
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel LC-TQ-MS/MS method for quantifying mefenamic acid-NDSRI (N-nitroso drug substance-related impurity) in mefenamic acid tablet and pediatric suspension dosage forms: a comparative study with a cost-effective white, green, and blue UPLC method†
Srinivas Nakkaa,
Naresh Kumar Katarib,
Siva Krishna Muchakayala ac,
Sreekantha Babu Jonnalagadda*b and
Surendra Babu Manabolu Surya*a
ac,
Sreekantha Babu Jonnalagadda*b and
Surendra Babu Manabolu Surya*a
aDepartment of Chemistry, School of Science, GITAM Deemed to be University, Hyderabad-502329, India. E-mail: smanabol@gitam.edu
bSchool of Chemistry & Physics, College of Agriculture, Engineering & Science, University of KwaZulu-Natal, Westville Campus, P Bag X 54001, Durban 4000, South Africa. E-mail: Jonnalagaddas@ukzn.ac.za
cAnalytical Research and Development, Catalent Pharma Solutions, 1100 Enterprise Drive, Winchester, Kentucky-40391, USA
First published on 21st January 2025
Abstract
The current research presents novel LC-TQ-MS/MS and cost-effective UPLC methods intended for the accurate quantification of mefenamic acid-N-nitroso drug substance-related impurity-NDSRI (N-MFA) in mefenamic acid (MFA) tablet and pediatric suspension dosage forms. The acceptable intake of N-MFA is derived from the TD50 (Median Toxic Dose-50%) value of N-nitroso diphenylamine. The analytical separation was achieved for the UPLC method using an XBridge BEH Shield RP18 Column (150 × 3.0 mm, 3.5 μm), employing 0.1% formic acid and acetonitrile as mobile phases in a gradient mode. A flow rate of 0.7 mL min−1 was set for the pump, and the detection wavelength was 230 nm. LC-TQ-MS-MS analysis was accomplished utilizing a Xevo TQ-XS mass spectrometer with electrospray ionization in the positive mode. N-MFA quantification was carried out using multiple reaction monitoring (MRM) as the mass transition: m/z 271.13 → 241.14. The proposed methods were successfully validated following ICH guidelines and are capable of quantifying at 0.01 ng mL−1 at an MFA concentration of 1 mg mL−1 for the LC-MS/MS method and at 0.036 μg mL−1 at an MFA concentration of 10 mg mL−1 for UPLC. The validated methods exhibited excellent linearity over the concentration ranges of 0.01 to 125 ppm for LC-MS/MS and 0.036 μg mL−1 to 1.250 μg mL−1 (3.6 to 125 ppm) for UPLC, with a Pearson correlation coefficient of 0.999. The method's trueness, in terms of accuracy recoveries, ranged from 90.43% to 101.34% for LC-MS/MS and 92.12% to 101.21% for UPLC. The applicability of these methods was successfully verified through the analysis of commercial samples of MFA formulations. Additionally, the sustainability and eco-friendliness of the method were assessed using whiteness and greenness metrics, including RGB12 (Red-Gree-Blue) tools, GAPI (Green Analytical Procedure Index), analytical eco-scale, AGREE (Analytical GREEnness), AGREEprep (Analytical GREEnnesspreparation), and BAGI (Blue Applicability Grade Index). Both methods can be used to determine N-MFA content in mefenamic acid formulation dosage based on the availability of instruments LC-TQ-MS/MS and UPLC in R&D (Research and Development) and quality control laboratories.
Introduction
Regulatory agencies have underscored the significance of predicting toxicity and precisely measuring the levels of nitrosamine contaminants in certain pharmaceuticals. These impurities, which include nitrosamines derived from active pharmaceutical ingredients, are referred to as Nitrosamine Drug Substance Related Impurities (NDSRIs). Nitrosamines, or N-nitrosamines, are molecules characterized by their N-nitroso functional group. They are classified as a group of concern due to their status as human carcinogens, with evidence showing that long-term intake above acceptable levels can increase cancer risks.These NDSRI substances are categorized into one of three “Cohort-of-Concern” (CoC) groups: aflatoxin-like, alkyl-azoxy, and N-nitroso. “CoC” denotes a collection of highly potent mutagenic compounds requiring special attention due to potential health risks. The focus of pharmaceutical regulatory authorities has shifted from common nitrosamines to NDSRIs because of their potential carcinogenicity and their presence in drug products at unacceptable levels.1–6 As a result, regulatory authorities recalled various drug products worldwide. Furthermore, the detection and quantification of NDSRIs are of significant concern to analytical researchers.7–11
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a class of medications that relieve pain and inflammation, including in renal transplants and chronic non-cancer pain in children.12,13 About 30 million people worldwide use NSAIDs every day, according to a report by Medical News Today. The worldwide market for NSAIDs was valued at USD 15.58 billion in 2019 and is envisioned to reach USD 24.35 billion by 2027.
Mefenamic acid (MFA) is chemically known as 2-[(2,3-dimethylphenyl)amino]benzoic acid, with the chemical formula C15H15NO2 and a molecular weight of 241.28 g mol−1. MFA is classified as an anthranilic acid (phenolate) NSAID that exhibits both antipyretic and analgesic properties. This medication treats moderate pain and inflammation associated with osteoarthritis, post-operative pain, menstrual pain, rheumatoid arthritis, toothaches, and severe back and muscle pain. Additionally, it is also used to manage menorrhagia.14,15
To our knowledge, this is the first UPLC-MS/MS and environmentally friendly UPLC method for quantifying the target compound N-MFA. As a result, there are no existing methods available to compare with the proposed UPLC-TQ-MS/MS and the cost-effective green UPLC method. Various regulatory agencies, including the US FDA and Health Canada, have proposed that the acceptable intake (AI) value for N-MFA is 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng per day.16,17 The interim control limit for N-MFA is 62.4 ppm, which can be derived from the AI value of the impurity and the maximum daily dose of the MFA. Hence, simple and highly sensitive UPLC-MS/MS and UPLC methods have been developed for the accurate quantification of N-MFA in MFA drug formulations, including tablets and pediatric suspensions.
000 ng per day.16,17 The interim control limit for N-MFA is 62.4 ppm, which can be derived from the AI value of the impurity and the maximum daily dose of the MFA. Hence, simple and highly sensitive UPLC-MS/MS and UPLC methods have been developed for the accurate quantification of N-MFA in MFA drug formulations, including tablets and pediatric suspensions.
The current study aimed to assess genotoxicity using (Q)-SAR (Quantitative Structure–Activity Relationship) models and to develop new LC-TQ-MS/MS and UPLC methods for quantifying N-MFA in both the MFA drug substance and its formulation dosage forms. Recent regulatory guidelines demonstrate that setting the quantitation limit (QL) at 30% is considered reasonably good and acceptable. If the values consistently fall below the QL across all process validation batches and corresponding stability samples (both accelerated and long-term), it is acceptable to proceed with skip lot testing. UPLC can detect the N-MFA impurity below the 30% control limit (∼19 ppm). Therefore, this cost-effective UPLC method is proposed alongside the LC-TQD-MS/MS for use in quality control laboratories. Both methods have been successfully validated regarding the QL, linearity, precision, accuracy, and robustness. The sustainability and eco-friendliness of the UPLC method were evaluated using greenness and whiteness assessment metrics, including GAPI, the analytical eco-scale, AGREE, AGREEprep, BAGI, and RGB12 tools.18–20 The results indicated that the proposed method exhibits a high level of greenness and whiteness. Fig. 1 illustrates the chemical structures of MFA, N-DPA (N-nitroso diphenylamine), and N-MFA. The applicability of these methods was successfully verified through the analysis of commercial samples of APIs and dosage forms of MFA.
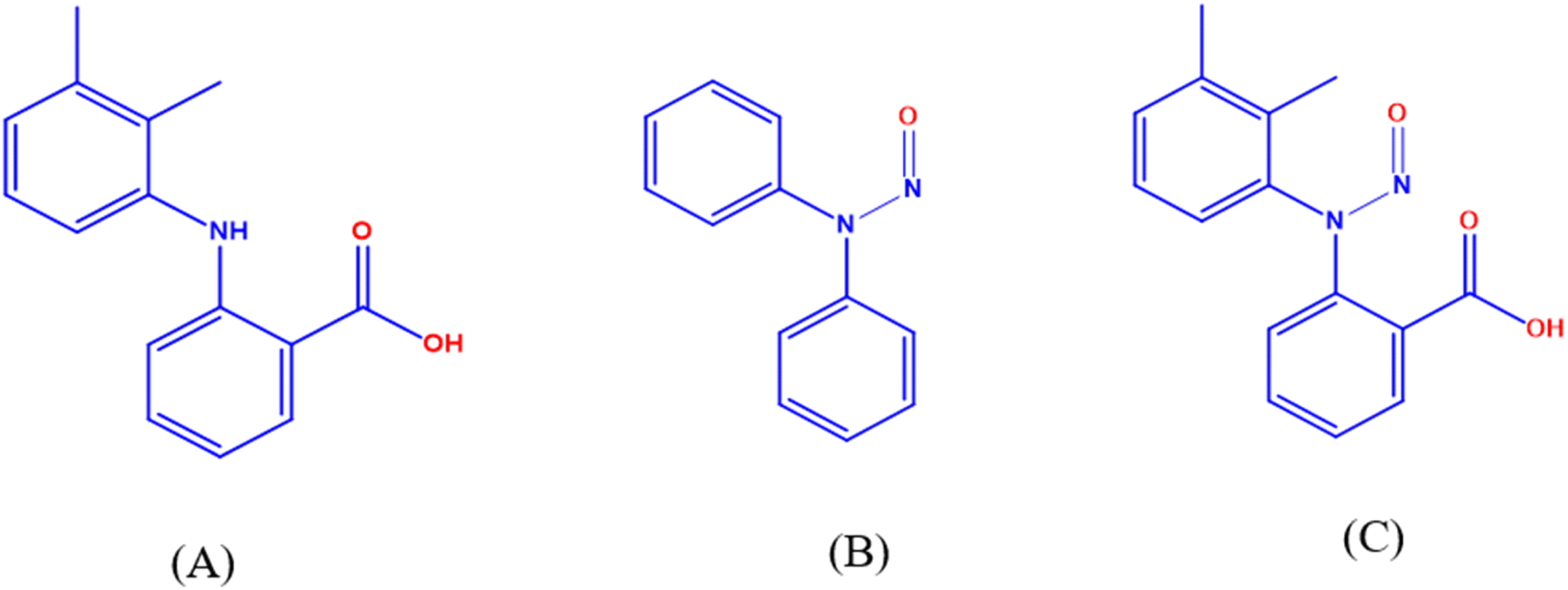 | ||
| Fig. 1 Structures of (A) MFA, (B) N-DPA, and (C) N-MFA. | ||
Materials and method
Instrument and software
The chromatographic investigation was accomplished on the Waters H class UPLC system, which includes a PDA-photo diode array detector model (L20UPD127A) modules, an Auto Sampler Manager with a Flow-Through Needle (FTN-L20FTP352G), Quaternary Solvent Manager (QSM-L20QSP358A) (Waters Corporation, USA). For the LC-TQ-MS/MS method (MRM analysis) a Waters Xevo TQ-XS MS system equipped with an ESI ion source was utilized. A Mettler-Toledo (Model: XPE205, Columbus, Ohio, USA) analytical balance was employed to weigh the samples and the impurity standard. The analysis utilized high-purity water obtained from the Millipore Milli-Q purification system (Bedford, MA, USA). Centrifuge (Model: 5810R, Hemburg, Germany) from Eppendorf. ChemDraw Professional 15.0 sketched MFA, N-DPA, and N-MFA molecular structures. The two primary (Q)-SAR methodologies employed were the Derek Nexus (version 6.2.0, featuring the Derek KB 2022 1.0 knowledge base) and the Sarah Nexus (version 3.2.0, which includes the Nexus version 1.9 and the Sarah Model 2022.1). Target Lynx software v 4.2 was used to compute all LC-TQ-MS/MS acquisition and process parameters.Reagents
The reference standard of MFA (purity = 99.63%) and MFA-NDSRI (purity = 99.23%) were gift samples obtained from M/S Integrum Lifesciences (Hyderabad, India). NMR and MS spectra have been used to characterize the analytical reference standards (Fig. S1 and S2†). Three different brands of commercially existing MFA tablets with a label claim of 500 mg and MFA pediatric suspension (100 mg/5 mL) were obtained from a local pharmacy. LC-MS grade acetonitrile and formic acid were acquired from Honeywell (Honeywell International (India) Private Limited, Hyderabad, India). Millex-GV hydrophilic PVDF (polyvinylidene fluoride) 0.22 μm syringe filters (Burlington, Massachusetts, USA) have been obtained from Millipore.Mobile phase preparation
The mobile phase was made by adding 1.0 mL of formic acid to 1000 mL of water, followed by sonication for degassing.Diluent preparation
A mixture consisting of 0.1% formic acid in water and acetonitrile in the ratio of 20:80 v/v.
MFA-NDSRI impurity standard diluted stock solution preparation
A stock solution of MFA-NDSRI, with a concentration of 0.5 mg mL−1, was prepared by dispersing the required quantity of impurity standard with diluent. A diluted stock solution with a concentration of 2000 ng mL−1 was then obtained by further diluting this solution in the diluent, which was subsequently labeled as a diluted stock solution.Sample preparation
For the LC-TQ-MS-MS analysis, a 1 mg mL−1 solution was prepared by accurately weighing about 10 mg of MFA API (Active Pharmaceutical Ingredient) and for UPLC 100 mg of MFA API into a 10 mL volumetric flask, then dissolving in 5 mL of diluent and sonicating until completely dissolved. Following the equilibration of the flask to room temperature, the volume was adjusted to the calibration mark with the addition of a diluent. Subsequently, the prepared solution was filtrated through a Millex-GV hydrophilic PVDF syringe filter, having a pore size of 0.22 μm.Formulation tablet sample preparation
Grind ten tablets of three different commercially available MFA brands of dosage formulations separately into a fine powder employing a mortar and pestle. Transfer an amount equivalent to 10 mg of MFA API for LC-TQ-MS-MS and 100 mg of MFA API for UPLC in a 10 mL volumetric flask. Add 5 mL of diluent and sonicate for 10 minutes to dissolve. Allow the flask to equilibrate to room temperature before making up to the mark with diluent. Place the sample solution in the centrifuge and spin it at 4800 rpm for 15 minutes. Filter the resulting supernatant through a Millex-GV hydrophilic PVDF syringe filter having a pore size of 0.22 μm.Pediatric suspension sample preparation
Agitate the MFA pediatric suspension, with the label claim 100 mg/5 mL. Transfer an amount equivalent to 10 mg (0.5 mL) of the MFA suspension for LC-TQ-MS-MS and 100 mg (5 mL) of the MFA suspension for UPLC in a 10 mL volumetric flask. Add 3 mL of diluent and sonicate for 10 minutes to ensure dissolution. Following the equilibration of the flask to room temperature, the volume was adjusted to the calibration mark with the addition of a diluent. Place the sample solution in the centrifuge and spin it at 4800 rpm for 15 minutes. Filter the resulting supernatant through a Millex-GV, hydrophilic PVDF syringe filter with a pore size of 0.22 μm.Results and discussion
(Q)-SAR prediction data
The ICH M7(R1)21 guideline recognizes the predictions of (Q)-SAR as a vital tool in the characterization of hazards. In this instance, genotoxicity could be assessed based on the warning structures of substances utilized in toxicological studies. Two harmonizing (Q)-SAR methodologies are commonly employed to ascertain the results of the bacterial reverse mutation test: one is based on expert rules, and the other on statistical analysis. The molecular structures of N-MFA and MFA were put into Nexus 2.5.0, utilizing the ICH categorization model for predictive analysis. N-MFA falls under class 3 with a CoC; Table 1 illustrates the predicted results.| Structure | ICH M7 class | Coherent of concern | Derek prediction | Sarah prediction | Experimental data | Similar to API | Overall in silico |
|---|---|---|---|---|---|---|---|
 |
Class-3 | Yes (N-nitroso compound) |  (plausible) (plausible) |
 |
Carc: unspecified | Alert(s) not found in API | Positive |
| Ames: unspecified |
Diphenylamine is a precursor compound that can produce various nitrosamines, including N-MFA. The TD50 is a crucial parameter for evaluating the toxicity of substances and guiding the safe usage of drugs. The TD50 values are instrumental in establishing metrics, including the AI, Threshold of Toxicological Concern (TTC), and Permitted Daily Exposure (PDE), which are critical for assessing human exposure. For N-MFA, structure similarity was observed with N-DPA. Thus, the AI value for N-MFA was derived from the TD50 value of NDPA, with an AI value of 78000 ng per day. The specification limit for N-nitroso impurity is established based on the specific drug substance's maximum daily dosage (MDD). MDD value of MFA was established as 1250 mg, starting with an initial dose of 500 mg, followed by subsequent doses of 250 mg every six hours. Therefore, the N-MFA should be controlled below 62.4 ppm ((78000 ng per day)/(1250 mg per day)) as per regulatory guidelines. For ease of calculation, 62.5 ppm is set as the specification limit.
Optimization of the chromatographic UPLC method
The main objective of the research was to develop a clear, specific, reliable, and accurate RP-UPLC method for measuring the N-MFA content in MFA drug substances, tablets, and pediatric suspension formulations. The chromatographic conditions are primarily determined by the physicochemical characteristics of the analyte, which play a crucial role in the method's development. This includes the selection of the mobile phase, stationary phase, flow rate, gradient program, detection wavelength, injection volume, column oven temperature, and the concentrations of standards and samples. Generally, the separation of compounds with similar structural characteristics poses significant challenges.The initial development method involved preparing a mixed solution with MFA and N-nitroso MFA concentrations equivalent to 1 mg mL−1, we selected 230 nm (Fig. S3†) as the detection wavelength for our study to quantify N-MFA content in MFA formulation dosage. The analyte's retention time and peak shape depend on the column length, particle size, and internal diameter. In the column scouting studies, various C18 columns with different column lengths, including Hypersil BDS C18 (150 × 4.6 mm, 3 μm), Symmetry C18 (100 × 4.6 mm, 3.5 μm), and Sunfire C18 column (50 × 4.6 mm, 3.5 μm) were utilized to evaluate the separation. Concurrent with the mobile phase selection studies, various buffers were employed. These included 10 mM ammonium acetate, 20 mM potassium phosphate monobasic at different pH levels, 4.5 and 3.0, and 0.1% orthophosphoric acid, combined with acetonitrile in differing ratios to form mobile phases A and B. While using Hypersil BDS C18 (150 × 4.6 mm, 3 μm) column in 10 mM ammonium acetate and 0.1% formic acid buffers with acetonitrile showed poor peak shapes of MFA and N-MFA. The poor resolution between MFA and N-MFA peaks on a Symmetry C18 (100 mm × 4.6 mm, 3.5 m) column was observed in 20 mM potassium phosphate monobasic buffers at pH (4.5 and 3.0) and 0.1% formic acid buffer. To improve separation, a range of column oven temperatures from 30 °C to 45 °C was evaluated with an incremental increment of 5 °C and finalized the column oven temperature as 40 °C hence no effect on the separation above 40 °C. Using the Waters XBridge BEH Shield RP18 Column (150 × 3.0 mm, 3.5 μm), both MFA and N-MFA peaks were distinctly separated with excellent resolution when judged to other conditions, utilizing 0.1% formic acid and acetonitrile as aqueous and organic mobile phases respectively. Furthermore, to attain a lower Limit of Quantitation (LOQ), the injection volume was set at 50 μL. The results from the column and the mobile phase selection investigations are summarized in Table S1.†
The finalized chromatographic method parameters are summarized as follows: Waters XBridge BEH Shield RP18 Column (150 × 3.0 mm, 3.5 μm), 0.1% formic acid and acetonitrile as mobile phase-A and B with flow rate 0.7 mL min−1, column oven temperature set at 40 °C, the chromatographic peaks detection wavelength at 230 nm, with injection volume 50 μL and gradient elution as follows time (min)/% mobile phase B: 0.0/10, 2.0/10, 4.0/80, 8.0/80, 8.1/10, and 10.0/10.
Development and optimization of mass spectrometry conditions
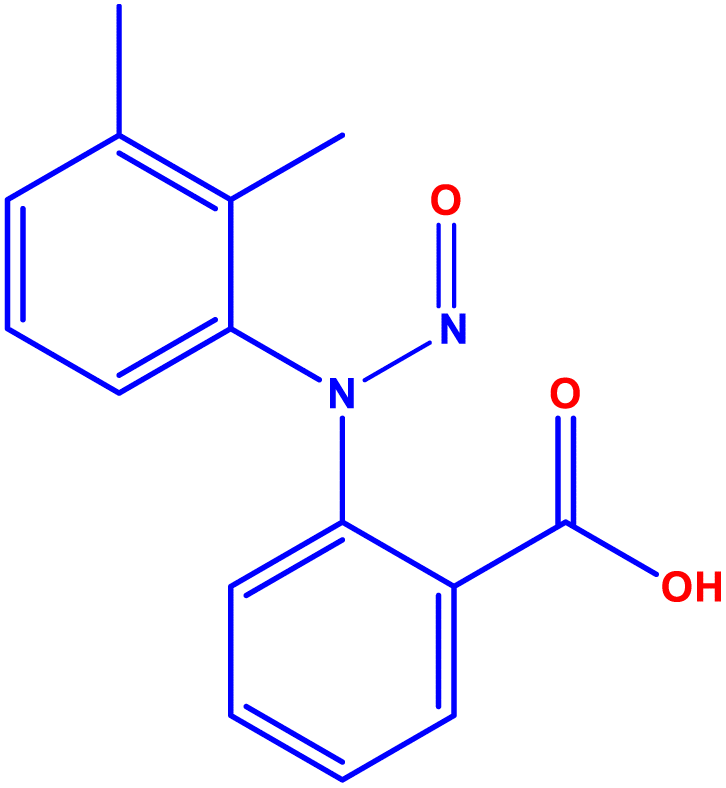
To develop an accurate quantification of N-MFA in MFA tablets and pediatric suspensions, optimizing mass parameters is a decisive step. We intended to attain a sensitive, precise, and accurate method. For N-MFA quantification, the MRM technique was utilized. For initial mass conditions optimization by injecting the 1000 ng mL−1 concentration of N-MFA into mass spectrometry using infusion mode to identify the prominent product ion in electron spray ionization in positive ion mode (m/z 271.13 → 241.14). Due to the pump flow rate being set at 0.7 mL min−1 and the recommended desolvation gas temperature and gas flow being 450 °C and 900 L per Hr, hence performed the analysis with the values mentioned earlier. Employing the Intellistart software, which is built into Waters mass lynx software, to fine-tune MS/MS parameters, including cone voltage, capillary voltage, and collision energy values with different mass transitions. Intellistart identified three major MRM transitions, including m/z 271.13 → 241.14, m/z 271.13 → 223.12, and m/z 271.13 → 208.10 (Fig. 2). After careful optimization of cone voltage and collision energies finalized, the quantifier ion transition as m/z 271.13 → 241.14 and the qualifier ion transition as m/z 271.13 → 223.12 (Fig. S4†). A mass spectrometer was installed, a 50 μL loop, which improves the peak shape when injecting the samples containing a high percentage of organic solvents. The highest intensity of quantifier and qualifier ions are detected by using the below MS/MS optimized conditions: cone voltage is 18 V, capillary voltage is 2.48 kV, collision energy is 8 V and 18 V for qualifier and quantifier respectively, desolvation gas temperature is 450 °C, and gas flow 900 L h−1. In the MS/MS method, the events program flow state was directed from 0 to 5.4 min set to mass to detect the N-MFA and the flow state from 5.5 to 10 minutes to avoid the high concentration of MFA contamination triggered by formulation tablet and pediatric suspension samples 1 mg mL−1 concentration. The optimized MS/MS parameters are shown in Table 2.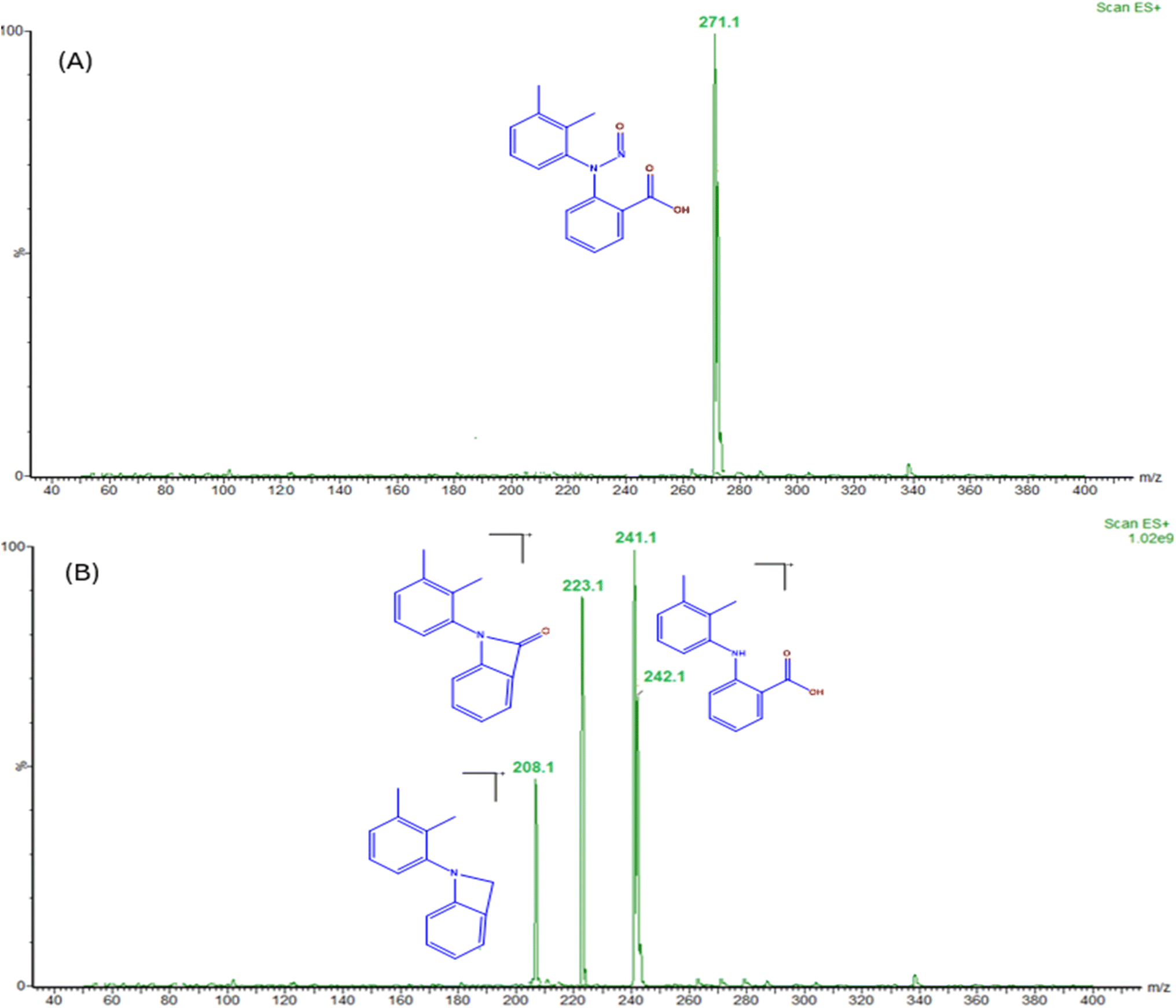 | ||
| Fig. 2 (A) Mass spectrum of NMFA, and (B) MS/MS fragmentation pattern of N-MFA. | ||
| Compound | Precursor ion (m/z) | Product ion (m/z) | Retention window (min) | Collision energy (V) | Cone voltage (V) | Capillary voltage (kV) | |
|---|---|---|---|---|---|---|---|
| Qualifier | Quantifier | ||||||
| NMFA | 271.132 | — | 241.144 | 4.51 ± 0.25 | 8 | 18 | 2.48 |
| 223.124 | — | 4.51 ± 0.25 | 18 | 18 | 2.48 | ||
Optimization of diluent for the extraction of N-MFA
Due to the limited solubility of N-MFA in water, a larger volume of organic solvent combined with formic acid was required to achieve complete dissolution. Simultaneously, various compositions of 0.1% formic acid in water and acetonitrile were evaluated. The results indicated that a 20:80 v/v mixture of 0.1% formic acid in water and acetonitrile yielded the most favorable conditions for dissolving all components and enhancing the quality of chromatographic peak shapes. The retention times for N-MFA and MFA eluted at 4.49 minutes and 6.99 minutes, respectively.
Filter evaluation study
For the accurate analysis of any drug product testing, a suitable and compatible syringe filter must be selected to ensure the accuracy of the analytical outcome. Furthermore, it shall be assured that any of the filters chosen do not absorb any peaks of interest and that any extraneous peaks are absent. The filtration study was compared to the unfiltered centrifuged solution of MFA, with the peak areas of N-MFA comparable. The Millex GV PVDF filters with a pore size of 0.22 μm did not retain N-MFA, indicating that the filter is pertinent to the present study.Robustness study
As per the revised ICH Q2(R2) (Validation of analytical procedures)22 method validation guidelines, the robustness evaluation can be included as a key component of the development data for an analytical procedure. This evaluation provides critical insights into the method's reliability and performance under varying conditions, ensuring that the procedure can consistently deliver accurate and reproducible results. The robustness of the proposed LC-MS/MS method was assessed by varying the concentration of formic acid (%), flow rate (mL min−1), column temperature (°C), desolvation gas flow (L h−1), and desolvation temp (°C). The sample solution spiked with N-MFA at the specification level of 62.5 ppm was injected into the UPLC at a concentration of 10 mg mL−1 to evaluate the method's robustness for tablet dosage forms. The optimized concentration of formic acid was adjusted by ±0.01% from the concentration of 0.1%, the flow rate was changed by ±0.2 mL min−1 from the nominal pump flow rate of 0.7 mL min−1, ±2 °C modified the column oven temperature (°C) from the set 40 °C, desolvation gas flow (L h−1) was altered by ±50 L per Hr from the gas flow of 450 L h−1 and desolvation temp (°C) was changed by ±50 °C from the 450 °C.The robustness of the proposed UPLC method was assessed by varying the concentration of formic acid (%), flow rate (mL min−1), and column temperature (°C), and the detection wavelength was shifted by ±2 nm from the established 230 nm. The LC-MS/MS and UPLC methods were developed to ensure consistency, with the absolute percentage difference in N-MFA impurity content remaining below 0.16% and 0.08% under all nominal conditions for the LC-TQ-MS/MS and UPLC methods. The method robustness data is provided in Table S2.†
Method validation
The present proposed LC-TQ-MS/MS and UPLC methods have been fully validated by ICH Q2R2, USP 〈1225〉 guidelines,23 and published journals,24–26 following the method validation parameters, such as specificity, limit of detection, limit of quantitation, linearity, precision, accuracy, robustness, and solution stability. Table 3 presents the results of the validation study, demonstrating that the method is specific, precise, linear, and accurate.| Validation parameter | Typical acceptable criteria | Results | |
|---|---|---|---|
| LC-TQ-MS/MS | UPLC | ||
| Specificity | There should be no interference from the diluent and placebo | No interference | No interference |
| Limit of detection (LOD) | Concentration at LOD | 0.005 ng mL−1 | 0.013 μg mL−1 |
| S/N value (≥3) | 5.9 | 4.5 | |
| Limit of quantitation (LOQ) | Concentration at LOQ | 0.01 ng mL−1 | 0.035 μg mL−1 |
| S/N value (≥10) | 16.9 | 11.3 | |
| LOQ precision | Content (ppm) (n = 6, %RSD < 10) | 2.35 | 3.05 |
| Method precision | Content (ppm) (n = 6, %RSD < 5, mean ± SD) | 0.85, 62.57 ± 0.03 | 1.02, 62.59 ± 0.09 |
| Horwitz ratio (Hr < 1) | 0.48 | 0.68 | |
| Intermediate precision | Content (ppm) (n = 12 of MP& IP %RSD <5, mean ± SD) | 0.98, 62.58 ± 0.04 | 1.04, 62.55 ± 0.07 |
| Horwitz ratio (Hr < 1) | 0.59 | 0.79 | |
| Linearity | Range | 0.01–125 ng mL−1 | 0.035–1.25 μg mL−1 |
| Slope | 39.1917 | 3.9789 | |
| Intercept | 15.6498 | −9.9192 | |
| Pearson coefficient (>0.99) | 0.9989 | 0.9990 | |
| Accuracy in pure MFA drug substance (%) (n = 3, average percentage) | The level at LOQ mean ± SD | 91.98 ± 0.94 | 94.08 ± 1.82 |
| The level at 62.50 ppm mean ± SD | 100.15 ± 1.52 | 99.84 ± 1.59 | |
| The level at 125.0 ppm mean ± SD | 99.86 ± 1.47 | 101.21 ± 1.21 | |
| Accuracy in formulated MFA (%) (n = 3, average percentage) | The level at LOQ mean ± SD | 90.43 ± 0.87 | 92.12 ± 1.13 |
| The level at 62.50 ppm mean ± SD | 99.17 ± 1.32 | 99.87 ± 0.58 | |
| The level at 125.0 ppm mean ± SD | 101.34 ± 0.49 | 100.25 ± 0.90 | |
| Solution stability | Peak area (0 Hr to 24 Hrs, % difference with initial <10.0) | 2.14 | 1.35 |
System suitability
The system suitability test (SST) is crucial in chromatographic methods. It serves to minimize the risk of critical factors affecting analytical test results. The acceptance criteria for the SST solution are the percentage Relative Standard Deviation (%RSD) of N-MFA peak area (≤5.0), Tailing factor (0.8 to 2.0), resolution between MFA and N-MFA peaks (≥2.0), Theoretical plates (>5000), and signal-to-noise ratio (S/N) ≥10. The obtained results are 1.29 and 1.48 (%RSD of N-MFA peak area) for LC-MS/MS and UPLC methods, respectively, 1.12 (Tailing factor), 10.35 (Resolution between MFA and N-MFA), 53665 (Theoretical plates of N-MFA peak), and signal to noise ratio (S/N) 256:1 for LC-TQ-MS/MS and 108:1 for UPLC. The typical UPLC chromatogram for the SST solution is depicted in Fig. S5.†
Specificity
The current analytical method's specificity is demonstrated by the liquid chromatography system's capability to differentiate between the diluent, placebo, MFA, N-MFA, and N-MFA spiked at the specification level. Injected each 10 μL for LC-MS/MS and 50 μL for UPLC of diluent, placebo, N-MFA standard solution, and sample solution of MFA. The analysis showed that no co-eluting peaks were apparent at the retention time (tR) of N-MFA, allowing for the specific and accurate estimation of N-MFA in MFA tablet and pediatric suspension formulations. Overlay UV spectra of N-MFA and MFA are shown in Fig. S4,† and the typical LC-MS/MS and UPLC chromatograms indicating the specificity of the method can be found in Fig. 3 and 4, respectively.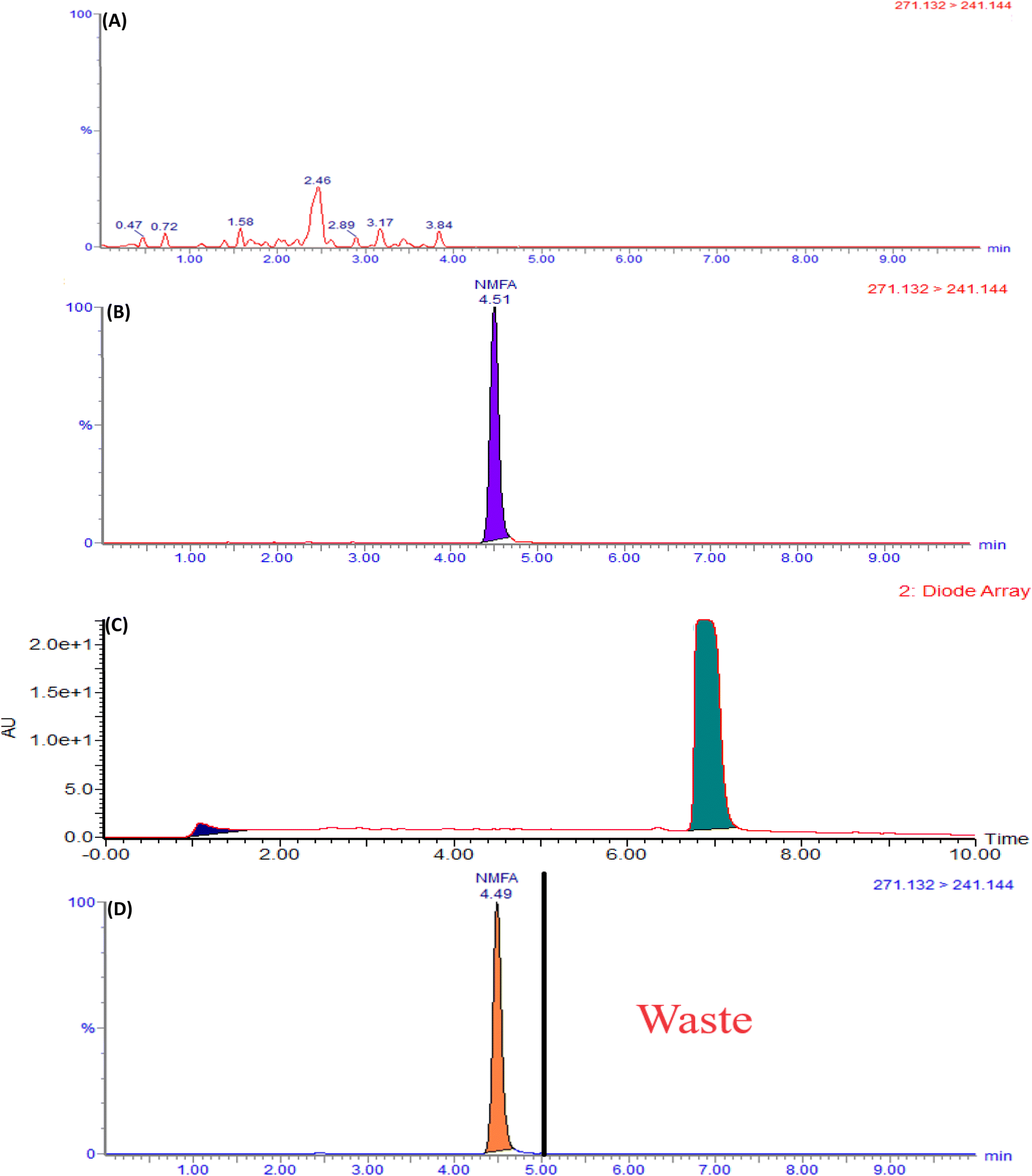 | ||
| Fig. 3 Typical MRM chromatograms of (A) placebo, (B) N-MFA standard at the specification level, and (C) UV spectra of N-MFA spiked to the MFA sample at the specification level, and (D) MRM of N-MFA spiked to the MFA sample at the specification level. | ||
 | ||
| Fig. 4 Typical chromatograms of (A) blank, (B) placebo, (C) N-MFA standard at specification level, and (D) N-MFA spiked to the MFA sample at specification level. | ||
Determination of detection limit (DL) and quantitation limit (QL)
The DL and QL for N-MFA were established using a signal-to-noise ratio (S/N) of 3 for DL and 10 for QL, respectively, by injecting standard solutions with known concentrations. The repeatability of the QL was determined by injecting the N-MFA six times into the liquid chromatograph. The %RSD value was calculated and found to be 1.89. The determined values for DL and QL of N-Nitroso MFA were 0.005 ng and 0.01 ng for LC-MS/MS and 0.013 μg mL−1 and 0.035 μg mL−1 for UPLC, respectively. Table 3 summarizes the results obtained.Linearity
The linearity of the LC-TQ-MS/MS method was examined across a concentration range of 0.01 to 125 ng mL−1, which translates to 0.01 to 125 ppm of N-MFA relative to a sample concentration of 1 mg mL−1. The results of the Pearson correlation coefficient (r), slope (a), and intercept (b) are obtained from the least squares linear regression equation Y = aX + b. For N-MFA, the regression equation is Y = 39.1917X + 15.6498, with a Pearson coefficient of 0.9989. For the UPLC method, linearity was examined across a concentration range of 0.035 to 1.250 μg mL−1, translating to 3.60 ppm to 125 ppm of N-MFA relative to a 10 mg mL−1 sample concentration. For N-MFA, the regression equation is Y = 3.9789X − 9.9192, with a Pearson coefficient of 0.9990. Table 3 summarizes the estimated results, showing a strong correlation between peak areas and N-MFA concentrations.Precision
Method precision/reproducibility was efficiently established by injecting six replicate preparations of MFA spiked with N-MFA at the specification level (62.5 ppm). The method's precision was assessed by the %RSD of the N-MFA, which should be ≤5%, and the Horwitz ratio (Hr) value, which should be ≤1. The Hr is a parameter indicating the tolerability of analytical methods in terms of precision and reproducibility. Additionally, different analysts assessed intermediate precision on different days, determining the %RSD for N-MFA content in six spiked sample solutions at the specified level. The precision results for both methods are summarized in Table 3.Accuracy and recovery study
The accuracy of the method was evaluated by making duplicate preparations at three distinct concentration levels: including LOQ (0.01 ng mL−1 for LC-MS/MS and 0.035 μg mL−1 for UPLC), 100% (62.5 ppm), and 200% (125 ppm), for both pure and dosage form samples of MFA employing the standard addition method. The individual recoveries obtained for the MFA drug substance and its dosage forms ranged from 90.43% to 101.34% % for LC-TQ-MS/MS and 92.12% to 101.21% for UPLC; the results are shown in Table 3.Solution stability
The stability of the N-MFA solution was confirmed by spiking it to 100% of the specification level (62.5 ppm), maintaining it at room temperature, and administering regular injections in both pure MFA and its dosage form. During a 24 hour study, the peak area of the N-MFA impurity was measured at intervals between 1.5 and 10.0 hours, compared to a freshly prepared standard solution. There were no significant changes detected in the peak area of N-MFA. The absolute percentage difference in the N-MFA peak area between the initial measurement and after 24 hours is 2.14 for the LC-TQ-MS/MS method and 1.35 for the UPLC method, respectively. This result confirms that the N-MFA in the solution remains persistent for at least 24 hours.Method applicability (analysis of MFA tablet formulation samples and pediatric suspension)
Pediatric suspensions must be safe and effective for treating various infections in children. Children are more vulnerable to genotoxic impurities than adults due to their higher cell division rates, longer lifespans, and still-developing immune systems. Therefore, pediatric suspensions must be high quality to minimize the risk of children being exposed to genotoxic impurities (NDSRIs). The validated LC-TQ-MS/MS and UPLC methods have been utilized to quantify the potentially genotoxic, carcinogenic, and cohort of concern properties of N-MFA in three distinct commercially available tablet and pediatric suspension formulations. Table S3† summarizes the N-MFA content in three tablet dosage formulations and one pediatric suspension formulation, with the calculated results falling below the 30% interim control limit (∼19 ppm).Assessment of greenness and whiteness profile for the developed method
The greenness of the newly developed LC-TQ-MS/MS and UPLC methods were assessed using published journals27–29 and modern tools, including GAPI, AGREE, AGREEPrep, analytical eco-scale, and BAGI, to analyze the data.Analytical eco-scale tool
The Analytical Eco-scale, a semi-quantitative method developed by Koen Van Aken,30 assesses the environmental impact of chemical processes. It assigns penalty points for factors that reduce the process's greenness—such as hazardous reagents, waste generation, or high energy consumption—deducting from a base score 100. The final score indicates whether the approach is ideally green or acceptable. Hazard Penalty Points (HPP) are calculated for chemicals and reagents by multiplying the number of hazard symbols by the signal word. Total Penalty Points (TPP) are then determined by multiplying the HPP by the Amount Penalty Points (APP). Formic acid and acetonitrile have 3 and 2 hazard symbols, respectively, with the signal word “Danger.” Their corresponding HPP values are 6 and 4. The APP values for acetonitrile in the current method are “1” and “2,” based on the volume used for LC-MS and UPLC analysis. Consequently, the TPP value for the organic solvent acetonitrile is 8. The penalty point for occupational hazards is 3. The penalty points assigned for the sonicator, waste generated (over 10 mL), and waste management (no treatment) are 1, 5, and 3, respectively. Due to its implementation, 27 and 25 penalty points for LC-MS/MS and UPLC methods have been assigned to these methods. According to the Analytical Eco-Scale, these methods fall into the green region with a score of 73 and 75. Table S4† summarizes the penalty points of the analytical eco-scale.Green analytical procedure index (GAPI) assessment tool
In 2018, Płotka-Wasylka introduced the GAPI31 tool. GAPI uses a pictogram with red, yellow, and green colors to indicate high, medium, and low environmental impacts, respectively. The sensitive LC-TQ-MS/MS method shows that pictograms 2, 3, 6, 8, 9, and 12 are green, while pictograms 4, 5, 10, and 11 are yellow, and pictograms 1, 7, 13, 14, and 15 are red, indicating both qualitative and quantitative impacts. Similarly, the cost-effective UPLC method reveals that pictograms 2, 3, 8, 9, and 12 are green, pictograms 4, 5, 10, and 11 are yellow, and pictograms 1, 6, 7, 13, 14, and 15 are red, also indicating both qualitative and quantitative impacts. An illustration of the GAPI pictogram is shown in Fig. 5.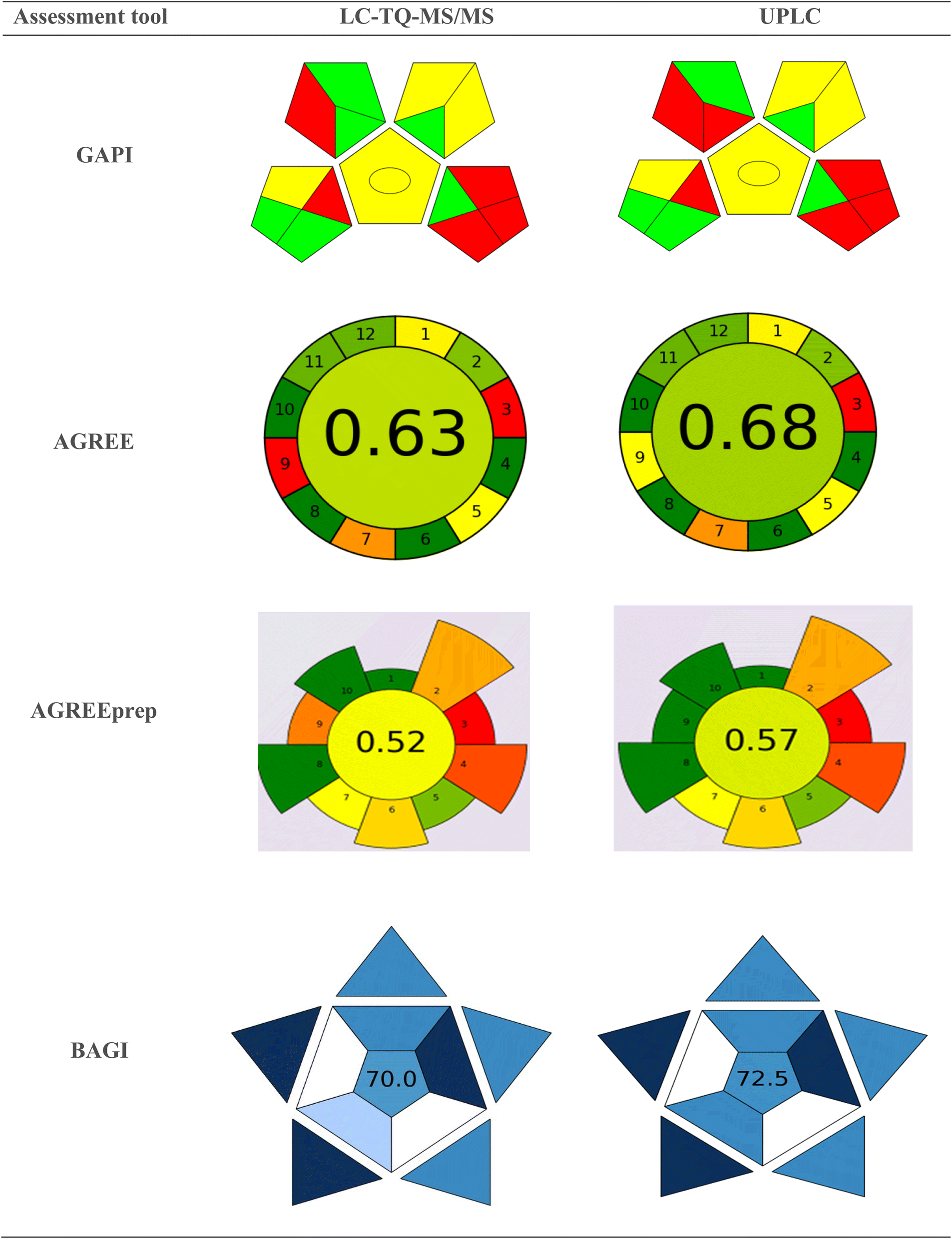 | ||
| Fig. 5 Pictograms of GAPI, AGREE, AGREEPrep, and BAGI for the proposed LC-TQ-MS/MS and UPLC methods. | ||
Analytical GREEnness (AGREE) and AGREEprep assessment tool
The AGREE tool, developed by the Gdańsk University of Technology in Poland, assesses a process's greenness based on 12 principles of green chemistry. AGREE is represented as a circular pictogram divided into 12 segments, each representing a distinct green principle. In analytical procedures, sample preparation is a vital step toward achieving analytical greenness.32 AGREEprep introduces the first metric specifically for sample preparation, quantifying the process's technical aspects and pinpointing improvement areas. It comprises ten sequential assessment steps corresponding to the ten green sample preparation (GSP) principles.33 The AGREE and AGREEprep scores for the LC-TQ-MS/MS method are 0.63 and 0.52, respectively, while for UPLC, they are 0.65 and 0.57. The corresponding pictograms are shown in Fig. 5. Table S5† outlines the principles of GAPI and AGREE, the selected method condition values, and their respective scores.Blue applicability grade index (BAGI)assessment tool
The BAGI tool was developed by Natalia Manousi, Wojciech Wojnowski, Justyna Płotka-Wasylka, and Victoria Samanidou.34 BAGI acts as a complementary metric to green metrics, which primarily emphasize the environmental impact of analytical methods, including solvent safety, waste generation, energy consumption, and more. Blue, represented by the RGB model, symbolizes the index, positioning it as an addition to the established green metrics tools. The BAGI and green metrics can be integrated to assess an analytical method's performance and environmental sustainability comprehensively. This tool evaluates ten key attributes: the type of analysis, the simultaneous estimation of analytes, the sample throughput per hour, the reagents and materials used, the required instruments, the concurrent treatment of samples, the need for preconcentration, the level of automation, the sample preparation procedure, and volume. It is a unique measure for assessing a method's feasibility in analytical chemistry. The scoring system ranges from 25 to 100, with a higher BAGI evaluation score indicating greater practicality of the method. Fig. 5 presents the BAGI index pictogram for the developed methods, showing an impressive score of 70.0 for LC-TQ-MS/MS and 72.5 for UPLC methods. These results highlight the outstanding practicality and significance of the techniques.The whiteness assessment tool RGB12 for the LC-TQ-MS/MS and UPLC methods
In 2021, Nowak et al. demonstrated that White Analytical Chemistry (WAC) elaborates on the 12 principles of GAC. The WAC tool was created as part of a broader initiative to encourage safer and more sustainable practices in analytical chemistry. The Whiteness Assessment tool RGB12 assesses the overall whiteness of an analytical method by combining the colors of three distinct groups: red, green, and blue.35 Using the RGB 12 model, this work offers a novel approach for quantifying the studied NDSRI by merging the principles of sustainable development with those of analytical chemistry. The colors assigned to each group represent distinct attributes: red signifies analytical efficiency (R1: Application Scope, R2: LOD and LOQ, R3: Precision, R4: Accuracy), green represents a commitment to green chemistry principles (G1: Reagents Toxicity, G2: Reagents and Waste Quantity, G3: Energy Consumption, G4: Direct Impacts), and blue indicates the potential for economic growth (B1: Cost Efficiency, B2: Time Efficiency, B3: Requirements, B4: Operational Simplicity).The proposed LC-TQ-MS/MS and UPLC methods obtained a remarkable score of 108.8 and 100% in the red color criterion, suggesting their capacity to precisely and accurately detect NDSRI impurity. Moreover, both methods demonstrate impressive environmental friendliness scores of 96.7% for LC-MS/MS and 94.2% for UPLC, as indicated by the green color. This highlights their strong commitment to minimizing environmental impact and waste, emphasizing their dedication to eco-friendliness. Furthermore, the efficiency of both methods is highlighted by their impressive scores of 87.3% for LC-TQ-MS/MS and 96.7% for UPLC in the blue color group. This underscores the methods' effectiveness in optimizing time and cost, emphasizing their enhanced efficiency and economic viability. Fig. S6† illustrates the remarkable whiteness attained by the developed LC-TQ-MS/MS and UPLC methods, achieving an impressive score of 97.6 and 96.9%, respectively. The RGB12 software report provides thorough details on the method's accomplishments, which align with the 12 principles of WAC.
Conclusion
In conclusion, two interdependent (Q)-SAR tools were utilized to assess and classify N-MFA as a Class-3 category, designated as a “CoC” which suggests potential genotoxicity. Diphenylamine is a precursor that can lead to the formation of N-MFA. The acceptable intake (AI) of N-MFA was derived from N-DPA, and the determined AI for N-MFA is 78000 ng; hence the specification limit is 62.4 ppm. Proposed novel LC-TQ-MS/MS and cost-effective UPLC methods are proposed to accurately quantify N-nitroso mefenamic acid (N-MFA) in mefenamic acid (MFA) tablets and pediatric suspension dosage forms. The proposed method has been successfully validated in compliance with ICH guidelines. The developed methods were further applied to commercially available mefenamic acid formulations to determine accurate and precise quantification of N-MFA. For routine quality control laboratories, this cost-effective UPLC method can be used to detect 30% of the interim control limit (∼19 ppm). Furthermore, sustainability and eco-friendliness were evaluated using greenness and whiteness assessment metrics, including the analytical eco-scale, GAPI, AGREE, AGREEprep, BAGI, and RGB12 tools. The results indicated that the method proposed exhibits a high level of greenness and whiteness. Both methods can be used to determine N-MFA content in mefenamic acid formulation dosage based on the availability of instruments LC-TQ-MS/MS and UPLC in R&D and quality control laboratories.
Data availability
The data that supports the findings of this study are available in the ESI of this article.†Author contributions
Srinivas Nakka: conceptualization, methodology, investigation, validation, software, writing – original draft, drawing – chemical structures and green chemistry tools. Naresh Kumar Katari: data-curation, visualization, review, and editing, Siva Krishna Muchakayala: formal analysis, visualization, green chemistry tools, review, and editing, Sreekantha Babu Jonnalagadda: resources, data-curation, review, and editing, and Surendra Babu Manabolu Surya: resources, review and editing, and supervision. Resources and project administration.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors Surendra Babu MS and S. Nakka acknowledge GITAM deemed to be University for research facilities and Jonnalagadda SB and Katari NK acknowledge University of KwaZulu-Natal for funding.References
- I. W. Ashworth, O. Dirat, A. Teasdale and M. Whiting, Org. Process Res. Dev., 2020, 9, 1629–1646 CrossRef
.
- U. Holzgrabe, J. Pharmaceut. Sci., 2023, 112, 1210–1215 CrossRef CAS
- S. Horne, M. D. Vera, L. R. Nagavelli, V. A. Sayeed, L. Heckman, D. Johnson, D. Berger, Y. Yip, C. L. Krahn, L. O. Sizukusa, N. F. M. Rocha, R. N. Bream, J. Ludwig, G. A. Keire and G. Condran, J. Pharmaceut. Sci., 2023, 5, 1166–1182 CrossRef
- M. Justin, S. Jörg and S. Christoph, J. Pharmaceut. Sci., 2023, 5, 1161–1162 Search PubMed
- B. Tuesuwan and V. Vongsutilers, J. Pharmaceut. Sci., 2021, 110, 3118–3128 CrossRef CAS
- W. Wichitnithad, S. Nantaphol, K. Noppakhunsomboon, W. Thitikornpong and P. Rojsitthisak, Talanta, 2023, 254, 124102 CrossRef CAS
- FDA, Pfizer Expands Voluntary Nationwide Recall to Include All Lots of CHANTIX® (Varenicline) Tablets Due to N-Nitroso Varenicline Content, 2021, https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/pfizer-expands-voluntary-nationwide-recall-include-four-additional-lots-chantixr-varenicline-tablets, accessed 8 March 2024 Search PubMed.
- FDA, Lupin Pharmaceuticals, Inc. Issues Voluntarily Nationwide Recall of All Irbesartan Tablets and Irbesartan and Hydrochlorothiazide Tablets Due to Potential Presence of N-Nitrosoirbesartan Impurity, 2021, https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/lupin-pharmaceuticals-inc-issues-voluntarilynationwide-recall-all-irbesartan-tablets-and-irbesartan, accessed 8 March 2024 Search PubMed.
- FDA, Pfizer Voluntary Nationwide Recall of Lots of ACCUPRIL® (Quinapril HCl) Due to N-Nitroso-quinapril Content, 2022, https://www.fda.gov/safety/recallsmarket-withdrawals-safety-alerts/pfizer-voluntary-nationwide-recall-lots-accuprilr-quinapril-hcl-due-n-nitroso-quinapril-content, accessed 8 March 2024 Search PubMed.
- FDA, Sandoz, Inc. Issues Nationwide Recall of 13 Lots of Orphenadrine Citrate 100 mg Extended-Release Tablets Due to Presence of a Nitrosamine Impurity, 2022, https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/sandoz-inc-ssues-nationwide-recall-13-lots-orphenadrine-citrate-100-mg-extendedrelease-tablets-due-recall-announcement, accessed 8 March 2024 Search PubMed.
- Health Canada, Pfizer recalls Inderal-LA (Propranolol Hydrochloride) Capsules Due to a Nitrosamine Impurity, 2022, https://www.recalls-rappels.canada.ca/en/alert-recall/pfizer-recalls-inderal-propranolol-hydrochloride-capsules-due-nitrosamine-impurity, accessed 8 March 2024 Search PubMed.
- C. Eccleston C, T. E. Cooper, E. Fisher, B. Anderson and N. M. Wilkinson, Cochrane Database Syst. Rev., 2017, 8, CD012537 Search PubMed
- K. Sridharan and S. Shah, Int. J. Risk Saf. Med., 2023, 34, 379–386 Search PubMed
- C. Nevio, Clin. Pharmacol., 2013, 6, 289–305 Search PubMed
- A. Husain, P. Ahuja, A. Ahmad and S. A. Khan, Med. Chem., 2016, 12, 585–591 CrossRef CAS
- Determining Recommended Acceptable Intake Limits for N-Nitrosamine Impurities in Pharmaceuticals: Development and Application of the Carcinogenic Potency Categorization Approach, https://www.fda.gov/drugs/spotlight-cder-science/determining-recommended-acceptable-intake-limits-n-nitrosamine-impurities-pharmaceuticals Search PubMed.
- Nitrosamine Impurities in Medications: Established Acceptable Intake Limits, https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/established-acceptable-intake-limits.html Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- A. Mostafa, RSC Adv., 2024, 14, 19658–19679 RSC
- M. Branković, Anal. Methods, 2023, 15, 6631–6642 RSC
- ICH M7(R1) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. Current Step 4 Version Dated 31 March 2017, https://www.database.ich.org/sites/default/files/M7_R1_Guideline.pdf Search PubMed.
- ICH Q2(R2) International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Topic Q2(R1): Validation of Analytical Procedures: Text and Methodology, 2005, https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1_Guideline.pdf Search PubMed.
- USP General Chapter 〈1225〉 Validation of Compendial Procedures, USP-43-NF38, 8166, United States Pharmacopeial Convention, Rockville, Maryland, USA, 2020 Search PubMed.
- S. Lorenza, J. L. Benedé and A. Chisvert, RSC Adv., 2023, 13, 2963–2971 RSC
- S. Nakka, S. K. Muchakayala and S. B. Manabolu Surya, Sustainable Chem. Pharm., 2024, 38, 101495 CrossRef CAS
- S. Nakka, N. K. Katari, S. K. Muchakayala, B. Jonnalagadda and S. B. Manabolu Surya, ACS Omega, 2024, 9, 8773–8788 CrossRef CAS PubMed
- Z. Mlinarić, L. Turković, I. Babić, T. Silovski, N. K. Glavač and M. Sertić, RSC Adv., 2024, 14, 32876–32882 RSC
- S. B. Gopalaiah and K. Jayaseelan, RSC Adv., 2024, 14, 22169–22184 RSC
- K. H. Michael, M. B. Osama and F. A. Ahmed Emad, Sustainable Chem. Pharm., 2024, 38, 101463 CrossRef
- K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 CrossRef PubMed
- J. Płotka-Wasylka, Talanta, 2018, 181, 204–209 CrossRef PubMed
- F. Pena-Pereira, W. Wojnowski and W. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS
- W. Wojnowski, M. Tobiszewski, F. Pena-Pereira and E. Psillakis, Trends Anal. Chem., 2022, 149, 116553 CrossRef CAS
- M. Natalia, W. Wojciech, P. W. Justyna and S. Victoria, Green Chem., 2023, 25, 7598–7604 RSC
- P. M. Nowak, R. Wietecha-Posłuszny and J. Pawliszyn, TrAC, Trends Anal. Chem., 2021, 138, 116223 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08425j |
| This journal is © The Royal Society of Chemistry 2025 |