
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenome mining of albocandins A–E from Streptomyces sp. YINM00030†
Zhou-Tian-Le Zhang‡
a,
Zhen Ren‡b,
Xiaoyu Sua,
Tian-Peng Xiea,
Mengzhuo Yia,
Hao Zhou *a,
Min Yin*a and
Zhong-Tao Ding*ac
*a,
Min Yin*a and
Zhong-Tao Ding*ac
aKey Laboratory of Functional Molecules Analysis and Biotransformation of Universities in Yunnan Province, Yunnan Characteristic Plant Extraction Laboratory, School of Chemical Science and Technology, School of Medicine, Yunnan University, Kunming, Yunnan 650500, PR China. E-mail: yinmin@ynu.edu.cn; haozhou@ynu.edu.cn; ztding@ynu.edu.cn
bSchool of Agriculture and Life Sciences, Kunming University, Kunming, Yunnan 650214, PR China
cCollege of Traditional Chinese Medicine, Yunnan University of Chinese Medicine, Kunming, Yunnan 650500, PR China
First published on 21st January 2025
Abstract
The natural products of 2,5-diketopiperazines have attracted considerable attention due to their potent pharmacological activities. Guided by genome mining techniques, five albonoursin analogues, designated as albocandins A–E (1–5), were isolated from Streptomyces sp. YINM00030, an actinomycete sourced from the rhizosphere soil of medicinal plants. The structures and absolute configurations of these compounds were elucidated through comprehensive spectroscopic analyses, HRESIMS, electronic circular dichroism (ECD) calculations, and single-crystal X-ray diffraction analyses. The biosynthetic pathway of these compounds were proposed. The investigation of biological activity showed that albocandins C and D exhibited cytotoxic activity against five human cancer cell lines (HL-60, A549, SMMC-7721, MDA-MB-231, SW480) in vitro with IC50 values ranging from 3.50 to 32.66 μM.
Introduction
Natural products of 2,5-diketopiperazine are predominantly derived from actinomycetes and fungi. These compounds are not only prevalent in nature but are also frequently generated as degradation products of polypeptides. Diketopiperazines typically exhibit remarkable pharmacological activities, including antibacterial,1 antitumor,2,3 antiviral,4 immunomodulatory,5 and antioxidant effects.6 Since the groundbreaking synthesis of the cyclic dipeptide cyclo(Gly–Gly) by Curtius et al. in 1888, numerous derivatives of diketopiperazine have been documented.7 Diketopiperazines possess several distinctive structural features. Firstly, their core structure comprises an internal lactam six-membered ring formed through the condensation of two α-amino acids via peptide bonds; this conformationally constrained heterocyclic scaffold confers stability against proteolytic degradation. Secondly, the rigid backbone can mimic a preferred peptide conformation while accommodating constrained amino acids within its structure without exhibiting undesirable physical or metabolic properties associated with peptides. Additionally, this six-membered ring contains two hydrogen bond donors and acceptors that facilitate interactions with various receptors. There are up to six positions available for introducing diverse substituents and four positions that allow for stereochemical modulation, thereby significantly enhancing compound diversity. Collectively, these structural attributes render diketopiperazines particularly appealing candidates for drug discovery.7,8 Currently, a wide range of active diketopiperazines have been extensively investigated as lead compounds, and several drugs featuring diketopiperazines as the core structure have been successfully employed in practical applications. For instance, Tadalafil is utilized for the treatment of benign prostatic hyperplasia and pulmonary hypertension;9 GSK221149A serves as an oxytocin receptor blocker for alleviating premature labor pains;10 gliotoxin is currently under evaluation due to its immunosuppressive properties in selectively ex vivo removing immune cells responsible for tissue rejection;11 and bicozamine acts as a food additive used to prevent diarrhoea in cattle and pigs.12 Additionally, ambewelamides, verticillin, and phenylahistin have demonstrated diverse antitumor activities.3,13,14There are two pathways for the production of cyclic dipeptides in nature: non-enzymatic synthesis and enzymatic synthesis. The enzymatic synthesis encompasses the non-ribosomal peptide synthetase (NRPS) pathway and the cyclodipeptide synthase (CDPS) pathway.15,16 The NRPS pathway utilizes a variety of amino acids, including natural amino acids and their derivatives, as substrates to generate cyclic dipeptide.15 In contrast, the CDPS pathway employs small synthases, known as CDPSs, which consist of 200–300 amino acid residues and exclusively utilize aminoacyl-tRNA (aa-tRNA) loaded natural amino acids as substrates. Notably, this enzyme does not require ATP for activating amino acid molecules. Additionally, the CDPS gene cluster typically includes several post-modification enzymes, such as oxidoreductases, methyltransferases, and cytochrome P450 enzymes, which are involved in the further modification of cyclized products to ultimately form active diketopiperazines.16 Recently, arginine-containing cyclodipeptide synthases (RCDPSs), which are evolutionarily distinct from CDPS, have been exclusively identified within the fungal kingdom.17
In this study, we performed genome mining of a Streptomyces strain (Streptomyces sp. YINM00030) isolated from soil samples collected at the Chinese medicinal herb garden of Kunming University in China. A gene cluster potentially involved in the biosynthesis of novel diketopiperazines via the CDPS pathway was identified in this strain. Several proteins within this gene cluster exhibit moderate similarity to key enzymes AlbA, AlbB, and AlbC found in the biosynthetic gene cluster responsible for albonoursin production.18 One tRNA-dependent cyclodipeptide synthase, likely participating in cyclizing two amino acids into a lactam six-membered ring, shares 55% sequence identity with AlbC. The other two enzymes potentially involved in the oxidation of cyclodipeptides to yield the final product exhibit 67% and 64% sequence identity with AlbB and AlbA, respectively. No genes similar to albD were identified within the whole genome. Therefore, we hypothesize that this gene cluster may be responsible for producing novel diketopiperazine natural products. Guided by our genome mining results, we selected various media for fermentation studies on YINM00030 and successfully discovered five new diketopiperazine compounds named albocandins A–E (1–5). These compounds are based on the cyclo(L-Phe-L-Leu) structure and exhibit varying degrees of oxidation at different positions. Activity study demonstrated that compounds 3 and 4 exhibited potent inhibitory effects against lung cancer (A-549) and colon cancer (SW480), respectively. Additionally, cyclo(L-Phe-Δ-Leu) (6), a potential intermediate in the biosynthetic pathway of albonoursin, was also isolated. It has been reported that the cell-free extract of Streptomyces albulus KO-23 can convert compound 6 to albonoursin.19 A recent investigation revealed that co-expression of AlbA and AlbB leads to the formation of filamentous protein polymers, which play a catalytic role in the final production of albonoursin.20 However, no isolation of albonoursin was achieved from the fermentation products of strain YINM00030. These findings suggest that three core genes, alcA, alcB, and alcC, potentially contribute to novel diketopiperazines biosynthesis in Streptomyces sp. YINM00030. Further exploration into their biosynthetic mechanisms will provide new possibilities for expanding the structural diversity of these compounds.
Results and discussion
Taxonomy analysis of Streptomyces sp. YINM00030
The neighbor-joining phylogenetic tree of the 16S rRNA gene sequences showed that strain YINM00030 formed a cluster with Streptomyces candidus NBRC 12846T (99.66% similarity) (Fig. 1). The maximum-likelihood tree and maximum-parsimony tree also showed it clustered with strain Streptomyces candidus NBRC 12846T under high bootstrap values (Fig. S1 and S2†). The RAxML neighbor-joining phylogenomic tree demonstrated that YINM00030 formed a cluster with strain Streptomyces candidus NBRC 12846T under the 100 bootstrap values (Fig. S3†). Phylogenetic analysis of strain YINM00030 indicates that it is a member of the genus Streptomyces.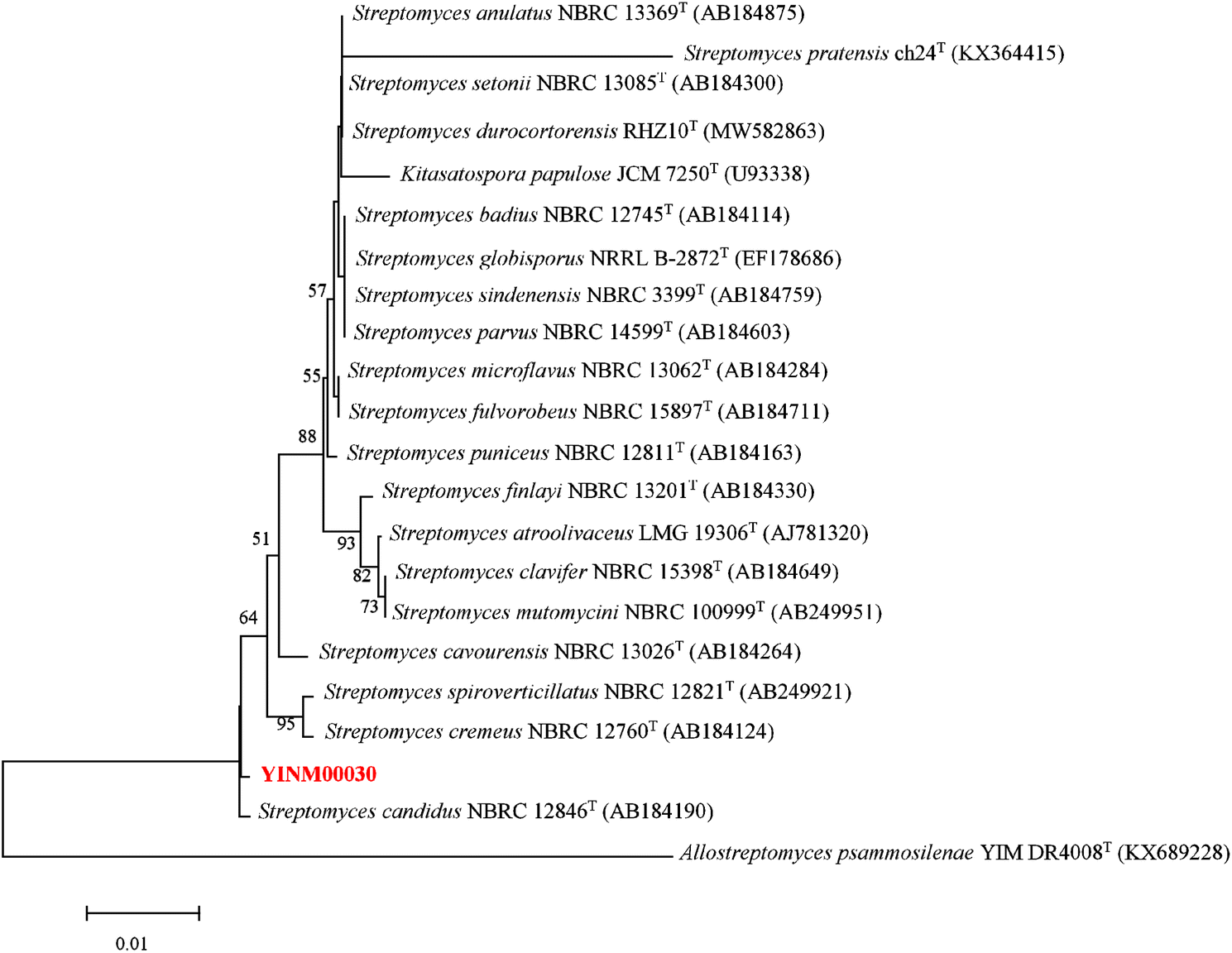 | ||
| Fig. 1 The neighbor-joining phylogenetic tree of strain YINM00030 and its closest relatives from the genus Streptomyces based on 16S rRNA genes. Bootstrap values (>50%) based on 1000 resamplings are given at the nodes. Allostreptomyces psammosilenae YIM DR4008T (accession no. KX689228) was used as outgroup. Bar, 0.01 substitutions per nucleotide position. | ||
Genome mining of CDPS gene cluster
A gene cluster, with three core genes alcA, alcB and alcC, was identified with the assistance of antiSMASH 7.0 software21 (Fig. S4†). AlcC, a member of the cyclodipeptide synthase, shares 55% sequence identity with AlbC in the albonoursin gene cluster, which is responsible for cyclizing phenylalanine and leucine to form cyclo(L-Phe-L-Leu).18 AlcA, belonging to cyclic dipeptide oxidase, exhibits 64% sequence identity with AlbA in the albonoursin gene cluster. Previous research has demonstrated that AlbA is responsible for the stepwise α, β dehydrogenation of cyclo(L-Phe-L-Leu) into the final product albonoursin. Initially, cyclo(L-Phe-L-Leu) undergoes transformation into cyclo(ΔPhe-L-Leu), followed by conversion into cyclo(ΔPhe-ΔLeu). In addition to efficiently oxidizing cyclo(L-Phe-L-Leu) to cyclo(ΔPhe-ΔLeu), AlbA can also dehydrogenate various different cyclic dipeptides, with aromatic residues and hydrophobic residues being superior substrate.19 The sequence identity between AlcB and AlbB is 67% (Table S1†). Recent studies have indicated that AlbB is essential for AlbA activity, and the filamentous protein polymers formed by co-expression of AlbA and AlbB can catalyze the final formation of albonoursin.20 Consistent with the albonoursin gene cluster, there is overlap between the end of the alcA gene and the start of the alcB gene. No similar gene to albD was identified in strain YINM00030, and the other genes in the albocandin gene cluster are unrelated to those in the albonoursin gene cluster. The difference between the newly identified albocandin gene cluster and reported albonoursin cluster imply that host strain YINM00030 may have potential to produce novel cyclodipeptides.Isolation of compounds guided by genome mining
Five different media were used for the fermentation of strain YINM00030. Six albonoursin analogs, including five newly discovered ones, were extracted from 20# medium ferments as anticipated (Fig. 2).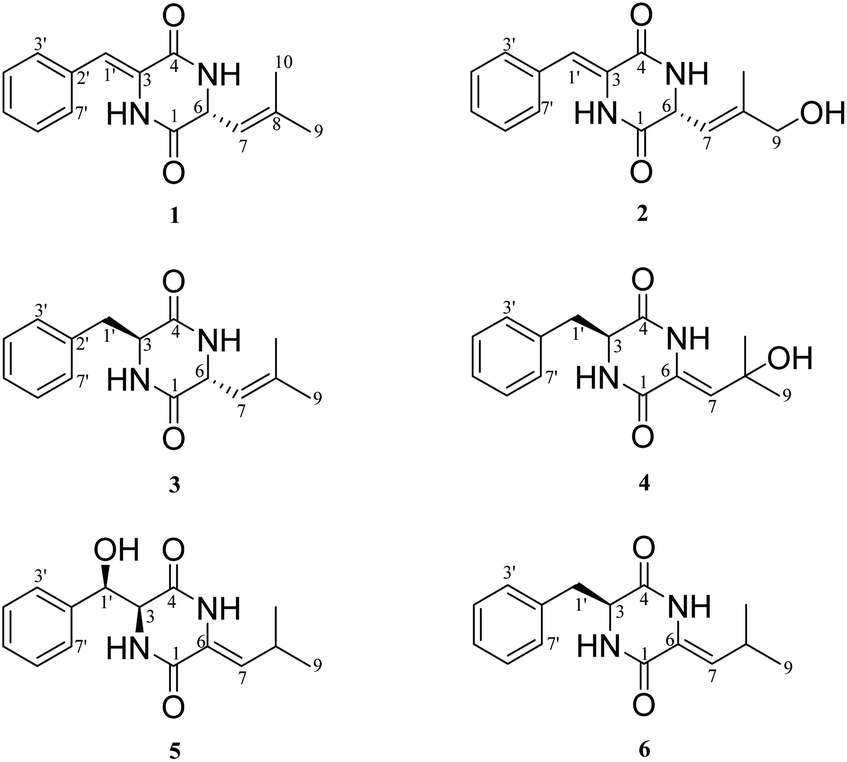 | ||
| Fig. 2 Structures of compounds 1–6. | ||
Compound 1 was obtained as white crystalline powder, and the molecular formula C15H16N202 was determined by its HRESIMS (m/z 257.1288 [M + H]+, calcd for C15H17N202, 257.1285), indicating nine degrees of unsaturation. The 13C NMR and DEPT spectral data (Table 1) allowed the classification of 15 carbon resonances into five nonprotonated carbons (δC 165.4, 159.9, 141.2, 133.0, and 125.7), eight methines (δC 129.6, 129.6, 129.0, 128.7, 128.7, 120.1, 116.6, and 54.6), and two methyls (δC 26.0, 18.7). Further analysis of the 1H–1H COSY and HMBC spectra facilitated the elucidation of the planar structure of compound 1 (Fig. 3). An α,β-unsaturated phenylalanine residue was revealed by HMBC correlations from H-1′ to C-4 and C-3′, from H-3′/7′ to C-5′, from H-4′/6′ to C-2′, and COSY correlations of H-3′/H-4′/H-5′/H-6′/H-7′. Meanwhile, another β,γ-unsaturated leucine residue was identified by the HMBC correlations from H3-9 and H3-10 to C-7 and C-8, from H-6 to C-1 and C-8, together with 1H–1H COSY correlation between H-6 and H-7. Finally, the connection of two fragments to form a diketopiperazine ring was supported by the HMBC correlations from H-1′ to C-1 and H-6 to C-4. Consequently, the planar structure of compound 1 was established.
| No. | 1a | 2b | 3c | 4c | 5c | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| a Measured at 400 MHz in CDCl3.b Measured at 400 MHz in methanol-d4.c Measured at 600 MHz in methanol-d4. | ||||||||||
| 1 | 165.4, C | — | 168.4, C | — | 171.4, C | — | 159.8, C | — | 162.8, C | — |
| 3 | 125.7, C | — | 127.4, C | — | 58.6, CH | 4.23, t (4.2) | 57.9, CH | 4.41, t (4.3) | 63.4, CH | 4.31, d (3.4) |
| 4 | 159.9, C | — | 162.9, C | — | 171.0, C | — | 166.8, C | — | 166.3, C | — |
| 6 | 54.6, CH | 4.94, d (9.0) | 55.2, CH | 4.96, d (9.3) | 53.7, CH | 3.28, d (9.3) | 126.7, C | — | 125.0, C | — |
| 7 | 120.1, CH | 5.28, d (9.0) | 120.5, CH | 5.51, m | 120.9, CH | 4.93, d (9.3) | 123.8, CH | 5.49, s | 127.2, CH | 5.26, d (10.4) |
| 8 | 141.2, C | — | 144.3, C | — | 141.5, C | — | 73.4, C | — | 25.7, CH | 2.26, m |
| 9 | 26.0, CH3 | 1.82, s | 67.4, CH2 | 3.99, s | 26.3, CH3 | 1.68, s | 30.6, CH3 | 1.09, s | 22.6, CH3 | 0.63, d (6.6) |
| 10 | 18.7, CH3 | 1.82, s | 14.3, CH3 | 1.81, s | 18.6, CH3 | 1.42, s | 31.0, CH3 | 1.27, s | 22.4, CH3 | 0.79, d (6.6) |
| 1′ | 116.6, CH | 6.99, s | 118.1, CH | 6.90, s | 41.5, CH2 | 2.99, dd (13.7, 4.7) | 41.6, CH2 | 3.01, m | 76.3, CH | 5.03, d (3.4) |
| 3.26, dd (13.6, 3.9) | 3.25, m | |||||||||
| 2′ | 133.0, C | — | 134.7, C | — | 137.2, C | — | 136.1, C | — | 139.4, C | — |
| 3′/7′ | 128.7, CH | 7.41, overlap | 130.3, CH | 7.47, overlap | 132.3, CH | 7.25, m | 131.7, CH | 7.16, m | 128.5, CH | 7.19, m |
| 4′/6′ | 129.6, CH | 7.45, overlap | 130.2, CH | 7.42, overlap | 130.3, CH | 7.33, overlap | 129.8, CH | 7.26, overlap | 129.0, CH | 7.15, overlap |
| 5′ | 129.0, CH | 7.37, overlap | 129.8, CH | 7.33, overlap | 129.2, CH | 7.33, overlap | 128.6, CH | 7.24, overlap | 129.2, CH | 7.15, overlap |
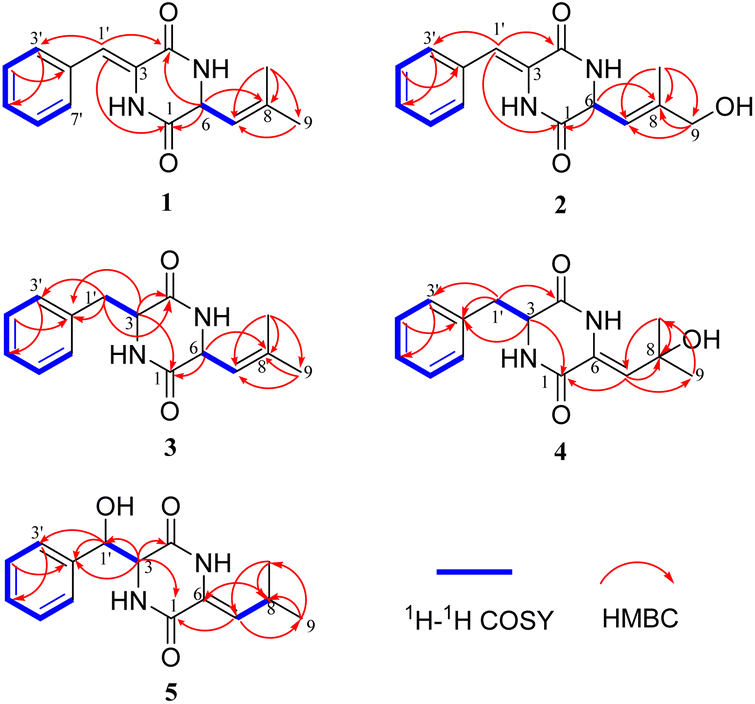 | ||
| Fig. 3 COSY and key HMBC correlations of compounds 1–5. | ||
Fortunately, a single crystal of 1 was obtained from MeOH/H2O mixture, and a single-crystal X-ray diffraction experiment using Cu Kα radiation was performed (Fig. 4). This result further confirmed the planar structure of 1 and its stereoconfiguration was determined. Additionally, the absolute configuration of 1 was established as 6R, through a computational approach that compared the experimental ECD spectrum with the TDDFT-calculated ECD spectrum (Fig. 5), and named as albocandin A.
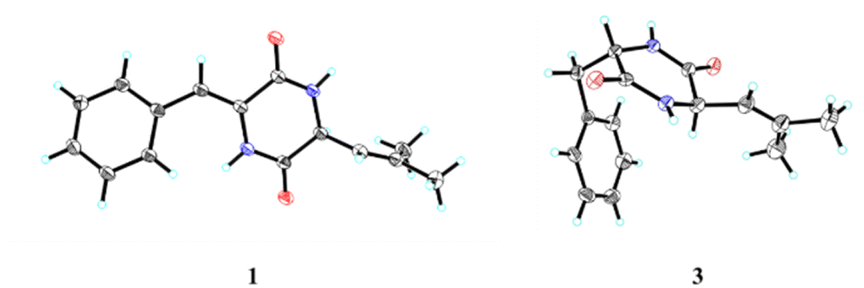 | ||
| Fig. 4 X-ray crystal structures of 1 and 3. | ||
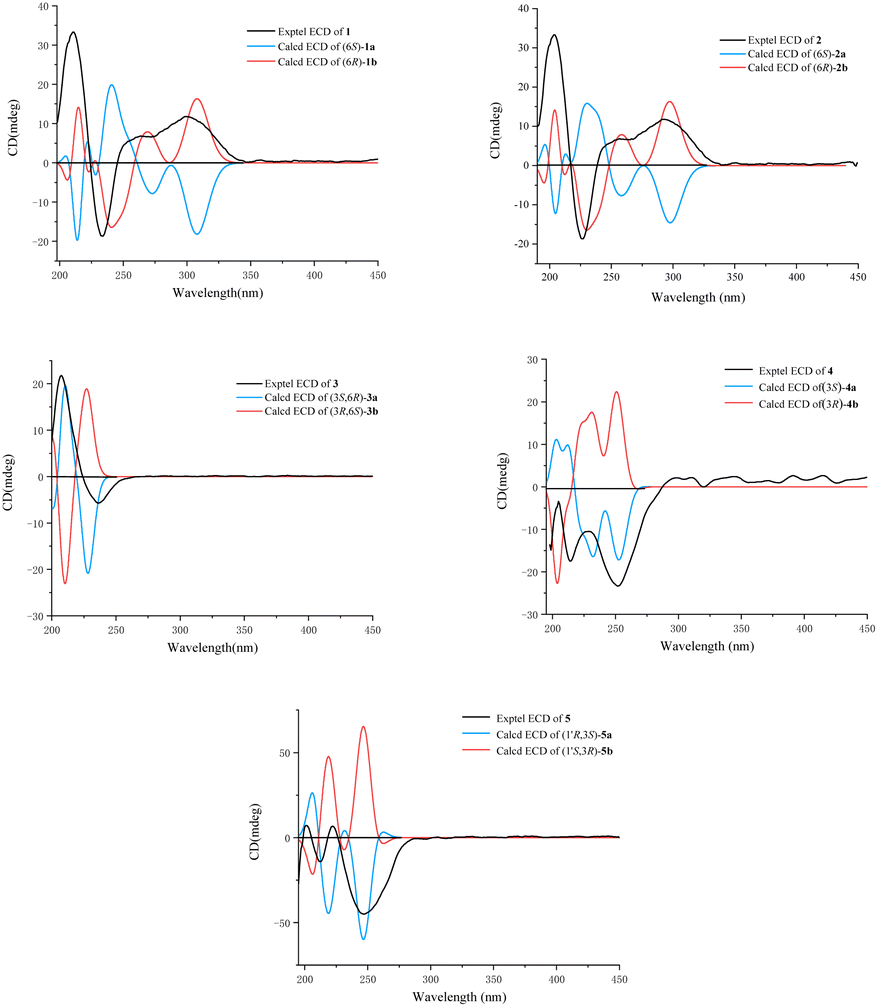 | ||
| Fig. 5 Experimental and calculated ECD spectra of 1–5 (in MeOH). | ||
Albocandin B (2) was isolated as faint yellow amorphous powder with a molecular formula of C15H16N203 deduced from its HRESIMS (m/z 273.1232 [M + H]+, calcd for C15H17N203, 273.1234), indicating nine degrees of unsaturation. Comparing the NMR data (Table 1) of 1 and 2, a high degree of similarity was observed, indicating that 2 and 1 share a similar molecular skeleton. The primary difference between compounds 2 and 1 lies in the presence of an additional oxymethylene (δH 3.99, δC 67.4) and the absence of a methyl signals in 2, which was supported by the HMBC correlations from H-9 and H-10 to C-7 and C-8. Moreover, spectroscopic analysis, including MS and 2D NMR (Fig. 3), confirmed the planar structure of 2. To clarify the absolute configuration of 2 by comparing the experimental ECD spectrum with calculated ECD spectrum of two possible isomers (6R-2 and 6S-2) (Fig. 5), the result showed the ECD curve of 6R-2 was better consistent with the measured curve. Therefore, the absolute configuration of 2 was also unambiguously determined to be 6R.
Albocandin C (3) was isolated as white crystalline powder and designated with the molecular formula C15H18N202 based on the HRESIMS analysis (m/z 259.1438 [M + H]+, calcd for C15H18N202, 259.1441) with eight degrees of unsaturation. The NMR spectral similarities between 3 and 1 suggested a structural resemblance, except that the olefin double bond between C-3 and C-1′ in 1 was reduced to a saturated bond in 3, which was supported by the chemical shift values of C-3 and C-1′ in 3 (Table 1). Detailed analysis of the key correlations in the 1H–1H COSY and HMBC spectra (Fig. 3) of compound 3 confirmed this deduction and further elucidated its planar structure. The stereoconfiguration of 3 was defined as shown by the X-ray single crystal diffraction analysis (Fig. 4), then its absolute configuration was elucidated by the ECD data fitting experiment (Fig. 5). Consequently, the structure of 3 was identified as (3S,6R)-3-benzyl-6-(2-methylpropenyl)-piperazine-2,5-dione.
Albocandin D (4) was obtained as a white amorphous powder, and its molecular formula was determined as C15H18N203 on the basis of HRESIMS (m/z 275.1389 [M + H]+, calcd for C15H19N203, 275.1390), corresponding to eight degrees of unsaturation. Its 1D NMR data (Table 1) resembled those of cyclo(L-Phe-Δ-Leu) (6),19 except that a methane was absent and an oxygenated quaternary carbon at C-8 (δC 73.4) was present in 4. Combined with the molecular mass information, 4 was identified to be a hydroxylated analogue of 6. Further analyses of NMR data, including COSY and HMBC experiments (Fig. 3), yielded the planar structure of 4. As shown in Fig. 5, the calculated ECD spectrum for (3S)-4 matched well with the experimental ECD spectrum, which elucidated the absolute configuration of 4 as 3S.
Albocandin E (5), a faint yellow amorphous powder, its molecular formula was deduced as C15H18N203 based on its HRESIMS (m/z 273.1233 [M − H]− calcd for C15H17N203, 273.1245), indicating eight degrees of hydrogen deficiency. The NMR data and its molecular formula indicated the presence of a highly similar skeleton as compound 6 except for the appearance of an additional hydroxy group in 5.19 The location of the hydroxyl at C-1′ in 5 was deduced by the COSY correlation between H-3 and H-1′ and HMBC correlations from H-1′ to C-2′ and -3′ (Fig. 3). The planar structure of 5 was confirmed by the detailed 2D NMR analysis. To determine the absolute configuration of 5 by comparing the experimental ECD spectrum with calculated ECD spectrum of all the four possible isomers, and the calculated ECD curve of the (3S,1′R)-enantiomer showed the identical cotton effects (CEs) as the experimental ECD curve for 5 (Fig. 5 and S43†). Therefore, the absolute configuration of 5 was established as 3S, 1′R.
Cytotoxicity assay
In the cytotoxicity assay, compounds 1–5 were evaluated using the MTS method to confirm their efficacy against human myeloid leukemia (HL-60), lung cancer (A-549), liver cancer (SMMC-7721), breast cancer (MDA-MB-231) and colon cancer (SW480) in vitro.22 Compound 4 exhibited significant cytotoxic activity against all five cancer cell lines, particularly demonstrating potent inhibitory effects on HL-60 and A-549 with IC50 values of 3.50 and 5.83 μM, respectively. Compound 3 displayed moderate cytotoxicity against HL-60, A-549, SMMC-7721, and SW480 cell lines, with IC50 values of 22.32, 21.60, 16.79, and 32.66 μM, respectively (Table 2). The remaining compounds did not show any inhibitory activities at a concentration of 40 μM.| Compound | IC50 ± SD, μM | ||||
|---|---|---|---|---|---|
| HL-60 | A549 | SMMC-7721 | MDA-MB-231 | SW480 | |
| a Positive control.b The IC50 values were expressed as means ± SD (n = 3) from three independent experiments. | |||||
| 3 | 22.32 ± 0.53 | 21.60 ± 1.21 | 16.79 ± 0.61 | — | 32.66 ± 1.57 |
| 4 | 3.50 ± 0.16 | 5.83 ± 0.25 | 23.33 ± 0.60 | 22.79 ± 0.48 | 14.22 ± 0.80 |
| Cisplatina | 2.92 ± 0.15 | 11.95 ± 0.25 | 5.71 ± 0.35 | 19.28 ± 0.30 | 20.14 ± 0.65 |
The proposed biosynthetic pathway of compounds 1–6
Compounds 1–6 are analogues of albonoursin, and based on the reported albonoursin biosynthetic mechanism,18–20 we propose a putative biosynthetic pathway for the newly discovered compounds in this study. Firstly, AlcC is responsible for cyclizing phenylalanine and leucine to form cyclo(L-Phe-L-Leu). Subsequently, AlcA and AlcB catalyze cyclo(L-Phe-L-Leu) to yield compounds 1, 3 and 6. Finally, compounds 2, 4 and 5 are presumed to be generated by hydroxylases located outside the gene cluster (Fig. 6). | ||
| Fig. 6 Proposed biosynthetic pathway of albocandins A–E (1–5), alcC: tRNA-dependent cyclodipeptide synthase. alcA: TTA leucine codon; alcB: nitroreductase family protein. | ||
Experimental
General experimental procedures
UV-vis spectra were recorded using a Shimadzu UV-2550 PC spectrometer (Shimadzu Co., Ltd., Tokyo, Japan). ECD spectra were recorded using a Chirascan circular dichroism spectrometer (Applied Photophysics Ltd., UK). The specific rotations were measured using a JASCO P-1020 digital polarimeter (Horiba, Tokyo, Japan). NMR spectra were recorded on a Bruker Avance-400 MHz instrument and a Bruker Avance-600 MHz instrument with cryogenic probe (Bruker, Karlsruhe, Germany) using tetramethylsilane as the internal standard. HRESIMS data were obtained on an Agilent G3250AA (Agilent, Santa Clara, CA, USA). The preparative HPLC was performed on an Agilent 1260 series equipped with a DAD detector and a Zorbax SB-C18 (250 × 9.4 mm, 5 μm) column. Silica gel (200–300 mesh, Qingdao Marine Chemical Group Co., Qingdao, China), Lichroprep RP-18 gel (Merck, Darmstadt, Germany), and Sephadex LH-20 (GE Healthcare Bio-Science AB, Uppsala, Sweden) were used for column chromatography (CC). Analytical thin-layer chromatography (TLC) was carried out using GF 254 plates (Qingdao Marine Chemical Group Co., Qingdao, China) and visualized by spraying with anisaldehyde-H2SO4 reagent.Microorganism
Strain YINM00030 was isolated from fresh soil samples collected from the Chinese medicinal herb garden in Kunming University, Yunnan Province, China. The soil sample suspensions were inoculated on M1 medium (asparagine 1 g, glycerol 10 g, K2HPO4·H2O 1 g, MgSO4·7H2O 0.05 g, CaCO3 0.3 g, vitamin complex and trace salt solution 1 mL, H2O up to 1000 mL, pH = 7.2) supplemented with nystatin and nalidixic acid (50 mg mL−1 each), and incubated at 28 °C for up to a month. The isolate was subsequently picked out and purified three times on ISP2 plates with nystatin and nalidixic acid before being preserved in ISP2 slants and 20% (v/v) glycerol tubes stored at 4 °C and −80 °C respectively for further use.Genome sequencing and albocandin gene cluster identification
Genomic DNA isolation, 16S rRNA gene amplification and sequencing were conducted following previously described method.23 The purified strain YINM00030 was inoculated into 100 mL tryptic soy broth (TSB) medium (casein tryptone 17 g, soy peptone 5 g, sodium chloride 5 g, D-glucose 2.5 g, dipotassium phosphate 2.5 g, H2O up to 1000 mL, pH = 7.3) at 28 °C with 200 rpm vigorous shaking for 24 h. The mycelium of YINM00030 was then preserved with dry ice and sent to Majorbio (Shanghai, China) for complete genome sequencing. Following sequencing with PacBio and Illumina HiSeq Technologies, the albocandin biosynthetic gene cluster was identified using antiSMASH 7.0.21Fermentation
For small-scale fermentation, the strain YINM00030 was activated in ISP2 medium at 28 °C for a week. Then, the fresh strain was inoculated into 500 mL Erlenmeyer flasks with 100 mL ISP2 liquid medium and cultured for 3–4 days at 28 °C and 200 rpm. Next, 5.0 mL of the seed culture was transferred into 500 mL Erlenmeyer flasks containing 100 mL of 5 different types of fermentation media (9#, 17#, 18#, 19# and 20#, detailed data not show), respectively. The optimal fermentation condition was determined by detecting of the crude exacts at 5, 10 and 15 days by high performance liquid chromatograph (HPLC) and thin layer chromatography (TLC). For large-scale fermentation, the seed culture of strain YINM00030 was prepared as described above. Then, 5.0 mL of the seed cultures were transferred into 500 mL Erlenmeyer flasks containing 100 mL of 20# fermentation medium (glycerol 50 g, corn meal 25 g, primary yeast 5 g, H2O up to 1000 mL, pH = 7.0), and cultured for 10 days at 28 °C and 200 rpm (27 L).Isolation and identification of the secondary metabolites
The fermentation broth of strain YINM00030 (27 L) was extracted with ethyl acetate (EtOAc) three times, and the solvent was removed under vacuum to obtain the EtOAc extract (30 g). The crude extracted was separated into five fractions (Fr.1–Fr.5) by column chromatograph on silica gel, eluting stepwise with a CH2Cl2–MeOH system (CH2Cl2, CH2Cl2–MeOH = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, CH2Cl2–MeOH = 10:1 v/v, CH2Cl2–MeOH = 5:1 v/v, MeOH). Fr.2 was fractionated by LiChroprep RP-18 column, and eluted stepwise with a MeOH–H2O system (20% MeOH, 40% MeOH, 60% MeOH, 80% MeOH and 100% MeOH) to afford Fr.2-1 to Fr.2-5. Fr.2-2 was isolated by a silica gel column with a petroleum ether-ethyl acetate gradient (5:1 v/v, 2:1 v/v, 1:1 v/v) to obtain Fr.2-2-1 to Fr.2-2-3. Fr.2-2-2 was further divided into three parts (Fr.2-2-2-1 to Fr.2-2-2-3) by a Sephadex LH-20 column with MeOH. Then, Fr.2-2-2-2 was separated by HPLC using a Zorbax SB-C18 column (250 × 9.4 mm, 5 μm; the mobile phase is 65% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 300 nm) to afford 1 (15.6 mg, tR = 7.5 min). Fr.2-1 was divided into five parts (Fr.2-1-1 to Fr.2-1-5) by a Sephadex LH-20 column with MeOH. Compound 3 (4.3 mg, tR = 19.6 min) and 4 (2.5 mg, tR = 21.0 min) were obtained from Fr.2-1-3 by HPLC purification (Zorbax SB-C18 column: 250 × 9.4 mm, 5 μm; the mobile phase is 40% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 220 and 254 nm). Fr.2-1-4 was fractionated by a silica gel column with a petroleum ether-ethyl acetate gradient (2:1 v/v, 1:1 v/v, 1:3 v/v, 1:5 v/v) to afford Fr.2-1-4-1 to Fr.2-1-4-4. Fr.2-1-4-3 was separated by HPLC using a Zorbax SB-C18 column (250 × 9.4 mm, 5 μm; the mobile phase is 55% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 310 nm) to give 5 (3.2 mg, tR = 6.3 min). Compound 2 (8.3 mg, tR = 7.8 min) were obtained from Fr.2-1-4-4 by HPLC purification (Zorbax SB-C18 column: 250 × 9.4 mm, 5 μm; the mobile phase is 50% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 290 nm).
:1 v/v, CH2Cl2–MeOH = 10:1 v/v, CH2Cl2–MeOH = 5:1 v/v, MeOH). Fr.2 was fractionated by LiChroprep RP-18 column, and eluted stepwise with a MeOH–H2O system (20% MeOH, 40% MeOH, 60% MeOH, 80% MeOH and 100% MeOH) to afford Fr.2-1 to Fr.2-5. Fr.2-2 was isolated by a silica gel column with a petroleum ether-ethyl acetate gradient (5:1 v/v, 2:1 v/v, 1:1 v/v) to obtain Fr.2-2-1 to Fr.2-2-3. Fr.2-2-2 was further divided into three parts (Fr.2-2-2-1 to Fr.2-2-2-3) by a Sephadex LH-20 column with MeOH. Then, Fr.2-2-2-2 was separated by HPLC using a Zorbax SB-C18 column (250 × 9.4 mm, 5 μm; the mobile phase is 65% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 300 nm) to afford 1 (15.6 mg, tR = 7.5 min). Fr.2-1 was divided into five parts (Fr.2-1-1 to Fr.2-1-5) by a Sephadex LH-20 column with MeOH. Compound 3 (4.3 mg, tR = 19.6 min) and 4 (2.5 mg, tR = 21.0 min) were obtained from Fr.2-1-3 by HPLC purification (Zorbax SB-C18 column: 250 × 9.4 mm, 5 μm; the mobile phase is 40% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 220 and 254 nm). Fr.2-1-4 was fractionated by a silica gel column with a petroleum ether-ethyl acetate gradient (2:1 v/v, 1:1 v/v, 1:3 v/v, 1:5 v/v) to afford Fr.2-1-4-1 to Fr.2-1-4-4. Fr.2-1-4-3 was separated by HPLC using a Zorbax SB-C18 column (250 × 9.4 mm, 5 μm; the mobile phase is 55% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 310 nm) to give 5 (3.2 mg, tR = 6.3 min). Compound 2 (8.3 mg, tR = 7.8 min) were obtained from Fr.2-1-4-4 by HPLC purification (Zorbax SB-C18 column: 250 × 9.4 mm, 5 μm; the mobile phase is 50% MeOH; the flow rate is 3.0 mL min−1; detection wavelength 290 nm).
Albocandin A (1): white crystalline powder; melting point: 200.1 °C; [α]25D 5.1 (c 0.10, MeOH); λmax(logε) 230 (2.12), 298 (2.19); 1H and 13C NMR data, Table 1; HRESIMS m/z 257.1288 [M + H]+ (calcd for C15H17N202, 257.1285).
Albocandin B (2): faint yellow amorphous powder; [α]25D −1.7 (c 0.10, MeOH); λmax(logε) 240 (2.21), 300 (2.42); 1H and 13C NMR data, Table 1; HRESIMS m/z 273.1232 [M + H]+ (calcd for C15H17N203, 273.1234).
Albocandin C (3): white crystalline powder; melting point: 196.0 °C; [α]25D 1.6 (c 0.10, MeOH); λmax(logε) 250 (1.98); 1H and 13C NMR data, Table 1; HRESIMS m/z 259.1438 [M + H]+ (calcd for C15H19N202, 259.1441).
Albocandin D (4): white amorphous powder; [α]25D −11.0 (c 0.10, MeOH); λmax(logε) 250 (1.87); 1H and 13C NMR data, Table 1; HRESIMS m/z 275.1389 [M + H]+ (calcd for C15H19N203, 275.1390).
Albocandin E (5): faint yellow amorphous powder; [α]25D −5.4 (c 0.10, MeOH); λmax(logε) 250 (2.17); 1H and 13C NMR data, Table 1; HRESIMS m/z 273.1233 [M − H]− (calcd for C15H17N203, 273.1245).
:1, v/v). C15H16N2O2, M = 256.30, a = 5.4192(5) Å, b = 15.6201(14) Å, c = 16.4094(14) Å, α = 70.796(4)°, β = 87.714(4)°, γ = 83.915(4)°, V = 1304.3(2) Å3, T = 150.(2) K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 4, μ(Cu Kα) = 0.709 mm−1, 28645 reflections measured, 4795 independent reflections (Rint = 0.1328). The final R1 values were 0.0849 (I > 2σ(I)). The final wR(F2) values were 0.2330 (I > 2σ(I)). The final R1 values were 0.1318 (all data). The final wR(F2) values were 0.2697 (all data). The Flack parameter was 0.0032(12). The goodness of fit on F2 was 1.093 (CCDC 2355054†).:1, v/v). C15H18N2O2, M = 258.31, a = 6.1179(4) Å, b = 8.0879(6) Å, c = 27.871(2) Å, α = 90°, β = 90°, γ = 90°, V = 1379.08(17) Å3, T = 100 K, space group P212121, Z = 4, μ(Cu Kα) = 0.671 mm−1, 21497 reflections measured, 2913 independent reflections (Rint = 0.0482). The final R1 values were 0.0558 (I > 2σ(I)). The final wR(F2) values were 0.1523 (I > 2σ(I)). The final R1 values were 0.0584 (all data). The final wR(F2) values were 0.1556 (all data). The Flack parameter was 0.17(16). The goodness of fit on F2 was 1.068 (CCDC 2355055†).
, Z = 4, μ(Cu Kα) = 0.709 mm−1, 28645 reflections measured, 4795 independent reflections (Rint = 0.1328). The final R1 values were 0.0849 (I > 2σ(I)). The final wR(F2) values were 0.2330 (I > 2σ(I)). The final R1 values were 0.1318 (all data). The final wR(F2) values were 0.2697 (all data). The Flack parameter was 0.0032(12). The goodness of fit on F2 was 1.093 (CCDC 2355054†).:1, v/v). C15H18N2O2, M = 258.31, a = 6.1179(4) Å, b = 8.0879(6) Å, c = 27.871(2) Å, α = 90°, β = 90°, γ = 90°, V = 1379.08(17) Å3, T = 100 K, space group P212121, Z = 4, μ(Cu Kα) = 0.671 mm−1, 21497 reflections measured, 2913 independent reflections (Rint = 0.0482). The final R1 values were 0.0558 (I > 2σ(I)). The final wR(F2) values were 0.1523 (I > 2σ(I)). The final R1 values were 0.0584 (all data). The final wR(F2) values were 0.1556 (all data). The Flack parameter was 0.17(16). The goodness of fit on F2 was 1.068 (CCDC 2355055†).ECD calculations
Conformational analyses were conducted using random searching in CONFLEX software, employing the MMFF94S force field with an energy cutoff of 5.0 kcal mol−1.24 The ECD spectra for each conformer were computed using the TDDFT methodology at the B3LYP/6-31 G(d,p) level of theory, with methanol as the solvent, utilizing the polarizable continuum solvent model (PCM) implemented in the Gaussian 09 program.25 The calculated ECD spectra of the conformers were subsequently combined using Boltzmann weighting. These processes were executed with the SpecDis 1.64 software.26Cytotoxicity assay
The cytotoxicities of compounds 1–5 against five human cancer cell lines (breast cancer MCF-7, colon cancer SW480, hepatocellular carcinoma SMMC-7721, lung cancer A549, and myeloid leukemia HL-60) were evaluated in vitro by the MTS method.22 Cisplatin (DDP) was used as the positive control.Conclusions
In recent years, our research has focused on investigating Chinese herbal-related actinomycetes with the potential for producing novel natural products. Genome mining for Streptomyces sp. YINM00030 has revealed its capability to produce novel diketopiperazine compounds. By exploring various culture conditions, we have successfully isolated six diketopiperazines, five of which were newly identified as albocandins. Albocandins C (3) and D (4) demonstrated significant cytotoxic activity against a range of tumor cells, prompting further exploration into their pharmacological mechanisms. This study exemplifies how genome mining can facilitate the discovery of secondary metabolites in actinomycetes and contributes to the diversification of diketopiperazines, establishing a new foundation for research and application due to their diverse activities.Data availability
Strain YINM00030 had been deposited at the Yunnan University. The sequence of the albocandin biosynthetic gene cluster in this strain has been deposited in GenBank under the accession number PP965795. The experimental data supporting this article have been included in the ESI.†Author contributions
M. Y., H. Z. and Z. D. designed the study, carried out the data analysis and wrote the manuscript. Z. Z., and Z. R. carried out the experiments and participated in data analysis. X. S., T. X., and M. Y. participated in data analysis. All authors have read and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (82160674 for M. Y., 22267001 for Z. D.), Yunnan Revitalization Talent Support Program (YNWR-QNBJ-2019-031 for M. Y., YNWR-QNBJ-2020-096 for Z. R.), Natural Science Foundation of Yunnan Province (202201AS070007 and 202002AA100007 for M. Y., 202201AT070225 for H. Z. and 202101BA070001-035 for Z. R.), Yunnan Key Laboratory of Stomatology, The Affiliated Stomatology Hospital of Kunming Medical University (2022YNKQ005 for M. Y.).References
- H. Kohn, W. Widger and C. D. Targets, Infect. Disord., 2005, 5, 273–295 CAS.
- K. Kanoh, S. Kohno, J. Katada, Y. Hayashi, M. Muramatsu and I. Uno, Biosci. Biotechnol. Biochem., 1999, 63, 1130–1133 CrossRef CAS.
- D. E. Williams, K. Bombuwala, E. Lobkovsky, E. D. de Silva, V. Karunaratne, T. M. Allen, J. Clardy and R. J. Andersen, Tetrahedron Lett., 1998, 39, 9579–9582 CrossRef.
- P. Waring and J. Beaver, Gen. Pharmacol., 1996, 27, 1311–1316 CrossRef CAS PubMed.
- G. D. Chen, Y. R. Bao, Y. F. Huang, D. Hu, X. X. Li, L. D. Guo, J. Li, X. S. Yao and H. Gao, Fitoterapia, 2014, 92, 252–259 CrossRef CAS.
- Y. Aniya, I. I. Ohtani, T. Higa, C. Miyagi, H. Gibo, M. Shimabukuro, H. Nakanishi and J. Taira, Free Radical Biol. Med., 2000, 28, 999–1004 CrossRef CAS.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS.
- C. Balachandra, D. Padhi and T. Govindaraju, ChemMedChem, 2021, 16, 2558–2587 CrossRef CAS PubMed.
- G. N. Maw, C. M. Allerton, E. Gbekor and W. A. Million, Bioorg. Med. Chem. Lett., 2003, 13, 1425–1428 CrossRef CAS PubMed.
- J. Liddle, M. J. Allen, A. D. Borthwick, D. P. Brooks, D. E. Davies, R. M. Edwards, A. M. Exall, C. Hamlett, W. R. Irving, A. M. Mason, G. P. McCafferty, F. Nerozzi, S. Peace, J. Philp, D. Pollard, M. A. Pullen, S. S. Shabbir, S. L. Sollis, T. D. Westfall, P. M. Woollard, C. Wu and D. M. Hickey, Bioorg. Med. Chem. Lett., 2008, 18, 90–94 CrossRef CAS.
- H. Hof and C. Kupfahl, Mycotoxin Res., 2009, 25, 123–131 CrossRef CAS PubMed.
- A. Magyar, X. Zhang, F. Abdi, H. Kohn and W. R. Widger, J. Biol. Chem., 1999, 274, 7316–7324 CrossRef CAS PubMed.
- M. Chu, I. Truumees, M. L. Rothofsky, M. G. Patel, F. Gentile, P. R. Das, M. S. Puar and S. L. Li, J. Antibiot., 1995, 48, 1440–1445 CrossRef CAS.
- K. Kanoh, S. Kanoh, J. Katada, J. Takahashi and I. Uno, J. Antibiot., 1999, 52, 134–141 CrossRef CAS.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC.
- M. Gondry, L. Sauguet, P. Belin, R. Thai, R. Amouroux, C. Tellier, K. Tuphile, M. Jacquet, S. Braud, M. Courçon, C. Masson, S. Dubois, S. Lautru, A. Lecoq, S. Hashimoto, R. Genet and J. L. Pernodet, Nat. Chem. Biol., 2009, 5, 414–420 CrossRef CAS.
- D. A. Yee, K. Niwa, B. Perlatti, M. Chen, Y. Li and Y. Tang, Nat. Chem. Biol., 2023, 19, 633–640 CrossRef CAS.
- S. Lautru, M. Gondry, R. Genet and J. L. Pernodet, Chem. Biol., 2002, 9, 1355–1364 CrossRef CAS.
- H. Kanzaki, S. Yanagisawa and T. Nitoda, J. Antibiot., 2000, 53, 1257–1264 CrossRef CAS.
- M. P. Andreas and T. W. Giessen, Nat. Commun., 2024, 15, 3574 CrossRef CAS PubMed.
- K. Blin, S. Shaw, H. E. Augustijn, Z. L. Reitz, F. Biermann, M. Alanjary, A. Fetter, B. R. Terlouw, W. W. Metcalf, E. J. N. Helfrich, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2023, 51, W46–W50 CrossRef CAS.
- D. Beleford, R. Rattan, J. Chien and V. Shridhar, J. Biol. Chem., 2010, 285, 12011–12027 CrossRef CAS PubMed.
- T. Liu, Z. Ren, W. X. Chunyu, G. D. Li, X. Chen, Z. T. Zhang, H. B. Sun, M. Wang, T. P. Xie, M. Wang, J. Y. Chen, H. Zhou, Z. T. Ding and M. Yin, Front. Microbiol., 2022, 13, 831174 CrossRef PubMed.
- K. Otsuki, M. Zhang, A. Yamamoto, M. Tsuji, M. Tejima, Z. S. Bai, D. Zhou, L. Huang, C. H. Chen, K. H. Lee, N. Li, W. Li and K. Koike, J. Nat. Prod., 2020, 83, 3584–3590 CrossRef CAS.
- R. Guo, P. Zhao, X. Q. Yu, G. D. Yao, B. Lin, X. X. Huang and S. J. Song, Org. Chem. Front., 2021, 8, 953–960 RSC.
- G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D, 2D NMR and HRESIMS spectra of new compounds, along with other details. CCDC 2355054 and 2355055. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra08447k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |