harakawa, T. Ina, T. Mitsudome, N. Tomotsu and S. Yamazoe, Organometallics, 2019, 38, 4497–4507 CrossRef CAS.
- W. Huang, B. Li, Y. Wang, W. Zhang, L. Wang, Y. Li, W.-H. Sun and C. Redshaw, Catal. Sci. Technol., 2011, 1, 1208–1215 RSC.
- W. Zhang and S. Luo, Chem. Commun., 2022, 58, 12979–12982 RSC.
- W. Zhang, L. Zhang and S. Luo, J. Am. Chem. Soc., 2023, 145, 14227–14232 CrossRef CAS.
- K. Zherikova and N. Morozova, J. Struct. Chem., 2012, 53, 761–767 CrossRef CAS.
- N. Ronaghi, D. M. Fialho, C. W. Jones and S. France, J. Org. Chem., 2020, 85, 15337–15346 CrossRef CAS.
- G. Smitha, S. Patnaik and C. S. Reddy, Synthesis, 2005, 711–713 CAS.
- M. Yang, Y. Guo, X. P. Zhang, H. Sun, Y. Wang, W. Zhang, Y. Wu, Y. Jian and Z. Gao, Asian J. Org. Chem., 2022, 11, e202100701 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08083a |
| ‡ J. Wu and M. Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |