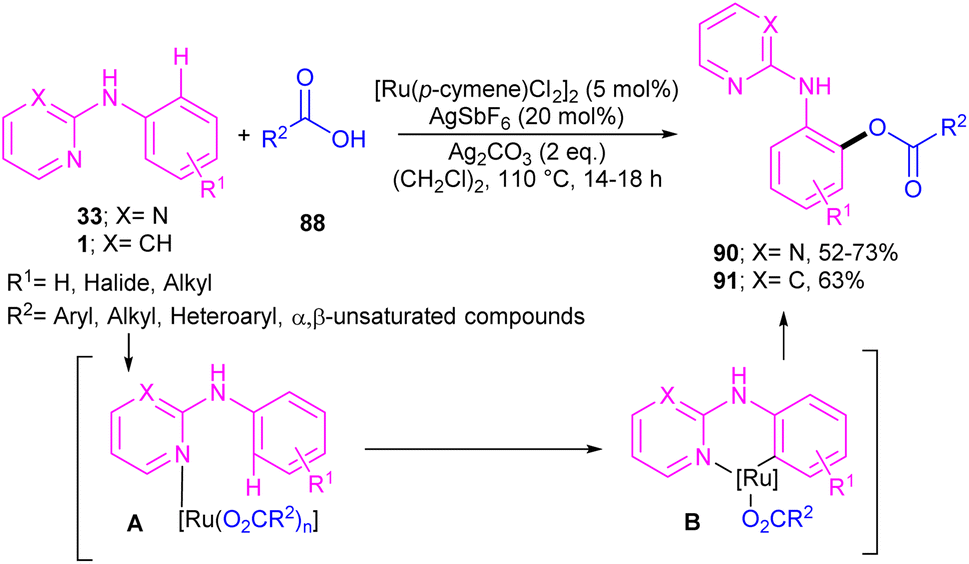DOI:
10.1039/D4RA08384A
(Review Article)
RSC Adv., 2025,
15, 1134-1151
Transition metal-catalyzed cross-coupling reactions of N-aryl-2-aminopyridines
Received
27th November 2024
, Accepted 6th January 2025
First published on 14th January 2025
Abstract
Due to the presence of the pyridyl directing group, N-aryl-2-aminopyridines can quickly form stable complexes with metals, leading to cyclization and functionalization reactions. A large number of N-heterocycles and nitrogen-based molecules can be easily constructed via this direct and atom-economical cross-coupling strategy. In this review, we have highlighted the transformations of N-aryl-2-aminopyridines in the presence of various transition metal catalysts, such as palladium, rhodium, iridium, ruthenium, cobalt and copper.
 Fatemeh Doraghi | Fatemeh Doraghi received her bachelors and masters degrees in organic chemistry from Shahid Chamran University, Ahvaz in 2008 and 2014, respectively. She received her PhD degree in organic chemistry from University of Tehran, Tehran in 2022. During her PhD research, she focused on the development of new synthetic methodologies via transition metal as well as metal-free oxidative coupling reactions. She is currently employed as a researcher at the Endocrinology and Metabolism Research Center, Tehran University of Medical Sciences, Tehran, Iran. |
 Lina Rezainia | Lina Rezainia was born in 2002 in Tehran, Iran. She received her bachelors degree in pure chemistry from the University of Tehran, Iran in 2024, and graduated with honors. Before receiving her degree, she started doing research in organic synthesis, as well as polymer chemistry. |
 Mohammad Hossein Morshedsolouk | Mohammad Hossein Morshedsolouk is currently a second-year polymer chemistry student at the University of Tehran. He obtained his bachelors degree in practical chemistry from Shahid Beheshti University. Presently, he is affiliated with the Research Center for Membrane Technology at the University of Tehran. Last year, he worked at the Endocrinology and Metabolism Clinical Sciences Institute at Tehran University of Medical Sciences, where his research focused on the synthesis of various organic compounds. Currently, he is engaged in the study of polymeric membranes and exploring approaches to enhance their performance. |
 Hamed Navid | Hamed Navid was born in 1991 in West Azerbaijan Province, Iran. He received his masters degree in organic chemistry from University of Tehran, Tehran in 2020. His thesis was on the assembly of indole cores through a palladium-catalyzed metathesis of Ar–X σ-bonds. He is currently employed as a manager by Azin Polymer Asia Company, in Tehran, Iran. At the moment he is focused on the development of new polymer compounds. |
 Bagher Larijani | Bagher Larijani was born in 1961 in Iran. He obtained his MD from Tehran University, in 1986. His current research interests focus on diabetes. |
 Mohammad Mahdavi | Mohammad Mahdavi was born in 1981 in Tehran, Iran. He received his M.Sc. from the University of Tehran in 2008 and his PhD degree in medicinal chemistry from the Tehran University of Medical Science, Tehran, Iran, in 2016. Since August 2017, he has been with the Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran where he is an Assistant Professor. His current research interests focus on the synthesis of heterocyclic compounds with biological activities. |
1. Introduction
Transition metal-catalyzed directing group-assisted heteroannulations of amines with other coupling partners have emerged as a powerful and efficient tool.1–5 N-Heterocyclic compounds are the main nuclei in functional materials,6,7 pharmaceuticals8–11 and natural products.12–14 Consequently, various straightforward and affordable routes have been developed for synthesizing bioactive N-heterocycles. Among them, nitrogen-directing group-assisted substrates, leading to ortho-selective C–H activation are very popular. The nitrogen auxiliary groups form a variety of stable metal complexes and are then easily removed or undergo further transformations.
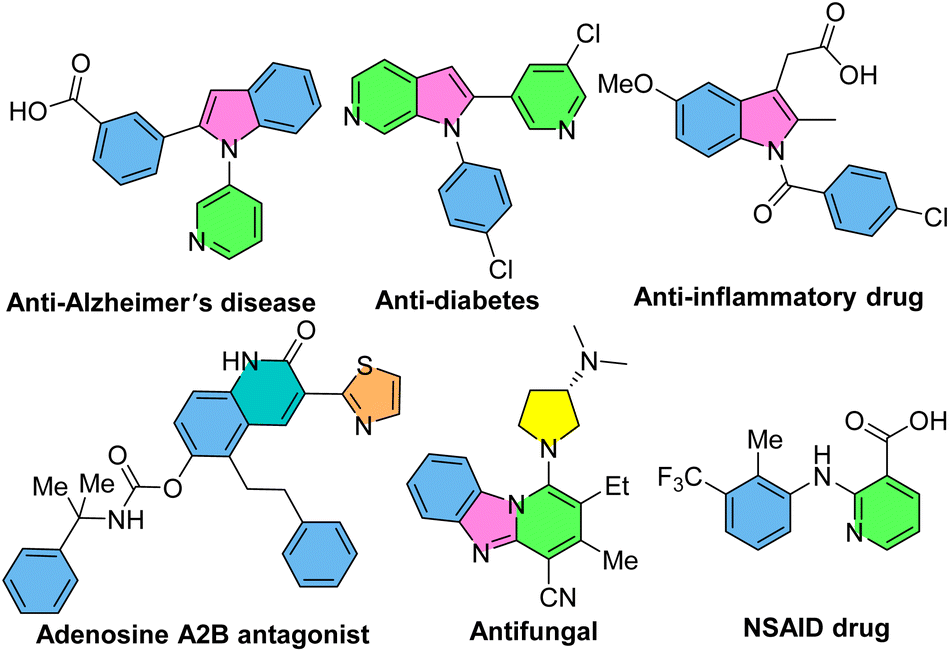
N-Aryl-2-aminopyridines, which are readily prepared by the coupling of anilines with 2-bromopyridines,15–17 are extensively used as substrates in the context of C–H activation. Owing to the presence of the pyridyl as a directing group, N-aryl-2-aminopyridines can easily incorporate into the chelation-assisted C–H bond functionalization. Various transition metals, such as palladium, rhodium, iridium, ruthenium, cobalt and copper can be involved in the formation of five-, six- and seven-membered complex intermediates with the nitrogen-pyridyl group through the coordination and C–H bond activation process. A diverse range of bioactive N-heterocycles, including indoles, carbazoles, quinolinones, benzimidazoles, indolinones, imidazopyridines and functionalized N-aryl-2-aminopyridines can be obtained via annulation or functionalization of N-aryl-2-aminopyridines.18–23 Some of biologically active compounds containing these valuable cores are depicted in Scheme 1.
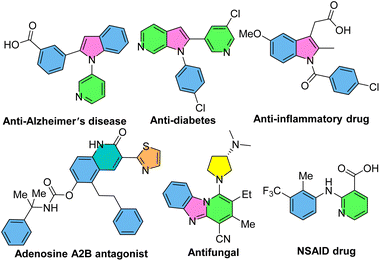 |
| | Scheme 1 Some pharmaceutically important nitrogen-containing compounds. | |
Considering the importance of N-aryl-2-aminopyridine derivatives, we have highlighted the developments related to these synthons. In this context, we have discussed the annulation and functionalization of N-aryl-2-aminopyridine in the presence of transition metal catalysts. We have categorized this review based on the metal type to provide a better insight into the behaviour of each metal catalyst in the presence of N-aryl-2-aminopyridines.
2. Transition metal-catalyzed cross-coupling of N-aryl-2-aminopyridines
2.1. Pd-catalyzed cross-coupling of N-aryl-2-aminopyridines
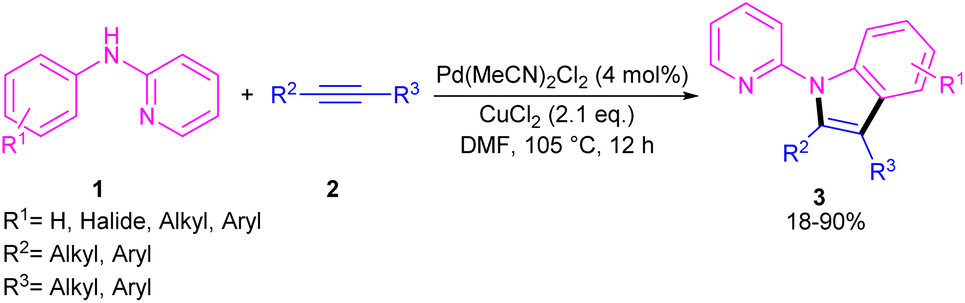
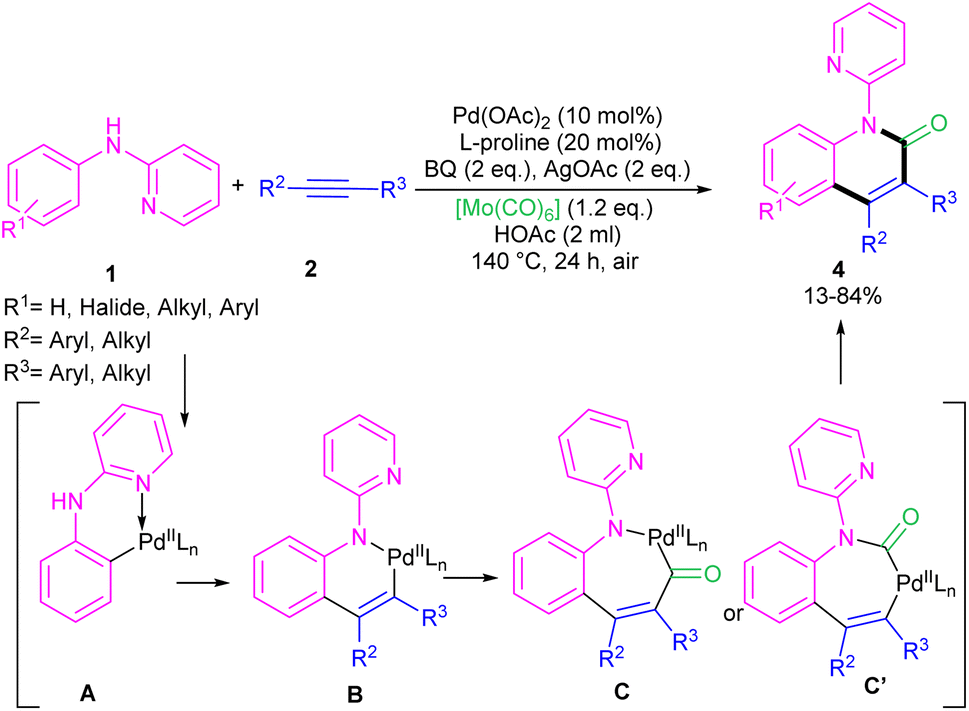
2.1.1. Annulation. In 2011, Li's research group performed a study on the Pd(II)-catalyzed synthesis of N-(2-pyridyl)indole frameworks 3 through the annulation of N-aryl-2-aminopyridines 1 with internal alkynes 2 (Scheme 2).24 In this synthetic method, 4 mol% of Pd(MeCN)2Cl2 as a catalyst, 1.2 equiv. of CuCl2 and O2 atmosphere as the oxidants were used in DMF solvent. It should be noted that this palladium(II) system did not prove effective for the reaction of N-aryl-2-aminopyridines with acrylates towards N-(2-pyridyl)quinolones, as these products were achieved in the presence of a Rh(III) catalyst. However, in Wu's laboratory, N-(2-pyridyl)quinolones were well synthesized from the annulation of N-aryl-2-aminopyridines 1, internal alkynes 2 and Mo(CO)6 as a CO source through a (3 + 2 + 1)-cycloaddition process (Scheme 3).25 The currently accepted mechanism involved the initial C–H activation of substrate 1 with Pd(II), followed by the insertion of an alkyne to produce intermediate B. Subsequent coordination and insertion of CO to B led to intermediate C or C′, which upon reductive elimination gave product 4. The generated Pd(0) was re-oxidized by BQ and AgOAc to the active Pd(II) species.
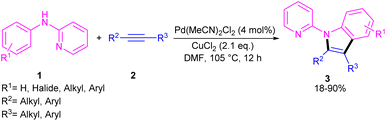 |
| | Scheme 2 Pd(II)-catalyzed annulation of N-aryl-2-aminopyridine and alkynes. | |
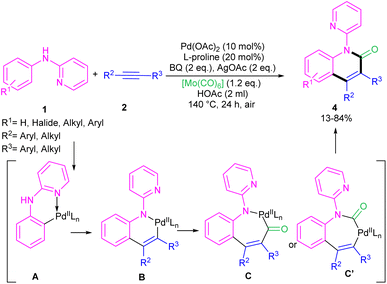 |
| | Scheme 3 Pd(II)-catalyzed annulation of N-aryl-2-aminopyridine, alkynes and Mo(CO)6. | |
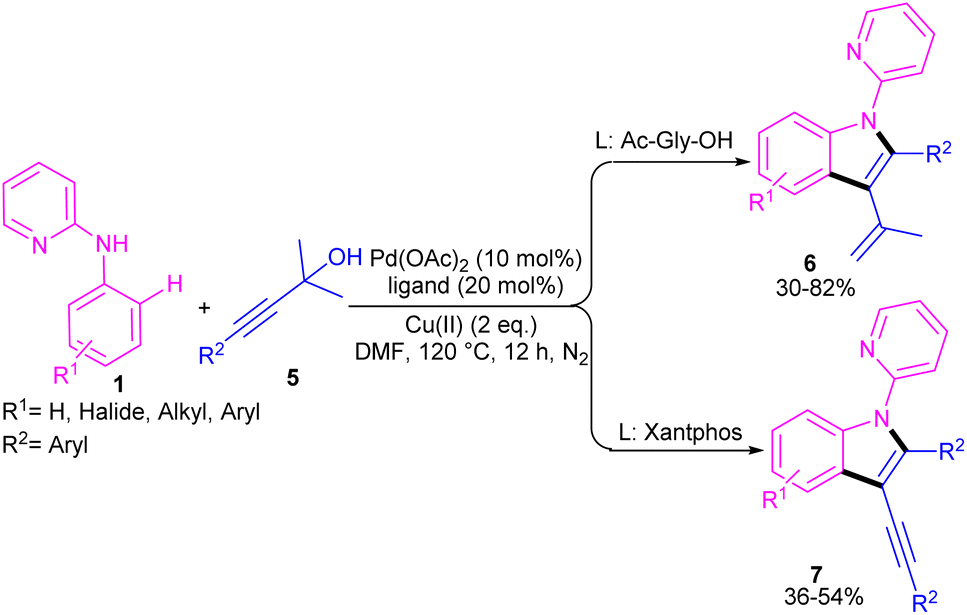
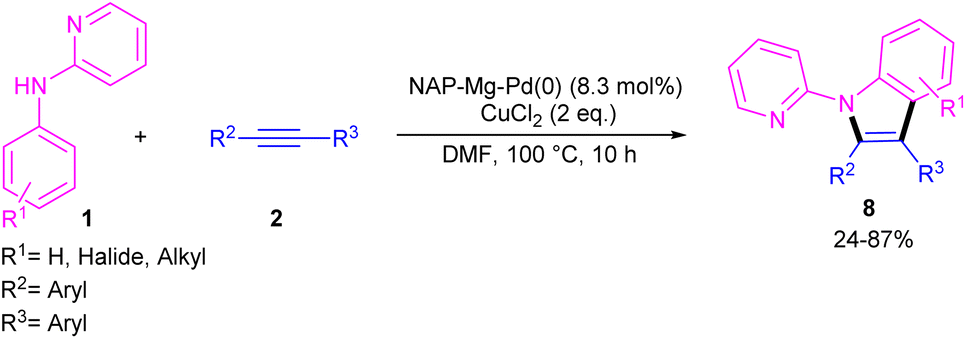
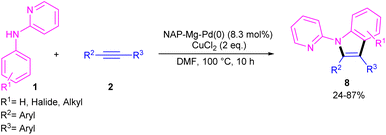
In 2021, Zhou's group reported the assembly of N-pyridoindoles 6, 7 by using N-aryl-2-aminopyridines 1 and propargylic alcohols 5 (Scheme 4).26 The catalytic system involved Pd(OAc)2 as a catalyst, Cu(OAc)2 or CuSO4 as an oxidant, Ac-Gly-OH or xantphos as a ligand. Other metal catalysts, such as Co(OAc)2·4H2O, Ni(OAc)2·4H2O, [RuCl2(p-cymene)]2 and [Cp*RhCl2]2 were not workable as much as Pd(OAc)2. The indole products were isolated in moderate to high yields (30–82%). Another Pd-catalyzed cyclization reaction between N-aryl-2-aminopyridines and alkynes access to indoles was reported by Hu and co-workers.27 PdCl2 as a catalyst, Cu(TFA)2·xH2O as an oxidant, and DTBP as a co-oxidant efficiently catalyze this (3 + 2)-cycloaddition process. Nano magnesium oxide can stabilize Pd(0) leading to a heterogeneous nanocatalyst for the C–H activation/annulation of N-aryl-2-aminopyridines 1 with alkynes 2 (Scheme 5).28 As an oxidant, CuCl2 was more practical than the Ag salts, TBHP or O2 atm. Many N-(2-pyridyl)indole products 8 were isolated in good yields by simple operation. Besides, N-aryl-2-aminoquinoline and N-aryl-2-aminopyrimidine were also yielded the products in 24% and 77%, of yields, respectively. The nanocatalyst was then investigated for its recyclability, which efficiently catalyze the reaction for four cycles without significant loss of catalytic activity. So, it can be concluded that this nanocatalyst acts better than the palladium homogeneous catalysts.
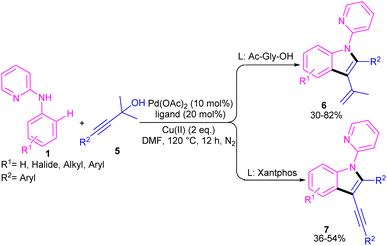 |
| | Scheme 4 Pd(II)-catalyzed annulation of N-aryl-2-aminopyridines with propargylic alcohols. | |
 |
| | Scheme 5 NAP–Mg–Pd(0) catalyzed oxidative coupling of N-aryl-2-aminopyridines with alkynes. | |
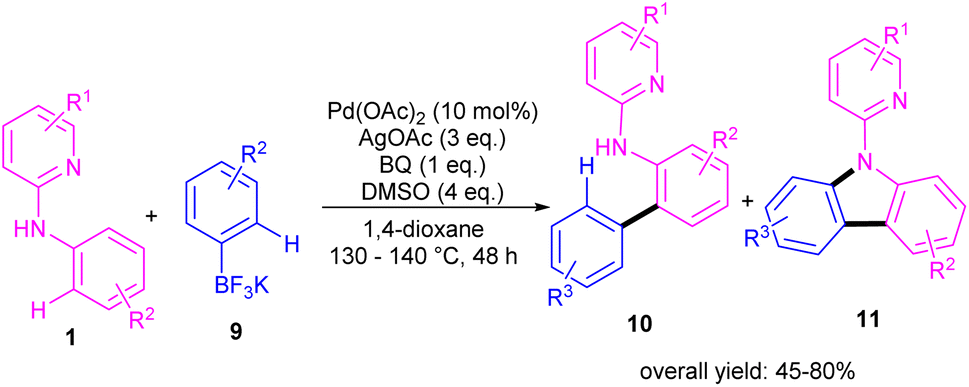
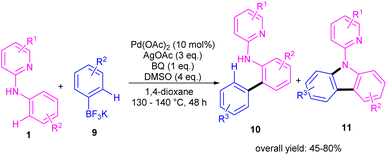
During an investigation of carbazole synthesis by means of a Pd(II)-catalyzed, one-pot reaction, Wu's group applied a procedure to N-aryl-2-aminopyridines 1 to achieve 9-(pyridin-2-yl)-9H-carbazoles 11 in moderate to high yields (Scheme 6).29 The reaction involved N-aryl-2-aminopyridines and potassium aryltrifluoroborates in the presence of a Pd(OAc)2 catalyst, AgOAc oxidant and benzoquinone (BQ) ligand. Using starting materials with different functional groups resulted in different ratios of products 10 and 11, indicating the low selectivity of this reaction. The BQ ligand in collaboration with Ag(I) had an important role in not only the transmetalation but also reductive elimination.
 |
| | Scheme 6 Pd(II)-catalyzed cross-coupling of N-phenylpyridin-2-amines with potassium aryltrifluoroborates. | |
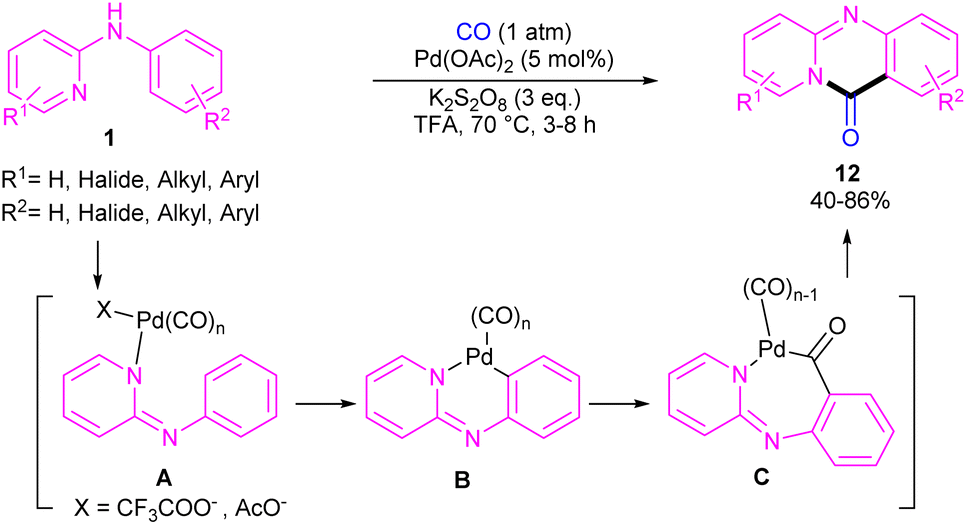
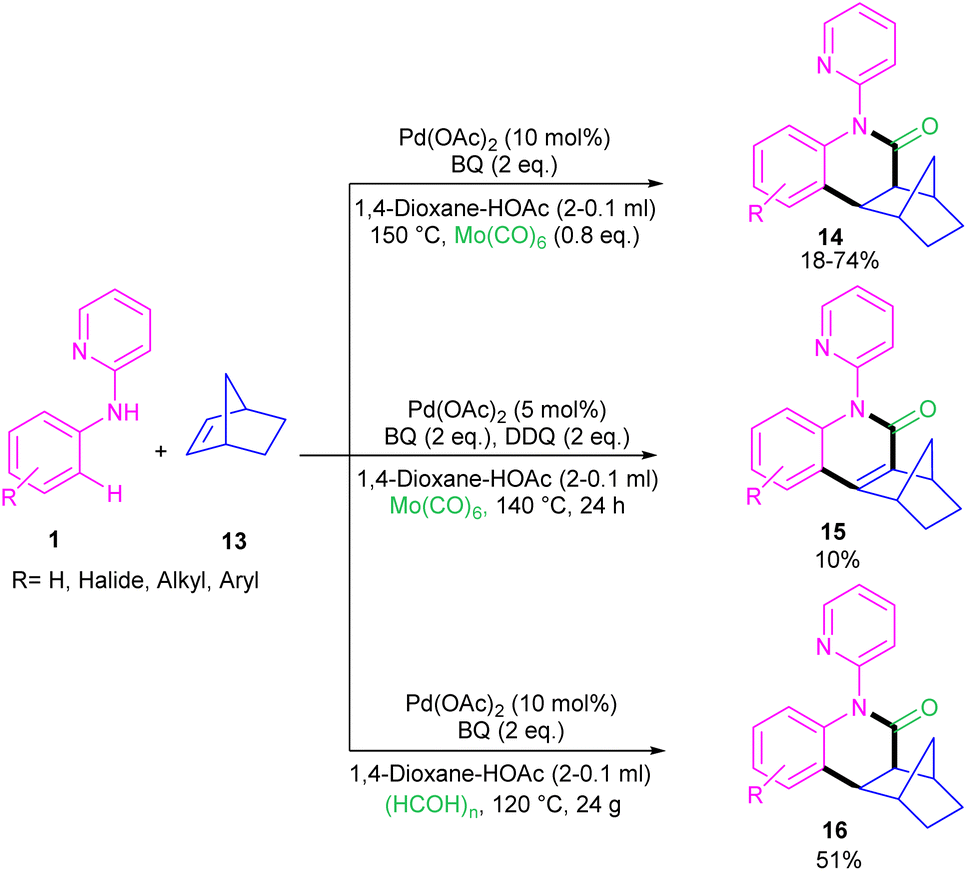
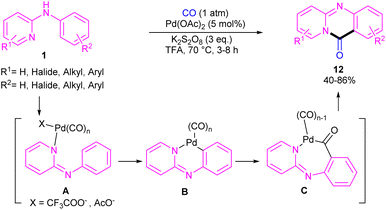
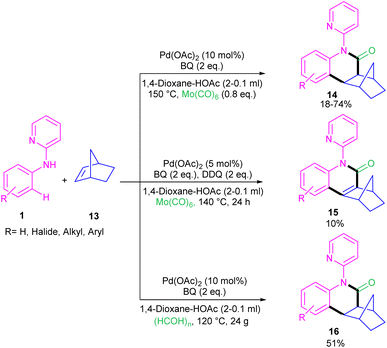
For preparing 11H-pyrido[2,1-b]quinazolin-11-ones 12, Zhu et al. used Pd(OAc)2 as a catalyst and CO gaseous to perform carbonylative intramolecular cyclization of N-aryl-2-aminopyridines 1 (Scheme 7).30 The pyridyl nitrogen acted not only as a directing group for the metal but also as an intramolecular nucleophile. The reaction was found to proceed through intermediate A, B and C. The Pd(II) complex A was generated from the chelation of the nitrogen with the CO ligated Pd(II), which upon electrophilic cyclopalladation on the phenyl ring gave intermediate B via the C–H bond cleavage. After that, palladacycle C was formed via migratory insertion of a coordinated CO into the aryl–Pd bond, which after reductive elimination liberated the final product 12. Later, Wu and co-workers reported the synthesis of 5-(pyridin-2-yl)-hexahydro-7,10-methanophenanthridin-6(5H)-ones from N-phenylpyridin-2-amine 1 and norbornene 13 as substrates using Pd(OAc)2 as a catalyst and Mo(CO)6 as a solid CO source (Scheme 8).31 Overall catalytic cycle involved Pd(II) insertion into the aromatic C–H bond, alkene coordination/insertion, CO coordination/insertion, and final reductive elimination. The carbonylation/cyclization reaction was also carried out by changing the BQ to DDQ as an oxidant or using paraformaldehyde as a CO source. However, DDQ can lead to the over oxidized product 15.
 |
| | Scheme 7 Pd(II)-catalyzed C(sp2)–H pyridocarbonylation of N-aryl-2-aminopyridines. | |
 |
| | Scheme 8 Pd(II)-catalyzed carbonylative C–H activation of N-aryl-2-pyridinamines with norbornene. | |
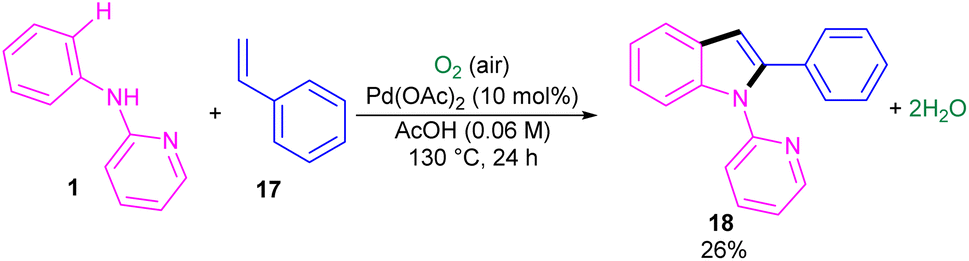
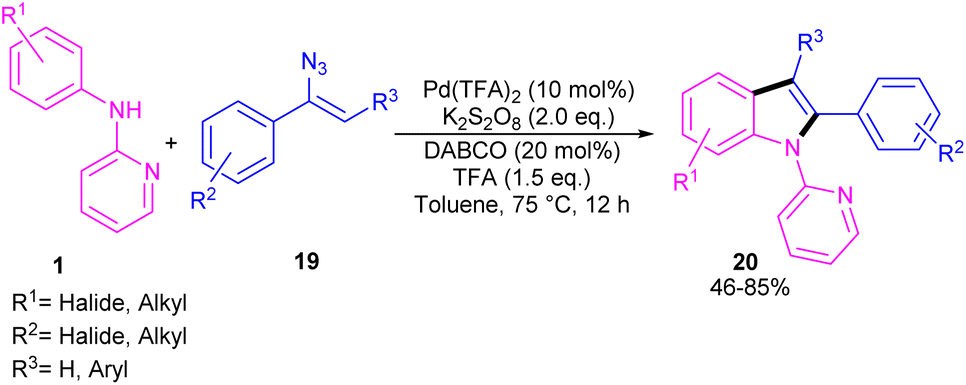
The first report on the incorporation of olefins in intramolecular cyclization without the need of any additional directing group was performed by Maiti and his research team in 2014 (Scheme 9).32 N-Phenyl-2-aminopyridine 1 showed moderate reactivity in this reaction, resulting in 26% of yield of product 18. In 2018, Wang, Cui and co-workers designed a strategy to achieve 2-arylindoles 20 via palladium-catalyzed reaction of N-phenyl-2-aminopyridine 1 with (1-azidovinyl)-benzene 19 (Scheme 10).33 The mechanism started with a concerted metalation/deprotonation (CMD) process to access the palladacycle A. This intermediate could be moved through two pathways. In path I, the six-membered intermediate A underwent migratory insertion with vinyl azide 19, and a subsequent elimination of TFA to yield another six-membered palladacycle F. Oxidative cyclization towards the indoline G, followed by the elimination of HN3 furnished indole 20. Meanwhile, K2S2O8 oxidized Pd(0) to the active Pd(II) species to restart the next catalytic cycle. In path II, vinyl azide 19 transformed to 2H-azirine 19′ by the help of DABCO. Then, migratory insertion into palladacycle A, and subsequent reductive elimination generated intermediate C. The generated Pd(0) species could be reoxidized to Pd(II) species. At last, C was subjected to an intramolecular nucleophilic addition to form intermediate D, which by further deamination delivered indole 20 (Scheme 11). It should be noted that intermediates C and D were determined by HRMS analysis, confirming the second pathway. In addition, KIE study (KH/KD = 1.4) indicated that the C–H bond cleavage is involved in the rate-determining step.
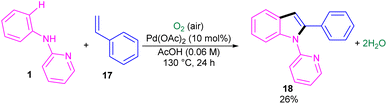 |
| | Scheme 9 Pd(II)-catalyzed cyclization of anilines with vinyl azides. | |
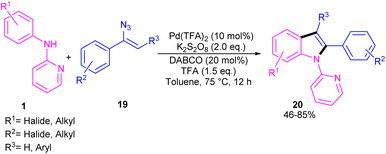 |
| | Scheme 10 Pd(II)-catalyzed cyclization of anilines with vinyl azides. | |
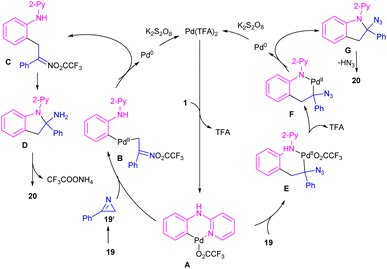 |
| | Scheme 11 Plausible mechanism for Pd(II)-catalyzed cyclization of anilines with vinyl azides. | |
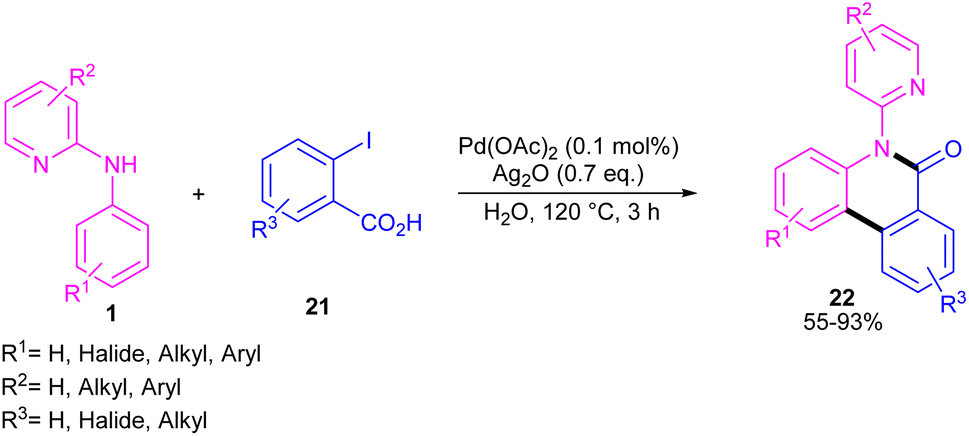
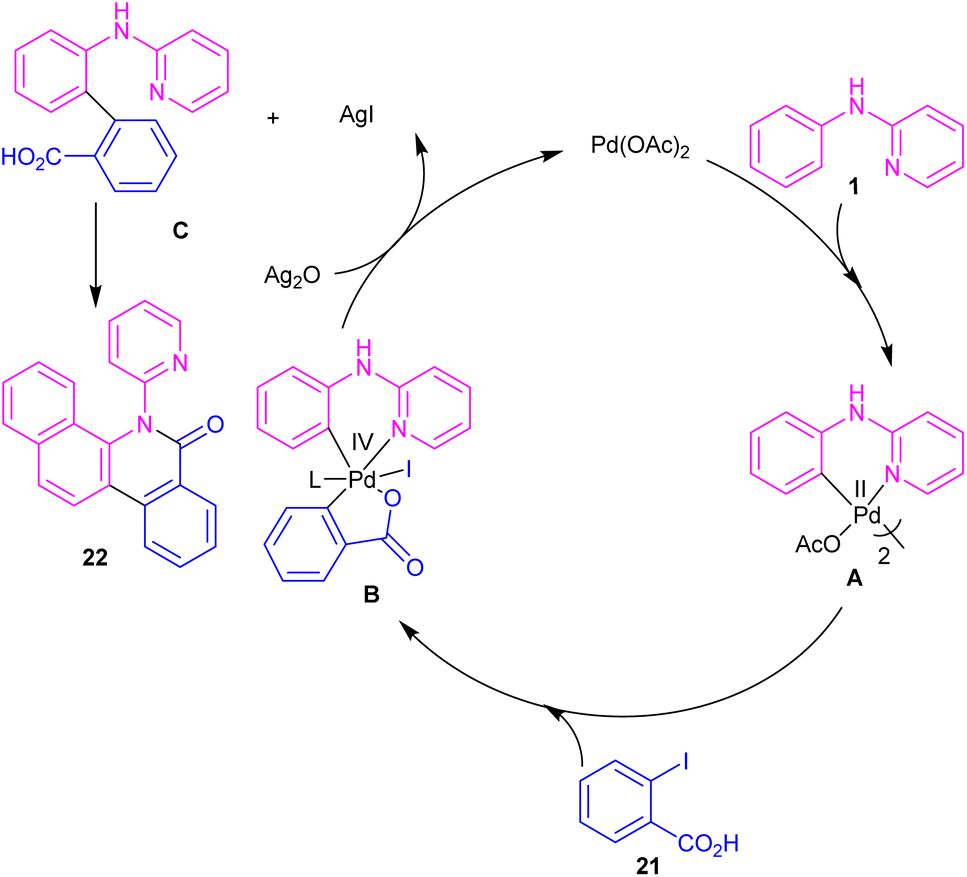
N-Aryl-2-aminopyridines 1 and 2-iodobenzoic acids 21 can react together in the presence of 0.1 mol% of Pd(OAc)2 and 0.7 equiv. of Ag2O in H2O as a green solvent to construct phenanthridinone scaffolds 22 (Scheme 12).34 The use of methyl 2-iodobenzoate and iodobenzene instead of 2-iodobenzoic acids did not result in any product, indicating the crucial role of the acid unit in the cyclization process. The isolation of the palladium intermediate I was successful, which in the reaction with 2-iodobenzoic acid led to the desired product in 91% yield. So, the reaction was found to move through the coordination of Pd(II) to the N-atom of N-aryl-2-aminopyridine, followed by C–H activation to form a six-membered palladacycle dimer A. In the next step, carboxylate-assisted oxidative addition to the metal center to render a Pd(IV) intermediate B. Then, reductive elimination of B in the presence of Ag(I) furnished the ortho-arylated product C. Further intramolecular acylation gave phenanthridinone 22 (Scheme 13).
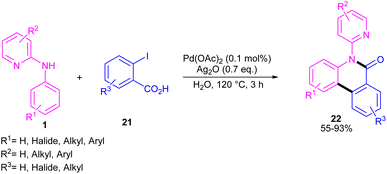 |
| | Scheme 12 Pd(II)-catalyzed cyclization of N-aryl-2-aminopyridines with 2-iodobenzoic acids. | |
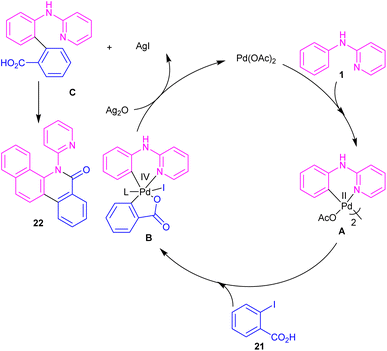 |
| | Scheme 13 Catalytic cycle for Pd(II)-catalyzed cyclization of N-aryl-2-aminopyridines with 2-iodobenzoic acids. | |
2.1.2. Functionalization.
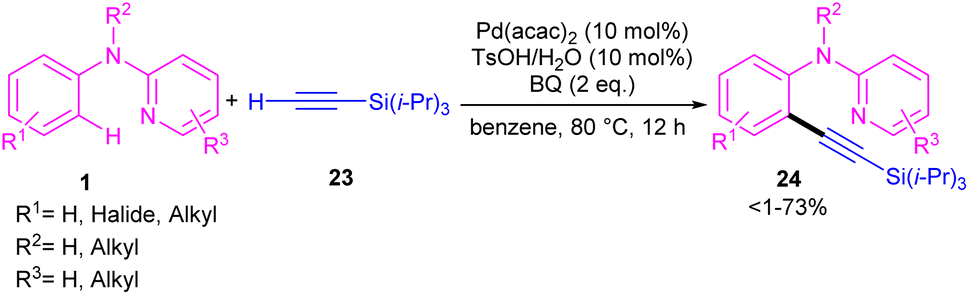
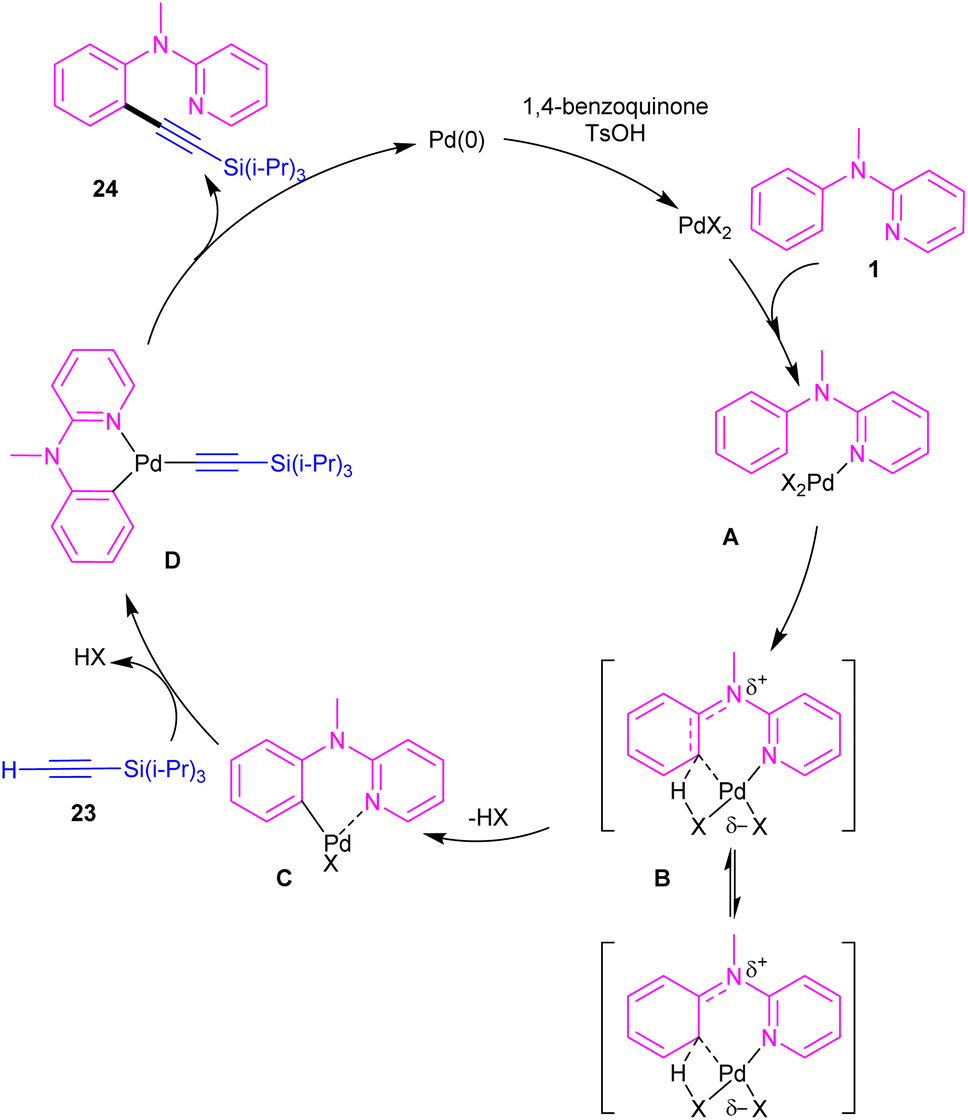
2.1.2.1. Alkynylation. Chang and co-workers investigated the reaction of N-aryl-2-aminopyridines 1 with (triisopropylsilyl)acetylene 23 in the presence of the palladium catalysts (Scheme 14).35 Several palladium salts were explored [Pd(OAc)2, Pd(acac)2]; Pd(acac)2 proved to be the most effective, in 100 mol% at 80 °C and in benzene as solvent. The KIE experiments indicated that C(sp2)–H bond cleavage is related to the rate-determining step. To determine the role of the nitrogen of N-aryl-2-aminopyridine in the catalytic pathway, the authors replaced N-aryl-2-aminopyridine with N-methyldiphenylamine and 2-benzylpyridine as coupling reactants, resulting in no product. So, the reaction was proposed to carry out through intermediate A, followed by electrophilic metalation pathway to generate the arylpalladium π-complex B. It was found that the nitrogen linker between phenyl and 2-pyridyl moieties could stabilized B via equilibrating with B′. The conversion of B to the six-membered palladacycle C, and subsequent ligand exchange by acetylene afforded the arylpalladium acetylide intermediate D, which under reductive elimination delivered the alkynylated product 24. The Pd(0) could be oxidized to Pd(II) in the presence of BQ and TsOH (Scheme 15).
