
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the capabilities of 2-alkynyl aryl/benzyl azides: synthesis approaches for indoles, quinolines, and their derivatives via transition metal catalysis
Seyedmohammad Hosseininezhad
a,
Sina Pirani Ahmad Abad
a and
Ali Ramazani
 *ab
*ab
aThe Organic Chemistry Research Laboratory (OCRL), Department of Chemistry, Faculty of Science, University of Zanjan, Zanjan 45371-38791, Iran. E-mail: hossinichemistry1397@gmail.com; aliramazani@gmail.com; aliramazani@znu.ac.ir
bThe Convergent Sciences & Technologies Laboratory (CSTL), Research Institute of Modern Biological Techniques (RIMBT), University of Zanjan, Zanjan 45371-38791, Iran
First published on 14th January 2025
Abstract
In recent research, quinoline and indole structures have gained recognition for their significant clinical relevance and effectiveness. These compounds are known for their wide-ranging pharmacological effects, which include anticancer, antibacterial, antifungal, antiviral, and anti-inflammatory properties. Researchers have successfully implemented a variety of innovative synthetic strategies, leading to the creation of numerous compounds that display fascinating biological activities in diverse fields. This has sparked growing interest in developing quinoline and indole-based analogues, given their impressive variety of biological effects. Over the past few years, new, efficient, and more accessible synthetic techniques—such as green chemistry and microwave-assisted synthesis—have been introduced to produce a diverse array of quinoline and indole structures. This development reflects an expanding area of interest in both academic and industrial settings, making it easier to investigate their biological capabilities. In this review, we examine the intriguing transformations of 2-alkynyl aryl and benzyl azide derivatives into indoles and quinolines, emphasizing the role of metal catalysts such as Au, Cu, Rh, Pd, and Ag, from 2011 to 2024. We showcase the variety of substrates involved, highlight notable advancements in this area of research, and address the limitations faced by chemists. Additionally, we offer insights into the mechanisms driving these important reactions, aiming to enhance understanding and inspire future work in this dynamic field.
 Seyedmohammad Hosseininezhad | Seyedmohammad Hosseininezhad was born in Zanjan, Iran, in 1997. He holds a BSc degree in Pure Chemistry from Payam Noor University (PNU) in 2020 as a first-ranked student. In 2023, he completed his MSc degree in Organic Chemistry with a GPA of 4 out of 4 from Babol Noshirvani University of Technology (BNUT). After graduating with MSc degree, he immediately joined prof. Ramazani's group at the University of Zanjan (ZNU) as a research assistant to increase his knowledge in the field of organic synthesis. Hosseininezhad's research activities are focused on CO and CO2 conversion, catalysts, and organic synthesis. Throughout his career, he has authored or co-authored over 15 scientific paper and book chapter. He is the main collaborator in a joint international project between the Chinese Academy of Sciences and the Iran National Science Foundation (The Silk Road Scientific Fund (SRSF)). He is also an international referee in some international journals and conferences. In addition, he is a member of the scientific core of organic chemistry at Farhangian University and a student member of the Royal Society of Chemistry (RSC). Google Scholar: https://scholar.google.com/citations?user=M1ZAuSUAAAAJ%26hl=en%26oi=ao. |
 Sina Pirani Ahmad Abad | Sina Pirani Ahmad Abad obtained his BSc in Pure Chemistry from Payam Noor University (PNU) in 2022. He then earned his MSc in Organic Chemistry with a GPA of 4.0 out of 4.0 from the University of Zanjan (ZNU) in 2024, under the supervision of Prof. Ali Ramazani and Prof. Ali Morsali. His research focuses on Nanochemistry, specifically Metal–Organic Frameworks (MOFs) and their medical applications, as well as Medicinal Chemistry and organic synthesis. https://scholar.google.com/citations?user=qh9k8G8AAAAJ%26hl=en. |
 Ali Ramazani | Professor Dr Ali Ramazani completed his PhD under the supervision of Professor Issa Yavari in the Department of Chemistry at the Tarbiat Modares University (TMU) in Tehran, Iran. He currently works as a Full Professor in Chemistry at the University of Zanjan in Zanjan, Iran. His studies are focused on organic synthesis, medicinal chemistry and nanochemistry and he has published more than 600 papers and patents. He is an Editorial Board Member of the international Journal Nanochemistry Research. He has received several national and international awards, including the 2013 Khwarizmi International Award, several top-cited author awards and best-paper awards from leading ISI Journals, Best Researcher Awards, and the Best Lecturer Awards at the University of Zanjan. Google Scholar: https://scholar.google.co.in/citations?user=XDWRu9gAAAAJ%26hl=en. |
1 Introduction
In recent decades, heterocyclic compounds have garnered significant interest for various reasons, with their biological activity and their application as drug molecules being particularly noteworthy. Additionally, these compounds are recognized for their presence in a wide array of functional molecules, natural products, organic materials, and pharmaceuticals.1–4 Quinolines represent a significant class of nitrogen-containing heterocyclic aromatic compounds. Recently, researchers have increasingly focused on quinolines due to their extensive array of biological activities and their diverse range of applications. Quinoline is predominantly derived from several primary sources, including petroleum, the processing of coal, the preservation of wood, and the extraction of oil from shale. Additionally, derivatives of quinoline are found in various natural products, particularly in alkaloids. This versatile heterocyclic structure is prevalent in numerous areas, including natural products, pharmaceuticals, agrochemicals, functional materials, and as ligands in transition metal catalysts (Fig. 1).5–7 | ||
| Fig. 1 Compounds that exemplify the quinoline framework demonstrate various properties. | ||
Indole is a prevalent heterocyclic structure commonly encountered in nature. It holds an important position in the formulation of a diverse array of dyes, fragrances, pharmaceuticals, and agricultural chemicals (Fig. 2). Consequently, the synthesis of the indole core and its subsequent functionalization represent critical areas of research within heterocyclic chemistry. This topic has garnered the interest of synthetic organic chemists for more than 150 years.8
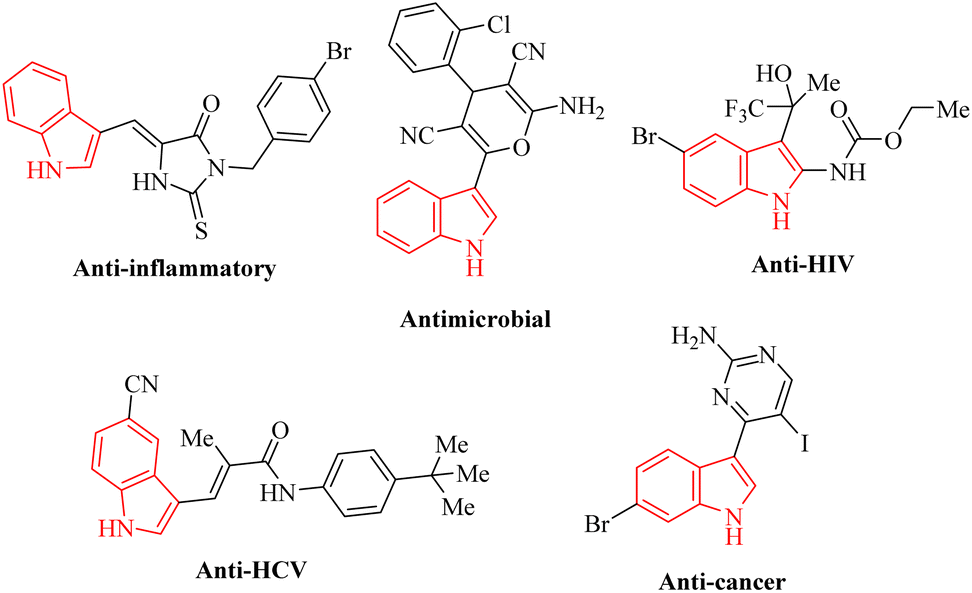 | ||
| Fig. 2 Compounds that illustrate the indole structure exhibit a diverse range of properties. | ||
In addition, cross-coupling reactions catalyzed by transition metals that utilize C–H activation present an alternative approach to conventional synthetic methods. Recently, these reactions have gained significant importance for the formation of C–C and C–N bonds.5–18
Au catalysis has become a convenient method for creating diverse and complex molecules.19,20 These catalysts have demonstrated significant utility in activating π systems, including alkynes and allene derivatives, for nucleophile addition.21 Much of the synthetic chemistry in this field is closely associated with the Lewis acidic behavior of electrophilic Au species.22–26 Apart from its acidic properties, Au can act as an electron donor, which helps stabilize intermediate cationic species and enhances reaction pathways that cannot be promoted by other Lewis acids.27 In the Au(I)-catalyzed reaction between an alkyne and an azide, the dual reactivity of the pi acid/electron donor is emphasized. This process involves nucleophilic azide addition, leading to the generation of an alpha-imino Au carbene A, followed by the Au-assisted expulsion of N2 (Fig. 3I).21 Toste and colleagues harnessed this reactivity pattern to create an Au(I)-catalyzed intramolecular acetylenic Schmidt reaction. In this process, a homopropargylazide B is transformed into a pyrrole C (Fig. 3II).28 Recent studies have extensively examined Au-catalyzed tandem reactions, where Au-carbenoid species29–35 act as key intermediates in reactions involving diyne derivatives,36–39 enyne derivatives,40–42 and alkyne derivatives with N-oxides,43–48 as well as in the rearrangement of 1,2-propargylic ester derivatives.49,50 However, the in situ generation of α-imino Au carbenoids from azide derivatives and alkyne derivatives remains largely unexplored.51–65 Specifically, alkyne derivatives can act as sources of carbenes when they react with nucleophiles that have a leaving group, under the catalysis of Au or Pt (Fig. 3III).43,51 Alkyne derivatives have various applications in the transformation of organic compounds2 and can also act as starting materials for producing alpha-oxo and alpha-imino Au carbenes, which participate in common carbene reactions like 1,2-migration, X–H bond insertion, ylide formation, cyclopropanation, and more.66 When using 2-alkynyl arylazide derivatives as substrates, the resulting Pd carbene can participate in cross-coupling reactions to synthesize polysubstituted indoles,67–72 as illustrated in Fig. 3IV. While the formation of Pd carbene from alkynes is documented.73–75 Complexes of Ag(I) are commonly employed as stoichiometric oxidants for oxidizing a range of organic and inorganic substrates. Numerous studies have utilized the complexes of Ag(I) as catalysts in oxidation and group-transfer reactions.76–82 Recent research has demonstrated that Ag species display notable catalytic activities, acting as a transition metal catalyst.76,83–96 Ag catalysts, when viewed as transition metals, are generally seen as less efficient and not as effective as other late transition metals.97 Cyclization of 2-alkynylbenzyl azide catalyzed by Ag and Au have been shown in Fig. 3V and VI. Using Cu catalysts is significantly more attractive for forming Cu carbene intermediates or activating alkyne species due to their lower cost, reduced toxicity, and greater availability.98 Common transformations in this field encompass alkynylation,99–103 cycloaddition,104–108 allene formation,109–113 among various others.114–118
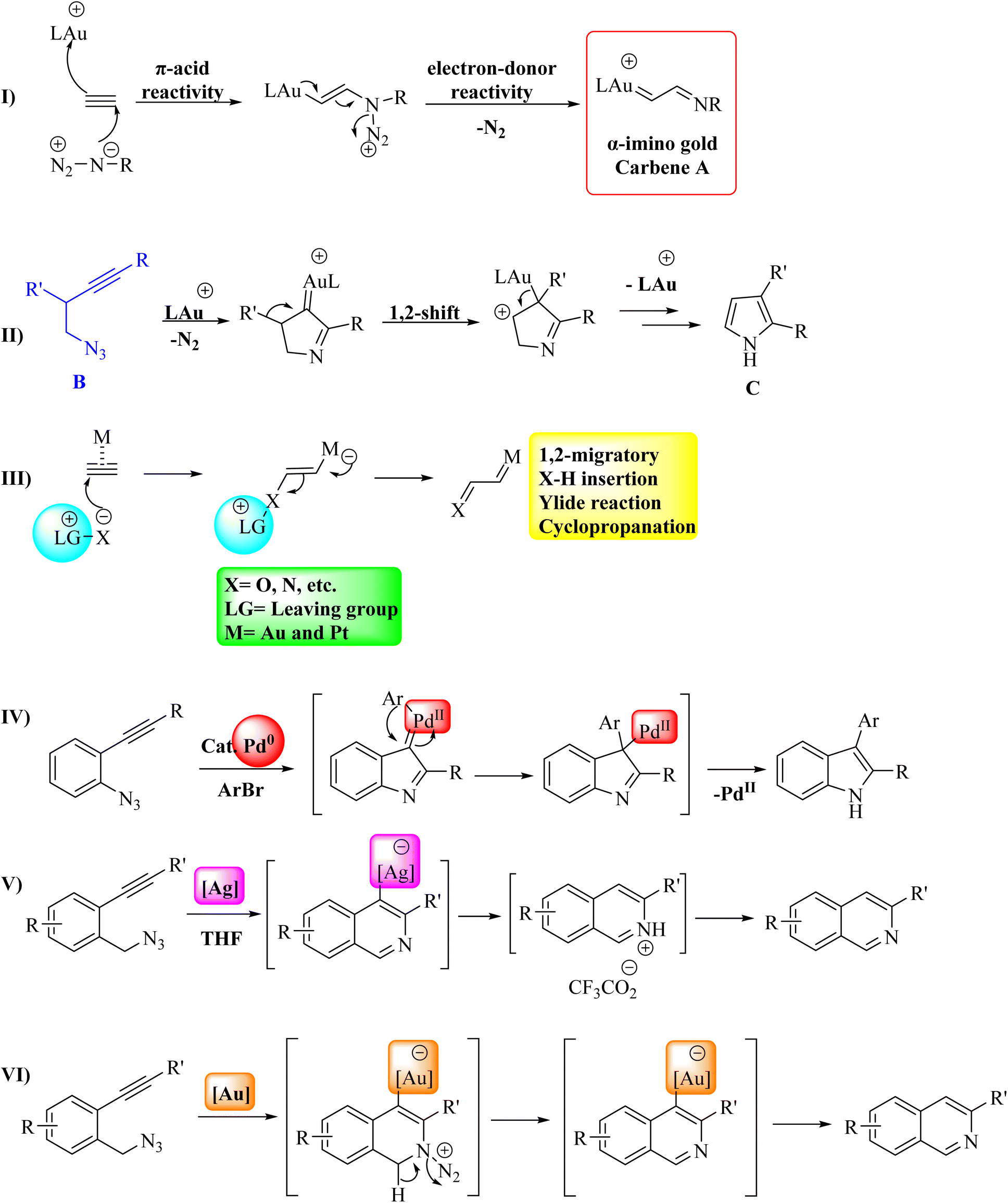 | ||
| Fig. 3 (I) The design of synthetic methods to capture α-imino gold carbene intermediates, (II) Au(I)-catalyzed intramolecular acetylenic Schmidt reaction, (III) alkynes as sources for metal carbene formation, (IV) the suggested method for synthesizing indole catalyzed by Pd, (V) cyclization of 2-alkynylbenzyl azide by Ag catalyst. (VI) Cyclization of 2-alkynylbenzyl azide by Au catalyst. | ||
Given the widespread use of 2-alkynyl aryl/benzyl azide derivatives in organic transformations, we explore the fascinating conversions of 2-alkynyl aryl and benzyl azide derivatives into indole derivatives and quinoline derivatives, focusing on the use of metal catalysts like Au, Cu, Rh, Pd, and Ag. Also, we highlight the diverse range of substrates used, significant breakthroughs in the field, and the challenges faced by chemists. Furthermore, we delve into the mechanisms behind these important reactions to improve understanding and inspire future exploration in this vibrant area of research.
2 2-Alkynylaryl azides
2.1 Synthesis of indoles
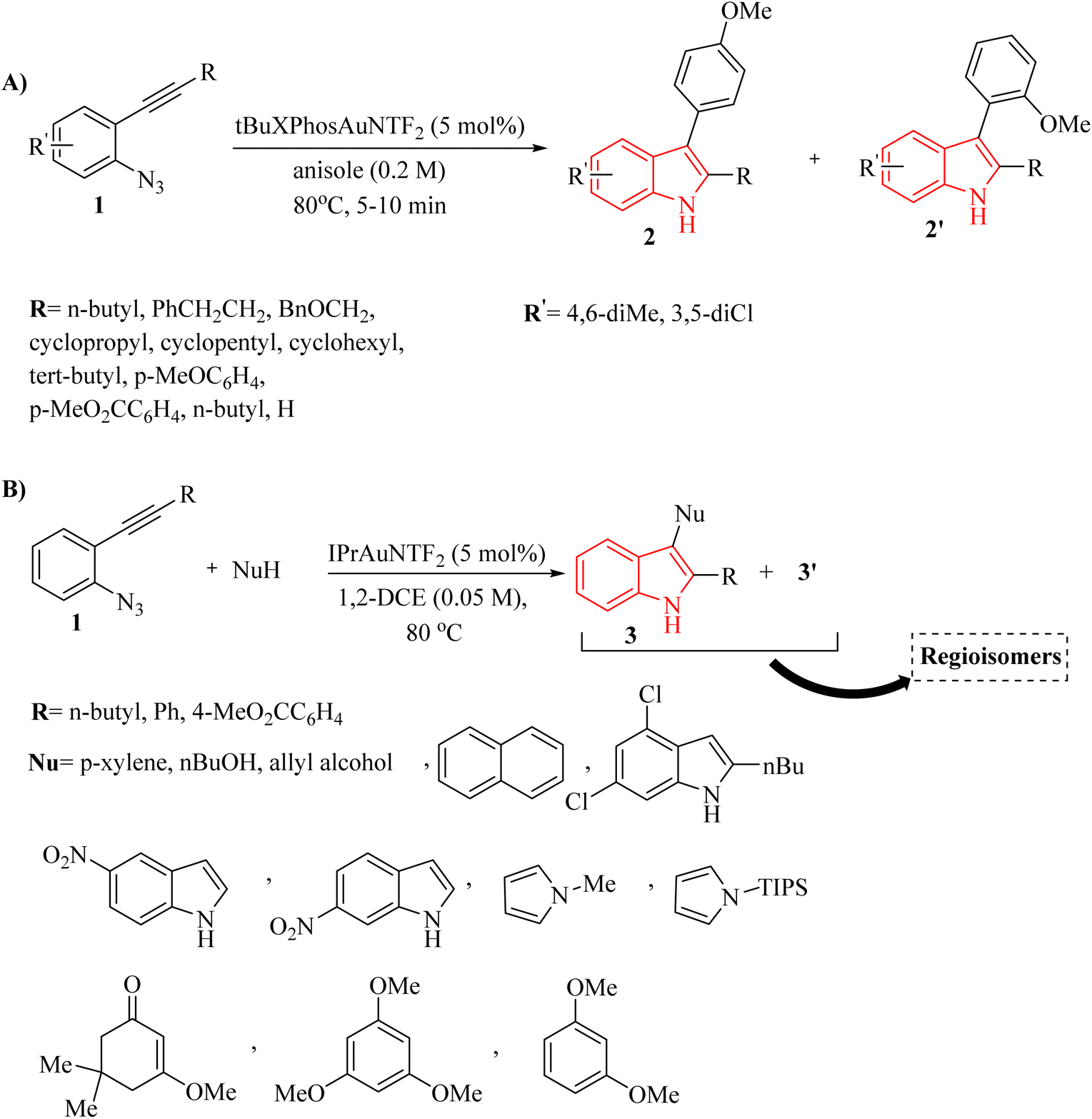 | ||
| Scheme 1 (A) The range of o-azidoarylalkyne substrates 1, (B) the range of various nucleophiles. | ||
A mechanism underlying this transformation is depicted in Scheme 2. The formation of II/III at a higher temperature (e.g., 80 °C) is facilitated while its precursor (I) at a lower temperature (e.g., −20 °C) may persist. Then, it plays a role by reacting with nucleophile derivatives. This reaction is carried out via an SN2′ process. When the reaction is proceeded via II/III, it showed greater regioselectivity due to the short bond of Au–C length. It should be noted that Au–C bond length in I longer than the bond of Au–C in II/III.119
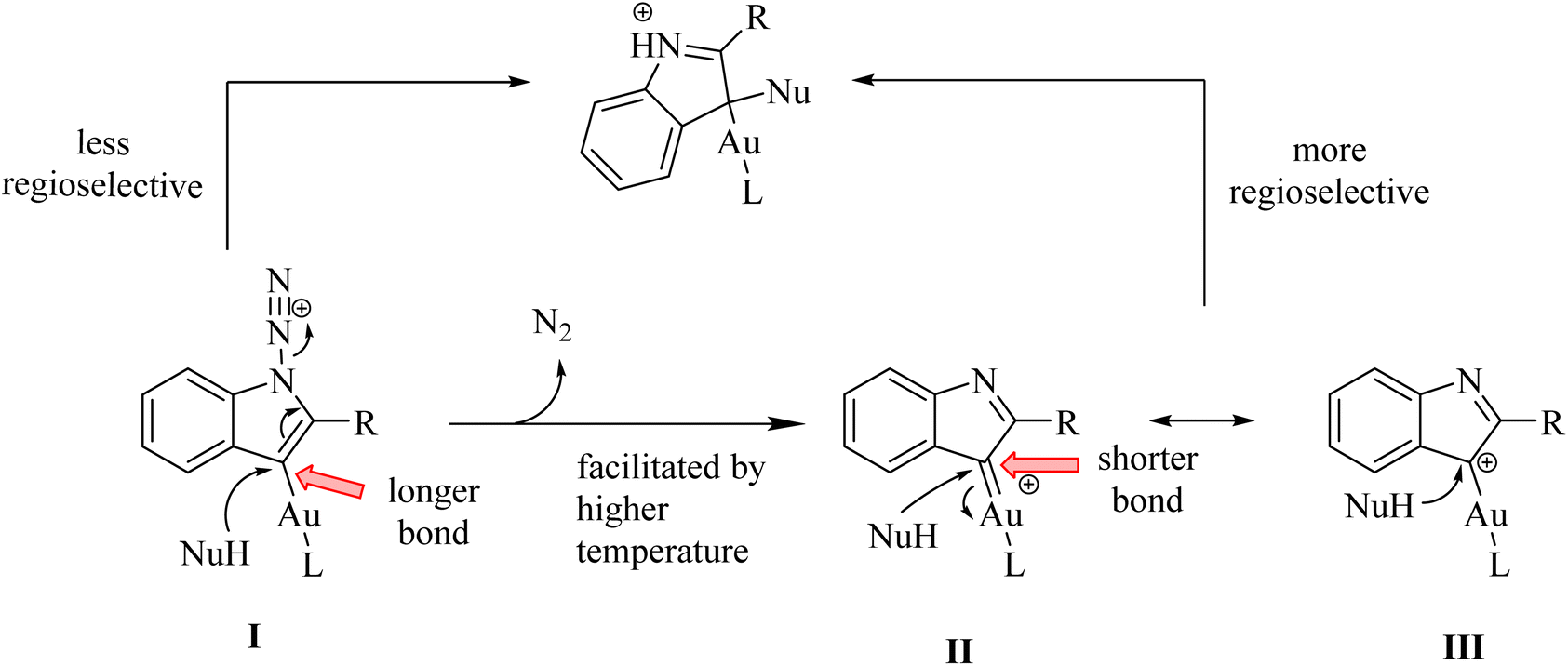 | ||
| Scheme 2 The inverse correlation between regioselectivities and reaction temperatures. | ||
In 2011, Wetzel and Gagosz120 reported a novel Au(I)-catalyzed reaction for the conversion of 2-alkynyl arylazide derivatives 4 into indolin-3-one derivatives 6 (Scheme 3A) and 3-substituted indole derivatives 7 (Scheme 3B). This reaction, conducted in 1,2-DCE at 50–60 °C, was demonstrated rapid and efficient performance, while also exhibiting tolerance towards various functional groups. Notably, the potential to generate indolin-3-one derivatives containing two vicinal asymmetric quaternary carbon canters is of particular interest due to its prospective utility in the synthesis of pseudoindoxyl alkaloid derivatives. The desired products gave with impressive yields of up to 99%. When allylic alcohol derivatives with substitutions at the C3-position were employed, an extra asymmetric canter was formed. When phenol was tested, it did not lead to the formation of the desired product. In an unexpected result, even the poorly nucleophilic tert-butanol formed the corresponding product. The desired reaction had some challenges such as (1) difficulty in isolating expected products during reactions, (2) because of steric effects there was moderate selectivity in Claisen products, and (3) inability to observe intermediates via the reaction monitoring. There were inadequate studies on reaction mechanisms for type of nucleophiles as well as a limited understanding of selectivity in Claisen rearrangements.
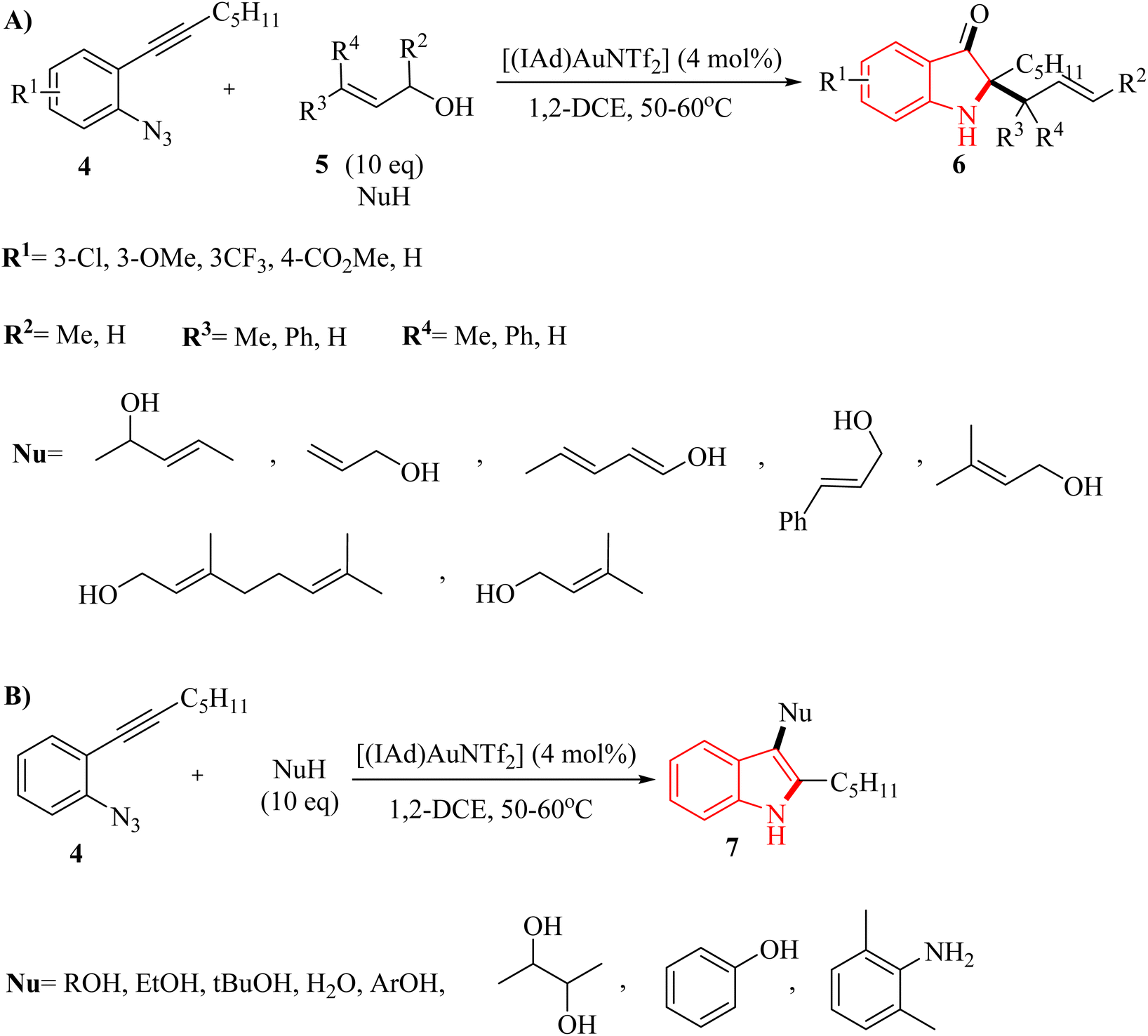 | ||
| Scheme 3 (A) Range of substrates of 2-alkynyl arylazide derivatives 4 and 5, (B) generation of 3-substituted indole derivatives 7. | ||
A mechanism underlying this transformation is depicted in Scheme 4. The Au(I) complex can activate the alkyne moiety in compound I. After the extrusion of N2, this activation may result in the formation of an intermediate alpha-imino Au carbene, denoted as compound II. The species undergoes a nucleophilic addition, resulting in the formation of a product labelled as III. Indole 7 could be generated from compound V through two distinct pathways. First, it can form via iminium intermediate IV using a prototropy/demetalation sequence. Alternatively, it can also be produced directly from compound V through a protodemetalation/tautomerization sequence. Through a direct insertion of II into the Nu–H bond, the intermediate V is generated. In the following, when an allylic alcohol is acted as the nucleophile, compound 6 is formed, which could be explained by a Claisen rearrangement of compound VI. Compound 6 could also be generated through an alternative pathway, starting from IV and proceeding via intermediate IX.120
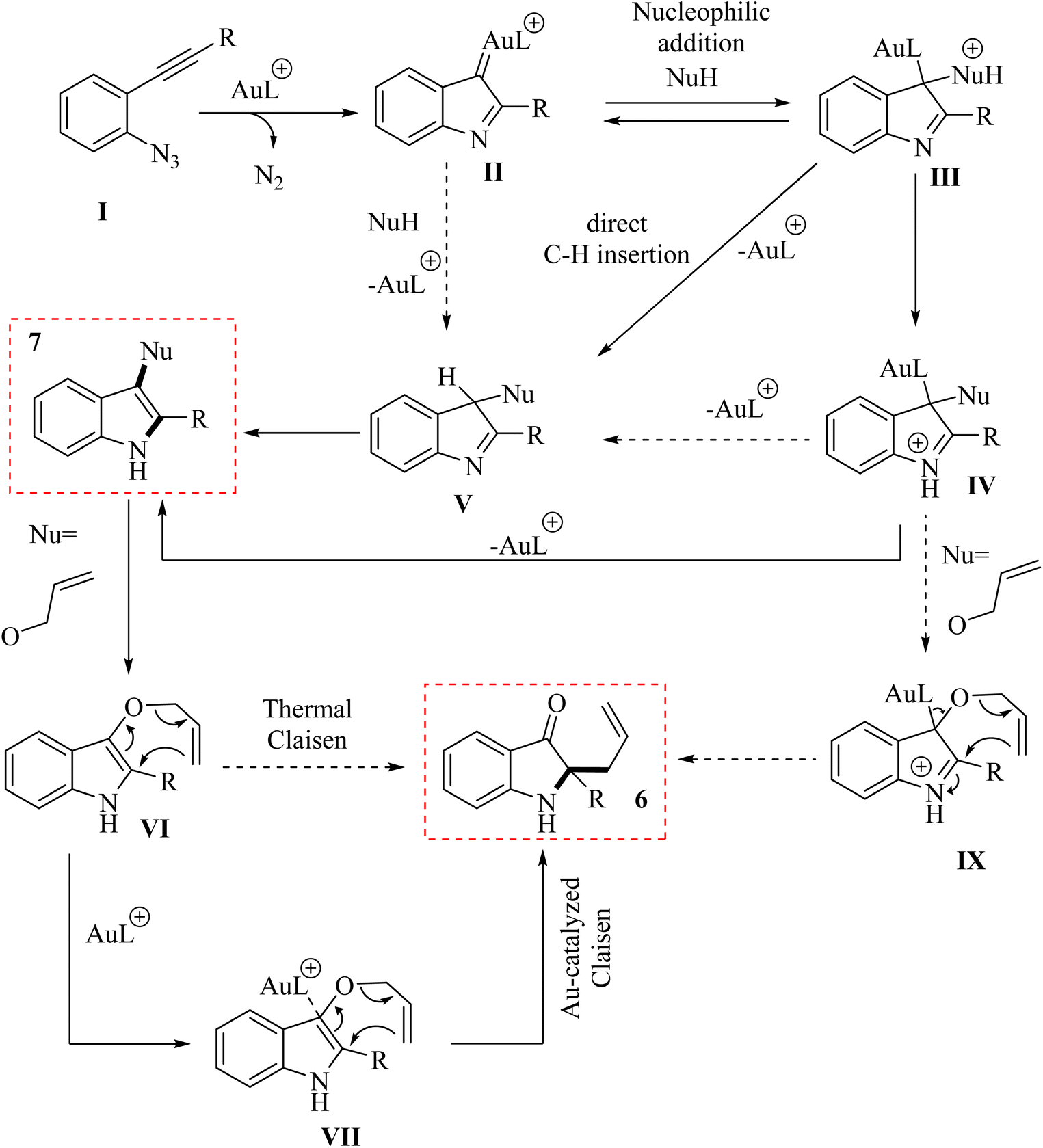 | ||
| Scheme 4 A mechanism for the Au-catalyzed 4 into indole derivatives (7 and 6). | ||
Zhang et al.121 introduced a study detailing an effective Au-catalyzed (XPhosAuNTf2) chemoselective synthetic pathway to produce 4H-1,3-dioxine derivatives 10 (Scheme 5A) and indole derivatives 11 (Scheme 5B). This process involved the tandem annulation of 8 with aldehyde derivatives 9. Notably, the reactions exhibited high efficiency and precise control over product chemoselectivity when various 8 were used in conjunction with desired Au catalyst in DCE at 0–60 °C. In the case of 8, the desired products generated in 34–75% yields. Nevertheless, when cyclopropanecarbaldehyde, 4-(2-azidophenyl)but-3-yn-1-ol, and benzaldehyde were employed as substrates, they generated a blend of decomposition products that remained unidentified, even after analyzing the crude mixture with 1H NMR. Authors observed that the 10 was not produced when starting material (ArN3), which contained 3,5-dichloro-substituted groups on the aryl ring, underwent the standard reaction conditions. In the case of 11, the desired products gave in 54–94% yields. Benzaldehyde was also investigated; however, it yielded a blend of decomposition products that remained unidentified. There were some gaps regarding the reaction, such as (1) inadequate understanding of product chemoselectivity based on substituent groups, (2) limited investigation of alpha-imino Au carbenoids from azide derivatives and alkyne derivatives. It should be noted that decomposition products were observed with certain substrates and conditions.
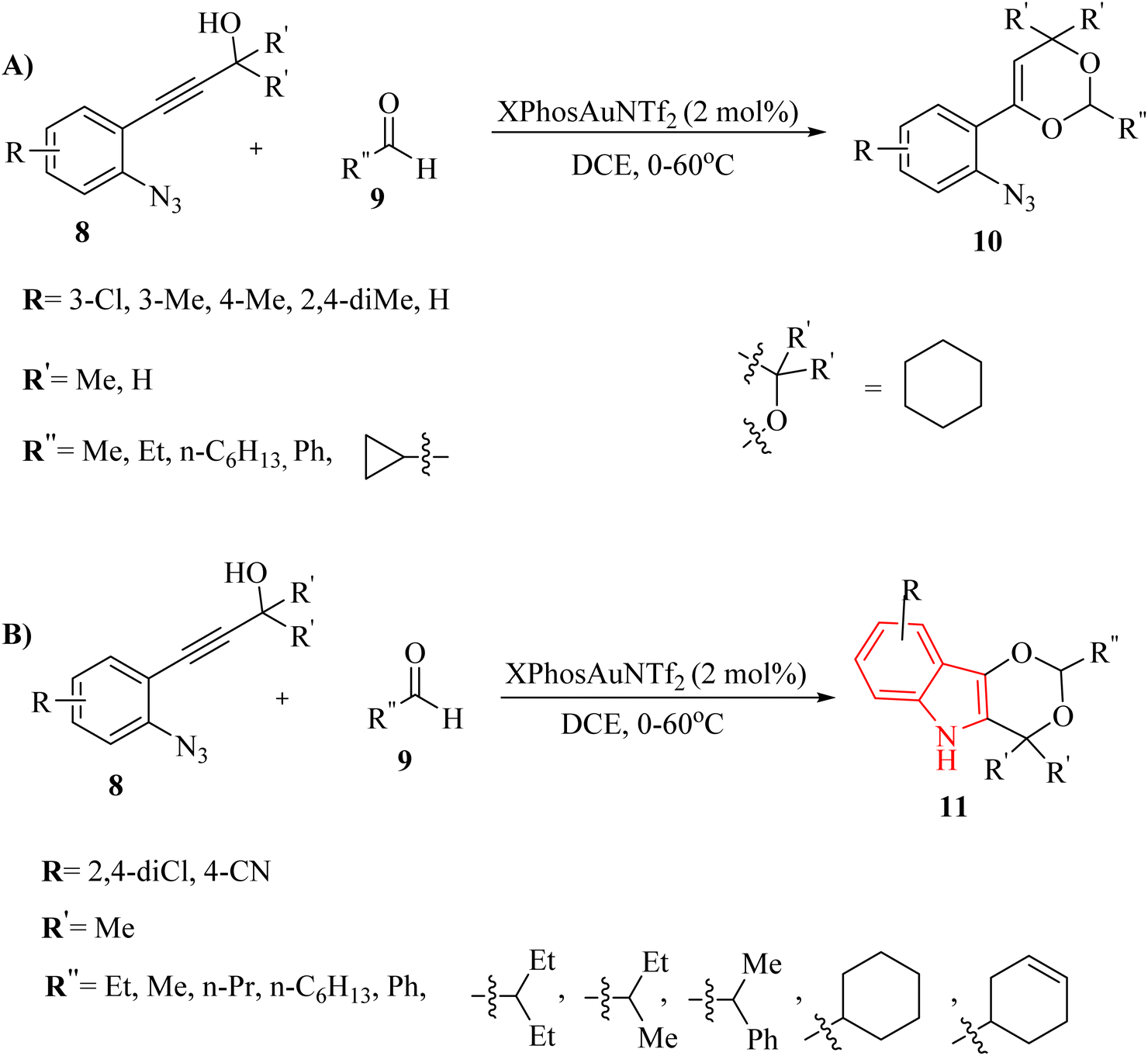 | ||
| Scheme 5 (A) Generation of dioxine derivatives 10, (B) generation of dioxino-indole derivatives 11. | ||
A proposed mechanism underlying this transformation is depicted in Scheme 6. This process may entail the activation of compound 8 by coordinating with an Au catalyst through its alkyne moiety. Subsequently, compound 9 attacks to form complex I, which then undergoes intramolecular alkoxylation to yield II. Finally, a protodemetalation step leads to the formation compound 10. When 8 is used as the substrate the intermediate IV is produced in situ. This intermediate is subsequently trapped by 9, resulting in the formation of V. Following intramolecular alkoxylation of V, the tricyclic ring–Au complex VI is emerged. Finally, a subsequent protodemetalation step leads to the production of compound 11.121
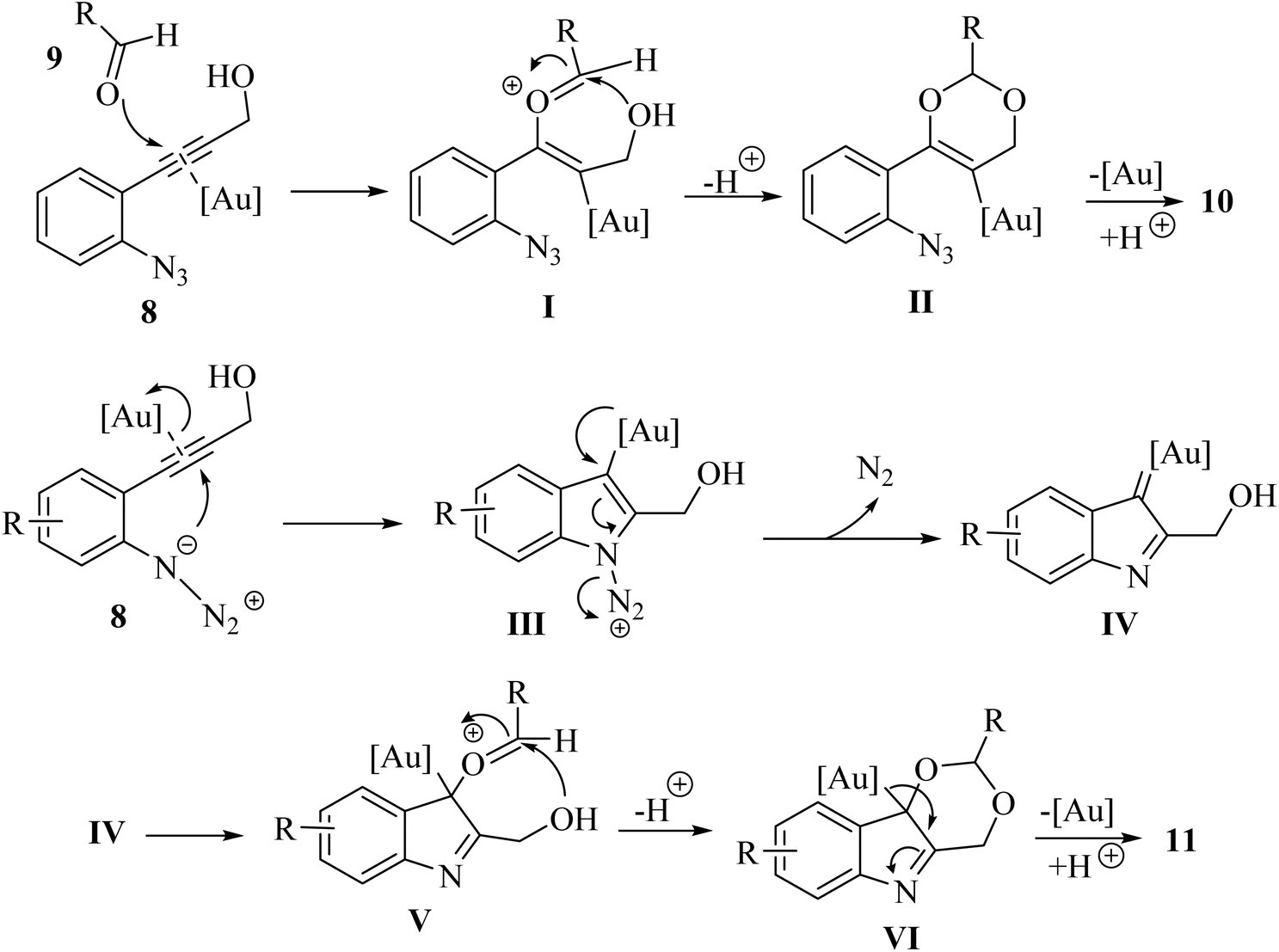 | ||
| Scheme 6 A proposed mechanism for the reaction catalyzed by Au between compounds 8 and 9. | ||
Zhang et al.122 introduced a notable discovery in the field of organic synthesis. Authors illustrated an unforeseen Au-catalyzed (tBuXPhosAuNTf2) method for producing indole derivatives through the ring-opening reaction of 2-alkynyl arylazide derivatives 12 with oxygen-containing heterocycles 13, in the presence of methanesulfonic acid in 1,4-dioxane at 60 °C (Scheme 7). This innovative approach exhibited a remarkable tolerance for a diverse range of substrates of 12, which can be conveniently prepared in just two steps from readily available materials. Furthermore, the Au-catalysed tandem reactions proved to be practical, as they did not necessitate inert or moisture-free conditions. The corresponding products generated in 28–95% yields. However, when using substrates containing a cyclopropyl group or a terminal alkyne moiety, and reacting compound 12 with oxetane, tetrahydrofuran, or 2-phenyloxirane, only a mixture of unknown side products was obtained. These products could not be identified. There were some challenges regarding the reaction, such as (1) some substrates were yielded unknown side products, (2) inert and moisture-free conditions were not always practical, and (3) the reaction was required specific conditions for optimal yields. It should be noted that the mechanism of Au-catalyzed reactions remains speculative.
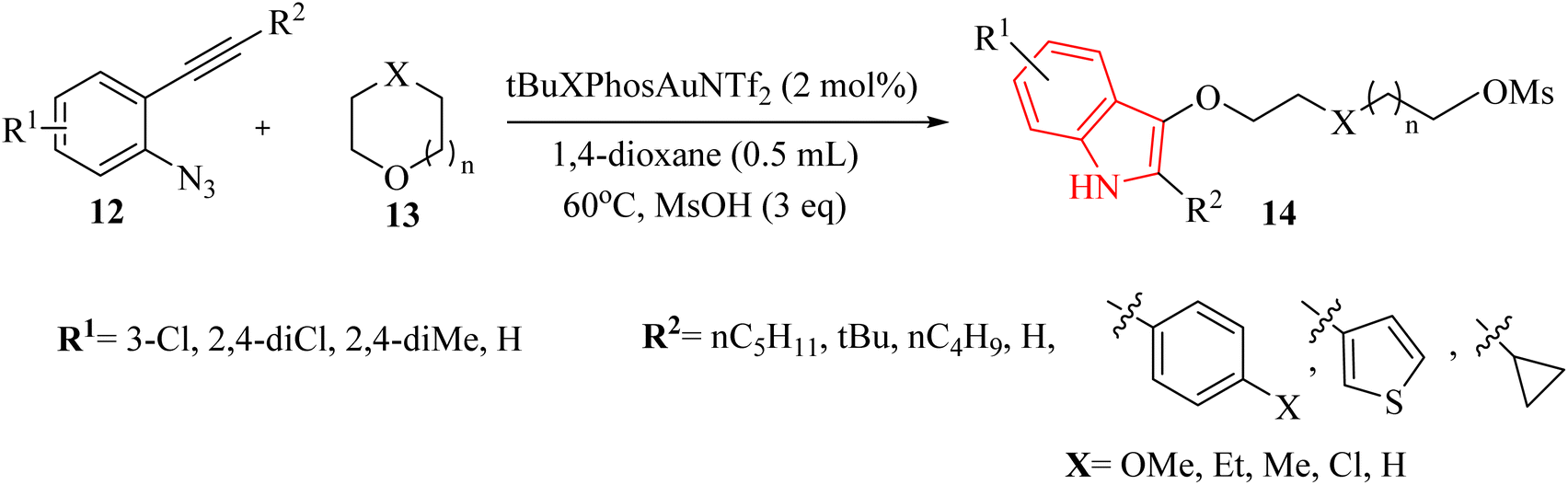 | ||
| Scheme 7 Scope of 2-alkynyl arylazide derivatives 12 and oxygen-containing heterocycles 13. | ||
A mechanism underlying this transformation is depicted in Scheme 8. In this process, 12 is activated by coordinating it with an Au catalyst. The azide attacks the alkyne, forming intermediate I. Subsequently, alpha-imino Au carbene II is generated after N2 expulsion, facilitates by the Au catalyst. This intermediate is further attacked by 1,4-dioxane, resulting in the release of intermediate III. Finally, ring opening of III using MsOH is produced complex IV, which undergoes a protodemetalation step to yield 14.122
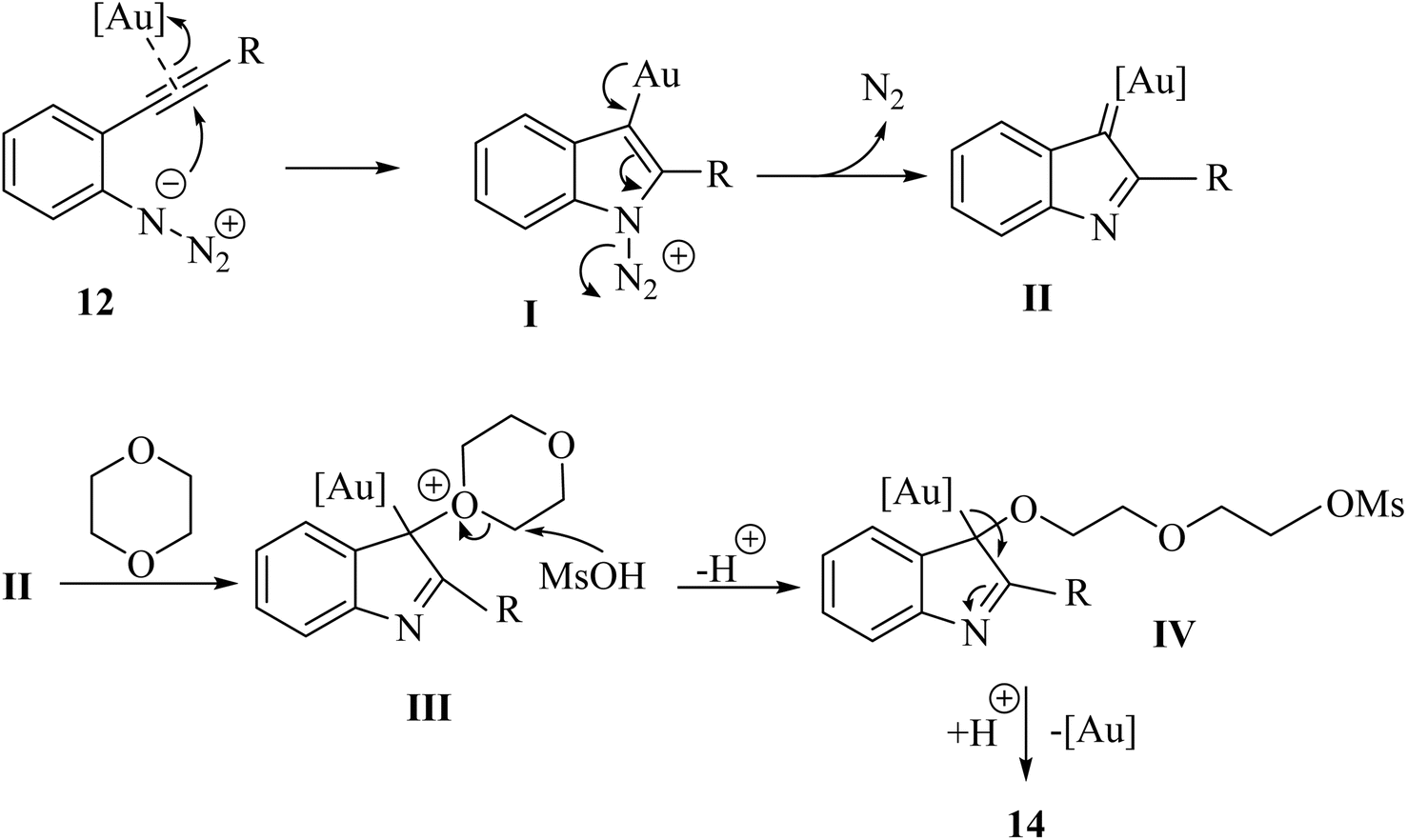 | ||
| Scheme 8 A mechanism for the Au-catalyzed ring-opening reactions of 12 with 13. | ||
Li et al.123 introduced a novel synthetic approach, catalysed by Au, for the efficient formation of aryl-fused carbazole derivatives 20. This method starts with 2-alkynyl arylazide derivatives 15 and alkyne derivatives 16 and progresses through a series of reactions (Scheme 9). The process includes cyclopropenation followed by an intramolecular reaction similar to the Friedel–Crafts reaction involving metal carbene and arene. This sequence was facilitated by two distinct Au carbene intermediates and occurred in a DCE/H2O mixture at a temperature of 60 °C. The results illustrated that this reaction can accommodate diverse azido alkyne derivatives (up to 85% yield) and alkyne derivatives (up to 70% yield). The tolerance of the Br group, which was typically incompatible with Pd-catalysed methodologies for carbazole synthesis, provided an opportunity to modulate the structure of related products through cross-coupling reactions, thereby enhancing their structural complexity. In the mentioned reaction aliphatic alkyne derivatives could not undergo the reaction. There were some challenges in the mentioned reaction, such as (1) the role of H2O in reactions was not fully understood, (2) inadequate investigation of aliphatic alkyne derivatives in reactions.
 | ||
| Scheme 9 Scope of azido alkyne derivatives 15 and alkyne derivatives 16. | ||
A mechanism underlying this transformation is depicted in Scheme 10. Initially, 15 is generated the alpha-imino Au carbene I, which could undergo cyclopropenation with an acetylene to yield II. Compound II in the presence of the desired catalyst is transformed into intermediate III (Au carbene). Subsequent IV is generated after an intramolecular metal carbene/arene Friedel–Crafts-type reactions, which after protonation and tautomerization then is converted to compound 17. In addition to the main reaction, a potential side reaction might be occurring, leading to the formation of compound X through the alkynyl C–H insertion of I. However, a control experiment revealed that pre-synthesized compound X did not convert into 17 under the given reaction conditions.123
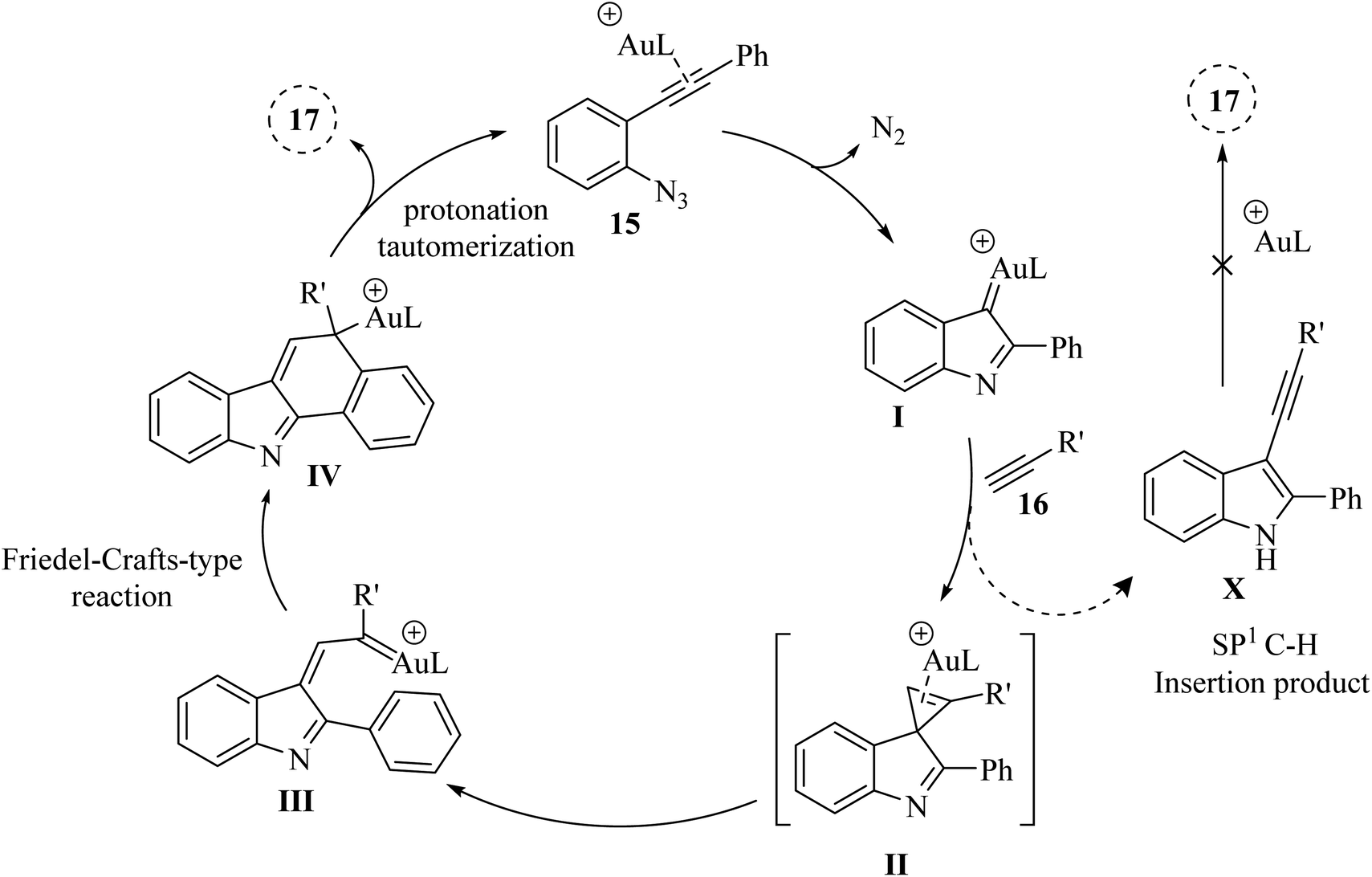 | ||
| Scheme 10 A mechanism for the cyclization reactions of 15 with 16 catalyzed by Au. | ||
In 2020, Li et al.124 developed a groundbreaking technique for synthesizing N1 and N2-indol-3-yl 1,2,3-triazole compounds (20 and 20′, respectively). This was achieved using an Au-catalyzed (IPrAuCl/AgNTf2) cascade reaction that combines ortho-alkynyl arylazide derivatives 18 with 1,2,3-triazole derivatives 19 (Scheme 11). The process took place in 1,2-DCE at 80 °C under Ar. During this reaction, an amino Au carbene intermediate was formed in situ and then captured by different triazole molecules. The reaction tends to preferentially undergo N1-selective nucleophilic attack, resulting in moderate to high selectivity between N1 and N2 products. There were some challenges regarding the reaction, such as (1) ineffective performance of type of Ag salts with respect to AgNTf2, and (2) temperature and solvent adjustments did not improve reaction yields.
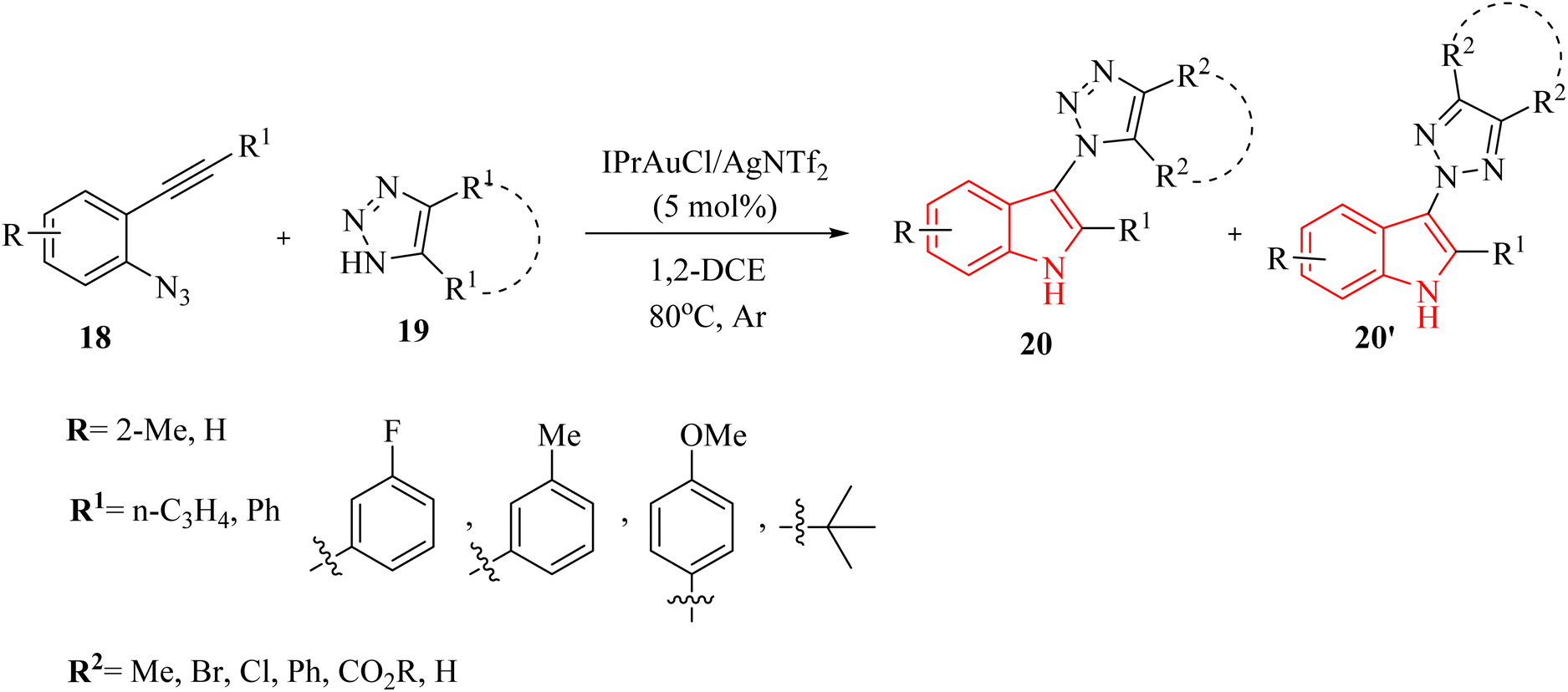 | ||
| Scheme 11 Scope of 2-alkynyl arylazide derivatives 18 and benzotriazole derivatives 19. | ||
A mechanism underlying this transformation is depicted in Scheme 12. Initially, 18 is generated the alpha-imino Au carbene I which could be intercepted by azole nucleophile derivatives. N1-selective nucleophilic attack is favored when compound 19 is used due to the relatively higher electron density at the N1 nitrogen with respect to the internal N2 nitrogen.124
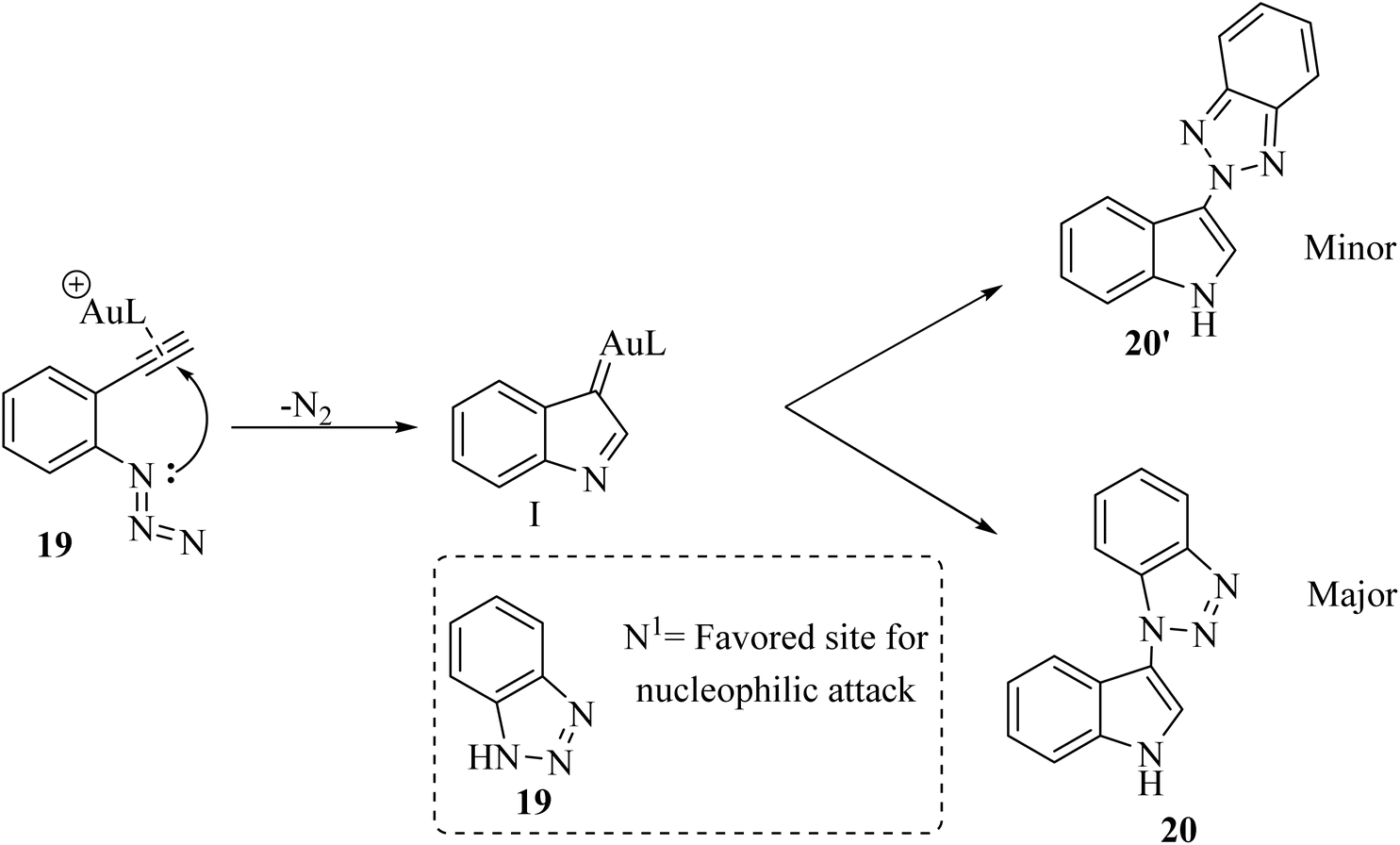 | ||
| Scheme 12 A proposed mechanism for the reaction of 18 with 19 catalyzed by Au. | ||
In 2023, Xie et al.125 devised an innovative method for directly creating alpha-(3-indolyl) ketone derivatives 23 (Scheme 13). This method involved an Au-catalyzed (tBuXPhosAuBF4) cascade reaction that combines 2-alkynyl aryl azide derivatives 21 with enecarbamate derivatives 22, which allowed for the umpolung (reversal of polarity) of the indole's 3-position. The process was highly efficient, utilizing a catalyst in DCE at 45 °C under a N2 atmosphere. This technique offered a straightforward and alternative pathway for the direct production of alpha-(3-indolyl) ketone derivatives. In the case of 21 derivatives, the desired products generated in 21–85% yields, as well as derivatives of 22 produced the desired products in 10–61% yields. In this study, various terminal alkynyl derivatives were evaluated. Interestingly, the transformation was compatible with both electron-donating and -withdrawing groups on the phenyl group, resulting in the corresponding products. In the case of the derivatives of 22, the experimental findings illustrated that the electronic characteristics of the substituents had a significant effect on the product yields. Notably, the presence of a potent electron-donating substituent (such as the methoxy group) markedly enhanced the reaction. In the case of the reaction limitations, there was inadequate evaluation of reaction scalability beyond 1.0 mmol scale as well as required to comparative studies with other catalytic systems. It should be noted that steric bulkiness affected reaction outcomes with certain substituents.
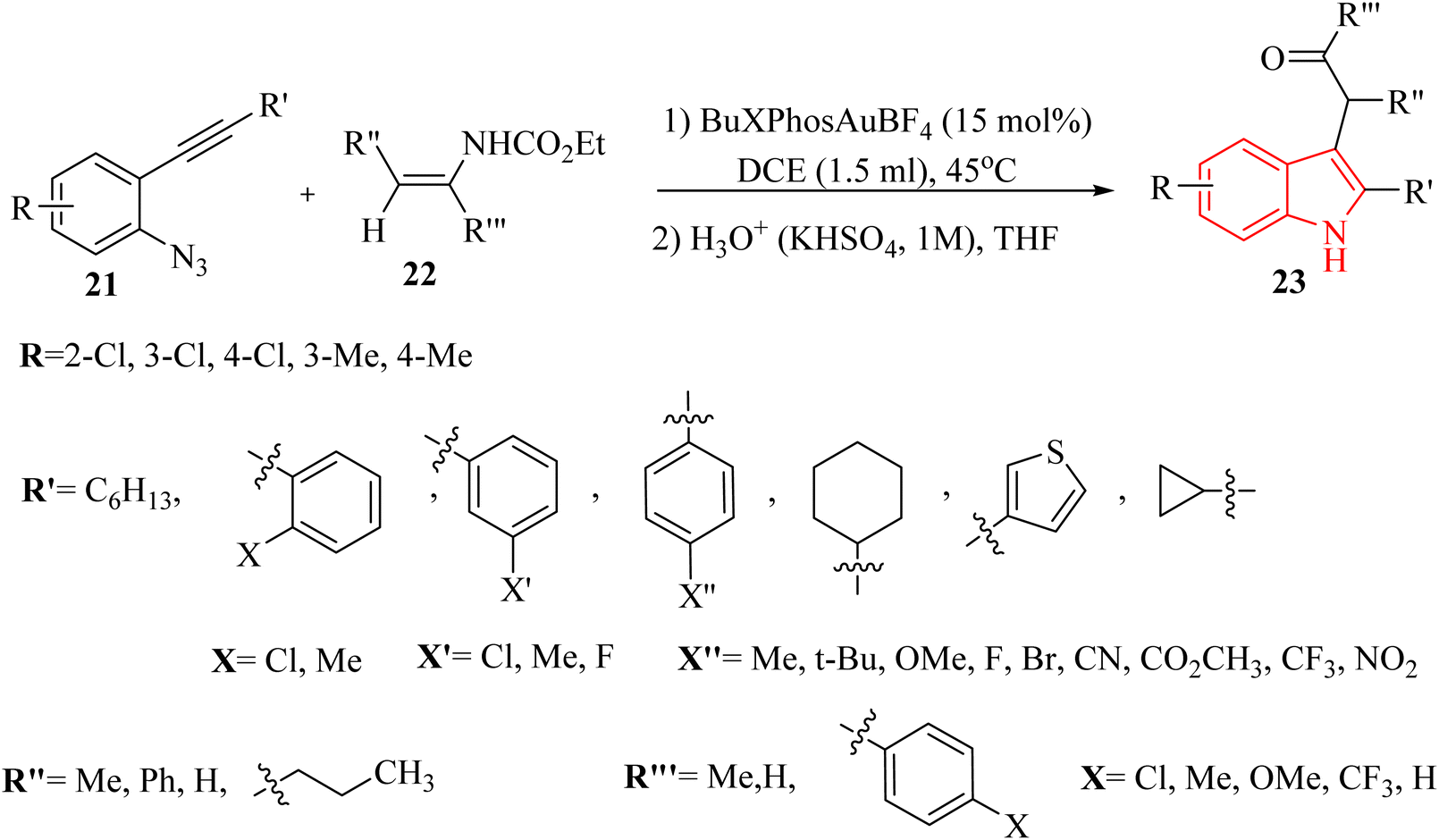 | ||
| Scheme 13 Scope of 2-(alkynyl)phenyl azide derivatives 21 and enecarbamate derivatives 22. | ||
In 2024, Zhu et al.126 reported innovative an Au-catalyzed method to synthesize indole derivatives 25 and indolinone derivatives 26 (Scheme 14). This was achieved by trapping Au carbene intermediates with nitriles and water in DCE at 110 °C. The critical intermediate, bata-sulfonamido-alpha-imino Au carbene, was produced via Au-catalyzed cyclization of N-(2-azidophenyl-ynyl)-methanesulfonamides. This intermediate then engaged in a formal [4 + 2] cascade annulation with nitrile derivatives and an intramolecular SN2′ type reaction with H2O, led to the formation of 25 and 26 with high yields. Additionally, the created 25 exhibited notable fluorescence characteristics. This study explored the applicability of Au-catalyzed [4 + 2]-annulation reactions involving diverse derivatives of 24 in conjunction with acetonitrile. According to this process, the substrate with an unreactive CN group which not involved in the annulation reaction, led to the corresponding product. In the following, using 5 equivalents of the nitrile in DCE, the corresponding products were obtained with a satisfactory yield. There were some limitations and challenges regarding the reaction, such as (1) the scalability of the technique was required further investigation, (2) limited evaluations on diverse nitrile substrates was presented, (3) mechanistic details of Au carbene intermediates were required more clarity, and (4) effects of types of solvents on reaction outcomes were not investigated.
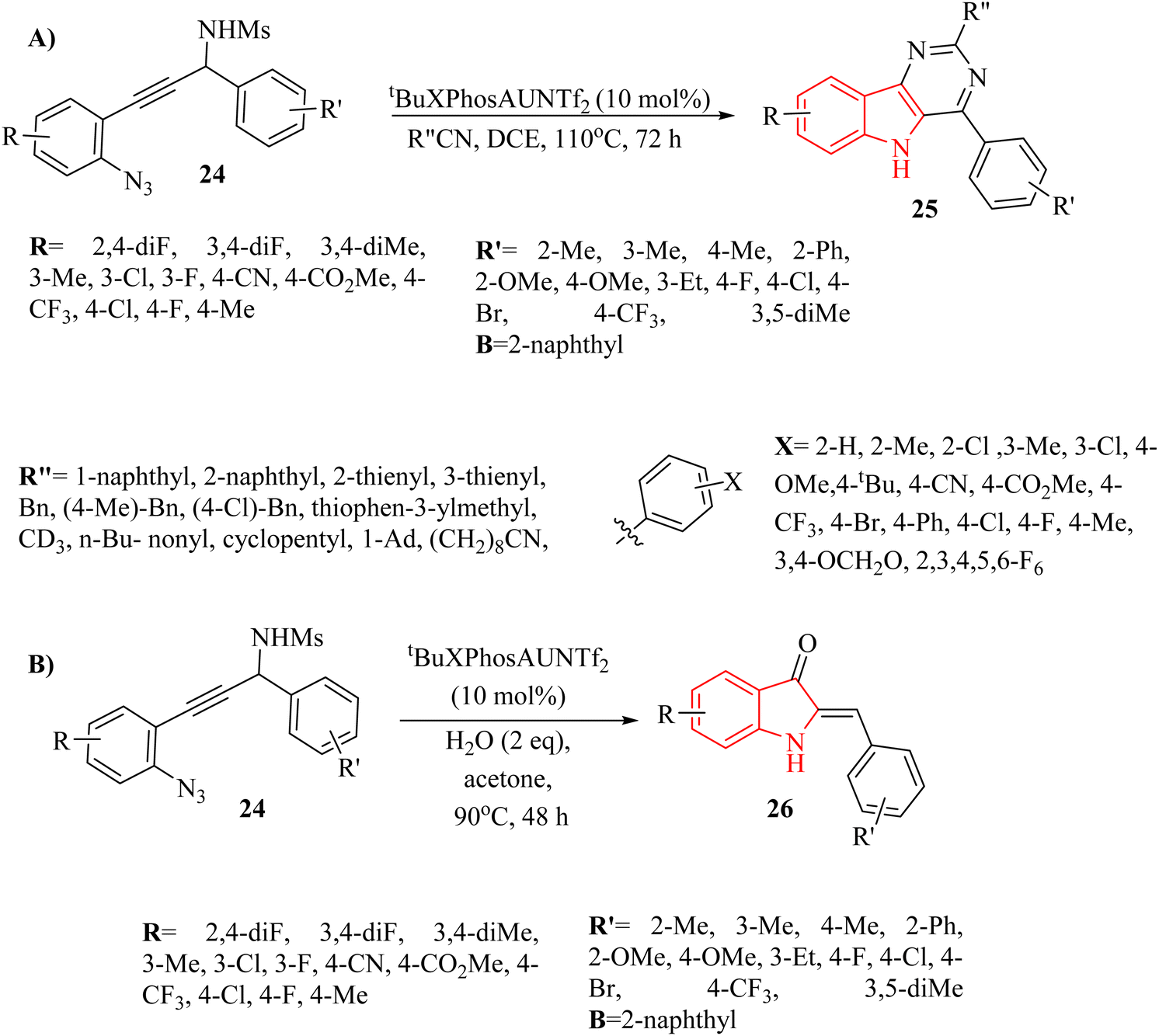 | ||
| Scheme 14 (A) Generation of indoles 25 and (B) 2-benzylideneindolin-3-one derivatives 26. | ||
A mechanism underlying this transformation is depicted in Scheme 15. Firstly, the Au catalyst is coordinated with the alkyne by π-coordination. Then, intermediate II is formed via the triggers a cyclization reaction involving an ionic nitrogen of the azide and the Au-activated alkyne. Next, III is formed via the back-bonding from the Au canter along with lose N2. After that, intermediate IV is generated from III in the presence of nitrile. In the following, product V is generated via the nucleophilic attack of the nitrogen of sulfonamide to the nitrile carbon. Lastly, compounds 25 is produced from V in the presence of H2O and O2 via the further aromatization of V. Also, compound VI could be formed from III which it could be trapped by H2O. In the following, it is converted to product VII (O–H insertion) and is regenerated the corresponding catalyst. Finally, product 26 is formed from the enol VII via the intramolecular SN2′ type reaction.126
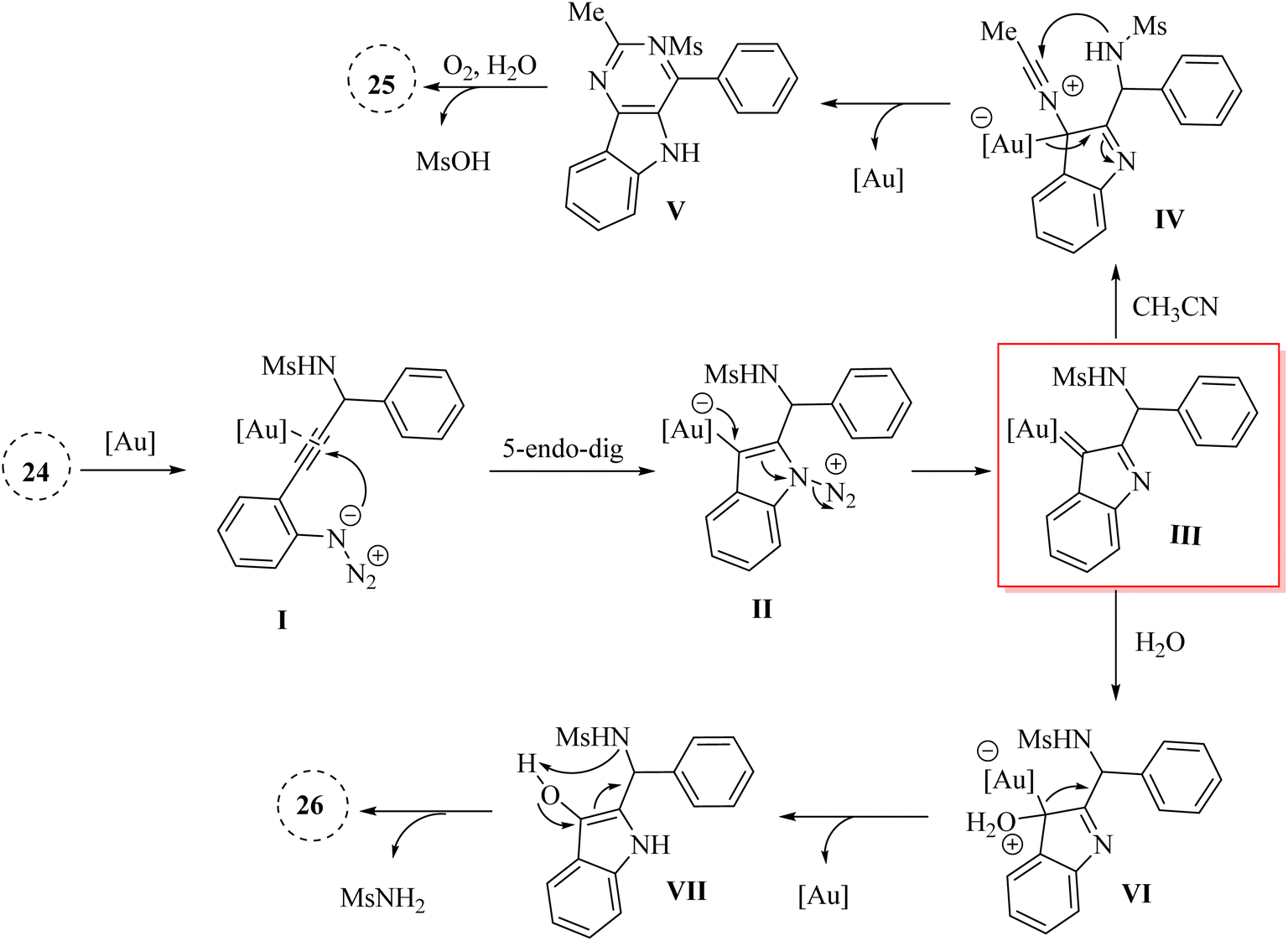 | ||
| Scheme 15 A mechanism for the reaction of 2-alkynyl arylazide derivatives 24 into indole derivatives 25 and indolinone derivatives 26 catalyzed by Au. | ||
In 2024, Wang et al.127 introduced a novel asymmetric catalysis approach using Au (t-BuXPhosAuSbF6) and L-proline. This method facilitated the direct creation of chiral 2,2-disubstituted 3-carbonyl indole derivatives 29 featuring quaternary carbon canters with stereochemistry, starting from 2-alkynyl arylazide derivatives 27 and ketone derivatives 28 (Scheme 16). The process involved generating key 2-phenyl-3H-indol-3-one intermediates and conducting an asymmetric Mannich reaction with H2O in DCM at room temperature, leading to satisfactory yields and outstanding enantioselectivity and diastereoselectivity. The desired catalysis reaction illustrated broad functional group tolerance across a wide range of the derivatives of 27. In addition, electron-donating groups substantially enhanced yields and significantly reduced reaction times. Conversely, electron-withdrawing groups resulted in longer reaction times and diminished yields. There were some challenges and limitations regarding the reaction, such as (1) inadequate investigations on reaction mechanisms and intermediates, (2) lack of comprehensive optimization for type of reaction conditions, (3) more diverse substrate applications were required in asymmetric synthesis, and (4) limited investigation of Au/chiral amine relay catalysis compatibility.
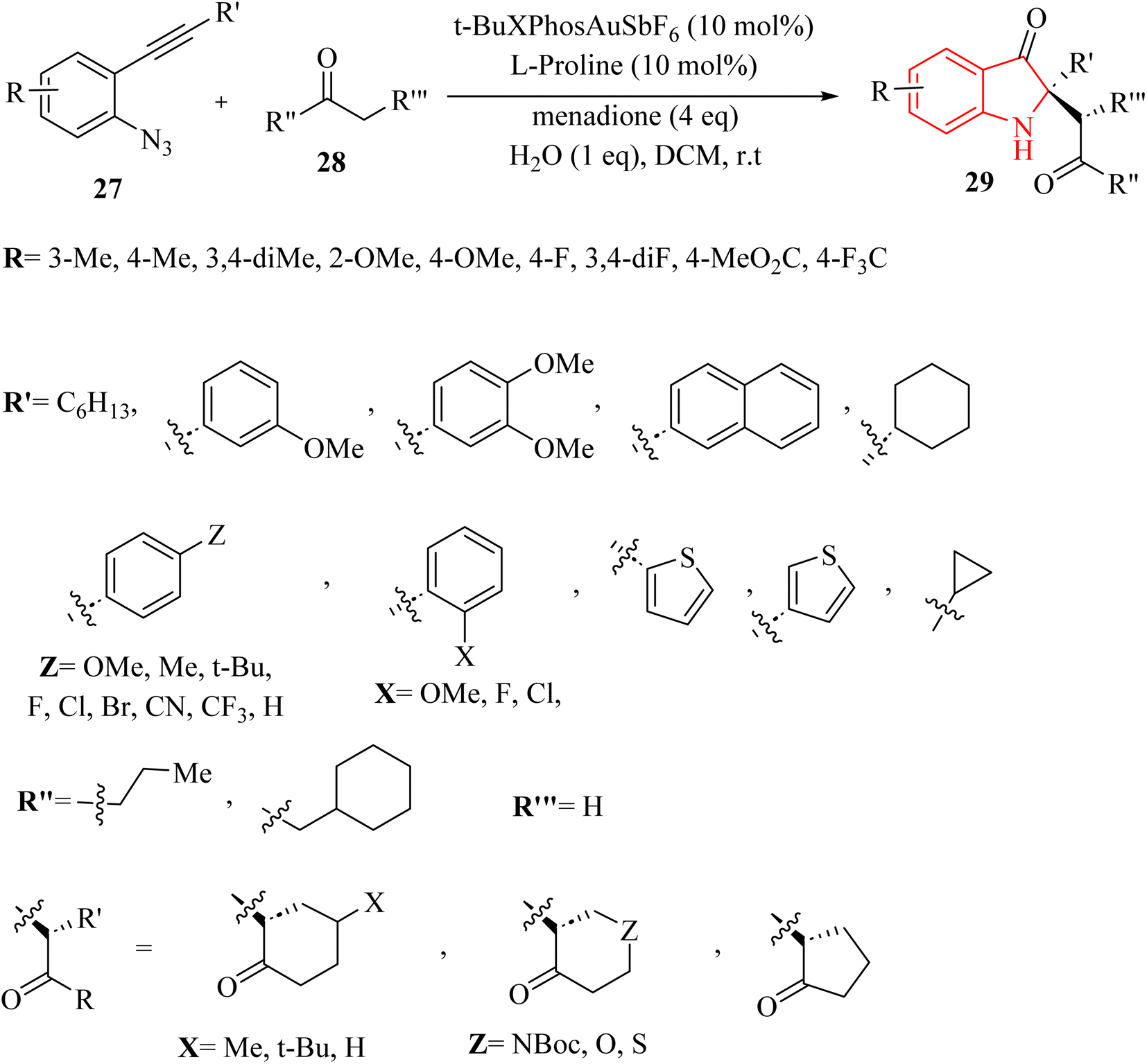 | ||
| Scheme 16 Synthesis of 3-carbonyl indole derivatives 29. | ||
A mechanism underlying this transformation is depicted in Scheme 17. Initially, the 2-alkynyl arylazide substrate 27 is activated by Au catalyst and is formed the Au-coordinated intermediate I. Next, compound II is formed from intermediate I via the cyclization. After that, complex III is formed from compound II along with lose of N2 and immediately trapped by H2O which this action is produced complex IV. Then, intermediate V is obtained from IV via the proton demetalation. In the following, intermediate VI is generated in the presence of the oxidant. In the next step, transition state VIII is formed via the reaction between enamine VII and imine intermediate VI which VII itself is obtained from via the reaction between proline and acetone. Finally, the corresponding product 29 is produced via the Mannich reaction.127
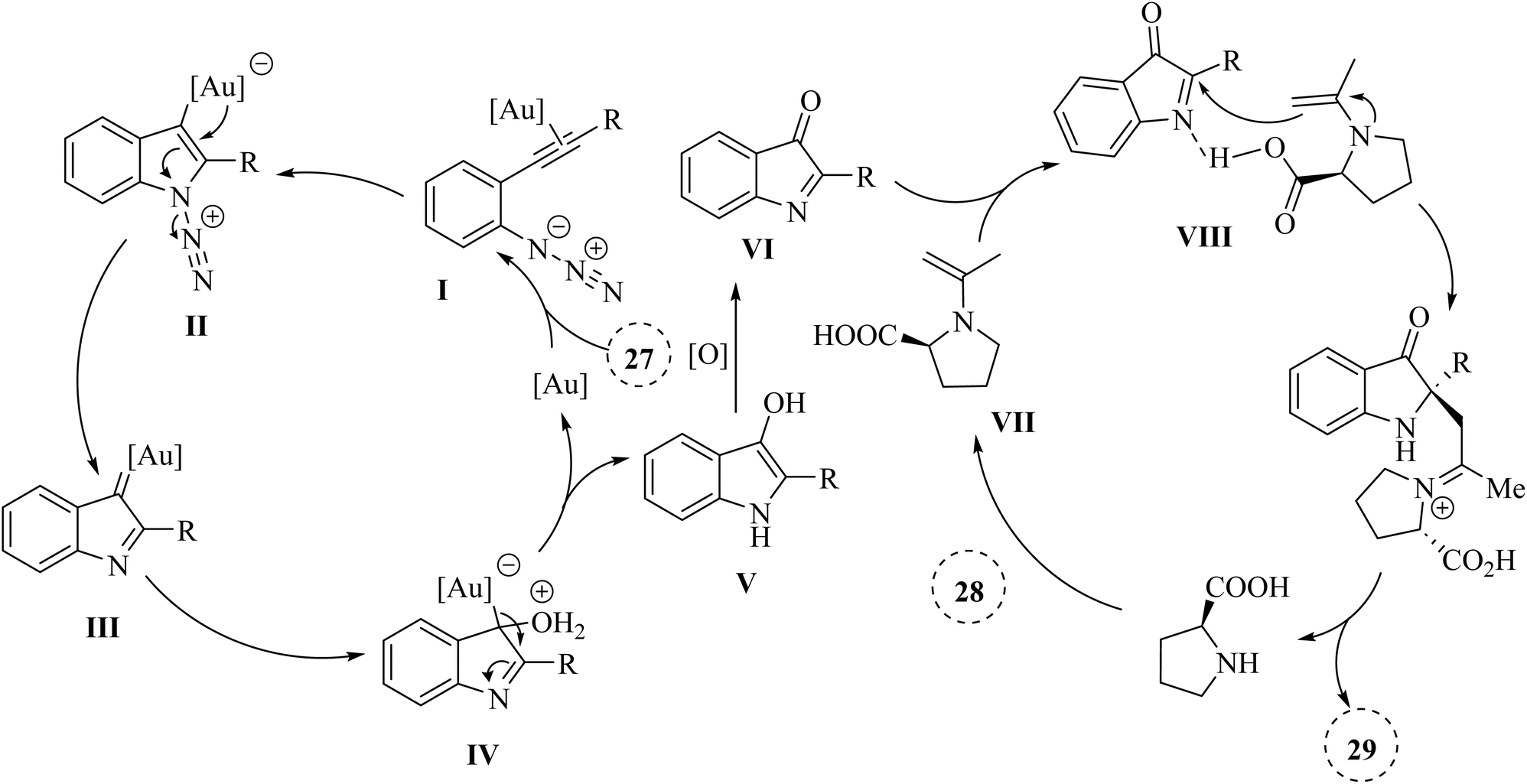 | ||
| Scheme 17 A mechanism for the reaction of 2-alkynyl arylazide derivatives 27 with ketone derivatives 28 catalyzed by Au. | ||
A novel strategy was developed for synthesizing aryl-annulated [c]carbazole derivatives via Au-catalyzed cascade cyclization of azido-diyne derivatives by Kawada and co-workers.128 When reacting with electron-rich benzene derivatives like anisole and xylene, benzo[c]carbazole derivatives 32 (Scheme 18A) were formed by functionalizing two benzene C–H bonds. Additionally, using N-boc-pyrrole and indole derivatives as coupling partners selectively produced heteroaryl-annulated carbazole derivatives—specifically pyrrolo[2,3-c]carbazole derivatives 33 (Scheme 18B) and indolo-carbazole derivatives 35 (Scheme 18C). The reaction involved an internal nucleophilic attack by the N3 on the nearby alkyne, resulting in the formation of an Au carbenoid species. The process was completed by the nucleophilic attack of arene derivatives on the carbenoid, followed by a 6-endo-dig cyclization of the introduced arene to the other alkyne. DFT calculations, competition experiments, and deuterium-labelling experiments provided evidence for the proposed reaction mechanism. Notably, a N,N′-dimethylated derivative of compound 35 showed both fluorescence and UV-vis-NIR spectral changes during electrolysis, demonstrating the potential usefulness of this reaction in materials chemistry. There were some limitations and challenges regarding the reactions, such as (1) separation of minor isomers from by-products was difficult, (2) electrophilic aromatic substitution was more likely than C–H insertion, (3) the relative reactivity of arene derivatives with N-Boc-pyrrole was unclear, (4) limited understanding of side reactions with indole derivatives, and (5) further investigation of electron-withdrawing groups on indole derivatives was required. It should be noted that deprotonation was not the rate-determining step for product formation.
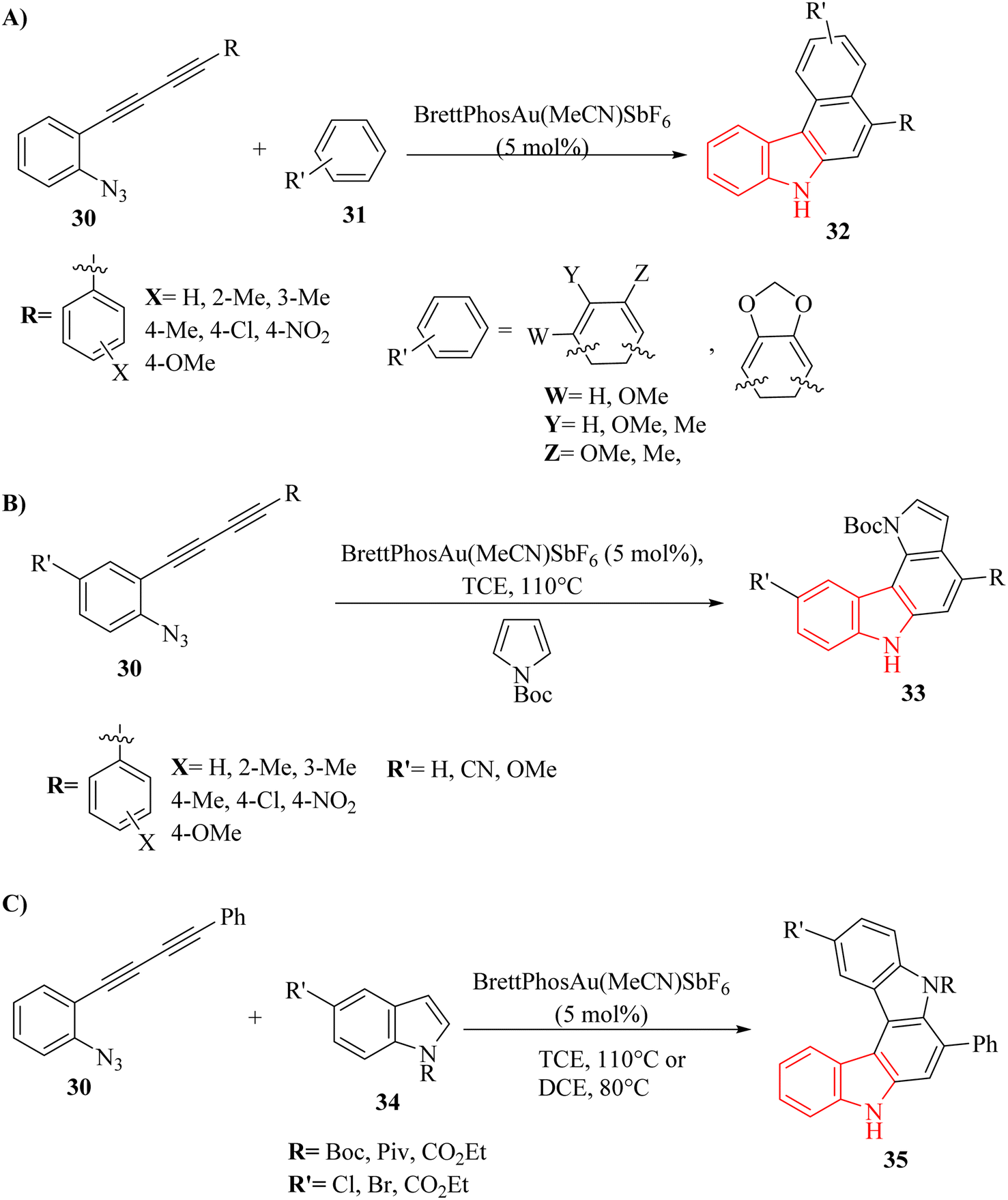 | ||
| Scheme 18 (A) Synthesis of carbazole derivatives 32, (B) synthesis of pyrrolo-carbazole derivatives 33, (C) synthesis of indolo-carbazole derivatives 35. | ||
Shen et al.129 documented an Au-catalyzed cascade cyclization resulted in the diverse creation of synthetically valuable derivatives of pyrrolo-indole 37 (up to 95% yield) (Scheme 19).
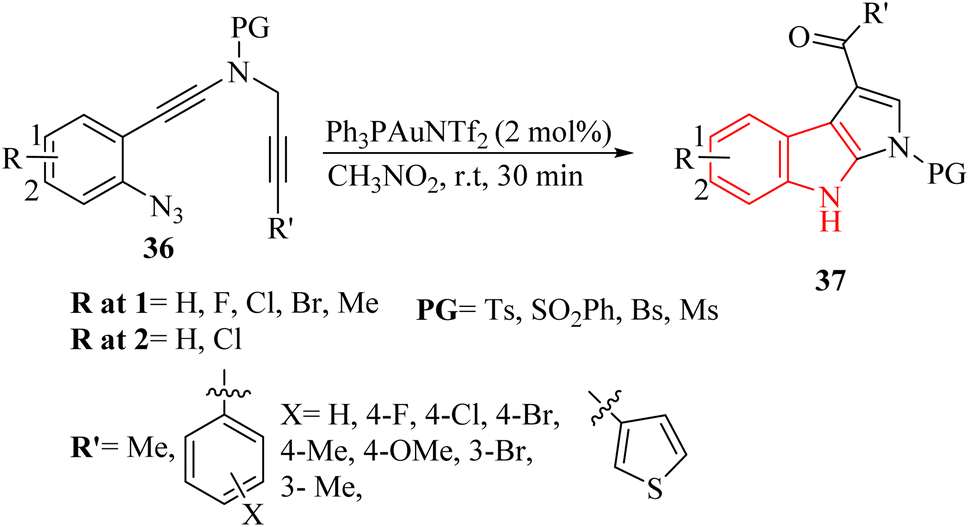 | ||
| Scheme 19 Synthesis of pyrrolo-indole derivatives 37. | ||
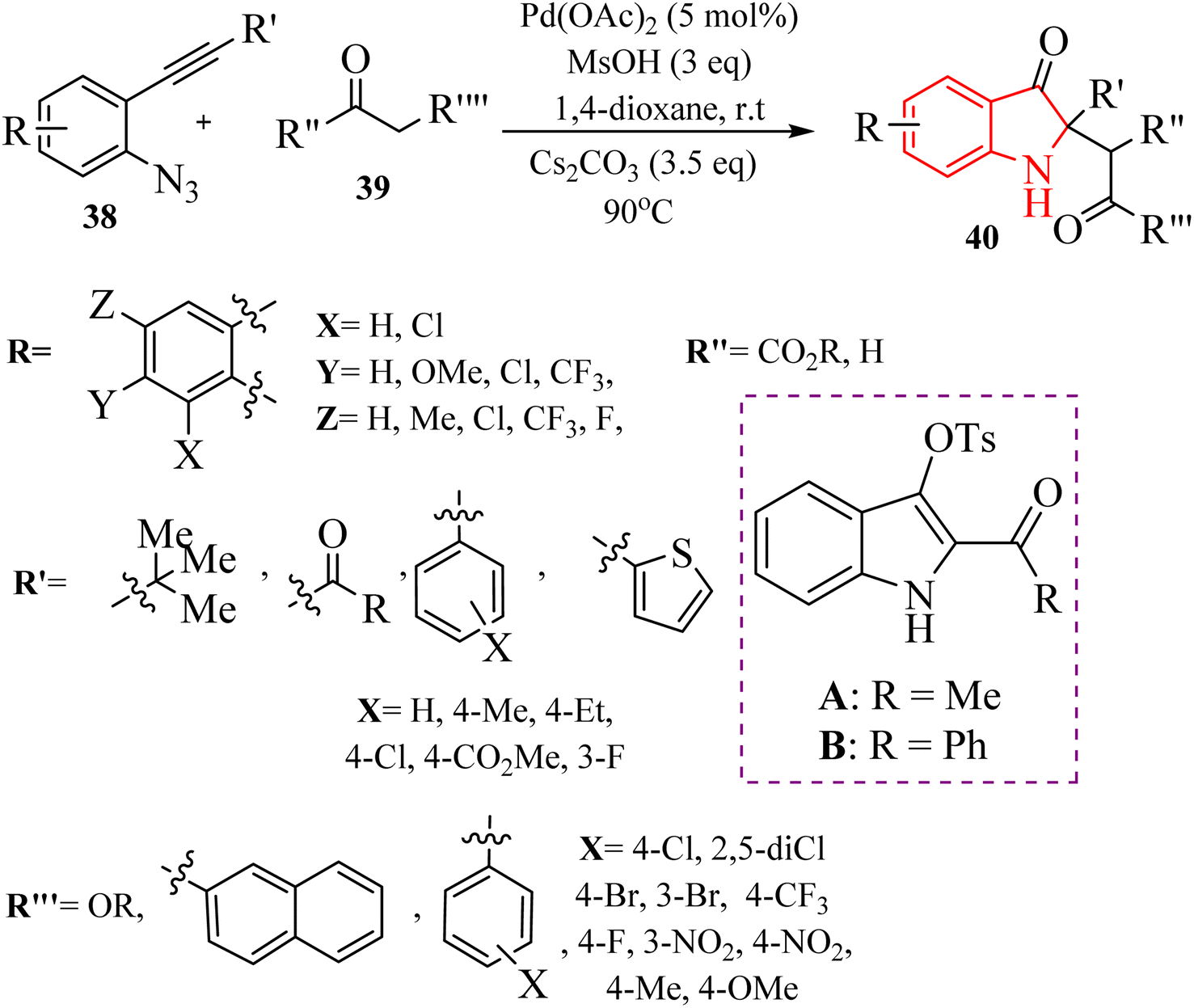 | ||
| Scheme 20 Synthesis of indolin-3-one derivatives 40. | ||
A mechanism underlying this transformation is depicted in Scheme 21. The activation process is produced intermediate I, coordinated with Pd. Subsequently, this intermediate undergoes cyclization, resulting in the formation of species II. Upon N2 release, Pd-carbenes III is formed, and then TsOH trapped them to yield complex IV. Next, intermediate V is generated from IV via the protodemetalation process. When exposed to heat and under basic conditions, V can experience a 1,3-sulfonyl shift to generate VI. Subsequently, reductive desulfonation leads to the formation of intermediate VII. Ultimately, the Mannich-type reaction involving ketone is resulted in the 40 formations.130
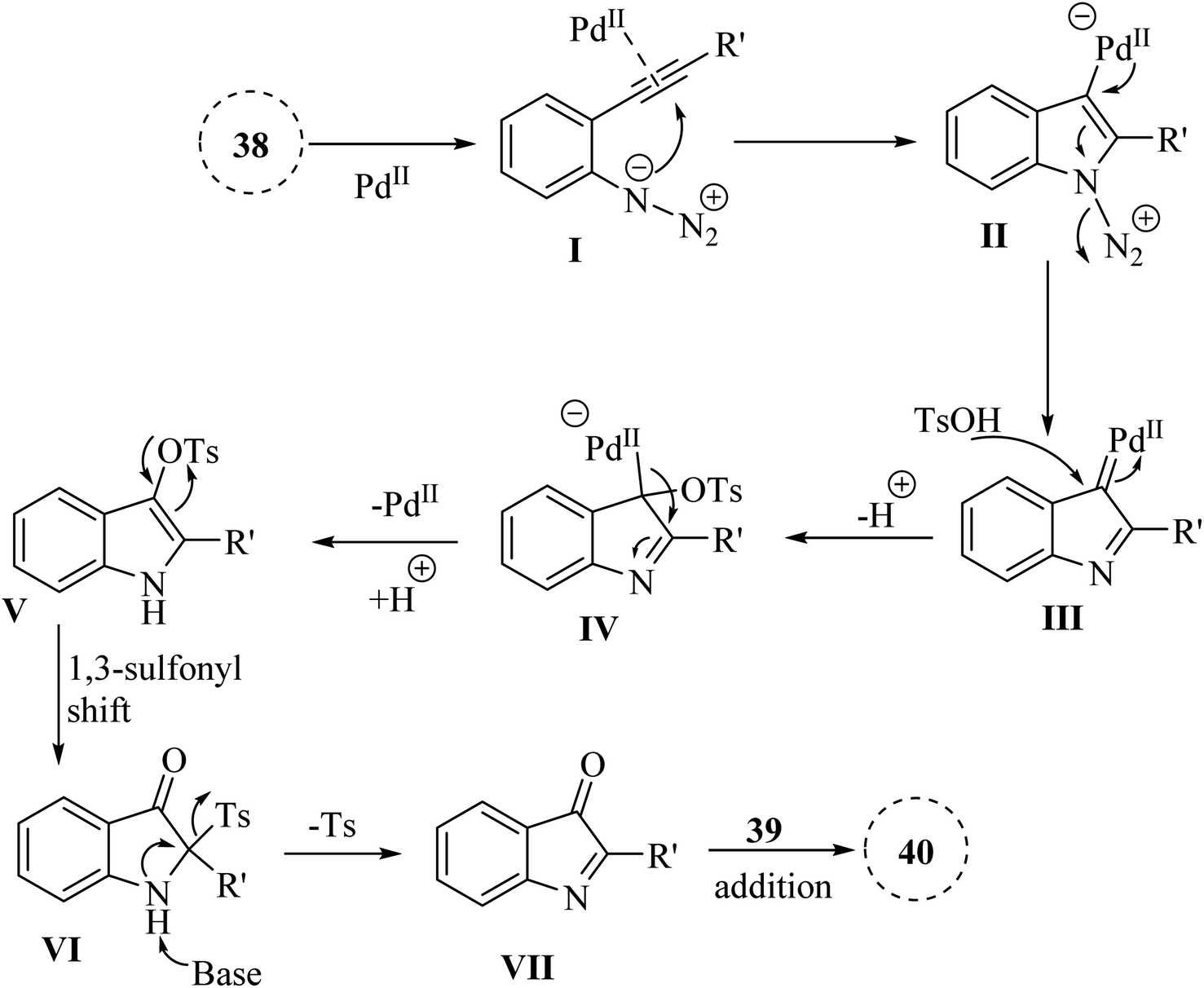 | ||
| Scheme 21 A mechanism for the one-pot reactions catalyzed by palladium involving 2-alkynyl arylazide derivatives 38. | ||
In 2024, Zhang et al.131 reported a highly effective technique for producing 1H-indole-3-sulfonate derivatives 42 through a series of consecutive reactions involving 2-alkynyl arylazide derivatives 41 and sulfonic acid derivatives, catalysed by Pd(II) in 1,4-dioxane at 25 °C (Scheme 22). The reactions were observed to proceed rapidly, with the most completing them within a more 10 minutes. The desired products generated in 82–96% yields when used MsOH in the reaction, and 4-methylbenzenesulfonic acid produced the desired products in 70–97% yields. Also, benzenesulfonic acid, 4-chlorobenzenesulfonic acid and, naphthalene-1-sulfonic acid were tested and generated the desired products in 87–95% yields. Authors investigation revealed that the reaction involving 41 with a terminal alkyne group was not very efficient. The crude mixture produced a blend of decomposition products that remained unidentified despite analysis using 1H NMR and TLC. The authors suggested that further investigation was needed to optimize the methodology for broader applications. These challenges underscored the need for a mild and efficient synthetic technique for 1H-indole-3-sulfonate derivatives.
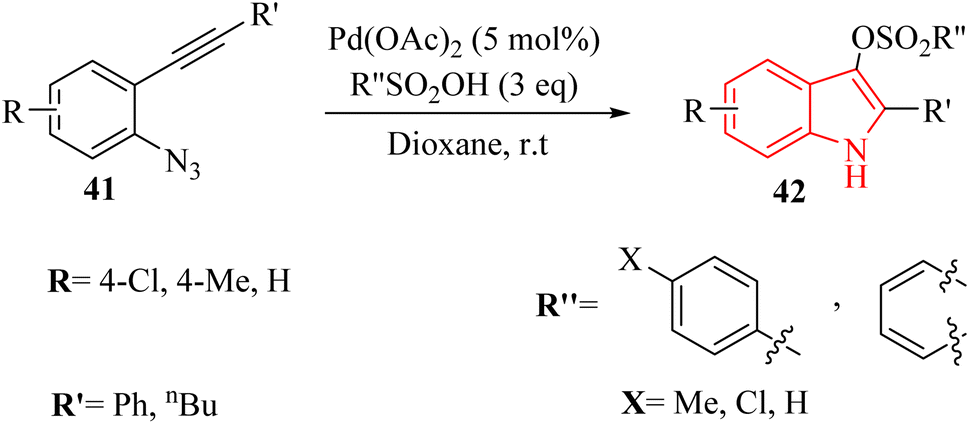 | ||
| Scheme 22 Synthesis of 1H-indole-3-sulfonate derivatives 42. | ||
A mechanism underlying this transformation is depicted in Scheme 23. Initially, compound 41 through coordination with a Pd catalyst at the triple bond, resulting in the formation of complex I. Then, complex II is formed through intramolecular amination of an N3 group with an activated alkyne. Next, IV is formed by trapping alpha-imino palladium carbene species C14 after the release of N2 using MsOH. Finally, the desired product 42 is formed through subsequent protodemetalation reactions.131
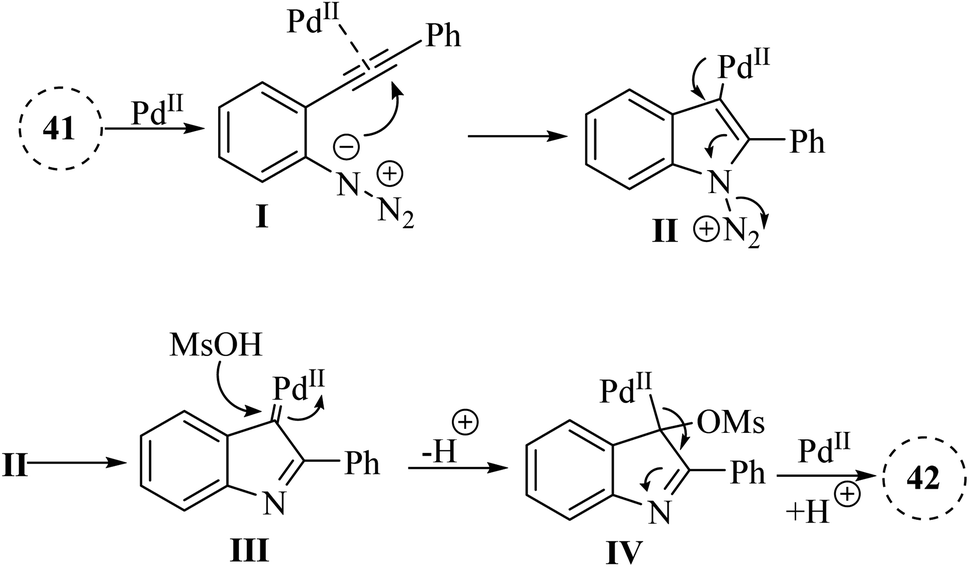 | ||
| Scheme 23 A mechanism for the reactions catalyzed by palladium involving 2-alkynyl arylazide derivatives 41. | ||
In 2018, Li et al.132 synthesized polyfunctional indolin-3-one derivatives (45, 47 and 48) through a Pd-catalysed one-pot insertion reaction of cyclic C-acylimines into C–C s-bonds reactions in the presence of Cs2CO3 in 1,4-dioxane at 90 °C (Scheme 24A and B). In the case of 2-alkynyl arylazide derivatives 43, it was found that the corresponding products were successfully obtained with yields of up to 96% (Scheme 24A). Furthermore, in the case of aryl ketone derivatives 46, the synthesized products demonstrated excellent yields ranging from 80% to 95% when utilizing beta-ketoester and beta-ketosulfone derivatives (Scheme 24B). However, when alpha-nitrocarbonyl and beta-ketophosphonate were employed in the reactions, lower product yields of 28% to 68% were observed. When alpha-bromocarbonyl was evaluated, the resulting mixture contained decomposition products that remained unidentifiable despite analysis using 1H NMR and TLC, indicating potential limitations in substrate compatibility. The study displayed that the yields of products could be affected by steric effects of the substrates. The research also noted that certain highly active carbon anions produced during the reaction may not led to insertion reactions due to insufficient stabilization.
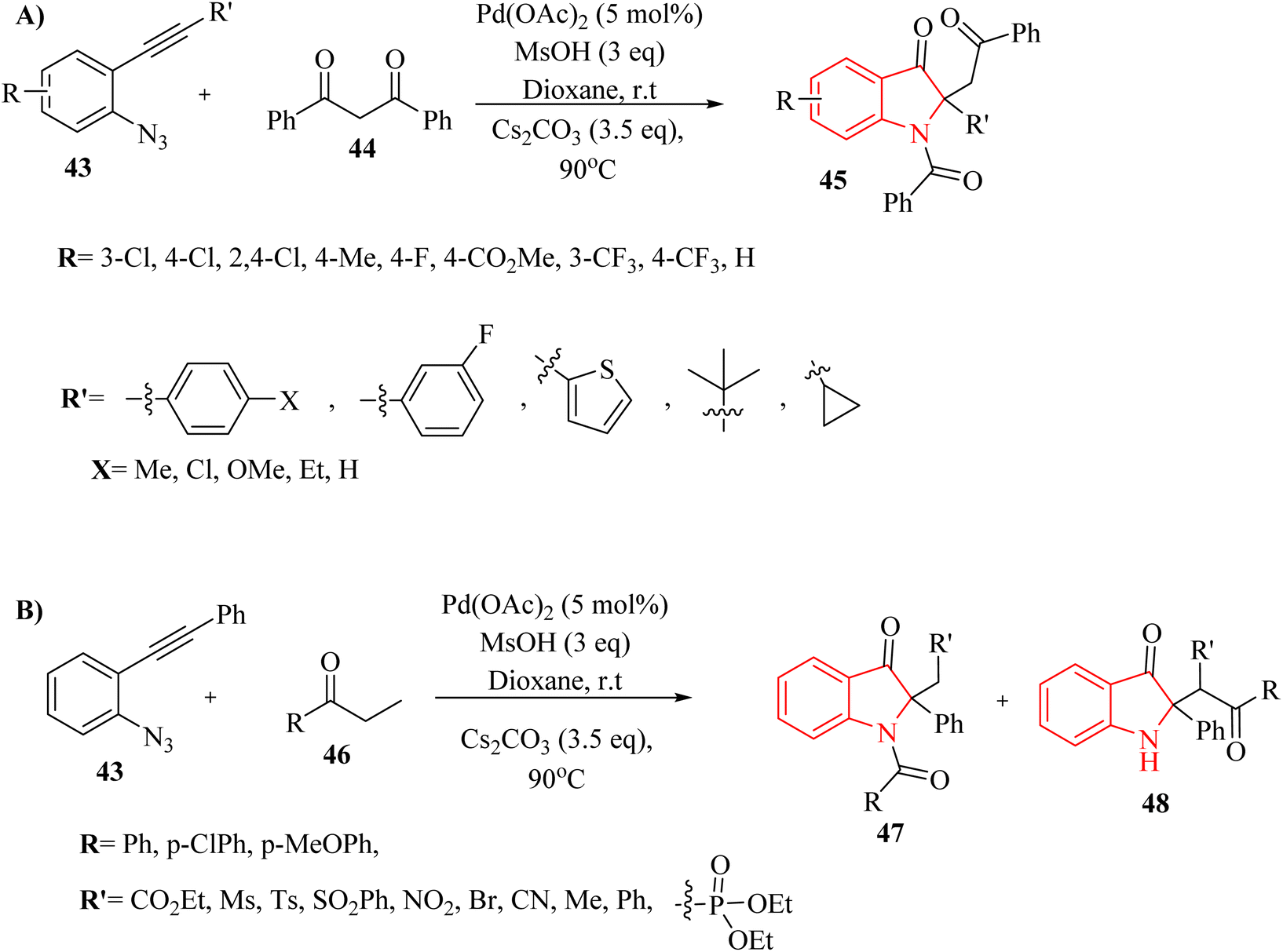 | ||
| Scheme 24 (A) Scope of the 2-alkynyl arylazide derivatives 43, (B) scope of the aryl ketone derivatives 46. | ||
A proposed mechanism underlying this transformation is depicted in Scheme 25. Compound 43 is activated by coordination of Pd catalyst with alkyne. This is formed a Pd-coordinated intermediate I. Then, species II is produced from I via an intramolecular cyclization. The alpha-imino Pd carbene III is formed by the release of N2. In the following, III is attacked by TsOH and is formed the complex IV. Following protodemetalation, the aryl sulfonate V is generated. Next, V is converted into the active intermediate VI via a 1,3-Ts shift step. After that, key intermediate VII is formed after subsequent reductive desulfonation of VI. In the next step, alkylated compound VIII is produced when VII undergoes Mannich-like addition. Complex IX is generated via an intramolecular attacking of N to C. Next, complex X is formed from IX via 1,3-acyl shift. Lastly, the desired product is produced after protonation process.132
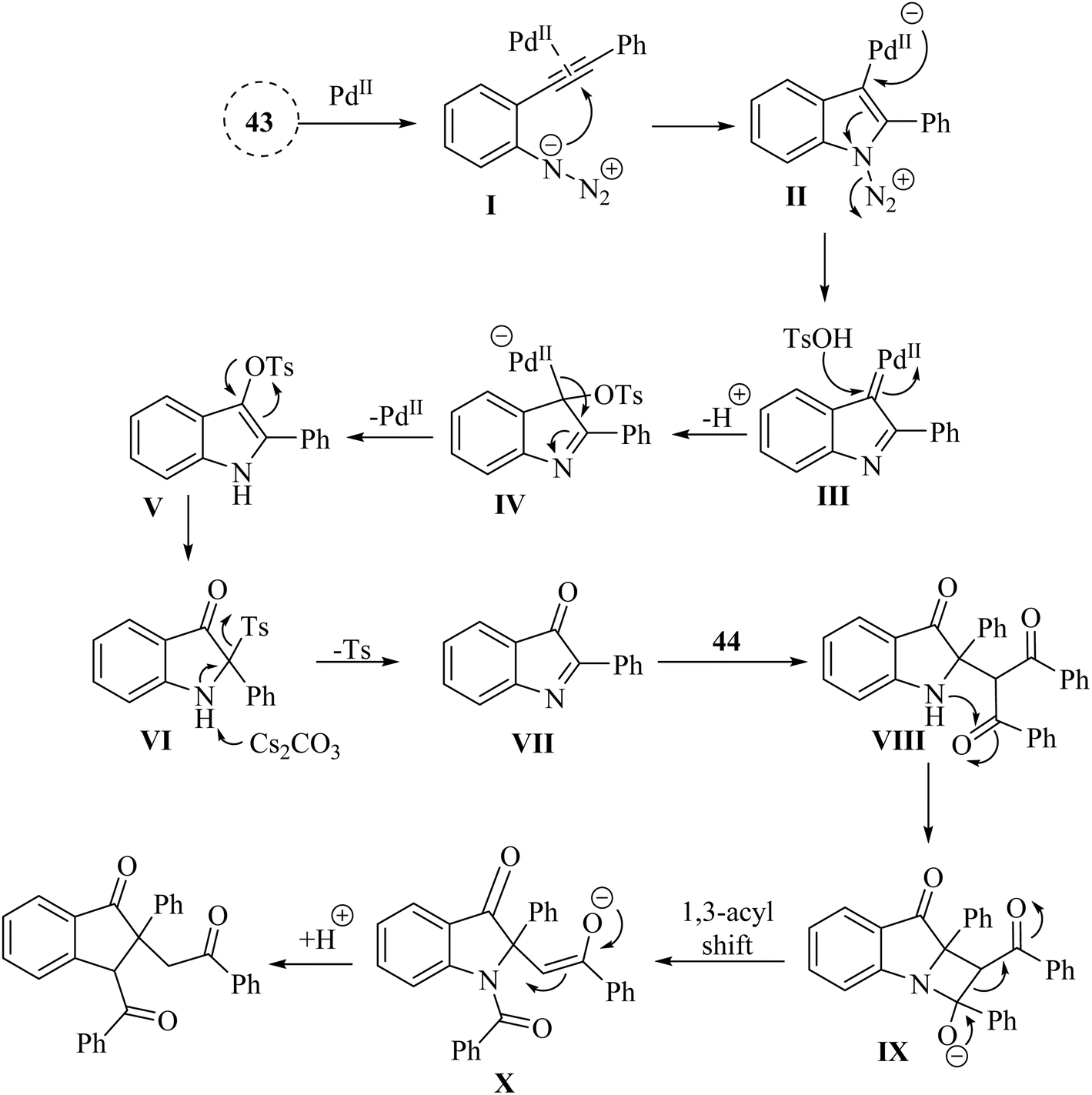 | ||
| Scheme 25 A mechanism for the Pd-catalyzed of 43. | ||
Yong et al.133 introduced a Pd-catalyzed one-pot two-step reaction of 2-alkynyl arylazide derivatives 49 and indole derivatives 50. The reactions involving 49, TsOH and 50 in the presence K2CO3 in 1,4-dioxane at 90 °C. In the case of the derivatives of 51, the desired products obtained with yields of up to 97% (Scheme 26). Nevertheless, no desired product was identified when 3-methyl-1H-indole was examined for its reactivity as a nucleophile (Scheme 26 X).
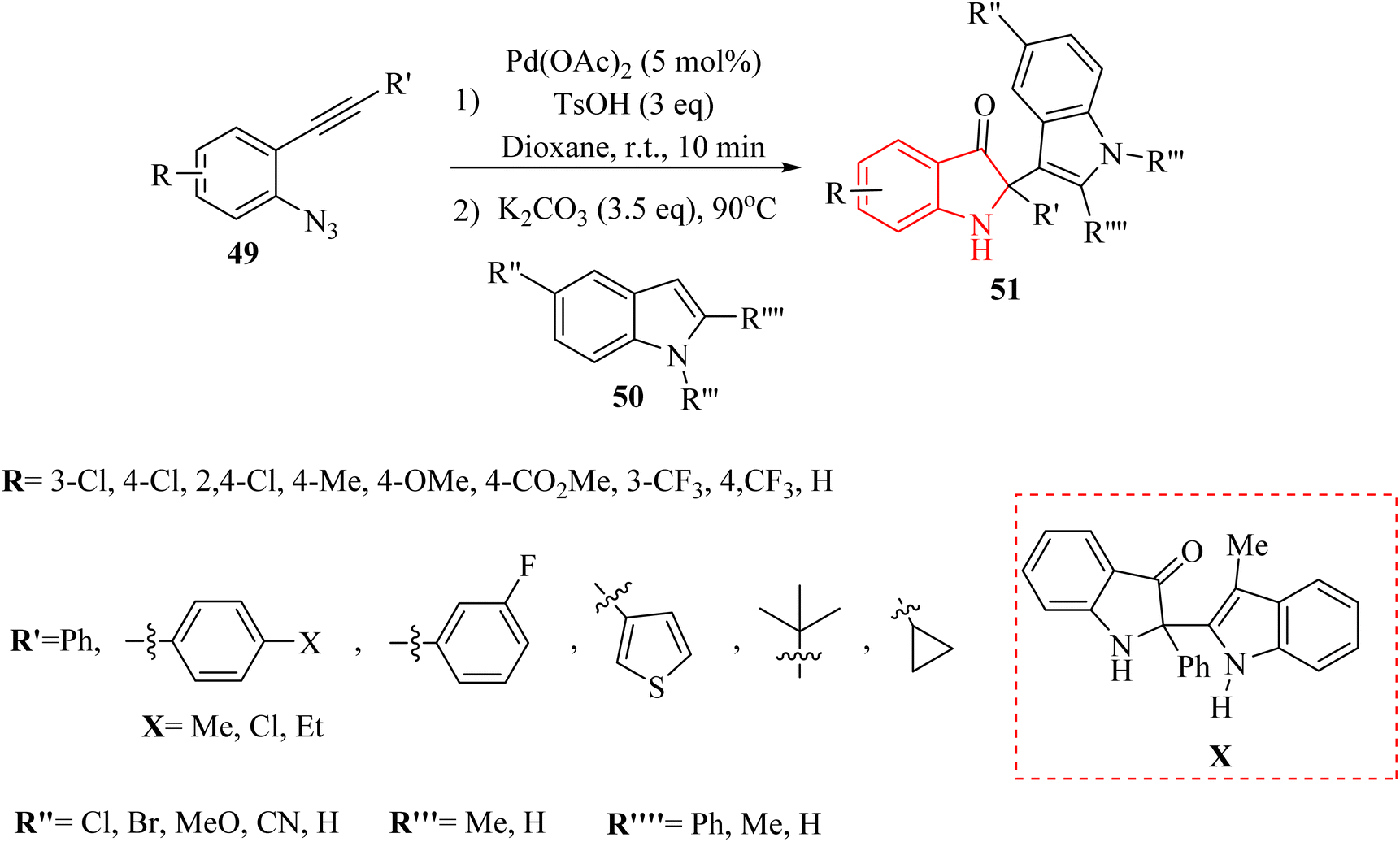 | ||
| Scheme 26 Synthesis of phenylindolin-3-one derivatives 51 catalyzed by Pd. | ||
A mechanism underlying this transformation is depicted in Scheme 27. Compound 49 is activated by coordination of Pd catalyst with alkyne. This is formed a Pd-coordinated I. Then, species II is produced from I via an intramolecular cyclization. The alpha-imino Pd carbene III is formed by the release of N2. In the following, III is attacked by TsOH and is formed the complex IV. Following protodemetalation, the aryl sulfonate V is generated. After 1,3-Ts shift of V and reductive desulfonation of VI, compound VII is generated. Finally, desired product 50 is formed after subsequent reductive desulfonation of VI which it is trapped by 51.133
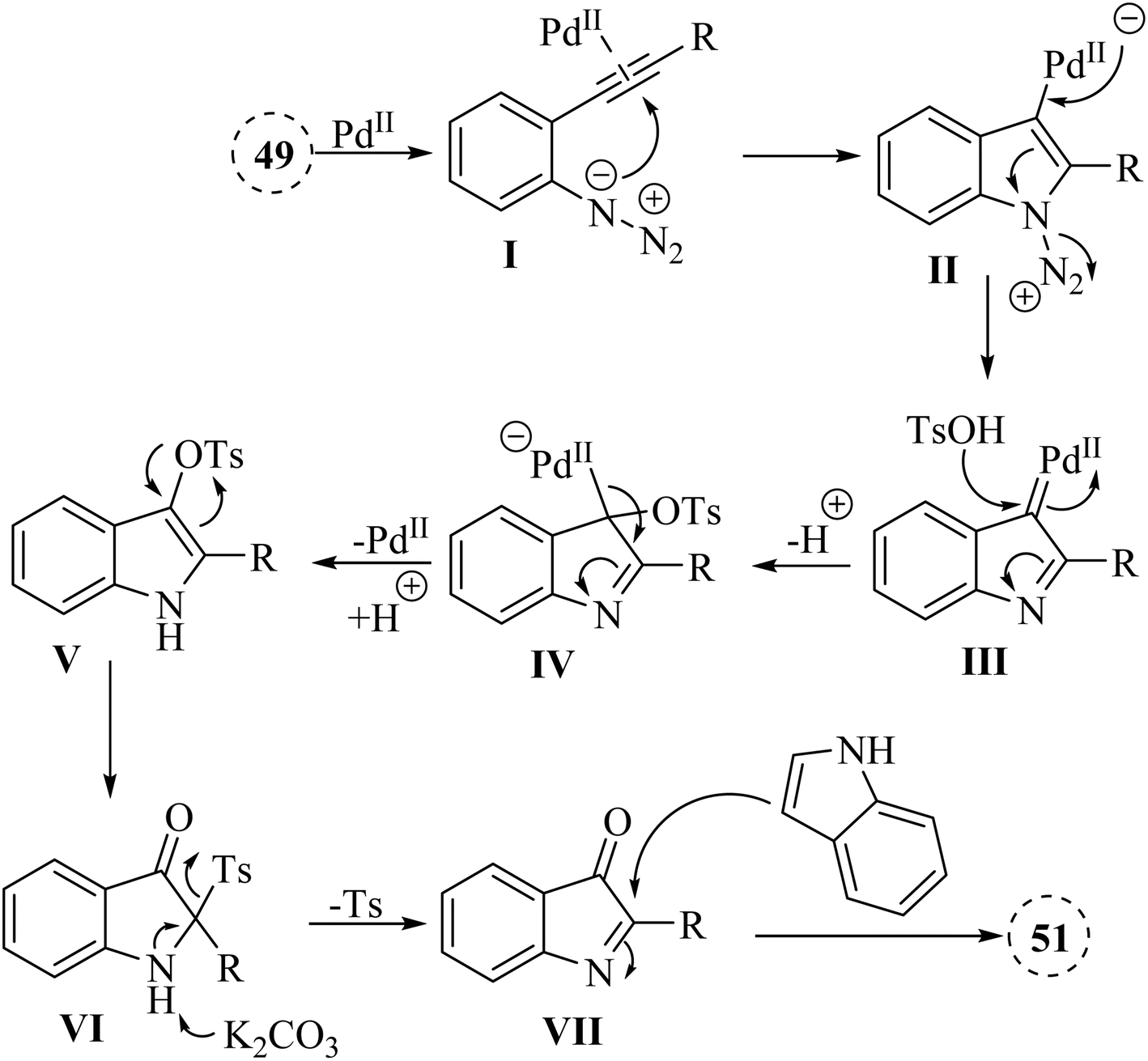 | ||
| Scheme 27 A mechanism for the one-pot reactions of 49 catalyzed by Pd. | ||
In 2021, Hu and colleagues134 developed an innovative and effective method to synthesize 3-thiocyanindole derivatives 53. This process involved a Pd-catalyzed bromination and cross-coupling reaction, assisted by NBS, of 2-alkynyl arylazide derivatives 52 with KSCN in a solvent mixture of EtOH and H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 90 °C (Scheme 28). The results demonstrated that this reaction can accommodate a wide range of structurally diverse 52 (up to 98% yield). A reaction involving a Br-containing substrate was conducted under standard conditions, yielding the corresponding product with a 71% yield after 4 hours. When authors extended the reaction time to 10 hours, they still obtained the desired product as the single product. The authors highlighted that existing thiocyanation reactions often face limitations such as harsh reaction conditions, which could be detrimental to sensitive substrates. It noted a narrow substrate scope, indicating that not all compounds could be effectively utilized in these reactions. Low product yields were also a concern, as many techniques did not generate adequate quantities of the corresponding thiocyanate products. These challenges underscored the necessity for developing a highly efficient and general thiocyanation technique that can accommodate a broader range of substrates.
:1) at 90 °C (Scheme 28). The results demonstrated that this reaction can accommodate a wide range of structurally diverse 52 (up to 98% yield). A reaction involving a Br-containing substrate was conducted under standard conditions, yielding the corresponding product with a 71% yield after 4 hours. When authors extended the reaction time to 10 hours, they still obtained the desired product as the single product. The authors highlighted that existing thiocyanation reactions often face limitations such as harsh reaction conditions, which could be detrimental to sensitive substrates. It noted a narrow substrate scope, indicating that not all compounds could be effectively utilized in these reactions. Low product yields were also a concern, as many techniques did not generate adequate quantities of the corresponding thiocyanate products. These challenges underscored the necessity for developing a highly efficient and general thiocyanation technique that can accommodate a broader range of substrates.
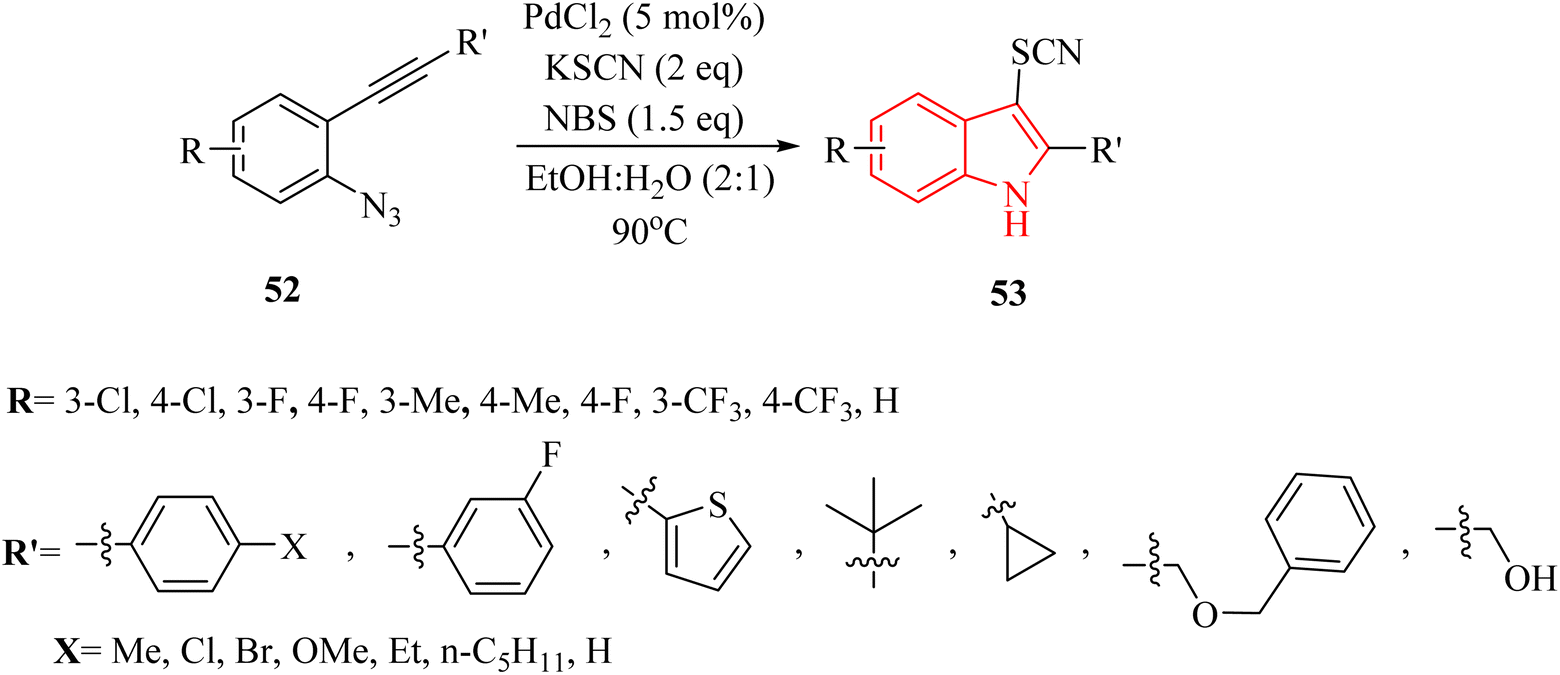 | ||
| Scheme 28 Synthesis of 3-thiocyanindole derivatives 53. | ||
A mechanism underlying this transformation is depicted in Scheme 29. Initially, compound 52 through coordination with a Pd catalyst at the triple bond, resulting in the formation of complex I. Then, complex II is formed through intramolecular amination of an N3 group with an activated alkyne. In the following, III is produced via a release of N2. Next, complex IV is formed from III when it is trapped by HBr. After that, intermediate V is generated after demetalation and protonation of IV. In the next step, VI is produced via an oxidative addition of V. Next, complex VII is generated via a ligand exchange with KSCN. Lastly, the corresponding product is obtained via a reductive elimination of VII.134
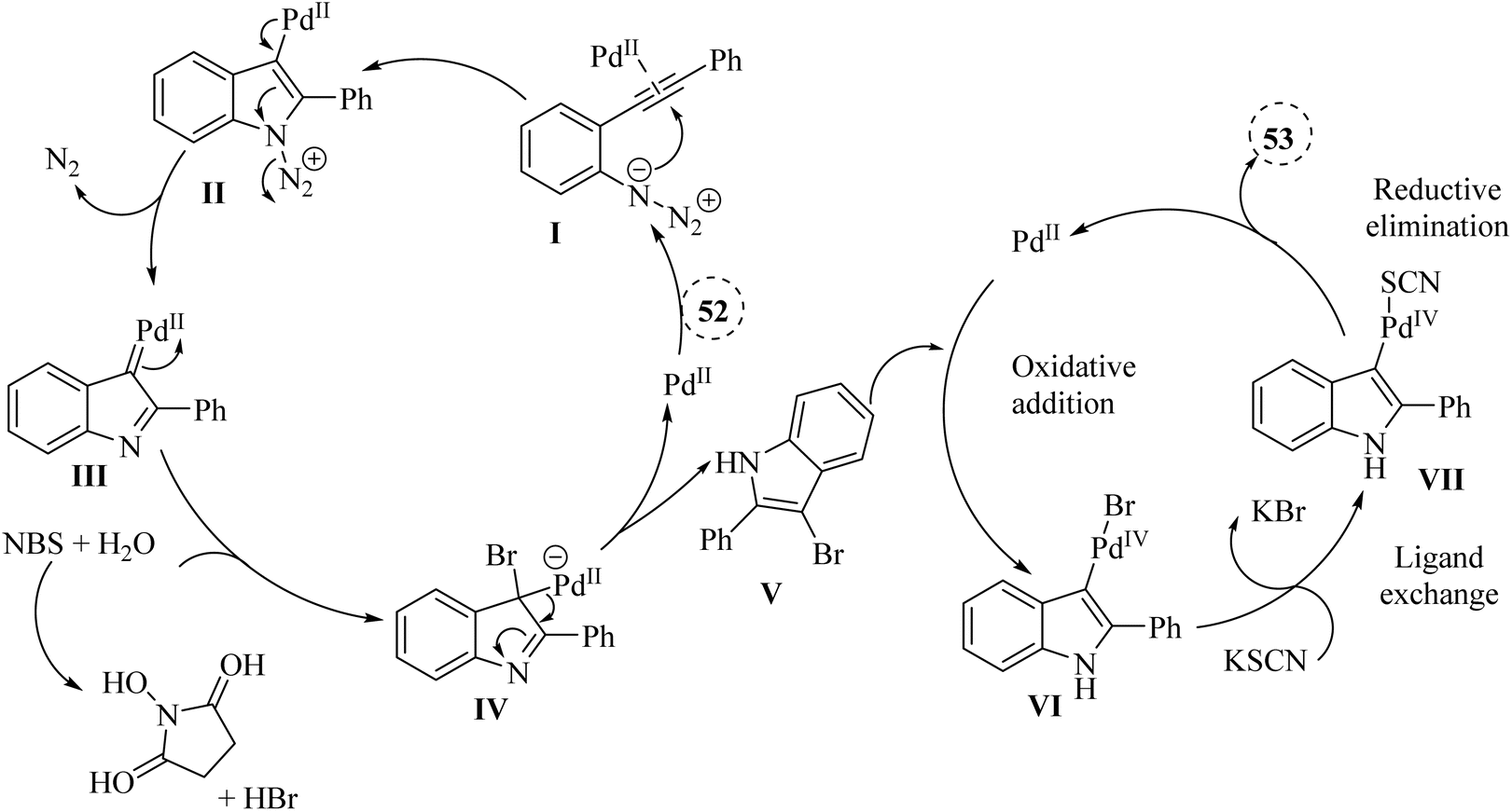 | ||
| Scheme 29 A mechanism for the Pd-catalyzed reactions of 52. | ||
In the year 2020, Li and colleagues135 outlined a new approach for producing N-fused 7 membered multi-functional polycyclic indoline-3-one derivative derivatives 55 by introducing cyclic C-acylimines into cyclic beta-diketone derivatives (56 or A) (Scheme 30). This process was carried out in the presence of Cs2CO3 at 90 °C. The reaction with the substrate containing an R′ = tert-butyl group was unsuccessful, and only the initial intermediate, 2-(tert-butyl)-1H-indol-3-yl 4-methylbenzenesulfonate, was able to be isolated. This likely occurred due to steric hindrance. Under standard reaction conditions, an attempt was made to carry out the insertion reaction with 56. Unfortunately, the corresponding insertion product was not generated (Scheme 30).
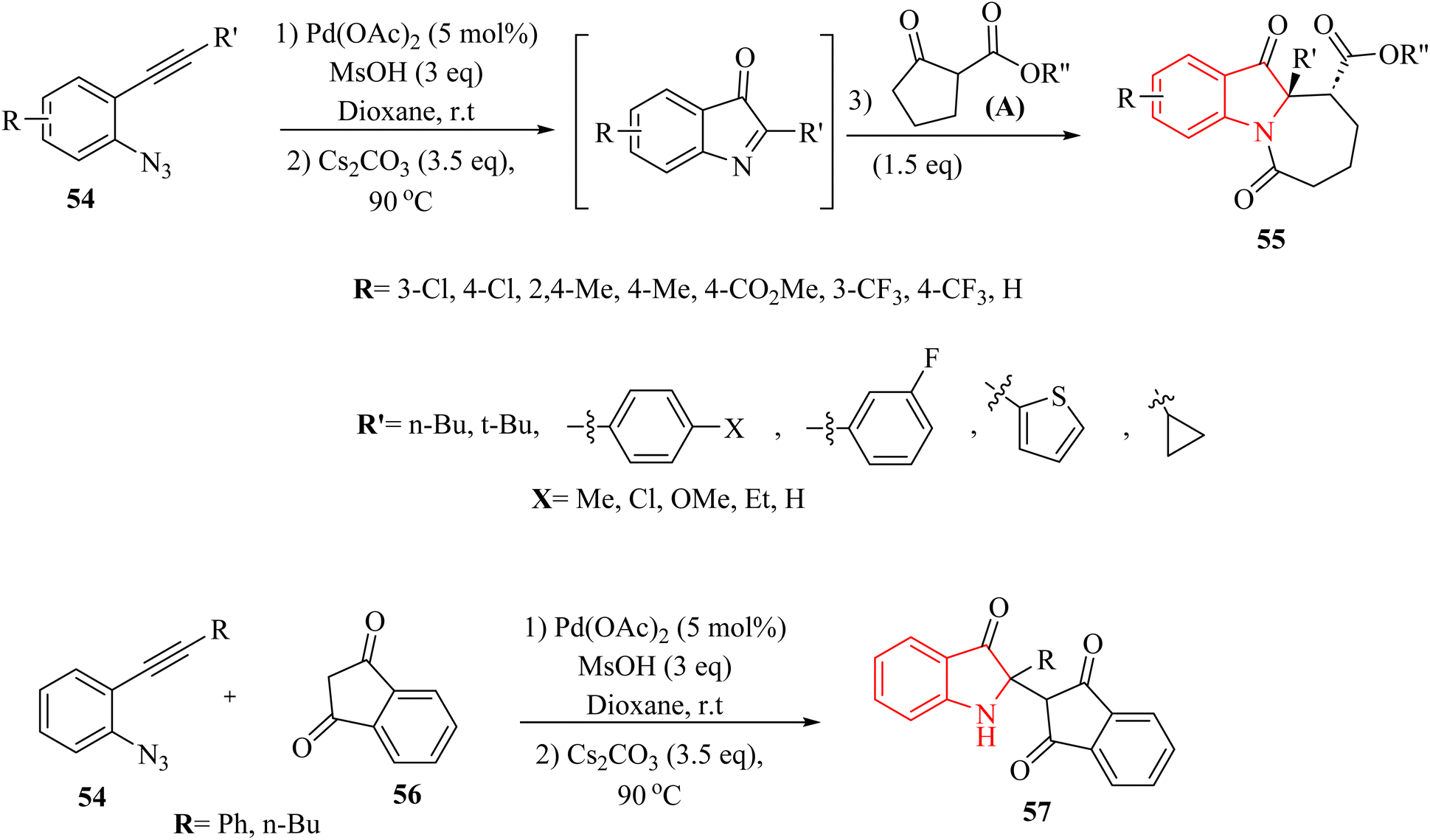 | ||
| Scheme 30 Synthesis of indoline-3-one derivative derivatives 55 and 3H-indol-3-ones 57. | ||
A mechanism underlying this transformation is depicted in Scheme 31. The activation process is produced intermediate I. Subsequently, this intermediate undergoes intramolecular cyclization of azide group, resulting in the formation of species II. Upon N2 release, Pd-carbene III is formed, and then TsOH is trapped them to yield complex IV. Next, intermediate V is generated from IV via the protodemetalation process. When exposed to heat and under basic conditions, VII is generated through a 1,3-sulfonyl shift of V and reductive desulfonation of VI. Subsequently, complex VIII is produced via a nucleophilic addition of A to VIII. Then, a four-membered ring complex IX is generated from VII via an intramolecular addition. Lastly, the corresponding product 55 is produced through protonation of complex X.135
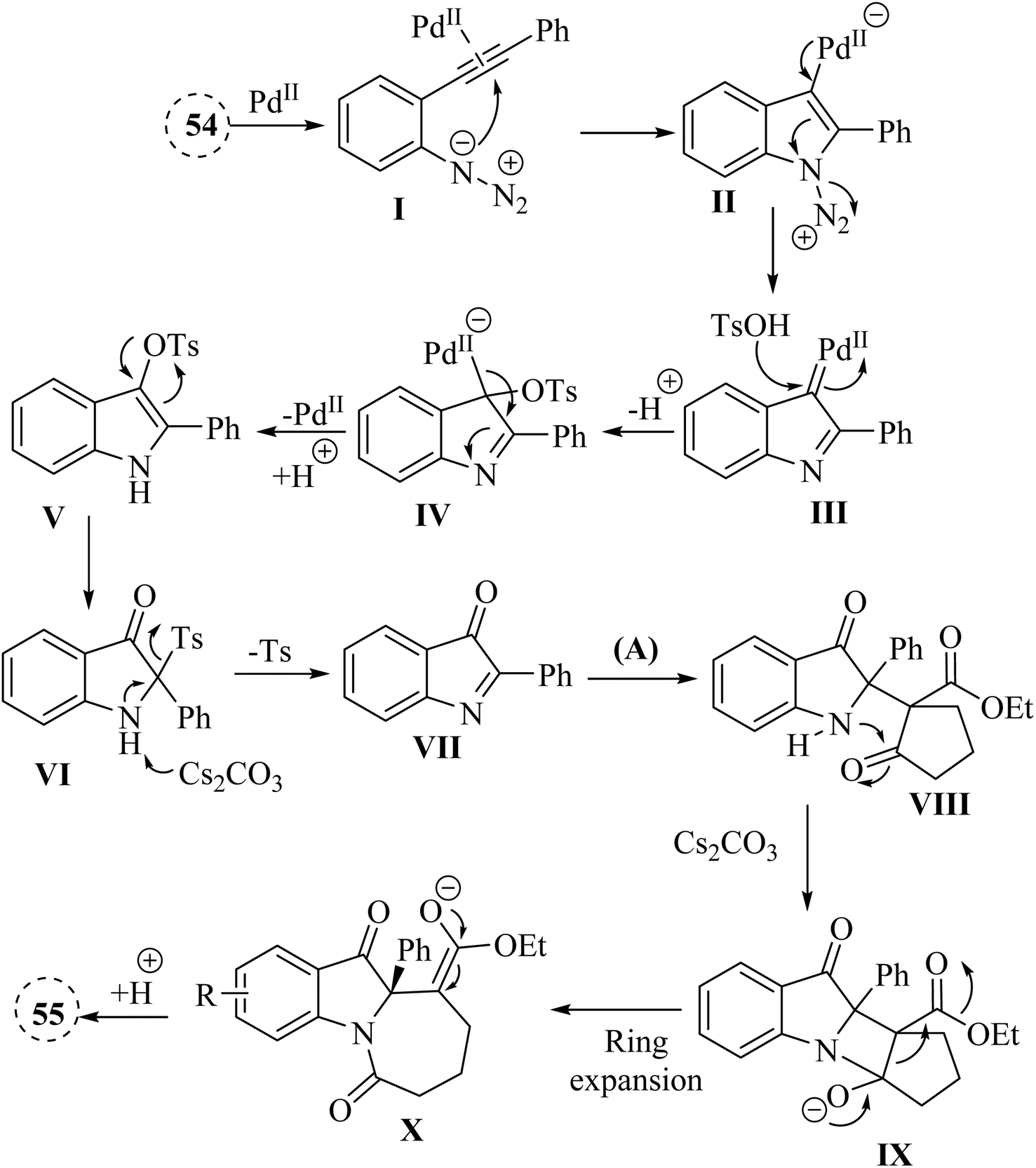 | ||
| Scheme 31 A mechanism for the reactions of 2-alkynyl arylazide derivatives 54 with cyclic beta-diketone derivatives (56 or A) catalyzed by Pd. | ||
Reactions 20, 24, and 30 are essentially similar but have differences that can be noted as follows. In the case of products, the reaction 20 focused on N-fused 7-membered rings, while the reaction 24 targeted polyfunctional indolin-3-one derivatives, and the reaction 30 emphasized C2-quaternary indolin-3-one derivatives. In the case of reaction mechanisms, the reaction 20 involved a straightforward insertion into cyclic β-diketone derivatives, the reaction 24 emphasized the insertion into carbon–carbon σ-bonds, and the reaction 30 highlighted a rearrangement of sulfonate derivatives, showcasing different mechanistic pathways. In the case of functional group tolerance, each method had varying degrees of functional group tolerance, with the reaction 24 illustrating a broader scope of substrates and conditions compared to the reaction 20 and 30. This consolidation not only emphasized the progress made in this area but also set the stage for future research exploring further applications and optimizations of these methodologies.130,132,135
In 2016, Zhou et al.136 reported the development of a Pd-catalyzed synthesis method for the production of polysubstituted indole derivatives 60. This method involved the coupling of aryl bromide derivatives 59 with 2-alkynyl arylazide derivatives 58 (Scheme 32). The reaction was conducted in the presence of LiOtBu, Pd2dba3, and dpppe in toluene at 100 °C for generated 60. The authors discussed challenges related to the low yields observed in the synthesis of indoles, particularly when using certain aryl bromide derivatives and azide derivatives as substrates. It should be noted that the reaction conditions need to be optimized, as variations in the loading of Pd catalysts and ligands significantly affect the yields.
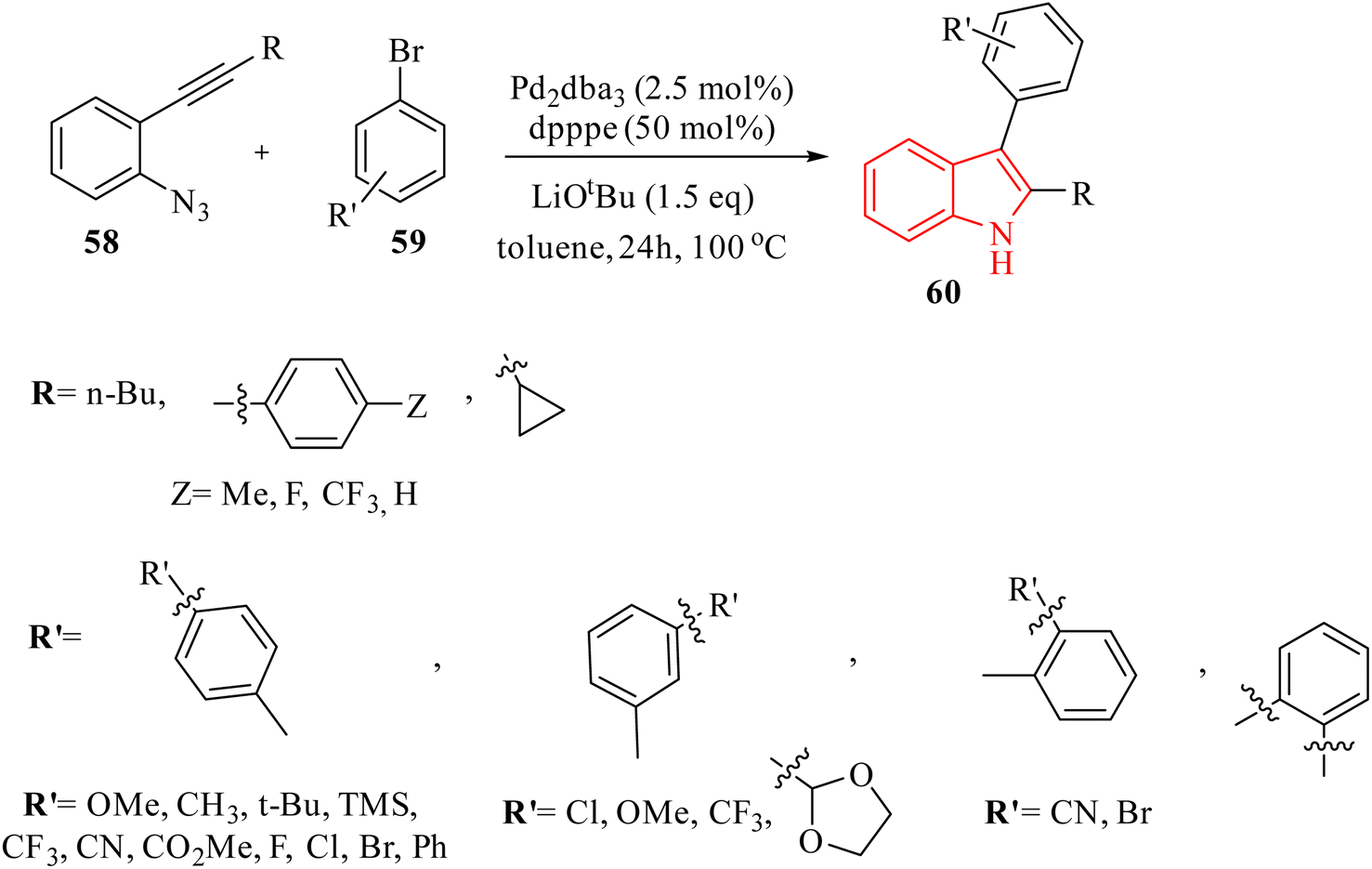 | ||
| Scheme 32 Synthesis of indoles 60. | ||
A mechanism underlying this transformation is depicted in Scheme 33. Two possible pathways were reported. In pathway a, oxidative addition of 59 to a Pd(0) catalyst is generated II. Then, compound 58 is produced III via in situ by a Staudinger reaction. After that, intermediate IV (5-endo-dig) is formed when the nitrogen of the phosphinimine group is attacked the aryl Pd(II)-activated triple bond nucleophilically. IV undergoes reductive elimination to form intermediate X, which is then transformed into the final product through hydrolysis. In pathway B, IV is transformed into VI via back electron donation from Pd. Following migratory insertion, VII is formed and then isomerized into intermediate VIII. Lastly, protonation of intermediate VIII is produced final product 60 and a Pd(II) species, which is then reduced to the Pd(0) catalyst by V. The authors also noted that the reaction mechanism was complex, requiring further investigation to fully understand the pathways involved.136
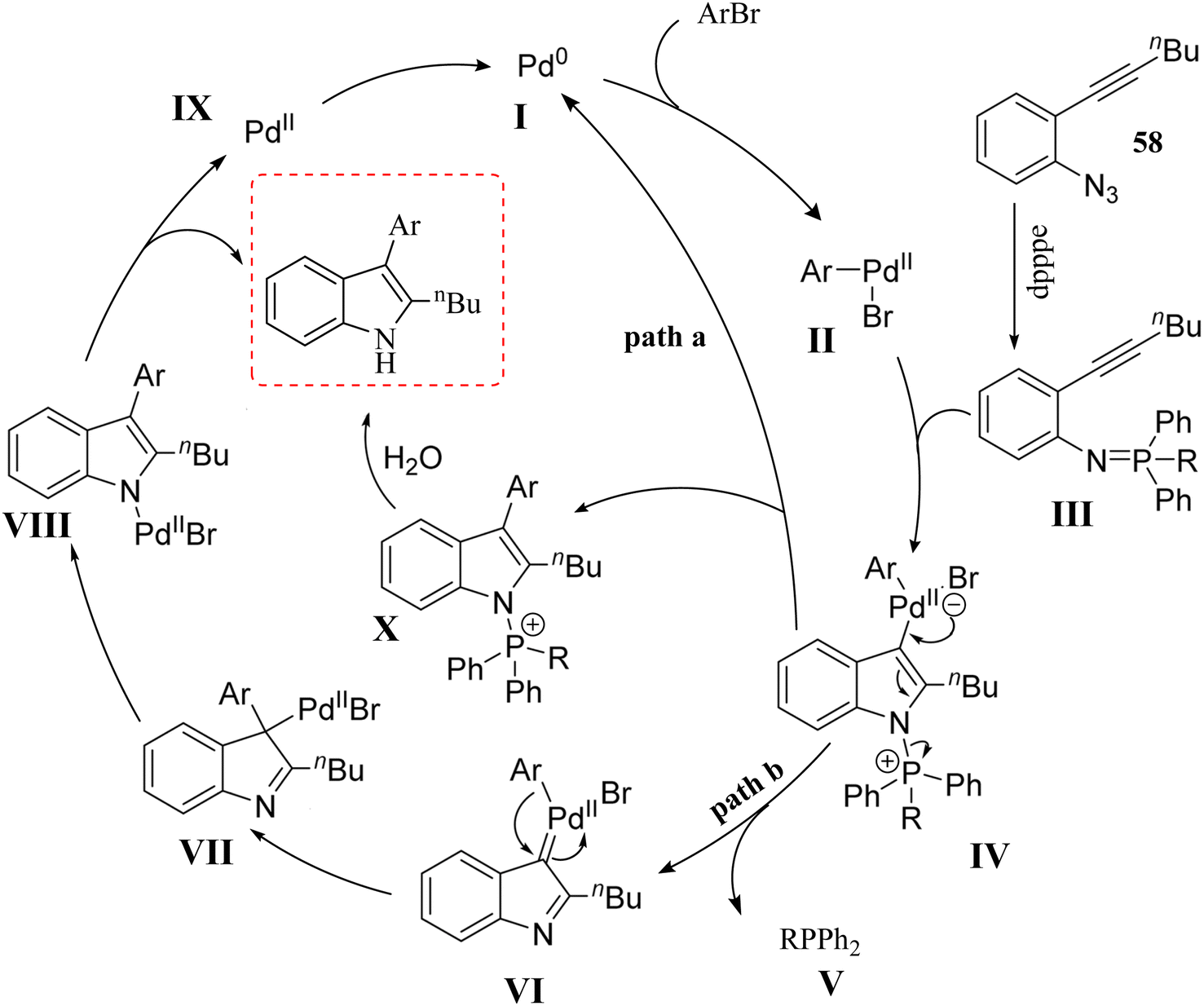 | ||
| Scheme 33 A mechanism for the reactions of 2-alkynyl arylazide derivatives 59 with aryl bromide 60 catalyzed by Pd. | ||
In 2021, Li and colleagues137 reported on the synthesis of functionalized pyrroloindoline derivatives 63 via a one-pot reaction catalysed by Pd(OAc)2. This reaction involved the use of thioacetamide derivatives 62 and 3H-indol-3-one derivatives, which were produced in situ from 2-alkynyl arylazide derivatives 61 when accompanied by Cs2CO3 in 1,4-dioxane at 90 °C (Scheme 34). The derivatives of 63 were generated in yields ranging from 56% to 84%. However, when substrates with different alkyl groups on the alkyne carbon (R′ = n-C6H13 or cyclohexane) were used, the desired products could not be obtained (Z). Instead, only the first-step product was observed. One challenge highlighted was the unstable properties of 3H-indol-3-one derivatives, which could have led to partial decomposition after column chromatography, complicating their use in further reactions. Also, existing synthetic techniques for pyrroloindoline derivatives often suffer from drawbacks such as the need for specific reagents, narrow substrate scope, and harsh reaction conditions, indicating a demand for more efficient synthetic strategies.
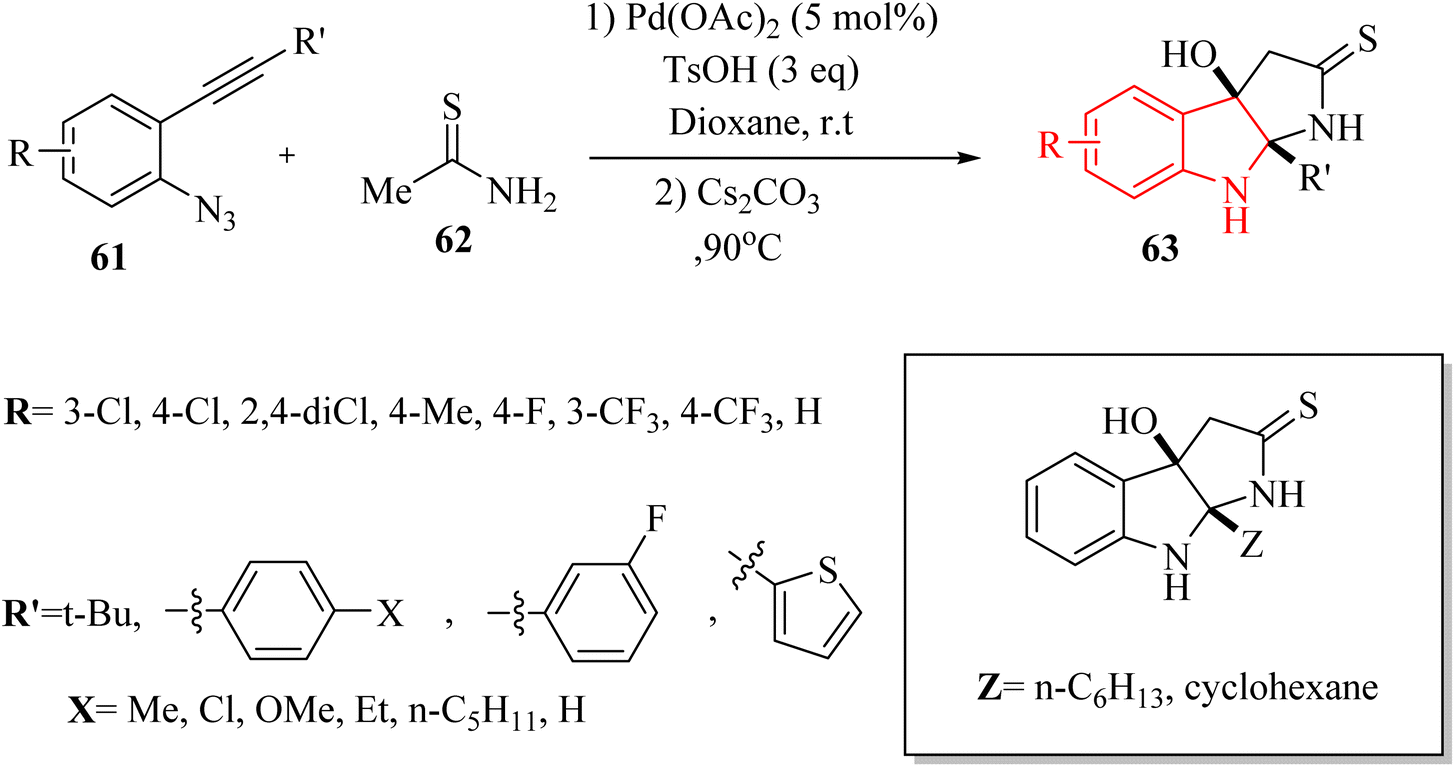 | ||
| Scheme 34 Synthesis of pyrroloindoline derivatives 63. | ||
A mechanism underlying this transformation is depicted in Scheme 35. To activation of 61, Pd-(OAc)2 is coordinated with the alkyne to generated I. In the following, it is attacked by azido group to formed complex III. Next, III is formed when N2 released from II. After that, IV is produced when III trapped by TsOH. Next, V is formed after demetalation and protonation of IV. Then, VII is produced following a 1,3-Ts shift of compound V and the desulfonation of compound VI, using Cs2CO3 at 90 °C. In the following, the carbon and/or nitrogen atom of 3 is attacked VII, resulting in the formation of VIII and/or IX, which then undergoes intramolecular cyclization to yield the 63 (pathway A). Next, VII is captured by the sulfur atom of three, resulting in the formation of X and/or XI, which subsequently cyclized to produce XII and/or XIII along with their resonance forms XIV and/or XV. Ultimately, a rearrangement of O and/or N yielded the 63 (pathway B).137
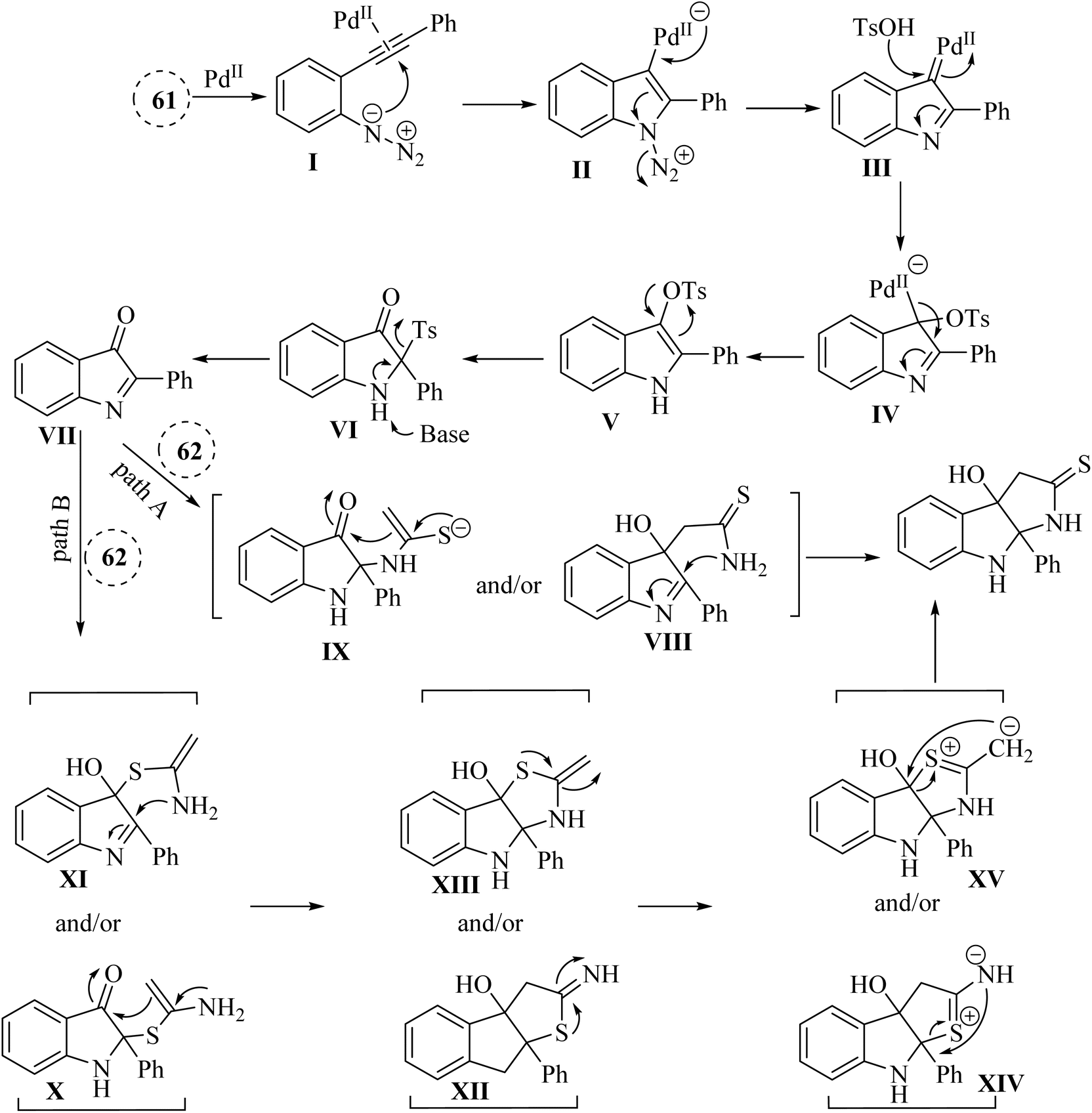 | ||
| Scheme 35 A mechanism for the reactions catalyzed by palladium involving 2-alkynyl arylazide derivatives 61 and thioacetamide derivatives 63. | ||
In 2020, Zhou and colleagues138 developed Pd-catalysed one-pot approach for synthesizing 3-hydroxy-2-oxindole derivatives 65 from in situ-generated 2-hydroxyl-indoline-3-one derivatives, derived from 2-alkynyl arylazide derivatives 64 in the presence of Cs2CO3 in 1,4-dioxane at 90 °C (Scheme 36). It should be noted that various groups tolerated. The resulting products gave in yields ranging from 51% to 93%. An acyloin rearrangement process occurred during the reactions. The authors displayed that further studies on the application of acyloin rearrangements are underway, suggesting a gap in understanding the broader implications and potential applications of this reaction process.
 | ||
| Scheme 36 Synthesis of 3-hydroxy-2-oxindole derivatives 65. | ||
A mechanism underlying this transformation is depicted in Scheme 37. Initially, activation of compound 64 is produced III, which could be captured by TsOH·H2O to form V. Then, VII is generated following a 1,3-Ts shift of V and a subsequent reductive desulfonation. Subsequently, the hydroxylation of TsOH·H2O crystal water or water is generated by acid-base neutralization to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds of VII leads to the formation of VIII. Ultimately, the corresponding product 65 is obtained via an acyloin rearrangement of VIII. The authors mentioned that the proposed mechanism for converting 1-azido-2-(phenylethynyl)benzene to 3-hydroxy-2-oxindole was speculative, indicating a need for more definitive mechanistic studies.138
N double bonds of VII leads to the formation of VIII. Ultimately, the corresponding product 65 is obtained via an acyloin rearrangement of VIII. The authors mentioned that the proposed mechanism for converting 1-azido-2-(phenylethynyl)benzene to 3-hydroxy-2-oxindole was speculative, indicating a need for more definitive mechanistic studies.138
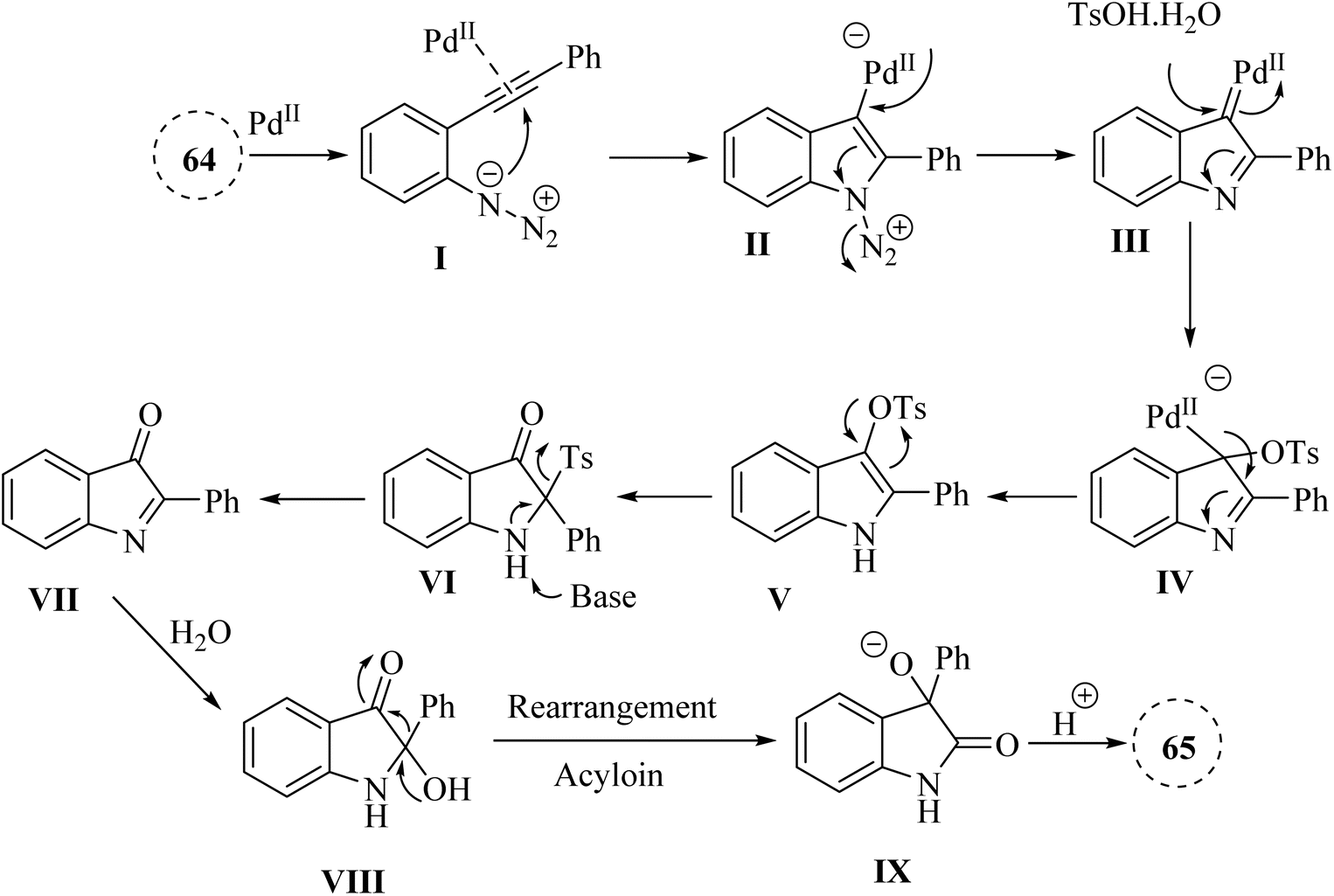 | ||
| Scheme 37 A mechanism for the reactions of 2-alkynyl arylazide derivatives 64 into 65 catalyzed by Pd. | ||
In 2020, Li et al.139 published a significant advancement in the synthesis of hybrid imidazo-indole derivatives 68 (Scheme 38). The study presented a pioneering approach for the rapid and effective production of these compounds through a Pd-catalysed one-pot annulation process. This process involved the reaction of thioureas 67 with 3H-indol-3-one derivatives, which were produced in situ from 2-alkynyl arylazide derivatives 66 in the presence of Cs2CO3 in 1,4-dioxane at 90 °C. The utilization of easily accessible starting materials further underscored the practical significance of this methodology. The study reported impressive product yields, with 66 products being obtained in the range of 56% to 97% yields, and 67 products achieved a remarkable 97% yield. However, it should be noted that 67 with aryl or bulky substituted groups did not yield the desired products, possibly due to steric effects. The authors were emphasized the importance of developing practical techniques for synthesizing imidazoloindoline derivatives, which remains underexplored.
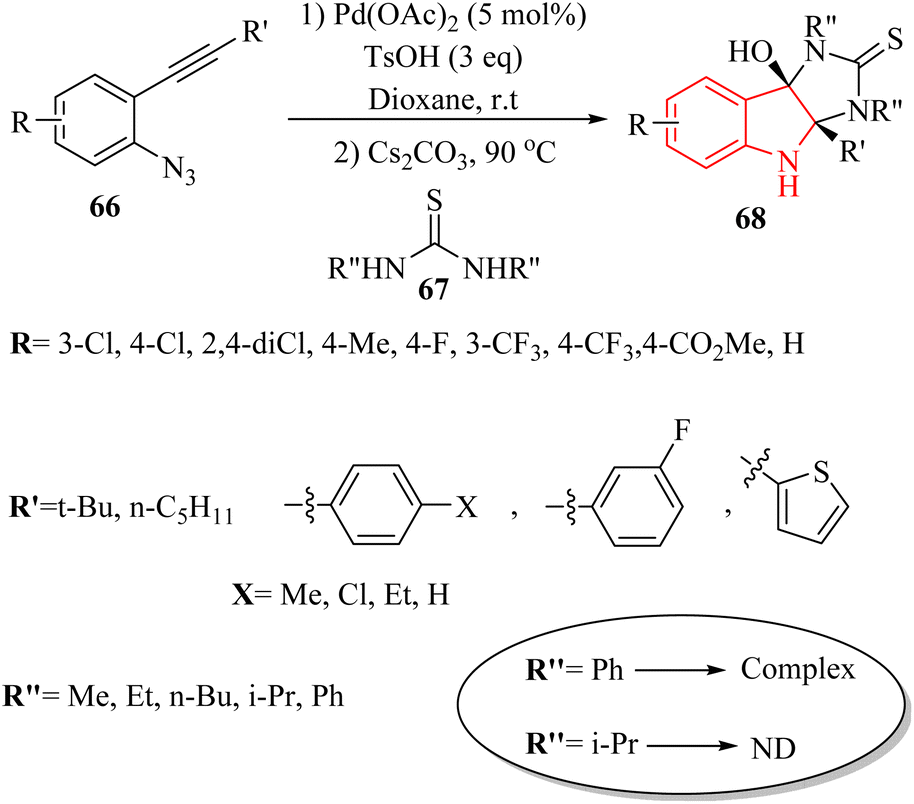 | ||
| Scheme 38 Synthesis of indole-2-thione derivatives 68. | ||
A mechanism underlying this transformation is depicted in Scheme 39. Compound 66 is transformed into VII through the capture of alpha-imino Pd carbene, followed by a 1,3-Ts shift and a reductive desulfonation sequence. Following this, intermediates VIII and/or IX is generated via the intermolecular amination of 67 with VII, which could then be further transformed into 68 through intramolecular amination cyclization (pathway A). Next, the sulfur atom of 67 is attacked the VII, resulting in the formation of X and/or XI. This is followed by an amination of intramolecular, producing XII and/or XIII and their resonance forms XIV and/or XV. Finally, the corresponding product is obtained through a final rearrangement step (pathway B).139
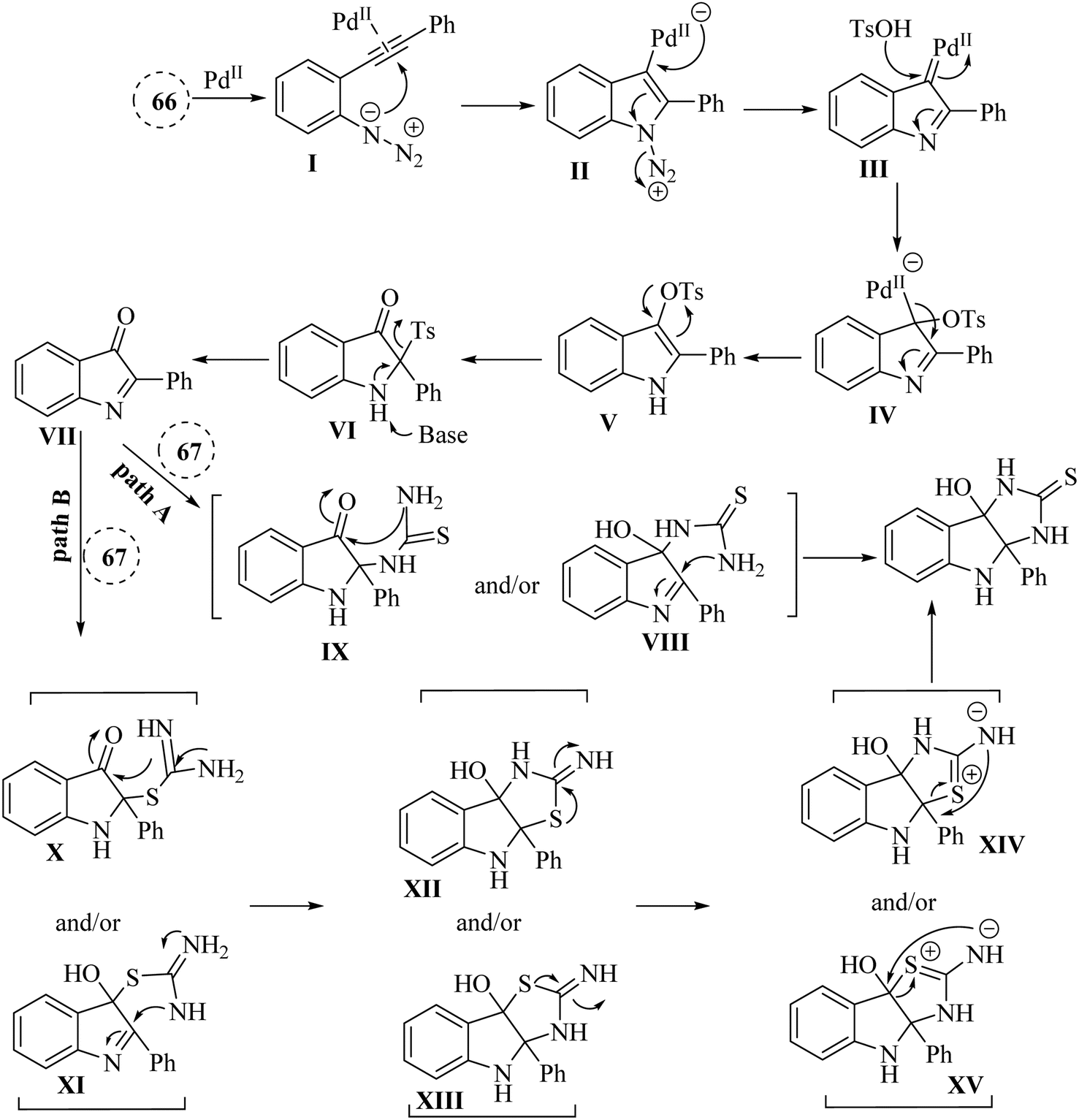 | ||
| Scheme 39 A mechanism for reactions of 2-alkynyl arylazide derivatives 66 and transformed into 68 catalyzed by Pd. | ||
 | ||
| Scheme 40 Synthesis of 3-haloindole derivatives 70. | ||
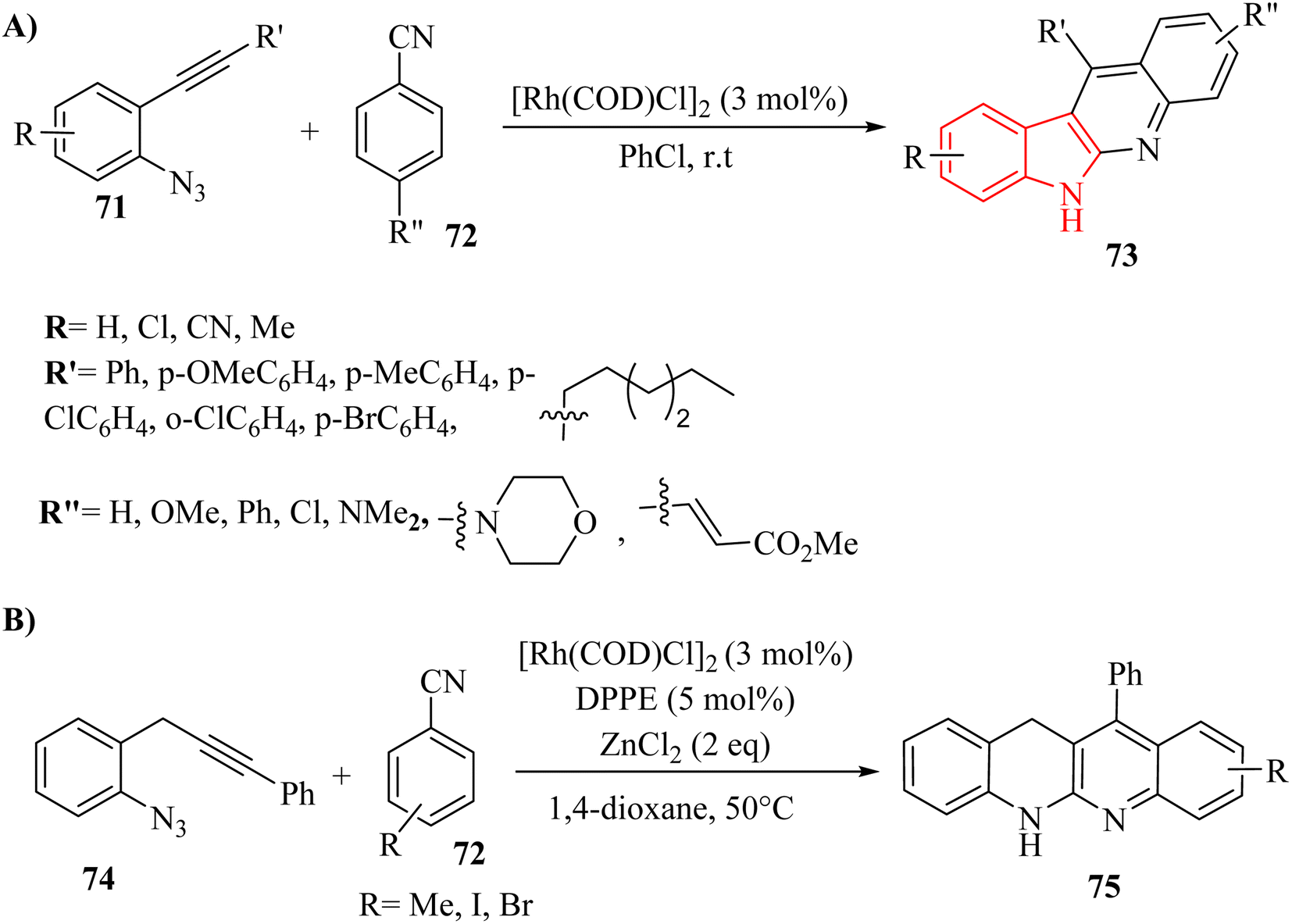 | ||
| Scheme 41 (A) Synthesis of indolo-quinolone derivatives 73, (B) synthesis of dibenzonaphthyridone derivatives 75. | ||
A mechanism underlying this transformation is depicted in Scheme 42. Firstly, intermediate I is formed via the coordination of compound 72 with [Rh(COD)Cl]2, which then interacted with compound 71 to produced II. Then, intermediate III is generated via the release of N2 in II. Next, III undergoes migratory insertion and the departure of rhodium and formed IV. Lastly, the desired product 73 is obtained from IV via an internal [4 + 2] cycloaddition reaction, followed by a 1,5-proton shift.141
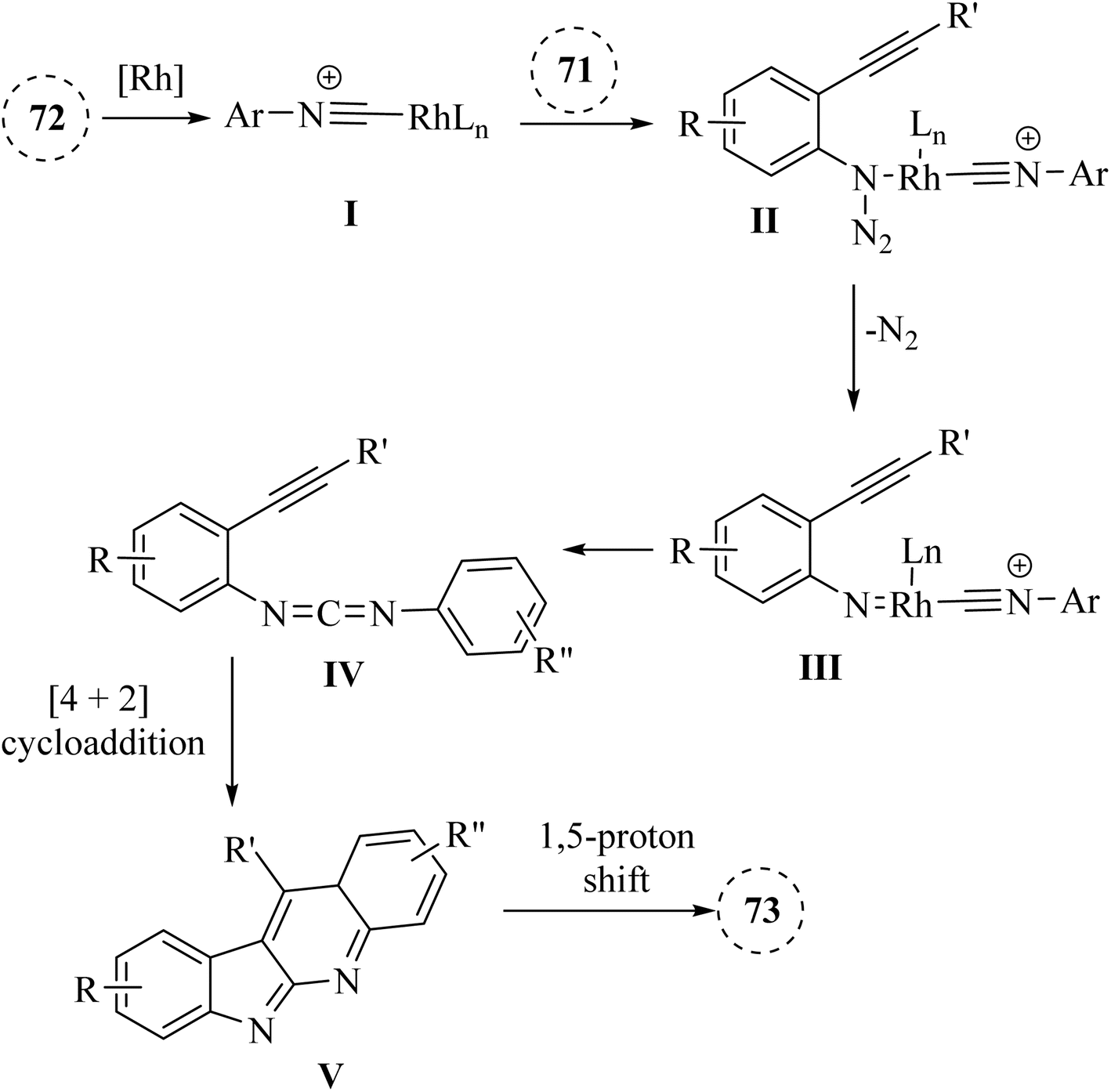 | ||
| Scheme 42 A mechanism for the reactions of 2-alkynyl arylazide derivatives 71 with arylisocyanide derivatives 72 catalyzed by Rh. | ||
Zhang and colleagues142 detailed a self-relay Rh(I)-catalyzed cascade reaction that systematically constructs two different sigma-donor/pi-acceptor ligands. This process results in the synthesis of intricate fused heterocyclic systems through relay-catalyzed nitrene transformations and a series of cyclization steps. This approach provided a straightforward and efficient strategy for assembling the pyrrolo-indole framework (78 and 80), which was an essential structural element present in many valuable natural products and pharmaceuticals (Scheme 43A and B). Moreover, authors evaluated the reactivity of isonitrile derivatives 77 and CO 79 (Scheme 43B). The findings demonstrated that isonitrile derivatives 77 exhibit greater reactivity compared to their isoelectronic counterpart 79, whether in coupling reactions with azide derivatives or in aza-Pauson–Khand-type cyclization. The introduction of two different s-donor/π-acceptor ligands resulted in relatively lower yields compared to using two isonitrile derivatives. Variation in the electronic nature or positions at the phenyl ring of aryl azide derivatives did not remarkably affect reactivity, indicating limited influence of substituents. Aliphatic substituents illustrated little reactivity in the transformation, suggesting a limitation in substrate scope. The presence of unexpected side reactions was common in late transition-metal-catalyzed nitrene transformations, which could complicate the reaction outcomes.
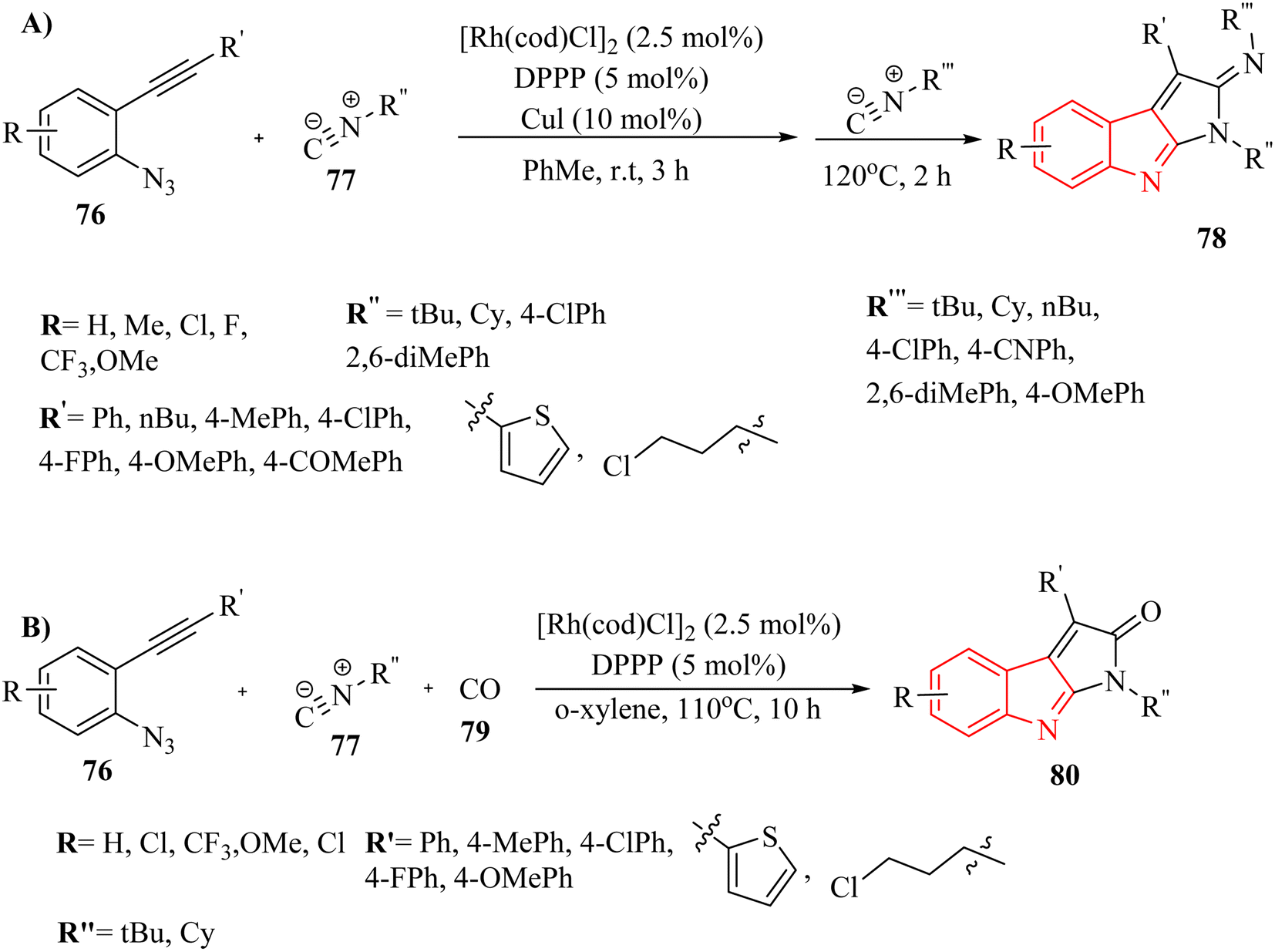 | ||
| Scheme 43 (A) Synthesis of pyrrolo-indole derivatives 78 with isonitriles 77, (B) synthesis of pyrrolo-indoles 78 with isonitriles 77 and CO 79. | ||
A mechanism underlying this transformation is depicted in Scheme 44. Initially, compound 76 and 77 are reacted and generated intermediate I along with release of N2. Subsequently, Intermediate I has the capacity to generate a new intramolecular C–C bond, resulting in the formation of rhodacycle II. This is occurred through π-complexation with Rh(I) followed by an oxidative cyclometalation step. Subsequently, the introduction of an additional sigma-donor/pi-acceptor ligand, such as RCN or CO, into the Rh–C bond of II is facilitated the creation of rhodium complex III. Finally, the targeted derivatives (78 and 80) were synthesized via a reductive elimination process, which concurrently restored the active Rh(I) catalyst.142
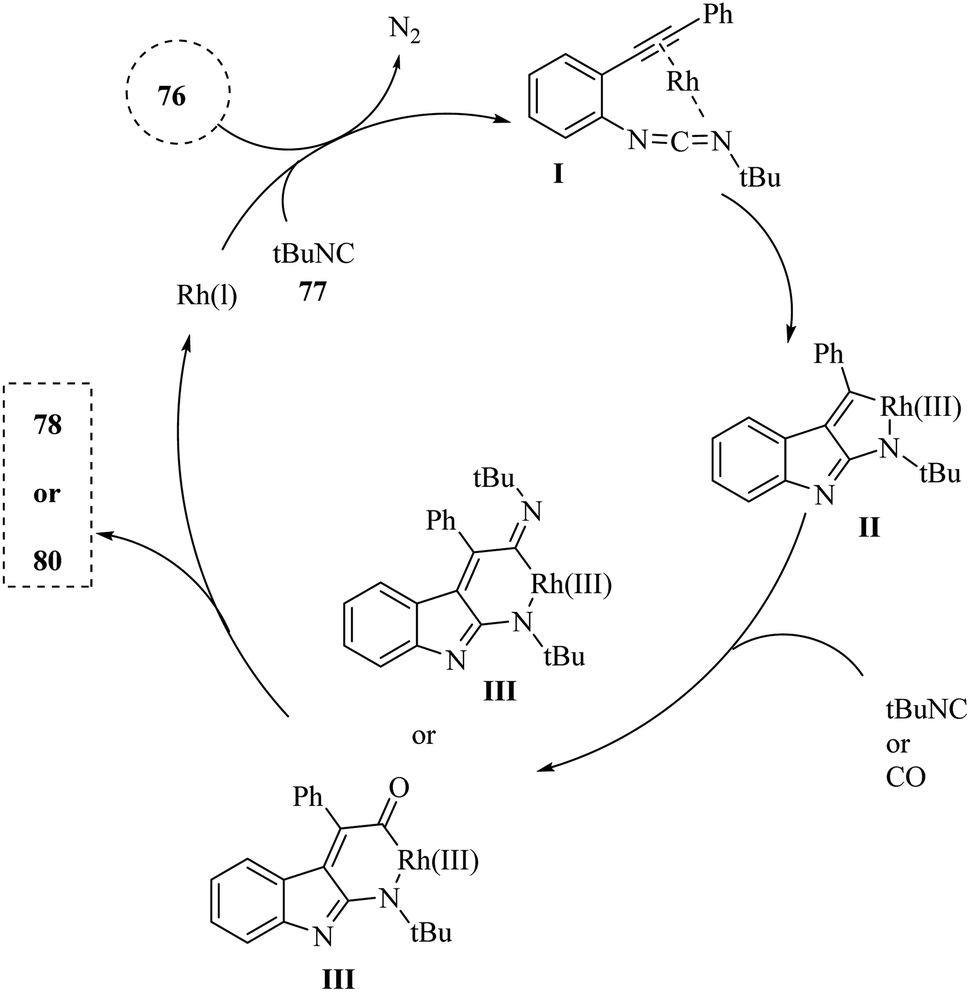 | ||
| Scheme 44 A mechanism for the reactions of 2-alkynyl arylazide derivatives 76 with isonitrile derivatives 77 or CO 79 catalyzed by Rh. | ||
2.2 Synthesis of quinolines
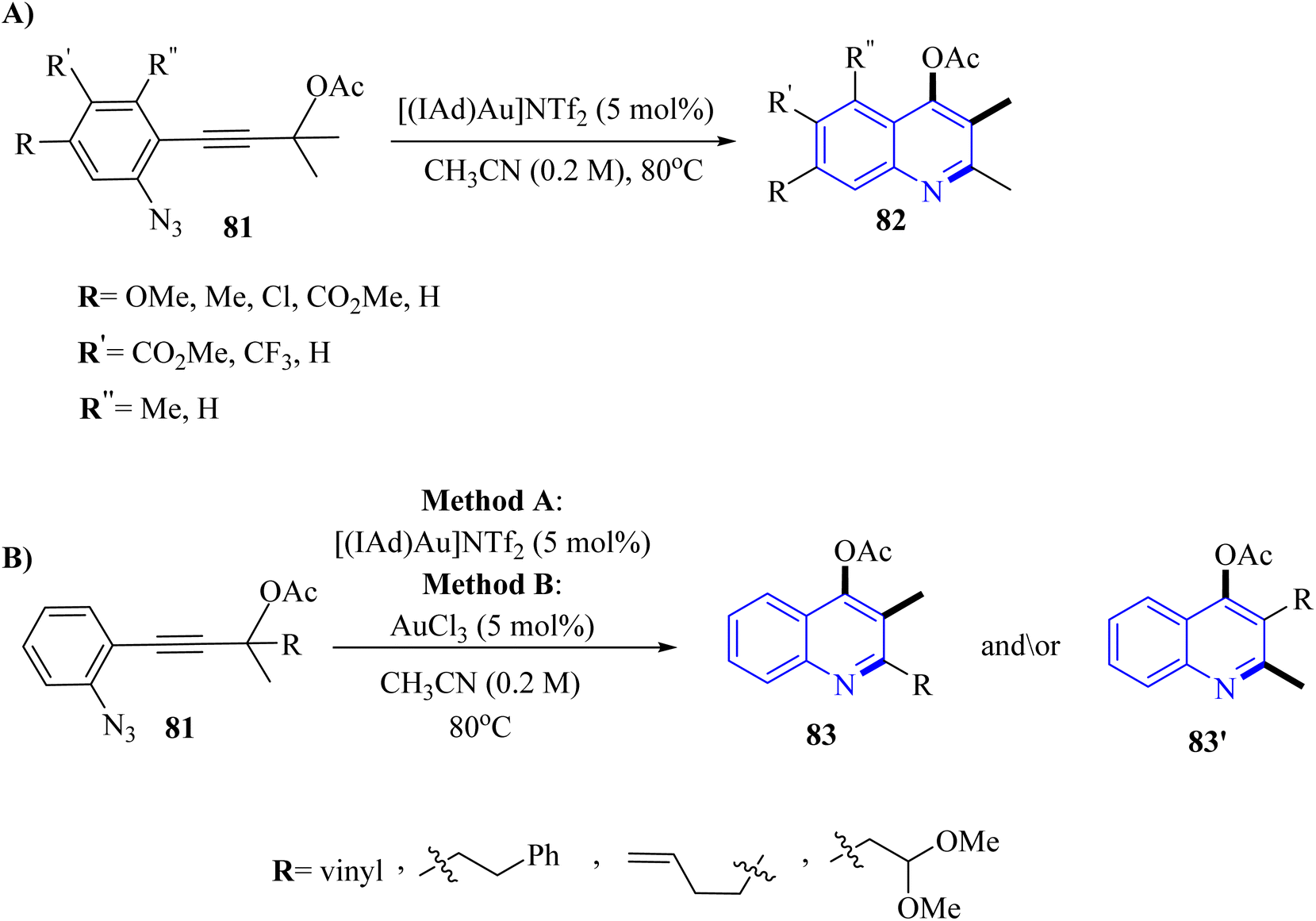 | ||
| Scheme 45 (A) Scope of aryl substitution, (B) scope of 1,2-group shift selectivity. | ||
A mechanism underlying this transformation is depicted in Scheme 46. Initially, when compound 81 is treated with an Au catalyst, substrate I is anticipated to experience a more favorable 1,3-acetoxy shift, leading to the generation of allene II. Next, the azide's nucleophilic addition to the Au-activated species. This resulted in the formation of a cyclized intermediate (referred to as intermediate III). Subsequently, via a 1,2-shift of the R′ group and the catalyst's regeneration, this intermediate is evolved into quinoline VI. Finally, cationic intermediate IV could be generated via 2 pathways. Path I: could be generated from III, and path II: could be produced through an Au carbenoid of type V (path II).143
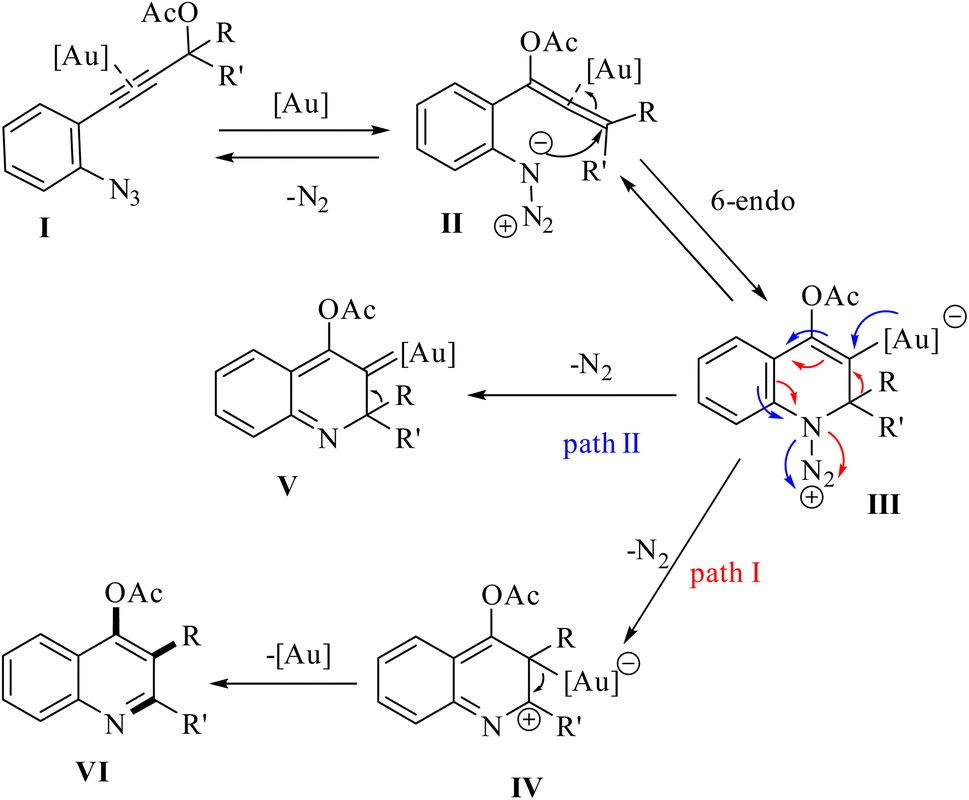 | ||
| Scheme 46 A mechanism for the gold-catalyzed transformation of 81 into polysubstituted quinolones. | ||
 | ||
| Scheme 47 Synthesis of pyrrolo-quinolin-1-one derivatives 85. | ||
2.3 Synthesis of 2-aminobenzophenones (bimetallic (Pd/Cu)-catalyzed transformation)
In the year 2021, Yang et al.144 presented a new and innovative process involving a single-step, three-part reaction of 2-alkynyl arylazide derivatives 86. The study was discussed the partial hydrolysis of the ester group under basic reaction conditions as a limitation, which affected the yield of the corresponding product. This process entailed the Pd-catalysed generation of 3-hydroxy-3-phenylindolin-2-one derivatives, followed by the hydrolysis of amide bonds and Cu-catalysed decarboxylation, resulting in the production of 2-aminobenzophenone derivatives 87 (Scheme 48). The reaction done when accompanied by Cs2CO3 and NaOH in 1,4-dioxane at 90 °C. The findings from this study revealed that the reaction was capable of accommodating a wide variety of structurally diverse 86, with yields of up to 90%. However, when the reaction was repeated using a substrate containing a steric hindered group (R′ = t-Bu), the desired product was not obtained, leading only to the formation of an acyloin rearrangement product with a yield of 76%. It should be noted that the reaction's yield was remarkably impacted by the use of a radical scavenger, indicating that a radical process might be involved, which complicates the reaction mechanism.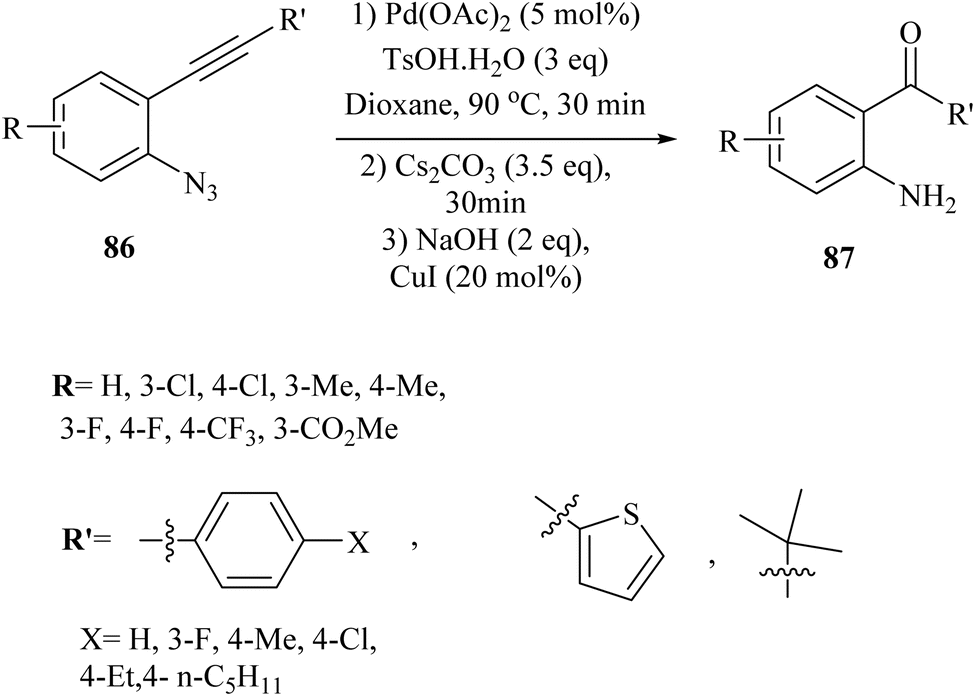 | ||
| Scheme 48 Synthesis of 2-aminobenzophenone derivatives 87. | ||
A mechanism underlying this transformation is depicted in Scheme 49. Initially, alpha-imino Pd carbene III is generated from 87 which is activated by Pd catalyst. Then, III is attacked by TsOH·H2O and produced complex IV. Next, intermediate V is obtained from a rearrangement of IV and also Pd catalyst is regenerated. After a 1,3-Ts shift process, VI is formed from V in the presence of Cs2CO3. After that, VII is produced via a reductive desulfonation of VI. Then, VIII is formed via a hydrolysis of CN bond of VII. In the following, an acyloin rearrangement to give IX. Next, X is formed via a hydrolysis of amide bond of IX. After that, XI is generated via a Cu-catalysed decarboxylation of X. Finally, it is oxidized to product 87.144
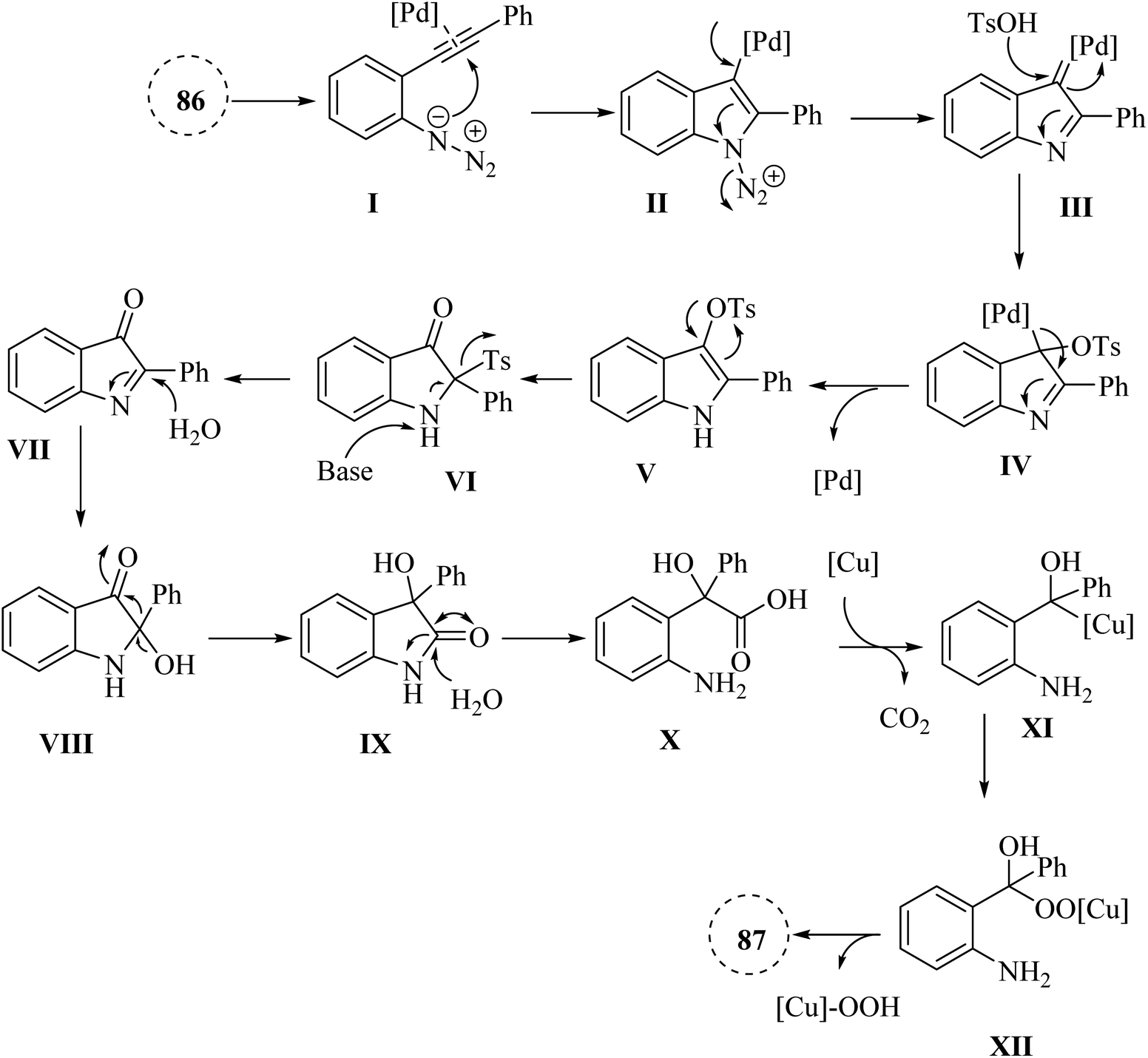 | ||
| Scheme 49 A mechanism for the reactions of 2-alkynyl arylazide derivatives 86 and transformed into 87 catalyzed by Pd and Cu. | ||
3. 2-Alkynylbenzyl azides
3.1 Synthesis of quinolines
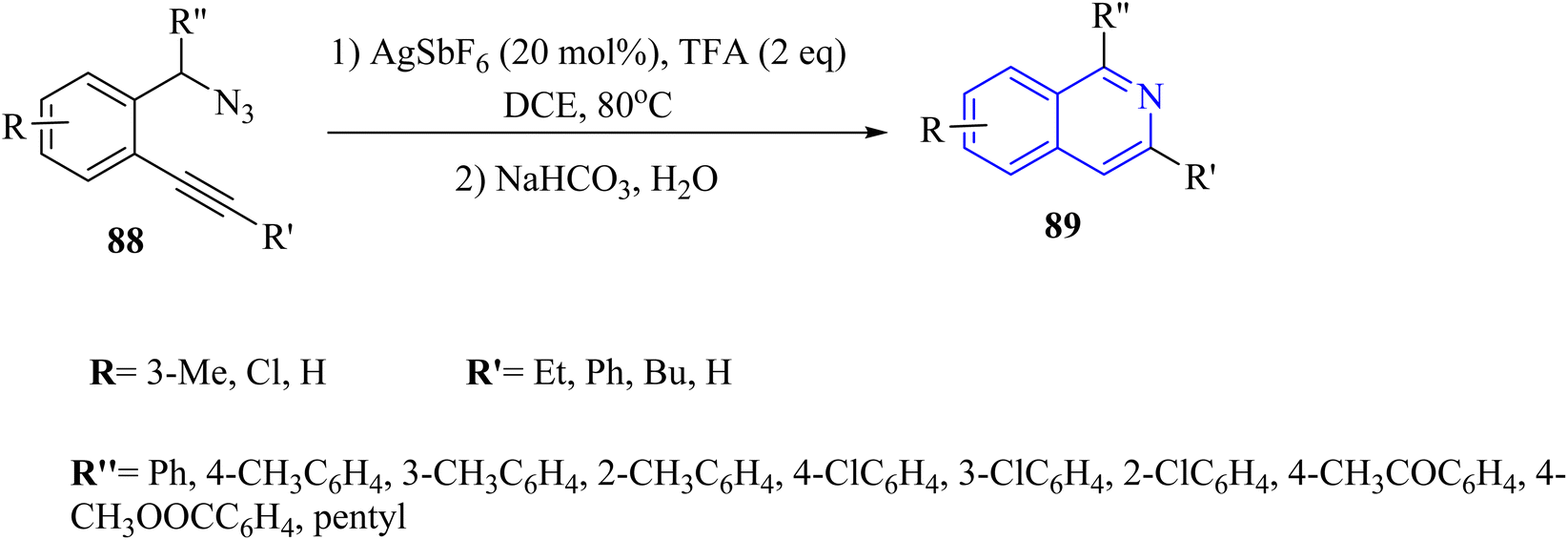 | ||
| Scheme 50 Synthesis of isoquinoline derivatives 89. | ||
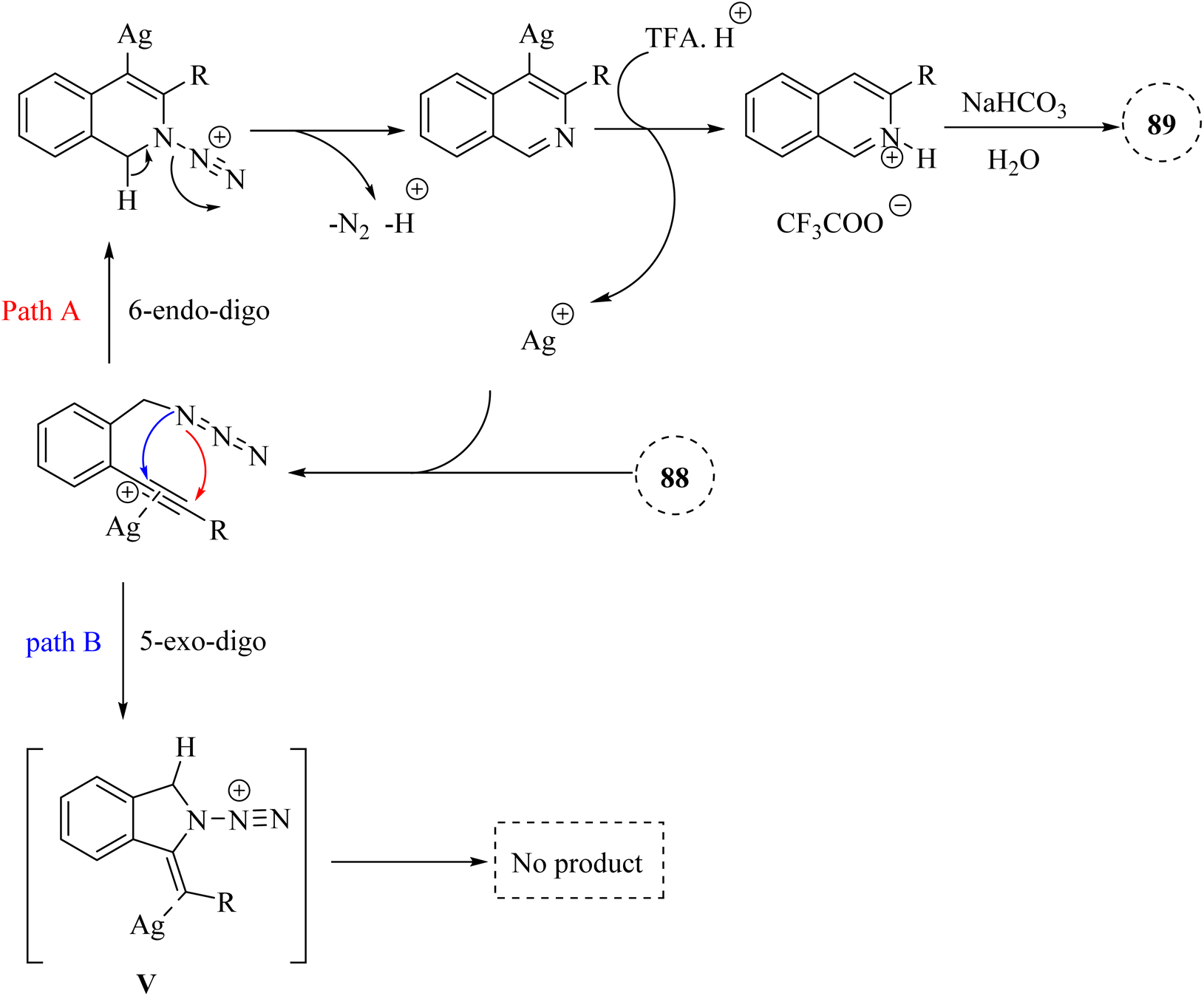 | ||
| Scheme 51 A mechanism for the reactions of 2-alkynyl benzyl azide derivatives 88 and their transformation into isoquinoline 89 catalyzed by Ag. | ||
A mechanism underlying this transformation is depicted in Scheme 51. The Ag catalyst is coordinated with the alkynyl group of 88 to forms complex I. Then, the nitrogen atom selectively is targeted the electron-deficient triple bonds through a 6-endodig cyclization process, resulting in the formation of II. Next, II is released N2 and H+, resulting in the formation of III. After that, III is reacted with TFA and H+, producing IV and regenerating the Ag catalyst for the next catalytic cycle. Lastly, the desired product 89 is obtained via the reaction between IV and saturated NaHCO3. The research did not extensively cover the mechanistic details of the cyclization process, which could be explored further.97
A method involving AgSCF3 and Na2S2O8 promoted trifluoromethylthiolation and cascade cyclization of ortho-propargyl arylazide derivatives (or ortho-alkynyl benzylazide derivatives 90) initiated by a C–C triple bond. This approach enabled the synthesis of valuable SCF3-substituted quinolone 91 (Scheme 52A) and isoquinoline 93 (Scheme 52B) compounds by forming one C(sp2)–SCF3 bond and one C–N bond in a single process.145 Trifluoromethylthio-substituted quinolone derivatives 91 were obtained in 47–89% yields from o-propargyl arylazide derivatives 90. The steric effect at the ortho position had minimal impact on this transformation. Overall, substituents with different electronic effects were well-compatible. The product with an ortho-Me group was detected in trace amounts due to the steric effect of the ortho substituent. Typically, this transformation illustrated comparable electronic and steric effects to those seen with opropargyl arylazide substrates. In this section, TLC analyses revealed an increase in byproducts, possibly resulting from the SCF3 radical's insufficiently selective addition to the C–C triple bond of the substrate. The substrate containing an alkyl group failed to produce the desired product. There were some challenges and limitations about the reaction such as (1) poor solubility was affected NMR spectrum acquisition, (2) inadequate acquisition time led to doublet peak in NMR, and (3) reaction system scalability was posed challenges in product yield.
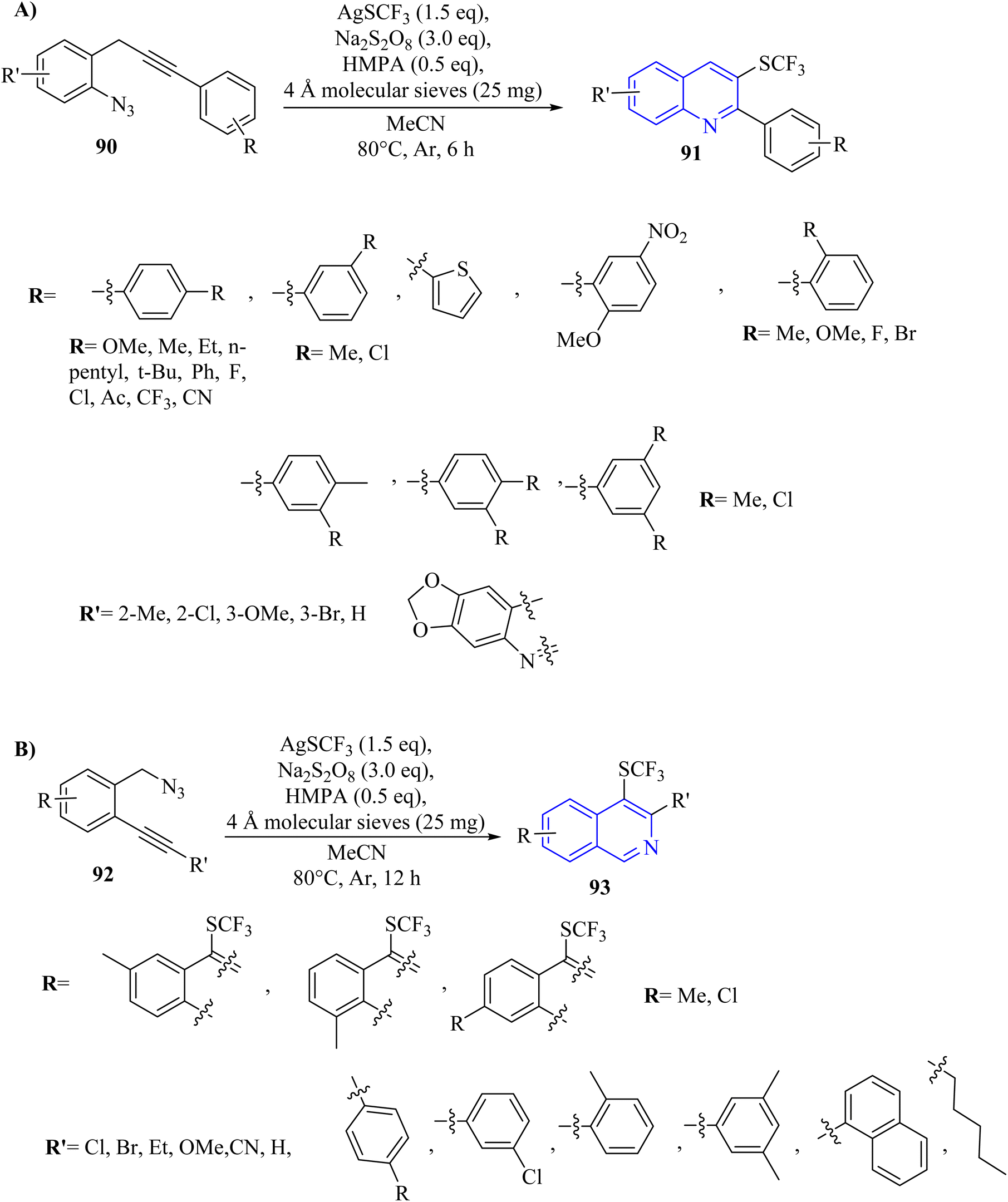 | ||
| Scheme 52 (A) Synthesis of quinolone 91, (B) synthesis of isoquinoline 93. | ||
A mechanism underlying this transformation is depicted in Scheme 53. The SCF3 radical is produced via the oxidation of AgSCF3 by Na2S2O8, which then added to the C–C triple bond of substrate 91, forming I. Then, radical anion intermediate II is generated from I via the subsequent 6-endo-dig cyclization process. In the following, II is released N2 and formed nitrogen radical intermediate III. Next, carbon radical intermediate IV is generated through a 1,3-radical migration of III, which is experienced an additional SET process, transitioning from IV to the Ag(II) species, resulting in the formation of the desired product 92. There is a need for further exploration of the reaction mechanisms involved, particularly the radical pathways that govern these transformations, as indicated by control experiments showing inhibition with specific additive.145
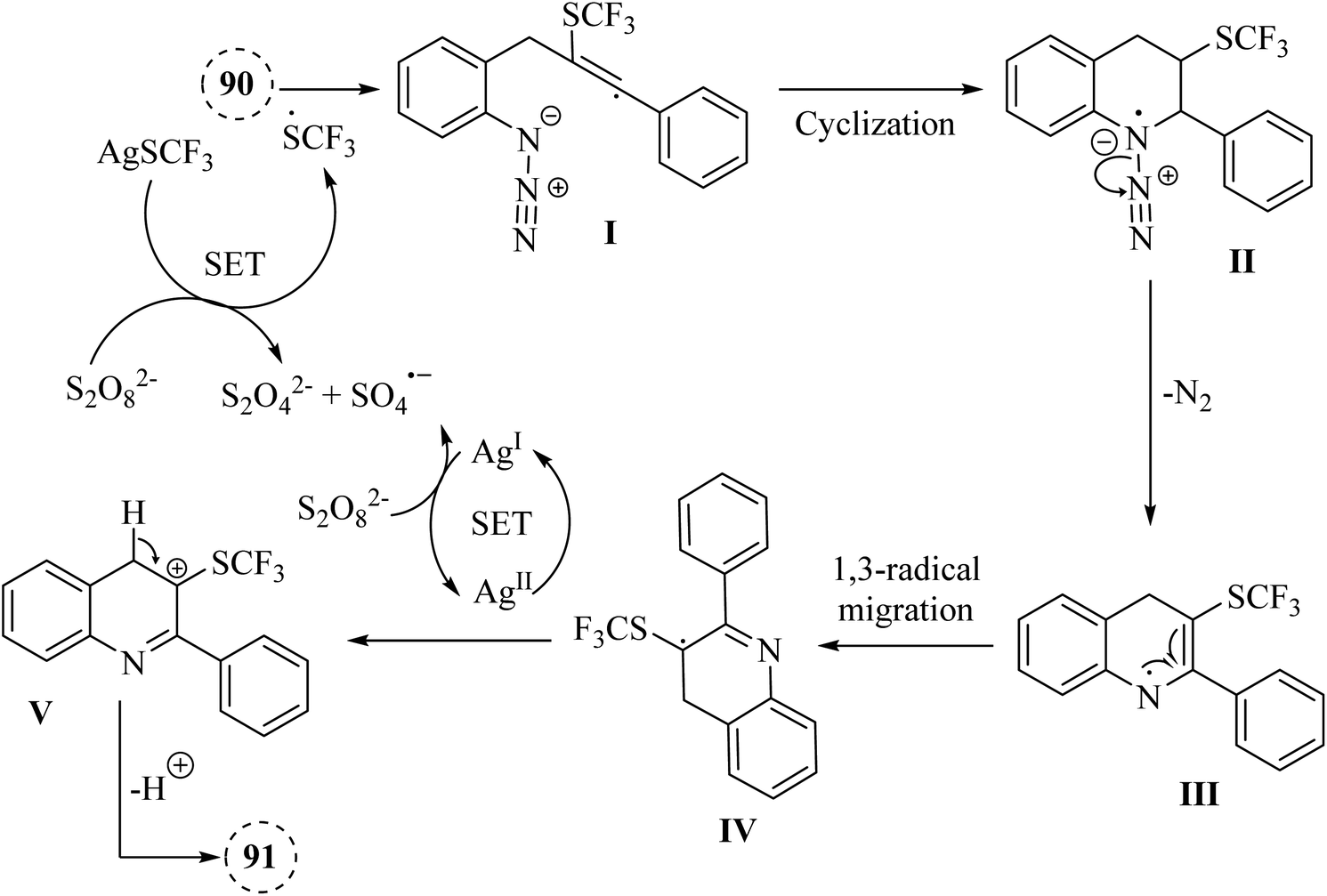 | ||
| Scheme 53 A mechanism for the AgSCF3 and Na2S2O8 promoted trifluoromethylthiolation and cascade cyclization of ortho-propargyl arylazide derivatives 90. | ||
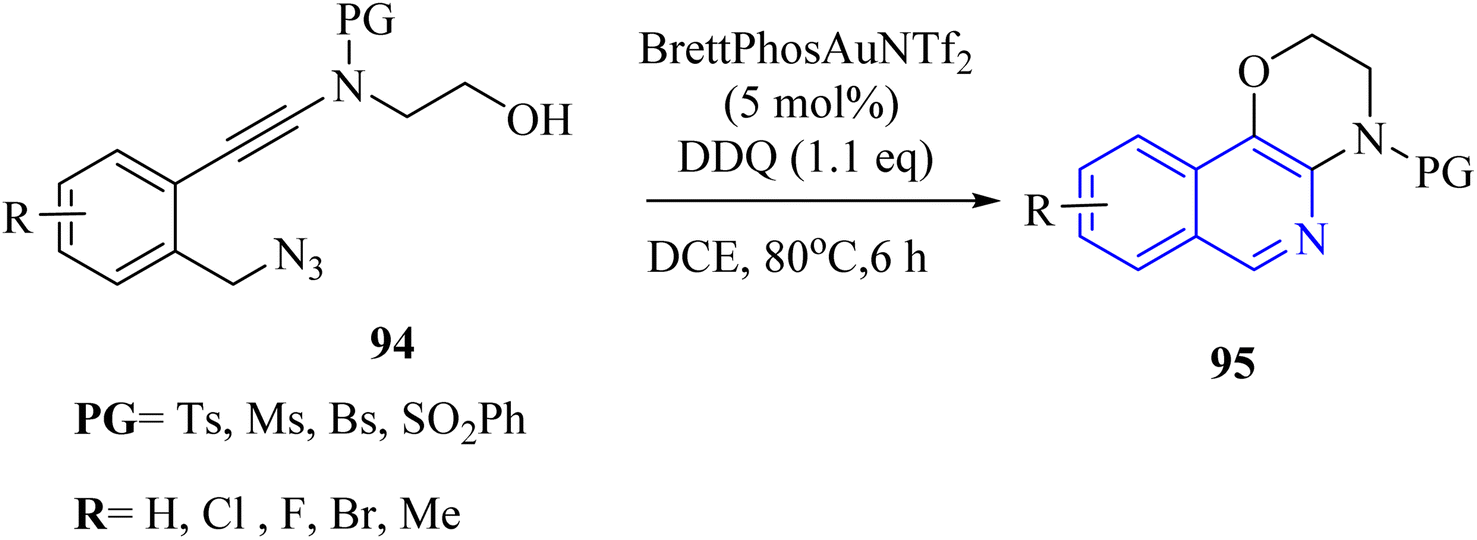 | ||
| Scheme 54 Synthesis of fused isoquinoline derivatives 95. | ||
A mechanism underlying this transformation is depicted in Scheme 55. The process began with the Au-catalysed nucleophilic addition of the azide to the alkyne group, which is then followed by the release of N2, resulting in the formation of the crucial alpha-imino Au carbene intermediate II. The next step is involved the OH group intramolecularly trapping the alpha-imino Au carbenoid, forming III. This intermediate then undergoes proton transfer, deauration, and dehydrogenative oxidation, ultimately producing the desired product 95.146
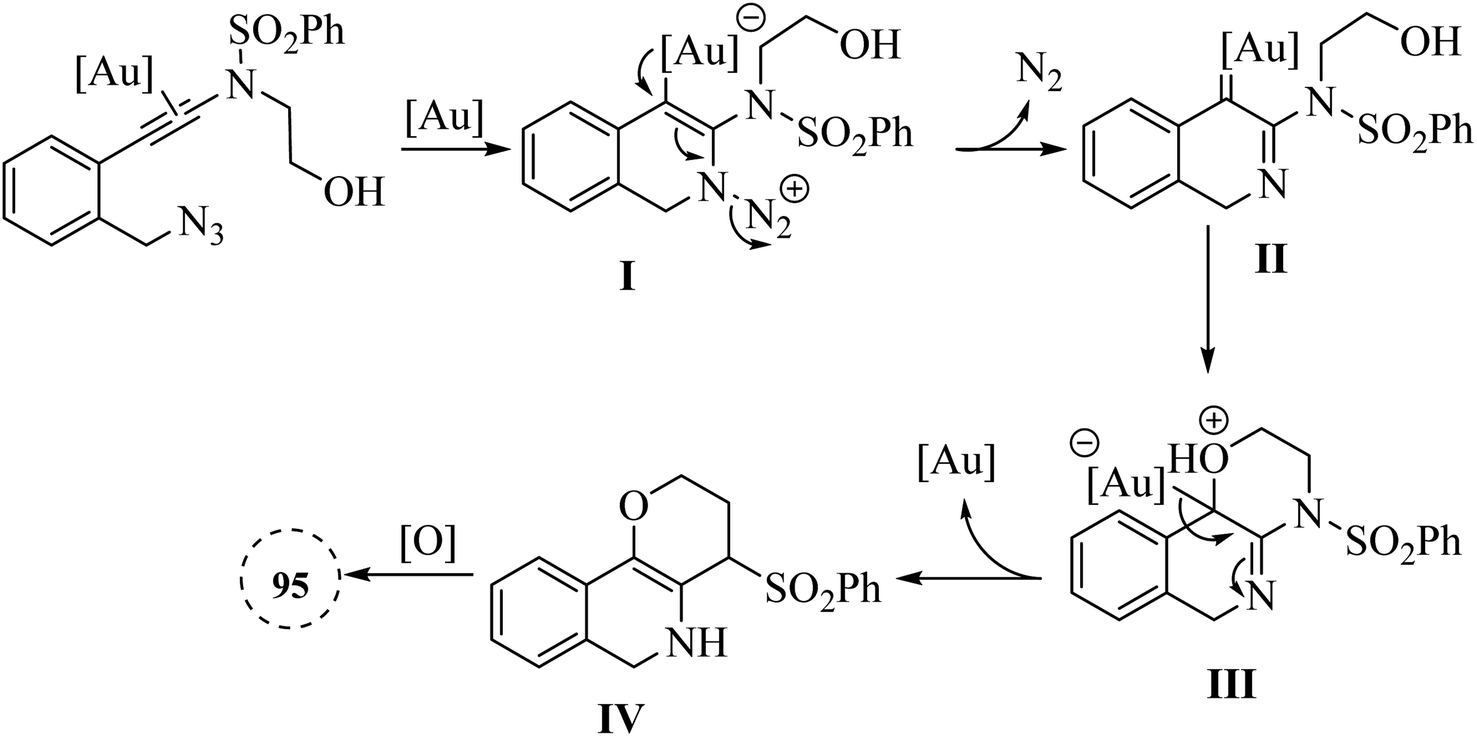 | ||
| Scheme 55 A mechanism for the reaction of ynamide derivatives 94 and their transformation into 95 catalyzed by Au. | ||
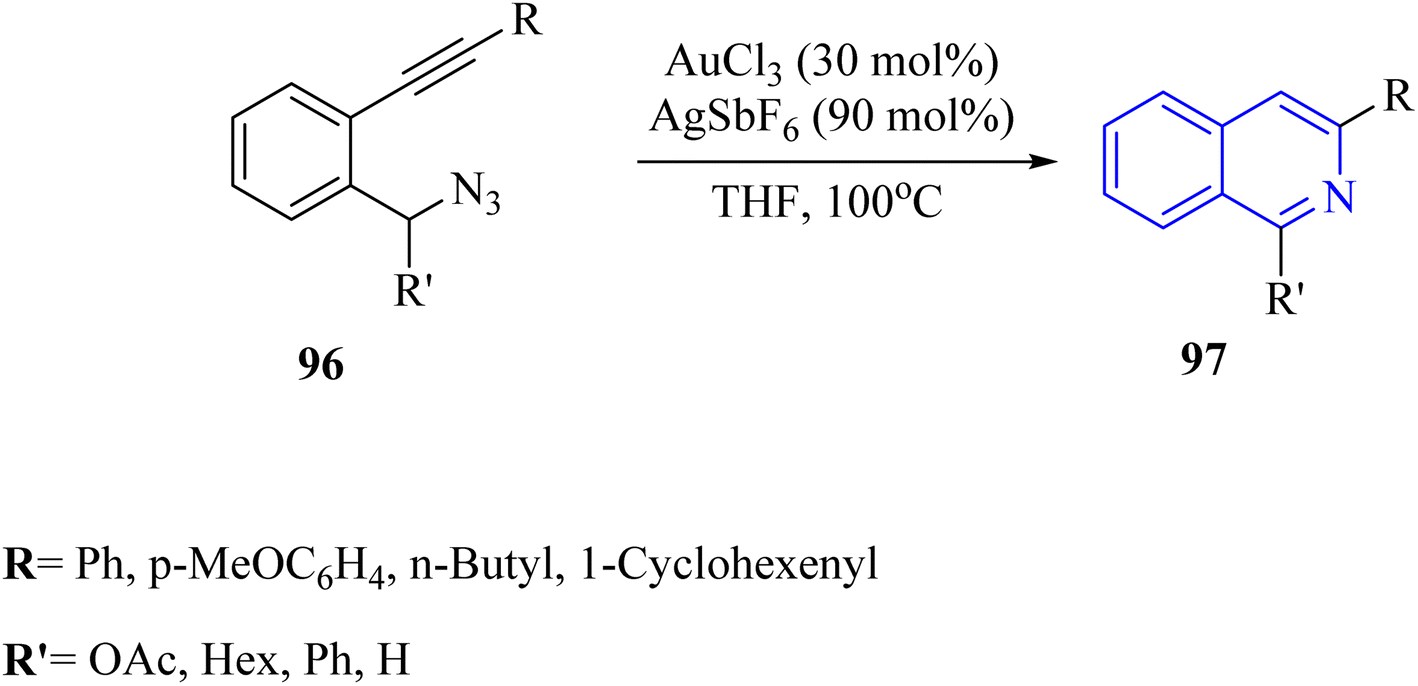 | ||
| Scheme 56 Synthesis of isoquinoline derivatives 97. | ||
A mechanism underlying this transformation is depicted in Scheme 57. First, the triple bond of compound 96 is coordinated with the Au catalyst, increasing the alkyne's electrophilicity and forming intermediate I. Then, the nitrogen atom is performed a nucleophilic attack on the electron-deficient alkyne, resulting in the formation of intermediate II. Compound III is generated via the removal of N2 and H+. The protonolysis of III then leads to the creation of the corresponding product 97 and restored the desired catalyst.147
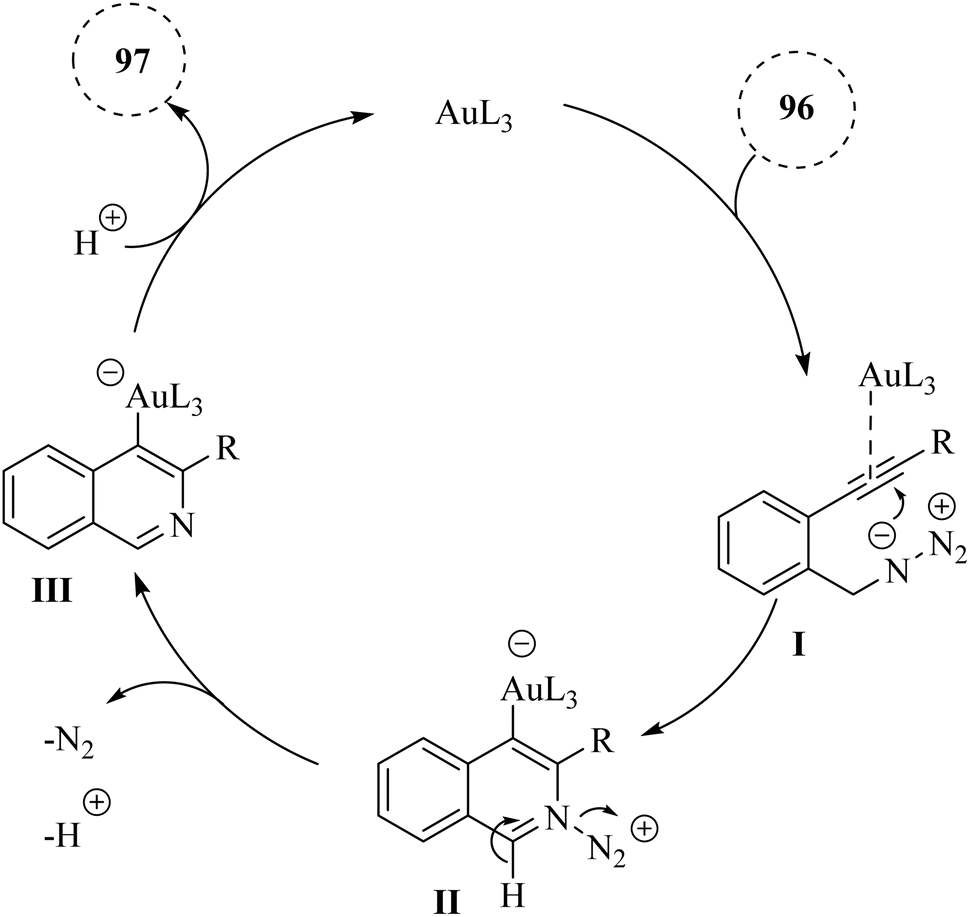 | ||
| Scheme 57 A mechanism for the reaction of 2-alkynyl benzyl azide derivatives 96 and their transformation into 97 catalyzed by Au and Ag. | ||
 | ||
| Scheme 58 Synthesis of bromoisoquinoline and bromoisoquinolone. | ||
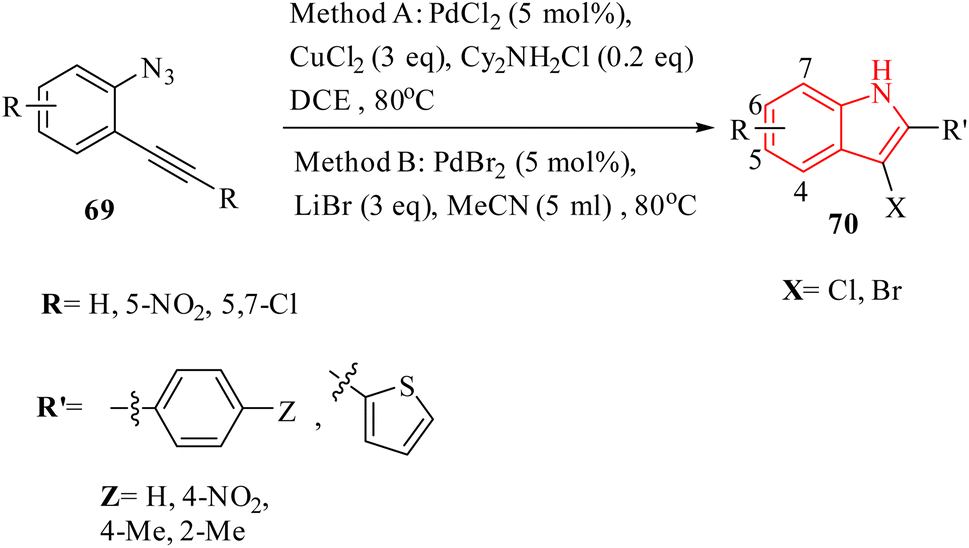
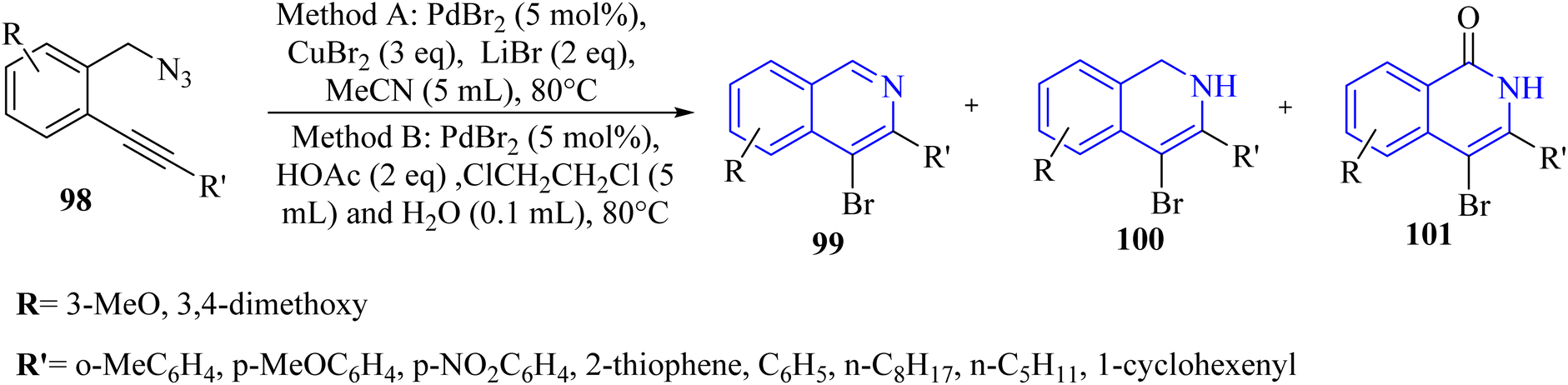
A novel and selective approach for synthesizing 4-haloisoquinoline derivatives 103 involved halopalladation cyclizations of alkyne derivatives with azide derivatives (Scheme 59).140 Using PdX2 (X = Cl or Br) and halide sources, various 2-alkynyl benzyl azide derivatives 102 efficiently underwent the halopalladation cyclization reaction, resulting in the formation of haloisoquinoline derivatives 103 (Scheme 59). Various derivatives of 102 were smoothly converted into the corresponding 103 derivatives (up to 77% yield) by treating them with CuCl2, PdCl2, and Cy2NCl (method A). The authors discovered that substrates featuring an electron-donating aryl group at the terminal alkyne were favorably accepted, whereas those containing a NO2 group were rendered inactive. The bromopalladation annulation of certain azide derivatives was performed under standard conditions B, which include CuBr2, PdBr2, and LiBr. There was a mention of ongoing efforts to extend the application of the developed protocol in organic synthesis, suggesting that its versatility and potential uses are not fully realized.
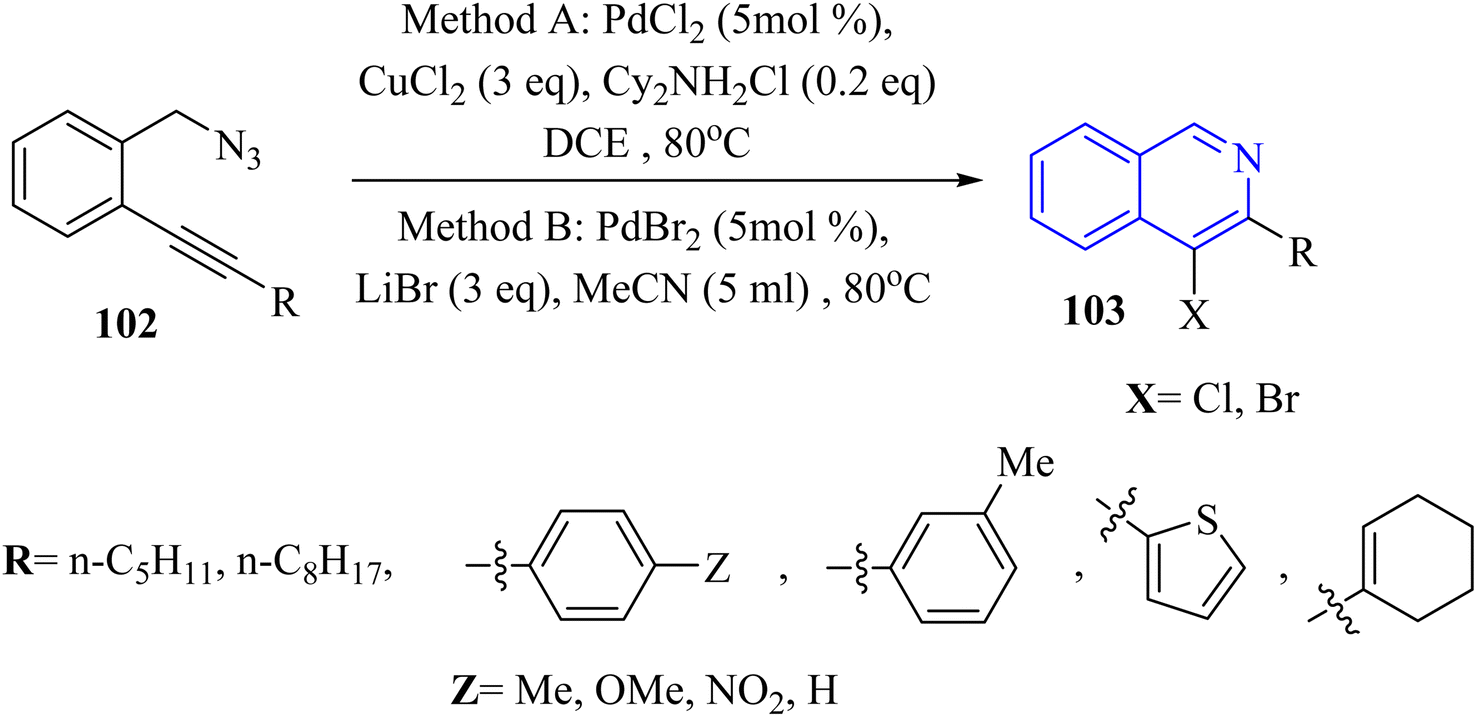 | ||
| Scheme 59 Synthesis of 4-haloisoquinoline derivatives 103. | ||
A mechanism underlying this transformation is depicted in Scheme 60. Initially, intermediate I is generated via the coordination of PdX2 with an alkyne. Then, intermediate II is formed from I by halopalladation of alkyne. Next, intermediate III is produced via the reaction of the N–N double bond with Pd within the II. The corresponding product, along with N2 and Pd(0) species, is formed through the reductive elimination and deprotonation of III. The active Pd(II)X2 species are reformed by oxidizing Pd(0) with CuX2, initiating a new catalytic cycle. It's also possible that the 103 is formed through a different mechanism: the halonium intermediate II′ is swiftly generated from I with the assistance of a Pd catalyst, subsequently undergoing addition and elimination processes to produce compound 103. The authors illustrated that further studies are needed to explore the mechanism of the halopalladation cyclization process, which remains an area of active investigation.140
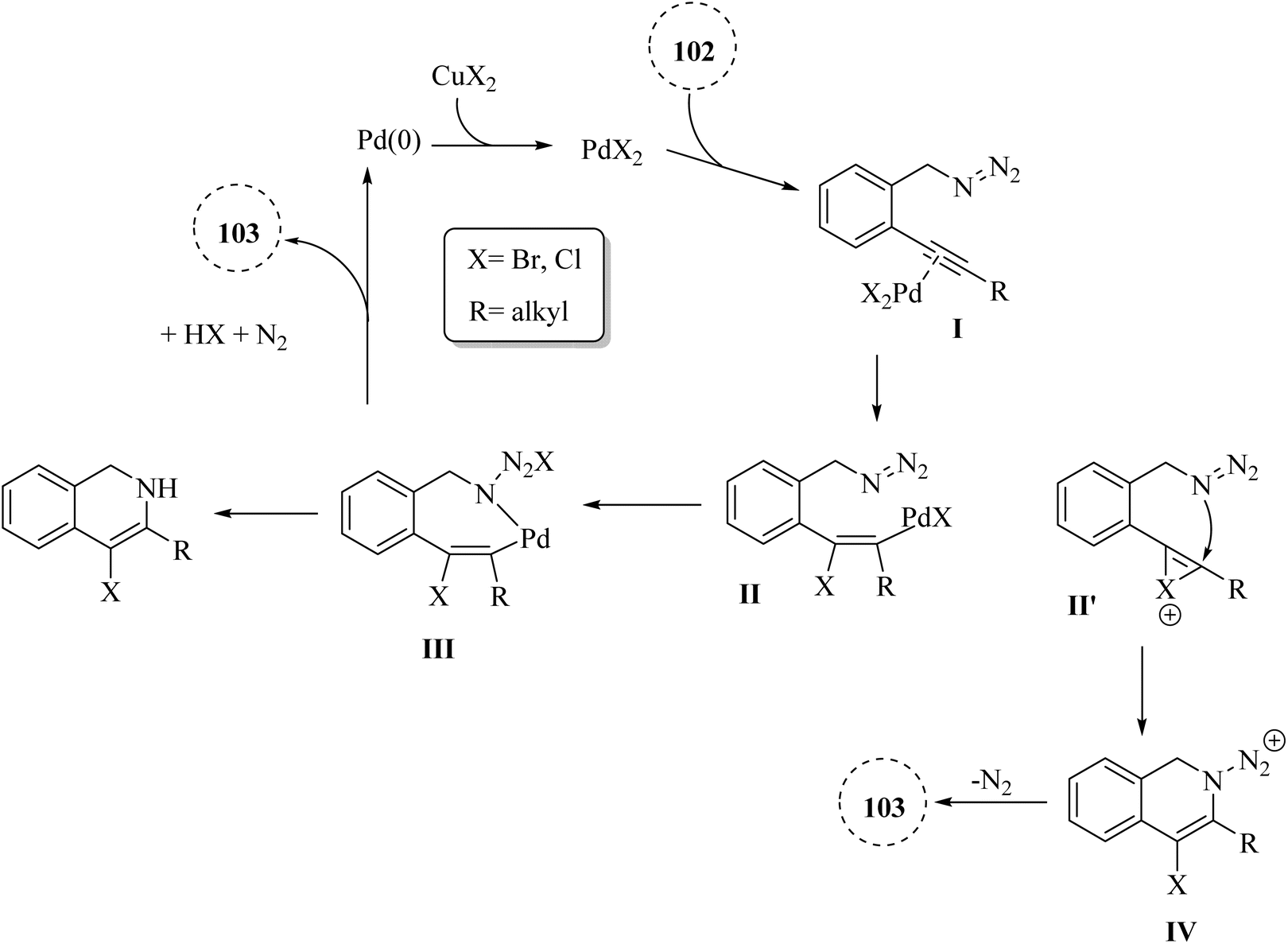 | ||
| Scheme 60 A mechanism for the reaction of 2-alkynyl benzyl azide derivatives 102 and their transformation into 103 catalyzed by Pd. | ||
In 2016, Zhou et al.136 reported the development of a Pd-catalyzed synthesis method for the production of polysubstituted isoquinoline derivatives 106. This method involved the coupling of aryl bromide derivatives 105 with 2-alkynyl benzylazide derivatives 104 (Scheme 61). The reaction was conducted in the presence of LiOtBu, Pd2dba3, and PPh3 in toluene at 100 °C for generated 106. The results of this process were significant, with the aryl bromide products being generated the 106 products yielding between 47% and 85%.
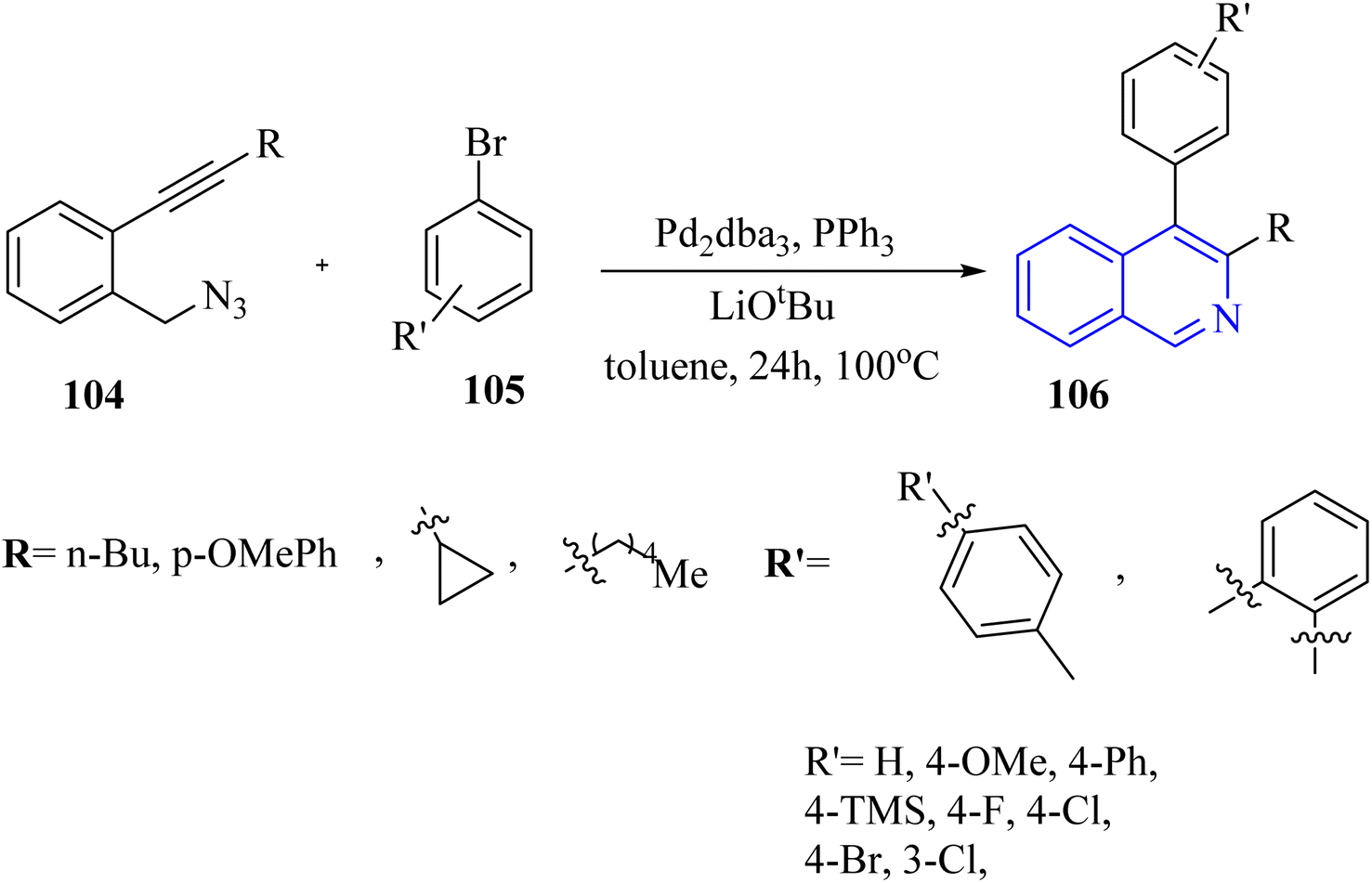 | ||
| Scheme 61 Synthesis of isoquinoline 106. | ||
4 Conclusions
Advances in various metal-catalyzed transformations in the synthesis of quinoline and indole derivatives emphasize the efficiency and versatility of these methods in organic synthesis, especially those involving Pd and Au as well as their bimetallic compounds. 2-Alkynyl benzyl azide derivatives and 2-alkynyl aryl azide derivatives were innovative substrates that can facilitate complex molecules by imposing significant functional groups and high yields.Reactions catalyzed by Au recognized for their ability to produce electrophilic intermediates and promote regioselective transformations, especially in the production of indole derivatives such as aryl-fused carbazole derivatives and indolinone derivatives. According to the explained mechanisms, it can be concluded that the Au carbene intermediates were very important and fundamental and showed the effect of the electronic properties of the substrate on the results.
On the other hand, the reactions catalyzed by Pd was showed high efficiency in the synthesis of polysubstituted isoquinoline derivatives and indole derivatives via electrocyclic processes. Also, the integration of two metals, Ag and Cu, has led to the development of the synthetic toolbox and thus the synthesis of complex heterocycles using the development of new methods.
By examining the studies, we can gain a better understanding of metal-catalyzed transformations and pave the way for future research aimed at synthesizing bioactive compounds and substances with various applications such as materials science and pharmaceuticals. It should be noted that the selectivity and efficiency of these synthetic strategies increases with the continuous exploration of reaction conditions, synthetic and mechanical pathways, and substrate Scope, and causes extensive innovations in the field of organic chemistry.
5 Future remarks
According to the studies and progress presented in this study on the synthesis of quinoline and indole derivatives vai metal-catalyzed transformations, several directions for further increase in the field of organic synthesis in the future can be imagined.First of all, it is necessary to explore metal catalysts beyond gold and palladium, because it creates new reaction pathways. Meanwhile, other transition metals, particularly Co and Ni, may offer unique reactivity profiles that are effective in the synthesis of complex heterocycles. On the other hand, as shown in some studies, the use of bimetallic catalytic systems promises synergistic effects that can increase the selectivity and efficiency of reactions.
Secondly, it should be considered that the principles of green chemistry should be prioritized in this synthetic methodology, such as the use of green solvents, green synthetic techniques and reducing waste, which can greatly increase the stability of these reactions. Also, in terms of synthetic techniques, we can mention the use of microwaves or synthesis with the help of ultrasonic waves, which reduces reaction time and increases efficiency, and also makes these processes more practical for industrial applications. Due to the mechanistic insight obtained from the current studies, it has paved the way for the rational design of new catalysts and reaction conditions. Future research could focus on computational modeling of reaction mechanisms to obtain prediction of results as well as optimization of conditions before conducting experimental studies. This insight could reduce trial and error and enable the development of new synthetic routes.
In the last stage, the research should move towards the investigation and biological evaluation of the synthesized compounds, because the investigation of the medicinal properties of new quinoline and indole derivatives leads to the discovery of new drugs. It should be noted that interdisciplinary collaboration with medicinal, organic chemists and biologists is necessary to convert these synthetic advances into practical applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Seyedmohammad Hosseininezhad: project administration, conceptualization, idea maker, writing – original draft, writing – review & editing Sina Pirani Ahmad Abad: writing – review & editing Ali Ramazani: project administration, investigation, conceptualization.Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Sharma, P. Kour and A. Kumar, J. Chem. Sci., 2018, 130, 1–25 CrossRef.
- S. Hosseininezhad and A. Ramazani, RSC Adv., 2024, 14, 278–352 RSC.
- S. Hosseininezhad and A. Ramazani, Arab. J. Chem., 2023, 16, 105234 CrossRef.
- B. Gao, B. Yang, X. Feng and C. Li, Nat. Prod. Rep., 2022, 39, 139–162 RSC.
- T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031–5040 CrossRef.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463–24476 RSC.
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC.
- R. Mancuso and R. Dalpozzo, Catalysts, 2018, 8, 458 CrossRef.
- J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef.
- S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544–6556 RSC.
- T. Liang, J. Li, Y. Liu, C. Wu, J. Wang, J. Lan and K. Y. Liu, Chin. J. Org. Chem., 2016, 36, 2619–2633 CrossRef.
- T. Guo, F. Huang, L. Yu and Z. Yu, Tetrahedron Lett., 2015, 56, 296–302 CrossRef.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466–478 CrossRef.
- Z. Shi and F. Glorius, Angew. Chem. Int. Ed., 2012, 51, 9220–9222 CrossRef PubMed.
- M. Platon, R. Amardeil, L. Djakovitch and J. C. Hierso, Chem. Soc. Rev., 2012, 41, 3929–3968 RSC.
- S. W. Youn and T. Y. Ko, Asian J. Org. Chem., 2018, 7, 1467–1487 CrossRef.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195–7210 CrossRef PubMed.
- A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 333–346 RSC.
- A. Wetzel and F. Gagosz, Angew. Chem. Int. Ed., 2011, 50, 7354–7358 CrossRef PubMed.
- A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766–1775 RSC.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef.
- Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS.
- A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed.
- A. S. K. Hashmi, Top. Organomet. Chem., 2007, 107, 3180–3211 CAS.
- D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III and F. D. Toste, et al., Nat. Chem., 2009, 1, 482–486 CrossRef CAS PubMed.
- D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS.
- Y. Wang, M. Zarca, L. Z. Gong and L. Zhang, J. Am. Chem. Soc., 2016, 138, 7516–7519 CrossRef CAS PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS.
- D. Qian and J. Zhang, Chem. Soc. Rev., 2015, 44, 677–698 RSC.
- Y. Wang, M. E. Muratore and A. M. Echavarren, Chem.–Eur. J., 2015, 21, 7332–7339 CrossRef CAS.
- F. Wei, et al., Sci. Bull., 2015, 60, 1479–1492 CrossRef CAS.
- H. S. Yeom and S. Shin, Acc. Chem. Res., 2014, 47, 966–977 CrossRef CAS.
- C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902–912 CrossRef CAS PubMed.
- Y. Wang, et al., Angew Chem. Int. Ed. Engl., 2013, 52, 7795–7799 CrossRef.
- P. Nösel, et al., J. Am. Chem. Soc., 2013, 135, 15662–15666 CrossRef PubMed.
- L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef PubMed.
- A. S. K. Hashmi, et al., Angew. Chem. Int. Ed., 2012, 51, 4456–4460 CrossRef PubMed.
- C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16–28 RSC.
- L. N. Dos Santos Comprido and A. S. K. Hashmi, Isr. J. Chem., 2013, 53, 883–891 CrossRef.
- C. Nieto-Oberhuber, S. López, E. Jiménez-Núñez and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 5916–5923 CrossRef PubMed.
- J. Xiao and X. Li, Angew. Chem. Int. Ed., 2011, 50, 7226–7236 CrossRef PubMed.
- Z. Xu, H. Chen, Z. Wang, A. Ying and L. Zhang, J. Am. Chem. Soc., 2016, 138, 5515–5518 CrossRef PubMed.
- L. R. Squire, J. Am. Chem. Soc., 2009, 132, 3258–3259 Search PubMed.
- L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2013, 132, 8550–8551 CrossRef PubMed.
- P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379–381 RSC.
- A. Mukherjee, et al., J. Am. Chem. Soc., 2011, 133, 15372–15375 CrossRef.
- N. Marion and S. P. Nolan, Angew. Chem. Int. Ed., 2007, 46, 2750–2752 CrossRef PubMed.
- D. Garayalde, E. Gómez-Bengoa, X. Huang, A. Goeke and C. Nevado, J. Am. Chem. Soc., 2010, 132, 4720–4730 CrossRef PubMed.
- P. W. Davies and M. Garzón, Asian J. Org. Chem., 2015, 4, 694–708 CrossRef.
- H. Jin, et al., Angew. Chem. Int. Ed., 2016, 55, 794–797 CrossRef.
- A. H. Zhou, et al., Chem. Sci., 2015, 6, 1265–1271 RSC.
- N. Li, T. Y. Wang, L. Z. Gong and L. Zhang, Chem.–Eur. J., 2015, 21, 3585–3588 CrossRef.
- C. Shu, et al., J. Am. Chem. Soc., 2015, 137, 9567–9570 CrossRef PubMed.
- L. Zhu, Y. Yu, Z. Mao and X. Huang, Org. Lett., 2015, 17, 30–33 CrossRef.
- S. K. Pawar, R. L. Sahani and R. S. Liu, Chem.–Eur. J., 2015, 21, 10843–10850 CrossRef PubMed.
- A. Prechter, G. Henrion, P. Faudot Dit Bel and F. Gagosz, Angew. Chem. Int. Ed., 2014, 53, 4959–4963 CrossRef PubMed.
- M. Garzon and P. W. Davies, Org. Lett., 2014, 16, 4850–4853 CrossRef PubMed.
- Y. Tokimizu, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2014, 16, 3138–3141 CrossRef.
- Z. Yan, Y. Xiao and L. Zhang, Angew. Chem., 2012, 124, 8752–8755 CrossRef.
- Y. Xiao and L. Zhang, Org. Lett., 2012, 14, 4662–4665 CrossRef.
- B. Lu, et al., Angew Chem. Int. Ed. Engl., 2009, 50, 8358–8362 CrossRef.
- L. Chaoqun and Z. Liming, Org. Lett., 2011, 13, 1738–1741 CrossRef.
- P. W. Davies, A. Cremonesi and L. Dumitrescu, Angew. Chem. Int. Ed., 2011, 50, 8931–8935 CrossRef PubMed.
- L. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689–6690 CrossRef.
- R. C. Larock, E. K. Yum and M. D. Refvik, J. Org. Chem., 1998, 63, 7652–7662 CrossRef.
- G. Battistuzzi, S. Cacchi and G. Fabrizi, Eur. J. Org. Chem., 2002, 16, 2671–2681 CrossRef.
- B. Z. Lu, et al., Org. Lett., 2006, 8, 3271–3274 CrossRef PubMed.
- S. Würtz, S. Rakshit, J. J. Neumann, T. Dröge and F. Glorius, Angew. Chem. Int. Ed., 2008, 47, 7230–7233 CrossRef.
- R. Nallagonda, M. Rehan and P. Ghorai, Org. Lett., 2014, 16, 4786–4789 CrossRef PubMed.
- I. Nakamura, G. B. Bajracharya, Y. Mizushima and Y. Yamamoto, Angew. Chem. Int. Ed., 2002, 41, 4328–4331 CrossRef.
- N. Monteiro, J. Gore and G. Balme, Tetrahedron, 1992, 48, 10103–10114 CrossRef.
- N. Monteiro and G. Balme, J. Org. Chem., 2000, 65, 3223–3226 CrossRef PubMed.
- Z. Li and C. He, Eur. J. Org. Chem., 2006, 4313–4322 CrossRef.
- Z. Li, D. A. Capretto, R. Rahaman and C. He, Angew. Chem. Int. Ed., 2007, 46, 5184–5186 CrossRef.
- Y. Cui and C. He, Angew. Chem. Int. Ed., 2004, 43, 4210–4212 CrossRef PubMed.
- T. B. Clark and K. A. Woerpel, J. Am. Chem. Soc., 2004, 126, 9522–9523 CrossRef CAS.
- Y. Cui and C. He, J. Am. Chem. Soc., 2003, 125, 16202–16203 CrossRef CAS.
- N. S. Josephsohn, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 4018–4019 CrossRef CAS PubMed.
- H. V. R. Dias, R. G. Browning and S. A. Polach, J. Am. Chem. Soc., 2003, 125, 9270–9271 CrossRef CAS.
- Y. Yamamoto, Chem. Rev., 2008, 108, 3199–3222 CrossRef CAS.
- J. M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS.
- M. A. Lvarez-Corral, M. Muñoz-Dorado and I. Rodríguez-García, Chem. Rev., 2008, 3174–3198 CrossRef.
- R. Umeda and A. Studer, Org. Lett., 2008, 10, 993–996 CrossRef CAS PubMed.
- H. Mandai, K. Mandai, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 17961–17969 CrossRef CAS PubMed.
- S. W. Youn and J. I. Eom, J. Org. Chem., 2006, 71, 6705–6707 CrossRef CAS PubMed.
- Y. C. Gae and C. Bolm, Org. Lett., 2005, 7, 4983–4985 CrossRef PubMed.
- C. G. Yang, N. W. Reich, Z. Shi and C. He, Org. Lett., 2005, 7, 4553–4556 CrossRef.
- X. Yao and C. J. Li, Org. Lett., 2005, 7, 4395–4398 CrossRef.
- N. T. Patil, N. K. Pahadi and Y. Yamamoto, J. Org. Chem., 2005, 70, 10096–10098 CrossRef PubMed.
- X. Yao and C. J. Li, J. Org. Chem., 2005, 70, 5752–5755 CrossRef.
- T. J. Harrison and G. R. Dake, Org. Lett., 2004, 6, 5023–5026 CrossRef.
- R. F. Sweis, M. P. Schramm and S. A. Kozmin, J. Am. Chem. Soc., 2004, 126, 7442–7443 CrossRef PubMed.
- C. Wei, Z. Li and C. J. Li, Org. Lett., 2003, 5, 4473–4475 CrossRef PubMed.
- Y. N. Niu, Z. Y. Yan, G. L. Gao, H. L. Wang, X. Z. Shu, K. G. Ji and Y. M. Liang, J. Org. Chem., 2009, 74, 2893–2896 CrossRef PubMed.
- K. Dong, M. Liu and X. Xu, Molecules, 2022, 27, 1–25 Search PubMed.
- B. V. Rokade, J. Barker and P. J. Guiry, Chem. Soc. Rev., 2019, 48, 4766–4790 RSC.
- J. N. Mo, J. Su and J. Zhao, Molecules, 2019, 24, 1216 CrossRef.
- V. Bisai, A. Suneja and V. K. Singh, Angew. Chem. Int. Ed., 2014, 53, 10737–10741 CrossRef.
- Q. Chen, et al., Angew. Chem. Int. Ed., 2016, 55, 5286–5289 CrossRef CAS.
- P. Maity, H. D. Srinivas and M. P. Watson, J. Am. Chem. Soc., 2013, 133, 17142–17145 CrossRef PubMed.
- F. Zhou, et al., J. Am. Chem. Soc., 2013, 135, 10994–10997 CrossRef CAS.
- S. Guo, P. Dong, Y. Chen, X. Feng and X. Liu, Angew. Chem., 2018, 130, 17094–17098 CrossRef.
- C. Zhang, et al., J. Am. Chem. Soc., 2012, 134, 9585–9588 CrossRef CAS PubMed.
- T. Hashimoto, Y. Takiguchi and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 11473–11476 CrossRef CAS PubMed.
- F. L. Hong, et al., J. Am. Chem. Soc., 2019, 141, 16961–16970 CrossRef CAS PubMed.
- Z. L. Liu, et al., Angew. Chem. Int. Ed., 2019, 58, 16538–16542 CrossRef CAS.
- Y. Kondo, K. Nagao and H. Ohmiya, Chem. Commun., 2020, 56, 7471–7474 RSC.
- F. Zhong, Q. Xue and L. Yin, Angew. Chem., 2020, 132, 1578–1582 CrossRef.
- Y. Huang, J. Del Pozo, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2018, 140, 2643–2655 CrossRef CAS PubMed.
- D. W. Gao, Y. Xiao, M. Liu, Z. Liu, M. K. Karunananda, J. S. Chen and K. M. Engle, ACS Catal., 2018, 8, 3650–3654 CrossRef CAS PubMed.
- H. Y. Jung, X. Feng, H. Kim and J. Yun, Tetrahedron, 2012, 68, 3444–3449 CrossRef CAS.
- H. Y. Jung and J. Yun, Org. Lett., 2012, 14, 2606–2609 CrossRef CAS.
- P. Liu, Y. Fukui, P. Tian, Z. T. He, C. Y. Sun, N. Y. Wu and G. Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700–11703 CrossRef.
- D. W. Gao, Y. Gao, H. Shao, T. Z. Qiao, X. Wang, B. B. Sanchez, J. S. Chen, P. Liu and K. M. Engle, Nat. Catal., 2020, 3, 23–29 CrossRef PubMed.
- C. A. M. Brokowski, Nat. Chem., 2015, 7, 38–44 CrossRef PubMed.
- B. Lu, Y. Luo, L. Liu, L. Ye, Y. Wang and L. Zhang, Angew. Chem., Int. Ed. Engl., 2011, 50, 8358 CrossRef PubMed.
- A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354–7358 CrossRef PubMed.
- X. Zhang, X. Sun, H. Fan, P. Li, C. Lyu and W. Rao, Eur. J. Org Chem., 2016, 25, 4265–4268 CrossRef.
- X. Zhang, X. Sun, H. Fan, C. Lyu, P. Li, H. Zhang and W. Rao, RSC Adv., 2016, 6, 56319–56322 RSC.
- N. Li, X. L. Lian, Y. H. Li, T. Y. Wang, Z. Y. Han, L. Zhang and L. Z. Gong, Org. Lett., 2016, 18, 4178–4181 CrossRef PubMed.
- T. Li, B. L. Chen, L. L. Zhu and Z. Chen, Tetrahedron Lett., 2020, 61, 151851 CrossRef.
- J. M. Xie, Y. L. Zhu, Y. M. Fu, C. F. Zhu, L. J. Cheng, Y. E. You, X. Wu and Y. G. Li, Org. Lett., 2023, 25, 421–425 CrossRef PubMed.
- Y. L. Zhu, Y. F. Dong, S. R. Wang, Y. G. Li, X. Wu and L. W. Ye, Org. Lett., 2024, 26, 631–635 CrossRef PubMed.
- W. B. Wang, J. C. Lu, H. Bai, Y. M. Fu, L. J. Cheng, C. F. Zhu, Y. G. Li and X. Wu, Org. Lett., 2024, 26, 1792–1796 CrossRef PubMed.
- Y. Kawada, S. Ohmura, M. Kobayashi, W. Nojo, M. Kondo, Y. Matsuda, J. Matsuoka, S. Inuki, S. Oishi, C. Wang and T. Saito, Chem. Sci., 2018, 9, 8416–8425 RSC.
- W. B. Shen, Q. Sun, L. Li, X. Liu, B. Zhou, J. Z. Yan, X. Lu and L. W. Ye, Nat. Commun., 2017, 8, 1748 CrossRef PubMed.
- X. Zhang, P. Li, C. Lyu, W. Yong, J. Li, X. Pan, X. Zhu and W. Rao, Adv. Synth. Catal., 2017, 359, 4147–4152 CrossRef CAS.
- X. Zhang, P. Li, C. Lyu, W. Yong, J. Li, X. Zhu and W. Rao, Org. Biomol. Chem., 2017, 15, 6080–6083 RSC.
- P. Li, W. Yong, R. Sheng, W. Rao, X. Zhu and X. Zhang, Adv. Synth. Catal., 2019, 361, 201–207 Search PubMed.
- W. Yong, P. Li, R. Sheng, W. Rao and X. Zhang, ChemistrySelect, 2018, 3, 11696–11699 CrossRef CAS.
- G. Hu, P. Li, Z. Zhou, F. Yang, S. Xu, H. Fan, X. Zhao and X. Zhang, New J. Chem., 2021, 45, 3828–3832 RSC.
- P. Li, R. Sheng, Z. Zhou, G. Hu and X. Zhang, Eur. J. Org Chem., 2020, 14, 2146–2152 Search PubMed.
- Q. Zhou, Z. Zhang, Y. Zhou, S. Li, Y. Zhang and J. J. Wang, Org. Chem., 2017, 82, 48–56 CrossRef CAS PubMed.
- P. Li, F. Yang, G. Hu and X. J. Zhang, Org. Chem., 2021, 86, 10360–10367 CrossRef CAS PubMed.
- Z. Zhou, Y. Xu, B. Zhu, P. Li, G. Hu, F. Yang, S. Xu and X. Zhang, New J. Chem., 2020, 44, 20303–20307 RSC.
- P. Li, B. Zhu, Y. Xu, Z. Zhou, G. Hu, F. Yang, S. Xu and X. Zhang, Org. Chem. Front., 2020, 7, 3480–3485 RSC.
- H. P. Zhang, S. C. Yu, Y. Liang, P. Peng, B. X. Tang and J. H. Li, Synlett, 2011, 07, 982–988 Search PubMed.
- M. Yang, T. Liu, Y. Gong, Q. W. Ai and Y. L. Zhao, Org. Chem. Front., 2022, 9, 4453–4459 RSC.
- Z. Zhang, F. Xiao, B. Huang, J. Hu, B. Fu and Z. Zhang, Org. Lett., 2016, 18, 908–911 CrossRef PubMed.
- C. Gronnier, G. Boissonnat and F. Gagosz, Org. Lett., 2013, 15, 4234–4237 CrossRef PubMed.
- F. Yang, S. Xu, H. Fan, X. Zhao and X. Zhang, Eur. J. Org Chem., 2021, 32, 4555–4558 CrossRef.
- Y. F. Qiu, Y. J. Niu, X. Wei, B. Q. Cao, X. C. Wang and Z. J. J. Quan, Org. Chem., 2019, 84, 4165–4178 CrossRef PubMed.
- Y. Pan, G. W. Chen, C. H. Shen, W. He and L. W. Ye, Org. Chem. Front., 2016, 3, 491–495 RSC.
- Z. Huo and Y. Yamamoto, Tetrahedron Lett., 2009, 50, 3651–3653 CrossRef.
- H. P. Zhang, H. Y. Li and H. F. Xiao, J. Chem. Res., 2013, 37, 556–558 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |