DOI:
10.1039/D4RA07393B
(Paper)
RSC Adv., 2025,
15, 1311-1322
Non-metal cocatalyst CNT-modified emerging g-C3N4 for enhanced treatment of waste drilling fluid filtrate†
Received
15th October 2024
, Accepted 9th January 2025
First published on 15th January 2025
Abstract
Loading with non-metal cocatalysts to regulate interfacial charge transfer and separation has become a prominent focus in current research. In this study, g-C3N4/CNT composites loaded with non-metallic cocatalysts were prepared through in situ pyrolysis using urea and CNTs. Various characterization techniques, including transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS), photoelectrochemical (PEC) analysis, fluorescence lifetime spectroscopy (TRPL), electron paramagnetic resonance spectroscopy (ESR), and photoluminescence (PL) spectroscopy, were employed to analyze the sample's microstructure, phase composition, elemental chemical states, and photoelectronic properties. The photocatalytic performance of the g-C3N4/CNT composite in the degradation of actual drilling hydraulic filtrate was evaluated under simulated sunlight irradiation, with the degradation results assessed through COD and chroma measurements. The findings indicate that in g-C3N4/CNT composites loaded with non-metal cocatalysts, the photocatalytic performance improves significantly as the CNT content increases. At a CNT mass fraction of 6.0%, the composite achieves optimal performance, with a COD degradation rate of 0.3742 h−1 and a chroma degradation rate of 81%. These results offer valuable insights into enhancing photocatalytic treatment of waste drilling hydraulic filtrate using g-C3N4-based non-metallic cocatalysts.
1. Introduction
Drilling fluid is crucial in the drilling technology, and as the number and depth of unconventional wells increase, the composition of high-performance drilling fluids has become more complex, diverse, and multifunctional.1–3 It is widely recognized that high-performance drilling fluids heavily rely on the incorporation of various chemical treatment agents.4,5 However, the chemicals in waste drilling fluid have resulted in significant ecological pollution. For example, complex chemical treatment agents are a direct cause of the extremely high chemical oxygen demand (COD), deep chromaticity, and high turbidity in drilling fluids.6–8 Therefore, how to reduce COD and other emission indicators of waste drilling fluid has become the biggest challenge. Furthermore, the development of green and environmentally friendly photocatalysts has become a prominent topic in current research.
As an emerging semiconductor photocatalyst, graphitic carbon nitride (g-C3N4) has garnered significant attention for its straightforward synthesis, broad light absorption spectrum, and consistent photocatalytic efficacy.9–11 However, the practical application of g-C3N4 is impeded by challenges such as significant charge recombination, short photogenerated charge transfer pathways, and a deficiency in surface catalytic active sites.12–14 Currently, to address the problems faced by the photocatalyst g-C3N4, researchers have employed various strategies to enhance its photocatalytic efficiency. These include modifying its morphology,15 tuning the band gap,16 constructing heterojunctions,17–19 and incorporating cocatalysts.20,21 Cocatalysts are indispensable and of pivotal importance for regulating surface charge to participate in catalytic reactions.11 Generally, noble metals such as Au, Ag and Pt are used as cocatalysts for g-C3N4.22 The difference the Fermi level of the between metal cocatalyst and g-C3N4 can stimulate charge flow, resulting in Fermi level balance, which then produces energy band bending and a Schottky barrier.23–25 The formation of the Schottky barrier can effectively inhibit the backflow of photogenerated electrons, thereby empowering more photoelectrons to participate in photocatalytic reactions.26–28 However, despite their ability to significantly enhance the photodegradation efficiency of g-C3N4, the scarcity of noble metals remains a key barrier to their widespread use in practical applications. Fortunately, noble metal-based co-catalysts, such as transition metal phosphide (CoP)29 and sulfide (PtS2),30 as well as non-metal carbon-based materials like graphene (GO)31 and carbon nanotubes (CNT),32 similarly form Schottky barriers to enhance charge separation efficiency on g-C3N4. For example, GO as a co-catalyst for g-C3N4, increasing the rate of hydrogen production from a methanol aqueous solution under visible light irradiation (λ > 420 nm) by 3.07 times compared to pure g-C3N4.33 Furthermore, photoluminescence spectra and transient photocurrent response measurements confirmed that even a small amount of graphene-modified g-C3N4 can significantly enhance visible light catalytic activity. However, compared to CNT co-catalysts, GO exhibits poorer conductivity and stability.34 Clearly, selecting CNT as the co-catalyst for g-C3N4 not only extends the light absorption range and promotes stable separation of photogenerated charges, but also eliminates the need for noble metals.35,36
Herein, the g-C3N4/CNT composite was synthesized in situ using a mixture of pyrolyzed urea and CNTs. The micromorphology, structural components, and optical properties of the composites, along with the generation and migration pathways of photogenerated charges, were thoroughly analyzed using various characterization techniques. In addition, the COD and chromaticity values of the actual waste drilling fluid filtrate were used to evaluate the photodegradation performance of the g-C3N4/CNT composite, achieving a COD removal rate of 95.0%. Finally, the mechanism of the non-metallic co-catalyst CNT in the photocatalytic system was proposed, with the aim of providing insights into the degradation of waste drilling fluids.
2. Experimental methods
2.1 Chemicals
Analytically pure urea, carbon nanotubes (CNTs), concentrated nitric acid (HNO3), concentrated sulfuric acid (H2SO4), silver sulfate (Ag2SO4), silver sulfate (HgSO4), potassium dichromate (K2Cr2O7), sodium sulfate (Na2SO4), and ethanol (C2H5OH) were purchased from Shanghai Aladdin Company. Meanwhile, deionized water was used throughout the experiment.
2.2 Preparation of photocatalysts
2.2.1 Preparation of modified CNTs37. 1.0 g of CNT was added to 100 mL of deionized water and ultrasonicated for 30 minutes. After high-speed centrifugation, the solid was transferred to 100 mL of concentrated HNO3 and stirred magnetically for 6 hours. The mixture was then rinsed with a large volume of water, followed by suction filtration and washing until neutral. The remaining black solid was collected and dried in an oven at 50 °C for 12 h to obtain the modified CNTs.
2.2.2 Preparation of g-C3N4/CNT composites. Place 300.0 mg of modified CNT and 10.0 g of urea in a mortar and grind thoroughly. Transfer the mixture to a crucible, wrap tightly with aluminum foil, and pyrolyze in a tube furnace. The facile synthesis of g-C3N4/CNT composites is shown in Scheme 1. Initially, raise the temperature from 25 °C to 250 °C at a rate of 2 °C min−1 and maintain this temperature for 1 hour. Then, increase the temperature to 350 °C and hold for 2 h, followed by a further increase to 550 °C, maintaining this temperature for another 2 h. After cooling to room temperature, collect the solid, centrifuge, wash with deionized water 3 to 4 times, and dry at 60 °C for 12 h. The obtained product is designated as g-C3N4/3.0 wt% CNT. Vary the CNT content to prepare additional samples, and synthesize pure g-C3N4 without adding CNT.
 |
| | Scheme 1 Synthesis of g-C3N4/CNT composites. | |
2.3 Characterization
X-ray powder diffraction (XRD) was performed using a German Bruker D8 Advance X-ray diffractometer with a Cu Kα radiation source. The microscopic morphology and elemental composition were examined using a Japanese JEOL JEM-F200 transmission electron microscope (TEM), a high-resolution transmission electron microscope (HR-TEM) combined with an energy dispersive spectrometer (EDS). A Thermo Scientific Phenom ProX scanning electron microscope from the Netherlands was used for scanning electron microscopy. X-ray photoelectron spectroscopy (XPS) was conducted with a US-based Thermo Scientific K-Alpha instrument to analyze the surface electronic state and composition. Fourier transform infrared spectroscopy (FTIR) was carried out with a US Thermo Fisher Scientific Nicolet iS20. The fully automatic specific surface area and porosity analysis (BET) was performed using an American Quantachrome Autosorb IQ instrument. Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) was tested using the UV-2500 model from Japan's Shimadzu Corporation. Fluorescence spectrum (PL) measurements were conducted with the F-7000 DC-0506 model from Hitachi High-Technologies Corporation (excitation wavelength: 320 nm), and the time-resolved photoluminescence (TRPL) was measured using an FLS980 spectrometer from Edinburgh Instruments, UK. Photoelectrochemical measurements were carried out using a CHI660E electrochemical workstation from Shanghai Chenhua Instrument Company. The electron spin resonance spectrometer (ESR) used was the EMX-PLUS model from German Bruker Company, and the xenon lamp was a PLS-SXE 300 model from Beijing Perfect Light Technology Company. Chemical oxygen demand (COD) and chromaticity tests were conducted using the GNST-900 water quality analyzer from China Suijing Environmental Protection Technology Company.
2.4 Photocatalytic tests
2.4.1 Photocatalytic degradation experiment of waste drilling fluid. A 50 mg sample was added to a centrifuge tube containing 100 mL of waste drilling filtrate, dispersed evenly, and then transferred to a quartz reactor placed on a magnetic stirrer. The reactor was connected to a 30 °C constant-temperature circulating cooling system to maintain a stable temperature. The mixture was kept in the dark for 1 h to allow the adsorption–desorption equilibrium between the sample and the waste drilling fluid to be reached. A 300 W xenon lamp was used as the light source for illumination. Samples of 1.5 mL were taken from the suspension every hour, centrifuged at high speed, and the supernatant was collected for testing.
2.4.2 Chromaticity and COD test experiment. Firstly, the chromaticity of the waste drilling fluid was tested on the GNST-900 water quality analyzer to evaluate the degradation add 1 mL of the supernatant to a 100 mL volumetric flask for dilution. For testing, prepare two digestion tubes: into the first, add 0.2 mL of the diluted sample, 1.8 mL of distilled water, and 2 mL of pre-prepared reagents (details in ESI†); into the second tube (reference), add 2 mL of distilled water and 2 mL of the same pre-prepared reagents. Place both digestion tubes into a digestion instrument and digest at 165 °C for 20 min. After cooling to room temperature, measure the COD using a water quality analyzer.
3. Results and discussion
3.1 Microstructure and elemental composition
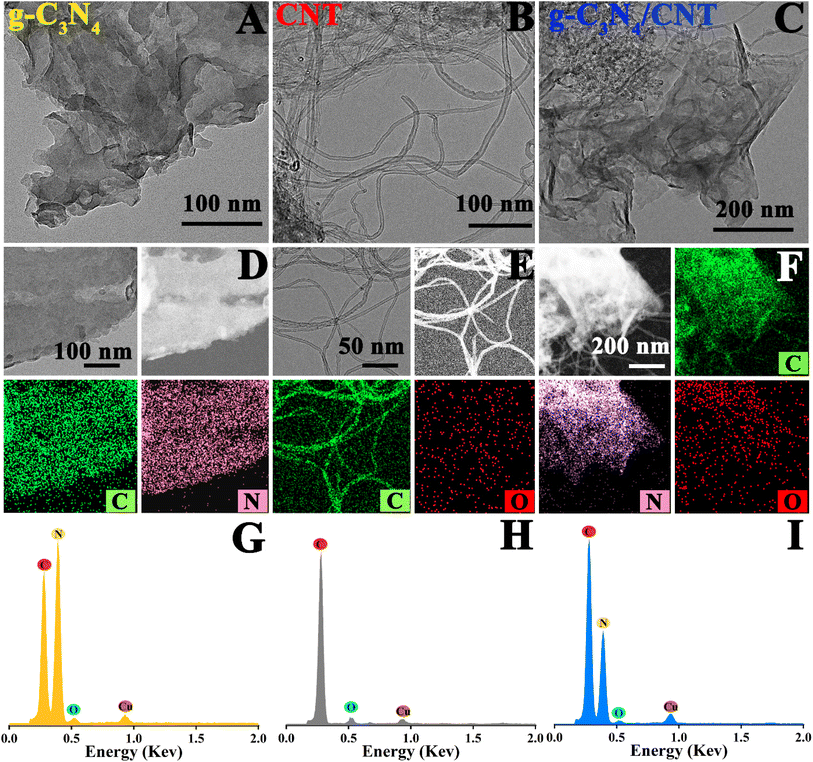
Fig. 1 shows the SEM image of the as-prepared sample. In Fig. 1A, the g-C3N4 exhibits a nanosheet stacking structure. The corresponding elemental distribution diagram in Fig. 1D reveals that the material is composed of C and N elements, as further confirmed by the EDS spectrum in Fig. 1G. Likewise, Fig. 1B illustrates that the CNT has a nanotube structure, with the elemental distribution diagram in Fig. 1E showing it is made up of C and O elements, which is corroborated by the EDS spectrum. For the g-C3N4/6.0 wt% CNT composite, Fig. 1C demonstrates that the nanosheets and nanotubes form a composite structure. The corresponding elemental distribution diagram, shown in Fig. 1F, indicates the composite consists of C, N, and O elements, which is confirmed by the EDS spectrum. Furthermore, the presence of Au, introduced during the testing process, is also detected. These findings confirm the successful preparation of g-C3N4/6.0 wt% CNT composites through the in situ pyrolysis of urea and modified carbon nanotubes.
 |
| | Fig. 1 SEM images of (A–C) g-C3N4, CNT and g-C3N4/CNT; the corresponding elemental mappings (D–F); and the EDS spectrum (G–I). | |
Fig. 2 presents the TEM images of the prepared samples. As shown in Fig. 2A, g-C3N4 exhibits a porous nanosheet structure, which aligns with previous literature reports.12 In Fig. 2D, the elemental mapping of the g-C3N4 nanosheets reveals that they are composed of C and N elements, while Fig. 2G displays the EDS spectrum, supporting this conclusion. Fig. 2B shows the image of CNT nanotubes with a diameter of approximately 10 nm. In combination with the elemental mapping in Fig. 2E and the EDS in Fig. 2H, it is evident that the composite is primarily composed of C and O elements. Fig. 2C depicts the composite prepared via pyrolysis of urea and modified CNT, where the close interaction between the two materials forming the nanocomposite is clearly visible. Fig. 2F provides an elemental mapping that shows the distribution of C, N, and O elements. The elemental composition is confirmed by the EDS spectrum, with the Cu element originating from the supporting copper mesh. In summary, the g-C3N4/6.0 wt% CNT composite, successfully prepared by pyrolysis, demonstrates strong interfacial contact, which facilitates the transport and migration of photogenerated carriers. This allows more photogenerated charges to participate in the photodegradation of drilling hydraulic filtrate, thereby significantly enhancing the photocatalytic performance of g-C3N4.
 |
| | Fig. 2 TEM images of (A–C) g-C3N4, CNT and g-C3N4/CNT; the corresponding elemental mappings (D–F); and the EDS spectrum (G–I). | |
3.2 The crystal structure and chemical states
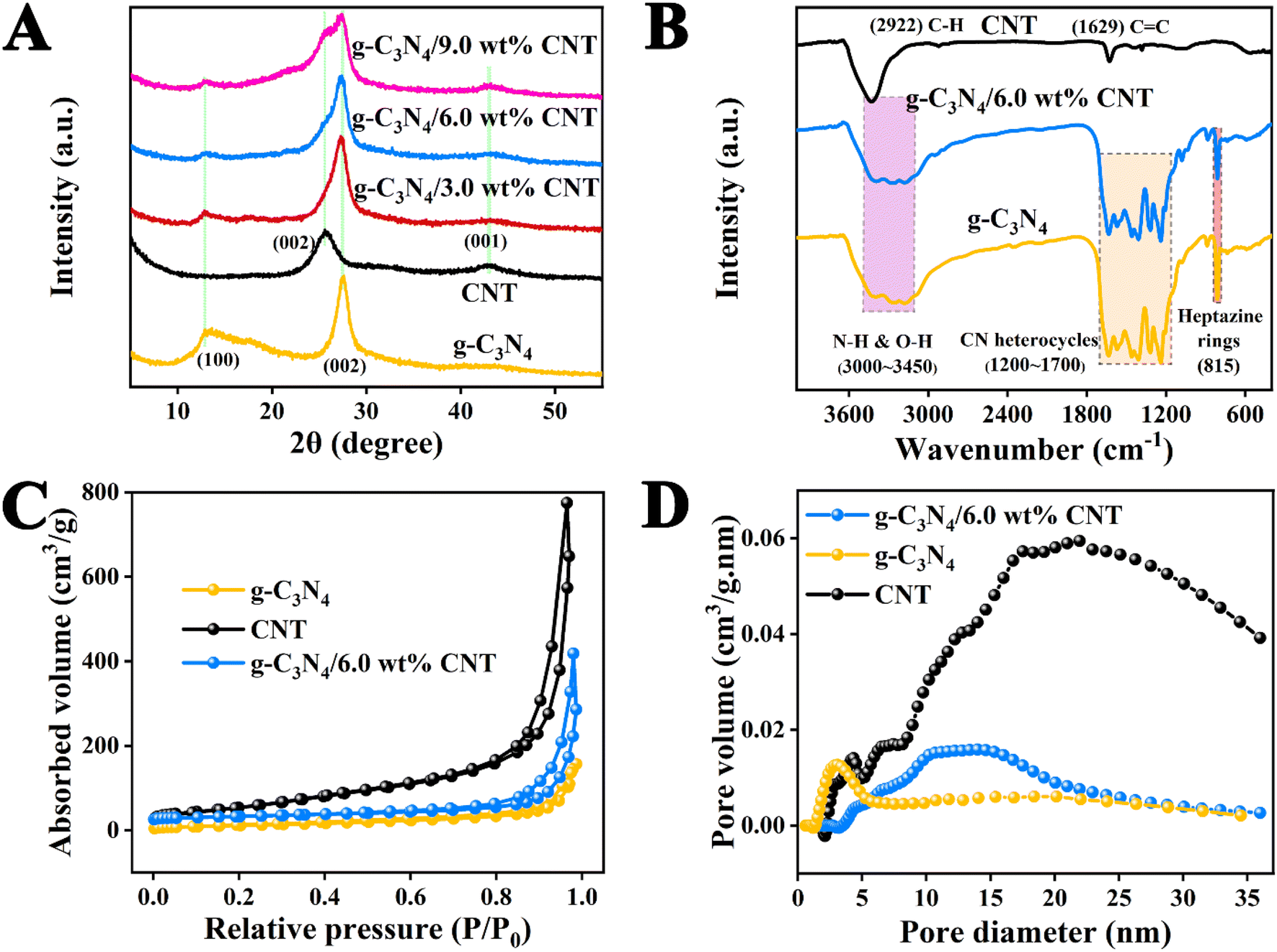
The XRD analysis of the samples provides key insights into the crystalline structure of the materials involved in Fig. 3A. The diffraction peaks at 2θ = 13.20° and 27.54° for pure g-C3N4 nanosheets correspond to the (100) and (002) planes, indicating the intra-layer repeating units of tri-s-triazine and the interlayer stacking of conjugated aromatic rings, respectively.38 For pure CNTs, the broad peaks observed at 25.87° and 43.06° are associated with the (002) and (001) planes, reflecting the typical structure of carbon nanotubes.39 The XRD patterns of the g-C3N4/CNT composites show both the diffraction peaks characteristic of g-C3N4 and the broad peaks of CNTs. As the proportion of CNT increases in the composites, the intensity of these diffraction peaks becomes more pronounced, confirming the formation of a g-C3N4/CNT complex. This suggests that the pyrolysis method effectively creates a composite material while maintaining the crystalline properties of both g-C3N4 and CNTs.
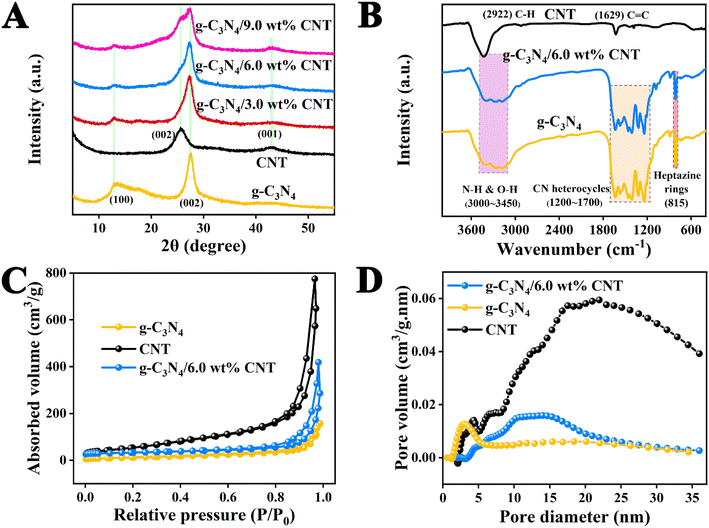 |
| | Fig. 3 (A) The XRD patterns of samples; (B) the FTIR spectra of g-C3N4, CNT and g-C3N4/6.0 wt% CNT; (C) N2 adsorption–desorption isotherms and (D) pore volume of samples. | |
The functional groups and bonding information in each sample were analyzed using FT-IR spectroscopy, as shown in Fig. 3B. In the pure g-C3N4 sample, the peak at 808 cm−1 is attributed to the characteristic vibration of the tri-s-triazine unit, while the peak at 1200–1700 cm−1 corresponds to the stretching vibrations of the CN heterocycle.25 Additionally, the broad peak between 3000 and 3650 cm−1 is associated with the stretching vibrations of terminal N–H and O–H groups. For monomeric CNTs, the peaks at 2922 cm−1 and 1629 cm−1 correspond to the stretching vibrations of C–H bonds and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, respectively.37 Notably, the functional groups in the g-C3N4/CNT composite align well with those of the pure materials, and the characteristic peaks of each monomer are present. These results confirm that the functional groups of both components are retained in the g-C3N4/CNT composite, providing essential conditions for enhancing the photocatalytic efficiency of g-C3N4.
C bonds, respectively.37 Notably, the functional groups in the g-C3N4/CNT composite align well with those of the pure materials, and the characteristic peaks of each monomer are present. These results confirm that the functional groups of both components are retained in the g-C3N4/CNT composite, providing essential conditions for enhancing the photocatalytic efficiency of g-C3N4.
To further evaluate the microstructural changes in the g-C3N4/6.0 wt% CNT composite, we conducted N2 adsorption–desorption analysis on g-C3N4, CNT, and g-C3N4/6.0 wt% CNT samples. The results are illustrated in Fig. 3C and D, while the specific surface area, pore structure, and pore size characteristics are summarized in Table S1.† According to the classification established by the International Union of Pure and Applied Chemistry (IUPAC), the sample displays a V-shaped adsorption–desorption characteristic curve and a distinct H3-type hysteresis loop.12 As detailed in Table S1,† the specific surface area, pore volume, and pore diameter of g-C3N4 are smaller than those of CNT; however, the incorporation of the cocatalyst in the g-C3N4/6.0 wt% CNT composite enhances these properties. These findings confirm that an increased number of reaction sites contributes to the improved photocatalytic efficiency of g-C3N4.
X-ray photoelectron spectroscopy (XPS) was used to investigate the surface electronic states and elemental composition of the g-C3N4, CNT, and g-C3N4/CNT samples. Among them, the percentage contents of C, N, and O elements in each sample are shown in S4.† In Fig. 4A, the XPS spectra of the g-C3N4/CNT composite reveals the presence of C 1s, N 1s, and O 1s peaks, which match the elemental composition of pure g-C3N4. This confirms the results obtained from element mapping and EDS in SEM and TEM. Fig. 4B shows the high-resolution C 1s spectra for both pure g-C3N4 and the g-C3N4/CNT composite. The binding energies for pure g-C3N4 are observed at 284.8 eV and 288.2 eV, corresponding to the C–C and NC–N bonds, respectively.40 In the g-C3N4/CNT composite, additional fine peaks are observed at binding energies of 283.6 eV, 284.9 eV, 286.5 eV, 288.2 eV, and 289.4 eV, which correspond to C–C, C–OH, C–O, NC–N, and O–CO groups, respectively.41 This suggests that the introduction of CNTs influences the chemical environment of carbon atoms in the composite. Fig. 4C presents the high-resolution N 1s spectra, indicating three types of nitrogen bonding configurations: N–CN, N–C, and C–NH2 or C–NH, in both pure g-C3N4 and the g-C3N4/CNT composite.42 For pure g-C3N4, the binding energies for N1s are 398.1 eV, 398.9 eV, and 400.7 eV, while in the g-C3N4/CNT composite, these shift slightly to 398.4 eV, 400.0 eV, and 400.9 eV, indicating a change in the electronic environment due to the formation of the composite.43 The shifts in the binding energies of N1s in the g-C3N4/CNT composite compared to pure g-C3N4 indicate electron loss suggesting that electron redistribution occurs during the formation of the composite. This change in electronic configuration implies that the surface elements in the g-C3N4/CNT composite are more stable, enhancing the g-C3N4 interface properties, which is favorable for applications such as the degradation of organic pollutants.
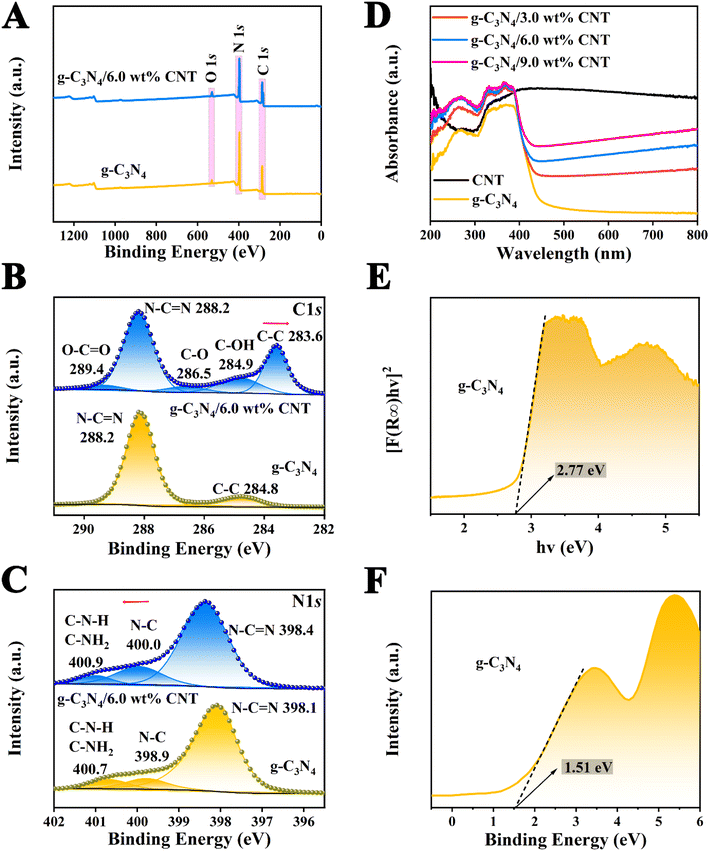 |
| | Fig. 4 XPS spectra of g-C3N4 and g-C3N4/6.0 wt% CNT: (A) the XPS survey spectrum analysis map; (B) C 1s fine spectrum and (C) N 1s fine spectrum; (D) the UV-vis spectra of samples; the bandgap energy diagram of g-C3N4 (E) and (F) the valence band (XPS) spectra. | |
3.3 Optical properties and band structure analysis
The UV-vis DRS spectra of the prepared samples are presented in Fig. 4D, showing that the absorption band edge of each sample is around 450 nm. While CNT, as a conductive material, has a weak light response, it acts as a light absorption enhancer by extending photon lifetimes and improving the light absorption capacity of g-C3N4. As the CNT content increases, the absorption band edge of the g-C3N4/CNT composite prepared by pyrolysis undergoes a slight red shift, thereby broadening the spectral response range of g-C3N4. Notably, the absorption band edge of the g-C3N4/6.0 wt% CNT composite is approximately 460 nm. Additionally, the band gap value (Eg) of g-C3N4 can be estimated using the Kubelka–Munk function:| | |
[F(R∞)hν]1/n = A(hv − Eg)
| (1) |
In the formula, the R∞ reflectivity (percentage form), Planck's constant h (eV s), optical frequency v (s−1) and a constant A are involved, respectively. According to literature reports, the value of n for g-C3N4 is 4.12 As shown in Fig. 4E, the calculated band gap of g-C3N4 is approximately 2.77 eV. Additionally, Fig. 4F illustrates that the valence band (VB) energy level of g-C3N4, determined via XPS analysis, is 1.51 eV. Finally, based on the equation: ECB = EVB − Eg. ECB, EVB and Eg represent the conduction band value, valence band value, and band gap value, respectively, it can be deduced that the conduction band (CB) of g-C3N4 is −1.26 eV.
3.4 Degradation of COD in drilling hydraulic filtrate
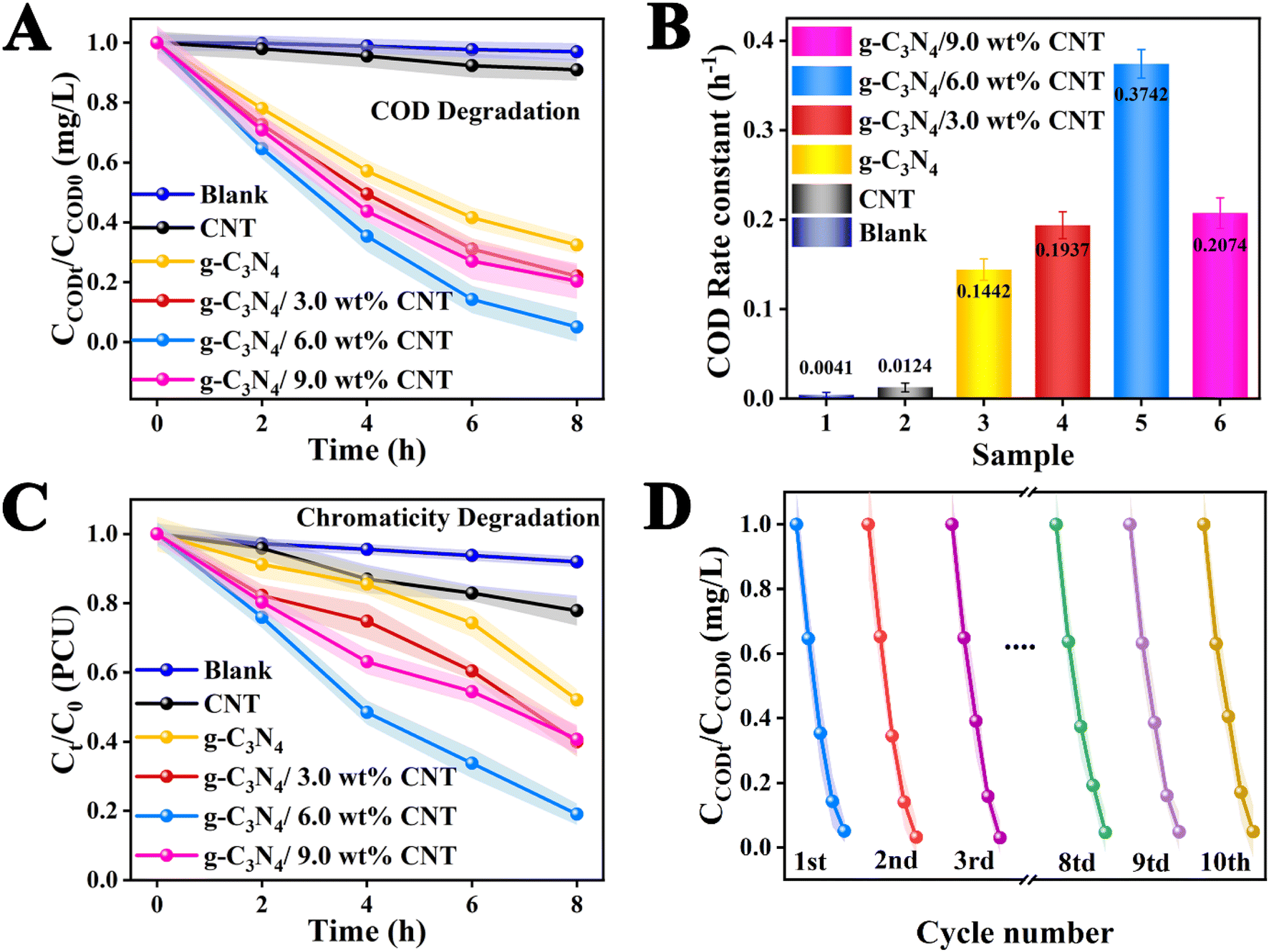
The results of the degradation of waste drilling hydraulic filtrate for each sample under xenon lamp irradiation for 8 h are presented in Fig. 5. The experimental results clearly indicate that the degradation of waste drilling hydraulic filtrate without a catalyst demonstrates its high stability, showing no degradation effect from the co-catalyst CNT. Furthermore, the degradation performance of the pure component g-C3N4 is suboptimal. It's encouraging to note that the photocatalytic degradation efficiency of the g-C3N4/CNT composite, produced via thermal decomposition, greatly exceeds that of pure g-C3N4. Moreover, the photocatalytic performance shows an initial increase followed by a decrease as the co-catalyst CNT loading changes. This decline may be attributed to excessive CNT loading, which a possible reason is the light shielding effects of the co-catalyst in the photocatalytic system.44,45 It is significant to note that the optimal co-catalyst CNT content is 6.0 wt%, achieving a degradation rate of 95%, which indicates superior photocatalytic performance. Meanwhile, as illustrated in the COD degradation rate bar chart in Fig. 5B, the photodegradation rates of COD for each sample are as follows: g-C3N4/6.0 wt% CNT (0.3742 h−1) > g-C3N4/9.0 wt% CNT (0.2074 h−1) > g-C3N4/3.0 wt% CNT (0.1937 h−1) > g-C3N4 (0.1442 h−1). The degradation rate for g-C3N4/6.0 wt% CNT is 2.37 times greater than that of pure g-C3N4. The findings suggest that the integration of g-C3N4 and CNT markedly improves photocatalytic efficiency by facilitating electron–hole separation, enhancing light absorption, increasing the number of reaction sites, and promoting the generation of reactive oxygen species. Simultaneously, the chromaticity of the drilling hydraulic filtrate was analyzed, as depicted in Fig. 5C. The degradation rates for chromaticity are as follows: g-C3N4/6.0 wt% CNT (81% PCU) > g-C3N4/3.0 wt% CNT (61% PCU) > g-C3N4/9.0 wt% CNT (60% PCU) > g-C3N4 (48% PCU) > CNT (22% PCU). Furthermore, the optimal g-C3N4/6.0 wt% CNT sample underwent cyclic stability testing in COD degradation. As shown in Fig. 5D, ten cycles were conducted under identical conditions, demonstrating good stability with minimal change in the degradation rate. Meanwhile, the used catalyst was analyzed via XRD and TEM, as shown in Fig. S1 and S2.† The XRD results indicated relative stability, and the TEM images revealed no significant changes in the microstructure, demonstrating that the catalyst exhibited relative stability. Overall, during repeated use under illumination, the electric field interface formed by the co-catalyst CNT remains intact, allowing for consistent generation of electron–hole pairs (e−/h+) necessary for COD degradation, with no signs of diminished redox ability.
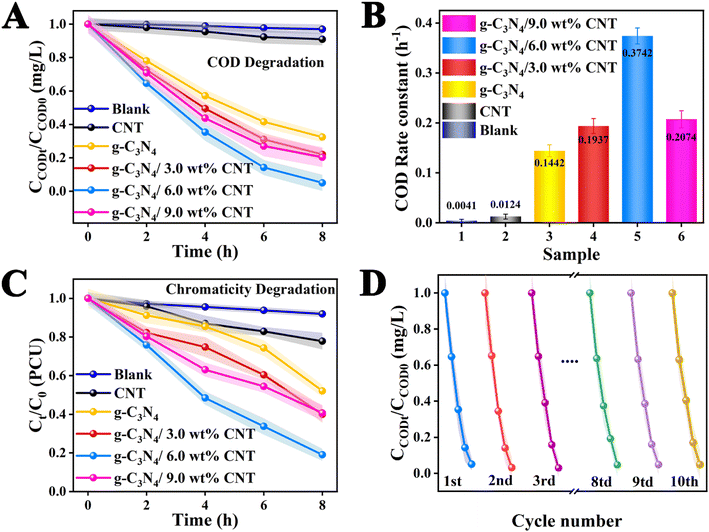 |
| | Fig. 5 (A) The COD of the actual waste drilling filtrate photodegradation using each sample during 8 h under simulate sun light irradiation and (B) the corresponding photodegradation rate constants; (C) chromaticity removal rate of actual waste drilling press filtrate and (D) the cyclic tests. | |
3.5 Photoinduced charge separation analysis
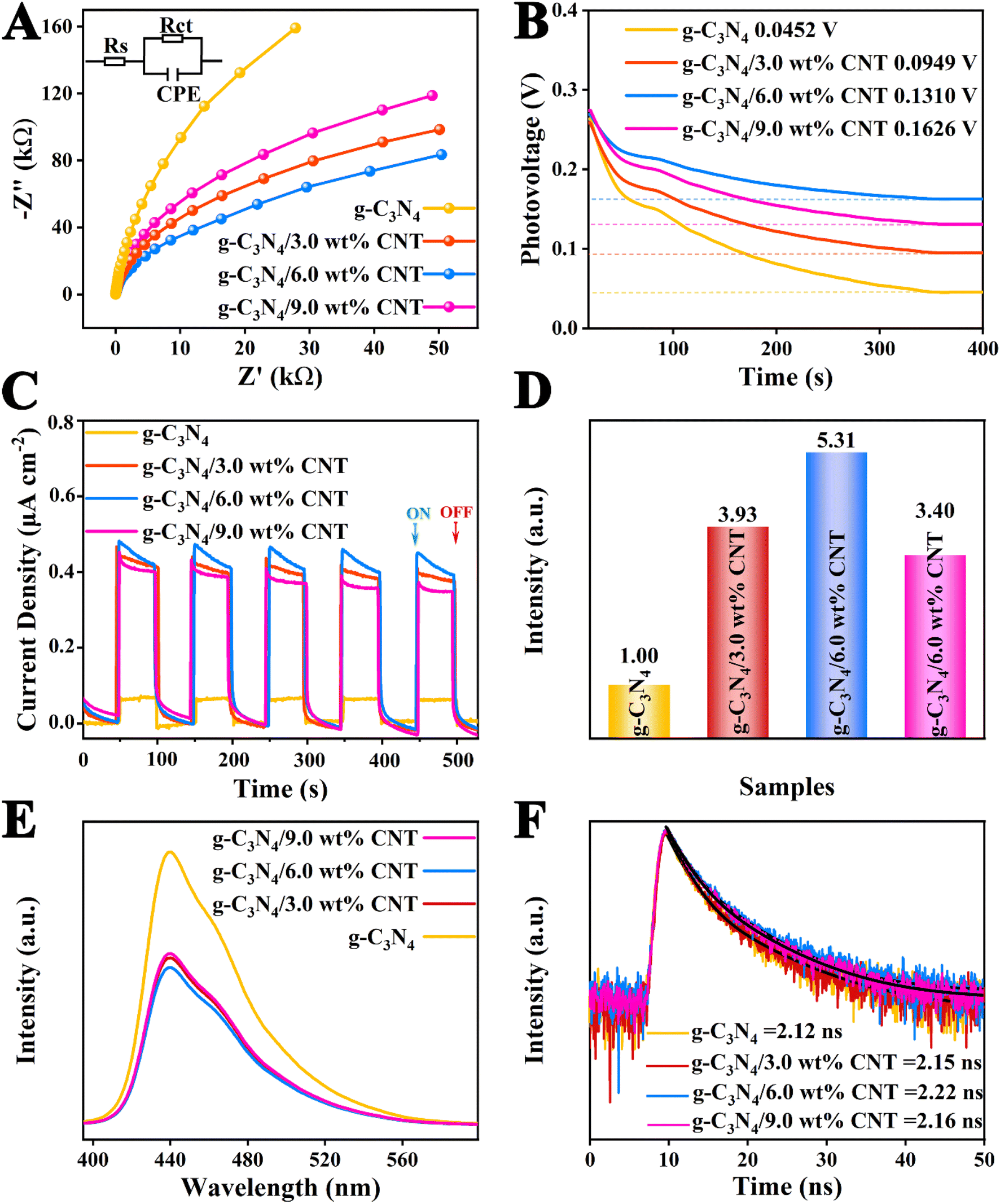
The electrochemical impedance spectroscopy (EIS) is illustrated in Fig. 6A and Table S3.† The order of the radius size in the Nyquist plot is as follows: g-C3N4/6.0 wt% CNT < g-C3N4/3.0 wt% CNT < g-C3N4/9.0 wt% CNT < g-C3N4. Generally, a smaller radius in the Nyquist plot indicates lower charge transport resistance, facilitating the migration and separation of electron–hole pairs.46 Additionally, the open circuit voltages for each sample, shown in Fig. 6B, are 0.0452 V, 0.0949 V, 0.1310 V, and 0.1626 V, respectively. Subsequently, the transient photocurrent measurements were conducted to evaluate the separation and migration efficiency of photogenerated charges for each sample, as depicted in Fig. 6C, demonstrating good repeatability and stability. To better assess the effect of the co-catalyst on the electric field intensity of g-C3N4, the photocurrent curves over 100 seconds were integrated (see Fig. S1†), yielding charge densities of 5.03 μA cm−2 (g-C3N4), 26.74 μA cm−2 (g-C3N4/9.0 wt% CNT), 38.43 μA cm−2 (g-C3N4/3.0 wt% CNT), and 38.43 μA cm−2 (g-C3N4/6.0 wt% CNT). The relatively electric field strength for each sample can be calculated using the following empirical formula:47where Fs represents the relatively electric field strength, Vs is the surface voltage, ρ is the surface charge density. The calculated relatively electric field strengths are presented in Fig. 6D, indicating that the relative electric field intensity initially increases and then decreases with the amount of loaded co-catalyst CNT. This suggests that the non-metallic co-catalyst CNT significantly alters the interface electric field of g-C3N4, effectively separating electrons and holes, thereby enhancing the photocatalytic reaction rate. Moreover, to investigate the recombination efficiency of electron–hole pairs in the heterojunction composites, photoluminescence (PL) testing was performed under an excitation wavelength of 320 nm.10 Generally, the intensity of the PL spectrum indicates a faster recombination of carriers and shorter lifetimes. The PL spectral intensities for each sample are shown in Fig. 6E, further revealing that the g-C3N4/CNT composite samples exhibit superior electron–hole pair separation and electron transition processes compared to pure g-C3N4. Finally, time-resolved photoluminescence (TRPL) spectra were measured to assess the average lifetimes of the photoelectrons. From Fig. 6F and Table S3,† the average lifetimes for each sample are 2.12 ns, 2.15 ns, 2.22 ns, and 2.16 ns, indicating that the loaded co-catalyst CNT improves the electron lifetimes at the interface of g-C3N4, corroborating the findings from the UV-vis DRS spectra regarding the co-catalyst CNT. In summary, the interface-coupled non-metallic co-catalyst CNT can create unique carrier transport pathways, enhancing interfacial charge transfer efficiency, resulting in a greater availability of oxidative holes in the valence band and reductive electrons in the conduction band to participate in photocatalytic reactions.
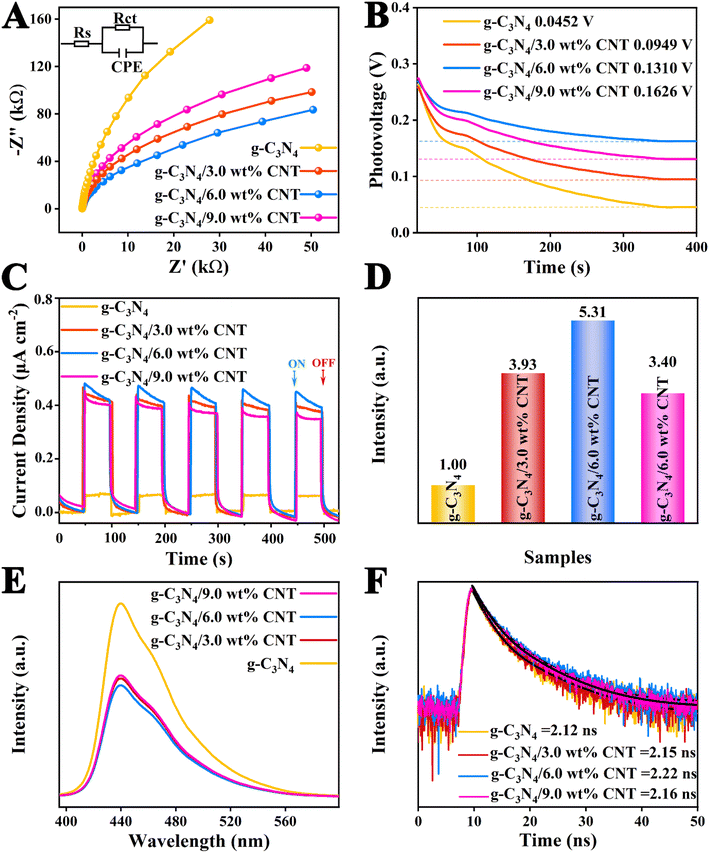 |
| | Fig. 6 (A) The Nyquist plots of samples, (B) the open circuit voltage of the samples, and (C) the corresponding transient photocurrent responses; (D) the relative electric field intensity of each sample and (E) the PL spectra; (F) TRPL decay curves. | |
3.6 Photocatalytic reaction mechanism
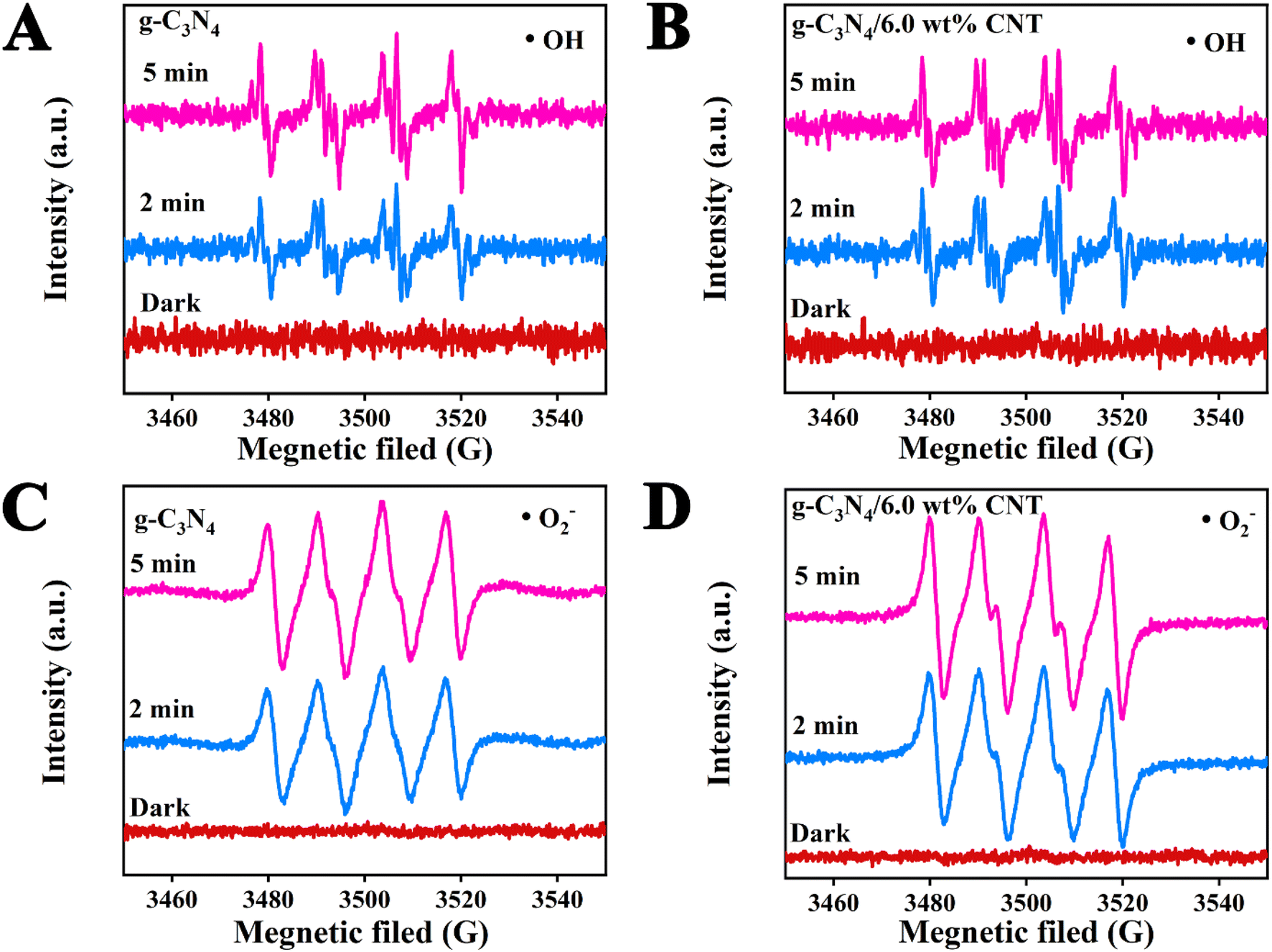
To investigate the impact of the cocatalyst CNT on the photodegradation of active species ˙OH and ˙O2− in waste drilling hydraulic filtrate using g-C3N4, DMPO (5,5-dimethyl-1-pyrroline N-oxide) electron spin resonance (ESR) spectroscopy was employed to detect the formation of ˙OH and ˙O2− in the photoreaction system after 2 and 5 minutes of visible light irradiation (Fig. 7). Fig. 7A and B illustrate the ˙OH radical signal intensities for g-C3N4 and g-C3N4/6.0 wt% CNT at different exposure times (2 and 5 minutes), with no detectable signal in the dark. The data clearly show that the peak value of ˙OH remains constant for both samples, indicating that ˙OH is not the primary active species during the photodegradation process. Fig. 7C and D present the ˙O2− radical signal intensities for g-C3N4 and g-C3N4/6.0 wt% CNT under similar conditions. A strong DMPO-˙O2− signal is observed for both materials after light exposure, with no signal detected in the dark. As exposure time increases, the intensity of the ˙O2− signal strengthens. Fig. 7D further highlights that the ˙O2− signal is more pronounced in g-C3N4/6.0 wt% CNT compared to pure g-C3N4, suggesting that CNT doping enhances the generation of reactive oxygen species. Results show no free radical signals were observed in the absence of light, confirming that illumination is crucial for the generation of ˙OH and ˙O2− radicals. Prolonged exposure to light significantly boosts free radical production, underscoring the importance of illumination. Furthermore, g-C3N4/CNT composites exhibit superior efficiency compared to pure g-C3N4, likely due to CNT loading enhancing electron transfer or minimizing electron–hole recombination.
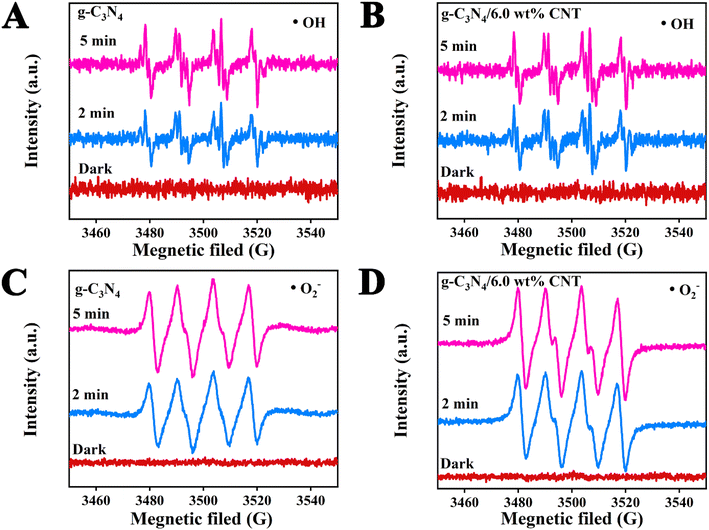 |
| | Fig. 7 The ESR spectral analysis of active species ˙OH (A and B) and ˙O2− (C and B) during photodegradation processes. | |
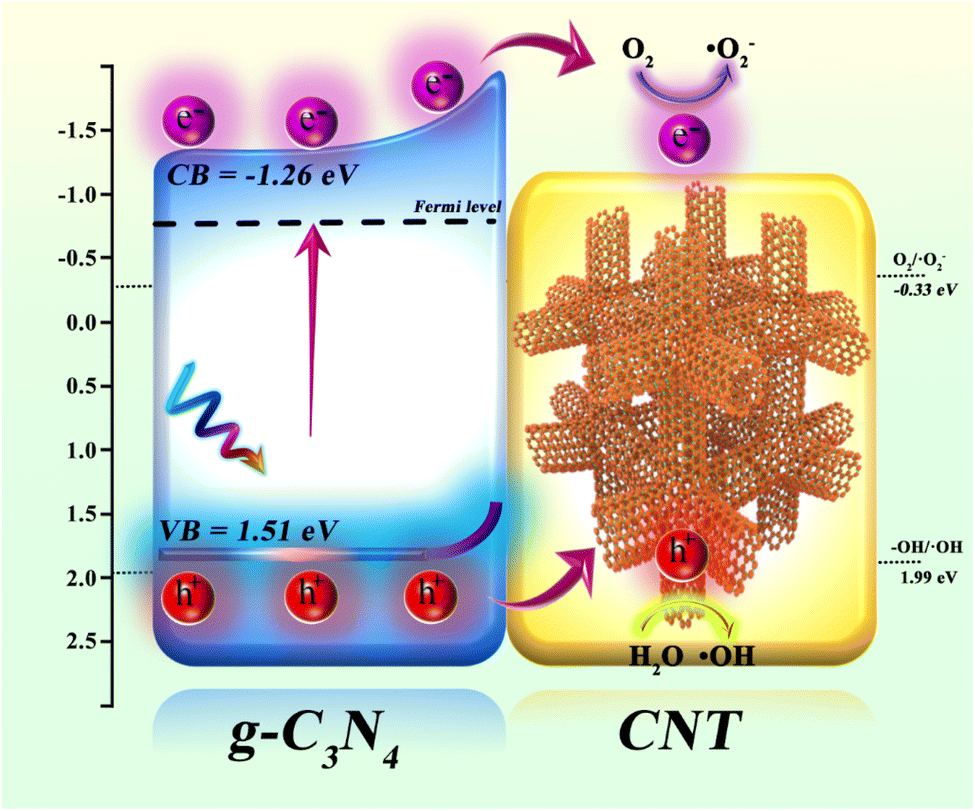
Using UV-vis DRS, XPS-VB, and ESR spectroscopy, the valence band (VB) and conduction band (CB) potentials of the synthesized g-C3N4, along with the active species (˙OH and ˙O2−) involved in the photocatalytic reaction, were determined. The photocatalytic mechanism of CNT-supported g-C3N4 is depicted in Fig. 8. Photogenerated electron–hole pair formation: when g-C3N4 is illuminated, electrons in the valence band (VB = 1.51 eV) are excited to the conduction band (CB = −1.26 eV), generating photoelectrons (e−) in the CB and leaving holes (h+) in the VB. Electron transfer: the excited photoelectrons migrate along the conduction band of g-C3N4 and are rapidly transferred via the conductive CNT. Due to the excellent conductivity of CNT, electrons are efficiently transferred to its surface, reducing electron–hole recombination and extending the lifetime of photogenerated carriers. Generation of reactive oxygen species: on the CNT surface, transferred photoelectrons react with dissolved O2 to form superoxide radicals (˙O2−) via the reduction of O2 (O2/˙O2− = −0.33 eV). These radicals actively participate in oxidative degradation reactions, breaking down organic pollutants. Role of photogenerated holes: simultaneously, the photogenerated holes in g-C3N4 react with H2O to produce hydroxyl radicals (˙OH), a process with an oxidation potential of 1.99 eV (–OH/˙OH). These highly oxidative ˙OH radicals effectively degrade organic pollutants in waste drilling fluids. Synergistic effect: the combination of g-C3N4 and CNT significantly enhances electron transfer and hole separation. As an electron transport carrier, CNT lowers the electron–hole recombination rate in g-C3N4, thereby boosting photocatalytic efficiency. Photogenerated electrons reduce O2 to generate ˙O2− radicals, while holes oxidize water to produce ˙OH radicals. These radicals act synergistically in degrading organic pollutants in waste drilling fluids. In conclusion, the g-C3N4/CNT composite material markedly improves photocatalytic degradation of organic pollutants in waste drilling fluids through enhanced electron transfer, efficient free radical generation, and the synergistic interaction between g-C3N4 and CNT.
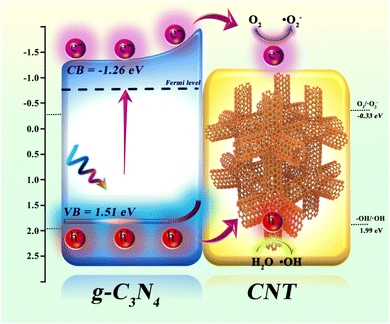 |
| | Fig. 8 The schematic of the proposed mechanism of g-C3N4/CNT composites for actual waste drilling filtrate under simulate sun light irradiation. | |
4. Conclusion
In conclusion, this paper successfully synthesized a g-C3N4/CNT composite by loading the co-catalyst CNT through thermal decomposition. The sample's micromorphology, optical properties, and catalytic mechanism were thoroughly analyzed. The g-C3N4/6.0 wt% CNT composite exhibited outstanding performance in the photodegradation of waste drilling hydraulic filtrate, achieving a COD photodegradation rate 2.37 times higher than that of pure g-C3N4 and a chromaticity removal rate of 81%. This enhanced performance is primarily attributed to the incorporation of the CNT co-catalyst, which improved electron transfer rates, increased light absorption capacity, exposed more active sites, and optimized the band structure of g-C3N4. These factors collectively enhanced the interfacial synergistic effect, inhibited the recombination of photogenerated charges, and strengthened the redox ability of photogenerated carriers, thereby significantly improving the photocatalytic efficiency.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.52274008). National key R & D projects (No. 2019YFA0708303). Science and Technology Cooperation Project of the CNPC-SWPU Innovation Alliance (No. 2020CX040102, 2020CX040201). The Joint Funds of the National Natural Science Foundation of China (No. U23A2026). CNPC's Key Applied Science and Technology Project Research on Shale Gas Scale Stimulation as well as Exploration and Development Technology (Project No. 2023ZZ21), Project of PtroChina Southwest Oil&Gasfied company “Research and Application of Effective Drilling Technology for Deep Shale Gas Horizontal Wells” (Project No. 20230302-08).
References
- X. Hu, X. Zhao and Y. Ke, J. Mol. Liq., 2019, 296, 111885 CrossRef CAS.
- S. Z. Razali, R. Yunus, S. Abdul Rashid, H. N. Lim and B. Mohamed Jan, Renew. Sustainable Energy Rev., 2018, 90, 171–186 CrossRef CAS.
- X. Liu, M.-C. Li, B. Liao, S. Liu, K. Lu, K. Lv, J. Sun, C. Liu, C. Mei and Q. Wu, Chem. Eng. J., 2023, 475, 146372 CrossRef CAS.
- J. L. Suter, P. V. Coveney, R. L. Anderson, H. C. Greenwell and S. Cliffe, Energy Environ. Sci., 2011, 4, 4572 RSC.
- X. Huang, J. Sun, H. Li, R. Wang, K. Lv and H. Li, Engineering, 2022, 11, 101–110 CrossRef CAS.
- Y. Bai, X. Shi, P. Wang, L. Wnag, K. Zhang, Y. Zhou, H. Xie, J. Wang and L. Ye, Chem. Eng. J., 2019, 356, 34–42 CrossRef CAS.
- N. H. Alias, J. Jaafar, S. Samitsu, N. Yusof, M. H. D. Othman, M. A. Rahman, A. F. Ismail, F. Aziz, W. N. W. Salleh and N. H. Othman, Chemosphere, 2018, 204, 79–86 CrossRef CAS PubMed.
- G. Li, T. An, J. Chen, G. Sheng, J. Fu, F. Chen, S. Zhang and H. Zhao, J. Hazard. Mater., 2006, 138, 392–400 CrossRef CAS PubMed.
- P. Ren, T. Zhang, N. Jain, H. Y. V. Ching, A. Jaworski, G. Barcaro, S. Monti, J. Silvestre-Albero, V. Celorrio, L. Chouhan, A. Rokicinska, E. Debroye, P. Kustrowski, S. Van Doorslaer, S. Van Aert, S. Bals and S. Das, J. Am. Chem. Soc., 2023, 145, 16584–16596 CrossRef CAS PubMed.
- D. Gu, Y. Wang, Z. Liang, Y. Dou, Z. Xu, J. Zheng, Y. Sun, F. Ding and Y. Gao, RSC Adv., 2023, 13, 15302–15310 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- Y. Bai, D. Gu, Z. Chen, J. He, L. Wu, D. Li and X. Shi, ACS Appl. Nano Mater., 2024, 7, 17339–17350 CrossRef CAS.
- H.-x. Zhong, Q. Zhang, J. Wang, X.-b. Zhang, X.-l. Wei, Z.-j. Wu, K. Li, F.-l. Meng, D. Bao and J.-m. Yan, ACS Catal., 2018, 8, 3965–3970 CrossRef CAS.
- X. Guo, J. Yang, Y. Wei, L. Wang and J. Pi, ACS Appl. Nano Mater., 2022, 5, 4230–4240 CrossRef CAS.
- L. Chen, Y. Wang, S. Cheng, X. Zhao, J. Zhang, Z. Ao, C. Zhao, B. Li, S. Wang, S. Wang and H. Sun, Appl. Catal., B, 2022, 303, 120932 CrossRef CAS.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Y. Bai, L. Ye, L. Wang, X. Shi, P. Wang, W. Bai and P. K. Wong, Appl. Catal., B, 2016, 194, 98–104 CrossRef CAS.
- Z. Ding, X. Li, C. Kang, S. Yan, D. Zhao, H. Cai, S.-Y. Zhang and Y.-J. Zeng, Chem. Eng. J., 2023, 473, 145256 CrossRef CAS.
- N. Arif, Y. Ma, M. N. Zafar, M. Humayun, M. Bououdina, S. Y. Zhang, Q. Zhang, X. Yang, H. Liang and Y. J. Zeng, Small, 2023, 20, 2308908 CrossRef PubMed.
- Z. Xia, C. Chen, X. Qi, Q. Xu, H. Tang and G. Liu, Adv. Sustainable Syst., 2022, 7, 2200134 CrossRef.
- J. Zhao, B. Fu, X. Li, Z. Ge, B. Ma and Y. Chen, ACS Appl. Energy Mater., 2020, 3, 10910–10919 CrossRef CAS.
- Q. Zhang, J. Deng, Z. Xu, M. Chaker and D. Ma, ACS Catal., 2017, 7, 6225–6234 CrossRef CAS.
- M. Li, Q. Pan, M. Xiao and J. Xiong, RSC Adv., 2022, 12, 2603–2611 RSC.
- Y. Dou, Y. Gao, D. Gu, J. Zheng, B. Zhang, X. Li, M. He, Q. Zou, T. Liao, F. Ding, Y. Sun and Z. Xu, Langmuir, 2023, 39, 17947–17958 CrossRef CAS PubMed.
- Y. Gao, J. Lin, Q. Zhang, H. Yu, F. Ding, B. Xu, Y. Sun and Z. Xu, Appl. Catal., B, 2018, 224, 586–593 CrossRef CAS.
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. Li, J. Xie, C. R. A. Catlow and J. Tang, Adv. Energy Mater., 2018, 8, 1801084 CrossRef.
- Q. Zhang, X. Liu, M. Chaker and D. Ma, ACS Mater. Lett., 2021, 3, 663–697 CrossRef CAS.
- Y. Deng, C. Feng, L. Tang, Y. Zhou, Z. Chen, H. Feng, J. Wang, J. Yu and Y. Liu, Chem. Eng. J., 2020, 393, 124718 CrossRef CAS.
- Y. Pan, K. Sun, Y. Lin, X. Cao, Y. Cheng, S. Liu, L. Zeng, W.-C. Cheong, D. Zhao, K. Wu, Z. Liu, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, Nano Energy, 2019, 56, 411–419 CrossRef CAS.
- B. Lin, Y. Zhou, B. Xu, C. Zhu, W. Tang, Y. Niu, J. Di, P. Song, J. Zhou, X. Luo, L. Kang, R. Duan, Q. Fu, H. Liu, R. Jin, C. Xue, Q. Chen, G. Yang, K. Varga, Q. Xu, Y. Li, Z. Liu and F. Liu, Mater. Horiz., 2021, 8, 612–618 RSC.
- Y. Lu, B. Ma, Y. Yang, E. Huang, Z. Ge, T. Zhang, S. Zhang, L. Li, N. Guan, Y. Ma and Y. Chen, Nano Res., 2017, 10, 1662–1672 CrossRef CAS.
- X. Liu, W. Yang, L. Chen, Z. Liu, L. Long, S. Wang, C. Liu, S. Dong and J. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 4463–4472 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- Q. Zhu, Z. Xu, B. Qiu, M. Xing and J. Zhang, Small, 2021, 17, 2101070 CrossRef CAS PubMed.
- G. Bi, J. Wen, X. Li, W. Liu, J. Xie, Y. Fang and W. Zhang, RSC Adv., 2016, 6, 31497–31506 RSC.
- P. N. Nguyen, T. Q. N. Tran, K. H. Le, D. T. Khong, H. P. Pham, Q. V. Dang, Q. H. Tran, T. M. Nguyen and N. Nguyen Dang, RSC Adv., 2024, 14, 31036–31046 RSC.
- W. Wang, C. Lu, Y. Ni and Z. Xu, Appl. Catal., B, 2013, 129, 606–613 CrossRef CAS.
- Z. Wang, Z. Wang, X. Zhu, C. Ai, Y. Zeng, W. Shi, X. Zhang, H. Zhang, H. Si, J. Li, C. Z. Wang and S. Lin, Small, 2021, 17, e2102699 CrossRef PubMed.
- J. Sun, Y. Huang, H. Li, Y. Qiu, J. Zhang, K. Hu, J. Li and Y. Ding, Environ. Technol. Innovat., 2021, 24, 101815 CrossRef CAS.
- J. Jiang, B. Zheng, W. Yu, X. Wu, R. Mi, Z. Huang, Y.-g. Liu, M. Fang and X. Min, J. Alloys Compd., 2023, 968, 171707 CrossRef CAS.
- C. Ge, L. Wang, G. Liu and R. Wu, J. Alloys Compd., 2019, 775, 647–656 CrossRef CAS.
- Y. Wang, L. Liu, T. Ma, Y. Zhang and H. Huang, Adv. Funct. Mater., 2021, 31, 2102540 CrossRef CAS.
- Y. Yang, M. Qiu, Q. Qi, F. Chen, J. Chen, Y. Liu and L. Yang, ACS Appl. Nano Mater., 2020, 3, 10296–10302 CrossRef CAS.
- B. Ma, J. Zhao, Z. Ge, Y. Chen and Z. Yuan, Sci. China Mater., 2019, 63, 258–266 CrossRef.
- K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS PubMed.
- Y. Dou, Z. Liang, Z. Xu, Y. Wang, J. Zheng, D. Ma and Y. Gao, ACS Appl. Nano Mater., 2023, 6, 12537–12547 CrossRef CAS.
- X. Shi, W. Dai, Y. Bai, G. Luo, X. a. Dong, Q. Ren and L. Ye, Appl. Catal., B, 2024, 343, 123523 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab