
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTaylor dispersion analysis and release studies of β-carotene-loaded PLGA nanoparticles and liposomes in simulated gastrointestinal fluids†
Roman M. Fortunatus a,
Sandor Baloga,
Flávia Sousaab,
Dimitri Vanheckea,
Barbara Rothen-Rutishausera,
Patricia Taladriz-Blanco*a and
Alke Petri-Fink*ac
a,
Sandor Baloga,
Flávia Sousaab,
Dimitri Vanheckea,
Barbara Rothen-Rutishausera,
Patricia Taladriz-Blanco*a and
Alke Petri-Fink*ac
aAdolphe Merkle Institute, University of Fribourg, 1700 Fribourg, Switzerland. E-mail: alke.fink@unifr.ch; patricia.taladrizblanco@unifr.ch
bDepartment of Pharmaceutical Technology and Biopharmacy, Groningen Research Institute of Pharmacy, University of Groningen, 9713 AV Groningen, The Netherlands
cDepartment of Chemistry, University of Fribourg, 1700 Fribourg, Switzerland
First published on 13th January 2025
Abstract
β-Carotene (βC), a natural carotenoid, is the most important and effective vitamin A precursor, known also for its antioxidant properties. However, its poor water solubility, chemical instability, and low bioavailability limit its effectiveness as an orally delivered functional nutrient. Nanoparticle encapsulation improves βC's bioaccessibility by enhancing its stability and solubility. This study compares two formulations, i.e. βC-loaded poly(lactic-co-glycolic acid) (PLGA) NPs and liposomes before and after exposure to simulated gastrointestinal fluids using various methods such as Taylor dispersion analysis (TDA), cryo-transmission electron microscopy, dynamic light scattering (DLS), and nanoparticle tracking analysis (NTA). TDA, a microfluidic technique, proved more effective than DLS and NTA in determining nanoparticle size in simulated gastrointestinal fluids. This highlights TDA's potential for assessing nanoparticle colloidal stability in simulated gastro-intestinal fluids, crucial for evaluating encapsulated bioactives' bioavailability. High-performance liquid chromatography (HPLC) revealed that PLGA nanoparticles incorporate and preserve βC more effectively during long-term storage compared to liposomes. Adding ascorbic acid significantly reduced degradation in simulated gastrointestinal fluids. Release studies showed that liposomes released 52% of βC after 36 hours, while PLGA nanoparticles released only 9% over 168 hours. These results provide valuable insights for selecting an appropriate βC nanocarrier for oral delivery based on desired release rates.
Introduction
Beta-carotene (βC), a potent antioxidant and precursor of vitamin A, is a vital bioactive component.1 As an antioxidant, βC scavenges free radicals in the body, thereby reducing oxidative stress and the risk of modern chronic civilization diseases such as cancer and cardiovascular diseases.2 βC contributes significantly to daily vitamin A intake, providing up to 35% of daily requirements in industrialized countries3 and as much as 80%4 in emerging economies. Vitamin A deficiency (VAD) remains a widespread health issue affecting 33.3% of preschool children and 15.3% of pregnant women globally,5 with the highest rates in sub-Saharan Africa (48%) and South Asia (44%). In 2019, VAD affected 334 million (95% CI = 253.00–433.74) children and adolescents across 165 low- and middle-income countries.6 Vitamin A deficiency (VAD) primarily results from inadequate consumption of vitamin A-rich foods to meet the body's physiological requirements. While meat and dairy products are the main sources of preformed vitamin A in many Western populations, a growing number of vegans7 increasingly depend on pro-vitamin A carotenoids, particularly βC, as a low-cost vitamin A source.8Despite βC's potential health benefits, its efficacy in food and nutraceutical applications is limited by poor water solubility, chemical instability, lipophilic character, and low bioavailability.9 To overcome these limitations, nanotechnology-based formulations have been employed to encapsulate, protect, and enhance the delivery of βC by improving stability and biocompatibility.10–13 Despite extensive research on the encapsulation of βC within colloidal nanoparticles (NPs),14,15 there remains a notable lack of studies focused on accurately characterizing the colloidal stability of these NPs and their encapsulated bioactives in complex gastrointestinal environments. After nanoencapsulation, it is essential to monitor the physicochemical properties of nanocarriers under gastrointestinal conditions to evaluate their stability and behaviour. However, the complex nature of gastrointestinal fluids16 poses a significant challenge, including (i) characterizing the physicochemical properties of nanocarriers, (ii) accurately quantifying the βC released from the nanocarriers in the gastrointestinal medium, where it is susceptible to degradation,17 while controlling the subsequent release of βC, and (iii) preserving the chemical stability of βC upon release in the gastric fluids. These challenges make it difficult to accurately assess and predict the behaviour of nanocarriers in the digestive environment.18
In this study, poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) and liposomes were investigated as model biocompatible βC nanocarriers and their colloidal stability in simulated gastric and intestinal fluids, hereafter referred to as simulated gastrointestinal fluids (SGIF), was assessed by Taylor dispersion analysis (TDA) alongside complementary techniques, including nanoparticle tracking analysis (NTA), cryo-transmission electron microscopy (Cryo-TEM), and dynamic light scattering (DLS). While DLS is a well-established technique for measuring NP size (distribution)19,20 TDA offers a distinct advantage over DLS and NTA. Using TDA, we were able to assess the size of both PLGA NPs and liposomes in 20% SGIF without the need for NP purification. TDA, which determines NPs' size in laminar flow through optical absorption,21–23 is a suitable technique for analyzing nanoformulations in complex matrices, as it is less sensitive to polydispersity.24 TDA has also been successfully applied in pharmaceutical (nano)formulations, allowing the resolution of multicomponent samples.25–27
In addition, our study overcomes the challenge associated with the chemical instability of βC in gastric fluids by using ascorbic acid (AA), leveraging the well-documented role of AA in preserving βC stability.28–30 By incorporating SGIF with an optimal concentration of AA, we successfully monitored the release of βC from the formulation into SGIF with precision.
PLGA and liposomes have been approved by regulatory authorities such as the World Health Organization (WHO), the Food and Drug Administration (FDA), and the European Medicines Agency (EMA).31,32
Results and discussion
Characterization of the PLGA NPs and liposomes in SGIF
Preliminary characterization of the unloaded PLGA NPs and liposomes (herein referred to as NPs) before βC encapsulation and their subsequent release in simulated gastric fluid (SGF), simulated intestinal fluid (SIF), and water were carried out using TDA, DLS, and Cryo-TEM. Results obtained by TDA in water showed a hydrodynamic diameter (referred to as size) of 171 ± 9 nm for PLGA NPs and 124 ± 6 nm for liposomes (Fig. 1). Complementary data from DLS and NTA were consistent with the TDA results (Table 1, Fig. S1D and E†), indicating that TDA is a suitable technique to determine the size of the NPs, as supported by previous literature findings.25,27 Cryo-TEM imaging revealed that both types of NPs had spherical shapes with smooth surfaces. TEM micrographs on negative stained NPs revealed PLGA NPs with a core diameter of 189 ± 63 nm (n = 1116). In contrast, the liposomes showed core diameters of 102 ± 20 nm (n = 270) (Fig. 1 and S1C†). It is important to note that imaging and processing stained soft nanomaterials can introduce artifacts leading to broad-size distributions.33–35 | ||
| Fig. 1 TDA analysis of empty PLGA NPs and liposomes to determine their hydrodynamic diameter under various conditions. Initially, the hydrodynamic diameter was determined using water as the eluent immediately after preparation. Subsequently, measurements were taken after resuspension in simulated gastric (SGF) or simulated intestinal fluid (SIF) at t = 0 h. For these measurements, water was used as the eluent for purified samples, while 20 vol% SGF or 20 vol% SIF was used as the eluent for non-purified samples. The hydrodynamic diameter was determined after fitting the data using the Taylor–Aris model.21–23 The results are expressed as average n = 5, ±standard deviation. ⋯ experimental data values — fitted data values. Both NPs were visualized by a cryo-transmission electron microscope, scale bar = 300 nm for PLGA NPs and 200 nm for liposomes. | ||
| Size DLS [nm] | Size TDA [nm] | Eluent | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PLGA NPs | Liposomes | PLGA NPs | Liposomes | |||||||
| a For DLS analyses, non-purified samples were diluted using Milli-Q water.b Samples were purified by centrifugation and resuspended in Milli-Q water.c For the sequential exposure NPs were incubated in simulated gastric fluid (SGF) for 2 h followed by simulated intestinal fluid (SIF) for 24 h. | ||||||||||
| Medium | 0 h | 24 h | 0 h | 24 h | 0 h | 24 h | 0 h | 24 h | ||
| Water | 201 ± 5 | — | 133 ± 2 | — | 171 ± 9 | — | 124 ± 6 | — | Water | |
| Pure SGF | Non-purifieda | 201 ± 5 | 200 ± 6 | 135 ± 2 | 129 ± 2 | 132 ± 3 | 135 ± 9 | 162 ± 8 | 87 ± 3 | Pure SGF |
| Purifiedb | 210 ± 3 | 206 ± 4 | 134 ± 2 | 140 ± 2 | 147 ± 4 | 179 ± 7 | 125 ± 5 | 110 ± 15 | Water | |
| 20 vol% SGF | Non-purifieda | 182 ± 4 | — | 126 ± 2 | — | 174 ± 13 | — | 119 ± 14 | — | 20 vol% SGF |
| Pure SIF | Non-purifieda | 205 ± 4 | 206 ± 5 | 2668 ± 9068 | 155 ± 17 | 108 ± 7 | 114 ± 21 | 71 ± 3 | 56 ± 6 | Pure SIF |
| Purifiedb | 221 ± 6 | 218 ± 6 | 137 ± 3 | 134 ± 2 | 166 ± 2 | 167 ± 36 | 115 ± 12 | 76 ± 11 | Water | |
| 20 vol% SIF | Non-purifieda | 182 ± 3 | — | 2738 ± 213 | — | 159 ± 21 | — | 109 ± 17 | — | 20 vol% SIF |
| Sequential exposurec | Non-purifieda | 418 ± 18 | 506 ± 490 | 1525 ± 3600 | 916 ± 2985 | 107 ± 5 | 125 ± 27 | 56 ± 1 | 58 ± 8 | Pure SGF/SIF |
| Purifiedb | 386 ± 29 | 354 ± 19 | 129 ± 1 | 167 ± 5 | 166 ± 8 | 166 ± 5 | 119 ± 26 | 101 ± 35 | Water | |
In pure SGF and SIF, the size of PLGA NPs measured by DLS remained consistent at 0 h and 24 h (201 ± 5 and 200 ± 6 in SGF, 205 ± 4, and 206 ± 5 in SIF, respectively). These sizes were comparable to those measured in water and in purified samples (i.e., samples dispersed in pure SGF and SIF, cleaned by centrifugation and resuspended in water), indicating that PLGA NPs are stable in SGF and SIF (Table 1 and Fig. S2†). Similarly, liposomes exhibited colloidal stability in pure SGF over 24 h (Table 1 and Fig. S2†). However, when liposomes were incubated in pure SIF, a size increase was observed by DLS (2668 ± 9068 nm, Table 1 and Fig. S2†) at 0 h, suggesting potential aggregation of the liposomes or biased results due to the contribution of SIF components to the measured sizes (Table 1 and Fig. S3†). At 24 h, the size of liposomes in pure SIF was similar to that obtained in water and purified samples, raising concerns about the reproducibility of the analyses (Table 1 and Fig. S2†). Ultimately, results for purified liposomes after incubation in SIF (Table 1 and Fig. S2†) indicated that the liposomes did not aggregate; instead, the SIF components likely affected the DLS measurements. It is worth noting that the scattering intensity of PLGA NPs was approximately five times higher than that of the liposomes (Fig. S3B†). Consequently, unlike the liposomes, the contribution of the SGIF components did not significantly affect the DLS results for PLGA NPs incubated in SGIF.
The potential of TDA in resolving the size of the NPs in SGIF was evaluated by comparing results to those obtained by DLS and NTA. DLS relies on intensity fluctuations of scattered light caused by particles' Brownian motion, which leads to a bias towards larger particle sizes due to the sixth power dependency of scattered light intensity on diameter.36 In contrast, TDA is less influenced by populations of larger NPs, providing a more balanced size assessment in mixed populations.24 This is primarily because TDA relies on absorbance rather than scattered light intensity, making it a more reliable method for determining the size of the NPs in complex media.
Initial experiments on the NPs dispersed in pure SGIF and using pure SGIF as the eluent led to bi-component Taylorgrams26 displaying two distinct contributions: a narrow band corresponding to the medium (Fig. S3D and E†) and a broad band corresponding to the NPs (Fig. S4†). Modeling (ESI†) allowed determining NP sizes in pure SGIF. This demonstrates TDA's advantage over DLS for measuring NP sizes in pure SGIF without the need for sample purification beforehand. To confirm the results, samples were purified by centrifugation and redispersed in water, observing that the contribution of the medium was suppressed (Table 1 and Fig. S4†). The obtained size values were comparable to those measured using pure SGIF as the eluent. Additional experiments evaluated the feasibility of characterizing NPs in pure SGIF. These experiments showed that measurements could be performed on non-purified samples using 20 vol% of SGF or SIF as the eluent buffer without much contribution of the SGIF to the Taylorgram (Fig. 1). Under these conditions and at 0 h, the sizes were comparable across different conditions: in water, in purified samples after SGIF exposure using water as the eluent, and in non-purified samples using 20 vol% of SGIF as the eluent (Fig. 1 and Table 1). These findings highlight the robustness of TDA in measuring NP size, even in complex media such as SGIF. TDA shows clear advantages over DLS, particularly for weakly scattering NPs, such as liposomes, in the presence of interfering medium components and particles. (Fig. S5†).
To mimic oral delivery, NPs were incubated in SGF for 2 h followed by SIF for 24 h (hereafter referred to as sequential exposure) and then characterized by DLS, TDA, and electron microscopy. (Fig. 2 and S6†). Using DLS, multiple size distribution peaks were observed for both types of particles sequentially exposed to SGF and then SIF before purification, with hydrodynamic diameters ranging from 418 to 1525 nm (green plot, Fig. S2†). This observation likely results from strong scattering effects caused by agglomeration induced by proteins and/or ions from the SGIF.37 After sequential exposure and purification, the size of the PLGA NPs decreased but remained larger than the size of the particles in pure water. Conversely, the size of the liposomes after sequential exposure and purification was comparable to the size measured in pure water (Table 1 and Fig. S2†). Contrarily, data obtained by TDA using water as the eluent (Fig. 2 and Table 1) showed that the size of both NPs after sequential exposure and purification was similar to the size measured in water (Fig. 1 and Table 1), indicating that the NPs are stable upon sequential exposure to SGIF. This stability might be attributed to steric and electrostatic stabilization.38 Steric stabilization was provided by the surfactant used during nanoparticle synthesis, while electrostatic stabilization was achieved through the negative zeta potential of both particles. Compared to the observations in pure SGF and SIF, TDA could not resolve the size of both NPs after sequential exposure when using non-purified samples, resulting in lower size values than those obtained in water and purified samples (Table 1 and Fig. S6†). The stability of these particles was confirmed through TEM or SEM imaging (Fig. 2), whereby their robustness in maintaining size and structural integrity upon sequential exposure to simulated gastrointestinal conditions was highlighted, and their potential effectiveness for oral delivery applications was emphasized.
 | ||
| Fig. 2 Taylorgrams of purified (A) PLGA NPs and (B) purified liposomes incubated in simulated gastric fluid (SGF) for 2 h followed by simulated intestinal fluid (SIF) for 24 hours, with water as the eluent. TEM images of UranyLess, stained purified liposomes, and SEM images of PLGA NPs display particles in pure SGF and pure SIF after 24 hours and sequential exposure. The hydrodynamic diameter was determined after fitting the data using the Taylor–Aris model.21–23 The results are expressed as average n = 5, ±standard deviation. ⋯ experimental data values — fitted data values. | ||
βC release studies
Before encapsulation, the antioxidant properties of βC and its chemical stability were assessed. The DPPH (2,2-diphenyl-1-picrylhydrazyl) scavenging activity decreased upon exposure to SGIF compared to the control, with the lowest value recorded in SGF (Fig. 3A and Table S1†). This reduction in antioxidant activity is likely due to the degradation of βC into its isomers, βC-epoxide and βC-diepoxide, which lack antioxidant properties, with this degradation being caused by the oxidative and acidic environments present in SGF.17 Additionally, the chemical stability of βC at 1 μg mL−1 was monitored over 2 h in SGF and 8 days in SIF, showing a decrease in βC concentration over time (Fig. 3B). To improve βC stability, ascorbic acid (AA) was added to the SGIF, as AA is known to prevent βC degradation over time.28,29,39 Three concentrations of AA (0%, 3%, and 5 m/v%) were tested in SGIF to determine the optimal concentration for preserving βC stability. After 2 hours, the highest βC concentration (0.8 μg mL−1) was observed in SGF containing 5 m/v% AA (Fig. 3B). This improvement in βC stability is likely due to AA reacting with reactive oxygen species (e.g., singlet oxygen, superoxide anions, and hydroxyl radicals) that could otherwise degrade βC.40 Conversely, adding 3 m/v% AA to SIF resulted in a significantly higher βC concentration over the 8 day incubation period (Fig. 3C). Notably, adding 5 m/v% AA to SIF resulted in a lower βC concentration over 8 days, potentially due to the pro-oxidant effect of AA; however, the precise mechanism behind this effect remains unclear.41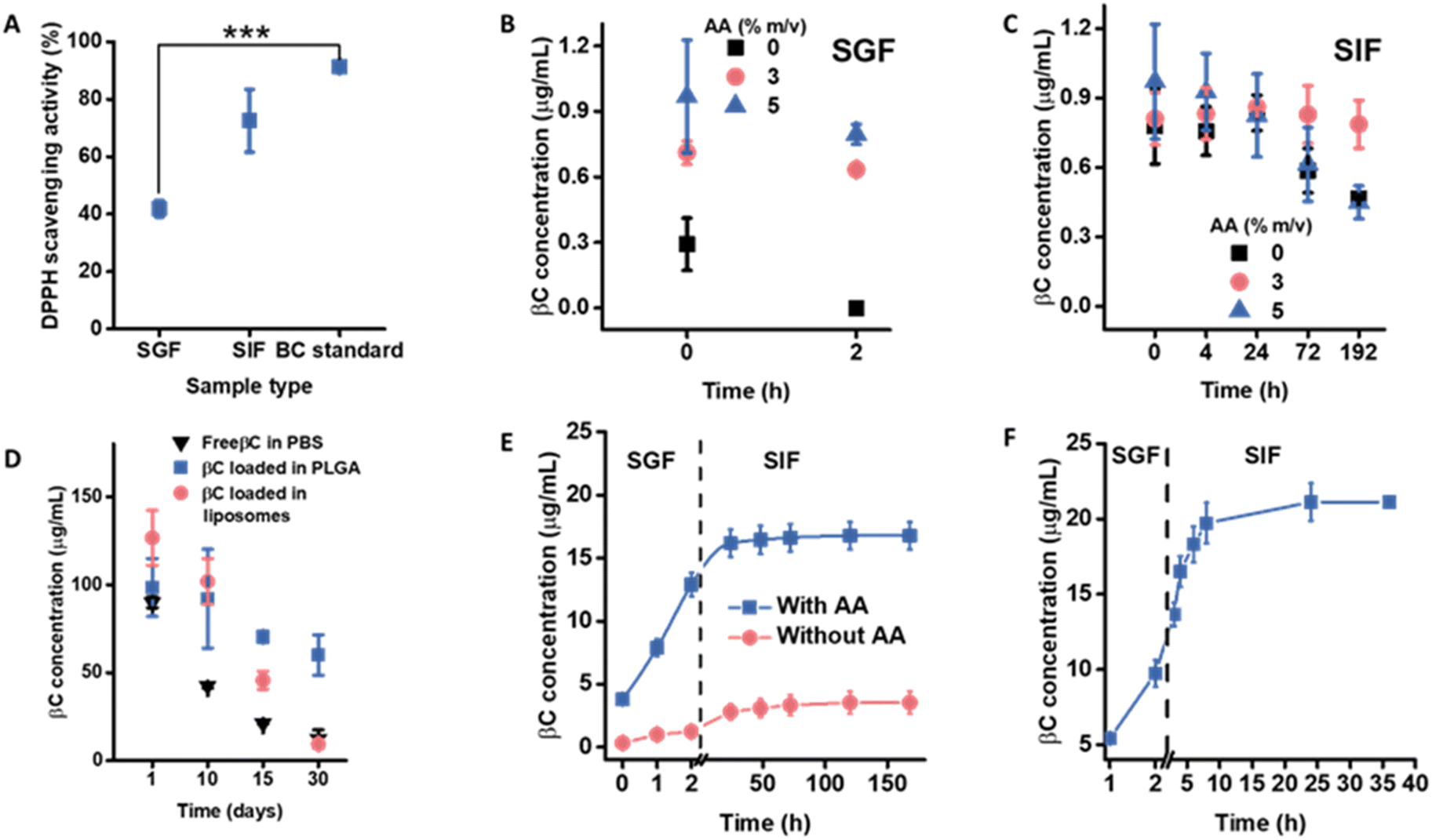 | ||
| Fig. 3 (A) Changes in βC activity (DPPH) in pure SGF and pure SIF. Control (βC standard) was the DPPH activity of βC standard (100 μg mL−1) in ethanol. Changes in βC concentration in (B) simulated gastric fluid (SGF) and (C) simulated intestinal fluid (SIF) in the presence or absence of ascorbic acid (AA). (D) Storage stability of encapsulated βC during storage in PBS for liposomes and water for PLGA NPs at 4 °C for 30 days. Results expressed as average n = 3, ±standard deviation. (E) βC release profiles from βC-loaded PLGA NPs in SGF and SIF with and without AA. (F) βC release profiles from βC-loaded liposomes in SGF and SIF with and without AA. | ||
Preliminary characterization of the βC-loaded NPs in water was monitored by DLS over 30 days. The results showed no significant changes in the size, polydispersity index, or zeta potential (Table S2†) of the NPs. These data were compared to those obtained for the empty NPs, revealing that βC encapsulation had no impact on these parameters over time (Table S2†), likely due to the low molecular weight of βC (0.5 kDa).42 The encapsulation efficiencies of βC within PLGA NPs and liposomes were determined using direct and indirect methods (see Experimental section for details). The indirect method yielded a maximum encapsulation efficiency (EE) of 90 ± 3% for PLGA NPs and 93 ± 2% for liposomes. The direct method yielded an EE of 60 ± 9% for PLGA NPs and 65 ± 11% for liposomes (Fig. S7†). Although this observation requires further study, the lower EE values for the direct method from both NPs were likely due to either inefficient extraction or overestimation in the indirect method, as the latter tends to overestimate EE.43,44 After encapsulation, the chemical stability of encapsulated βC without AA was monitored over a 30 day storage period at 4 °C (Fig. 3D). As shown in Fig. 3D, the concentration of βC decreased more rapidly in PBS alone than when βC was encapsulated in PLGA NPs (dispersed in water) and liposomes (dispersed in PBS). PLGA NPs showed a slower decrease in concentration than liposomes over the same storage period. This improved preservation of βC through encapsulation demonstrates the protective effect against degradative agents.11 The more substantial reduction in βC concentration within liposomes compared to PLGA NPs during storage is likely attributed to lipid hydrolysis and/or oxidation,45 which generates degradative compounds that break down the encapsulated βC. This is plausible since the liposomes used in this study contain soy lecithin, which includes unsaturated fatty acids prone to oxidation. Soy lecithin served as the liposome's primary ingredient, chosen for its cost-effectiveness as a phospholipid source in food applications.46 Additionally, liposomes with less densely packed lipid membranes exhibit higher membrane permeability, potentially increasing the exposure of βC to degradative species.47
Considering the improvement in βC stability in the presence of AA, 5 m/v% AA in SGF and 3 m/v% AA in SIF were added to study the release profile of βC-loaded NPs for sequential exposure in SGIF (Fig. 3E and F). βC released from PLGA NPs and liposomes without AA was also monitored and treated as the control. For PLGA NPs, a lower concentration of βC was detected throughout the release study in the SGIF without AA compared to the SGIF with AA. In the presence of AA, only 13 μg mL−1 (7 ± 1%) βC was released from PLGA NPs within SGF, and 17 μg mL−1 (9 ± 1% of the initial βC concentration in NPs, 188 μg mL−1) was released over the entire release study (Fig. 3E). The slow release profile of βC could be attributed to strong hydrophobic interactions between βC and the hydrophobic part of PLGA, i.e., the lactate chain.31 The characteristic slow-release profile of drugs or active pharmaceutical ingredients, such as βC, encapsulated in PLGA NPs, is widely acknowledged.48 SEM imaging of the PLGA NPs after the release in SGIF showed that particles remained stable (Fig. S8†). Upon purification and disintegration of the PLGA NPs, the concentration of βC was quantified, revealing that 80 ± 14% of βC remained unreleased.
In the case of liposomes, in the presence of AA, a steady release of βC was observed in SGF, with 10 μg per mL βC released after 2 h (25 ± 3%) and a faster release in SIF, with 21 μg mL−1 (52 ± 4% of the initial βC concentration, 40 μg mL−1) released after 36 h (Fig. 3F). The release rates of βC from liposomal systems were significantly faster in SIF compared to PLGA NPs. This is possibly due to the penetration of bile salts into the phospholipid bilayer, causing the swelling of liposome vesicles49 and a combination of enzymatic cleavages.50 Additionally, the pH of SIF was monitored throughout the release study, showing a decreasing trend over time. The reduction in pH indicated that the liposomal phospholipids were hydrolyzed,51 liberating free fatty acids (Fig. S9†). The concentration of βC released from liposomes in the SGIF without AA could not be quantified, likely due to degradation, resulting in values too low to be detected by HPLC.
Experimental
Materials
Poly(lactic-co-glycolic acid) PLGA 5004A (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) was kindly offered by Corbion-Purac Biomaterials (Holland, Netherlands). Acetone (HPLC grade, ≥99.8%), Pluronic-F127 (BioReagent grade), cholesterol (≥99%), and tetrahydrofuran (THF, HPLC grade, ≥99.9) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG 2000, >99%) was purchased from Avanti Polar Lipids (Birmingham, AL, USA). For high-performance liquid chromatography (HPLC) analysis, methanol (HPLC grade, ≥99.9%), βC standard (HPLC grade, ≥95%), both from Sigma-Aldrich (St. Louis, MO, USA) and methyl tert-butyl ether (MTBE, HPLC grade, ≥99%) from VWR International (Fontenay-sous-Bois, France) were used. For release studies, sodium chloride (NaCl, analytical grade, ≥99%), L-ascorbic acid (AA, HPLC grade, ≥99%), sodium hydroxide (NaOH, reagent grade, ≥98%), potassium dihydrogen phosphate (ACS reagent, ≥98.5%), sodium dodecyl sulfate (SDS, ACS reagent, ≥99%), 2,2-diphenyl-1-picrylhydrazyl (DPPH, GC grade, ≥99%), ethanol (HPLC grade, ≥99.8%), dichloromethane (HPLC grade, ≥99.8%), and hydrochloric acid (ACS reagent, 37%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pepsin (reagent grade) for simulated gastric fluid was obtained from Fisher Scientific (Loughborough, UK), while pancreatin and bile salts were both from porcine pancreas (8× USP specifications) for simulated intestinal fluid were purchased from Sigma-Aldrich (St. Louis, MO, USA). Triton X-100 (laboratory grade) and soy phospholipids (soy lecithin, ≥20% phosphatidylcholine) were purchased from Merck (Darmstadt, Germany), while phosphate-buffered saline (PBS) was purchased from Life Technologies Limited (Paisley, UK). Ultrapure water was prepared in-house with a conductivity of 0.055 μS cm−1 and a resistivity of 18.2 MΩ cm, using a Milli-Q station from Sartorius (Goettingen, Germany).
:50) was kindly offered by Corbion-Purac Biomaterials (Holland, Netherlands). Acetone (HPLC grade, ≥99.8%), Pluronic-F127 (BioReagent grade), cholesterol (≥99%), and tetrahydrofuran (THF, HPLC grade, ≥99.9) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG 2000, >99%) was purchased from Avanti Polar Lipids (Birmingham, AL, USA). For high-performance liquid chromatography (HPLC) analysis, methanol (HPLC grade, ≥99.9%), βC standard (HPLC grade, ≥95%), both from Sigma-Aldrich (St. Louis, MO, USA) and methyl tert-butyl ether (MTBE, HPLC grade, ≥99%) from VWR International (Fontenay-sous-Bois, France) were used. For release studies, sodium chloride (NaCl, analytical grade, ≥99%), L-ascorbic acid (AA, HPLC grade, ≥99%), sodium hydroxide (NaOH, reagent grade, ≥98%), potassium dihydrogen phosphate (ACS reagent, ≥98.5%), sodium dodecyl sulfate (SDS, ACS reagent, ≥99%), 2,2-diphenyl-1-picrylhydrazyl (DPPH, GC grade, ≥99%), ethanol (HPLC grade, ≥99.8%), dichloromethane (HPLC grade, ≥99.8%), and hydrochloric acid (ACS reagent, 37%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pepsin (reagent grade) for simulated gastric fluid was obtained from Fisher Scientific (Loughborough, UK), while pancreatin and bile salts were both from porcine pancreas (8× USP specifications) for simulated intestinal fluid were purchased from Sigma-Aldrich (St. Louis, MO, USA). Triton X-100 (laboratory grade) and soy phospholipids (soy lecithin, ≥20% phosphatidylcholine) were purchased from Merck (Darmstadt, Germany), while phosphate-buffered saline (PBS) was purchased from Life Technologies Limited (Paisley, UK). Ultrapure water was prepared in-house with a conductivity of 0.055 μS cm−1 and a resistivity of 18.2 MΩ cm, using a Milli-Q station from Sartorius (Goettingen, Germany).
Preparation of PLGA NPs and βC-loading
PLGA NPs were prepared using a modified nanoprecipitation method.52 Briefly, 20 mg of PLGA was dissolved in 1 mL of acetone and then gradually injected at a rate of 2 mL min−1 through a 21 G needle using a pumping system (New Era Pump System Inc., NY, U.S.A.) into a 20 mL solution of Pluronic F127 (0.1% w/v) under magnetic stirring at 360 rpm with an IKA RCT basic stirrer (IKA-Werke GmbH, Staufen, Germany). The dispersion was stirred for 3–4 h to evaporate the acetone. Subsequently, the NPs were washed by centrifugation at 12000g at 22 °C for 20 minutes using a Thermo Scientific XIR centrifuge (Waltham, MA, U.S.A.), and the pellet was re-suspended in 1 mL water. βC-loaded PLGA NPs were prepared by dissolving PLGA and 300 μg mL−1 of βC in acetone, following the same procedure described for empty PLGA NPs.
Preparation of liposomes and βC-loading
Liposomes were prepared using the ethanol injection method followed by extrusion using the procedure described by Liu et al.28 with slight modifications. Briefly, 40.3 mg of soy lecithin, 7.5 mg of DMG-PEG 2000, and 17.4 mg of cholesterol (molar ratio, 52:45:3) were dissolved in 1 mL of dichloromethane–ethanol mixture solution (1:2, v/v). Then, this mixture was injected dropwise into 4 mL of PBS (1×) solution with a 21 G syringe and maintained at 35 °C in a water bath while stirring for 20 minutes to obtain crude liposomes. Rotary evaporation at 40 °C with a stepwise vacuum at 300, 200, and 100 mbar (each for 30 minutes) was then carried out to remove the dichloromethane–ethanol mixture. Finally, the size of crude liposomes was reduced by passing them at least 11 times through a series of polycarbonate membranes, i.e., 0.8, 0.4, and 0.1 μm, using Avanti Mini-Extruder (Avanti Polar Lipids, Birmingham, USA). The βC-loaded liposomes were prepared following the same procedure described of empty liposomes by dissolving 250 μg of βC in the dichloromethane–ethanol mixture solution together with soy lecithin, DMG-PEG 2000 and cholesterol (52:45:3).
Physicochemical characterization
The hydrodynamic diameter and zeta potential of the NPs were determined by dynamic light scattering (DLS) using an Anton Paar Litesizer 500 particle analyzer (Anton Paar, Graz, Austria) operating with a 658 nm laser, measuring the autocorrelation function at a scattering angle of 175° and advanced cumulant model to get the hydrodynamic size. Zeta potential was obtained by applying the Smoluchowski approximation. Samples were appropriately diluted with Milli-Q water (pH = 6.0) before analysis. The concentration of the NPs, expressed as particles per mL, was obtained by nanoparticle tracking analysis (NTA) using a Nanosight NS500 (NanoSight Ltd, Minton Park, UK). Five measurements, each lasting 90 seconds, were conducted with a frame number of 1498 and particles per frame ranging from 20 to 60, using a 488 nm laser at 90° and 20 °C. The acquired data were processed with NTA 3.4.4 (2020) software (Malvern Panalytical Ltd, Enigma Business Park, UK). NPs were visualized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) using a Tescan Mira 3 SEM (Tescan, Brno, Czech Republic) and a FEI Tecnai Spirit TEM (Thermo Fisher, Waltham, MA, USA). For SEM, PLGA NPs were diluted with ultrapure water and placed on a glass slide mounted on the SEM stub with carbon tape. After drying, the NPs were sputter-coated with 3.5 nm of gold using a Cressington Sputter Coater 208 HR (Cressington Scientific Instruments, Chalk Hill, UK) and observed at 2 kV. For TEM, a dilute particle suspension was drop-casted onto a 300-mesh carbon-membrane-coated copper grid (Electron Microscopy Sciences, Hatfield, PA, USA). Excess NPs were blotted, subjected to uranyl-less negative staining (Electron Microscopy Sciences, Hatfield, PA, USA), and left to dry overnight at room temperature.53 TEM operation was at 120 kV, equipped with a CCD Olympus Veleta camera (Hachioji, Tokyo, Japan). For Cryo-TEM, four microliters of the sample were drop-casted on a glow-discharged Lacey 400-carbon film grid (Agar Scientific, UK). The glow discharging was performed by ELMO Glow Discharge System (Cordouan Technologies, France) for 15 s, 2 mV at 2 × 10−1 bar. The sample was then blotted for 4 seconds in automatic blotting mode and then plunged into liquid ethane (−180 °C) using an EM GP2 Automatic Plunge Freezer (Leica Microsystems, Wetzlar, Germany). The cryo-plunger chamber was maintained at >90% humidity and 20 °C. Cryo-TEM images were acquired using an FEI Eagle CCD camera (Thermo Fisher, Waltham, MA, USA) using the SerialEM acquisition software in low-dose mode.54 All additional image processing for SEM and TEM, including scale bar inclusion, contrast adjustments, noise reduction, and size distribution (automatic counting), were conducted using Fiji (ImageJ version 1.53).55 Segmentation of the UranyLess stained NPs and background was performed using the pixel classification workflow of Ilastik56 (https://www.ilastik.org/publications.html). Four images of PLGA NPs were combined into a stack image, while two images of liposomes were loaded into Ilastik for training and actual segmentation. The segmented stack images were exported into FIJI (ImageJ, National Institutes of Health, Bethesda, MD, USA)55 for automatic NP size analysis using a built-in size analysis package on the negative stained TEM micrographs. The diameter of the NPs was calculated from the area of each NP as diameter = 2 × √(area/π) using the negatively stained NPs.DPPH radical scavenging activity
The antioxidant activity of βC was measured following the method described by Basnet, et al.,57 with minor modifications. Briefly, 0.5 mL of ethanolic DPPH (2,2-diphenyl-1-picrylhydrazyl) solution (60 μM) was mixed with 0.5 mL of βC standard (100 μg per mL βC in SGIF). The reaction mixture was shaken and incubated for 30 minutes at room temperature in the dark. Absorbance was then measured at 520 nm using a V-670 spectrophotometer (Jasco Corporation, Tokyo, Japan). The antioxidant activity of βC was expressed as the percentage of DPPH scavenging activity, calculated using the following formula:whereby: Asample = absorbance of 100 μg per mL βC standard incubated in SGIF and DPPH, Ablank = absorbance of DPPH incubated in SGIF, and Acontrol = absorbance of DPPH incubated in ethanol or SGIF.
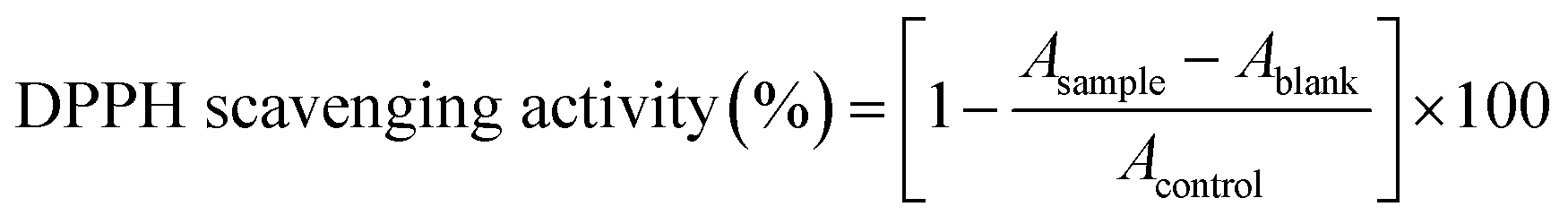
Taylor dispersion analysis (TDA)
TDA was used to determine the hydrodynamic diameter of empty and βC-loaded NPs incubated in the SGIF before and after the purification by centrifugation. To purify the particles incubated in SGIF, PLGA NPs were centrifuged at 12000 g for 15 minutes, while liposomes were centrifuged at 40000 g for 45 minutes. Following purification, the particles were resuspended in Milli-Q water. TDA measurements on purified samples were conducted using water as the eluent. For non-purified samples, measurements were performed using water, pure SGIF, or 20 vol% SGIF as the eluent, as specified in the figure captions. The eluent composition was identical to the medium used to resuspend the particles in all conditions. Time-resolved absorbance (hereafter referred to as Taylorgram) was followed by an ActiPix D100 UV-Vis area imaging detector with a sample rate of 20 Hz (Paraytec, York, UK) at room temperature. Samples were injected at 200 mbar for 0.2 minutes into a fused silica capillary (74.5 μm inner diameter, Polymicro Technologies, Phoenix, USA) under a continuous flow of eluent (90 mbar, for 20 minutes) using a capillary electrophoresis injection system (Prince 560 CE Autosampler, Prince Technologies B.V., Netherlands). The total length of the capillary was 120 cm, with a distance to the first window at 34 cm and a distance to the second window at 75 cm. Both windows were approximately 1 cm wide, and a band-pass filter with 214 nm center wavelength (Edmund Optics, York, UK) was used. The capillary was cleaned between samples by passing 0.1 M NaOH at 2500 mbar for 10 minutes, then rinsed with Milli-Q water at 2500 mbar for 10 minutes.
βC-release studies in SGIF
Two-stage consecutive in vitro release studies were conducted to mimic oral delivery in vivo, i.e., the release was studied in SGF for 2 h, followed by SIF for 36 h for liposomes and 8 days for PLGA NPs. The standardized INFOGEST model was followed for the composition of SGIF; however, enzyme concentration was used instead of enzyme activity. This concentration has been shown to produce results similar to those of the INFOGEST model.58,59 The SGF comprised 0.2 g NaCl, 0.32 g pepsin, and 0.7 mL concentrated HCl in 100 mL Milli-Q water, pH 2. The SIF comprised pancreatin (4.8 mg mL−1), porcine bile extract (10 mM), CaCl2 (0.15 mM), and 7.7 mL of 0.2 M NaOH in 100 mL in PBS, pH 6.8.49,60 SDS (5% w/v) was added to both media to maintain sink conditions and facilitate βC solubilization.The release of βC from βC-loaded PLGA NPs was studied by mixing the NPs with 1 mL of SGF (1:1 v/v) and incubated at 37 °C with continuous agitation at 100 rpm for 2 h. After one hour incubation, the mixture was centrifuged at 12000g for 20 minutes at 4 °C. The supernatant (900 μL) was collected, mixed with 100 μL of acetone, and injected into the HPLC system for analysis. The pellet was resuspended in 1 mL of SGF for an additional hour of incubation. After the SGF phase, the pellets were redispersed in 1 mL of SIF and incubated at 37 °C with continuous agitation at 100 rpm for 168 h as the release of a hydrophobic molecule like βC, from 50:50 PLGA is expected to be exceptionally slow.61 At pre-determined time intervals (2, 24, 48, 72, 120, and 168 h), samples were centrifuged at 12000g for 20 minutes at 4 °C. The collected pellet was resuspended in 1 mL of fresh SIF for every time interval, as shown in the SGF phase. For HPLC analysis, the supernatant was processed similarly to the SGF phase. The NPs were then dried in a conventional air oven (70 °C for 5 h), dissolved in THF, and analyzed by HPLC to determine the βC concentration remaining in the PLGA NPs at the end of the release study.
The release of βC from βC-loaded liposomes was studied following the procedure described by Xu et al.49 Liposomes were mixed with SGF (1:1 v/v, total volume of 3 mL) and incubated at 37 °C with shaking for 2 h. Afterward, the pH was adjusted to 7.5 with 1 M NaOH, and then the sample was mixed with an equal volume of SIF for another 36 h. The pH of the SIF was maintained at 7.5 by manually adding 0.1 M NaOH to neutralize free fatty acids (FFAs) released from lipid digestion. At specific intervals (1, 2, 3, 4, 6, 8, 14, 24, and 36 h), 200 μL of the sample was withdrawn and replaced with fresh SGF or SIF. The withdrawn samples were ultracentrifuged at 40000g for 45 minutes, and the supernatant was mixed with 300 μL of acetone before HPLC analysis. The pellets containing the liposomes were disintegrated by adding 280 μL of Triton X solution (10% v/v), followed by 300 μL of acetone, and injected into the HPLC system to quantify the βC concentration within the liposomes.
Quantification of βC by HPLC
βC was quantified using a Thermo Scientific UltiMate 3000 HPLC system (Waltham, MA, USA). The separation was performed on a Nucleosil reversed-phase C18 column (100 × 4.6 mm, pore size: 5 μm) from Sigma-Aldrich (Schnelldorf, Germany). The mobile phase consisted of methanol and MTBE (90:10). The isocratic method ran for 15 minutes at a 1 mL min−1 flow rate, with βC detection at 450 nm using a diode array detector.62 An injection volume of 20 μL was used, exhibiting a retention time of 8.5 minutes. To prevent light exposure, samples were stored in amber-colored vials. Assay performance was assessed for linearity, the limit of quantification, and the limit of detection, as reported in the International Conference on Harmonization guidelines.63 Encapsulation efficiency (EE) was determined using direct and indirect methods. For the indirect method, the concentration of encapsulated βC was determined by subtracting free βC in the supernatant from the total βC added to the formulation. PLGA NPs were centrifuged at 20000g for 30 minutes, and liposomes were centrifuged at 4000g for 20 minutes through 10K filters (Microsep Advance Centrifugal device). In the direct method, encapsulation efficiency (EE) was calculated as the ratio of the concentration of βC in disintegrated NPs to the initial βC concentration added during formulation.64
Statistical analysis
All mathematic fittings were performed on Mathematica (Wolfram Research, Inc., Mathematica, Version 14.0 of 2024, USA). The size of NPs was obtained by fitting the Taylorgrams,21–23 as described in the ESI.† Results are expressed as a mean ± standard deviation from a minimum of three independent experiments. Differences were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.Conclusions
In this study, the power of TDA as a suitable technique for determining the average size of NPs in SGIF was demonstrated through comprehensive characterization of βC-loaded PLGA NPs and βC-loaded liposomes. When compared to conventional DLS, TDA's efficacy in complex media was particularly noteworthy.The successful encapsulation of βC in both PLGA NPs and liposomes was achieved, with a significant advancement observed in the reduction of βC degradation through the strategic incorporation of AA at appropriate concentrations. This finding not only underscores the potential of AA as a potent stabilizing agent in harsh gastrointestinal environments but also opens new avenues for enhancing the bioavailability of orally administered compounds.
These results collectively represent a significant step forward in NP characterization and formulation stability, potentially advancing approaches to oral drug delivery systems and enabling more effective nutrient delivery, as well as enhancing functional food development.
Data availability
The data supporting this article have been included as part of the ESI.† All raw data used to create the presented figures and tables can be accessed in Zenodo via https://10.5281/zenodo.14415466.Author contributions
R. M. F. performed all the experiments included in this manuscript and analyzed the DLS, TDA, and HPLC data; was responsible for data interpretation and drafting of the manuscript. S. B. developed the mathematical script to analyze the TDA data and was responsible for data interpretation. D. M. assisted with the Cryo-TEM sample preparation and imaging. F. S. showed R. M. F. the synthesis of PLGA NPs and conceptualized the release studies. P. T. B. was responsible for the TDA conceptualization, data interpretation, supervision, and manuscript drafting. B. R. R. and A. P. F. supervised the project and revised the manuscript. All authors have approved the final version of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges support from the Swiss Excellence Scholarship through the State Secretariat for Education, Research and Innovation (ESKAS 2022.508) and the Adolphe Merkle Foundation.References
- T. Grune, G. Lietz, A. Palou, A. C. Ross, W. Stahl, G. Tang, D. Thurnham, S. A. Yin and H. K. Biesalski, J. Nutr., 2010, 140, 2268S–2285S CrossRef CAS
.
- J. Fiedor and K. Burda, Nutrients, 2014, 6, 466–488 CrossRef PubMed
- D. Weber and T. Grune, Mol. Nutr. Food Res., 2012, 56, 251–258 CrossRef CAS PubMed
- M. J. Haskell, Am. J. Clin. Nutr., 2012, 96, 1193S–1203S CrossRef CAS
- WHO, Global prevalence of vitamin A deficiency in populations at risk 1995–2005, WHO Global Database on Vitamin A Deficiency, World Health Organization, Geneva, 2009, available https://www.who.int/publications/i/item/9789241598019 Search PubMed
- P. Song, D. Adeloye, S. Li, D. Zhao, X. Ye, Q. Pan, Y. Qiu, R. Zhang, I. Rudan and Global Health Epidemiology Research, J. Glob. Health, 2023, 13, 04084 CrossRef
- C. Leitzmann, Am. J. Clin. Nutr., 2014, 100(suppl. 1), 496S–502S CrossRef CAS
- A. Harari, N. Melnikov, M. Kandel Kfir, Y. Kamari, L. Mahler, A. Ben-Amotz, D. Harats, H. Cohen and A. Shaish, Nutrients, 2020, 12(6), 1625 CrossRef CAS
- K. Gul, A. Tak, A. K. Singh, P. Singh, B. Yousuf, A. A. Wani and F. Yildiz, Cogent Food Agric., 2015, 1, 1018696 CrossRef
- C. Dima, E. Assadpour, S. Dima and S. M. Jafari, Compr. Rev. Food Sci. Food Saf., 2020, 19, 2862–2884 CrossRef PubMed
- M. Jalali-Jivan, H. Rostamabadi, E. Assadpour, M. Tomas, E. Capanoglu, M. Alizadeh-Sani, M. S. Kharazmi and S. M. Jafari, Adv. Colloid Interface Sci., 2022, 307, 102750 CrossRef CAS
- V. K. Maurya, A. Shakya, M. Aggarwal, K. M. Gothandam, T. Bohn and S. Pareek, Antioxidants, 2021, 10(3), 426 CrossRef CAS
- K. Sridhar, B. S. Inbaraj and B. H. Chen, Antioxidants, 2021, 10(5), 713 CrossRef CAS PubMed
- S. Bera, R. Mitra and J. Singh, Biotechnol. Genet. Eng. Rev., 2023, 1–57, DOI:10.1080/02648725.2023.2213988
- M. Pourmadadi, H. Ahmadi, M. Abdouss, A. Rahdar and S. Pandey, BioNanoScience, 2024, 14, 1832–1853 CrossRef
- J. Xue, C. Blesso and Y. Luo, Annu. Rev. Food Sci. Technol., 2023, 14, 1–33 CrossRef CAS PubMed
- W. Tao, X. Ye and Y. Cao, Food Sci. Hum. Wellness, 2021, 10, 370–374 CrossRef CAS
- X. Zhang, G. Chen, H. Zhang, L. Shang and Y. Zhao, Nat. Rev. Bioeng., 2023, 1, 208–225 CrossRef CAS
- J. Rodriguez-Loya, M. Lerma and J. L. Gardea-Torresdey, Micromachines, 2023, 15(1), 24 CrossRef PubMed
- N. Hondow, R. Brydson, P. Wang, M. D. Holton, M. R. Brown, P. Rees, H. D. Summers and A. Brown, J. Nanopart. Res., 2012, 14(7), 977 CrossRef
- R. Aris, Proc. R. Soc. Lond. A Math. Phys. Sci., 1956, 235, 67–77 Search PubMed
- G. I. Taylor, Proc. R. Soc. Lond. A Math. Phys. Sci., 1953, 219, 186–203 CAS
- G. I. Taylor, Proc. R. Soc. Lond. A Math. Phys. Sci., 1954, 225, 473–477 CAS
- D. A. Urban, A. M. Milosevic, D. Bossert, F. Crippa, T. L. Moore, C. Geers, S. Balog, B. Rothen-Rutishauser and A. Petri-Fink, Colloid Interface Sci. Commun., 2018, 22, 29–33 CrossRef CAS
- A. Ibrahim, R. Meyrueix, G. Pouliquen, Y. P. Chan and H. Cottet, Anal. Bioanal. Chem., 2013, 405, 5369–5379 CrossRef CAS
- C. Malburet, L. Leclercq, J. F. Cotte, J. Thiebaud, E. Bazin, M. Garinot and H. Cottet, Anal. Chem., 2022, 94, 4677–4685 CrossRef CAS
- C. Malburet, L. Leclercq, J. F. Cotte, J. Thiebaud, E. Bazin, M. Garinot and H. Cottet, Gene Ther., 2023, 30, 421–428 CrossRef CAS PubMed
- X. Liu, P. Wang, Y. X. Zou, Z. G. Luo and T. M. Tamer, Food Res. Int., 2020, 136, 109587 CrossRef CAS
- Y. Liu, Z. Hou, J. Yang and Y. Gao, J. Food Sci. Technol., 2015, 52, 3300–3311 CAS
- C. Qian, E. A. Decker, H. Xiao and D. J. McClements, Food Chem., 2012, 135, 1036–1043 CrossRef CAS
- P. Blasi, J. Pharm. Invest., 2019, 49, 337–346 CrossRef CAS
- E. M. Kim and H. J. Jeong, Chonnam Med. J., 2021, 57, 27–35 CrossRef CAS PubMed
- U. Baxa, in Characterization of Nanoparticles Intended for Drug Delivery, ed. S. E. McNeil, Springer New York, New York, NY, 2018, pp. 73–88, DOI:10.1007/978-1-4939-7352-1_8
- L. E. Franken, E. J. Boekema and M. C. A. Stuart, Adv. Sci., 2017, 4, 1600476 CrossRef PubMed
- L. Liu, W. Yu, J. Seitsonen, W. Xu and V. P. Lehto, Chemistry, 2022, 28, e202200947 CrossRef CAS
- C. M. Maguire, M. Rosslein, P. Wick and A. Prina-Mello, Sci. Technol. Adv. Mater., 2018, 19, 732–745 CrossRef CAS
- W. Dudefoi, H. Rabesona, C. Rivard, M. Mercier-Bonin, B. Humbert, H. Terrisse and M. H. Ropers, Food Funct., 2021, 12, 5975–5988 RSC
- H. T. Phan and A. J. Haes, J. Phys. Chem. C, 2019, 123, 16495–16507 CrossRef CAS
- L. Gu, Y. Su, M. Zhang, C. Chang, J. Li, D. J. McClements and Y. Yang, Food Res. Int., 2017, 96, 84–93 CrossRef CAS PubMed
- H. Morais, P. Rodrigues, V. Almeida, E. Forgács, T. Cserháti, L. Gomes and J. Oliveira, in Natural Products in the New Millennium: Prospects and Industrial Application, ed. A. P. Rauter, F. B. Palma, J. Justino, M. E. Araújo and S. P. dos Santos, Springer Netherlands, Dordrecht, 2002, pp. 155–163, DOI:10.1007/978-94-015-9876-7_16
- S. Pal and N. R. Jana, ACS Appl. Nano Mater., 2022, 5, 4583–4596 CrossRef CAS
- M. C. Pereira, L. E. Hill, R. C. Zambiazi, S. Mertens-Talcott, S. Talcott and C. L. Gomes, LWT–Food Sci. Technol., 2015, 63, 100–107 CrossRef CAS
- Y. Amini, S. Amel Jamehdar, K. Sadri, S. Zare, D. Musavi and M. Tafaghodi, Bio-Med. Mater. Eng., 2017, 28, 613–620 CAS
- J. J. Muso-Cachumba, G. Ruiz-Lara, G. Monteiro and C. d. O. Rangel-Yagui, Braz. J. Pharm. Sci., 2023, 59, e23365 CrossRef CAS
- J. Musakhanian, J. D. Rodier and M. Dave, AAPS PharmSciTech, 2022, 23, 151 CrossRef CAS PubMed
- J. Li, J. Nan, H. Wu, H. J. Park, Q. Zhao and L. Yang, Food Chem., 2022, 389, 132931 CrossRef CAS
- F. Song, G. Yang, Y. Wang and S. Tian, Innovative Food Sci. Emerging Technol., 2022, 81, 103155 CrossRef CAS
- F. Y. Han, K. J. Thurecht, A. K. Whittaker and M. T. Smith, Front. Pharmacol, 2016, 7, 185 Search PubMed
- T. Xu, J. Zhang, R. Jin, R. Cheng, X. Wang, C. Yuan and C. Gan, J. Sci. Food Agric., 2022, 102, 5759–5767 CrossRef CAS PubMed
- W. Liu, Y. Hou, Y. Jin, Y. Wang, X. Xu and J. Han, Trends Food Sci. Technol., 2020, 104, 177–189 CrossRef CAS
- C. Tan, Y. Zhang, S. Abbas, B. Feng, X. Zhang and S. Xia, Colloids Surf., B, 2014, 123, 692–700 CrossRef CAS PubMed
- P. J. Kennedy, F. Sousa, D. Ferreira, C. Pereira, M. Nestor, C. Oliveira, P. L. Granja and B. Sarmento, Acta Biomater., 2018, 81, 208–218 CrossRef CAS
- K. P. Cruz, B. F. C. Patricio, V. C. Pires, M. F. Amorim, A. Pinho, H. C. Quadros, D. A. S. Dantas, M. H. C. Chaves, F. R. Formiga, H. V. A. Rocha and P. S. T. Veras, Front. Chem., 2021, 9, 644827 CrossRef CAS PubMed
- D. N. Mastronarde, J. Struct. Biol., 2005, 152, 36–51 CrossRef PubMed
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed
- S. Berg, D. Kutra, T. Kroeger, C. N. Straehle, B. X. Kausler, C. Haubold, M. Schiegg, J. Ales, T. Beier, M. Rudy, K. Eren, J. I. Cervantes, B. Xu, F. Beuttenmueller, A. Wolny, C. Zhang, U. Koethe, F. A. Hamprecht and A. Kreshuk, Nat. Methods, 2019, 16, 1226–1232 CrossRef CAS PubMed
- P. Basnet, H. Hussain, I. Tho and N. Skalko-Basnet, J. Pharm. Sci., 2012, 101, 598–609 CrossRef CAS PubMed
- K. Cabezas-Teran, C. Grootaert, J. Ortiz, S. Donoso, J. Ruales, F. Van Bockstaele, J. Van Camp and T. Van de Wiele, Food Res. Int., 2023, 164, 112301 CrossRef CAS PubMed
- D. B. Rodrigues, C. Chitchumroonchokchai, L. R. B. Mariutti, A. Z. Mercadante and M. L. Failla, J. Agric. Food Chem., 2017, 65, 11220–11228 CrossRef CAS PubMed
- A. Brodkorb, L. Egger, M. Alminger, P. Alvito, R. Assuncao, S. Ballance, T. Bohn, C. Bourlieu-Lacanal, R. Boutrou, F. Carriere, A. Clemente, M. Corredig, D. Dupont, C. Dufour, C. Edwards, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. R. Mackie, C. Martins, S. Marze, D. J. McClements, O. Menard, M. Minekus, R. Portmann, C. N. Santos, I. Souchon, R. P. Singh, G. E. Vegarud, M. S. J. Wickham, W. Weitschies and I. Recio, Nat. Protoc., 2019, 14, 991–1014 CrossRef CAS PubMed
- C. Wischke and S. P. Schwendeman, Int. J. Pharm., 2008, 364, 298–327 CrossRef CAS PubMed
- A. Gomes, A. L. R. Costa, D. D. Cardoso, G. Nathia-Neves, M. A. A. Meireles and R. L. Cunha, Food Chem., 2021, 341, 128155 CrossRef CAS PubMed
- ICH-Q2B, EMA/CHMP/ICH/82072/2006, 1995 Search PubMed.
- F. Sousa, A. Cruz, P. Fonte, I. M. Pinto, M. T. Neves-Petersen and B. Sarmento, Sci. Rep., 2017, 7, 3736 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08138b |
| This journal is © The Royal Society of Chemistry 2025 |