
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis and in vitro and in silico studies of pyrano[3,2-c]quinoline-3-carboxylates as dual acting anti-cancer and anti-microbial agents and potential topoisomerase II and DNA-gyrase inhibitors†
Ashraf A. Aly *a,
Hisham A. Abd El-Nabya,
Essam Kh. Ahmeda,
Sageda A. Gedamya,
Kari Rissanenb,
Martin Niegerc,
Alan B. Brownd,
Michael G. Shehate,
Marwa M. Shaabanf and
Amal Attaf
*a,
Hisham A. Abd El-Nabya,
Essam Kh. Ahmeda,
Sageda A. Gedamya,
Kari Rissanenb,
Martin Niegerc,
Alan B. Brownd,
Michael G. Shehate,
Marwa M. Shaabanf and
Amal Attaf
aChemistry Department, Faculty of Science, Minia University, 61519 El-Minia, Egypt. E-mail: ashrafaly63@yahoo.com; ashraf.shehata@mu.edu.eg; hisham_minia@mu.edu.eg; essam.mohd@mu.edu.eg; sagedaali332@yahoo.com
bDepartment of Chemistry, University of Jyväskylä, P. O. Box 35, FIN-40014 Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi
cDepartment of Chemistry, University of Helsinki, P. O. Box 55, A. I. Virtasen aukio I, 00014 Helsinki, Finland. E-mail: martin.nieger@helsinki.fi
dDepartment of Chemistry and Chemical Engineering, Florida Institute of Technology, Melbourne, FL 32901, USA. E-mail: abrown@fit.edu
eDepartment of Microbiology, Faculty of Pharmacy, Alexandria University, Alexandria 21521, Egypt. E-mail: michael.shehat@alexu.edu.eg
fDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University, Alexandria 21521, Egypt. E-mail: maraw.mamdouh@alexu.edu.eg; amal.atta@alexu.edu.eg; missalex_pharma@yahoo.com
First published on 21st January 2025
Abstract
A microwave-assisted method was utilized to synthesize novel pyranoquinolone derivatives as dual acting topoisomerase II/DNA gyrase inhibitors with apoptosis induction ability for halting lung cancer and staphylococcal infection. Herein, the designed rationale was directed toward mimicking the structural features of both topoisomerase II and DNA gyrase inhibitors as well as endowing them with apoptosis induction potential. The absolute configuration of the series was assigned using X-ray diffraction analysis. Cytotoxic activity against NSCLC A549 cells showed that ethyl 2-amino-9-bromo-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (IC50 ≈ 35 μM) was the most potent derivative in comparison to the positive control Levofloxacin and was selected for further investigation to assess its selectivity (SI = 1.23). Furthermore, in vitro antibacterial screening revealed the potential activity of this bromo derivative against Staphylococcus aureus. Mechanistic studies showed that the aforementioned compound exhibited promising inhibitory activity against topoisomerase II (IC50 = 45.19 μM) and DNA gyrase (IC50 = 40.76 μM) compared to reference standards. In addition, the previous compound induced a A549 cell apoptosis by 38.49-fold and it also increased the total apoptosis by 20.4% compared to a 0.53% increase in the control. Docking simulations postulated its interactions and suggested well fitting into its molecular targets.
1. Introduction
Cancer is a fatal disease characterized by uncontrolled cell proliferation that invades surrounding tissue and is associated with a high mortality rate.1 The WHO's global cancer report predicts that the percentage of deaths from cancer will double in the next several years. As a result, many academics are now considered the development of novel effective anticancer medications, it has been considered to be an urgent requirement.2 Thus, research on heterocyclic cores possessing anticancer potential has attracted great interest worldwide.3Globally, lung cancer is the leading cause of cancer-related mortality.4 Adjuvant chemotherapy and antineoplastic agents are conventional treatments for advanced non-small cell lung cancer (NSCLC).5 Since topoisomerase enzymes are crucial for DNA metabolism, finding enzyme inhibitors is a key goal in the hunt for novel anticancer medications.6 They act by inhibiting topoisomerases from relegating DNA strands after cleavage, leading to DNA damage. The majority of anticancer agents are mainly directed toward DNA topoisomerase inhibition.7 The development of novel anticancer medications that specifically target topoisomerase II (Topo II) is a source of interest for medicinal chemists to overcome resistance and improve chemotherapeutic outcomes. For instance, etoposide is a significant chemotherapeutic drug inhibiting Topo II, which has been used to treat a variety of human malignancies for over 20 years and is still among the most often prescribed anticancer medications worldwide.8 Moreover, quinoline and pyran derivatives exhibit a variety of pharmacological activities, making them important pharmaceutically active heterocyclic compounds.9–11 They are important cores for creating new classes of structural entities used in cancer treatment (Fig. 1).9,12 The quinoline framework is essential for the development of anticancer drugs via a variety of mechanisms, including angiogenesis inhibition, apoptosis induction, growth inhibition by cell cycle arrest, and cell-migration disruption.13 In addition, drugs composed of quinolone and pyran rings targeting topoisomerase are currently widely used as frontline medications for the treatment of cancer (Fig. 1).14
 | ||
| Fig. 1 Reported quinoline-, pyran- and pyranoquinoline-based anticancer and antibacterial derivatives as potential inhibitors of topoisomerase II and DNA gyrase. | ||
Patients with cancer are at an increased risk of bacterial and antibiotic-resistant infections compared with healthy individuals.15 Particularly, lung cancer patients undergoing chemotherapeutic and surgical treatment are more likely to suffer from pulmonary complications caused mainly by bacterial infection. Regarding the bacterial spectrum of lung cancer patients, Staphylococcus aureus is one of the most frequently isolated organisms.16 Moreover, growing evidence indicates that staphylococcal infection is closely linked to the incidence and development of several cancer types, signifying that both inflammation and immunity play a certain role in the development of cancer; thus, there is an association between Staphylococcus and carcinogenesis.15,17 Consequently, bacterial infection may affect the growth of lung cancer by activating inflammatory signaling. Staphylococcus aureus lipoteichoic acid (LTA) induced a prominent increase in the cellular growth and proliferation of the NSCLC cell lines A549.18 Besides, Staphylococcus aureus infection is involved in the regulation of cancer cell metastasis.19
Molecular approaches incorporating pyran and quinoline moieties (Fig. 1) have revealed remarkable antimicrobial activity against Gram-negative and Gram-positive bacteria as well as against fungal pathogens.20–23 Particularly, quinolones are broad-spectrum antibacterial agents targeting DNA gyrase, a type II topoisomerase. DNA gyrase plays a vital role in transcription and bacterial DNA replication, making it an essential therapeutic target in antibacterial drug discovery. However, adverse effects and emerging drug resistance render the currently available quinolones less effective.24 Several quinoline entities (Fig. 1) with diverse scaffolds have been identified as DNA gyrase inhibitors, which could serve as good leads for the development of novel antibacterial agents.25,26 Additionally, various synthetic techniques have been utilized to develop new quinolones or to modify the quinolone scaffold with the aim of reducing toxicity and overcoming resistance.27,28 In addition, spiramycin, a pyran derivative, is a macrolide antibiotic with activity primarily against Staphylococcus aureus.29
As a follow-up to our work on the synthesis of biologically active-fused quinolones, and in light of the aforementioned activity of pyranes and quinolines, this study aims to combine the pyrane and quinoline scaffolds into a single molecule in addition to modifying the nature of the linked quinoline ring by diversifying the substitution, allowing us to assess the SAR among the investigated series in the quest of promising compounds regarding activity (Fig. 2). Herein, we report a novel series of pyrano[3,2-c]quinoline-3-carboxylates combating the staphylococcal infection-lung cancer interplay by inhibiting both DNA gyrase and topoisomerase II with apoptosis induction ability.
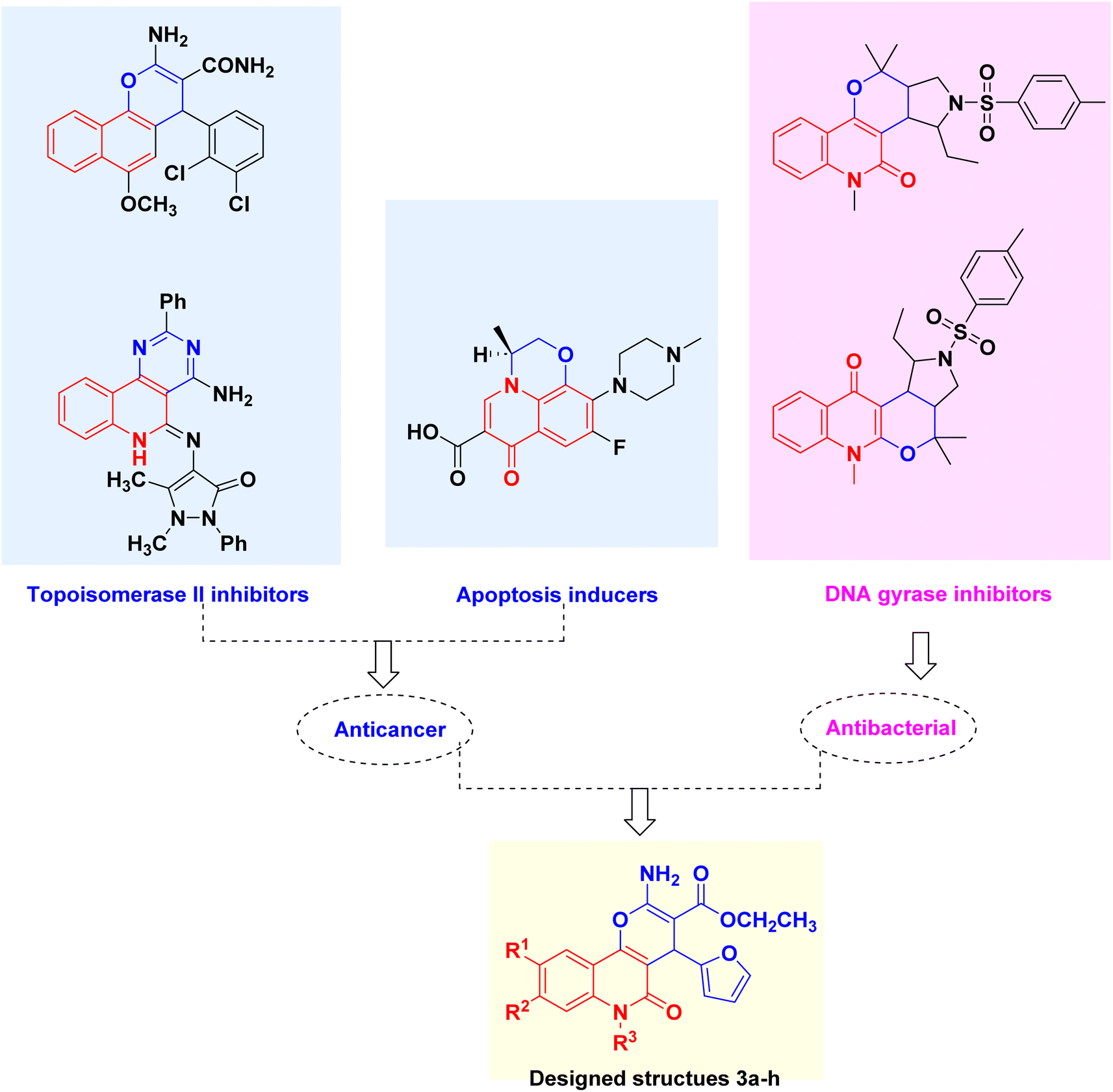 | ||
| Fig. 2 Lead Topo II inhibitors, apoptosis inducers, DNA gyrase inhibitors and design rationale of the multitarget pyranoquinolones 3a–h. | ||
2. Results and discussion
2.1. Chemistry
Pyrano[3,2-c]quinoline-3-carboxylate derivatives 3a–h were obtained via the reaction of 4-hydroxy-2-oxo-1,2-dihydroquinoline derivatives 1a–h with ethyl (E)-2-cyano-3-(furan-2-yl)acrylate (2) (Scheme 1). The reaction was performed under different conditions, such as ethanol/K2CO3 at reflux (Method I) and ethanol/K2CO3/microwave (Method II) (Scheme 1).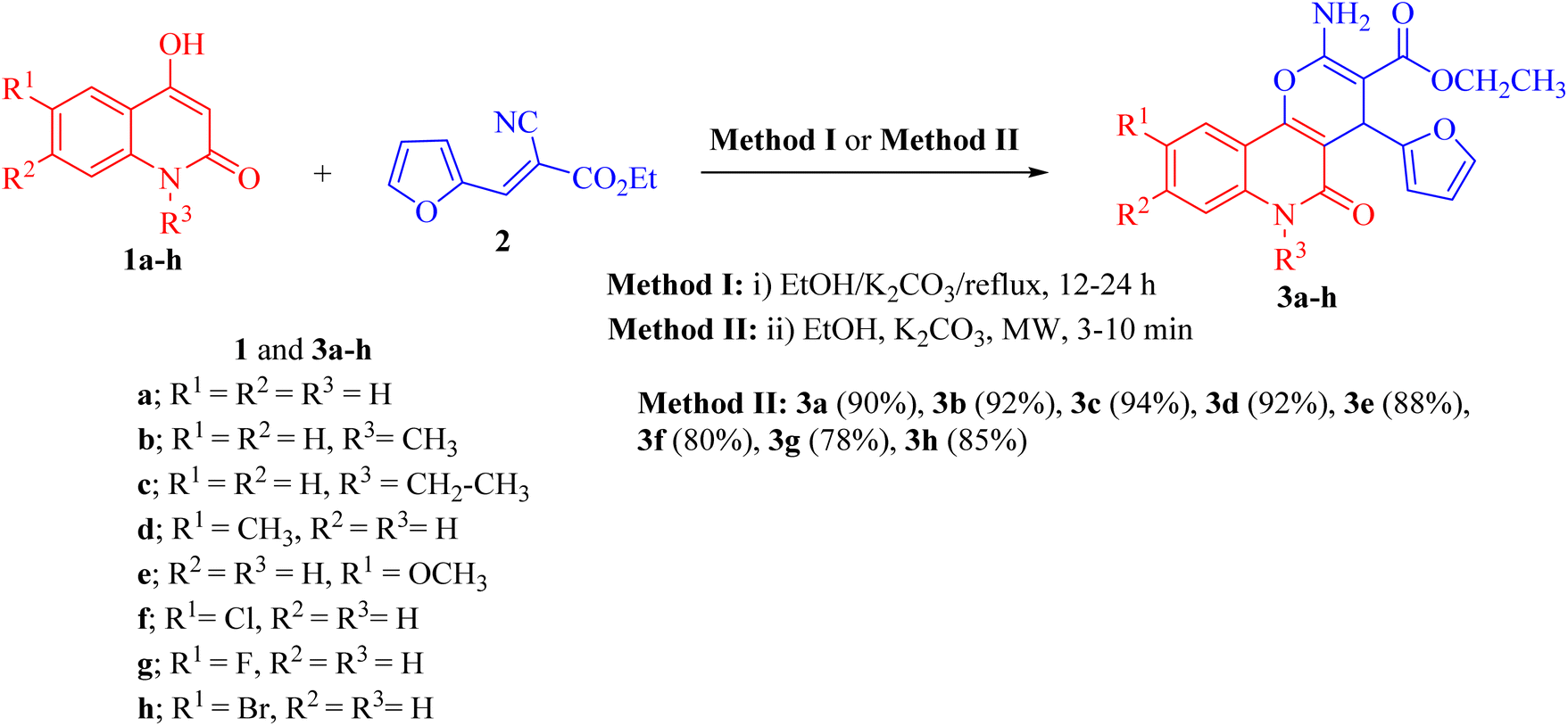 | ||
| Scheme 1 Synthesis of ethyl 2-amino-4-(furan-2-yl)-5-oxo-pyrano[3,2-c]quinoline-3- carboxylates 3a–h. | ||
Interestingly, when the reaction was carried out by Method II, the yields of products 3a–j were found to be excellent for some derivatives (75–94%) and also took a shorter time (Table 1). Method II showed that the reaction between 1a–h and 2 was completed faster and gave excellent yields (80–94%) compared with the conventional method (Method I).
| Compounds | Time (min or h) | Yield (%) | ||
|---|---|---|---|---|
| Method I (h) | Method II (min) | Method I | Method II | |
| 3a | 12 | 3 | 75 | 90 |
| 3b | 14 | 4 | 65 | 92 |
| 3c | 15 | 4 | 77 | 94 |
| 3d | 18 | 7 | 80 | 92 |
| 3e | 20 | 6 | 78 | 88 |
| 3f | 26 | 8 | 64 | 80 |
| 3g | 20 | 9 | 65 | 78 |
| 3h | 24 | 10 | 82 | 85 |
Under microwave irradiation, compounds 3a, 3b and 3c were formed completely in a short time (>5 min) and afforded yields of 90%, 92% and 94%, respectively. The best yield was obtained in the case of 3c (94%), and the reaction was completed in 4 min (Table 1). The 1H NMR spectrum of 3a showed the ethyl protons as a triplet for CH3 at δH = 1.21 (J = 7.1 Hz) and the CH2-ester at δH = 4.11 (ABX3, JAB = 14.2, JAX = 7.1 Hz; 1H) and δH = 4.08 ppm (ABX3, JAB = 14.2, JBX = 7.1 Hz; 1H). The NH2 protons resonated as a broad singlet at δH = 7.77 ppm, while the H-pyran appeared as a singlet at δH = 5.03 ppm. The three protons of furan appeared as a doublet at δH = 6.28 (J = 1.8 Hz), a double-doublet at δH = 6.07 (J = 2.0, 1.0 Hz) and a doublet at δH = 7.37 ppm (J = 0.8 Hz) for H-3′, H-4′ and H-5′, respectively. The 13C NMR spectrum confirmed the 1H NMR spectral data. As 13C NMR spectrum revealed the ethyl-ester carbon signals at δC = 14.3 (CH3) and 58.8 (CH2), while the CH-pyran appeared as a singlet at δC = 28.2 ppm. The two carbonyl carbon signals (C-3a and C-5) for the ester and quinolyl groups resonated at δC = 167.7 and 156.4 ppm, respectively. The carbon signal of pyran-2-C (C-2) in the 13C NMR spectrum appeared at δC = 160.6 ppm. Distinctive carbons are shown in Fig. 3. The NMR spectral data for compound 3a are shown in Table 2.
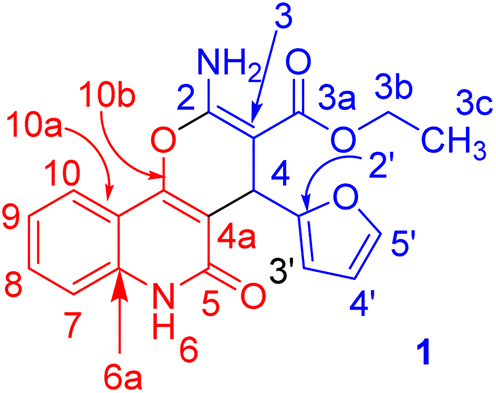 | ||
| Fig. 3 Distinctive carbons of compound 3a. | ||
| 1H NMR (DMSO-d6) | 1H–1H COSY | Assignment |
|---|---|---|
| 7.96 (d, J = 8.0 Hz; 1H) | 7.58, 7.30 | H-10 |
| 7.77 (b; 2H) | NH2 | |
| 7.58 (dd, J = 8.1, 7.4 Hz; 1H) | 7.96, 7.35, 7.30 | H-8 |
| 7.37 (d, J = 0.8 Hz; 1H) | H-5′ | |
| 7.35 (d, J = 8.3 Hz; 1H) | 7.58 | H-7 |
| 7.30 (dd, J = 7.8, 7.4 Hz; 1H) | 7.96, 7.58 | H-9 |
| 6.28 (d, J = 1.8 Hz; 1H) | 6.07 | H-3′ |
| 6.07 (dd, J = 2.0, 1.0 Hz; 1H) | 6.28 | H-4′ |
| 5.03 (s; 1H) | H-4 | |
| 4.11 (ABX3, JAB = 14.2, JAX = 7.1 Hz; 1H) | 1.21 | H-3b |
| 4.08 (ABX3, JAB = 14.2, JBX = 7.1 Hz; 1H) | 1.21 | H-3b |
| 1.21 (ABX3, JAX = JBX = 7.1 Hz; 3H) | 4.11, 4.08 | H-3c |
| 13C NMR (DMSO-d6) | Assignment |
|---|---|
| 167.7 | C-3a |
| 160.6, 160.1 | C-2, C-2′ |
| 156.4 | C-5 |
| 151.9 | C-10b |
| 141.0 | C-5′ |
| 137.7 | C-6a |
| 131.0 | C-8 |
| 121.9, 121.6 | C-9, C-10 |
| 115.30 | C-7 |
| 112.2, 110.2, 109.5 | C-3′, C-4′, C-10a |
| 105.0 | C-4a |
| 74.6 | C-3 |
| 58.8 | C-3b |
| 28.2 | C-4 |
| 14.3 | C-3c |
X-ray structure analysis proved the structure of 3a (Fig. 4) and was identified as ethyl 2-amino-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate.
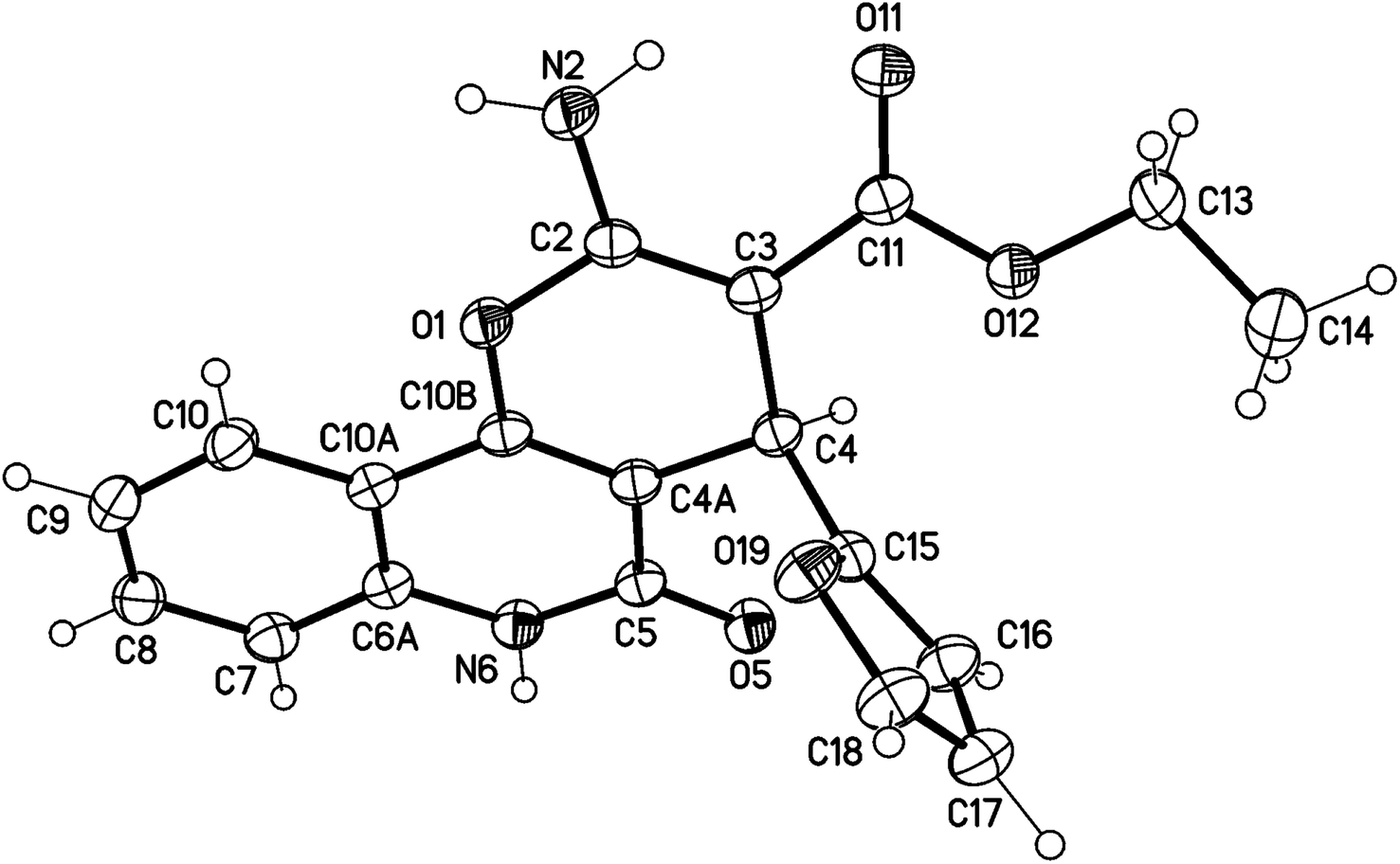 | ||
| Fig. 4 Molecular X-ray structure of compound 3a (displacement parameters are drawn at a 50% probability level). | ||
In compound 3b (Fig. 5), the 1H NMR spectrum revealed the CH3-ester as an ABX3, (JAX = JBX = 7.1 Hz) at δH = 1.21. The CH2 protons of the ester group appeared as non-equivalent two protons at δH = 4.12 (ABX3, JAB = 10.7, JAX = 7.1 Hz) and at δH = 4.06 ppm (ABX3, JAB = 10.7, JAX = 7.1 Hz). They have a COSY relationship with each other and with the CH3 protons of the ester group. The methyl (N–CH3) and CH-pyran (H-4) protons resonated at δH = 3.63 and 5.05 ppm. The three protons of furan appeared as a doublet at δH = 6.07 (J = 3.0 Hz) for H-4′ and a double-doublet at δH = 6.27 ppm (J = 3.0, 1.9 Hz) for H-3′. H-5′ of the furan molecule resonated as a doublet at δH = 7.36 (J = 0.8 Hz). The amino protons appeared in the 1H NMR spectrum at δH = 7.79. Table 3 illustrates the assigned δ values of the NMR spectral data for compound 3b.
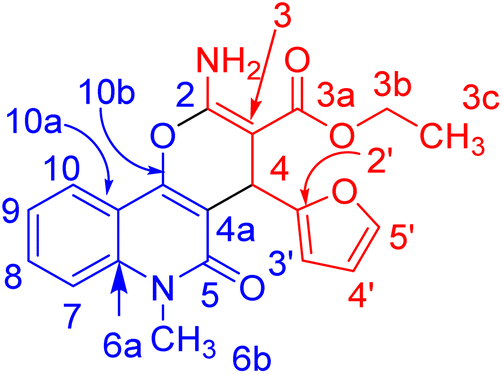 | ||
| Fig. 5 Distinctive carbons of compound 3b. | ||
| 1H NMR (DMSO-d6) | 1H–1H COSY | Assignment |
|---|---|---|
| 8.07 (dd, J = 7.9, 1.0 Hz; 1H) | 7.71, 7.39, 6.07 | H-10 |
| 7.79 (b; 2H) | NH-2a | |
| 7.71 (ddd, J = 8.5, 7.2, 1.3 Hz; 1H) | 8.07, 7.58, 7.39 | H-8 |
| 7.58 (d, J = 8.5 Hz; 1H) | 7.71, 7.39 | H-7 |
| 7.39 (dd, J = 7.7, 7.4 Hz; 1H) | 8.07, 7.71, 7.58 | H-9 |
| 7.36 (d, J = 0.8 Hz; 1H) | 6.27 | H-5′ |
| 6.27 (dd, J = 3.0, 1.9 Hz; 1H) | 7.36, 6.07 | H-3′ |
| 6.07 (d, J = 3.0 Hz; 1H) | 8.07, 6.27 | H-4′ |
| 5.05 (s; 1H) | H-4 | |
| 4.12 (ABX3, JAB = 10.7, JAX = 7.1 Hz; 1H) | 4.06, 1.21 | H-3b |
| 4.06 (ABX3, JAB = 10.7, JBX = 7.1 Hz; 1H) | 4.12, 1.21 | H-3b |
| 3.63 (s; 3H) | H-6b | |
| 1.21 (ABX3, JAX = JBX = 7.1 Hz; 3H) | 4.12, 4.06 | H-3c |
| 13C NMR (DMSO-d6) | Assignment |
|---|---|
| 167.6 | C-3a |
| 160.0, 159.9 | C-2, C-2′ |
| 156.4 | C-5 |
| 150.9 | C-10b |
| 141.0 | C-5′ |
| 138.5 | C-6a |
| 131.5 | C-8 |
| 122.0 | C-9, C-10 |
| 114.9 | C-7 |
| 112.8, 110.2, 108.9 | C-3′, C-4′, C-10a |
| 105.1 | C-4a |
| 74.6 | C-3 |
| 58.8 | C-3b |
| 29.3 | C-6b |
| 28.9 | C-4 |
| 14.3 | C-3c |
X-ray structure analysis proved the structure of 3b (Fig. 6) and was identified as ethyl 2-amino-4-(furan-2-yl)-6-methyl-5-oxo-5,6-dihydro-4H-pyrano [3,2-c]quinoline-3-carboxylate.
 | ||
| Fig. 6 Molecular structure of the 1st crystallographic independent molecule of compound 3b (displacement parameters are drawn at a 50% probability level). | ||
It is noteworthy that compounds 3a and 3b crystallized in a centrosymmetric space group (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2)) and the relative configuration was determined. Therefore, both enantiomers (R and S at C4 and C24, respectively) were present in a ratio of 1
(no. 2)) and the relative configuration was determined. Therefore, both enantiomers (R and S at C4 and C24, respectively) were present in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
The mechanism proposed for the formation of compounds 3a–h begins with a nucleophilic attack of the active CH-3 from 1a–h to the electrophilic carbon in 2 via Michael addition to produce intermediates 4a–h (Scheme 2). Further nucleophilic attack of the hydroxyl-lone pair in 4a–h then occurs to the electrophilic carbon in the nitrile group, forming intermediate 5a–h (Scheme 2). Finally, the aromatization of 5a–h gives the final products 3a–h (Scheme 2).
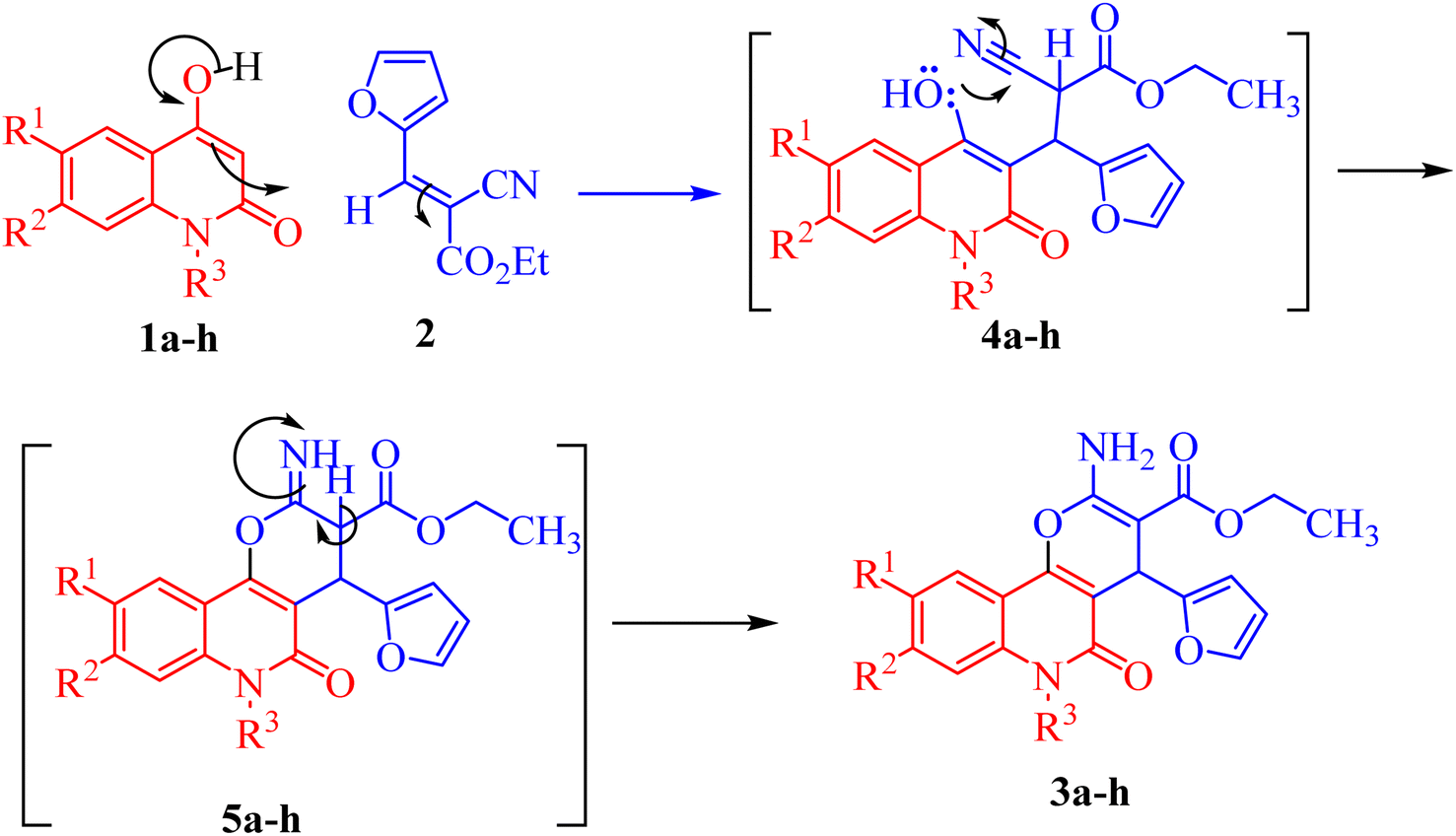 | ||
| Scheme 2 Mechanism describing the formation of compounds 3a–h. | ||
2.2. Biological screening
| Compound | Structures | A549 | A549 |
|---|---|---|---|
| IC50 (μg mL−1) | IC50 (μM) | ||
| 3a | 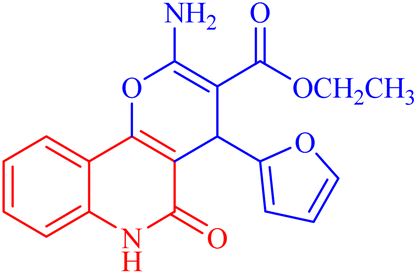 |
122.9 ± 3.6 | 348.8009 |
| 3b | 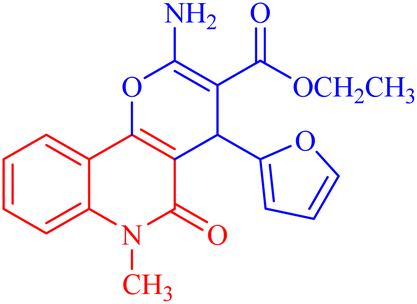 |
127.5 ± 1.1 | 440.8112 |
| 3c | 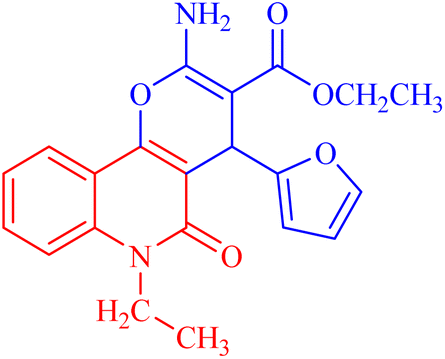 |
131.55 ± 3.2 | 417.9811 |
| 3d | 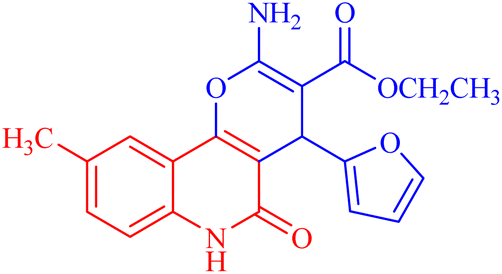 |
47.465 ± 3.06 | 116.194 |
| 3e | 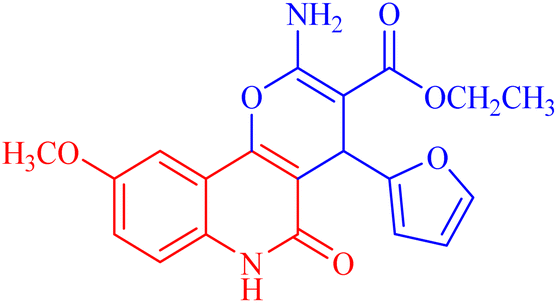 |
53.285 ± 0.815 | 139.3153 |
| 3f | 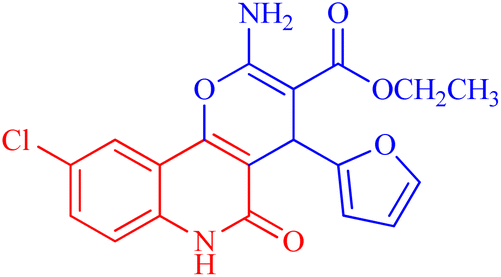 |
30.96 ± 1.47 | 78.56977 |
| 3g | 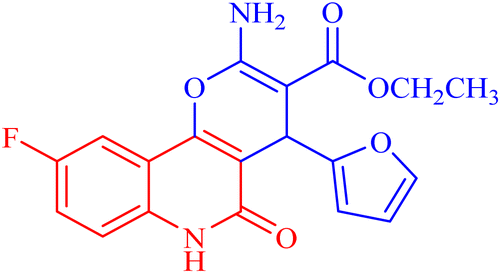 |
45.97 ± 0.87 | 125.6143 |
| 3h | 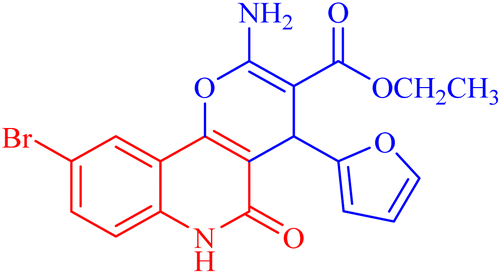 |
15.64 ± 1.02 | 35.10806 |
| Levofloxacin | — | 148.2 ± 1.42 | 410.10 |
Besides considering cytotoxicity against A549 cells, testing safety on normal cells WI38 and selectivity to cancer cells is the main evaluation factor of the studied pyranoquinolones. Interestingly, the most active derivative 3h exhibited lower cytotoxic activity against normal cells (IC50 = 43.28 μM) with a selectivity index of 1.23.
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 3h | 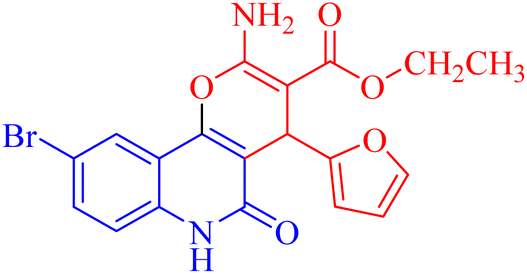 |
45.19 ± 3.37 |
| Etoposide | — | 34.99 ± 2.58 |
| Compound | Inhibition zone diameter (mm) DMSO control (8 mm) |
|---|---|
| 3a | 8 |
| 3b | 8 |
| 3c | 8 |
| 3d | 10 ± 0.5 |
| 3e | 9 ± 0.3 |
| 3f | 17.5 ± 0.85 |
| 3g | 18 ± 1 |
| 3h | 19 ± 1.1 |
| Compound | MIC (μg mL−1) | MIC (μM) |
|---|---|---|
| 3f | 78.125 | 201.983 |
| 3g | 78.125 | 210.954 |
| 3h | 39.062 | 90.580 |
| Spiramycin30 | 32 | 37.957 |
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 3h | 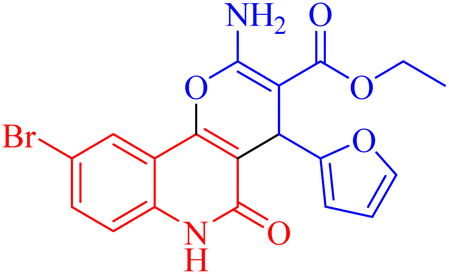 |
40.76 ± 0.64 |
| Levofloxacin | — | 31.34 ± 0.45 |
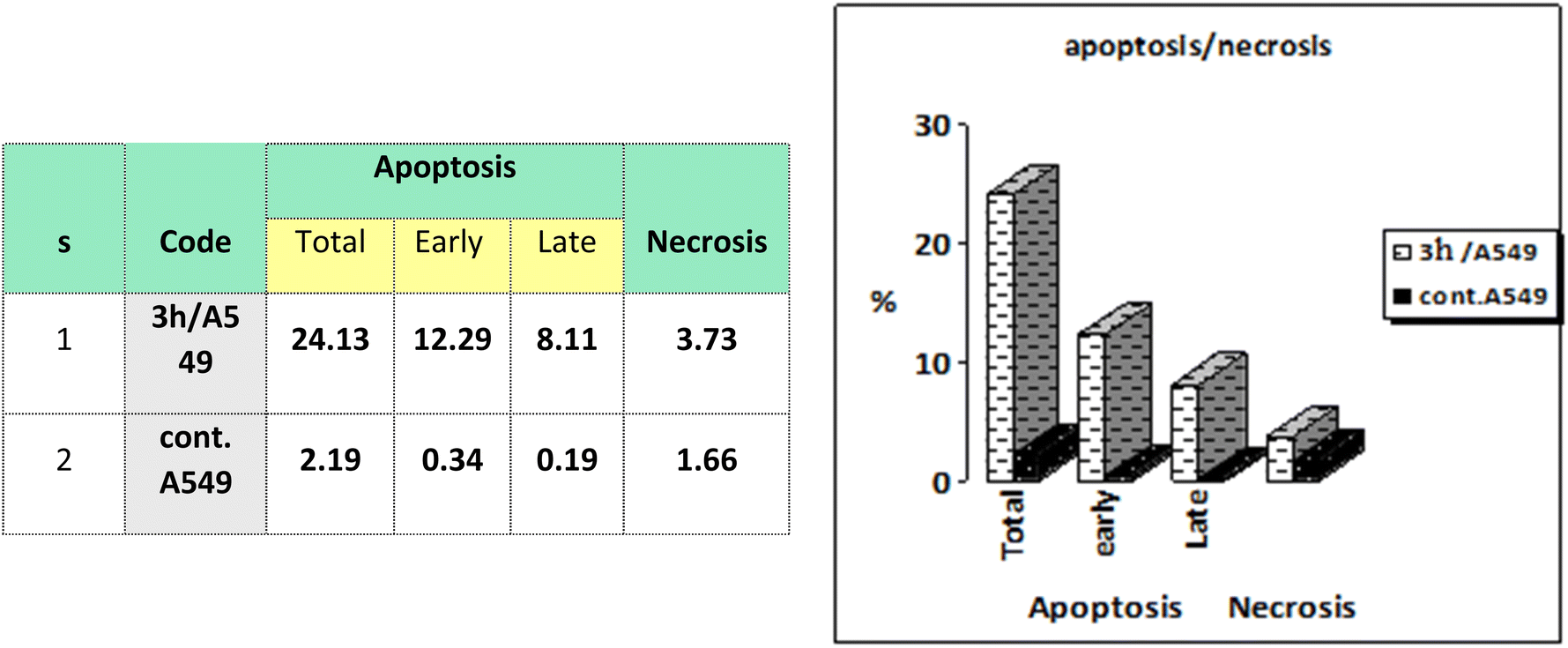 | ||
| Fig. 7 Representation of A549 cells treated with 3h and analyzed using flow cytometry after double staining of the cells with annexin-V FITC and PI. | ||
Cell cycle analysis is a crucial test that investigates the percentages of the cell population in each cell phase with cytotoxic substances after treatment. Lung epithelial cancer cells A549 were treated with compound 3h. It was subjected to DNA flow cytometry to determine at which cell cycle the cell proliferation was arrested. As shown in Fig. 8, the compound treatment significantly increased the cell population at the G1 phase by 63.39% compared to the control 57.02%. In comparison, the other phases did not significantly change.
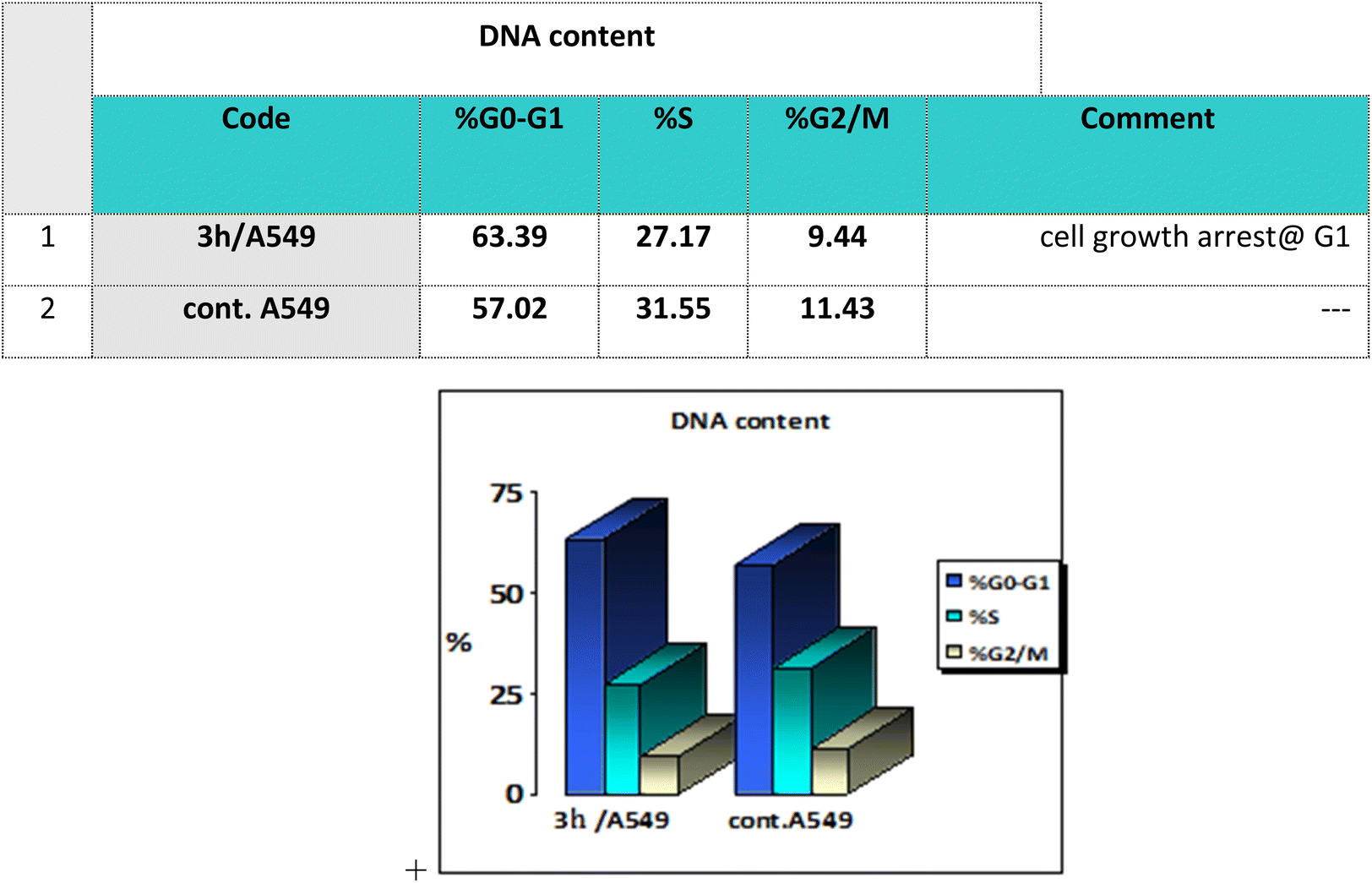 | ||
| Fig. 8 Bar representation of the percentage of cell population at each cell cycle G1, S and G2/M. | ||
2.3. Molecular docking
A computational study utilizing MOE 2019.10259 was employed to predict the binding scores and modes of the investigated derivatives 3a–h into the target proteins topoisomerase II (PDB: 5GWK31) and DNA gyrase (PDB: 2XCT32) compared to co-crystallized ligands etoposide and ciprofloxacin, respectively.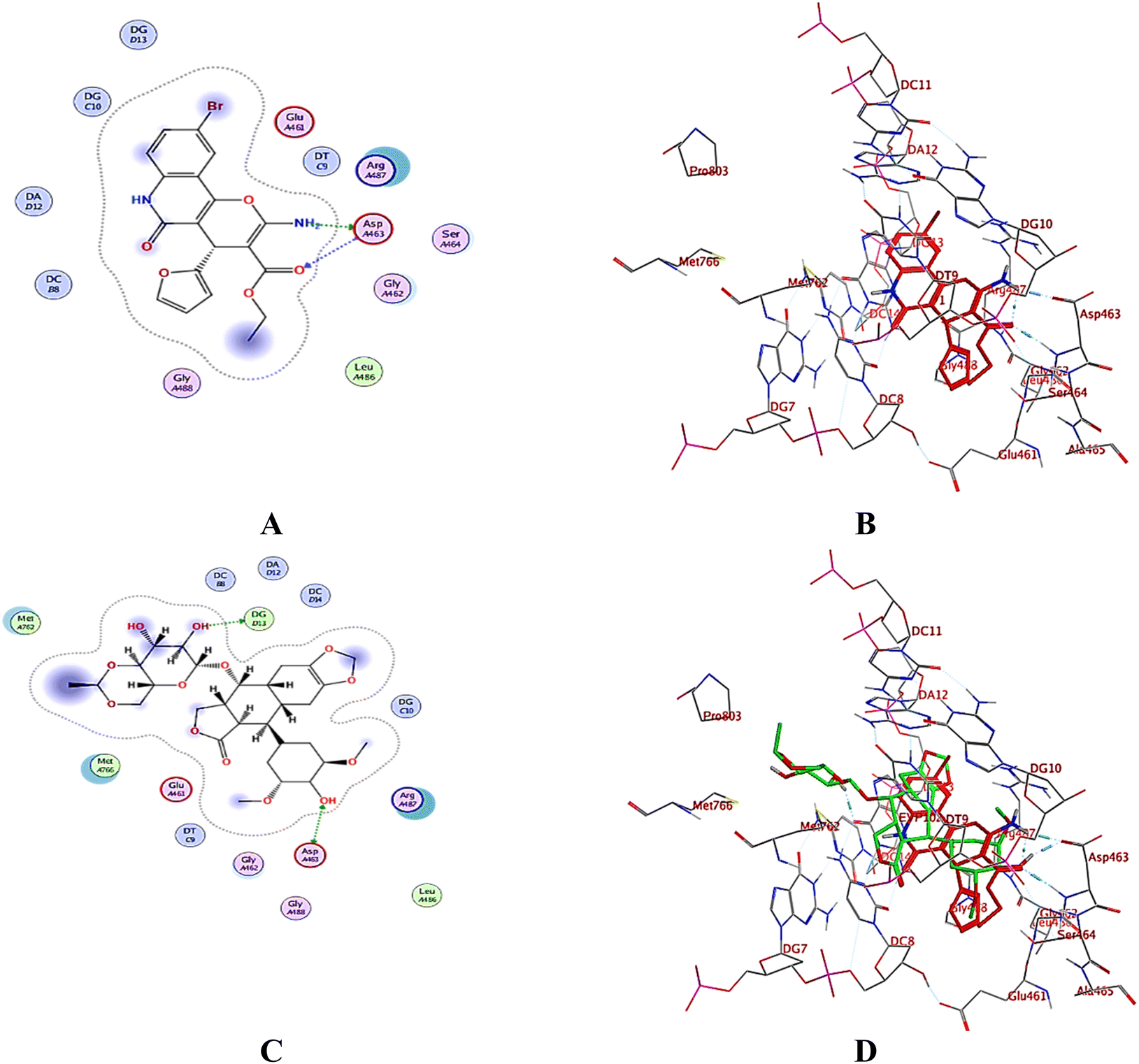

2.4. In silico drug likeness and molecular property prediction
Molecular property prediction is becoming a useful tool in the generation of molecules with the correct parameters to be useful drug candidates. About 40% of oral drugs fail in clinical trials because of their poor pharmacokinetic properties. Drug development involves the assessment of absorption, distribution, metabolism and excretion (ADME) increasingly earlier in the discovery process, at a stage when the considered compounds are numerous, but access to physical samples is limited. Thus, we applied computational methods to predict physical and molecular properties to assess their availability as useful drug candidates. Here, we present a new SwissADME web tool that provides free access to a pool of fast yet robust predictive models for physicochemical properties, pharmacokinetics, drug-likeness and medicinal chemistry friendliness, including in-house proficient methods such as iLOGP and Bioavailability Radar (Table 9).| ID | Ma | LogPb |
HBDc | HBAd | Nrotbe | TPSAf | MRg | LogSh |
Fi | GI absorption | BBBj | Pgpk substrate |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a M, molecular weight (dalton).b ilogP, octanol/water partition coefficient.c HBD, hydrogen bond donor.d HBA, hydrogen bond acceptor.e Nrotb, # of rotatable bonds.f TPSA, total polar surface area.g MR, molar refractivity.h ilogP, logarithm of compound aqueous solubility.i F, Abbott oral bioavailability score HIA%, human gastrointestinal absorption.j BBB permeant, blood–brain barrier penetration.k Pgp, permeability glycoprotein. |
||||||||||||
| 3a | 352.34 | 2.44 | 2 | 5 | 4 | 107.55 | 93.41 | −3.47 | 0.56 | High | No | No |
| 3b | 366.37 | 3.15 | 1 | 5 | 4 | 96.69 | 82.02 | −2.31 | 0.56 | High | No | No |
| 3c | 380.39 | 3.38 | 1 | 5 | 5 | 96.69 | 103.12 | −3.89 | 0.56 | High | No | No |
| 3d | 366.37 | 3.04 | 2 | 5 | 4 | 107.55 | 98.38 | −3.77 | 0.56 | High | No | Yes |
| 3e | 382.37 | 2.75 | 2 | 6 | 5 | 116.78 | 99.9 | −3.54 | 0.56 | High | No | Yes |
| 3f | 386.79 | 2.75 | 2 | 5 | 4 | 107.55 | 98.42 | −4.06 | 0.56 | High | No | No |
| 3g | 370.33 | 2.81 | 2 | 6 | 4 | 107.55 | 93.37 | −4.03 | 0.56 | High | No | No |
| 3h | 431.24 | 3.17 | 2 | 5 | 4 | 107.55 | 101.11 | −4.38 | 0.56 | High | No | No |
All the compounds had high GI absorption and obeyed the Lipinski rule of five. The overall results showed that the tested compound showed a very good drug-likeness pharmacokinetic and pharmacodynamic profile.
3. Conclusion
A series of new pyrano[3,2-c]quinoline-3-carboxylate derivatives 3a–h were designed and synthesized efficiently using a microwave. The structure of the products was confirmed using a combination of spectral techniques, including infra-red (IR), nuclear magnetic resonance (NMR), mass spectrometry (MS) and elemental analyses, in addition to X-ray structure analysis. The newly synthesized compounds were evaluated for their in vitro antiproliferative activity against lung epithelial cancer cells A549 using an MTT viability assay. Compound 3h showed the highest activity with an MIC of 15.14 μg ml−1 compared to the positive control Levofloxacin. The cell cycle arrest behavior detected by propidium iodide and the apoptosis induction was investigated. In addition, the newly synthesized compounds were evaluated for their in vitro antibacterial activity against the standard Staphylococcus aureus strain. Compound 3h showed the highest antibacterial activity with an MIC of 39.062 μg ml−1 using Levofloxacin as the standard reference drug. An in silico study was performed, including docking of the newly synthesized compounds into topoisomerase II and DNA-gyrase binding pockets, in addition to predicting their physicochemical and pharmacokinetic properties. In silico studies showed the ability of the synthesized members to bind to the topoisomerase II and DNA-gyrase. Our study revealed that compound 3h is a promising dual acting anticancer and antibacterial agent targeting topoisomerase II and DNA-gyrase with acceptable oral bioavailability and physicochemical and pharmacokinetic properties.4. Experimental
4.1. Chemistry
Melting points were measured in open capillaries using a Gallenkamp melting point apparatus (Weiss-Gallenkamp, Loughborough, UK) and were uncorrected. The IR spectra were recorded by applying the ATR technique (ATR = Attenuated Total Reflection) with an FT device (FT-IR Bruker IFS 88), Institute of Organic Chemistry, Karlsruhe University, Karlsruhe, Germany. The NMR spectra were measured in DMSO-d6 using a Bruker AV-400 spectrometer, 400 MHz for 1H, and 100 MHz for 13C. The chemical shifts are expressed in δ (ppm) versus internal tetramethylsilane (TMS) = 0 for 1H and 13C. The description of signals includes s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, and m = multiplet. The following abbreviations were used to distinguish between signals: Ar-H = aromatic-CH. Signals of the 13C NMR spectra were assigned with the help of DEPT90 and DEPT135 and were specified in the following way: + = primary or tertiary carbon atoms (positive DEPT signal), − = secondary carbon atoms (negative DEPT signal), and Cq = quaternary carbon atoms (no DEPT signal). Correlations were established using 1H–1H COSY, 1H–13C HSQC and HMBC experiments. Mass spectra were recorded using a FAB (fast atom bombardment) Thermo Finnigan Mat 95 (70 eV). The HRSM was recorded using LC/Q-TOF, 6530 (Aligent Technologies, Santa Clara, CA, USA) at the Natural Product Research lab, Faculty of Pharmacy, Fayoum University, Egypt. Elemental analyses were carried out at the Microanalytical Center, Cairo University, Egypt. TLC was performed on analytical Merck 9385 silica aluminum sheets (Kieselgel 60) with Pf254 indicator; TLCs were viewed at λmax = 254 nm.4.1.2.1. Method I. A mixture of 1a–h (1 mmol) and 2 (1 mmol, 0.138 g) in 40 ml absolute ethanol and anhydrous K2CO3 (1.5 mmol, 0.192 g) was stirred at room temperature for 12–24 h. The reaction was monitored through TLC analysis. The formed products 3a–h were filtered off and washed several times with H2O (150 ml) and with ether (30 ml). The obtained products were recrystallized from the stated solvents to afford pure compounds 3a–h.
4.1.2.2. Method II. The above method was repeated by exposing the reaction mixture to MW irradiation for 3–10 min. The reaction was refluxed in a Milestone Microwave Lab station at 120 °C for 3–10 min. All reactions were monitored by TLC with 1
:1 ethyl acetate/petroleum ether as an eluent and were carried out until the starting materials were completely consumed. After few minutes (Table 1), microwave irradiation was stopped, and the reaction mixture was analyzed by TLC.
4.1.2.2.1 Ethyl 2-amino-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3a). Compound 3a was obtained as pale brown crystals, (DMF/EtOH); yield: m.p. 270–272 °C; IR (KBr): νmax/cm−1 = 3460–3463 (NH2), 3325 (NH), 3100 (CH-Ar), 2975 (CH-Aliph), 1692 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ester), 1656 (CO, quinolone), 1578 (CC), 1437 (CH2), 1375 (CH3). NMR (see Table 2). MS (FAB, 3-NBA), m/z (%): 352.1 [M+] (18), 353.1 [M + 1] (22), 307.1 (100), 289.1 (35). Anal. calcd C19H16N2O5 (352.35): C, 64.77; H, 4.58; N, 7.95. Found: C, 64.85; H, 4.48; N, 8.10.
O, ester), 1656 (CO, quinolone), 1578 (CC), 1437 (CH2), 1375 (CH3). NMR (see Table 2). MS (FAB, 3-NBA), m/z (%): 352.1 [M+] (18), 353.1 [M + 1] (22), 307.1 (100), 289.1 (35). Anal. calcd C19H16N2O5 (352.35): C, 64.77; H, 4.58; N, 7.95. Found: C, 64.85; H, 4.48; N, 8.10.
4.1.2.2.2 Ethyl 2-amino-4-(furan-2-yl)-6-methyl-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3b). Compound 3b was obtained as pale brown crystals, (DMF/EtOH); yield: 92%; m.p. 260–262 °C. IR (KBr): νmax/cm−1 = 3275 (NH), 3050 (CH-Ar), 2928 (CH-Aliph), 1682 (C
O, ester), 1654 (CO, quinolone), 1614 (CC), 1463 (CH2), 1418 (N–CH3), 1375 (CH3). NMR (see Table 3). MS (FAB, 3-NBA), m/z (%): 366.1 [M]+ (35), 367.1 [M + 1] (45). HRSM [M + Na+] = calcd: 389.1114; found: 389.1124. Anal. calcd for C20H18N2O5 (366.37): C, 65.57; H, 4.95; N, 7.65. Found: C, 65.47; H, 4.85; N, 7.70.
4.1.2.2.3 Ethyl 2-amino-6-ethyl-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3c). Compound 3c was obtained as pale brown crystals, (DMF/EtOH), yield: 94%; m.p. 264–266 °C. IR (KBr): νmax/cm−1 = 3268 (NH), 3056 (CH-Ar), 2828 (CH-Aliph), 1767 (C
O, pyranone), 1655 (CO, ester), 1541 (CO, quinolone), 1485 (CH2), 1375 (CH3). 1H NMR (DMSO-d6, ppm): δH = 8.08 (dd, J = 7.9, 0.8 Hz, 1H; H-10), 7.76 (b, 2H; NH-2a), 7.64 (ddd, J = 8.3, 7.0, 1.00 Hz, 1H; H-8), 7.57 (d, J = 8.4 Hz, 1H; H-7), 7.34 (dd, J = 8.3, 7.5 Hz, 1H; H-9), 7.33 (bs; H-5′), 6.26 (dd, J = 3.0, 1.9 Hz, 1H; H-3′), 6.03 (d, J = 3.0 Hz, 1H; H-4′), 5.06 (s, 1H; H-4), 4.34–4.22 (m, 4H, 2×CH2), 1.27 (t, J = 7.1 Hz, 3H, H-3c), 1.20 (t, J = 7.0 Hz, 3H, H-6c). 13C NMR (DMSO-d6, ppm): δC = 167.6 (C-3a), 159.9, 159.9 (C-2, C-2′), 156.3 (C-5), 150.9 (C-10b), 141.0 (C-5′), 138.6 (C-6a), 131.4 (C-8), 122.1 (C-9, C-10), 114.9 (C-7), 112.8, 110.3, 108.9 (C-3′, C-4′, C-10a), 105.1 (C-4a), 74.6 (C-3), 58.9 (C-3b), 38.4 (C-6b), 28.9 (C-4), 14.3 (C-3c) 12.6 (C-6c). MS (FAB, 3-NBA), m/z (%): 379.06 [M+ − 1] (35), 382.54 [M + 2]+ (18), 267.07 (100). HRSM [M + Na+] = calcd: 403.1270; found: 403.1281. Anal. calcd for C21H20N2O5 (380.40): C, 66.31; H, 5.30; N, 7.36. Found: C, 66.35; H, 5.28; N, 7.46.
4.1.2.2.4 Ethyl 2-amino-4-(furan-2-yl)-9-methyl-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3d). Compound 3d was obtained as brown crystals (DMF); yield; 92%; m.p. 280–282 °C. IR (KBr): νmax/cm−1 = 3482–3435 (NH2), 3266 (NH), 3090 (CH-Ar), 2840 (CH-Aliph), 1737 (C
O, pyranone), 1695 (CO, ester), 1521 (CO, quinolone), 1415 (CH2), 1325 (CH3). 1H NMR (DMSO-d6, ppm): δH = 11.73 (s, 1H, NH), 7.96 (s, 1H; H-10), 7.77 (b, 2H, NH2), 7.58 (d, J = 8.1, 7.3 Hz, 1H, H-8), 7.35 (d, J = 0.8 Hz, 1H, H-5′), 7.38 (d, J = 8.2 Hz, 1H, H-7), 6.25 (d, J = 1.8 Hz, 1H, H-3′), 6.02 (d, J = 2.0 Hz, 1.0 Hz, 1H, H-4′), 5.06 (s, 1H, H-4), 4.09 (q, J = 7.05 Hz, 2H, H-3b), 2.24 (s, 3H; H-9a), 1.20 (t, J = 7.06 Hz, 3H, H-3c). 13C NMR (DMSO-d6, ppm): δC = 167.6 (C-3a), 160.6, 159.1 (C-2, C-2′), 156.3 (C-5), 151.9 (C-10b), 141.1(C-5′), 137.6 (C-6a), 131.0 (C-9), 122.6, 122.6 (C-8, C-10), 115.4 (C-7), 112.2, 110.2, 109.5 (C-3′, C-4′, C-10a), 105.0 (C-4a), 74.7 (C-3), 58.9 (C-3b), 28.22 (C-4), 20.3 (C-9a), 14.3 (C-3c). MS (FAB, 3-NBA), m/z (%): 366.46 [M]+ (50), 293.25 [M]+ (100), 253.22 (100), 224.24 (100). HRMS [M + Na+] = calcd: 389.1114; found: 389.1121. Anal. calcd for C20H18N2O5 (366.37): C, 65.57; H, 4.95; N, 7.65. Found: C, 64.57; H, 4.85; N, 7.61.
4.1.2.2.5 Ethyl 2-amino-4-(furan-2-yl)-9-methoxy-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3e). Compound 3e was obtained as brown crystals, (DMF/EtOH); yield: 88%; m.p. 390–292 °C. IR (KBr): νmax/cm−1 = 3432–3475 (NH2), 3268 (NH), 3056 (CH-Ar), 2828 (CH-Aliph), 1767 (C
O, pyranone), 1655 (CO, ester), 1541 (CO, quinolone), 1485 (CH2), 1375 (CH3). 1H NMR (DMSO-d6, ppm): δH = 12.23 (s, 1H, NH), 7.98 (s, 1H; H-10), 7.79 (b, 2H, NH2), 7.61 (d, J = 8.2 Hz, 7.4, 1H, H-8), 7.38 (d, J = 0.7 Hz, 1H, H-5′), 7.41 (d, J = 8.0 Hz, 1H, H-7), 6.27 (d, J = 1.8 Hz, 1H, H-3′), 6.05 (d, J = 2.1 Hz, 1.0, 1H, H-4′), 5.04 (s, 1H, H-4), 4.06 (q, J = 7.0 Hz, 2H, H-3b), 3.71 (s, 3H; H-9a), 1.18 (t, J = 7.0 Hz, 3H, H-3c).13C NMR (DMSO-d6, ppm): δC = 167.1 (C-3a), 161.6, 161.5 (C-2, C-5), 158.3 (C-9, C-10b), 154.9 (C-2′), 145.7 (C-5′), 135.0 (C-6a), 124.7 (C-7), 118.0 (C-10a), 114.9, 112.7, 110.7 (C-8, C-10, C-4′), 107.8 (C-3′), 103.1 (C-4a), 75.7 (C-3), 61.3 (C-3b), 56.9 (C-9a), 28.2 (C-4), 14.6 (C-3c). MS (FAB, 3-NBA), m/z (%): 382.15 [M + 1] (30), 175.93 [M]+ (45). HRMS [M + H+] = calcd: 383.1243; found: 383.1227. Anal. calcd for C20H18N2O6 (382.37): C, 62.82; H, 4.75; N, 7.33. Found: C, 62.72; H, 4.55; N, 7.53.
4.1.2.2.6 Ethyl 2-amino-9-chloro-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3f). Compound 3f was obtained as brown crystals, (DMF/CH3OH); yield: 80%; m.p. 310–312 °C. IR (KBr): νmax/cm−1 = 3432–3475 (NH2), 3222 (NH), 3010 (CH-Ar), 2812 (CH-Aliph), 1761 (C
O, pyranone), 1650 (CO, ester), 1540 (CO, quinolone), 1475 (CH2), 1335 (CH3). 1H NMR (DMSO-d6, ppm): δH = 12.42 (s, 1H, NH), 7.98 (d, J = 8.1 Hz, 1H, H-7), 7.81 (b, 2H, NH2), 7.41 (s, 1H, H-10), 7.37 (d, J = 0.9 Hz, 1H, H-5′), 7.32 (d, J = 8.1 Hz, 1H, H-8), 6.25 (d, J = 1.7 Hz, 1H, H-3′), 6.01 (d, J = 2.0 Hz, 1.0, 1H, H-4′), 5.02 (s, 1H, H-4), 4.02 (q, J = 7.04 Hz, 2H, H-3b), 1.16 (t, J = 7.03 Hz, 3H, H-3c). 13C NMR (DMSO-d6, ppm): δC = 167.9 (C-3a), 160.5, 160.3 (C-2, C-2′), 158.4(C-5), 155.0 (C-10b), 142.7 (C-5′), 135.2 (C-6a), 131.2 (C-9), 128.9, 126.6 (C-8, C-10), 122.6 (C-7), 114.2, 110.7, 108.7 (C-10a, C-4′, C-3′), 100.6(C-4a), 75.6 (C-3), 61.3 (C-3b), 28.5(C-4), 14.1 (C-3c). MS (FAB, 3-NBA), m/z (%): 386.07 [M + 1] (10), 366.57 [M]+ (15). HRMS [M + H+] = calcd: 387.0747; found: 387.0617. Anal. calcd for C19H15ClN2O5 (386.79): C, 59.00; H, 3.91; Cl, 9.17; N, 7.24. Found: C, 59.08; H, 3.95; Cl, 9.21; N, 7.30.
4.1.2.2.7 Ethyl 2-amino-9-fluoro-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3g). Compound 3g was obtained as pale brown crystals, (DMF/EtOH); yield: 78%; m.p. 302–304 °C. IR (KBr): νmax/cm−1 = 3442–3455 (NH2), 3261 (NH), 3052 (CH-Ar), 2820 (CH-Aliph), 1760 (C
O, pyranone), 1659 (CO, ester), 1549 (CO, quinolone), 1445 (CH2), 1355 (CH3). 1H NMR (DMSO-d6, ppm): δH = 12.45 (s, 1H, NH), 7.88 (d, J = 8.2 Hz, 1H, H-7), 7.81 (b, 2H, NH2), 7.37 (d, J = 0.8 Hz, 1H, H-5′), 7.20 (d, J = 8.2 Hz, 1H, H-8) 7.02 (s, 1H, H-10), 6.27 (d, J = 1.7 Hz, 1H, H-3′), 6.02 (d, J = 2.01 Hz, 1.0, 1H, H-4′), 5.04 (s, 1H, H-4), 4.03 (q, J = 7.01 Hz, 2H, H-3b), 1.14 (t, J = 7.01 Hz, 3H, H-3c). 13C NMR (DMSO-d6, ppm): δC = 167.6 (C-3a), 160.6, 160.1 (C-2, C-9), 156.3 (C-5), 152.9, 152.6 (C-10b, C-2′), 141.9 (C-5′), 135.7 (C-6a), 118.0 (C-10a), 115.7, (C-8), 112.6 (C-7), 111.4, 111.3 (C-10, C-4′), 106.0 (C-3′), 100.9 (C-4a), 74.7 (C-3), 59.9 (C-3b), 28.3 (C-4), 14.3 (C-3c). MS (FAB, 3-NBA), m/z (%): 372.84 [M + 2] (10), 179.25 [M]+ (85). HRMS [M + H+] = calcd: 371.1044; found: 371.1054. Anal. calcd for C19H15FN2O5 (370.34): C, 61.62; H, 4.08; F, 5.13; N, 7.56. Found: C, 61.60; H, 4.06; F, 5.12; N, 7.50.
4.1.2.2.8 Ethyl 2-amino-9-bromo-4-(furan-2-yl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carboxylate (3h). Compound 3h was obtained as brown crystals, (CHCl3/EtOH); yield: 85%; m.p. 320–322 °C. IR (KBr): νmax/cm−1 = 3462–3465 (NH2), 3330 (NH), 3000 (CH-Ar), 2965 (CH-Aliph), 1690 (C
O, ester), 1652 (CO, quinolone), 1574 (CC), 1433 (CH2), 1370 (CH3). 1H NMR (DMSO-d6, ppm): δH = 11.93 (s, 1H, NH), 7.98 (s, 1H, H-10), 7.81 (b, 2H, NH2), 7.37 (d, J = 0.8 Hz, 1H, H-5′), 7.56 (d, J = 8.2 Hz, 1H, H-7), 7.42 (d, J = 8.3 Hz, 1H, H-8), 6.18 (d, J = 1.8 Hz, 1H, H-3′), 6.03 (d, J = 2.0, 1.0 Hz, 1H, H-4′), 5.01 (s, 1H, H-4), 4.08 (q, J = 7.1 Hz, 2H, H-3b), 1.16 (t, J = 7.1 Hz, 3H, H-3c). 13C NMR (DMSO-d6, ppm): δC = 167.3 (C-3a), 160.5, 160.5 (C-2, C-2′), 158.4 (C-5), 155.0 (C-10b), 142.8 (C-5′), 138.2 (C-6a), 132.2 (C-8), 126.7 (C-10), 121.6, 121.5 (C-7, C-10a), 115.2, 110.7, 106.7 (C-9, C-4′, C-3′), 100.5 (C-4a), 75.5 (C-3), 61.3 (C-3b), 28.5 (C-4), 14.1 (C-3c). MS (FAB, 3-NBA), m/z (%): 431.64 [M]+ (30), 149.78 [M]+ (100). Anal. calcd for C19H15BrN2O5 (431.24): C, 52.92; H, 3.51; Br, 18.53; N, 6.50. Found: C, 53.02; H, 3.48; Br, 18.55; N, 6.40.
4.2. Biological screening
:100 in double strength cation adjusted Mueller–Hinton broth to reach 1-2x 106 CFU ml−1. After that, 100 ml of the prepared bacterial suspension was transferred with equal volume to the serially diluted compounds (100 μl) present in the 96 well plates. This leads to a final bacterial density in the wells of 5–10 × 105 CFU ml−1. Additional wells for broth growth positive and negative controls containing either only bacterial suspension or uninoculated broth were included in each test. The plates were then incubated at 37 °C for 18 h. The plates were read using a microplate reader (Biotek, USA), and the MIC was determined to be the lowest concentration that inhibited bacterial growth. For solvent control, the MIC of the used DMSO was determined using the same experimental settings, as described above. Each experiment was performed at least twice.CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Eleventh Edition. CLSI document M07. Wayne, PA: Clinical and Laboratory Standards Institute. 2018.
:100) and propidium iodide (PI)” at a concentration of 10 μg ml−1 in the dark for 30 min. The stained cells were then acquired by applying a Cytoflex FACS machine. Data were analyzed using cytExpert software.364.3. Molecular docking
The docking scores and binding modes of the newly synthesized compounds with the target proteins (DNA gyrase and topoisomerase II) were predicted by employing the MOE 2019.10259 software.4.4. Investigated derivative preparation
First, Chemdraw software was employed to draw the tested compounds, which were then imported to an optimized database, as previously discussed.374.5. Protein preparation
3D crystal structures of the target proteins (PDB: 5GWK31 and PDB: 2XCT32) were obtained from the protein data bank website,38 followed by structure preparation and optimization for established docking, as described in detail.35 Subsequently, docking of the assessed derivatives 3a–h to the target protein complex was carried out by applying the default docking protocol. For each docked structure, the pose with prominent amino acid interactions, the best docking score, and RMSD were selected and visualized.Data availability
Data will be made available on request. CCDC 2312660 (3a, https://www.ccdc.cam.ac.uk/structures/search?Ccdc=2312660&Author=Nieger&Access=referee) and 2312661 (3b, https://www.ccdc.cam.ac.uk/structures/search?Ccdc=2312661&Author=Nieger&Access=referee) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
A. A. Aly: conceptualization, writing, and editing; H. A. Abd El-Naby, E. K. Ahmed: supervision; S. A. Gedamy: methodology, writing the draft; M. Nieger, K. Rissanen: X-ray, writing draft; Alan B. Brown: editing; Marwa M. Shaaban, Amal Atta: biology, molecular modeling, writing and editing; Michael G. Shehat: biology.Conflicts of interest
The authors declare no conflict of interest.References
- A. A. Sharhan, A.-M. Ahmed Neema, S. A.-A. Rawaa and H. H. Kzar, South Asian Res. J. Pharm. Sci., 2023, 5, 186–191 CrossRef.
- C. Duan, M. Yu, J. Xu, B.-Y. Li, Y. Zhao and R. K. Kankala, Biomed. Pharmacother., 2023, 162, 114643 CrossRef CAS PubMed.
- M. H. A. Al-Jumaili, A. A. Hamad, H. E. Hashem, A. D. Hussein, M. J. Muhaidi, M. A. Ahmed, A. H. A. Albanaa, F. Siddique and E. A. Bakr, J. Mol. Struct., 2023, 1271, 133970 CrossRef CAS.
- J. A. Barta, C. A. Powell and J. P. Wisnivesky, Ann. Glob. Health, 2019, 22(85), 8 CrossRef.
- M. Nagasaka and S. M. Gadgeel, Expert Rev. Anticancer Ther., 2018, 18, 63–70 CrossRef CAS PubMed.
- C. O. Okoro and T. H. Fatoki, Int. J. Mol. Sci., 2023, 24, 2532 CrossRef CAS.
- A. Vaidya, S. Jain, A. K. Jain, B. Prashanthakumar, S. K. Kashaw and R. K. Agrawal, Med. Chem. Res., 2015, 24, 383–393 CrossRef CAS.
- E. Baldwin and N. Osheroff, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 363–372 CrossRef CAS PubMed.
- K. D. Upadhyay, N. M. Dodia, R. C. Khunt, R. S. Chaniara and A. K. Shah, ACS Med. Chem. Lett., 2018, 9, 283–288 CrossRef CAS PubMed.
- F. A. Almalki, Saudi Pharm. J., 2023, 31, 998–1018 CrossRef CAS PubMed.
- H.-G. Fu, Z.-W. Li, X.-X. Hu, S.-Y. Si, X.-F. You, S. Tang, Y.-X. Wang and D.-Q. Song, Molecules, 2019, 24, 548 CrossRef PubMed.
- M. Ramadan, M. Abd El-Aziz, Y. A. Elshaier, B. G. Youssif, A. B. Brown, H. M. Fathy and A. A. Aly, Bioorg. Chem., 2020, 105, 104392 CrossRef CAS.
- V. Pradhan, R. Kumar, A. Mazumder, M. M. Abdullah, M. Shahar Yar, M. J. Ahsan and Z. Ullah, Chem. Biol. Drug Des., 2023, 101, 977–997 CrossRef CAS.
- S. Grossman, C. W. Fishwick and M. J. McPhillie, Pharmaceuticals, 2023, 16, 261 CrossRef CAS PubMed.
- Y. Wei, E. Sandhu, X. Yang, J. Yang, Y. Ren and X. Gao, Microorganisms, 2022, 10(12), 2353 CrossRef CAS.
- M. Koslow, G. E. Shochet, A. Matveychuk, L. Israeli-Shani, A. Guber and D. Shitrit, J. Thorac. Dis., 2017, 9, 5300–5305 CrossRef.
- N. Gotland, M. L. Uhre, H. Sandholdt, N. Mejer, L. F. Lundbo, A. Petersen, A. R. Larsen and T. Benfield, Medicine, 2020, 99, e19984 CrossRef CAS PubMed.
- K. Hattar, C. P. Reinert, U. Sibelius, M. Y. Gökyildirim, F. S. B. Subtil, J. Wilhelm, B. Eul, G. Dahlem, F. Grimminger, W. Seeger and U. Grandel, Cancer Immunol., Immunother., 2017, 66, 799–809 CrossRef CAS.
- J. L. Qi, J. R. He, C. B. Liu, S. M. Jin, R. Y. Gao, X. Yang, H. M. Bai and Y. B. Ma, MedComm, 2020, 1, 188–201 CrossRef.
- S. Asghari, S. Ramezani and M. Mohseni, Chin. Chem. Lett., 2014, 25, 431–434 CrossRef CAS.
- M. Jayagobi, R. Raghunathan, S. Sainath and M. Raghunathan, Eur. J. Med. Chem., 2011, 46, 2075–2082 CrossRef CAS PubMed.
- B. S. D. Mathada and M. B. H. Mathada, Chem. Pharm. Bull., 2009, 57, 557–560 CrossRef CAS.
- R. Watpade, A. Bholay and R. Toche, J. Heterocycl. Chem., 2017, 54, 3434–3439 CrossRef CAS.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, MedChemComm, 2019, 10, 1719–1739 RSC.
- H. M. Abd El-Lateef, A. A. Elmaaty, L. M. Abdel Ghany, M. S. Abdel-Aziz, I. Zaki and N. Ryad, ACS Omega, 2023, 11(20), 17948 CrossRef PubMed.
- M. A. I. Elbastawesy, F. A. M. Mohamed, I. Zaki, M. I. Alahmdi, S. S. Alzahrani, H. A. Alzahrani, H. A. M. Gomaa and B. G. M. Youssif, J. Mol. Struct., 2023, 1278, 134902 CrossRef CAS.
- A. C. Spencer and S. S. Panda, Biomedicines, 2023, 11, 371 CrossRef CAS.
- T. Khan, K. Sankhe, V. Suvarna, A. Sherje, K. Patel and B. Dravyakar, Biomed. Pharmacother., 2018, 103, 923–938 CrossRef CAS.
- Meyler's Side Effects of Drugs, ed. J. K. Aronson, Elsevier, Oxford, 16th edn, 2016, pp. 470–471, DOI:10.1016/B978-0-444-53717-1.01465-7.
- L. Oliveira, H. Langoni, C. Hulland and P. Ruegg, J. Dairy Sci., 2012, 95, 1913–1920 CrossRef CAS.
- Y. R. Wang, S. F. Chen, C. C. Wu, Y. W. Liao, T. S. Lin, K. T. Liu, Y. S. Chen, T. K. Li, T. C. Chien and N. L. Chan, Nucleic Acids Res., 2017, 45, 10861–10871 CrossRef CAS PubMed.
- B. D. Bax, P. F. Chan, D. S. Eggleston, A. Fosberry, D. R. Gentry, F. Gorrec, I. Giordano, M. M. Hann, A. Hennessy, M. Hibbs, J. Huang, E. Jones, J. Jones, K. K. Brown, C. J. Lewis, E. W. May, M. R. Saunders, O. Singh, C. E. Spitzfaden, C. Shen, A. Shillings, A. J. Theobald, A. Wohlkonig, N. D. Pearson and M. N. Gwynn, Nature, 2010, 466, 935–940 CrossRef PubMed.
- B. Bhudevi, P. V. Ramana, M. Mudiraj and A. R. Reddy, Indian J. Chem., Sect. B, 2009, 48, 255–260 Search PubMed.
- E. A. Mohamed, M. M. Ismail, Y. Gabr and M. Abass, Chem. Pap., 1994, 48, 285–292 CAS.
- S. Chandrasekaran, A. Abbott, S. Campeau, B. L. Zimmer, M. Weinstein, J. Hejna, L. Walker, T. Kirn and R. Patel, J. Clin. Microbiol., 2018, 56(3), 10–128 CrossRef PubMed.
- M. S. Nafie, S. M. Kishk, S. Mahgoub and A. M. Amer, Chem. Biol. Drug Des., 2022, 99, 547–560 CrossRef CAS PubMed.
- J. Y. Al-Humaidi, M. M. Shaaban, N. Rezki, M. R. Aouad, M. Zakaria, M. Jaremko, M. Hagar and B. H. Elwakil, Life, 2022, 12, 1341 CrossRef CAS.
- https://www.rcsb.org/.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2312660 and 2312661. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06201a |
| This journal is © The Royal Society of Chemistry 2025 |