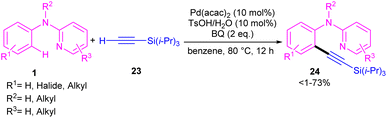 |
| | Scheme 14 Pd(II)-catalyzed reaction of N-aryl-2-aminopyridines with (triisopropylsilyl)acetylene. | |
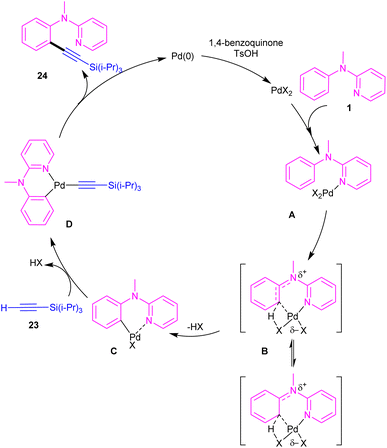 |
| | Scheme 15 Proposed mechanism for Pd(II)-catalyzed reaction of N-aryl-2-aminopyridines with (triisopropylsilyl)acetylene. | |
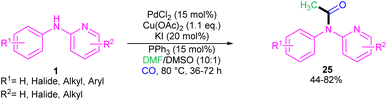 |
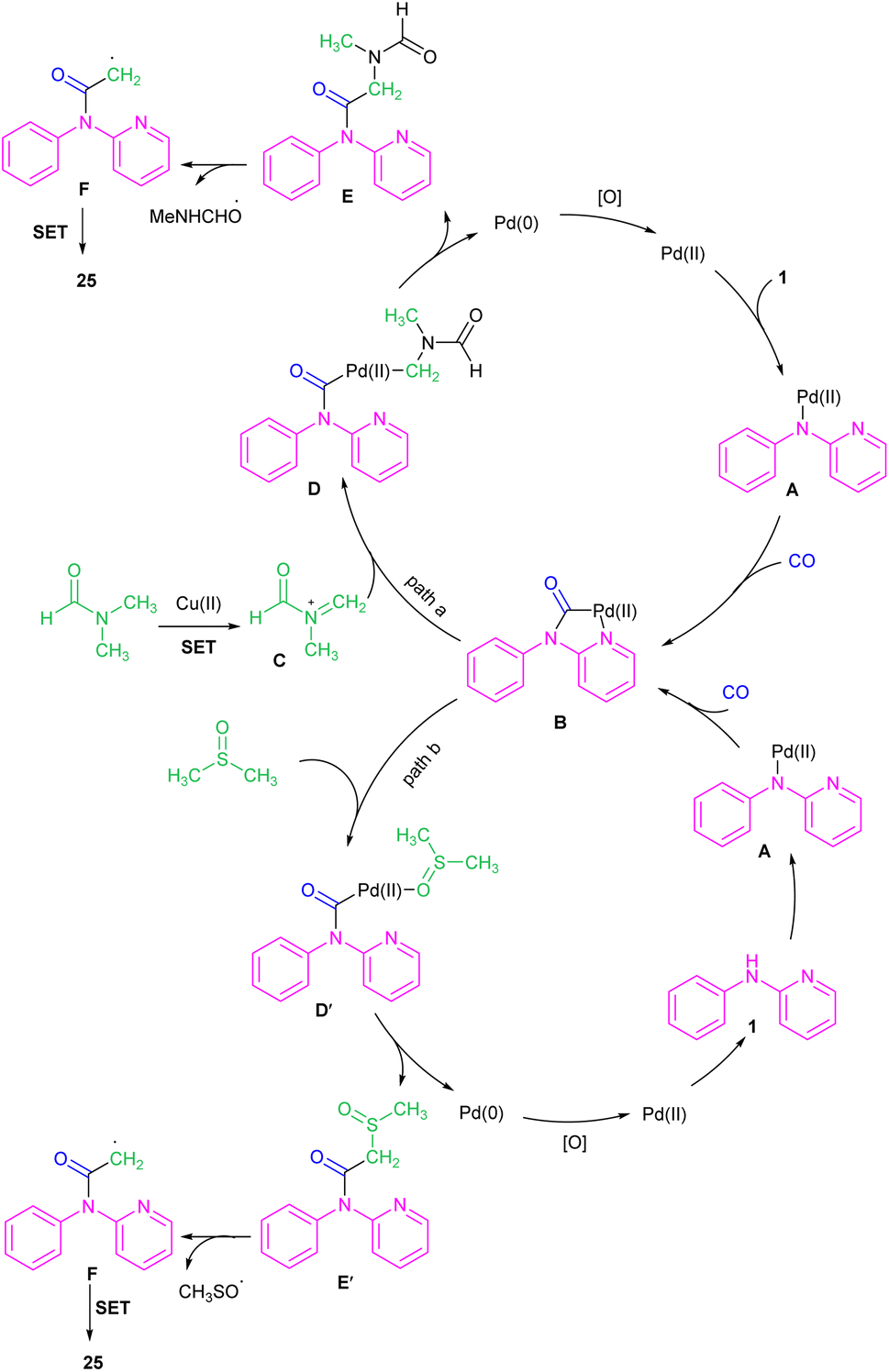
| | Scheme 16 Pd(II)-catalyzed acylation of N-phenyl-2-aminopyridines with DMF and CO. | |
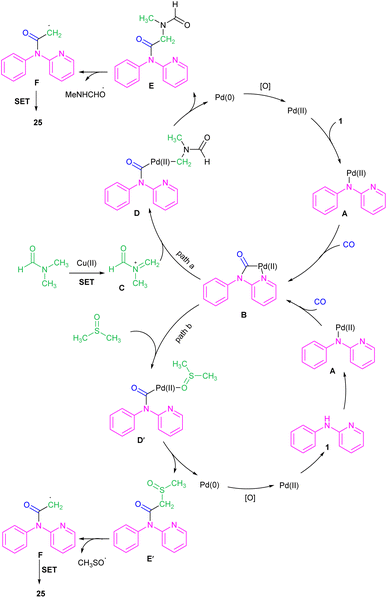 |
| | Scheme 17 Rational mechanism for Pd(II)-catalyzed acylation of N-phenyl-2-aminopyridines with DMF and CO. | |
2.2. Rh-catalyzed cross-coupling of N-aryl-2-aminopyridines
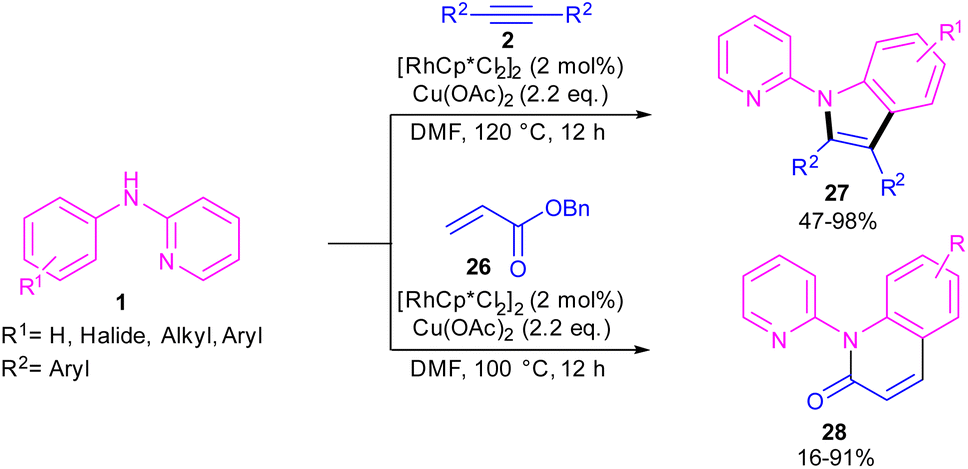
2.2.1. Annulation. In 2010, Li and co-workers reported an interesting method for producing N-(2-pyridyl)indoles 27 and N-(2-pyridyl)quinolones 28 through the annulation of two different reagents with N-aryl-2-aminopyridine 1 (Scheme 18).38 For this purpose, they treated N-aryl-2-aminopyridine 1 with alkynes 2 in the presence of 2 mol% of [RhCp*Cl2]2 to make N-(2-pyridyl)indoles 27 via (3 + 2)-cycloaddition process. In another transformation, 4 mol% of [RhCp*Cl2]2 and acrylates 26 as coupling partners were used to perform (3 + 3)-cycloaddition with N-aryl-2-aminopyridine 1 towards the synthesis of N-(2-pyridyl)quinolones 28. In addition, under these catalytic conditions, the reaction of styrene with N-aryl-2-aminopyridine led to mono- and disubstituted olefination products.
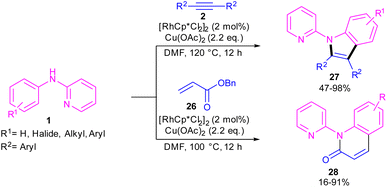 |
| | Scheme 18 Rh(III)-catalyzed C-annulation of N-aryl-2-aminopyridines with alkynes and acrylates. | |
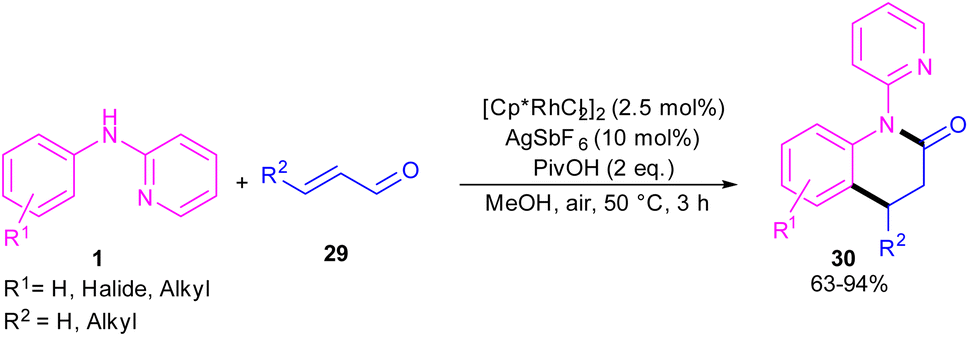
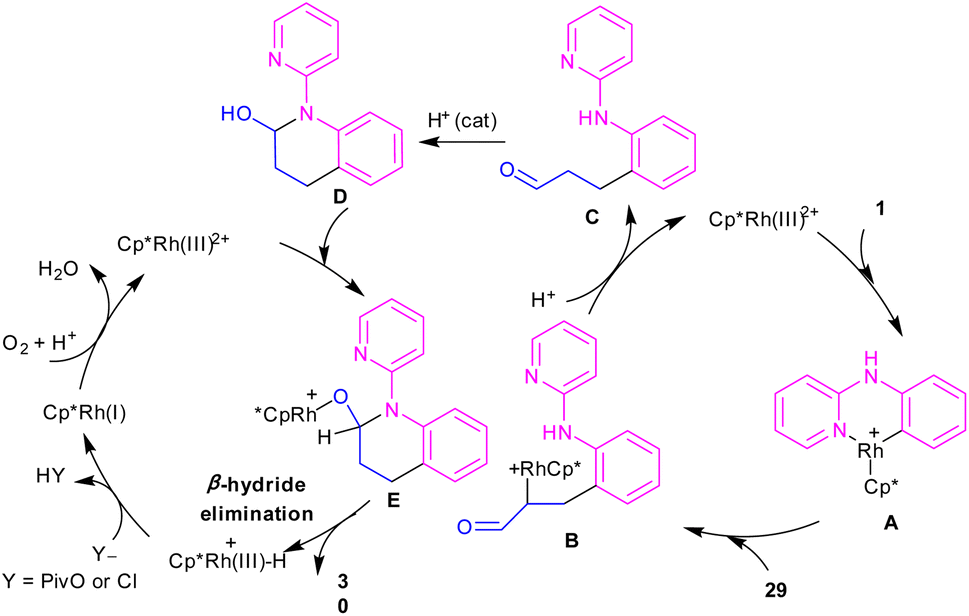
In 2017, Huang and co-workers developed Rh(III)-catalyzed C(sp2)–H functionalization/cyclization reaction of N-phenyl-2-aminopyridine and α,β-unsaturated aldehydes (Scheme 19).39 A series of dihydroquinolinones were well synthesized from both electron-rich and electron-poor N-aryl-2-aminopyridine substrates. According to the rhodium catalytic cycle in Scheme 20, pyridine-directed Rh(III) insertion in the α-C(sp2)–H bond of N-aryl-2-aminopyridine 1 to produce a six-membered rhodacycle A. Then, propenal 29 inserted into A to form intermediate B, which was protonated to generate intermediate C. Acid-catalyzed intramolecular nucleophilic addition of the amine onto the carbonyl group to yield α-hydroxyl tetrahydroquinoline D. Afterward, through a ligand exchange between Cp*Rh(III)2+ and D, a rhodium intermediate E was obtained, which underwent β-hydride elimination to afford the tetrahydroquinoline product 30 and Cp*Rh(III)+–H. The interaction of the anion Y− and Cp*Rh(III)+–H resulted in the loss of a HY molecule, followed by reductive elimination to Cp*Rh and subsequent oxidation to the catalytically active Cp*Rh2+ species.
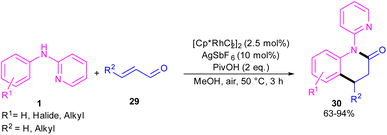 |
| | Scheme 19 Rh(III)-catalyzed C–H functionalization/cyclization reaction of N-arylpyridin-2-amines with α,β-unsaturated aldehydes. | |
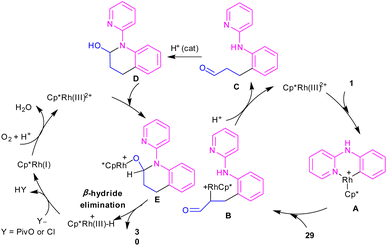 |
| | Scheme 20 Rh(III)-catalyzed C–H functionalization/cyclization reaction of N-arylpyridin-2-amines with α,β-unsaturated aldehydes. | |
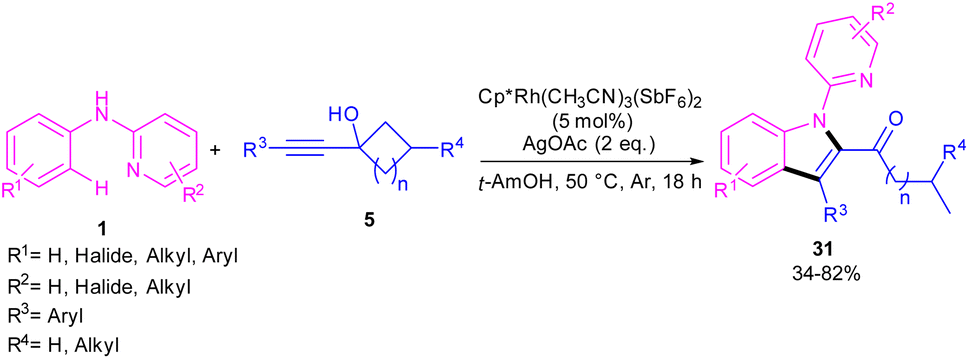
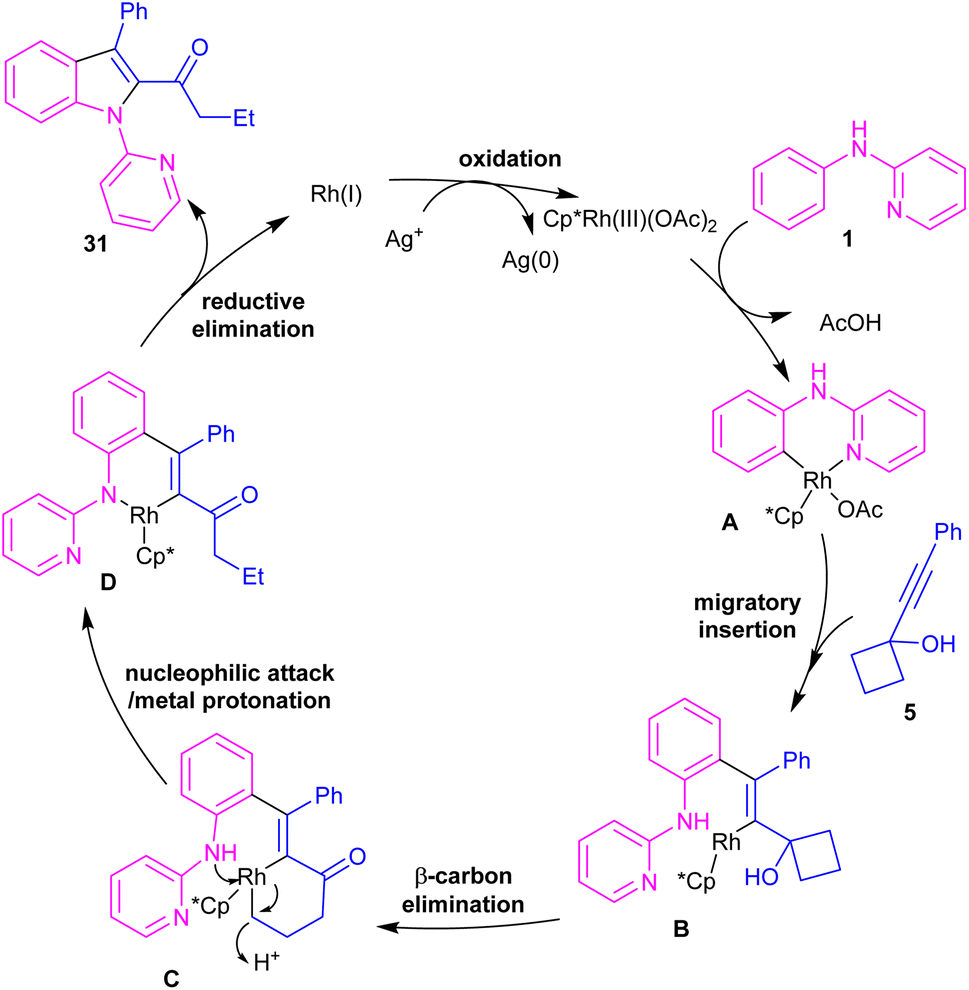
In the same year, Zeng's research team developed a rhodium catalytic strategy for the cross-coupling/cyclization between N-aryl-2-pyridinamines 1 and propargylic cycloalkanols 5 (Scheme 21).40 A series of 1,2,3-trisubstituted indoles 31 were synthesized in moderate to high yields. Evaluating various transition metals, including Pd(OAc)2, Cp*Co(CO)I2, (p-cymeneRuCl)2, [Cp*IrCl2]2, RhCl3, [Cp*RhCl2]2 and Cp*Rh-(CH3CN)3(SbF6)2 showed that only rhodium complexes have catalytic activity in this transformations, while others remained ineffective. A reasonable rhodium catalytic cycle involved the C–H activation of substrate 1 with the Rh(III) catalyst to form a six-membered rhodacycle A via a CMD process. Next, the migratory insertion of 5 to Rh(III) complex A afforded Rh(III) species B, which under a β-carbon elimination gave rhodacycle C. Intramolecular nucleophilic attack of the N-atom to Rh(III) and subsequent protonation rendered a Rh(III) complex D, followed by reductive elimination to yield 1,2,3-trisubstituted indole 31. The generated Rh(I) was then oxidized to the catalytically active Rh(III) species (Scheme 22). The synthetic application of this method was showed by the introduction of a carbonyl methylene group at the C7-position of indole and also the reduction of ketone to the alcohol moiety.
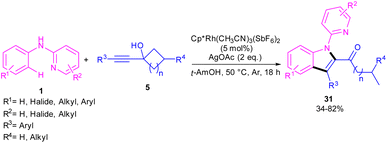 |
| | Scheme 21 Rh(III)-catalyzed cross-coupling/cyclization between N-aryl-2-pyridinamines and propargyl cycloalkanols. | |
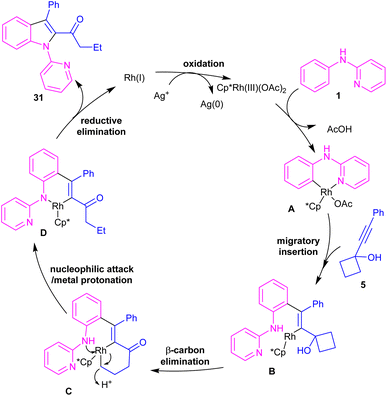 |
| | Scheme 22 Possible mechanism of Rh(III)-catalyzed cross-coupling/cyclization between N-aryl-2-pyridinamines and propargyl cycloalkanols. | |
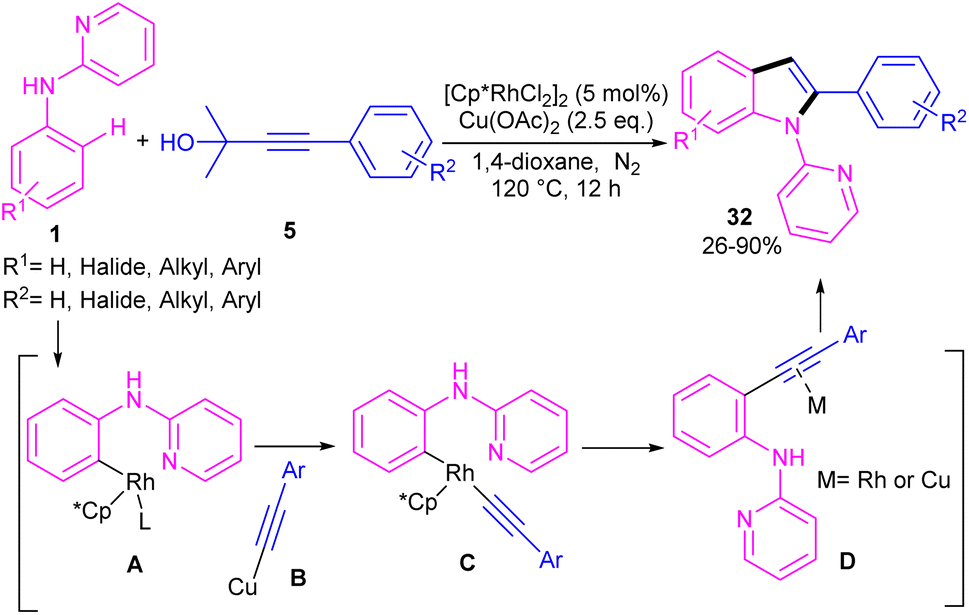
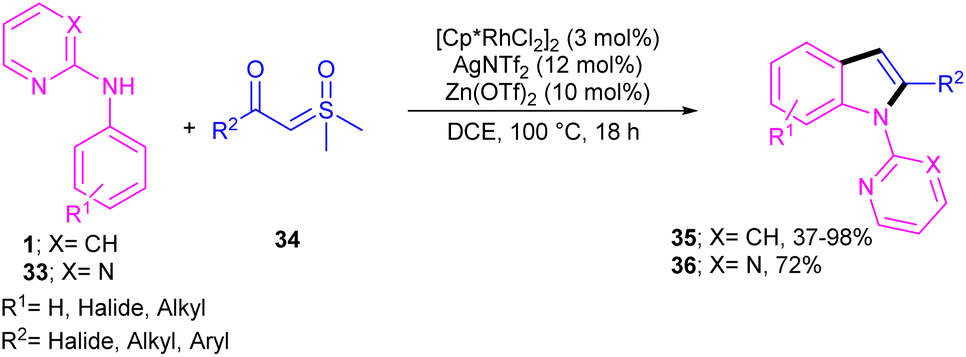
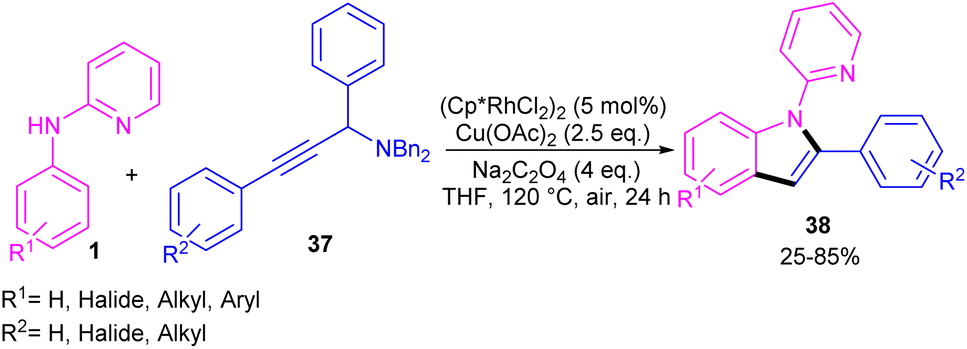
Propargylic alcohols can be a practical coupling partner in the reaction with N-aryl-2-aminopyridines to assemble indole derivatives (Scheme 23).41 5 mol% of Rh(III) and 2.5 equiv. of Cu(II) constitute an efficient catalytic system for the C–H activation and cascade annulation reaction. In this regard, ortho C(sp2)–H cleavage of 1 gave ruthenacycle A, which was then coordinated with the alkynyl copper species B to form an alkynyl rhodium C through transmetalation. Next, reductive elimination and subsequent annulation via nucleophilic attack of the amino group to the activated alkynyl moiety occurred in the presence of a Lewis acid (Rh or Cu salt) to deliver product 32. The generated Rh(I) was oxidized by Cu(II) to the active Rh(III) species to fulfill the catalytic cycle. None of metal catalysts including Ni(OAc)2·4H2O, Co(OAc)2·4H2O, [RuCl2(p-cymene)]2, and Pd(OAc)2 were workable. The same rhodium catalyst can also catalyze the annulation of N-aryl-2-aminopyridines 1 and N-aryl-2-aminopyrimidine 33 with sulfoxonium ylides 34 to provide indoles 35, 36 in up to excellent yields (Scheme 24).42 The indole synthesis involved C–H activation, coordination/insertion, and reductive elimination. The DMSO and H2O molecules were the only byproducts of this reaction. A similar rhodium catalytic system was reported for the reaction of N-aryl-2-aminopyridines 1 with propargylic amines 37 to assemble the indole products 38 (Scheme 25).43 A reaction mechanism similar to propargylic alcohols was suggested for this transformation, involving Rh-catalyzed C–H activation, alkyne insertion, and Lewis acid-promoted nucleophilic attack of the amino group to the activated alkynyl moiety towards intramolecular cyclization.
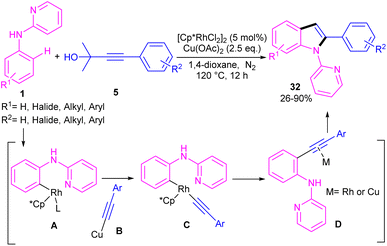 |
| | Scheme 23 Rh(III)-catalyzed annulation of N-phenyl 2-aminopyridine and propargylic alcohols. | |
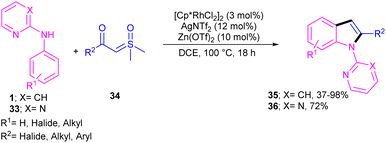 |
| | Scheme 24 Rh(III)-catalyzed annulation of N-phenyl 2-aminopyridine and sulfoxonium ylides. | |
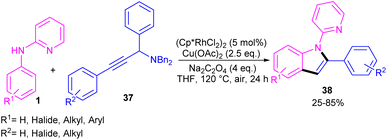 |
| | Scheme 25 Rh(III)-catalyzed annulation of N-phenyl 2-aminopyridine and propargylic amines. | |
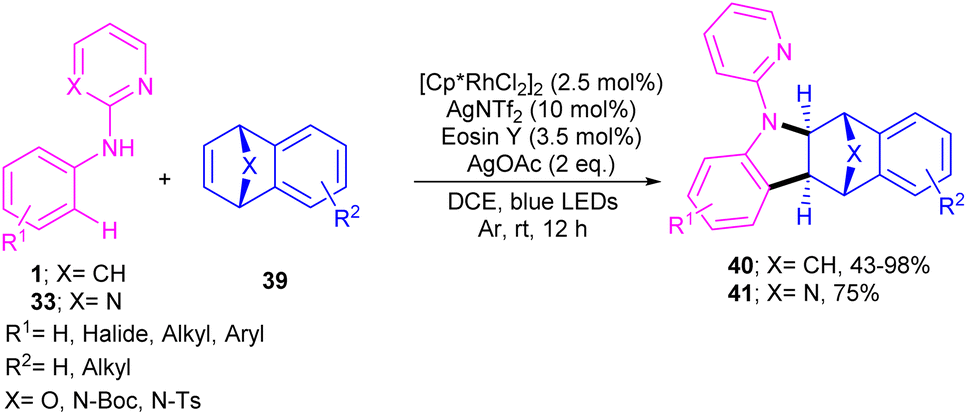
The cooperation of a rhodium catalysis and a photocatalysis can provide an efficient system for synthesizing a new library of bridged tetrahydro benzocarbazole scaffolds 40, 41 (Scheme 26).44 Various oxa- and aza-bicyclic alkenes were tolerated smoothly in the (2 + 3)-annulation reaction with N-aryl 2-aminopyridines 1 leading to bridged oxa-, aza-tetrahydro benzocarbazoles. The structure of bicyclic motif was intact in this reaction. Further aromatization under acidic conditions can provide the corresponding benzo[b]carbazoles via a ring-opening/hydration step. The method has the advantages of low catalyst loading, mild reaction conditions, broad functional groups tolerance and gram-scale synthesis. In addition, the removal of the directing group pyridine, acylation and Ullmann C–N coupling under copper catalysis were also carried out in this work.
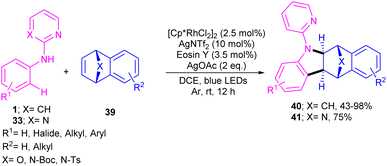 |
| | Scheme 26 Rh(III)-catalyzed C–H annulation of aromatic amines with bicyclic alkenes. | |
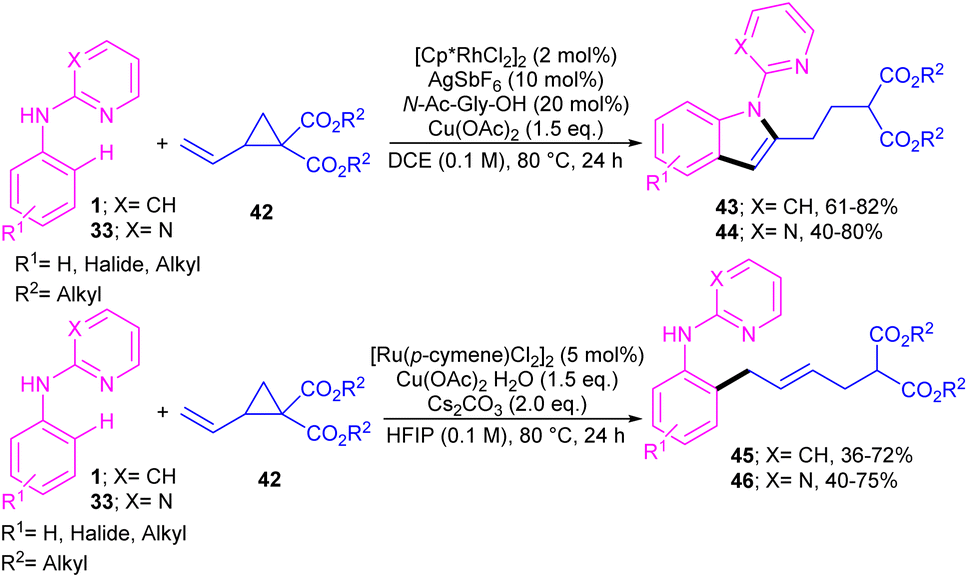
A series of N-phenyl 2-aminopyridines 1 and N-phenyl 2-aminopyrimidines 33 were investigated in the reaction with electron-deficient cyclopropanes 42 under metal catalysis systems (Scheme 27).45 Interestingly, when N-phenyl 2-aminopyridine used as a substrate, open-chain aniline products were obtained under ruthenium catalytic conditions. While these substrates did not reactive in the presence of a rhodium catalyst. Unlike, N-phenyl 2-aminopyrimidines were well tolerated in not only ruthenium-catalyzed reaction with cyclopropanes towards aniline products but also in rhodium-catalyzed annulation reaction with cyclopropanes towards indole frameworks. However, the replacement of NH-phenyl moiety with O-phenyl or S-phenyl group in the pyridine and pyrimidine substrates did not yield any products, indicating the important role of the nitrogen as a chelating group for the formation the metal complexes.
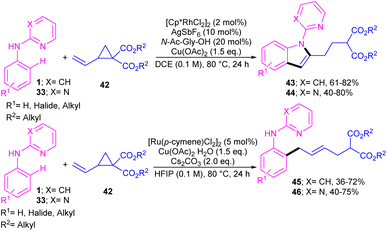 |
| | Scheme 27 Catalyst-controlled chemodivergent approach to access C2-substituted indoles and aniline derivatives. | |
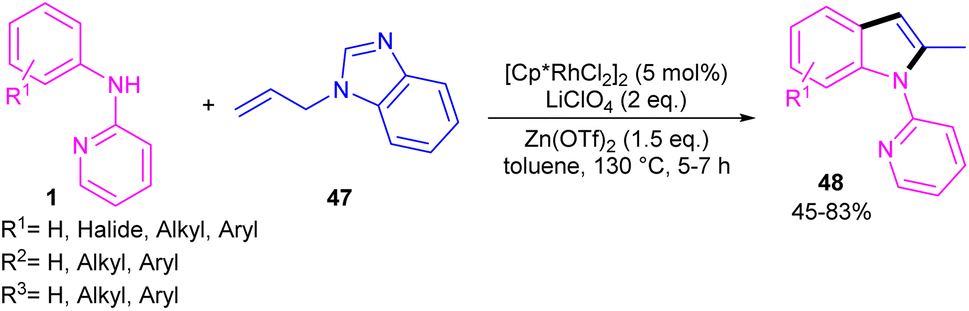
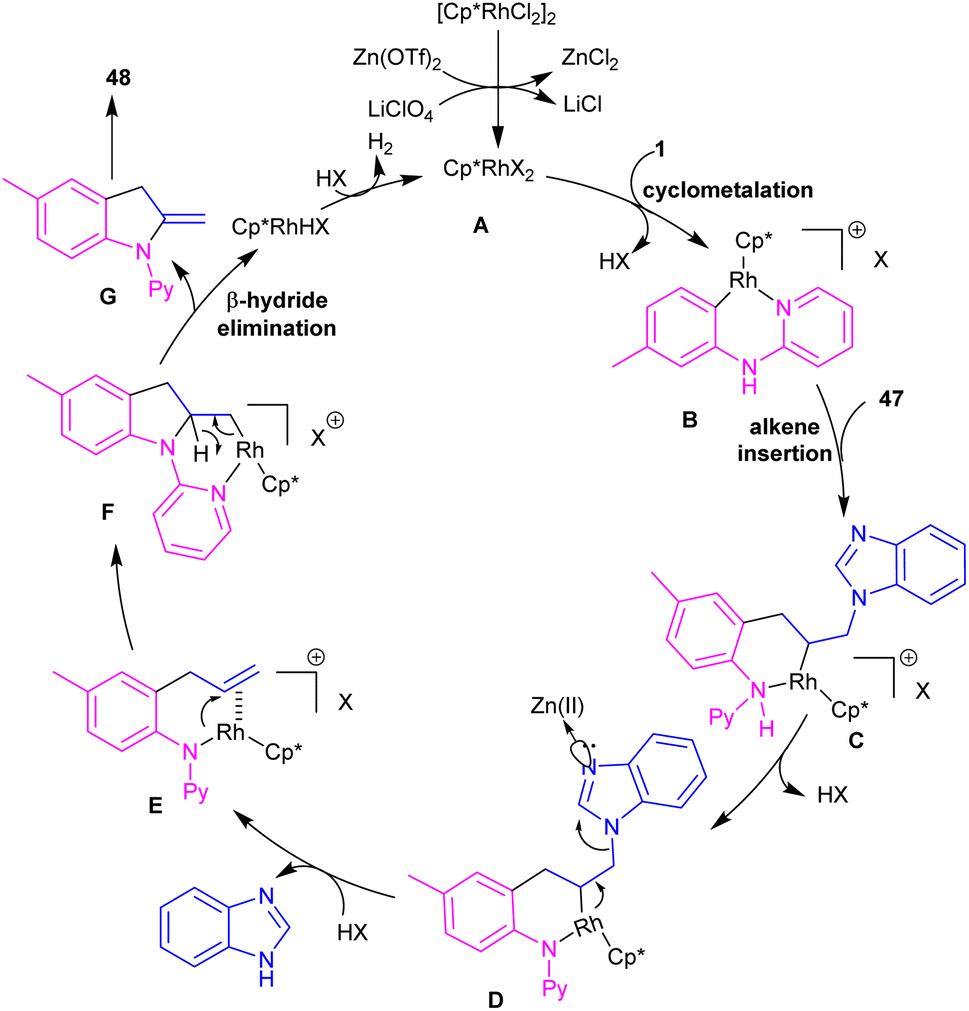
2-Methylindole derivatives can be achieved from the (3 + 2)-annulation of N-phenyl 2-aminopyridines and N-allylbenzimidazoles (Scheme 28).46 The method involved the cleavage of C–N bond of N-allylbenzimidazole to act as a 2C synthon participated in the intramolecular (3 + 2)-cycloaddition with N-phenyl 2-aminopyridines. Radical scavenger experiments by TEMPO and BHT demonstrated a non-radical pathway. The catalytic cycle involved irreversible cyclometalation of the active catalyst A with N-(p-tolyl)pyridine-2-amine 1, followed by migratory insertion of alkene 47 into the Rh–C bond. Next, the removal of benzimidazole gave intermediate E, followed by the addition of alkene to the Rh–N bond to form intermediate F. The β-hydride elimination of F, and subsequent aromatization delivered product 48 (Scheme 29). The utility of this method was demonstrated by removal of the pyridyl group, bromination, and acetylation reactions of indole as well as the gram-scale synthesis.
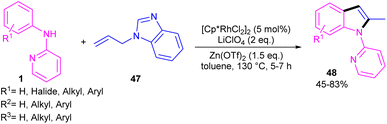 |
| | Scheme 28 Rh(III)-catalyzed (3 + 2)-annulation of N-phenyl 2-aminopyridine and N-allylbenzimidazole. | |
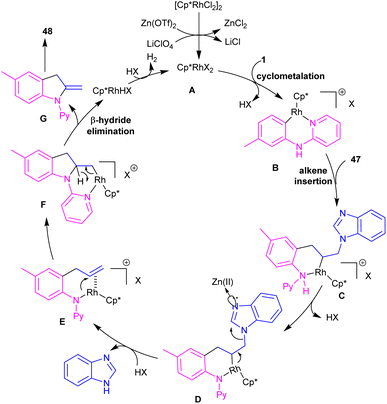 |
| | Scheme 29 Catalytic cycle for Rh(III)-catalyzed (3 + 2)-annulation of N-phenyl 2-aminopyridine and N-allylbenzimidazole. | |
2.2.2. Functionalization.
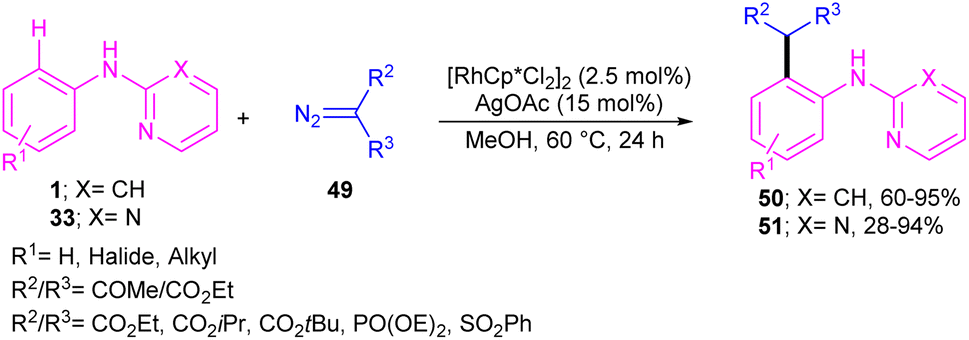
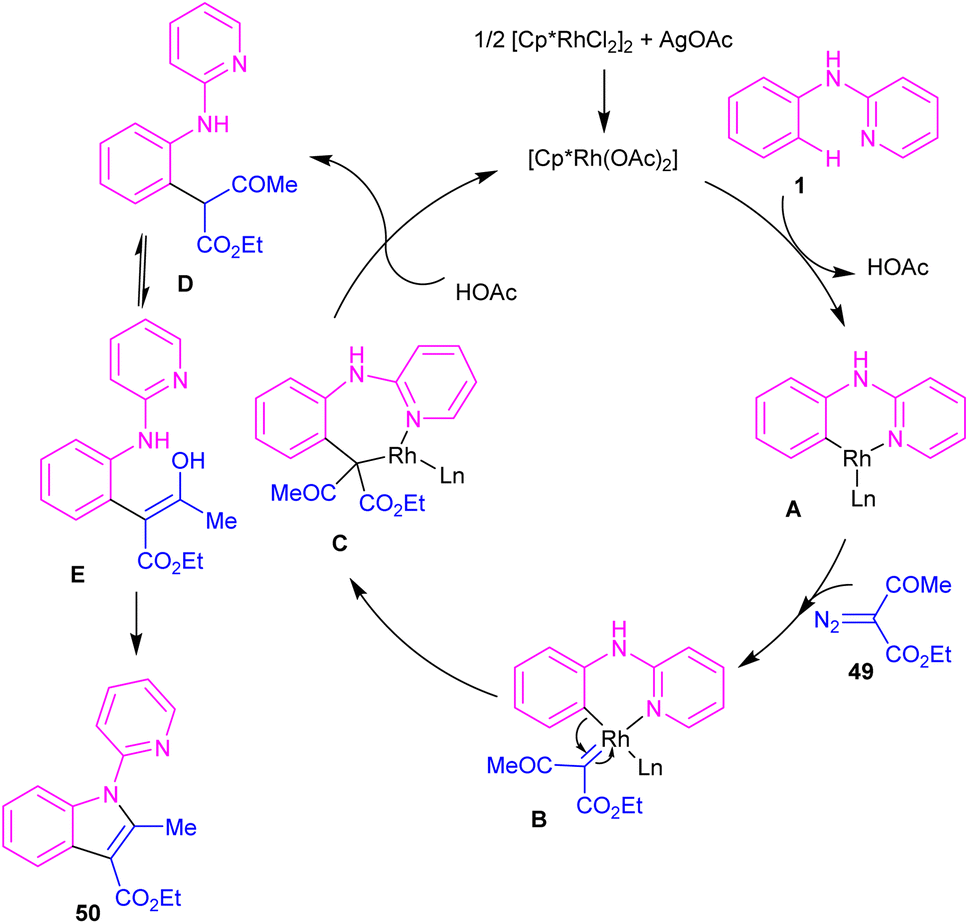
2.2.2.1. Alkylation. Kim and co-workers reported the cross-coupling of N-aryl-2-aminopyridines and N-aryl-2-aminopyrimidines with diazo compounds in moderate to excellent chemical yields in the presence of [RhCp*Cl2]2 (2.5 mol%) and AgOAc (15 mol%) in methanol (Scheme 30).47 Diazo malonates as coupling partners gave the alkylation products, while the indole products were isolated using diazo acetoacetates. In every case of alkylation, only monoalkylation product was observed, and it occurred exclusively in the C2-position. Late-stage functionalizations of the indole compounds at the C7-position, including alkylation, cyanation and amidation were also performed in this work. The mechanism of the reaction involved the generation of a six-membered rhodacycle A from 1 and the Rh(III), followed by the coordination of α-diazo compound 49 along with release of N2 to form the metal-carbenoid intermediate B. Migratory insertion in B rendered the seven-membered rhodacycle C, which was protonated to generate the ortho-alkylated product D and the reactive Rh(III) species. On the other hand, through a keto–enol tautomerization of D, intermediate E was obtained, which was then subjected to dehydration to afford indole 50 (Scheme 31).
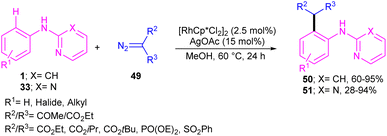 |
| | Scheme 30 Rh(III)-catalyzed alkylation of N-phenyl 2-aminopyridine with diazo compounds. | |
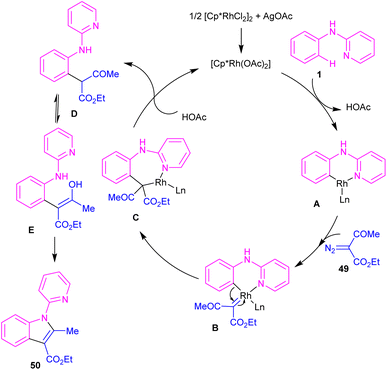 |
| | Scheme 31 Plausible catalytic pathway for Rh(III)-catalyzed alkylation of N-phenyl 2-aminopyridine with diazo compounds. | |
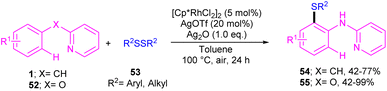 |
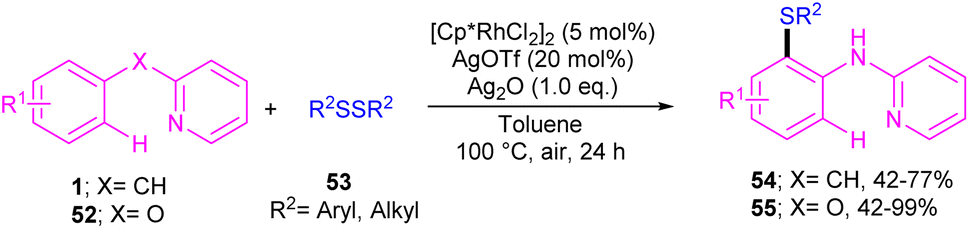
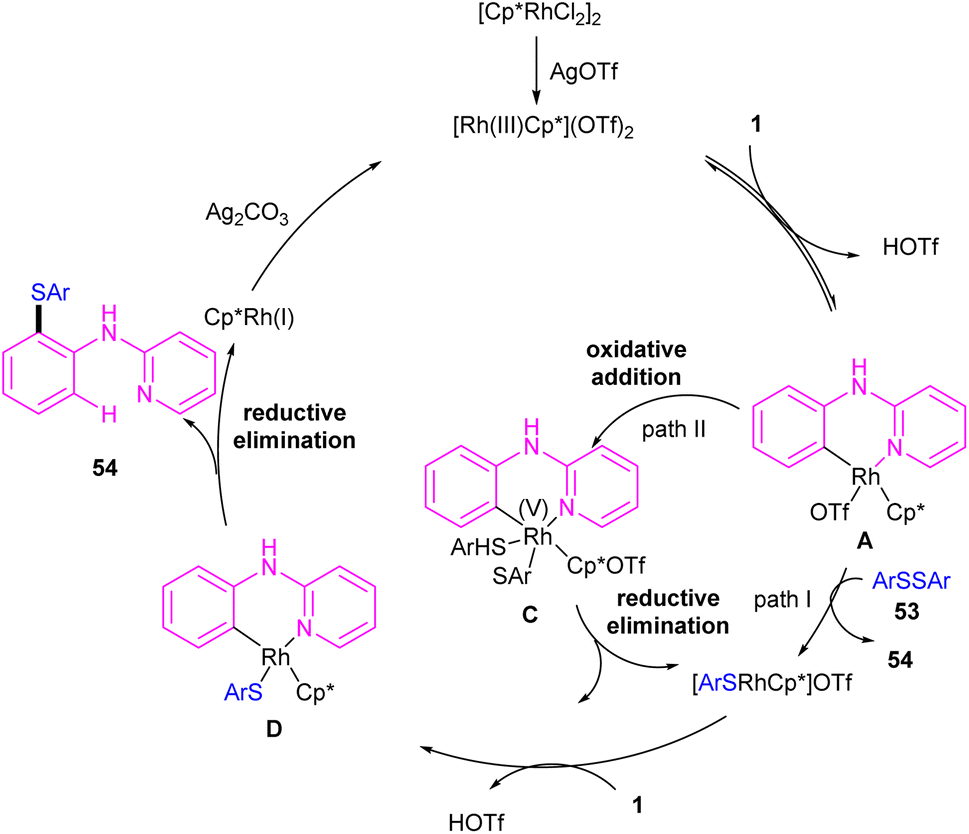
| | Scheme 32 Rh(III)-catalyzed annulation of N-aryl-2-pyridinamines with disulfides. | |
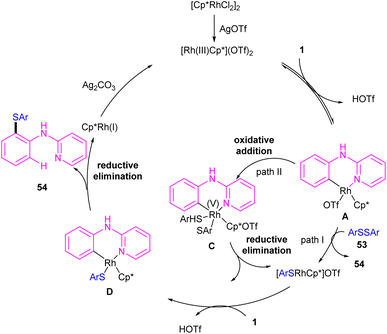 |
| | Scheme 33 Credible mechanism for Rh(III)-catalyzed annulation of N-aryl-2-pyridinamines with disulfides. | |
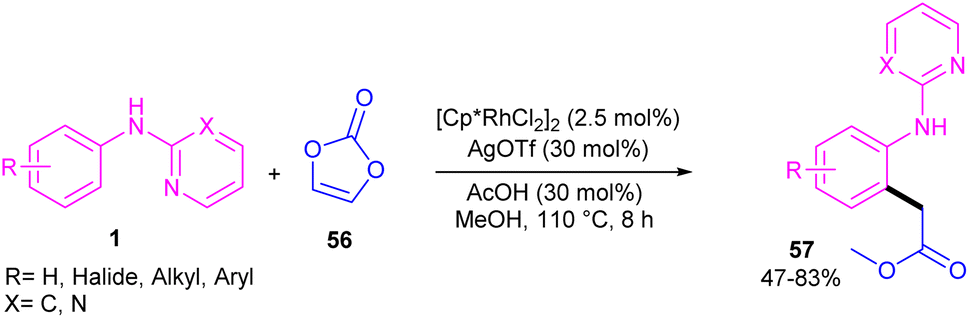
Regioselective C–H carboxymethylation of N-aryl 2-aminopyridines 1 with vinylene carbonate 56 can be carried out in the presence of a rhodium catalyst (Scheme 34).49 The transformation was not successful using other metal catalysts, such as ruthenium and iridium complexes. The catalytic procedure involved C–H activation, the vinylene carbonate insertion, and 1,2-Rh–C migration/decarboxylation. The easy removal of pyridine directing group was also possible under basic conditions. In addition, the large-scale synthesis the product (1.02 g, 70% yield) and the hydrolysis of the ester to acid group showed the utility of this method.
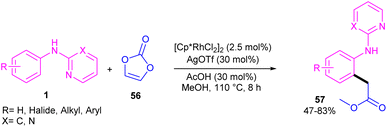 |
| | Scheme 34 Rh(III)-catalyzed C–H carboxymethylation of N-aryl-2-aminopyridines with vinylene carbonate. | |
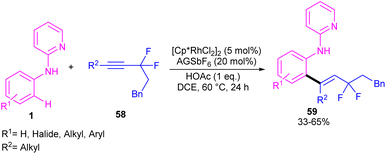 |
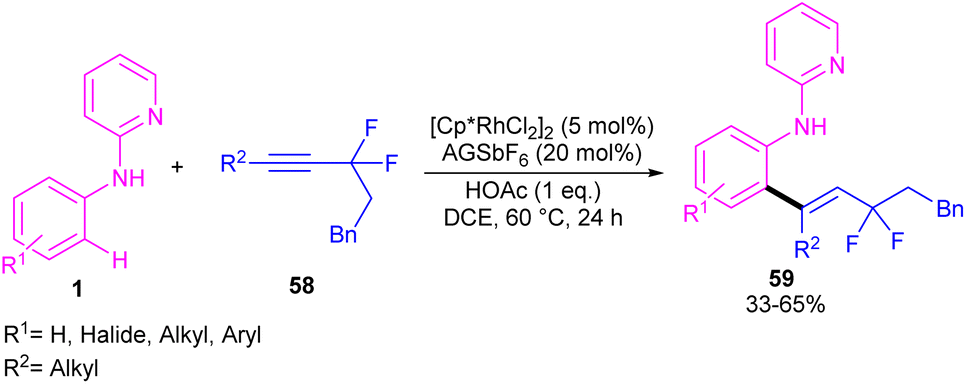
| | Scheme 35 Rh(III)-catalyzed C–H alkenylation of N-pyridylanilines and gem-difluoromethylene alkynes. | |
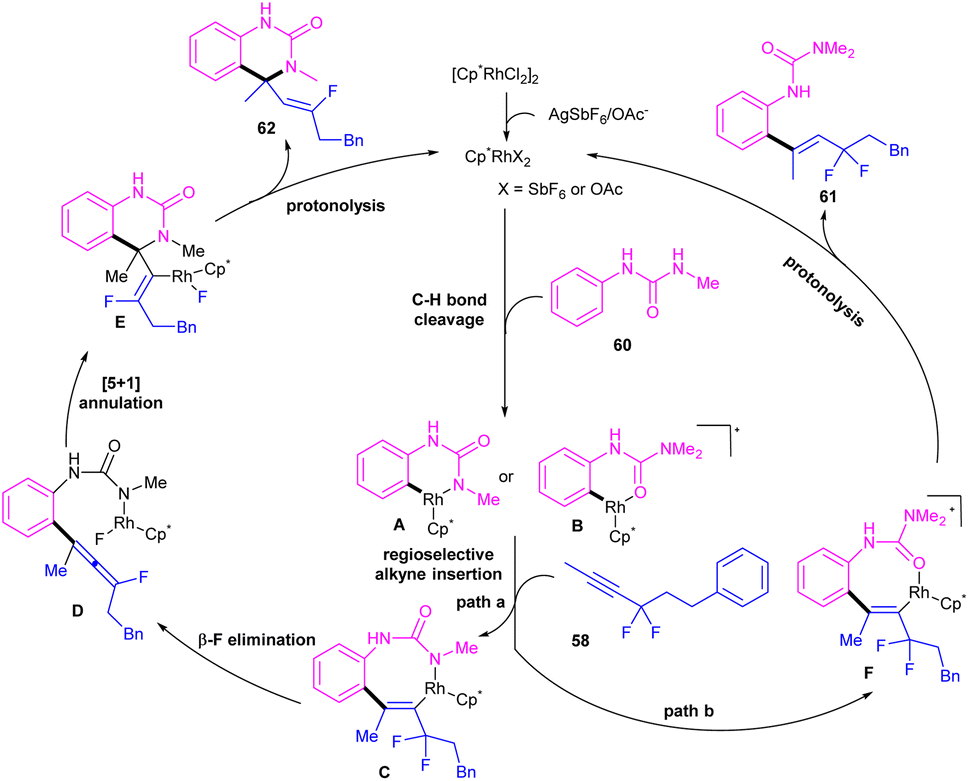
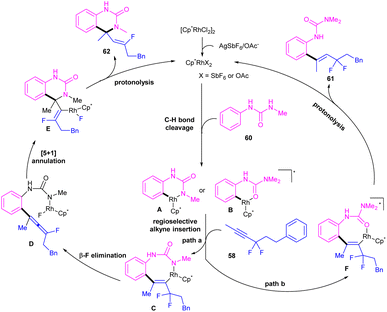 |
| | Scheme 36 Plausible mechanism for Rh(III)-catalyzed C–H alkenylation with N-pyridylanilines and gem-difluoromethylene alkynes. | |
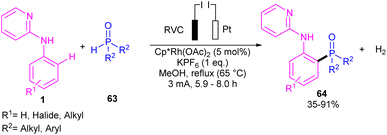 |
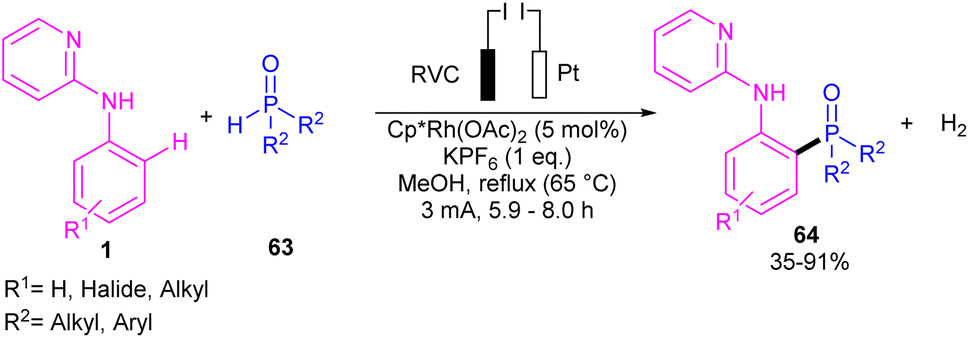
| | Scheme 37 Rh(III)-catalyzed C–H phosphorylation of N-aryl-2-aminopyridines with phosphorous reagents. | |
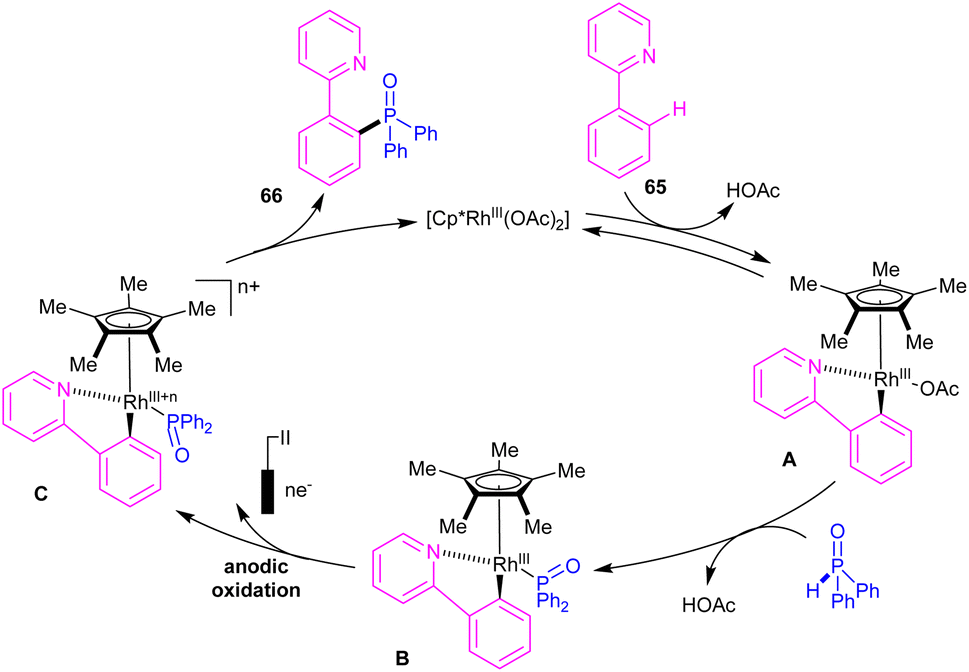
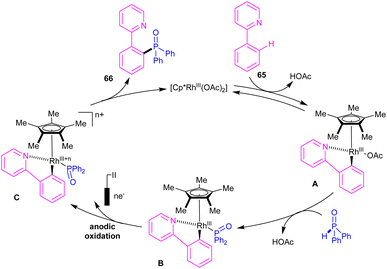 |
| | Scheme 38 Electrocatalytic cycle for Rh(III)-catalyzed C–H phosphorylation of N-aryl-2-aminopyridines with phosphorous reagents. | |
2.3. Ir-catalyzed cross-coupling of N-aryl-2-aminopyridines
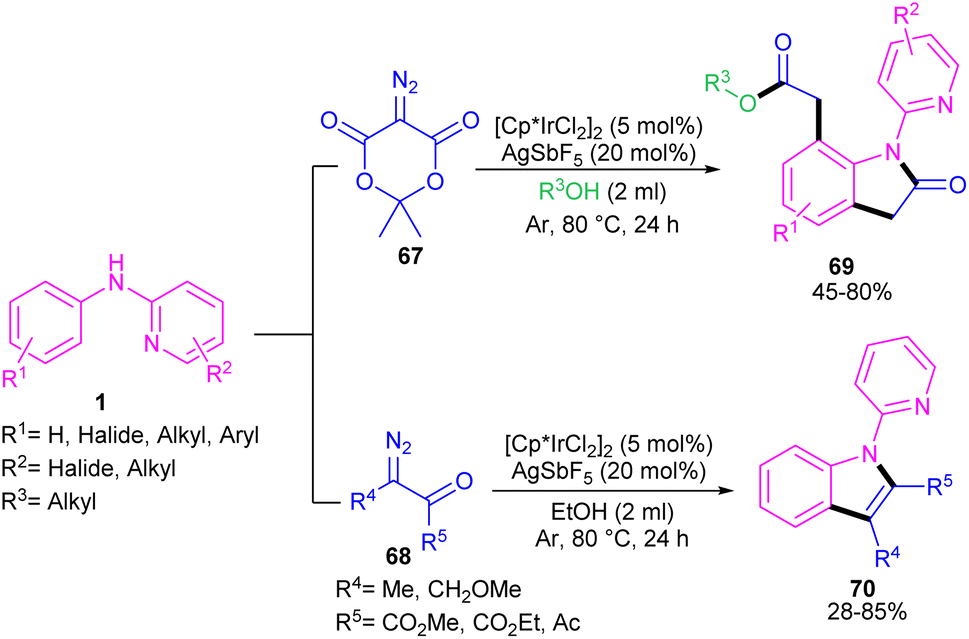
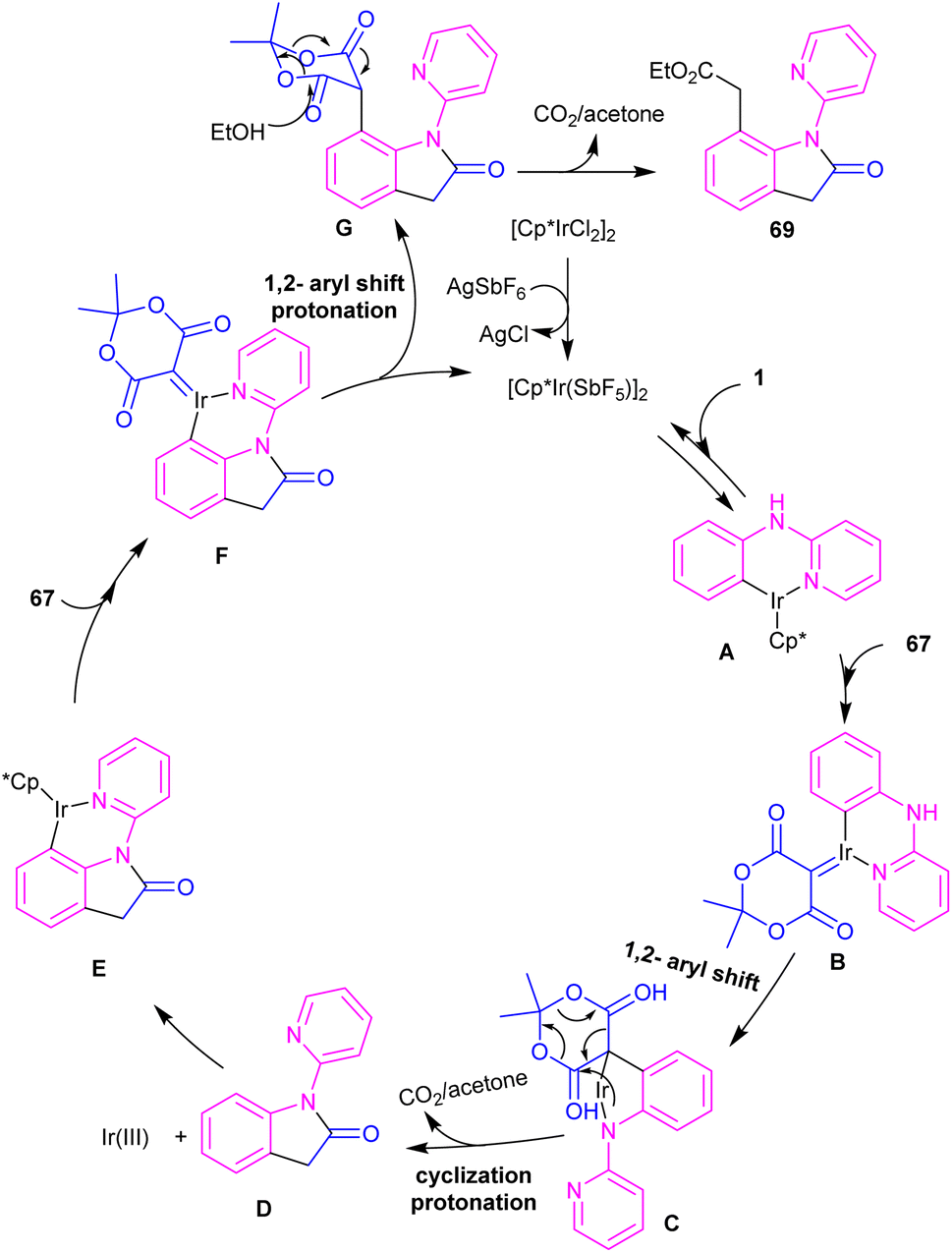
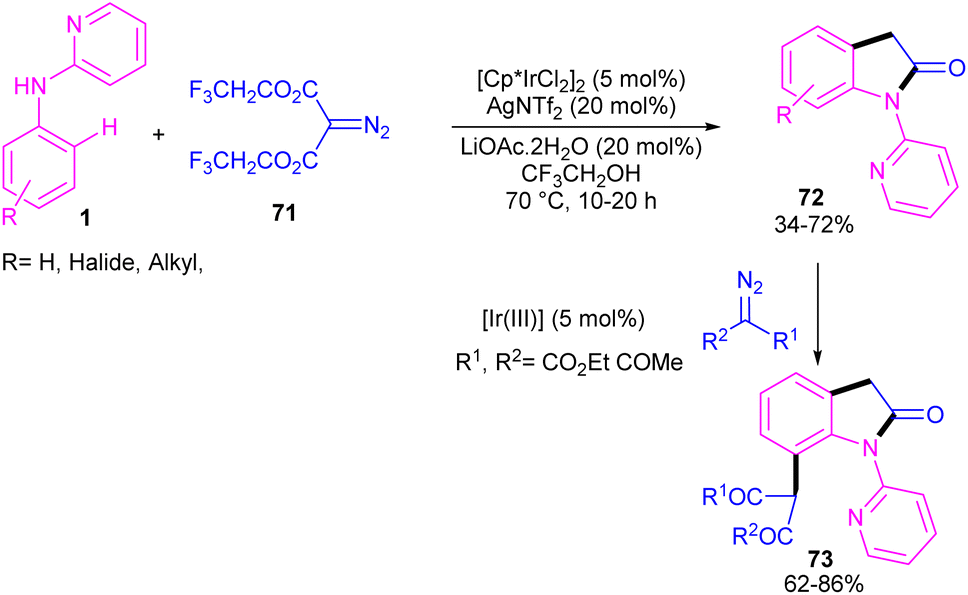
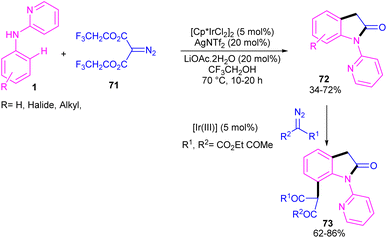
2.3.1. Annulation. Zeng and co-workers established an iridium(III) catalysis system for the annulation/functionalization of N-aryl-2-pyridinamines with diazo Meldrum's acid (Scheme 39).52 Various diazo compounds were investigated in the reaction with N-aryl-2-pyridinamines, in which 1,3-diketo-2-diazo compounds, and 1,3-ketoester-2-diazo compounds led to the corresponding indoles via mono selective ortho C–H activation. However, 1,3-diester-2-diazo compound and ethyl diazoacetate did not give any product. A possible mechanism was proposed for this carbenoid insertion/cyclization reaction, involving N-pyridyl coordination of 1 to the Ir(III), and subsequent phenyl C–H activation to form a six-membered Ir(III) complex A via a CMD process. Next, diazo compound 67 coordinated to the metal center of A, followed by denitrogenation to yield Ir–carbene B. After that, B underwent 1,2-aryl migratory insertion and cyclization/deprotonation to form N-(2-pyridyl)-2-indolone D. Finally, pyridyl-assisted the C(sp2)–H bond carbenoid insertion to obtain intermediate G, which nucleophilically attacked by an alcohol to provide the 7-substituted-2-oxindoles 69 with the release of acetone and CO2 (Scheme 40). Another work on the synthesis of oxindoles starting from N-phenyl-2-aminopyridine 1 with bis(2,2,2-trifluoroethyl) 2-diazomalonate 71 was described in the same year (Scheme 41).53 In this method, the same iridium(III) catalyst used by Zeng, could catalyze the assembly of oxindoles and 7-substituted-2-oxindoles. The general mechanism involved pyridyl-directed C–H activation, interaction of 71 with the Ir center to form iridium–carbene bond, migratory insertion, reductive elimination and the removal of CF3CH2H and CO2.
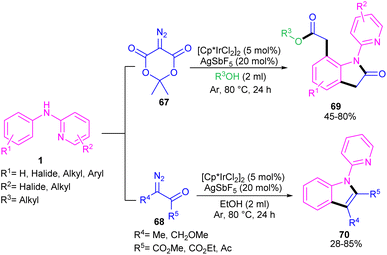 |
| | Scheme 39 Ir(III)-catalyzed annulation of N-aryl-2-pyridinamines with diazo Meldrum's acid. | |
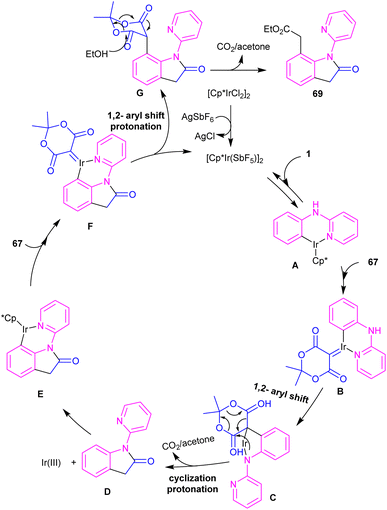 |
| | Scheme 40 Possible mechanism for Ir(III)-catalyzed annulation of N-aryl-2-pyridinamines with diazo Meldrum's acid. | |
 |
| | Scheme 41 Ir(III)-catalyzed annulation of N-phenyl-2-aminopyridine with bis(2,2,2-trifluoroethyl) 2-diazomalonate. | |
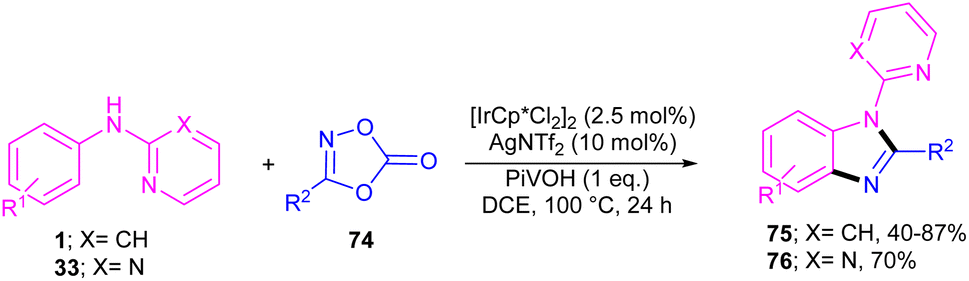
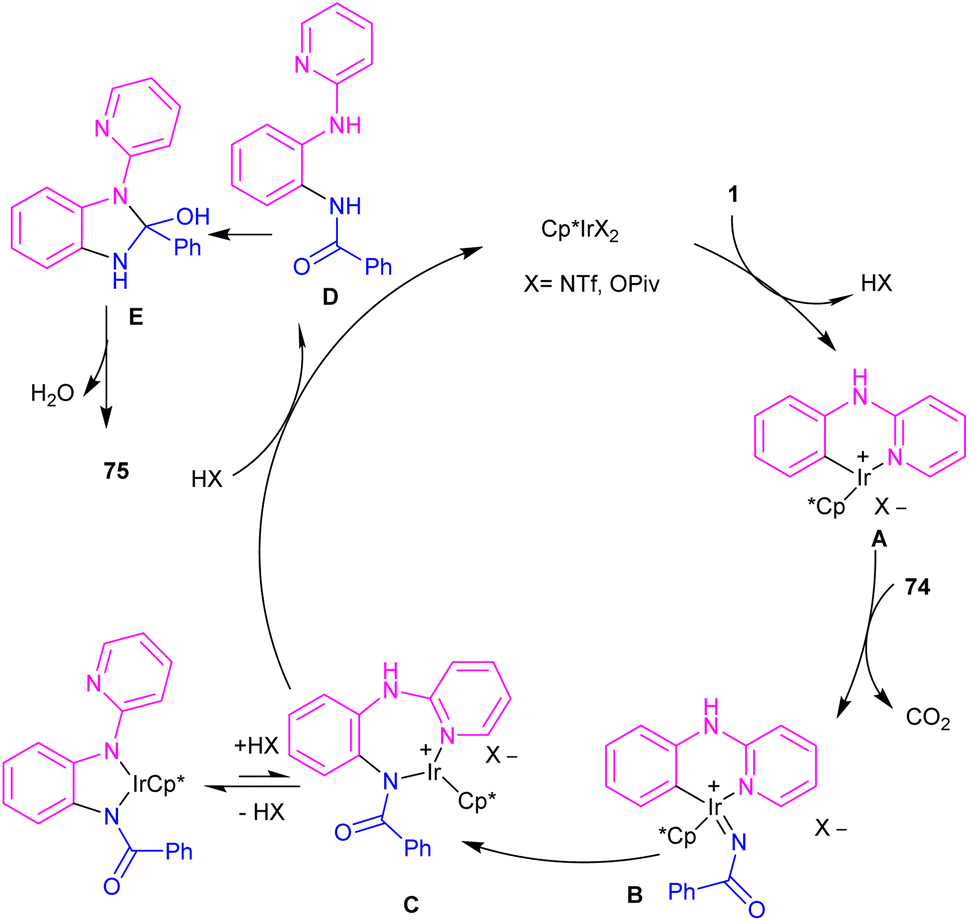
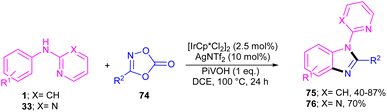
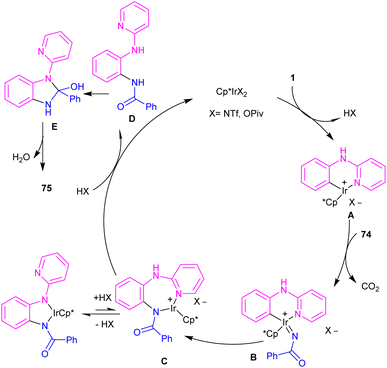
The first C–H activation and intermolecular coupling reaction access to benzimidazoles was reported by the Li group in 2017 (Scheme 42).54 The authors highlighted the importance of both [Cp*IrCl2]2 and a silver additive for this reaction. The reaction of N-aryl-2-pyridinamines and dioxazolones involved a redox-neutral mechanism and CO2 and H2O were the only byproducts. First, nitrogen-assisted C–H activation of 1 with the metal produced complex A, which further coordinated with 74 to yield a nitrenoid species B with the emission of CO2. Migratory insertion of nitrenoid into the Ir–aryl bond led to amidate C, which was protonated into an amidated intermediate D, together with regeneration of the Cp*Ir(III). Eventually, PivOH or Cp*Ir(III)-mediated intramolecular nucleophilic attack at the acyl group, followed by dehydration to furnish product 75 (Scheme 43).
 |
| | Scheme 42 Ir(III)-catalyzed C–H activation and amidation of aniline derivatives. | |
 |
| | Scheme 43 Rational mechanism for Ir(III)-catalyzed C–H activation and amidation of aniline derivatives. | |
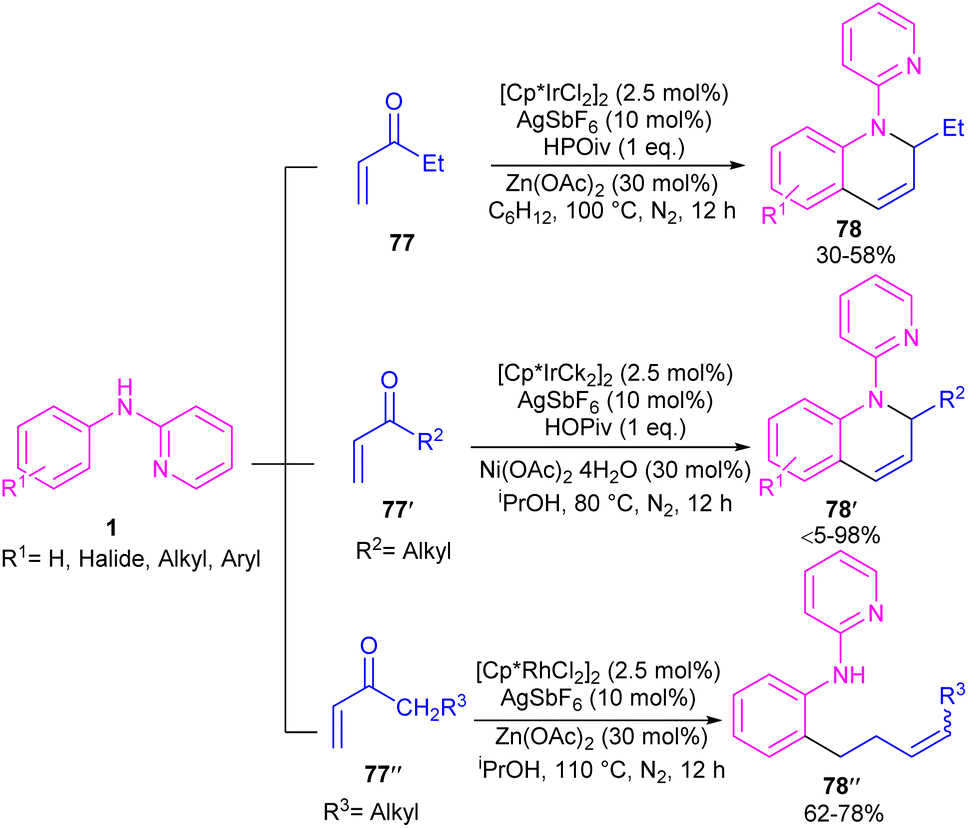
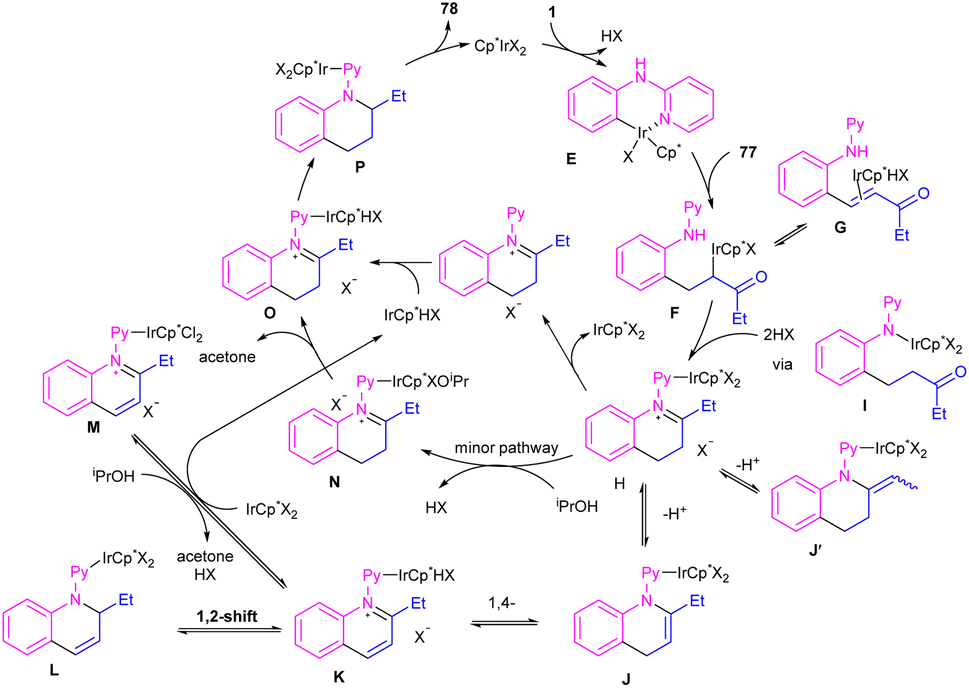
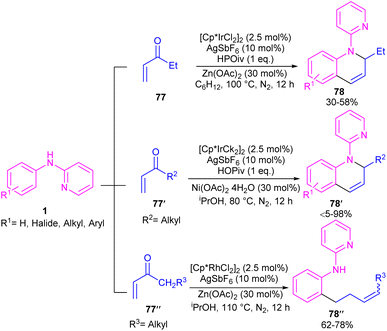
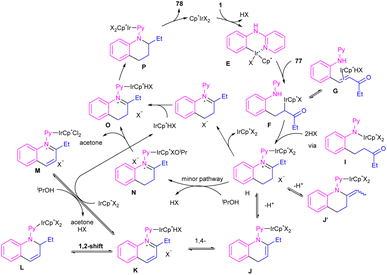
By controlling of the type of the transition metal catalyst, Li co-workers were able to isolate two different products from the reaction of N-aryl 2-aminopyridines with enones (Scheme 44).55 In their work, iridium-catalyzed reaction of N-aryl-2-aminopyridines and enones can produce tetra- or di-hydroquinoline products, while a rhodium catalysis system including these two starting materials led to the alkylation products. It should be noted that using different iridium catalytic conditions could also lead to tetrahydroquinolines or 1,2-dihydroquinolines. It was shown that iridium catalyze the reaction through intermediate A, which under acidic media could be converted to intermediate B. Subsequent transfer hydrogenation in B provided the cyclized products. Whereas, intermediate A directly afforded the alkylation product 78′′ under rhodium catalysis. A plausible mechanism was proposed for generating 1,2-dihydroquinolines. First, after the generation of iridacycle E via C–H activation of aniline 1 by metal, coordination and insertion of enone 77 into the Ir–Ar bond occurred to obtain the Ir(III) alkyl complex F in equilibrium with hydride G. After that, F was protonated and cyclized in the presence of Lewis acid Zn(II) or Ni(II). The obtained iminium ion H was then subjected to a ligand exchange with isopropoxide and subsequent β-H elimination to produce the Ir(III) hydride O. Next hydride attack to O provided amine P, which was converted to product 78 after a ligand dissociation. However, in the major pathway, reversible deprotonation of H gave enamines J or J′. Intermediate J can be converted to K, and L via reversible 1,4- and 1,2-hydride insertion. Through a ligand dissociation–association in the metal center of H and K, intermediate M was obtained, which can be reduced by iPrOH into H. Finally, disproportionation of a dihydroquinoline (J/L), and subsequent reduction of the quinolinium K delivered product 78 (Scheme 45).
 |
| | Scheme 44 Ir(III) and Rh(III)-catalyzed annulation and alkylation of N-aryl 2-aminopyridines with enones, respectively. | |
 |
| | Scheme 45 Plausible mechanism for Ir(III)-catalyzed annulation of N-aryl 2-aminopyridines with enones. | |
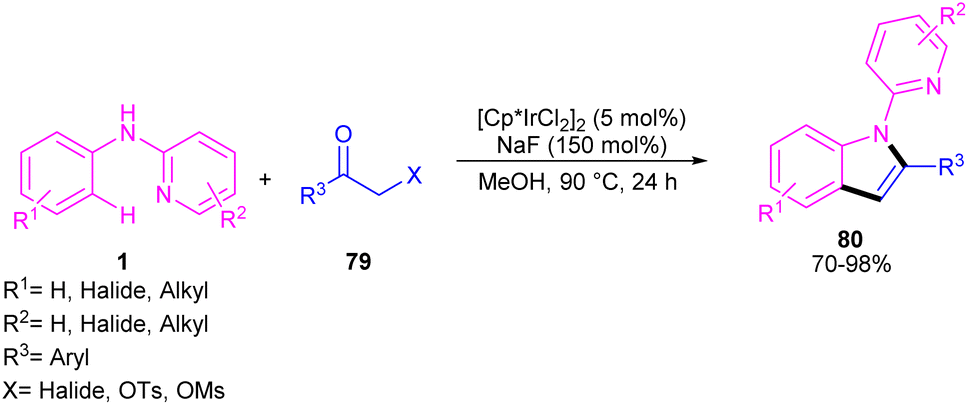
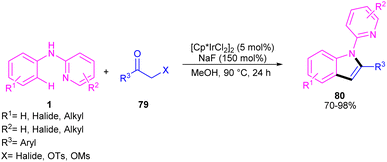
The indole synthesis can be achieved from the annulation of N-aryl-2-aminopyridines 1 with α-chloro ketones 79 under iridium catalysis (Scheme 46).56 The reaction was not proceeded in the presence of [Ru(p-cymene)Cl2]2 and Cp*Co(CO)I2, while led to 15% of chemical yield using [RhCp*Cl2]2. The transformation did not significantly influence by the electronic properties of substituents at the both benzene and pyridyl ring of N-aryl-2-aminopyridines, where the all products were obtained in high to excellent yields. Moreover, a wide range of α-chloro ketones with aryl, naphthyl, and heteroaryl moieties were compatible.
 |
| | Scheme 46 Ir(III)-catalyzed C–H annulation of N-phenyl-2-aminopyridines with α-chloro ketones. | |
2.3.2. Functionalization.
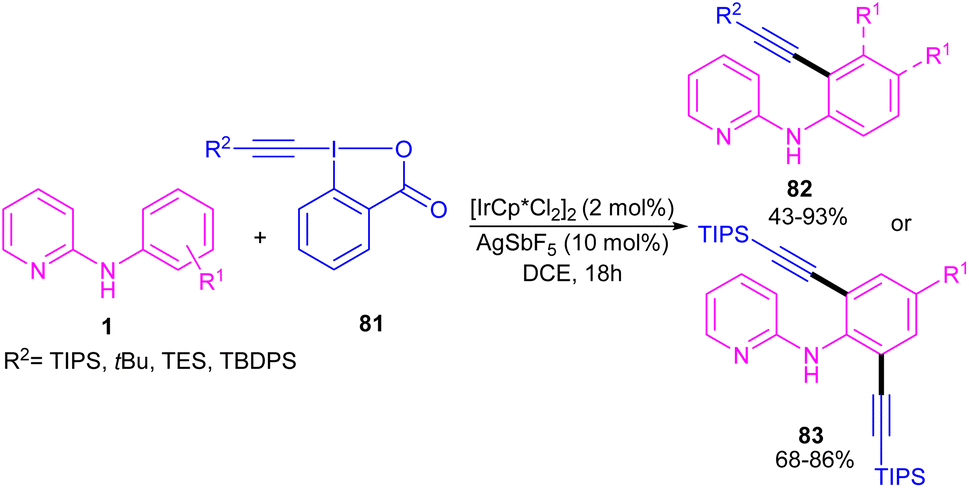
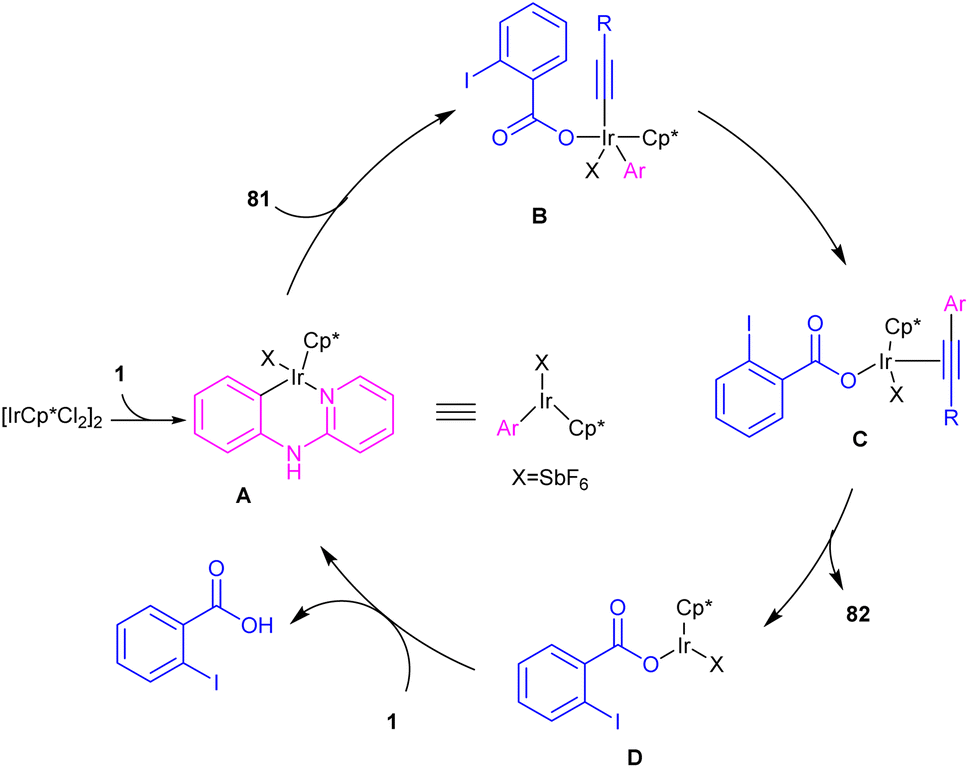
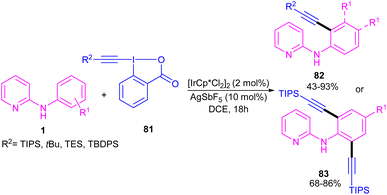
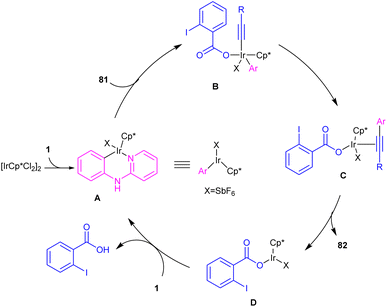
2.3.2.1. Alkenylation. In 2016, N-phenyl-2-aminopyridines were investigated for the alkynylation in the presence of a hypervalent iodine alkyne (TIPS-EBX) as a coupling partner (Scheme 47).57 Iridium(III) could efficiently catalyze the ortho-C–H alkynylation of N-phenyl-2-aminopyridines. Selectivity of mono alkynylation can be better achieved using TIPS-EBX compared to other alkyne sources. However, ortho- and meta-substituted benzene ring led to selective mono-alkynylation, while para-substituted benzene gave dialkynylated products. The reaction mechanism involved the formation of an active cationic Ir(III) species from the ligand exchange between [IrCp*Cl2]2 and AgSbF6, followed by the coordination of metal center with 1 to produce a six-membered iridacycle A. Oxidative addition of R-EBX 81 to A gave Ir(V) intermediate B, which under reductive elimination afforded Ir(III) alkyne C. Another C–C reductive elimination led to Rh(III) benzoic species D and the coupling product 82. Subsequently C–H activation of 1 resulted in the active iridacycle A and 2-iodobenzoic acid (Scheme 48). KIE experiment suggested the C–H activation is the rate-determining step.
 |
| | Scheme 47 Ir(III)-catalyzed C–H alkynylation of N-phenyl-2-aminopyridines. | |
 |
| | Scheme 48 Catalytic cycle for Ir(III)-catalyzed C–H alkynylation of N-phenyl-2-aminopyridines. | |
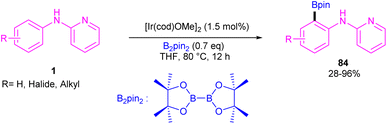 |
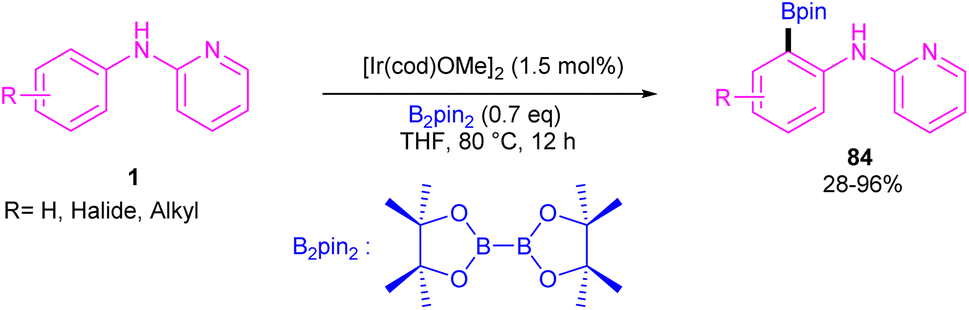
| | Scheme 49 Ir(III)-catalyzed C–H borylation of 2-anilinopyridines and 2-phenoxypyridines. | |
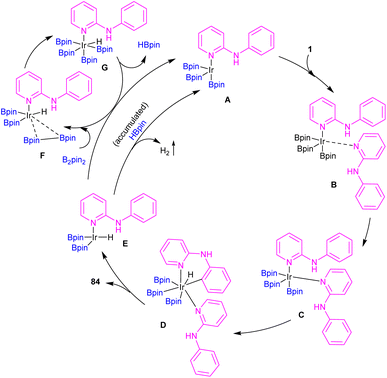 |
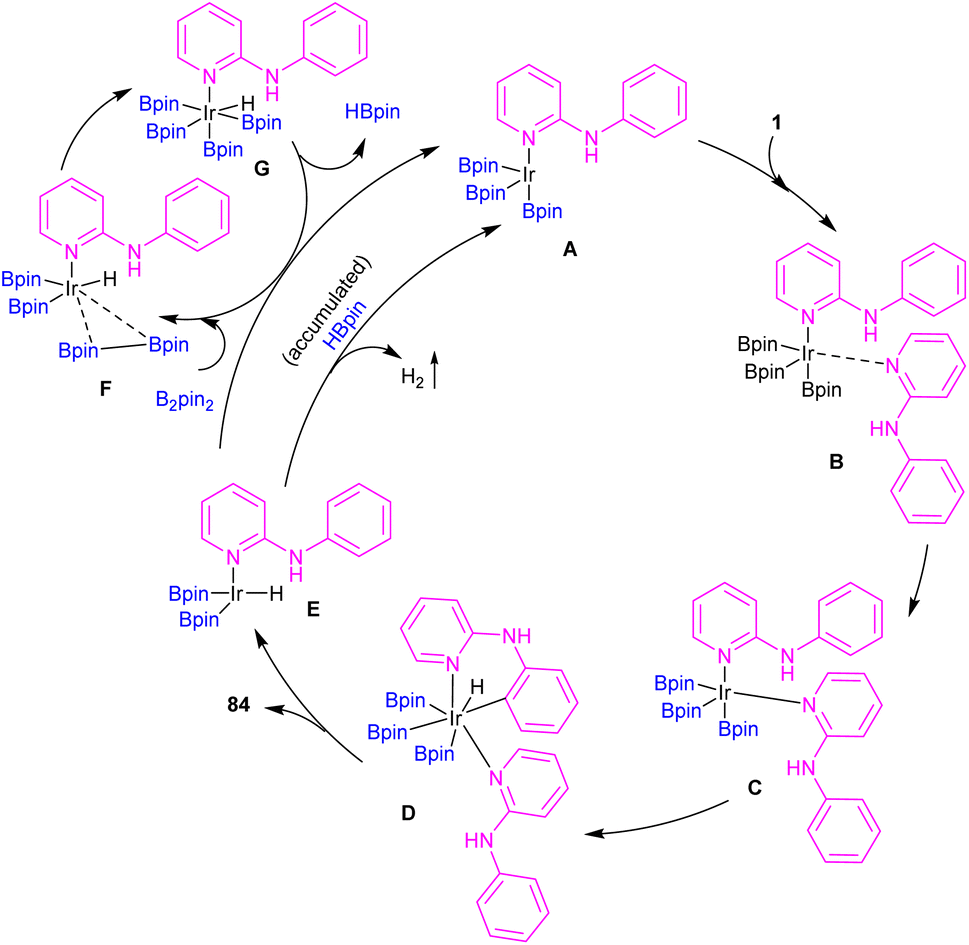
| | Scheme 50 Catalytic cycle for Ir(III)-catalyzed C–H borylation of 2-anilinopyridines and 2-phenoxypyridines. | |
2.4. Ru-catalyzed cross-coupling of N-aryl-2-aminopyridines
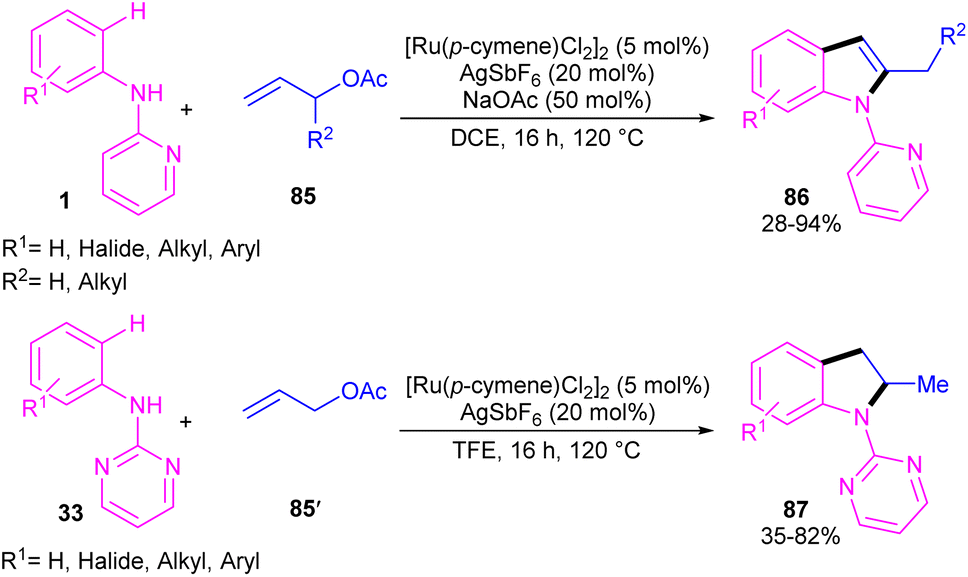
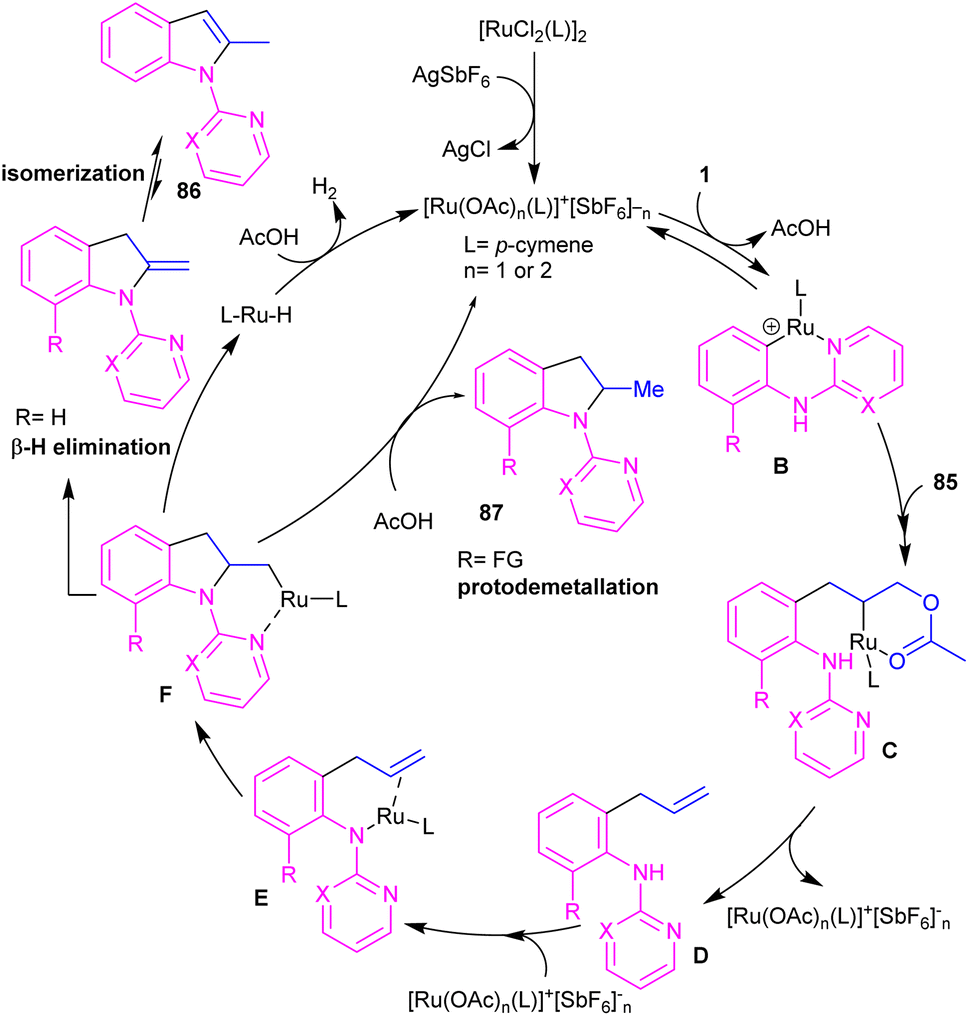
2.4.1. Annulation. A cascade C–H allylation/oxidative annulation of N-aryl-2-aminopyridines or N-aryl-2-aminopyrimidines with electron-deficient alkenes was reported by Jana and his team in 2018 (Scheme 51).59 In the case of N-aryl-2-aminopyridines as a substrate, β-hydride elimination from the σ-alkyl-Ru intermediate led to 2-methylindoles, while for N-aryl-2-aminopyrimidine substrates, due to the suppression of β-hydride elimination by steric effect in interaction with the pyrimidine group which stabilized the alkyl-Ru species through coordination, a protodemetalation occurred in lieu of β-hydride elimination. The authors proposed a mechanism for the Ru(II)-catalyzed cascade reaction as shown in Scheme 52. It was proposed that ortho C–H activation of 1 with the active ruthenium complex generated intermediate B, which underwent migratory insertion of alkene to obtain an alkyl ruthenium intermediate C. Then, β-acetoxy elimination gave allylation intermediate D. Further coordination of the Ru with the nitrogen and subsequent alkene insertion to the allyl group resulted in intermediate F. In the case of ortho-unsubstituted N-aryl-2-aminopyrimidines, F underwent a rapid β-hydride elimination and double bond isomerization to yield product 86. Whereas, for the ortho-substituted N-aryl-2-aminopyrimidines 33, the protodemetalation in the protic solvent, like TFE, afforded product 87. The generated ruthenium hydride species in this step was converted to the active catalyst via the interaction of H2 gas with AcOH. Indole derivatives can also be achieved through ruthenium-catalyzed annulation of N-aryl-2-aminopyridines with α-carbonyl sulfoxonium ylides.60 However, [IrCp*Cl2]2 and [RhCp*Cl2]2 resulted in very low yield of product, and Cp*Co(CO)I2 did not workable.
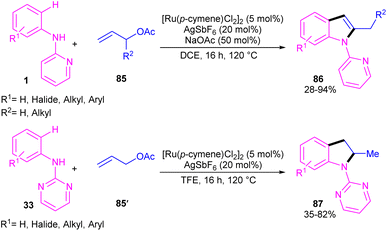 |
| | Scheme 51 Ru(II)-catalyzed C–H allylation/oxidative annulation of N-aryl-2-aminopyridines or N-aryl-2-aminopyrimidines with electron-deficient alkenes. | |
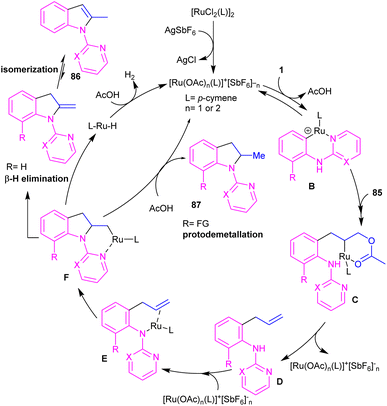 |
| | Scheme 52 Plausible mechanism for Ru(II)-catalyzed C–H allylation/oxidative annulation of N-aryl-2-aminopyridines or N-aryl-2-aminopyrimidines with electron-deficient alkenes. | |
2.4.2. Functionalization.
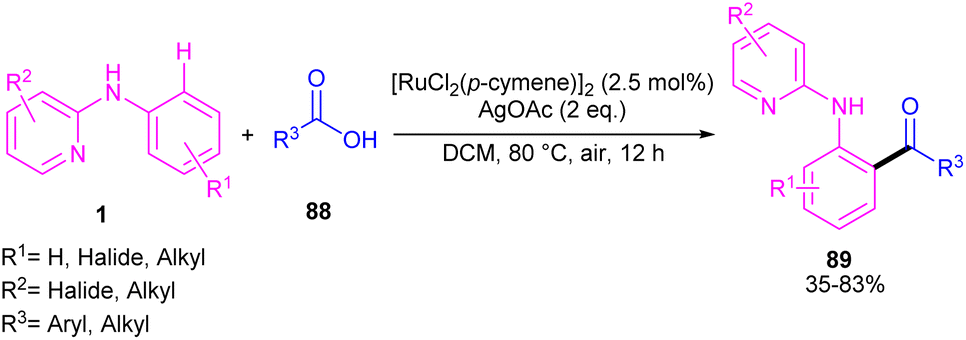
2.4.2.1. Acylation and acetoxylation. Li and co-workers described ortho-acylation of N-(2-pyridyl)-anilines 1 with arylglyoxylic acids 88 under ruthenium catalysis (Scheme 53).61 N-(2-Pyridyl)-anilines bearing electron-donating and aryl and heteroaryl glyoxylic acids electron-withdrawing groups at the both pyridine and benzene ring were evaluated, in which electron-rich substrates showed better reactivity than electron-deficient ones. In the case of arylglyoxylic acids, a certain order was not realized, while heteroarylglyoxylic acids like 2-thiopheneglyoxylic acid and 2-furanglyoxylic acid led to slightly lower products. Overall reaction mechanism involved pyridine-assisted direct C(sp2)–H bond activation via CMD pathway, acid coordination, decarboxylation, and reductive elimination. The ruthenium/silver catalysis was found to be workable for acylating N-aryl-2-aminopyridine or N-aryl-2-aminopyrimidines (Scheme 54).62 Carboxylic acids were selected as acylating reagents, which were coordinated with the metal center of complex A, followed by the reductive elimination towards the acylated product 90, 91.
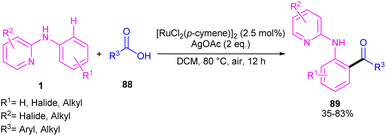 |
| | Scheme 53 Ru(II)-catalyzed C–H acylation of aniline derivatives with α-oxocarboxylic acids. | |
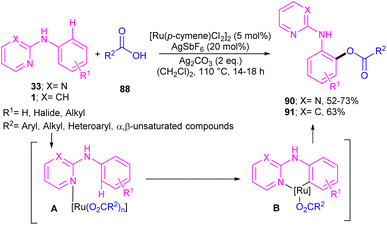 |
| | Scheme 54 Ru(II)-catalyzed C–H acylation of N-aryl-2-aminopyridines or N-aryl-2-aminopyrimidines with carboxylic acids. | |
2.5. Co-catalyzed cross-coupling of N-aryl-2-aminopyridines
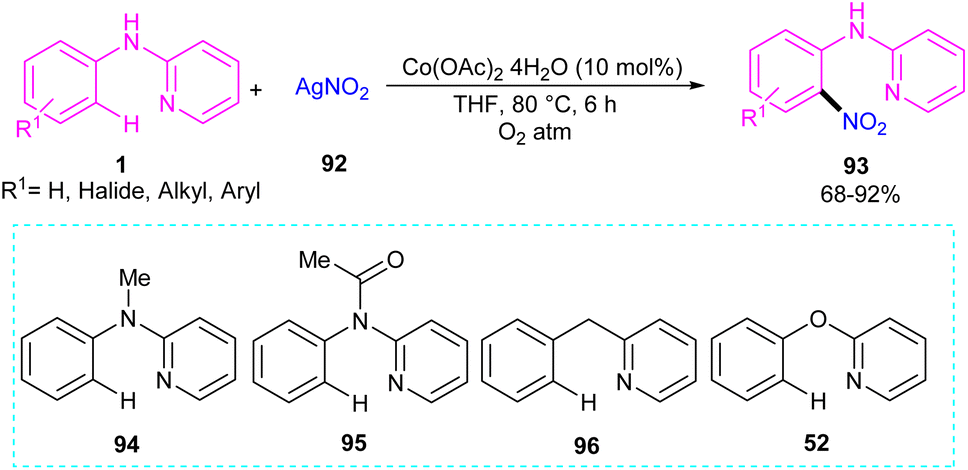
2.5.1. Nitration. An efficient nitration method for N-phenyl-2-aminopyridine 1 was described by Das's group in 2017 (Scheme 55).63 By using an Co(OAc)2 catalyst, nitration of N-phenyl-2-aminopyridine with AgNO3 was achieved in good yields (70–92%). N-Thiophene-2-aminopyridine was also nitrated under cobalt catalysis in 68% of yield. The cobalt acetate can also catalyze the methoxylation of N-phenyl-2-aminopyridine using methanol as both a coupling partner and solvent. Mechanistic experiments were performed, and these allowed the authors to conclude that the reaction proceeded through a radical pathway and the C–H activation was not the rate-determining step. Interestingly, none of the substrates 94, 95, 96, and 52 were not compatible which proved the vital role of the NH in the coordination chemistry with the metal. The mechanism of the reaction started with the generation of reactant complex (RC) from the active catalyst Co(III)(OAc)2(NO2) and N-aryl-2-aminopyridine. Through a PCET reaction, a proton in RC transferred to acetate, leading to the reduction of Co(III) reduced to Co(II). Due to the high energy barrier for the nitro group transfer from Co(II) to phenyl group, acetic acid group exchanged with an acetate ion to form a Co(III) complex 12. The transfer of nitro functional group from I2 gave complex 13. In I3, H-transfer reaction offered intermediate I4. Further proton transfer to the imino-functional group from AcOH provided intermediate I5. Finally, complex I5 converted to the nitrated product after the release of Co(OAc)2 (Scheme 56).
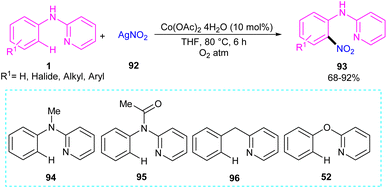 |
| | Scheme 55 Co(II)-catalyzed regioselective C(sp2)–H bond nitration of N-phenyl-2-aminopyridine. | |
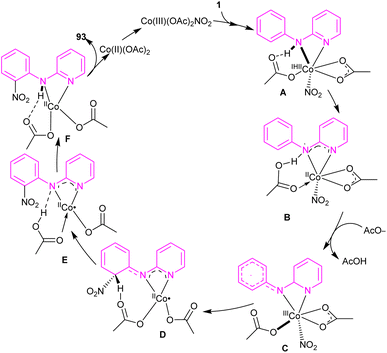 |
| | Scheme 56 Catalytic cycle for Co(II)-catalyzed regioselective C(sp2)–H bond nitration of N-phenyl-2-aminopyridine. | |
2.6. Cu-catalyzed cross-coupling of N-aryl-2-aminopyridines
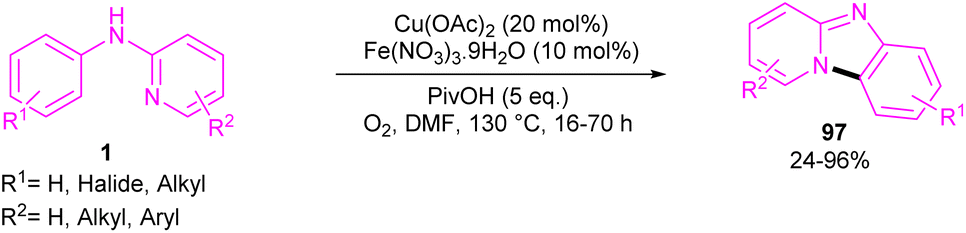
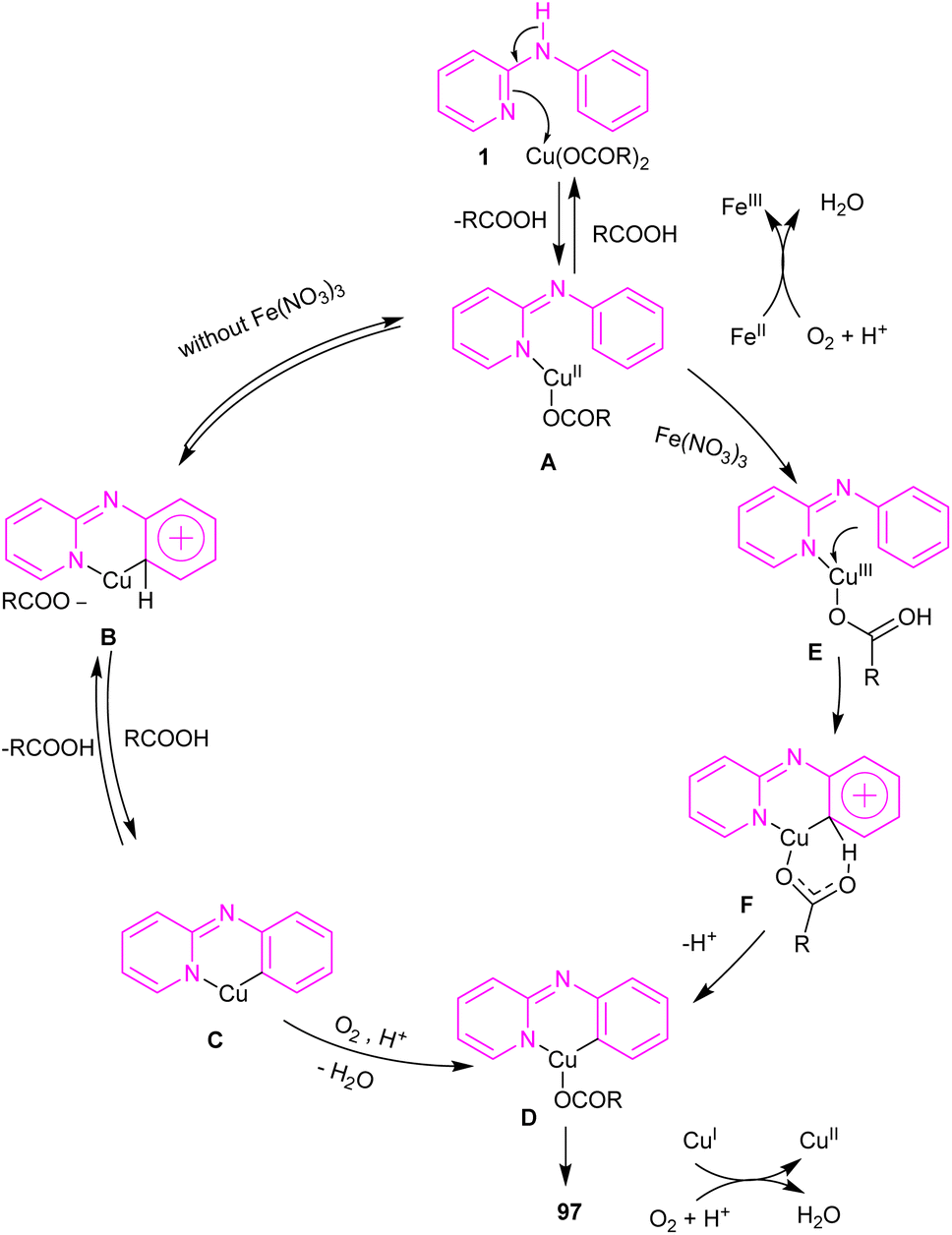
2.6.1. Annulation. An intramolecular C–H amination of N-phenyl-2-aminopyridine derivatives was established using Cu(OAc)2 as a catalyst (Scheme 57).64 The pyridinyl nitrogen was found to be both a directing group and a nucleophile. Screening of this transformation in the presence of Fe(NO3)3·9H2O and without it, showed the significant role of Fe(NO3)3·9H2O in increasing of the reaction efficiency. The iron(III) helped in forming the electrophilic Cu(III) species, leading to the subsequent SEAr reaction. In this context, two possible pathways were proposed in the presence and absence of iron(III). In the first case, the Cu(II) complex A was oxidized to the more electrophilic Cu(III) intermediate E, followed by sequentially electrophilic substitution, and elimination of the aromatic proton to yield intermediate D. Reductive elimination then took place before protonation to furnish pyrido[1,2-a]benzimidazoles 97. However, without Ir(III) salt, the Cu(II) complex A underwent an electrophilic aromatic substitution to render the Cu(II) intermediate C. After that, C was protonated before the oxidation process. The generated active intermediate D was then converted to product 97 via reductive elimination. The oxidation of Cu(I) to Cu(II) was carried out by O2 (Scheme 58).
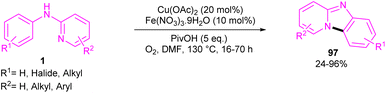 |
| | Scheme 57 Cu(II)-catalyzed intramolecular C–H amination of N-phenyl-2-aminopyridine. | |
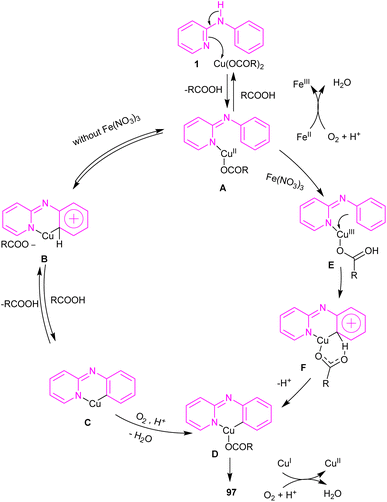 |
| | Scheme 58 Proposed reaction pathways for Cu(II)-catalyzed intramolecular C–H amination of N-phenyl-2-aminopyridine. | |
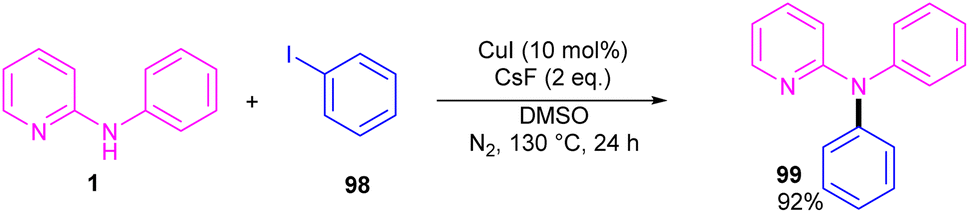
2.6.2. Arylation. Copper-catalyzed N-arylation of N-phenyl-2-aminopyridine 1 was reported by Ribas and Güell in 2014 (Scheme 59).65 The C–N cross-coupling was carried out in the presence of 10 mol% of CuI and 2 equiv. of CsF in DMSO. This method has presented a ligand-free Ullmann-type C–N bond coupling. DMSO served both as a solvent and ligand, overcoming the need for any external auxiliary ligands. A wide range of amines exhibited good compatibility in this methodology.
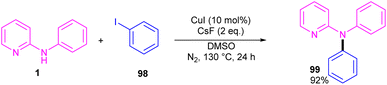 |
| | Scheme 59 Cu(I)-catalyzed N-arylation of N-phenyl-2-aminopyridine. | |
3. Conclusions
In this review, we presented developments on N-aryl-2-aminopyridines reactions under transition metal catalysis systems. As shown in this context, the nitrogen atom in the pyridyl group structure can form a series of more stable complexes with metals, leading to annulation or functionalization of N-aryl-2-aminopyridines. Various transition metal catalysts, such as Pd, Rh, Ir, Ru, Co and Cu salts can efficiently catalyze the transformations involving N-aryl-2-aminopyridines. Despite the great developments, these synthetic methods still face some disadvantages, such as high temperatures, the use of external oxidants and additives for the C–H activation reactions. Hence, designing greener and milder procedures in this field is highly desirable. In particular, the functionalization of N-aryl-2-aminopyridines still requires a lot of work.
Considering the sustainability and effectiveness of photocatalysis and electrocatalysis systems, it is worth mentioning that the combination of these two catalytic systems with metal containing systems can constitute an efficient multiple catalytic system for transforming N-aryl-2-aminopyridines.
We hope that this review will provide new insights into the chemistry and transformations of N-aryl-2-aminopyridines to design more practical and sustainable methods in the field of N-heterocyclic chemistry.
Data availability
The data supporting the findings of this study are available within the article.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef PubMed.
- S. Sunny and R. Karvembu, Adv. Synth. Catal., 2021, 363, 4309–4331 CrossRef.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef PubMed.
- J. Jeon, C. Lee, I. Park and S. Hong, Chem. Rec., 2021, 21, 3613–3627 CrossRef PubMed.
- O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef.
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- H. Song, E. Pietrasiak and E. Lee, Acc. Chem. Res., 2022, 55, 2213–2223 CrossRef PubMed.
- A. Amin, T. Qadir, P. K. Sharma, I. Jeelani and H. Abe, Open Med. Chem. J., 2022, 16, 1–27 Search PubMed.
- A. Omar, Al-Azhar J. Pharm. Sci., 2020, 62, 39–54 CrossRef.
- T. Qadir, A. Amin, P. K. Sharma, I. Jeelani and H. Abe, Open Med. Chem. J., 2022, 16, 1–34 Search PubMed.
- D. Kumar and S. Kumar Jain, Curr. Med. Chem., 2016, 23, 4338–4394 CrossRef PubMed.
- E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390–2402 CrossRef.
- S. M. Umer, M. Solangi, K. M. Khan and R. S. Z. Saleem, Molecules, 2022, 27, 7586 CrossRef PubMed.
- C. Prandi and E. G. Occhiato, Pest Manage. Sci., 2019, 75, 2385–2402 CrossRef PubMed.
- K. W. Anderson, R. E. Tundel, T. Ikawa, R. A. Altman and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 6523–6527 CrossRef.
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef PubMed.
- S. Hostyn, G. Van Baelen, G. L. Lemiere and B. U. Maes, Adv. Synth. Catal., 2008, 350, 2653–2660 CrossRef.
- G.-D. Tang, C.-L. Pan and X. Li, Org. Chem. Front., 2016, 3, 87–90 RSC.
- A. Khadra, S. Mayer and M. G. Organ, Chem.–Eur. J., 2017, 23, 3206–3212 CrossRef PubMed.
- T. T. Deng, J. Huang, G. Lian, W. W. Sun and B. Wu, Asian J. Org. Chem., 2021, 10, 2880–2882 CrossRef.
- F. Tjosaas and A. Fiksdahl, J. Organomet. Chem., 2007, 692, 5429–5439 CrossRef.
- B. Ramesh and M. Jeganmohan, Org. Lett., 2017, 19, 6000–6003 CrossRef PubMed.
- Y. Luo, L. Guo, X. Yu, H. Ding, H. Wang and Y. Wu, Eur. J. Org Chem., 2019, 3203–3207 CrossRef.
- J. Chen, Q. Pang, Y. Sun and X. Li, J. Org. Chem., 2011, 76, 3523–3526 CrossRef CAS.
- J. Chen, K. Natte, A. Spannenberg, H. Neumann, M. Beller and X. F. Wu, Chem.–Eur. J., 2014, 20, 14189–14193 CrossRef CAS.
- Q. Li, M. Zhu, X. Yan, Y. Xia and X. Zhou, J. Organomet. Chem., 2021, 948, 121930 CrossRef CAS.
- W. Guo, W. Hu, X. Zhang, J. Huang, H. Wang, M. Xia and L. Tao, Asian J. Org. Chem., 2024, 13, e202300651 CrossRef CAS.
- P. V. Reddy, M. Annapurna, P. Srinivas, P. R. Likhar and M. L. Kantam, New J. Chem., 2015, 39, 3399–3404 RSC.
- J. H. Chu, P. S. Lin, Y. M. Lee, W. T. Shen and M. J. Wu, Chem.–Eur. J., 2011, 17, 13613–13620 CrossRef CAS.
- D. Liang, Y. He and Q. Zhu, Org. Lett., 2014, 16, 2748–2751 CrossRef CAS.
- J. Chen, K. Natte and X.-F. Wu, J. Organomet. Chem., 2016, 803, 9–12 CrossRef CAS.
- U. Sharma, R. Kancherla, T. Naveen, S. Agasti and D. Maiti, Angew. Chem., Int. Ed., 2014, 53, 11895–11899 CrossRef CAS PubMed.
- L. Jie, L. Wang, D. Xiong, Z. Yang, D. Zhao and X. Cui, J. Org. Chem., 2018, 83, 10974–10984 CrossRef CAS.
- X. Ding, L. Zhang, Y. Mao, B. Rong, N. Zhu, J. Duan and K. Guo, Synlett, 2020, 31, 280–284 CrossRef CAS.
- S. H. Kim, S. H. Park and S. Chang, Tetrahedron, 2012, 68, 5162–5166 CrossRef CAS.
- J. Xu, X. Li, S. Wang, C. Kang, G. Jiang and F. Ji, J. Org. Chem., 2023, 88, 6168–6175 CrossRef CAS PubMed.
- K. Yang, Q. Li, Z. Li and X. Sun, Chem. Commun., 2023, 59, 5343–5364 RSC.
- J. Chen, G. Song, C.-L. Pan and X. Li, Org. Lett., 2010, 12, 5426–5429 CrossRef CAS.
- Z.-J. Wu, K. L. Huang and Z.-Z. Huang, Org. Biomol. Chem., 2017, 15, 4978–4983 RSC.
- X. Hu, X. Chen, Y. Zhu, Y. Deng, H. Zeng, H. Jiang and W. Zeng, Org. Lett., 2017, 19, 3474–3477 CrossRef PubMed.
- X. Yan, R. Ye, H. Sun, J. Zhong, H. Xiang and X. Zhou, Org. Lett., 2019, 21, 7455–7459 CrossRef PubMed.
- Z. Shen, C. Pi, X. Cui and Y. Wu, Chin. Chem. Lett., 2019, 30, 1374–1378 CrossRef.
- S. He, X. Yan, Y. Lei, H. Xiang and X. Zhou, Chem. Commun., 2020, 56, 2284–2287 RSC.
- Y. Wang, D. Jia, J. Zeng, Y. Liu, X. Bu and X. Yang, Org. Lett., 2021, 23, 7740–7745 CrossRef.
- S. K. Keshri, S. Madhavan and M. Kapur, Org. Lett., 2022, 24, 9043–9048 CrossRef PubMed.
- P. Biswal, T. Nanda, N. Prusty, S. R. Mohanty and P. C. Ravikumar, J. Org. Chem., 2023, 88, 7988–7997 CrossRef PubMed.
- N. K. Mishra, M. Choi, H. Jo, Y. Oh, S. Sharma, S. H. Han, T. Jeong, S. Han, S.-Y. Lee and I. S. Kim, Chem. Commun., 2015, 51, 17229–17232 RSC.
- S. Yang, B. Feng and Y. Yang, J. Org. Chem., 2017, 82, 12430–12438 CrossRef PubMed.
- Q. Liu, Z. Ma, J. Zhang and X.-Q. Li, Org. Biomol. Chem., 2023, 21, 8320–8328 RSC.
- J. Yang, W. Shi, W. Chen, H. Gao, Z. Zhou and W. Yi, J. Org. Chem., 2021, 86, 9711–9722 CrossRef.
- Z. J. Wu, F. Su, W. Lin, J. Song, T. B. Wen, H. J. Zhang and H. C. Xu, Angew. Chem., 2019, 131, 16926–16930 CrossRef.
- S. Bai, X. Chen, X. Hu, Y. Deng, H. Jiang and W. Zeng, Org. Biomol. Chem., 2017, 15, 3638–3647 RSC.
- U. Karmakar, D. Das and R. Samanta, Eur. J. Org Chem., 2017, 2780–2788 CrossRef.
- J. Xia, X. Yang, Y. Li and X. Li, Org. Lett., 2017, 19, 3243–3246 CrossRef PubMed.
- X. Zhou, J. Xia, G. Zheng, L. Kong and X. Li, Angew. Chem., Int. Ed., 2018, 57, 6681–6685 CrossRef PubMed.
- X.-F. Cui, X. Qiao, H.-S. Wang and G.-S. Huang, J. Org. Chem., 2020, 85, 13517–13528 CrossRef PubMed.
- G.-D. Tang, C.-L. Pan and F. Xie, Org. Biomol. Chem., 2016, 14, 2898–2904 RSC.
- M. M. Mahamudul Hassan, B. Mondal, S. Singh, C. Haldar, J. Chaturvedi, R. Bisht, R. B. Sunoj and B. Chattopadhyay, J. Org. Chem., 2022, 87, 4360–4375 CrossRef.
- M. K. Manna, G. Bairy and R. Jana, J. Org. Chem., 2018, 83, 8390–8400 CrossRef PubMed.
- X.-F. Cui, Z.-H. Ban, W.-F. Tian, F.-P. Hu, X.-Q. Zhou, H.-J. Ma, Z.-Z. Zhan and G.-S. Huang, Org. Biomol. Chem., 2019, 17, 240–243 RSC.
- Q. Liu, J.-Y. Yong, J. Zhang, T. Ban and X.-Q. Li, Org. Biomol. Chem., 2022, 20, 6890–6896 RSC.
- T. Sarkar, S. Pradhan and T. Punniyamurthy, J. Org. Chem., 2018, 83, 6444–6453 CrossRef.
- D. Nageswar Rao, S. Rasheed, G. Raina, Q. N. Ahmed, C. K. Jaladanki, P. V. Bharatam and P. Das, J. Org. Chem., 2017, 82, 7234–7244 CrossRef.
- H. Wang, Y. Wang, C. Peng, J. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217–13219 CrossRef.
- I. Güell and X. Ribas, Eur. J. Org Chem., 2014, 3188–3195 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Lina Rezainia
b,
Mohammad Hossein Morshedsolouk
a,
Lina Rezainia
b,
Mohammad Hossein Morshedsolouk