
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient CH4 oxidation to C1/C2 oxygenates over cluster-dispersing Rh decorated ZSM-5†
Xin Zhanga,
Wenzhi Li *ab,
Jingting Jina,
Liqun Wanga,
Zhiheng Luc and
Yihang Jianga
*ab,
Jingting Jina,
Liqun Wanga,
Zhiheng Luc and
Yihang Jianga
aLaboratory of Clean Low-Carbon Energy, Department of Thermal Science and Energy Engineering, University of Science and Technology of China, Hefei 230023, PR China
bInstitute of Energy, Hefei Comprehensive National Science Center, Hefei 230031, PR China
cSchool of Earth and Environment, Anhui University of Science and Technology, Huainan 232001, PR China
First published on 15th January 2025
Abstract
Crafting highly dispersed active metal sites on catalysts is an optimal method for improving the catalytic reactivity and stability, as it would improve atomic utilization efficiency, enhance reactant adsorption and activation ability through unique geometric and electronic properties. In this study, two synthesis methods were employed (ammonia evaporation (AE) and the impregnation method (IM)) to load Rh species onto the ZSM-5 support in order to attain tunable dispersivity, during which a 1.25-fold increase in the total yield of liquid oxygenated products (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 433.33 μmol gcat−1 h−1) was achieved specifically over a Rh–ZSM-5-AE sample when the reaction was carried out at a loading level of 0.3 wt% and at 240 °C for half an hour. The results of the study revealed that this elevated productivity originated from the smaller size and higher degree of dispersion of Rh clusters on AE samples. It was demonstrated that the ammonia evaporation method would cause Si leaching and introduce a substantial number of –OH groups during the preparation process, which worked in coordination in altering the electronic structure of Rh species. Consequently, these modifications modified the disordered Rh precursor adsorption, which resulted in a more homogeneous distribution of Rh species, hence facilitating the activation of methane. This study offers a practical and constructive approach for improving the dispersion of Rh nanoclusters and designing strong metal–support interactions (SMSI).
433.33 μmol gcat−1 h−1) was achieved specifically over a Rh–ZSM-5-AE sample when the reaction was carried out at a loading level of 0.3 wt% and at 240 °C for half an hour. The results of the study revealed that this elevated productivity originated from the smaller size and higher degree of dispersion of Rh clusters on AE samples. It was demonstrated that the ammonia evaporation method would cause Si leaching and introduce a substantial number of –OH groups during the preparation process, which worked in coordination in altering the electronic structure of Rh species. Consequently, these modifications modified the disordered Rh precursor adsorption, which resulted in a more homogeneous distribution of Rh species, hence facilitating the activation of methane. This study offers a practical and constructive approach for improving the dispersion of Rh nanoclusters and designing strong metal–support interactions (SMSI).
Introduction
Converting methane directly into liquid oxygenated products such as methanol and acetic acid is an energy-efficient method with significant potential for various industrial applications.1–8 However, the solid tetrahedral shape of CH4 presents a high energy barrier for the activation of the first C–H bond (439 kJ mol−1), making it difficult to avoid unwanted overoxidation reactions (e.g., to CO, CO2).9–15 Currently, CH3OH is produced through steam reforming of CH4 to generate H2 and CO, followed by Fischer–Tropsch synthesis. And acetic acid is further produced by the carbonylation of methanol. These steps are complex, energy-intensive (high temperatures at 700–1000 °C), and require harsh reaction conditions (high pressures up to 15–40 atmospheres).16–19 Thus, it becomes crucial to explore a more direct and mild way to convert CH4 to C1/C2 oxygenates while maintaining high activity and selectivity.Inspired by the active tri-copper cluster site of particulate methane monooxygenase (pMMO) in nature, early studies primarily focused on modelling its function using zeolites with copper and iron, which convert methane to methanol using strong oxidants (O2 or N2O) under mild conditions.20–24 These systems generally require a pre-oxidizing step at high temperature to form the active Cu/Fe-oxo configurations. And methane is then converted to methoxy species through catalysis, generating Cu/Fe-oxygen reactive species. Finally, methanol is extracted by introducing water vapor.25–28 This method proved to be effective, but its low oxide yield limits the potential for application in large-scale industries. To solve the problem, H2O2 has been employed as an oxidizing agent, which has greatly improved the efficiency of methanol synthesis over a variety of heterogeneous catalysis. However, the rapid decomposition of H2O2 in methane-selective oxidation reactions reduces its efficiency, and the high consumption of H2O2 limits the practical application of H2O2-based processes for converting methane to methanol.29–32
The method of using reducing gases to promote selective methane oxidation shows great potential. Recently, the partial catalytic oxidation of methane to oxygenated compounds like CH3OH, HCOOH, and CH3COOH has been successfully achieved using platinum group metal-based catalysts with CO gas.33–37 Among the platinum-group metals, rhodium has attracted great attention due to its excellent oxidizing properties and the SMSI effect with various carrier materials. Preparing active sites with greater dispersion would optimize the use of precious metals. In recent decades, zeolites have been viewed as promising supports for achieving high dispersion of metal species, including single atoms, nanoclusters, and nanoparticles, owing to their huge specific surface area and regular microporous channels.38–40
In the preliminary experiments, we loaded Rh species onto ZSM-5 zeolites. The results indicated that the catalyst activity prepared by ammonia evaporation (AE) achieved a total yield of liquid-phase oxygenated products of 32433.33 μmol gcat−1 h−1, which greatly surpassed that of the impregnation method (IM) under identical reaction conditions. Additionally, the liquid-phase product (including CH3OH, HCOOH, and CH3COOH) selectivity exceeded 87%. To investigate the reasons for this difference, combining with TEM, H2-TPR, XPS and in situ DRIFTS experiments, we systematically investigate the morphology and electronic structure of Rh species loaded on two different catalyst carriers and to identify potential electron transfer pathways between the two catalysts. Furthermore, the in situ DRIFTS technique was utilized to study the desorption of OH from the carrier surface at varying temperatures, and potential pathways for CO2 generation in the reaction atmosphere were modelled.
Experimental
Catalysts preparation
Results and discussions
Structural characterization and analysis of catalysts
Transmission electron microscopy (TEM) was employed to analyse the structure and crystal form of the catalysts, aiming to assess the dispersion of Rh species. Rh generated more agglomeration on the surface of support seen over 0.3 Rh/ZSM-5-IM catalyst (Fig. 1A), and the particle size statistics indicated that the average particle size was 1.88 nm. In contrast, the TEM image (Fig. 1B) of the 0.3 Rh/ZSM-5-AE catalyst showed Rh nanoclusters with an average particle size of 1.47 nm and more homogeneous dispersion. When the loading was increased to 0.5 wt%, the Rh nanoclusters on catalyst carriers prepared by ammonia evaporation still exhibited better dispersion and smaller particle size compared to those prepared by impregnation (Fig. S2†).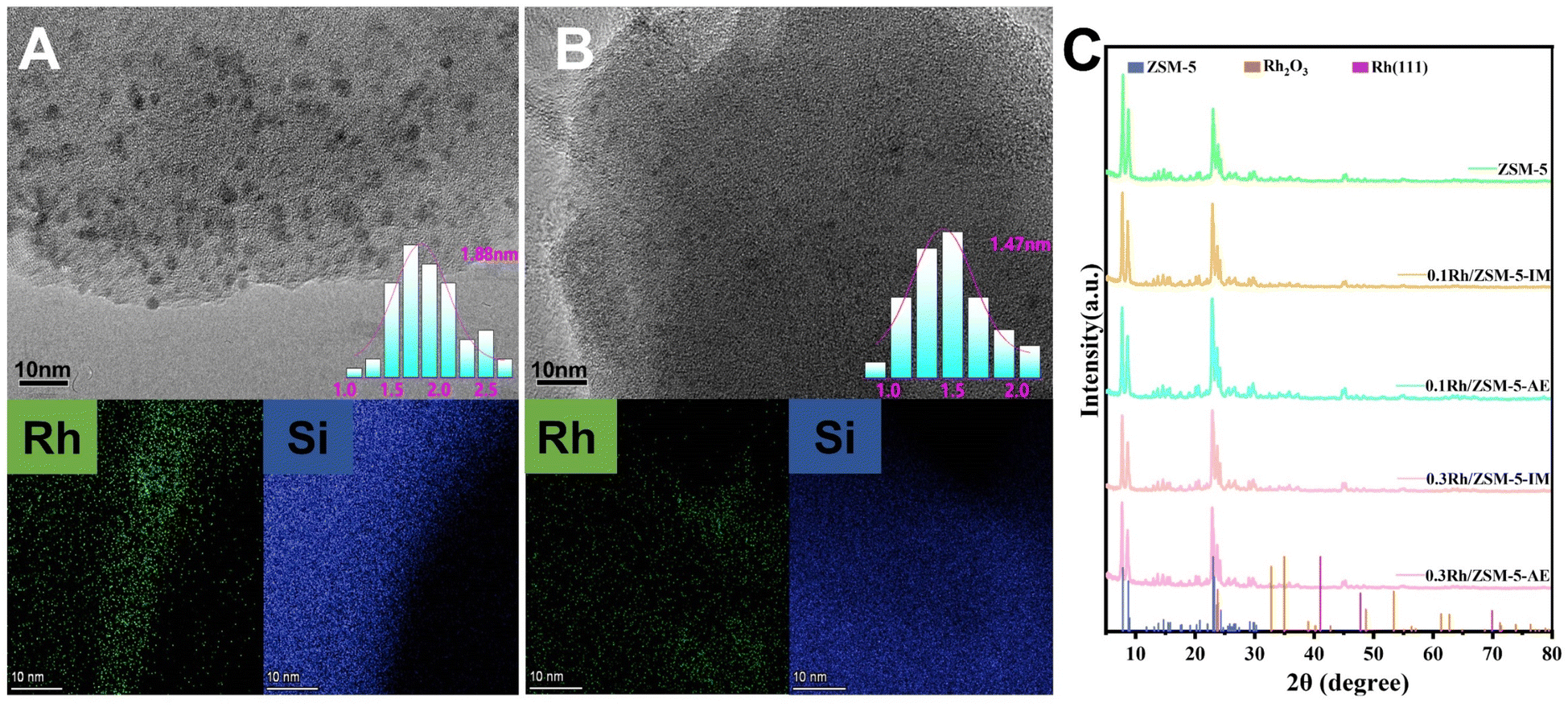 | ||
| Fig. 1 (A) TEM and EDS mapping images of 0.3 Rh/ZSM-5-IM and (B) 0.3 Rh/ZSM-5-AE. (C) XRD results of ZSM-5, 0.1 Rh/ZSM-5-IM, 0.1 Rh/ZSM-5-AE, 0.3 Rh/ZSM-5-IM, and 0.3 Rh/ZSM-5-AE. (After experimental testing, the catalysts prepared by both methods showed the highest activity at a loading level of 0.3 wt%. Therefore, 0.3 Rh/ZSM-5 was selected for characterization and analysis in this study.) (The scale bar in TEM images is 10 nm.) | ||
Fig. 1C showed the XRD patterns of catalysts with different loadings prepared by ammonia evaporation and impregnation methods, respectively. Remarkably, the XRD patterns of the four catalyst groups showed no peaks related to Rh species, especially when compared to the ZSM-5 carriers (Fig. S3†). This suggested that either Rh metal was widely distributed across the carriers or that the observed peaks resulted from the extremely small size of Rh nanoclusters and the low content of Rh metal.41
Fig. S3A† showed the XRD patterns of ZSM-5 carrier, 0.3 Rh/ZSM-5-IM and 0.3 Rh/ZSM-5-AE catalysts within the 2θ range of 5–10°. All three samples exhibited diffraction peaks around 7.8° and 8.88°, respectively, attributed to MFI-type zeolites. Further analyses indicated that the diffraction peaks of the 0.3 Rh/ZSM-5-IM catalyst closely aligned with those of the ZSM-5 carrier. This observation suggested that the impregnation method preserved the regular pore structure of the zeolite. In contrast, the diffraction peaks of the 0.3 Rh/ZSM-5-AE catalyst had evidently shifted to low-angle region. It was ascribed to the framework desilication in alkaline solution.42 Notably, despite this shift, the catalysts prepared using the ammonia evaporation method retained the characteristics of MFI-type zeolites and did not undergo any major structural changes.
EDS mapping images (Fig. 1) showed a uniform distribution of Rh species on catalyst carriers made using the AE method, unlike the larger Rh species found on those produced by impregnation. Fig. S4† presented the CO2 desorption profiles of the two catalysts, revealing desorption peaks around 450 °C due to the presence of strongly alkaline sites.43 It can be seen that the total basicity of the AE sample had increased compared to the IM sample. This might be due to the abundant hydroxyl groups on the catalyst surface of the 0.3 Rh/ZSM-5-AE catalyst after alkali treatment.44,45 The hydroxyl group may lead to a more homogeneous distribution of Rh species, hence facilitating the activation of methane. We employed in situ DRIFTS experiment to characterize the dehydration during the heating process of dried (uncalcined) 0.3 Rh/ZSM-5-AE and 0.3 Rh/ZSM-5-IM catalyst precursors, as illustrated in Fig. S5.† The intensity of the negative peaks around 3637 and 3745 cm−1 increased with rising temperature for both samples. Among them, the peak at 3637 cm−1 was attributed to the hydrogen-bonded OH group, whereas the peak around 3745 cm−1 corresponded to the isolated OH group.46–49 Upon further observation, it was observed that during dehydration, both peak intensities were significantly higher in the AE sample compared to the IM sample. This finding suggested that the introduction of ammonia into the system resulted in the generation of a substantial number of OH groups within the alkaline environment. Consequently, a greater quantity of hydroxyl species (surface-OH) was formed on the surface of the carrier. The surface-OH groups may be further converted to surface-O- groups. The evenly dispersed surface-O-groups can bind to the Rh species, resulting in more evenly dispersed Rh species on the carrier. In contrast, during the impregnation process, the Rh species only underwent limited interactions with the carriers, which resulted in significant agglomeration during subsequent calcination due to the limited number of surface-O groups. This may be the reason for the smaller Rh nanoclusters and more uniform dispersion in the catalysts prepared by ammonia evaporation.50 By analysing the yield plots (Fig. 3A and B), it was found that the formation of this difference coincided with the difference in activity of the yields.
XPS experiments were carried out to enhance the comprehension of the electronic structure of Rh species. The 3d5/2 spectra of Rh could be fitted to 309.05 eV and 308.75 eV, respectively (Fig. 2A). Based on the available literature and rhodium binding energy analyses,51,52 rhodium species in both catalysts were mainly present as Rh3+ and Rh4+. Further analysis demonstrated that the binding energy at the signal peak of rhodium species in the 0.3 Rh/ZSM-5-AE catalyst (308.75 eV) was lower than that observed at the signal peak of rhodium species in the 0.3 Rh/ZSM-5-IM catalyst (309.05 eV). For this purpose, the electronic structure of Si and Al species were explored. By fitting the Si 2p spectrum (Fig. 2B), the Si 2p spectrum showed that AE samples (102.95 eV) had a lower binding energy than IM samples (103.15 eV). However, fitting the Al 2p spectrum did not reveal any significant peak shift (Fig. S6†). Valence states of Rh (Fig. S7†) showed that the proportion of Rh3+ species in the IM samples (43.85%) were significantly lower than in the AE samples (75.83%), but the proportion of Rh0 species were higher than in the AE sample. In contrast, Rh0 species were nearly absent in the AE samples (Table S3†). The findings indicated that the Rh species in the AE sample experienced stronger electron transfer with the carrier. This electron transfer resulted in alterations in the valence condition of the Rh species, which were attributed to the supply of electrons from the Rh species to the Si species. In addition, after adequate mixing of the ammonia and the carrier, the alkaline environment partially dissolved the silicon on the carrier, forming defect sites,48,53 thereby enabling the Rh species to be embedded in the silicon defect sites. Such a procedure could also lead to alterations in the valence state of the Rh species. The aforementioned alterations in the electronic configuration of the Rh species were due to their interaction with the carrier, which was facilitated by the ammonia evaporation method. This interaction likely involved the introduction of –OH, which transformed the irregular binding of Rh with the carrier into a more regular and uniformed binding process, resulting in a more homogeneous distribution of Rh species with smaller size in the AE samples compared to the IM samples, thereby leading to a higher activity in the AE samples.
 | ||
| Fig. 2 (A) Rh 3d XPS spectra of 0.3 Rh/ZSM-5-IM and 0.3 Rh/ZSM-5-AE. (B) Si 2p XPS spectra of 0.3 Rh/ZSM-5-IM and 0.3 Rh/ZSM-5-AE. (C) H2-TPR curves of 0.3 Rh/ZSM-5-IM and 0.3 Rh/ZSM-5-AE. (D) CH4-TPD curves of 0.3 Rh/ZSM-5-IM and 0.3 Rh/ZSM-5-AE. | ||
Fig. 2C showed the experimental H2-TPR spectra of the 0.3 Rh/ZSM-5-AE and 0.3 Rh/ZSM-5-IM catalysts. The observed dual peaks at 70 °C were ascribed to a decrease in surface-reactive oxygen species.54 The two weak reduction peaks observed at approximately 260 °C and 380 °C for the 0.3 Rh/ZSM-5-IM catalysts were attributed to the reduction of RhxOy, which formed due to varying degrees of interaction between RhOx and the carrier.55–57 In contrast, the 0.3 Rh/ZSM-5-AE catalyst exhibited markedly intensive peak signals at approximately 290 °C and 370 °C compared to the 0.3 Rh/ZSM-5-IM catalyst. These increased signals in the AE sample could be originating from the stronger effect between Rh and the ZSM-5 support, which was induced by the introduction of –OH groups and the leaching of Si species (forming more defective sites) in the ammonia-involved alkaline environment during synthesis. During subsequent calcination, the rhodium precursor may populate these defective sites and interact with neighbouring Si to form –Si–O–Rh– configuration. This interaction altered the overall electronic structure of the material, making it more susceptible to reduction in the reducing environment. Consequently, this increased susceptibility led to higher H2 consumption.54 Since desilication decreased the Si/Al ratio and led to an increase in the number of acidic sites, NH3-TPD experiments were carried out on both 0.3 Rh/ZSM-5-IM and 0.3 Rh/ZSM-5-AE catalysts in order to assess the effect of alkali treatment on the acidity of the catalysts. As shown in Fig. S8,† both catalysts exhibited desorption peaks in the range of 100–250 and 350–500 °C, corresponding to the weak and strong acidic sites of the ZSM-5 carrier, respectively.58 The peak areas were proportional to the number of the acidic sites. Obviously, the AE sample had more acidic sites than the IM sample. This may be caused by the desilication of the AE sample during the alkali treatment process.59,60 The Si/Al ratio changes in 0.3 Rh/ZSM-5-AE, as presented in Table S1,† revealed that the partial dissolution of Si occurred in the zeolite framework during the ammonia evaporation process, ergo leading to the formation of defective sites. These defective sites may interact with the rhodium precursor ions to bounding, potentially influencing the morphology of the Rh species present.
Fig. 2D presented the CH4-TPD profiles of the two catalysts under investigation. Methane adsorption peaks were evident over both catalysts within the temperature range of 200 to 600 °C. Notably, the 0.3 Rh/ZSM-5-AE catalyst exhibited significantly greater adsorption peak intensity than that of the 0.3 Rh/ZSM-5-IM catalyst. The adsorption of CH4 was a crucial preliminary step for its subsequent activation and oxidation. The increased CH4 adsorption observed in the 0.3 Rh/ZSM-5-AE catalyst suggested a higher reactivity during the reaction process. This heightened reactivity was associated with a higher yield of oxygenated compounds, which can be attributed to the smaller and more homogeneous distribution of Rh species in the 0.3 Rh/ZSM-5-AE catalyst.
Catalytic properties of Rh/ZSM-5-IM and Rh/ZSM-5-AE
Methane oxidation experiments on Rh/ZSM-5-IM and Rh/ZSM-5-AE catalysts were carried out in an autoclave reactor, as shown in Fig. 3. Under identical reaction conditions, the liquid-phase products of Rh/ZSM-5-IM and Rh/ZSM-5-AE catalysts were CH3OH, HCOOH, and CH3COOH, while the gas-phase product was CO2. The study observed that at a loading level of 0.1 wt%, both catalysts exhibited low yields. However, a significant increase in yield was noted when the loading level was raised to 0.2 wt%, with the yield reaching its maximum at 0.3 wt%. The liquid-phase products of both catalysts showed volcano curves with loading, and the total yields reached a peak at a loading of 0.3 wt%. The 0.3 Rh/ZSM-5-AE catalyst produced a total liquid phase yield of 32433.33 μmol gcat−1 h−1 (Fig. 3B). Additionally, it was discovered that the AE samples demonstrated higher activity than the IM samples at equivalent loading levels. Specifically, the yield of CH3COOH was 15005.47 μmol gcat−1 h−1, which was the highest among the three products. Compared to the IM sample, the AE sample produced higher yields for all three products, particularly methanol and acetic acid.
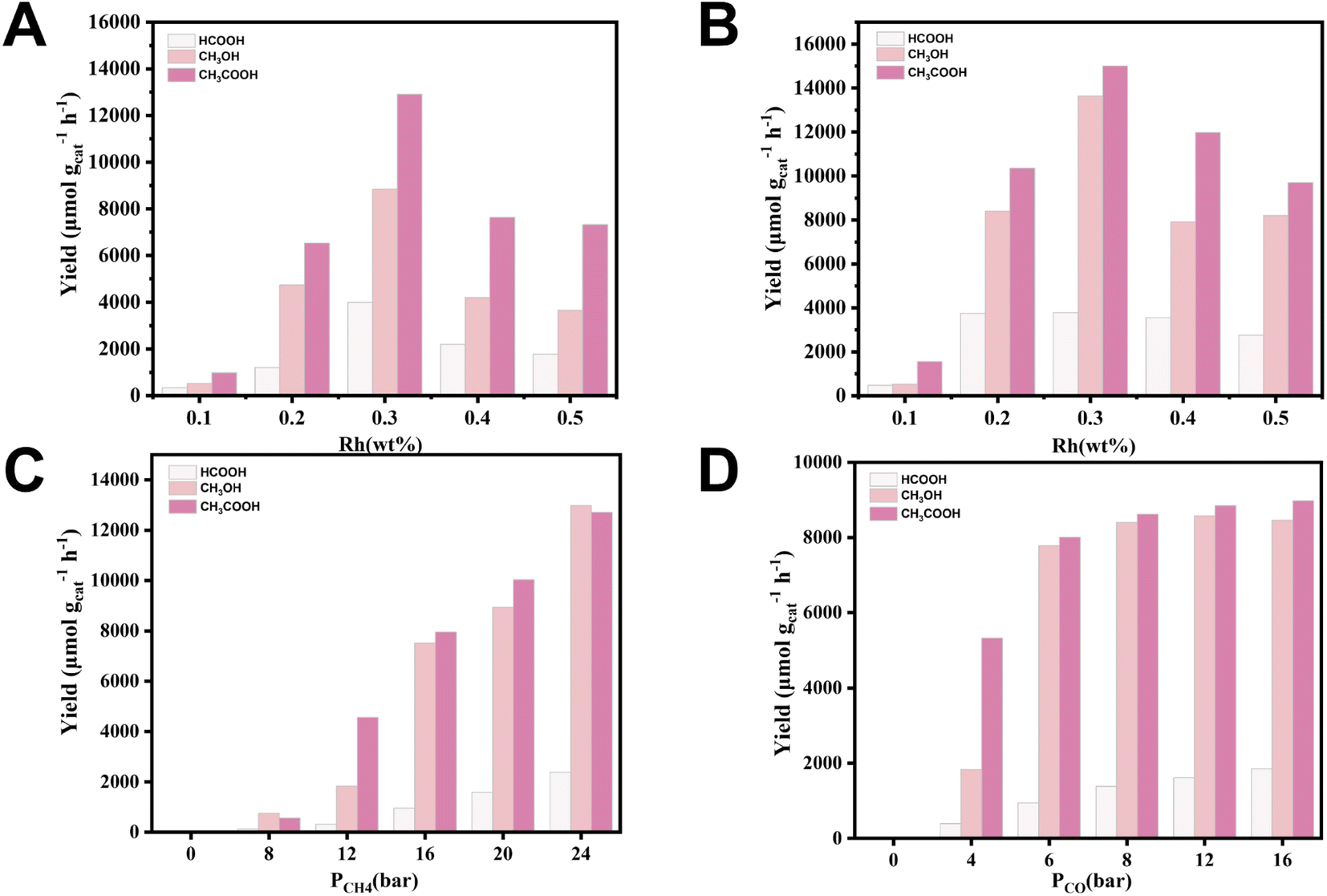 | ||
| Fig. 3 Effect of (A) different loadings for IM samples, (B) different loadings for AE samples (reaction conditions: PCH4 = 20 bar, PCO = 8 bar, PO2 = 3 bar, 20 mL water, 20 mg catalyst, 0.5 h at 240 °C); (C) different CH4 pressures for AE samples (reaction conditions: PCO = 5 bar, PO2 = 3 bar, 20 mL water, 20 mg catalyst, 0.5 h at 240 °C), and (D) different CO pressures for AE samples on the liquid-phase products (reaction conditions: PCH4 = 15 bar, PO2 = 3 bar, 20 mL water, 20 mg catalyst, 0.5 h at 240 °C). | ||
Within the experimental temperature, both catalysts exhibited low yields at 150 °C (Fig. S9†). However, yields significantly increased at 180 °C and peaked at 240 °C. After 240 °C, it is assumed that the liquid phase oxygenates become severely peroxidized, leading to a lower target products' yield. Fig. 3C demonstrated that the yield of each product increased with rising CH4 pressure. Fig. 3D revealed that the yield of formic acid increased with higher CO pressure, suggesting that the production of HCOOH was influenced by CO. The production rates of methanol and CH3COOH exhibited significant increases as the CO pressure increased from 0 bar to 8 bar. After this specific CO pressure, the methanol production rate stabilized, showing only minor fluctuations despite further increases in CO pressure. In contrast, the production rate of acetic acid continued to demonstrate a positive correlation with increasing CO pressure, suggesting that CO might played a predominating role in the generation of acetic acid. Our experiments demonstrated that no products formed in the water without CO involvement (Table S4†), indicating the necessity of CO for catalyst activation. At lower CO concentrations, the amount of CO was not sufficient, and therefore, the catalyst activation intensity was lower, which led to a lower CH3OH yield. Increasing the CO pressure promoted catalyst activation, significantly enhanced productivity. Fig. S10† showed the stability test of 0.3 Rh/ZSM-5-AE. The catalyst maintained a high yield after five cycles.
Due to the abundance of CO in the reaction atmosphere and the high reaction temperature, CO2 was inevitably detected at the end of the reaction, produced from both the oxidation of CO in the reaction atmosphere and the overoxidation of the liquid-phase products. Experiments had been conducted to verify this speculation. Only trace amount of CO2 was detected when N2 was used instead of CO, and there were also not any liquid-phase products generation (Table S4†). Yet when N2 was used instead of CH4, a large amount of CO2 was detected under the same reaction conditions (Fig. 4). To further explore the CO2 production pathway, the catalytic reaction properties were investigated using the in situ DRIFTS technique. Fig. 5A showed the infrared spectra of the catalyst under CH4 and O2 mixed atmosphere (CH4:O2 = 20:3) and mixed atmosphere of CH4, O2 and CO (CH4:O2:CO = 20:8:3), respectively. As seen from the spectra, hydroxyl peaks were formed on the catalyst surface (located at 3736 cm−1) in both reaction atmospheres.46–49 When the reaction atmosphere was only CH4 and O2, the signal of CO2 did not appear in the spectrum. This result was consistent with the extremely stable chemical nature of CH4, suggesting that these catalysts were not capable of catalytically oxidizing methane to form CO2 under forward mentioned conditions. Whereas, when the reaction atmosphere was switched to a mixture of CH4, CO, and O2, signals of both CO (located at 2112 and 2146 cm−1) and CO2 (located at 2310 and 2384 cm−1) species appeared.61,62 The above results indicated that CO can be activated on the catalyst and further converted to CO2, while the more stable CH4 was difficult to be activated by the catalyst to produce CO2. When these findings were combined with the experimental results, it could be inferred that the CO2 produced during the experiment came from both CH4 and CO, and the overoxidation of CO produced more CO2 than CH4.
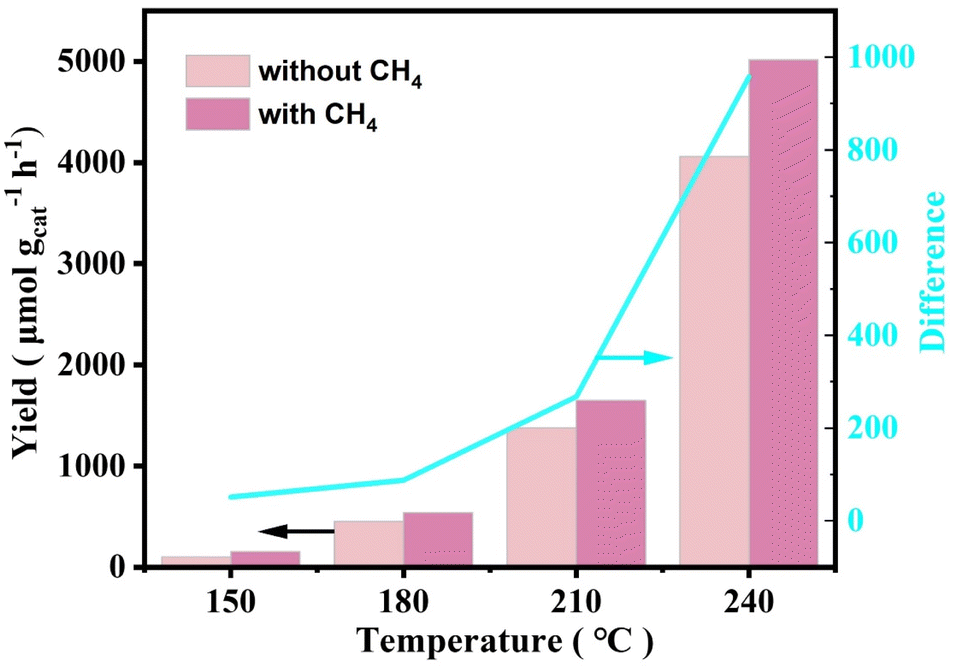 | ||
| Fig. 4 0.3 Rh/ZSM-5-AE temperature-dependent CO2 yield in CH4, O2, CO and N2, O2, CO gas atmospheres, respectively. The blue line corresponded to the right axis, indicating the difference in CO2 produced under two different reaction conditions. | ||
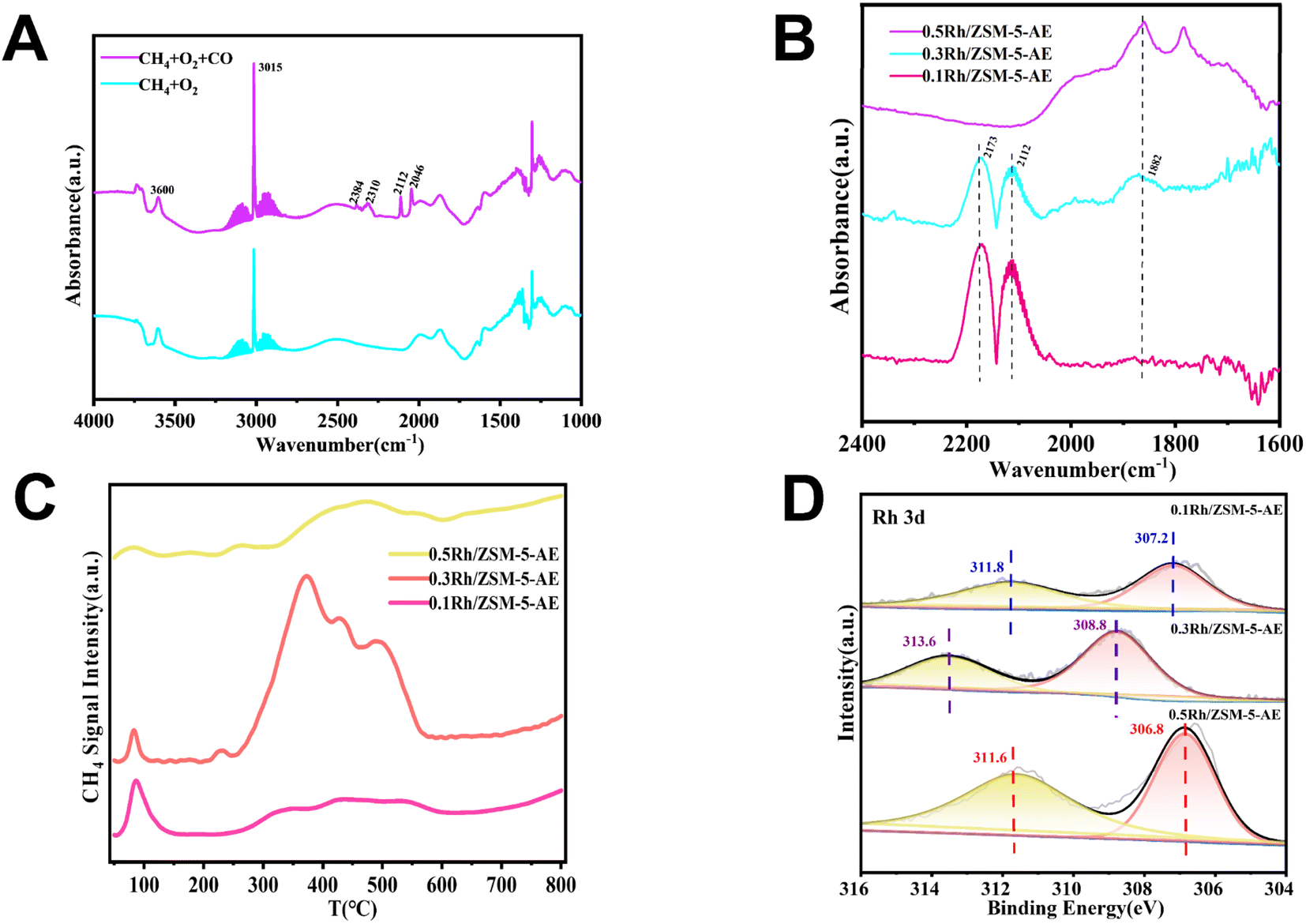 | ||
| Fig. 5 (A) In situ DRIFTS spectra of 0.3 Rh/ZSM-5-AE at 240 °C in CH4 + O2 atmosphere (20:3) and CH4 + O2 + CO (20:8:3) atmosphere, respectively. (B) In situ DRIFTS spectra of CO adsorption at 240 °C for AE samples with different loadings. (C) CH4-TPD curves of AE samples with different loadings. (D) Rh 3d XPS spectra of AE samples with different loadings. | ||
Reaction mechanism investigation
The yield results indicated that the catalytic reactivity of the catalysts synthesized through both techniques was closely linked to the size of the Rh nanoclusters, with both excessively small and large nanoclusters significantly impacting catalytic activity. At a loading of 0.1 wt%, the catalyst exhibited lower yields owing to the existence of fewer active sites. As the loading increased, the quantity of metal active sites also rose, resulting in a significant improvement in yield. However, with further increases in loading, the metals on the catalyst surface tended to agglomerate, leading to a reduction in reaction yield. Notably, the activity of AE samples was higher than that of IM samples at the same loading. TEM images (Fig. 1A, B and S1†) showed that the size of Rh species was smaller and more homogeneous in all AE samples than that in IM samples at the same loading. And no obvious Rh nanoclusters were found on the carrier surface when the loading amount was 0.1 wt%. With further elevation of loading, Rh nanoclusters appeared and the cluster size increased with the increase of loading (Fig. S2†). It could be seen that the catalyst activity presented a strong link with the particle size of the Rh species. Based on the above analyses, it was found that the catalysts synthesized using the ammonia evaporation method facilitated interactions between the Rh species and the carriers. This method introduced hydroxyl groups (–OH) and generated defective sites, which altered the disordered binding of the carriers and the active species in the IM samples. Consequently, this resulted in smaller Rh nanoclusters size and more homogeneous distributions. These changes led to improved atom utilization and subsequently increased reactivity.In order to further investigate the existing state of the loaded metal on the carrier, the CO as a probe molecule in combination with DRIFTS was employed. In an effort to negate the impact of CO, we collected the data after 30 minutes of CO exposure followed by 30 minutes of purging with Ar gas. As shown in Fig. 5B, after purging with Ar gas, the IR spectrogram of the 0.1 Rh/ZSM-5-AE catalyst displayed only peaks at 2173 and 2112 cm−1, indicative of the adsorption of CO molecules on single-atom rhodium molecules. In contrast, the IR spectrogram of the 0.3 Rh/ZSM-5-AE catalyst showed not only IR peaks at 2173 and 2112 cm−1 but also a wide peak center at 1882 cm−1, which was attributed to the bridged CO on rhodium nanoclusters.63,64 For the 0.5 Rh/ZSM-5-AE catalyst, a peak corresponding to the adsorption of CO molecules on rhodium nanoclusters was detected only at 1882 cm−1. The finding suggested a correlation between the Rh loading and the condition of Rh species atop the carrier surface. At a Rh loading of 0.1 wt%, no significant agglomeration was observed on the carrier surface, suggested that the Rh species primarily existed in a monatomic state. When the Rh loading was increased to 0.3 wt%, the Rh species were present both as single atoms and nanoclusters. Further increasing the Rh loading to 0.5 wt% resulted in the Rh species primarily forming clusters. This corresponded to the results shown in the TEM images.
The H2-TPR curves of Rh/ZSM-5-AE catalysts at various loadings levels were shown in Fig. 6. The peaks observed below 100 °C were attributed to the reduction of reactive oxygen species atop the carrier surface. Regarding the catalyst with a loading of 0.1 wt%, only weak reduction peaks were detected at 283 and 368 °C, which attributed to fewer Rh sites, resulting in less H2 consumption. As the loading increased to 0.3 wt%, more pronounced reduction peaks appeared at 293 and 371 °C, suggested that a larger number of oxidation state metal sites atop the carrier surface consumed significant amounts of H2. However, when the loading was further increased to 0.5 wt%, the peaks of reduction detected at 238 °C and 370 °C were much lower in intensity compared to the 0.3 wt% Rh/ZSM-5-AE catalyst. This indicated that with the further increase in loading, the metal sites on the catalyst surface transitioned from oxidation states to metallic states, again evidenced the significant agglomeration of the Rh species.
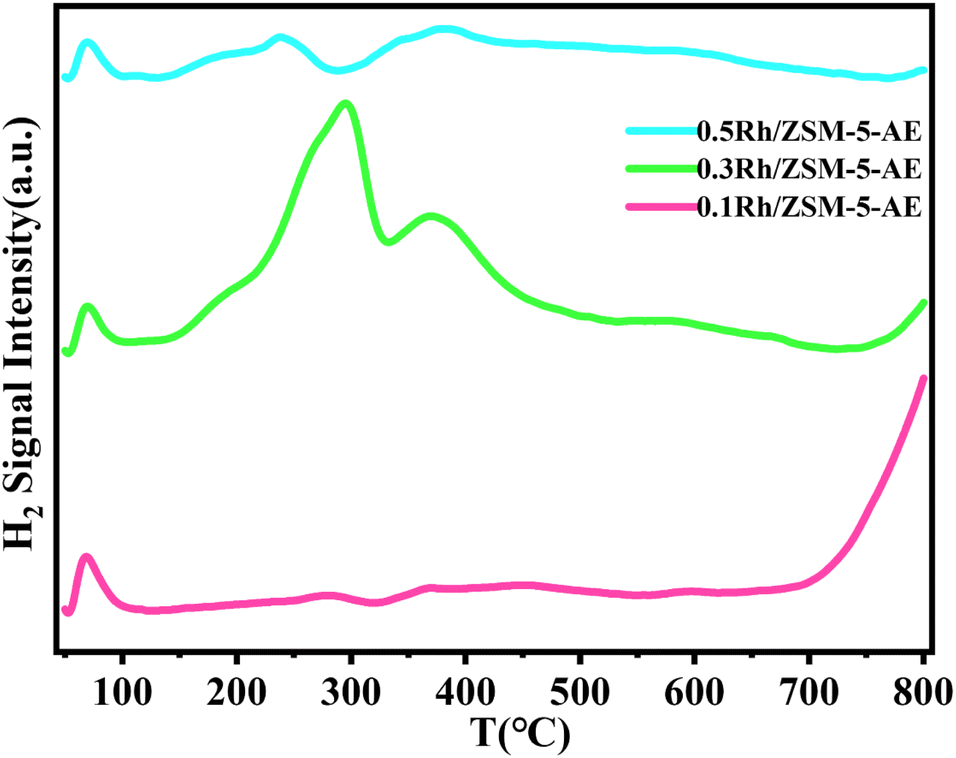 | ||
| Fig. 6 H2-TPR curves of AE samples with different loadings. | ||
The XPS experiment further corroborated the appealing conclusion from the electronic point of view. Fig. 5D showed the Rh 3d XPS patterns of Rh/ZSM-5-AE samples with varying loadings, illustrating the progression of binding energy as the loading increases. The binding energy of Rh species in the 0.1 Rh/ZSM-5-AE catalyst was 308.5 eV, which was attributed to the Rh3+ species. When the loading was increased to 0.3 wt%, the binding energy shifted to 308.8 eV, which was attributed to the mixed state with Rh3+ and Rh4+. And as the loading further increased to 0.5 wt%, the binding energy moved to 306.8 eV and the peak exhibited a substantial increase in intensity, which was attributed to the formation of Rh0 species in larger Rh nanoparticles.51,52 The state of Rh species gradually transitioned from the oxidation state to the metallic state as the loading was increased. When considering the yields, it had showed that the 0.5 Rh/ZSM-5-AE catalyst, which predominantly existed in the metallic state of Rh, demonstrated significantly higher activity compared to the 0.1 Rh/ZSM-5-AE catalyst, which primarily existed in the oxidation state of Rh. Furthermore, the 0.3 Rh/ZSM-5-AE catalyst, which featured a coexistence of both the metallic and oxidation states of Rh, exhibited the most elevated activity among the examined catalysts. The catalyst prepared through ammonia evaporation method demonstrated a further reduction in the size of Rh nanoclusters in contrast to the catalyst prepared through impregnation method. The reduction in the nanocluster size was accompanied with a significant alteration in the electronic structure of the Rh species, which was influenced by the interaction with the carrier. Specifically, the ammonia evaporation process facilitated the partial leaching of Si, leading to the formation of a mixed state of Rh3+ and Rh4+, which significantly enhanced the reactivity of the catalyst. The above conclusions indicated that the reactive center of activity of the catalyst was composed of Rh single atoms and Rh nanoclusters in the form of oxidation state in a specific range of size (1.4–1.8 nm, with the best size around 1.5 nm), and that altering the electronic structure of Rh and controlling the size of Rh nanoclusters will further improve the activity. This was supported by the CH4-TPD profiles of AE samples with varying loadings, as shown in Fig. 5C, which demonstrated that methane adsorption capacity peaked at a loading of 0.3 wt%, correlating with increased reactivity. Therefore, controlling the particle size of Rh species would enhance CH4 adsorption capacity, and eventually improve the overall catalytic activity.
In addition, an experiment using N2 instead of CH4 and 20 μL of CH3OH in 10 mL of H2O was conducted with all other reaction conditions unchanged. Upon the conclusion of the reaction, no HCOOH or CH3COOH was detected in the liquid solution. This indicated that CH3OH was likely not an intermediate in the formation of HCOOH and CH3COOH, nor was HCOOH likely an intermediate in the formation of CH3COOH. Instead, each compound probably followed an independent pathway. The yield plots in Fig. 3D demonstrated a positive correlation between the yields of the acetic acid and CO pressure. Therefore, a potential route for the generation of CH3COOH was proposed. As illustrated in Fig. 7, the methane (CH4) underwent activation by Rh species, resulting in the formation of methyl species, which served as the methyl source for CH3COOH. The carboxyl group in CH3COOH was sourced from carbon monoxide (CO). CO was inserted into Rh–CH3 through carbonylation, leading to the formation of Rh–COCH3 species. Subsequently, water (H2O) was dissociated on Rh species, producing OH* and H* species. The OH* species then combined with Rh–COCH3 species to form Rh–COOHCH3. Finally, Rh–COOHCH3 detached from the Rh species, yielding acetic acid (CH3COOH). We increased the overall activity by constraining the Rh species size and allowing the Rh species to interact with the carrier by ammonia evaporation.
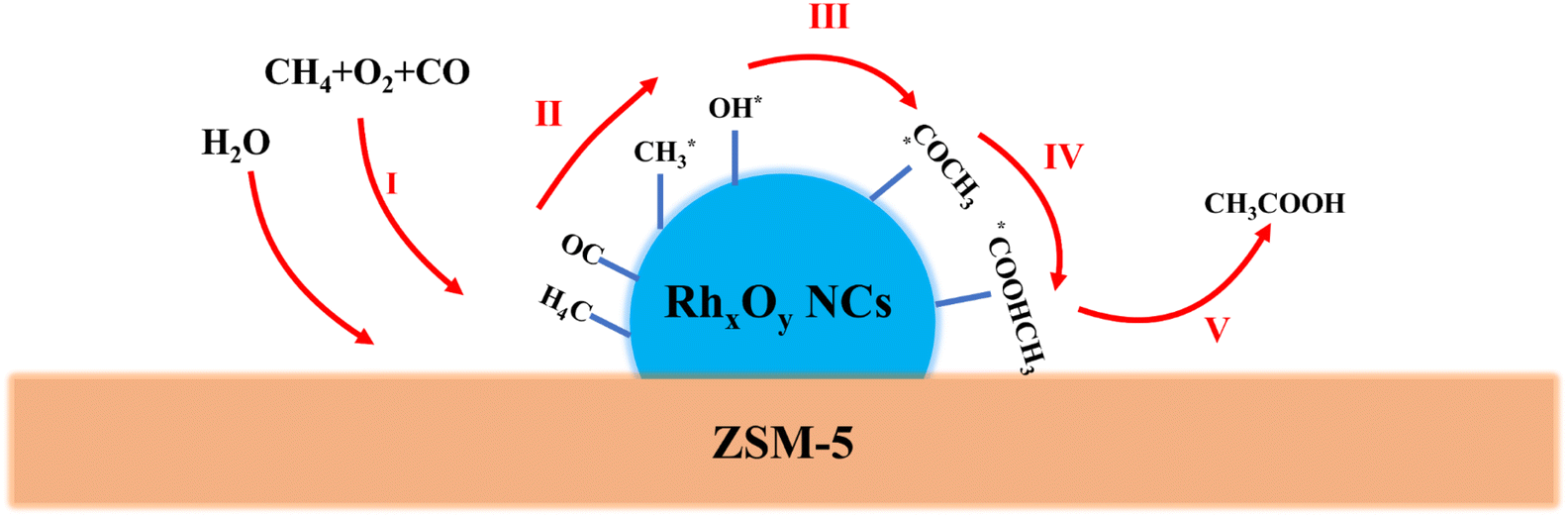 | ||
| Fig. 7 Schematic of one possible reaction pathway. | ||
Conclusions
In conclusion, Rh–ZSM-5 catalysts with specific particle size for efficient CH4 partial oxidation were prepared using the ammonia evaporation method instead of the conventional impregnation method. This approach aimed to achieve the oxidation of methane directly into oxygenated compounds, including methanol, formic acid, and acetic acid, facilitated by CO gas. During the ammonia evaporation, the dissolution of Si species produced defective sites and introduced enriched surface hydroxyl species, which enabled Rh ions to interact with the carriers. This interaction altered the electronic structure of the carrier-activated metal and modified the disordered Rh precursor adsorption form, resulting in a higher degree of dispersion and a more uniform particle size of Rh species. Consequently, this promoted CH4 adsorption and significantly increased the reactivity. Additionally, it was demonstrated that varying the Rh loading could alter the valence state of Rh species, as well as the size of the reactive sites, thereby significantly affecting the reactivity. Experimental investigations revealed that CO gas acts as a co-oxidant in the reaction, and the CO2 products originated from the overoxidation of both CO and CH4. These findings provide a valuable reference for designing carrier-active metal synergies and offer a feasible and constructive approach to designing catalysts based on noble metals for the oxidation of methane directly.Data availability
All relevant data are available from the authors on reasonable request.Author contributions
W. L. and X. Z. conceived and supervised the research. X. Z. designed and performed the experiments and data analysis. X. Z. and J. J. wrote the paper. All authors discussed the results and commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Research and Development Projects in Anhui Province (2022107020013), the Institute of Energy, Hefei Comprehensive National Science Center under Grant No. 21KZS219 and the Major Science and Technology Projects of Anhui Province (202003a05020022).Notes and references
- B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232–1237 CrossRef CAS PubMed.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2022, 123, 6359–6411 CrossRef PubMed.
- K. Chen, W. Li, G. Guo, C. Zhu, W. Wu and L. Yuan, ACS Omega, 2022, 7, 8536–8546 CrossRef CAS PubMed.
- Z. R. Chong, S. H. B. Yang, P. Babu, P. Linga and X.-S. Li, Appl. Energy, 2016, 162, 1633–1652 CrossRef.
- L. Brody, B. Mosevitzky Lis, A. Pérez Ortiz, M. Kosari, K. Vogt-Lowell, S. Portillo, R. Schomäcker, I. E. Wachs and F. Li, ACS Catal., 2024, 14, 13112–13125 Search PubMed.
- K. W. Yizengaw, T. A. Abay, D. W. Ayele and J.-C. Jiang, RSC Adv., 2022, 12, 23736–23746 RSC.
- P. Kumar, T. A. Al-Attas, J. Hu and M. G. Kibria, ACS Nano, 2022, 16, 8557–8618 CrossRef CAS PubMed.
- J. Niu, Z. Jin, J. Zhang, H. Liu, Y. Jin and J. Ran, Mol. Catal., 2023, 547, 113388 CrossRef CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- J. Peng, W. Liao, X. Fu, J. Li, S. Shang and Q. Zhang, Mol. Catal., 2024, 569, 114589 Search PubMed.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592–1602 RSC.
- X. Bu, J. Ran, J. Niu, Z. Ou, L. Tang and X. Huang, Mol. Catal., 2021, 515, 111891 CrossRef CAS.
- Z. Zhang, J. Li, T. Yi, L. Sun, Y. Zhang, X. Hu, W. Cui and X. Yang, Chin. J. Catal., 2018, 39, 1228–1239 CrossRef CAS.
- W. H. Saputera, G. Yuniar and D. Sasongko, RSC Adv., 2024, 14, 8740–8751 RSC.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- R. Horn and R. Schlögl, Catal. Lett., 2014, 145, 23–39 CrossRef.
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS PubMed.
- P. Kumar, P. Antal, X. Wang, J. Wang, D. Trivedi, O. F. Fellner, Y. A. Wu, I. Nemec, V. T. Santana, J. Kopp, P. Neugebauer, J. Hu, M. G. Kibria and S. Kumar, Small, 2024, 20, 2304574 CrossRef CAS PubMed.
- W. Wu, W. Li, M. Wu, H. Zhang, C. Zhu and Y. Jiang, RSC Adv., 2023, 13, 5393–5404 RSC.
- S. Arora and P. Gupta, Chem.–Asian J., 2024, 19, e202400282 CrossRef CAS PubMed.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- T. N.-M. Le, T. B. N. Le, P. T. Nguyen, T. T. Nguyen, Q. N. Tran, T. T. Nguyen, Y. Kawazoe, T. B. Phan and D. M. Nguyen, RSC Adv., 2023, 13, 15926–15933 RSC.
- T. Nakano, T. Abe, T. Matsumoto, K. Kimura, G. Nakamura, S. Hayami, Y. Shiota, K. Yoshizawa and S. Ogo, RSC Adv., 2022, 12, 12253–12257 RSC.
- E. M. Alayon, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Chem. Commun., 2012, 48, 404–406 RSC.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 138, 14–22 CrossRef CAS.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- C. Bo, L. Zhang, X. Liu, H. Chang, Y. Sun, X. Zhang, T. Tan and L. Piao, Nano Res., 2023, 17, 2473–2480 CrossRef.
- R. McVicker, N. Agarwal, S. J. Freakley, Q. He, S. Althahban, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Catal. Today, 2020, 342, 32–38 CrossRef CAS.
- W. Li, Z. Li, H. Shen, J. Cai, H. Jing, S. Xin, Z. Cao, Z. Xie, D. Li, H. Zhang and Z. Zhao, ACS Catal., 2024, 14, 10689–10700 CrossRef CAS.
- B. Yu, L. Cheng, J. Wu, B. Yang, H. Li, J. Xu, Y. Zhang, C. Pan, X.-M. Cao, Y. Zhu and Y. Lou, Energy Environ. Sci., 2024, 17, 8127–8139 RSC.
- T. Moteki, N. Tominaga and M. Ogura, ChemCatChem, 2020, 12, 2957–2961 CrossRef CAS.
- Y. Lv, J. Guo, C. Ding, Y. Yan, H. Chen, L. Ma, J. Wang, Y. Meng, Z. Ma, P. Liu and K. Zhang, Mol. Catal., 2023, 542, 113131 CrossRef CAS.
- T. Moteki, N. Tominaga, N. Tsunoji, T. Yokoi and M. Ogura, Chem. Lett., 2021, 50, 1597–1600 CrossRef CAS.
- R. J. Bunting, J. Thompson and P. Hu, Phys. Chem. Chem. Phys., 2020, 22, 11686–11694 RSC.
- T. Moteki, N. Tominaga and M. Ogura, Appl. Catal., B, 2022, 300, 120742 CrossRef CAS.
- X. Zhao, H.-H. Xu, L.-Y. Xu, C.-H. Wen, X.-D. Guo, C. Tang, W.-Z. Jia, M.-F. Luo and J. Chen, Mol. Catal., 2024, 561, 114193 Search PubMed.
- R. Gautam, N. Sharma, K. Saini and S. Saravanamurugan, Mol. Catal., 2024, 569, 114610 Search PubMed.
- Y. Guo, L. Zhao, M. Bi, B. Zhang, K. Guo, L. Miao, C. Cai, L. Chen, X. Shi and W. Cheng, Mol. Catal., 2023, 540, 113042 CrossRef CAS.
- R. State, M. Scurtu, A. Miyazaki, F. Papa, I. Atkinson, C. Munteanu and I. Balint, Arabian J. Chem., 2017, 10, 975–984 CrossRef CAS.
- R. Feng, X. Yan, X. Hu, Y. Wang, Z. Li, K. Hou and J. Lin, J. Porous Mater., 2018, 25, 1743–1756 CrossRef CAS.
- Y. Shi, Q. Gu, Y. Zhao, Y. Ren, B. Yang, J. Xu, Y. Zhang, C. Pan, Y. Zhu and Y. Lou, Chem. Eng. J., 2024, 485, 150093 CrossRef CAS.
- Y. Cui, B. Chen, L. Xu, M. Chen, C.-e. Wu, J. Qiu, G. Cheng, N. Wang, J. Xu and X. Hu, Fuel, 2023, 334, 126783 CrossRef CAS.
- B. Boukoussa, A. Hakiki, N. Bouazizi, A.-P. Beltrao-Nunes, F. Launay, A. Pailleret, F. Pillier, A. Bengueddach, R. Hamacha and A. Azzouz, J. Mol. Struct., 2019, 1191, 175–182 CrossRef CAS.
- S. M. Jung and P. Grange, Appl. Surf. Sci., 2004, 221, 167–177 CrossRef CAS.
- J. Zhang, T. a. Zhang, S. Feng and S. Lin, Arabian J. Chem., 2022, 15, 103718 Search PubMed.
- H. Yun, Y. Zhao, X. Kan and G. Li, Catal. Lett., 2022, 153, 364–377 CrossRef.
- A. Kondo, R. Kurosawa, J. Ryu, M. Matsuoka and M. Takeuchi, J. Phys. Chem. C, 2021, 125, 10937–10947 CrossRef CAS.
- X. Fu, Y. Liu, Q. Liu, Z. Liu and Z. Peng, Catalysts, 2022, 12, 276 CrossRef CAS.
- Y. Wang, Z. Song, D. Ma, H. Y. Luo, D. B. Liang and X. H. Bao, J. Mol. Catal. A:Chem., 1999, 149, 51–61 CrossRef CAS.
- R. Toyoshima, K. Ueda, Y. Koda, H. Kodama, H. Sumida, K. Mase and H. Kondoh, J. Phys. D Appl. Phys., 2021, 54, 204005 CrossRef CAS.
- J. Pang, M. Zheng, C. Wang, X. Yang, H. Liu, X. Liu, J. Sun, Y. Wang and T. Zhang, ACS Catal., 2020, 10, 13624–13629 CrossRef CAS.
- J. H. Lee, M. J. Kim, E. J. Lee, D.-W. Lee, C. H. Kim and K.-Y. Lee, Appl. Surf. Sci., 2022, 572, 151504 CrossRef CAS.
- T. Montini, A. M. Condò, N. Hickey, F. C. Lovey, L. De Rogatis, P. Fornasiero and M. Graziani, Appl. Catal., B, 2007, 73, 84–97 CrossRef CAS.
- K. Lejaeghere, V. Van Speybroeck, G. Van Oost and S. Cottenier, Crit. Rev. Solid State Mater. Sci., 2013, 39, 1–24 Search PubMed.
- M. Piumetti, M. Hussain, D. Fino and N. Russo, Appl. Catal., B, 2015, 165, 158–168 CrossRef CAS.
- F. Jing, X. Lv, Y. Pi, Y. Zhang, H. Xiang and S. Luo, Mol. Catal., 2025, 572, 114768 Search PubMed.
- F. Meng, Y. Wang, S. Wang and S. Wang, React. Kinet., Mech. Catal., 2016, 119, 671–683 CrossRef CAS.
- Y. Wei, P. E. de Jongh, M. L. M. Bonati, D. J. Law, G. J. Sunley and K. P. de Jong, Appl. Catal., A, 2015, 504, 211–219 CrossRef CAS.
- W. Zhou, Z. Ma, S. Guo, M. Wang, J. Wang, M. Xia, L. Jia, B. Hou, D. Li and Y. Zhao, Appl. Surf. Sci., 2018, 427, 867–873 CrossRef CAS.
- D. Wu, S. Zhou, C. Du, J. Li, J. Huang, H.-x. Shen, A. K. Datye, S. Jiang, J. T. Miller, S. Lin and H. Xiong, Nano Res., 2023, 17, 397–406 CrossRef.
- H. Wang, W. Xin, Q. Wang, X. Zheng, Z. Lu, R. Pei, P. He and X. Dong, Catal. Commun., 2022, 162, 106374 CrossRef CAS.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08602c |
| This journal is © The Royal Society of Chemistry 2025 |