
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of fused-ring systems and diarylmethane flavonoids via ortho-quinone methide intermediates†
Vidia A. Nuraini ab,
Valerio Falascab,
Daniel S. Wenholzbc,
David StC. Blackb and
Naresh Kumar*b
ab,
Valerio Falascab,
Daniel S. Wenholzbc,
David StC. Blackb and
Naresh Kumar*b
aStudy Program of Chemistry, Universitas Pendidikan Indonesia, Jl. Dr Setiabudhi 229, Bandung 40154, Indonesia. E-mail: v.nuraini@upi.edu
bSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: d.black@unsw.edu.au; n.kumar@unsw.edu.au; Tel: +61 2 9385 4698
cNoxopharm Limited, Chatswood, NSW, Australia
First published on 29th January 2025
Abstract
In this study, we investigated the ability of ortho-quinone methide (o-QM), an electron-poor diene, to undergo inverse electron-demand Diels–Alder (iEDDA) reaction with electron-rich dienophiles, resulting in fused-ring flavonoid systems. In addition, we explored the Michael-type addition using o-QM and various nucleophiles, providing access to diarylmethane products. The cycloaddition reactions proceeded in a highly regioselective way, depending on the charge distribution of the reacting partners. Overall, the electron-rich dienophiles or nucleophiles reacted with the intermediate o-QM to afford products in reduced to moderate yields. Electron-poor dienophiles or nucleophiles, on the other hand, failed to react with o-QM.
Introduction
Flavonoids, the most commonly studied phytoestrogens, are natural compounds distributed in plants and have been part of the human diet such as in fruits and vegetables.1 Different strategies have been developed to synthesize flavonoids, typically involving 2′-hydroxychalcones A as the intermediates. The chalcones can in turn be produced from the Claisen–Schmidt reaction between substituted acetophenone B and aromatic aldehyde C under basic or acidic conditions (Scheme 1a),2 the Friedel–Crafts reaction, which involves the treatment of phenols D with cinnamoyl chloride E (Scheme 1b),2,3 and the Heck coupling reaction, which combines aryl vinyl ketone F and iodobenzene G (Scheme 1c).2–4 As the building blocks, the 2′-hydroxychalcones A can be transformed either into flavanones H via intramolecular cyclization under basic5 or acidic conditions6 or flavones I through oxidative cyclization3 (Scheme 1). The oxidative rearrangement of chalcones also can lead to the formation of isoflavones.7 | ||
| Scheme 1 Synthetic approaches toward 2′-hydroxychalcones via Claisen–Schmidt reaction (a), Friedel–Crafts condensation (b), and Heck coupling reaction (c), as the precursors for flavonoid-class compounds. | ||
Many naturally-occurring flavonoids contain fused-ring systems. For example, 4′-hydroxyisorottlerin and isorottlerin isolated from the kamala tree Mallotus philippensis possess a fused dimethyl-2H-pyrane core.8 Hematoxylin, one of the main active components of Hematoxylum campechianum and Caesalpinia sappan, contains a 3,4-cycloalkyl fused benzopyran unit. This compound has been shown to demonstrate anti-inflammatory activity and inhibit human immunodeficiency virus 1 (HIV-1) integrase.9
Many ring systems can be formed via Diels–Alder reactions of a highly reactive intermediate known as ortho-quinone methide (o-QM). Several natural products (Fig. 1) such as dependensin 1,10 tocopherol 2,11 and hyperguinone B 3,12 were synthesized via cycloaddition of this intermediate.
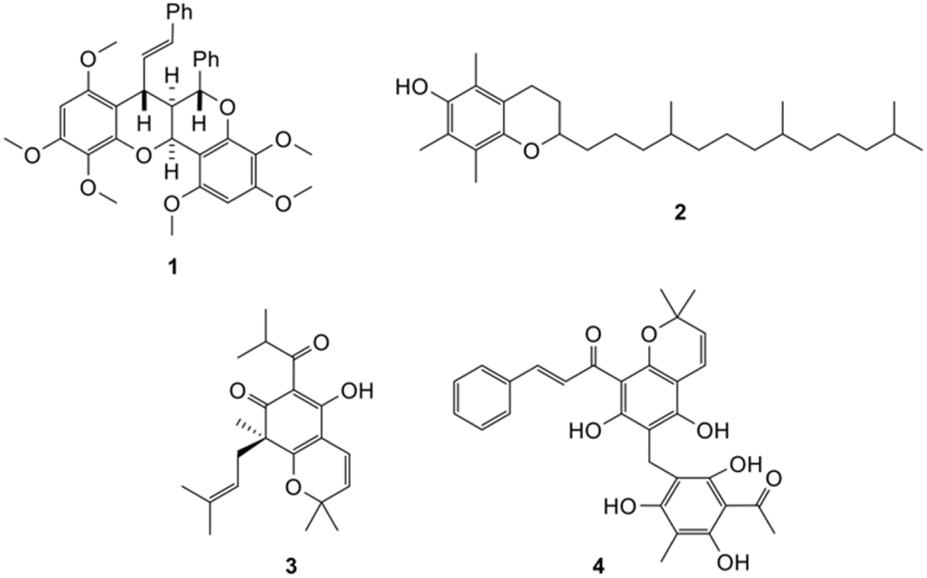 | ||
| Fig. 1 Selected examples of natural products synthesized from o-QM intermediate. | ||
Our group in particular has also utilized the o-QM intermediate for the synthesis of rottlerin 4.13 The Mannich reaction has been reported as a viable method to generate o-QMs intermediates for synthesizing fused-ring systems starting from phenolic structures such as phenol,14 p-cresol,15 naphthol,16 and isoflavonoid structures.17 However, this methodology has not been previously reported on a flavanone system. There are very few reports on the synthesis of fused-ring flavanone analogues,18,19 however, the o-QM strategy can be adapted for the synthesis of complex scaffolds based on flavanones. Increasing the structural complexity of flavonoid structures can enhance their specificity for biological targets.20,21 The prevalence and significant biological roles of fused-ring flavonoids in nature provide a strong rationale for the synthesis and evaluation of a broader range of previously unexplored fused-ring analogues.8,9 Furthermore, fused-ring structures are more likely to have improved solubility, lipophilicity and selectivity towards target receptors.20–22
o-QMs have been widely utilized for the construction of benzopyrans,23 chromenes,24 coumarins,25 and xanthenes,26 featured in many natural products. The reactivity of o-QMs can be explained by their resonance forms, where the neutral molecules are nonaromatic and resemble an α,β-unsaturated ketone, while the zwitterionic resonance structures are aromatic and highly polarized.27,28 Therefore, o-QMs have two reactive sites, and their ambiphilic nature makes them very useful in many organic syntheses.24,29 o-QMs are considered to be electron-poor dienes that can react with electron-rich dienophiles following an inverse electron-demand Diels–Alder mechanism.30,31 Besides reacting with 2π partners via cycloaddition, o-QMs can also react with nucleophiles through Michael addition, or alternatively undergo oxa-6π electrocyclization.27,28,32 In all three pathways, aromaticity is restored, providing a driving force for the reaction.23 In this study, we discuss the synthesis of fused-ring systems that have never been explored on 6-hydroxyflavanone 5, 7-hydroxyflavanone 6, dihydrodaidzein 7 as an attempt to replace the free hydroxyl groups via o-QM intermediates. We also investigated the formation of diarylmethane products as a result of nucleophilic addition to the o-QM derived from isoflavanone, dihydrodaidzein 7.
Results and discussion
Synthesis of Mannich base
Our approach for generating an o-QM was to use thermally-labile groups, such as the dimethylamine group, via the formation of Mannich bases. 6-Hydroxyflavanone 5 was refluxed with a highly reactive aminal electrophile, (bis(N,N-dimethylamino)methane) 8, in 1,4-dioxane for 24 h to give regioselectively the C5 N,N-dimethylaminomethyl-substituted flavanone 9 in 76% yield (Scheme 2a). Although there are two possible reactive sites in the 6-hydroxyflavanone 5, including at C5 and C7 positions, the Mannich reaction took place exclusively at the C5 position. The structure of the Mannich base 9 was unambiguously established from the 1H NMR spectrum, which showed the presence of two doublet signals at δH 7.02 ppm (d, J = 8.8 Hz, H7) and δH 6.92 ppm (d, J = 8.8 Hz, H8).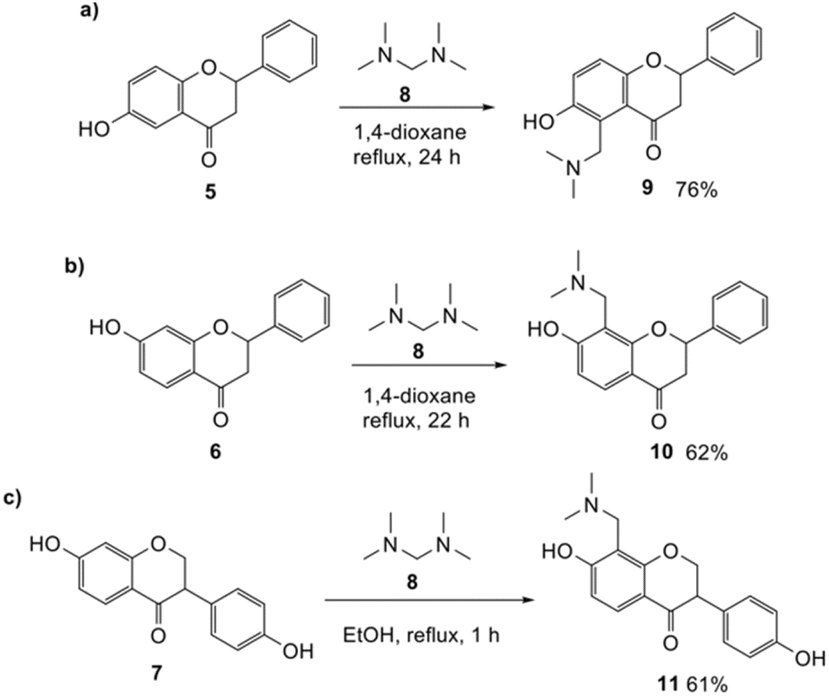 | ||
| Scheme 2 Regioselective Mannich reaction of 6-hydroxyflavanone 5 (a), 7-hydroxyflavanone 6 (b), and dihydrodaidzein 7 (c). | ||
The use of aminals in the Mannich reaction has several advantages, including shorter reaction times and reduced side product formation.33,34 Additionally, formaldehyde is not required, unlike in conventional Mannich reactions.34 In the case of 7-hydroxyflavanone 6, the resultant product was confirmed as C8-substituted aminomethyl analogue 10 (Scheme 2b). This result was in line with the Mannich reaction of 7-hydroxyisoflavone, which also led to the corresponding C8-substituted Mannich base.17 For dihydrodaidzein 7, the Mannich base was prepared by refluxing dihydrodaidzein 7 with aminal 8 in ethanol to give C8 substituted analogue 11 (Scheme 2c). The desired product 11 precipitated cleanly from the reaction mixture and was isolated as a white solid in moderate yield of 61%. Upon heating, these Mannich bases are proposed to form the o-QM intermediates via the loss of the dimethylamine moiety.
Diels–Alder reactions of o-QMs derived from 6-hydroxyflavanone
Our group previously worked on the o-QM reaction to synthesize fused-ring systems derived from phenoxodiol. The reaction was initially carried out by refluxing the Mannich base of phenoxodiol with ethyl vinyl ether as the dienophile in acetonitrile for 24 h. However, the reaction failed to give the expected product, and only the starting material was recovered. Changing the solvent to ethanol was also unsuccessful. While using the high boiling solvent, DMF did not yield a better outcome as the reaction mainly produced phenoxodiol and other degradation products. The reaction was then attempted by heating the Mannich base and ethyl vinyl ether in DMF at 140 °C using a pressure tube and an inert gas atmosphere for 24 h, resulting in the desired product in a low yield of 2%. Prolonged heating to 48 h increased the yield to 26%. Further attempts to improve the yield by extending the reaction time to 72 h did not give a better result. Hence, following this work, the cycloaddition reaction of 6-hydroxyflavanone was attempted by heating the mixture in DMF at a high temperature using a pressure vial under nitrogen gas.Heating a solution of Mannich base 9 in DMF using a sealed tube at 155 °C with excess dienophiles under a nitrogen atmosphere afforded the cycloadducts containing a tetrahydropyran core (Scheme 3) with yields varied depending on the dienophile reactivity (Table 1). Different categories of dienophiles were investigated including enol ethers, conjugated diene, and enaminoketones. It was observed that all ethers (cyclic and acyclic) react in a regiospecific manner, forming acetal-type products 13a–c (Table 1, entries 1–3) confirmed by the resonance of a downfield acetal proton at δH 5.25 ppm. The regioselectivity of Diels–Alder reactions can be explained based on the charge distribution of the reacting partners.35,36 The presence of the electron-donating oxygen atom in ether dienophiles increases the nucleophilicity of the β-carbon, thereby attacking the electrophilic methide carbon of o-QM 12. Therefore, the cycloadducts 13a–c were exclusively formed.
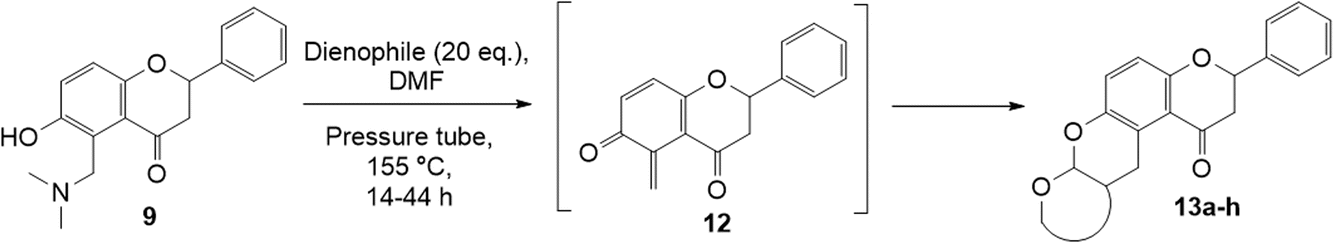 | ||
| Scheme 3 Cycloaddition reactions of Mannich base 9. | ||
| Entry | Dienophile | Time | Product | Yield [%] | Entry | Dienophile | Time | Product | Yield [%] |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  |
23 h | 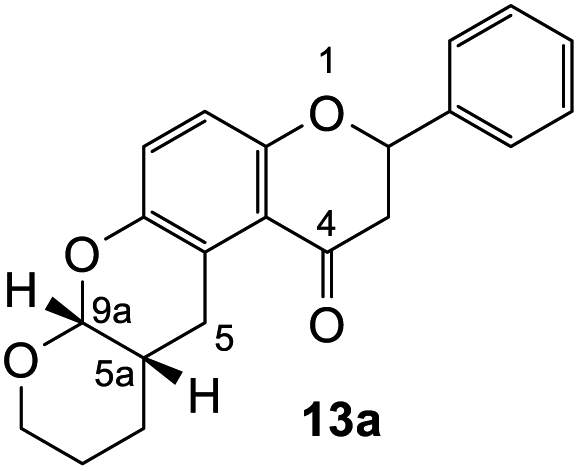 |
12 | 6 |  |
23 h | 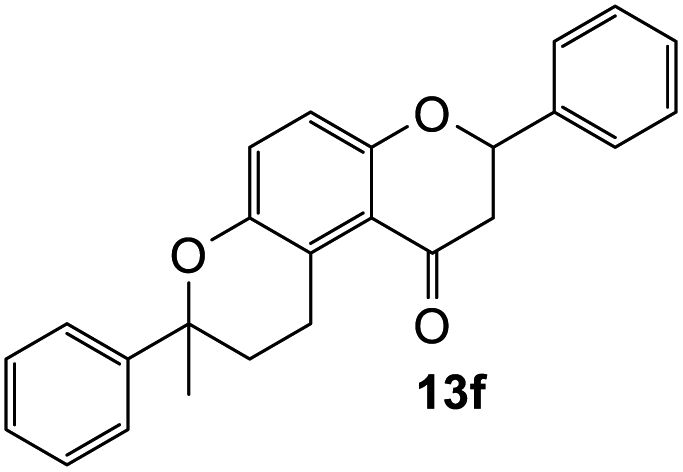 |
12 |
| 2 | 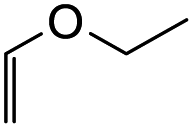 |
23 h | 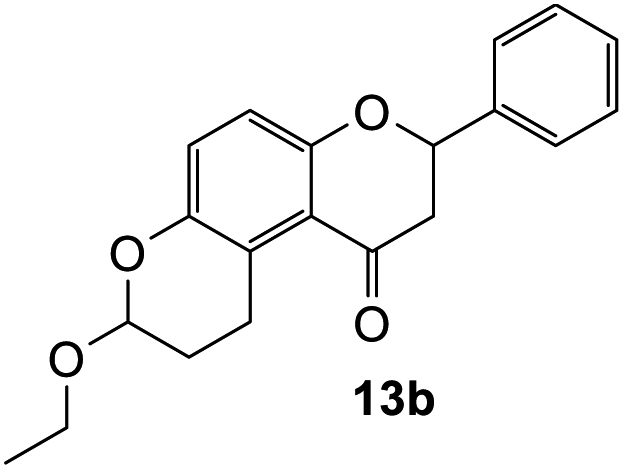 |
30 | 7 | 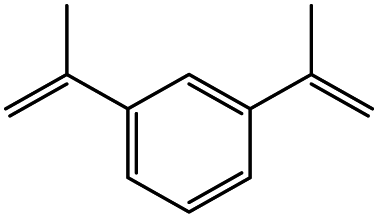 |
14 h | 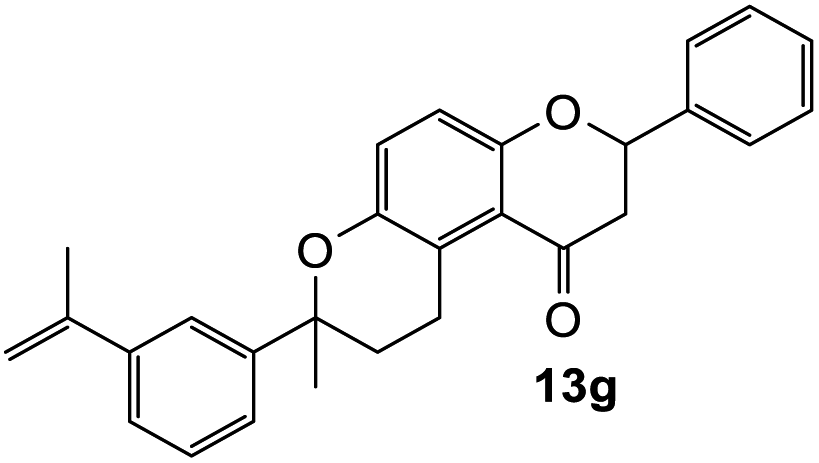 |
21 |
| 3 | 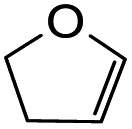 |
17 h | 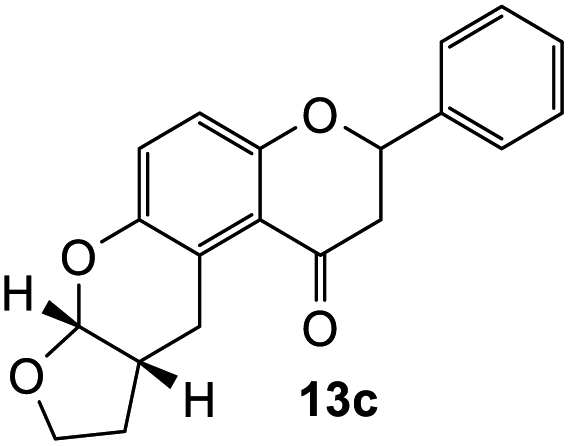 |
21 | 8 | 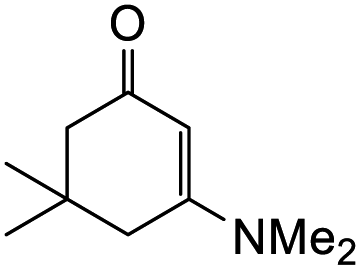 |
21 h | 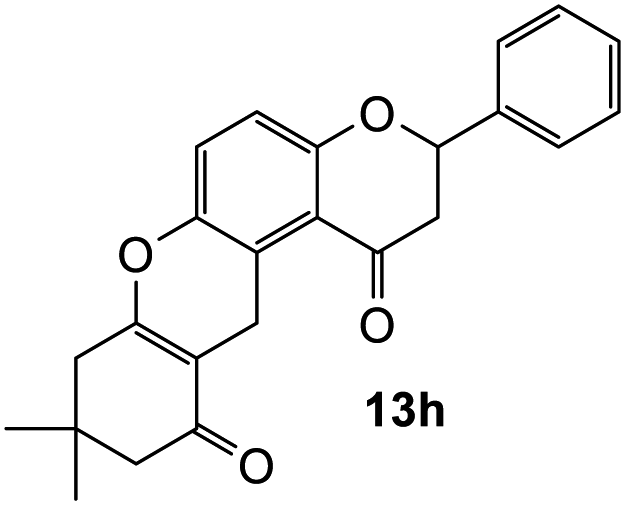 |
79 |
| 4 | 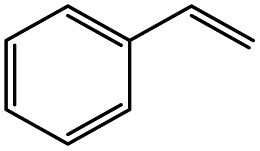 |
44 h | 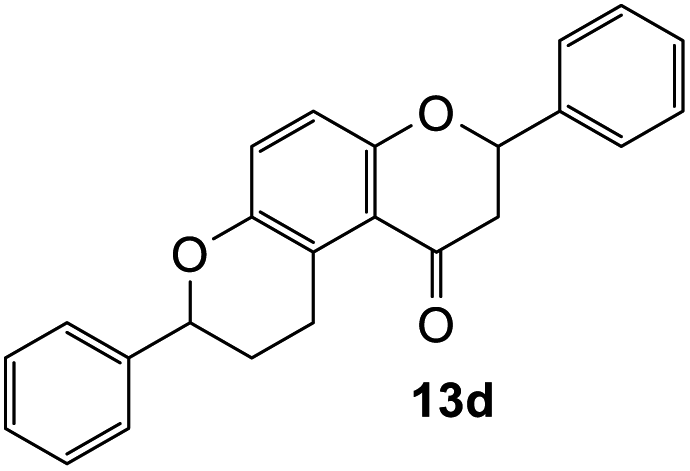 |
25 | 9 |  |
51 h |  |
0 |
| 5 |  |
24 h |  |
36 |
The fused-ring system 13a adopted a cis configuration, which was concluded from the value of the coupling constant (J) between the acetal proton H9a (δH 5.25 ppm, d, J = 2.6 Hz) and the bridgehead proton H5a. Additionally, an important NOE correlation was observed between these two protons. The relative stereochemistry of this adduct was in agreement with similar isoflavone cycloadducts as reported by Frasinyuk et al..17 As the Diels–Alder reactions enable the formation of new stereogenic centers, it is unavoidable to obtain diastereomeric mixtures unless the stereoselectivity is controlled. The diastereoisomerism caused the resonance of additional peaks for most of the carbon atoms in the 13C NMR spectrum of cycloadducts. These extra chemical shifts are shown in brackets in the 13C NMR data.
The reaction of o-QM 12 with p-methoxystyrene (36%), as expected, gave a higher yield of the cycloadduct than with styrene (25%) (Table 1, entries 4 and 5). This result indicated that the electron-rich dienophile favoured iEDDA reaction since the electron-donating substituent could raise the HOMO of the dienophile and thus reduce the activation energy.37 The o-QM 12 was also treated with α-methyl styrene and 1,3-diisopropenylbenzene, providing cycloadducts 13f (12%) and 13g (21%), respectively (Table 1, entries 6 and 7). The low yields of these two products could be due to the low nucleophilicity of the dienophiles, as both styrenes lack electron-donating groups. With a more electron-rich dienophile, 3-(dimethylamino)-5,5-dimethylcyclohex-2-en-1-one, the reaction with o-QM 12 generated the cycloadduct 13h in a higher yield of 79% (Table 1, entry 8). The reactivity of this dienophile is attributed to its ambiphilic properties, which combine the nucleophilicity of enamine with the electrophilicity of enone, leading to a push–pull nature of the double bond.24 The β-carbon atom attached to the electron-withdrawing group, carbonyl group in this case, exhibits considerable nucleophilicity, reversely, the α-carbon atom displays considerable electrophilicity.38 Hence, the β-carbon will attack the more electrophilic methide carbon of the o-QM 12, favouring the formation of the hemi-aminal intermediate. Subsequent elimination of dimethylamine results in product 13h. Attempts to use electron-poor benzofuran as the dienophile in the Diels–Alder reaction with o-QM 12, as expected, failed to give the anticipated product 13i, even after prolonged heating (Table 1, entry 9). This could be due to the inertness of benzofuran because of the high aromaticity, thus requiring a high energy barrier to dearomatize the five-membered ring.39,40
Diels–Alder reactions of o-QMs derived from 7-hydroxyflavanone
The Diels–Alder reaction of the 7-hydroxy Mannich base 10 proceeded through the o-QM 14 generated in situ, using similar conditions as described for the corresponding 6-substituted analogue 9 (Scheme 4). However, the reaction proved quite challenging, as the flavanone core readily underwent ring-opening to form the chalcone. Hence, the chalcone cycloadducts 16a–e were isolated in addition to the flavanone cycloadducts 15a–e in low yields (Table 2). In the literature, Diels–alder reactions on similar ring structures, such as dihydropyran and dihydrochromene favoured the ring-opened product, but could also induce ring-opening using PTSA as a catalyst.41–43 The formation of chalcone analogues was indicated by the appearance of two doublets at around δH 7.90 and 7.99 ppm with a large J value of 15.5 Hz, corresponding to the olefinic protons Hα and Hβ, respectively. A sharp singlet further downfield at δH 13.99 ppm indicating a hydroxy group provided further evidence that the chalcone structure had formed. It was hypothesized that the dimethylamine group, which was eliminated during the formation of the o-QM intermediate, facilitated the ring-opening reaction of the flavanone moiety.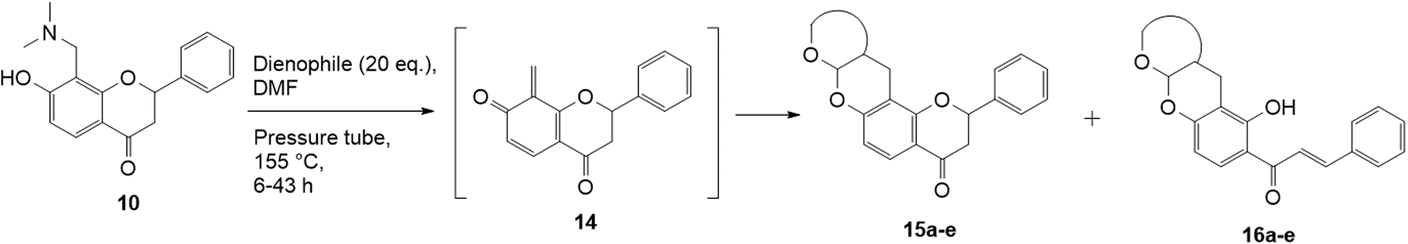 | ||
| Scheme 4 Cycloaddition reactions of Mannich base 10 with various dienophiles. | ||
| Entry | Dienophile | Time | Flavanone cycloadducts | Yield [%] | Chalcone cycloadducts | Yield [%] |
|---|---|---|---|---|---|---|
| 1 |  |
43 h | 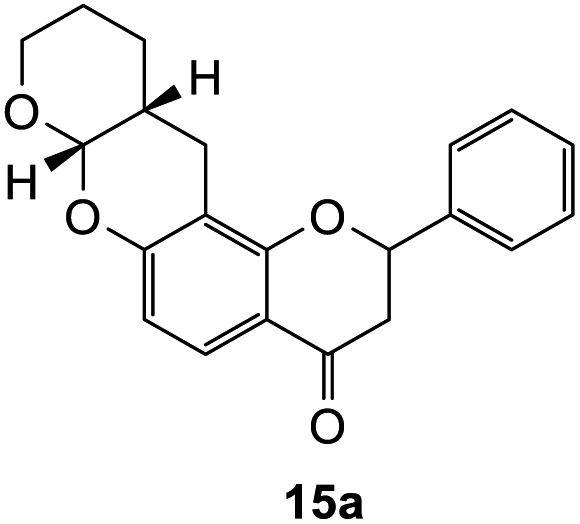 |
12 | 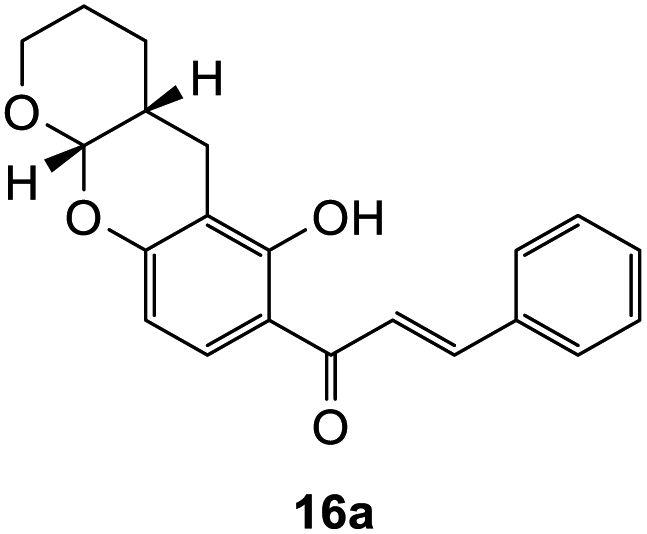 |
8 |
| 2 |  |
43 h | 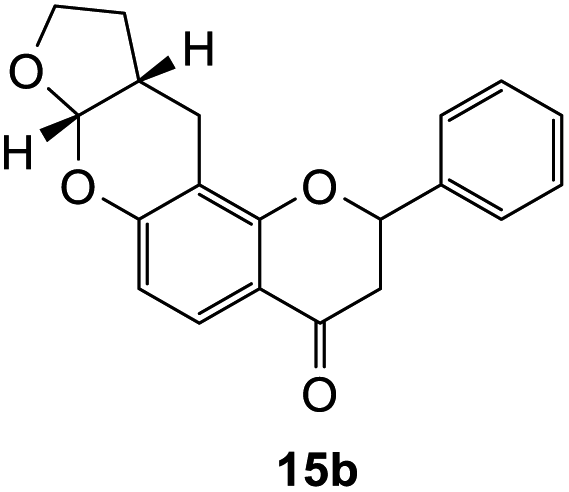 |
6 | 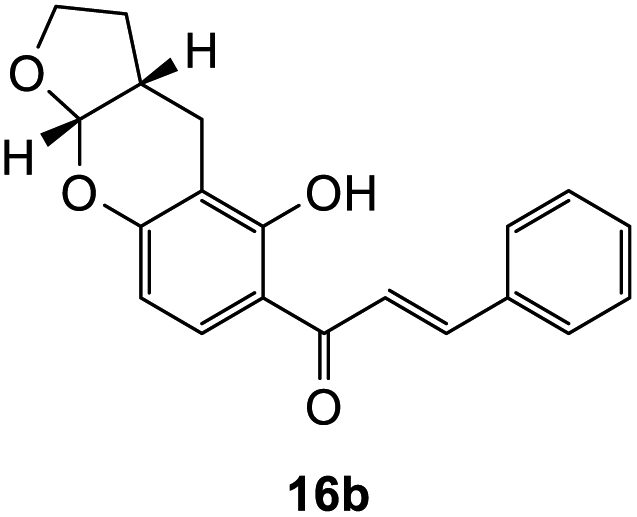 |
5 |
| 3 |  |
6 h | 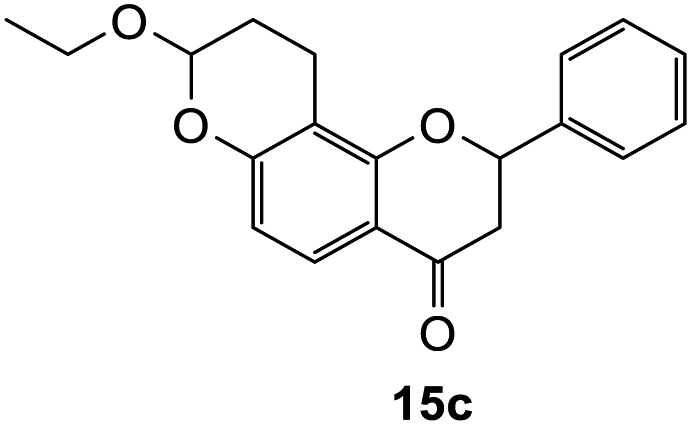 |
7 | 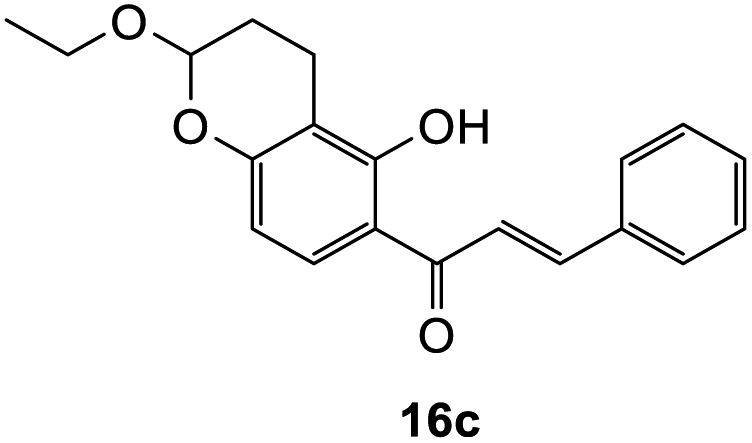 |
5 |
| 4 |  |
6 h | 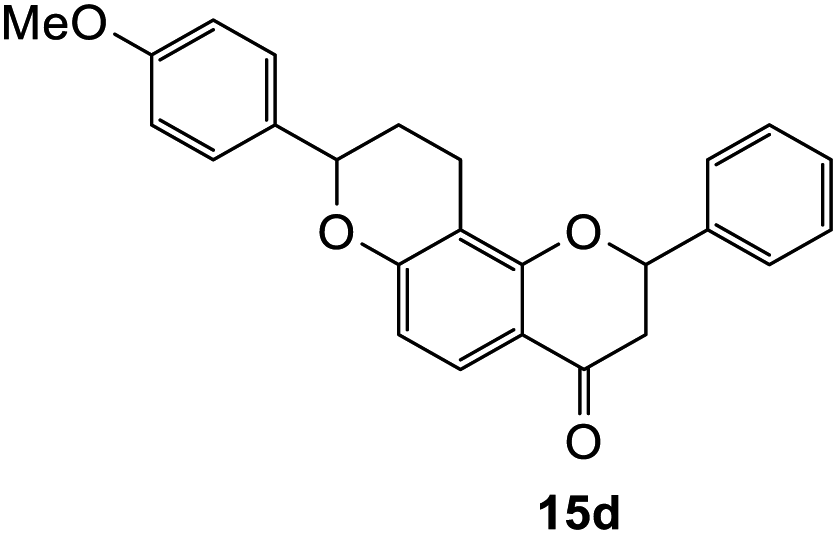 |
7 | 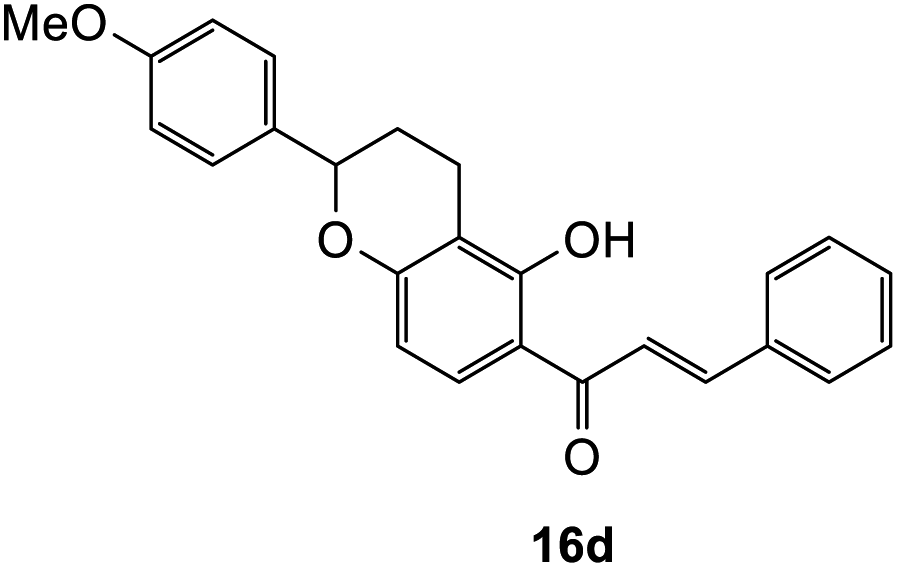 |
8 |
| 5 |  |
16 h | 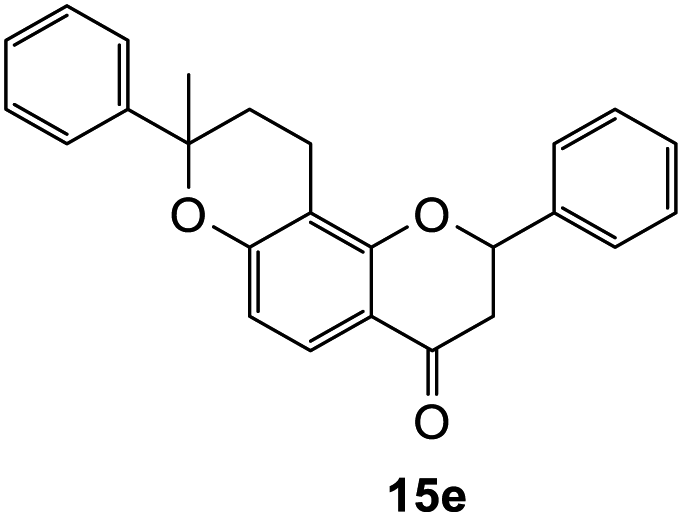 |
4 | 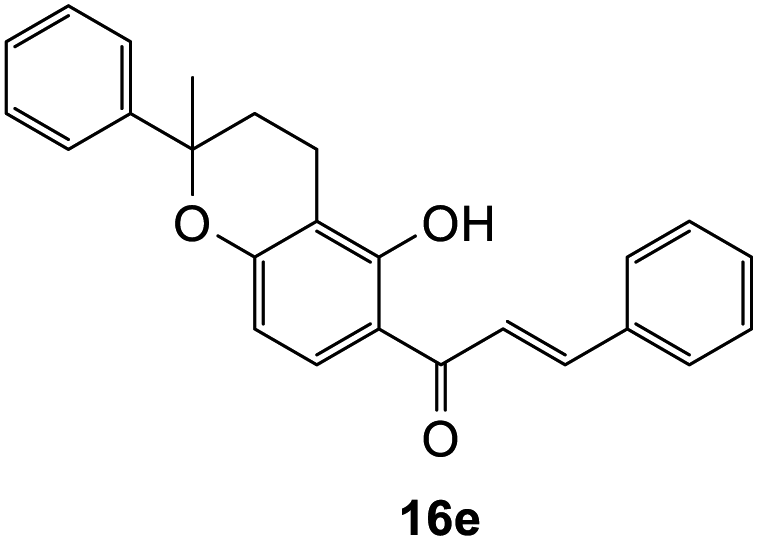 |
7 |
However, this ring-opening reaction did not occur in 6-hydroxy analogue. The intermediate in the ring-opening reaction involves the formation of phenoxide ion. In 6-hydroxy analogues, the para-hydroxy group destabilizes the phenoxide ion by the electron donation, making this a high-energy intermediate. Meanwhile, in the 7-hydroxy analogues, the hydroxy group is in the meta position to the phenoxide ion thereby avoiding such destabilization. Hence, the ring-opening reaction is more likely to occur in the 7-hydroxy analogues.
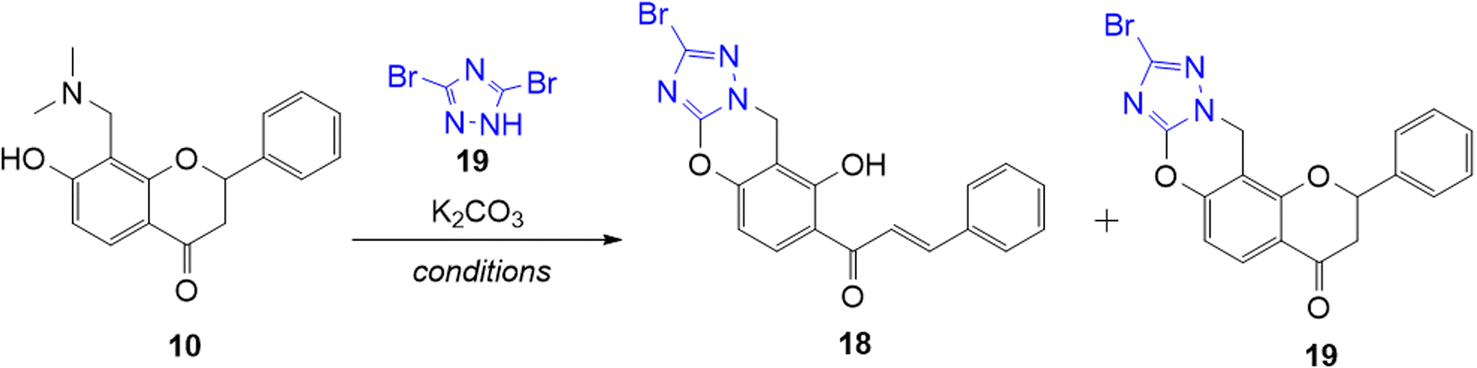
As previously mentioned, the o-QM intermediate can undergo Michael-addition pathway with nucleophiles. Hence, the Michael addition reaction of the Mannich base 10 was attempted using 3,5-dibromo-1,2,4-triazole 17. This triazole is an interesting nitrogen-based nucleophile because it allows the formation of fused nitrogen-containing heterocycles via aza-Michael addition reactions. Aza-Michael addition involves a nucleophile, specifically an amine such as triazole in this case as a Michael-donor and an electron-poor diene such as o-QM as a Michael-acceptor, providing a useful method for constructing new C–N bond.44
Initial attempts to obtain a successful Michael addition reaction between triazole 17 and o-QM precursor 14 were carried out by heating the reactants together in DMF in the presence of K2CO3 in a sealed tube for 4.5 h. The reaction led to complete consumption of the o-QM precursor 14, giving the ring-opened chalcone product 18 a low yield of 8%, while the anticipated ring-closed flavanone 19 product was not observed (Table 3, entry 1). Changing the solvent to N,N-dimethylacetamide (DMA) led to the isolation of the desired product 19 in 8% yield, together with the ring-opened analogue 18 in 58% yield (Table 3, entry 2). The use of a slightly less polar solvent, N-methylpyrrolidine (NMP) did not result in a better outcome as the ring-opened product 18 was formed in a higher yield of 21%, while only producing 3% yield of the desired product 19 (Table 3, entry 3). The cyclization of chalcone product 18 was then affected by treating it with concentrated HCl in MeOH/THF under reflux for 72 h to afford the flavanone product 19, which was purified by preparative HPLC (Scheme 5). However, the conversion was not complete and the reaction proceeded slowly, giving the flavanone product 19 in only 10%. The mechanism of the reaction is postulated to involve an aza-Michael addition reaction similar to that reported by Osipov et al.45 The alkylation takes place at the more nucleophilic N1(2) position rather than at N4 because of the presence of an adjacent atom with a lone pair of electrons (the α-effect). K2CO3 was needed to deprotonate the triazole, thereby facilitating attack of N1(2) at the electrophilic β-carbon of o-QM 14.
|
|||
|---|---|---|---|
| Entry | Conditions | 18 [% yield] | 19 [% yield] |
| 1 | Dienophile (1 eq.), DMF, K2CO3 (3 eq.), pressure tube, 150 °C, 4.5 h | 8 | Not isolated |
| 2 | Dienophile (1 eq.), DMA, K2CO3 (3 eq.), pressure tube, 150 °C, 4.5 h | 58 | 8 |
| 3 | Dienophile (1 eq.), NMP, K2CO3 (3 eq.), pressure tube, 150 °C, 3.5 h | 21 | 3 |
 | ||
| Scheme 5 Acid-catalyzed ring-closing reaction of chalcone 18. | ||
Regeneration of the aromaticity of the triazole moiety is believed to be the driving force of this reaction.
Synthesis of fused-ring isoflavanones and diarylmethane analogues
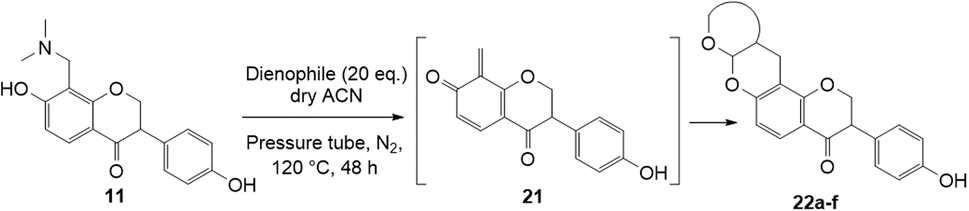
The synthesis of fused-ring systems derived from dihydrodaidzein was first attempted by following the same method as previously described for the flavanones. Heating the Mannich base 11 with 2,3-dihydrofuran in DMF afforded a product in 28% yield. Surprisingly, 1H NMR analysis of the product revealed the disappearance of the characteristic oxygenated methylene group of isoflavanone moiety at ca. δH 4.56 ppm and the methine proton at ca. δH 3.87 ppm. An additional methyl peak was observed instead at δH 1.69 ppm. The 13C NMR spectrum showed peaks for an oxygenated quaternary carbon at δC 90.4 ppm (C2) and a methyl group at δC 23.3 ppm (C3). Subsequently, the 2D HMBC analysis revealed that the methyl group was attached to a quaternary carbon (C2), and had three-bond length correlations with a carbonyl group at δC 198.9 ppm (C1). While the resonance of a doublet at δH 5.71 ppm (J = 4.1 Hz) corresponding to the acetal proton H6a and a multiplet at δH 2.69–2.77 ppm indicating a bridgehead proton H9a, confirmed that the cycloaddition reaction had taken place. Given these NMR data, the product was identified as the ring-opened analogue 20 (Scheme 6). In addition, HRMS identified a molecular mass of 357.1328, consistent with the protonated adduct of compound 20, with an expected mass of 357.1333.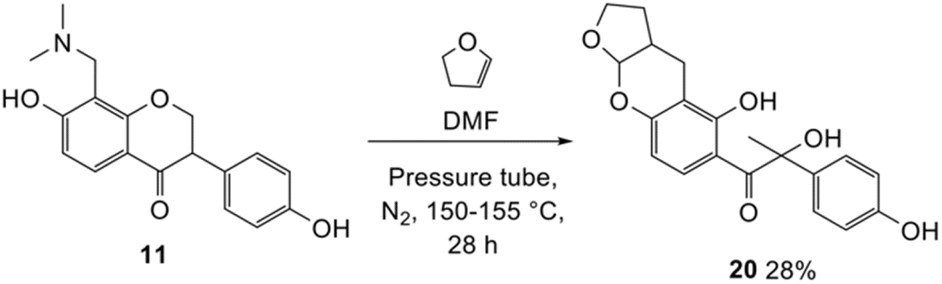 | ||
| Scheme 6 Formation of ring-opened analogue 20 through Diels–Alder reaction of Mannich base 11 with 2,3-dihydrofuran. | ||
Using the same condition, the cycloaddition reactions were further repeated for other dienophiles, such as 3,4-dihydro-2H-pyran, ethyl vinyl ether, and p-methoxystyrene. However, the reactions were unsuccessful, causing the formation of degradation products and the 1H NMR spectra could not be identified. Subsequent cycloaddition reaction of Mannich base with 2,3-dihydrofuran was attempted by heating the mixture at 120 °C in a pressure tube using dry acetonitrile as the solvent under the nitrogen atmosphere. These conditions led to the isolation of the desired product 22a in 18% yield after column chromatography.
Several test reactions were performed to explore the conditions required to form the ring-opened product. First, the Mannich base 11 was heated alone in DMF at a high temperature of 150–155 °C, which resulted in the cleavage of the dimethylamine group, forming the starting dihydrodaidzein 7 along with degradation products. This indicated that thermolysis of Mannich base 11, but not ring-opening, occurred under these conditions. In the second attempt, the Mannich base 11 was treated with 2,3-dihydrofuran in dry acetonitrile at 150–155 °C for 22 h. This reaction yielded both the expected product 22a and the ring-opened analogue 20, in a ratio of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Lowering the temperature to 120 °C and extending the reaction time to 50 h did not alter the outcome. In the final trial, the cyclized product 22a was heated alone in dry acetonitrile at 120 °C and monitored by TLC. At 48 h, the compound remained unchanged. However, after 69 h of heating, the adduct 22a began to degrade, as evidenced by the appearance of several spots on the TLC plate. Notably, the ring-opened product did not form during this trial. Given these observations, it can be concluded that the ring-opening reaction only occurred when both Mannich base 11 and dienophile, in this case 2,3-dihydrofuran, were heated together. The ring-opening of the isoflavanone core is postulated to involve a retro-Michael-type ring-opening to give the propenone intermediate under basic conditions.46 Subsequent nucleophilic addition by a water molecule generates the ring-opened product 20.
:1. Lowering the temperature to 120 °C and extending the reaction time to 50 h did not alter the outcome. In the final trial, the cyclized product 22a was heated alone in dry acetonitrile at 120 °C and monitored by TLC. At 48 h, the compound remained unchanged. However, after 69 h of heating, the adduct 22a began to degrade, as evidenced by the appearance of several spots on the TLC plate. Notably, the ring-opened product did not form during this trial. Given these observations, it can be concluded that the ring-opening reaction only occurred when both Mannich base 11 and dienophile, in this case 2,3-dihydrofuran, were heated together. The ring-opening of the isoflavanone core is postulated to involve a retro-Michael-type ring-opening to give the propenone intermediate under basic conditions.46 Subsequent nucleophilic addition by a water molecule generates the ring-opened product 20.
Hence, the cycloaddition reactions of Mannich base 11 were carried out in dry acetonitrile at a lower temperature of 120 °C (Scheme 7), providing the desired cycloadducts 22a–f in yields varying between 10 and 33% (Table 4). Similar to cycloaddition adducts derived from flavanones, the Diels–Alder reactions between the Mannich base 11 and vinyl ether-type dienophiles regioselectively provided the acetal-type products (Table 4, entries 1–3). With p-methoxystyrene (Table 4, entry 4), the reaction also proceeded in high regioselectivity, where the resulting product bore the phenyl ring at C8 rather than C9. These results again suggested that charge distribution of the reacting partners determined the regioselectivity of the reactions.
 | ||
| Scheme 7 Formation of cycloadducts 22a–f from Mannich base 11. | ||
| Entry | Dienophile | Time | Product | Yield [%] | Entry | Dienophile | Time | Product | Yield [%] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 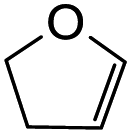 |
48 h | 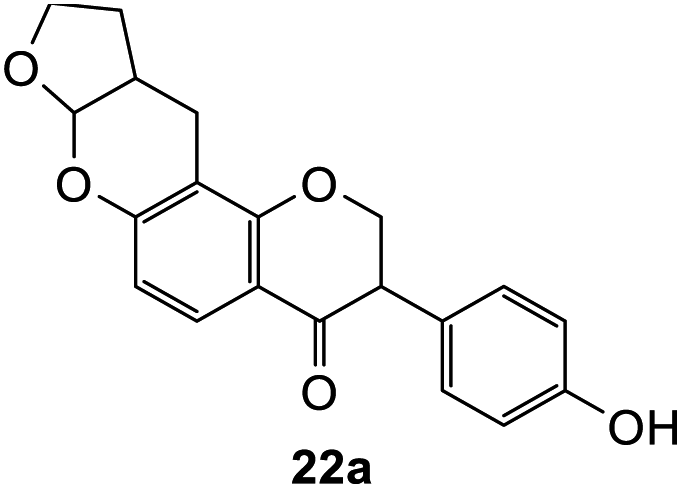 |
18 | 6 | 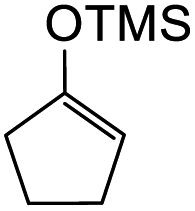 |
48 h | 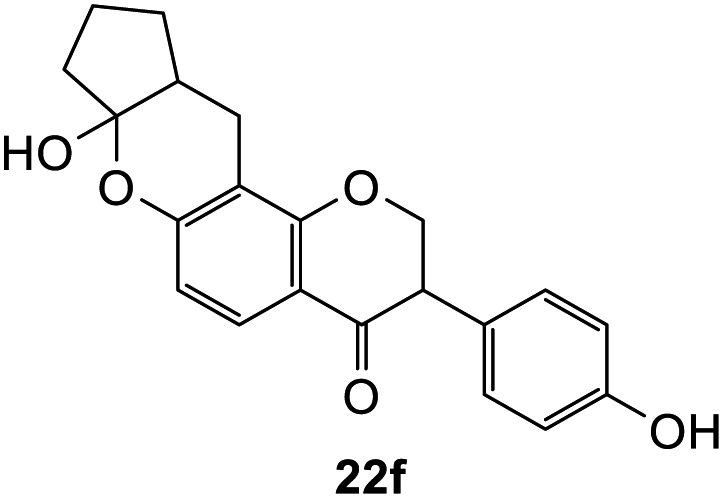 |
25 |
| 2 | 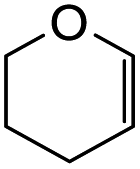 |
48 h |  |
13 | 7 | 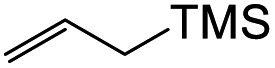 |
48 h |  |
0 |
| 3 | 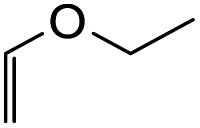 |
48 h | 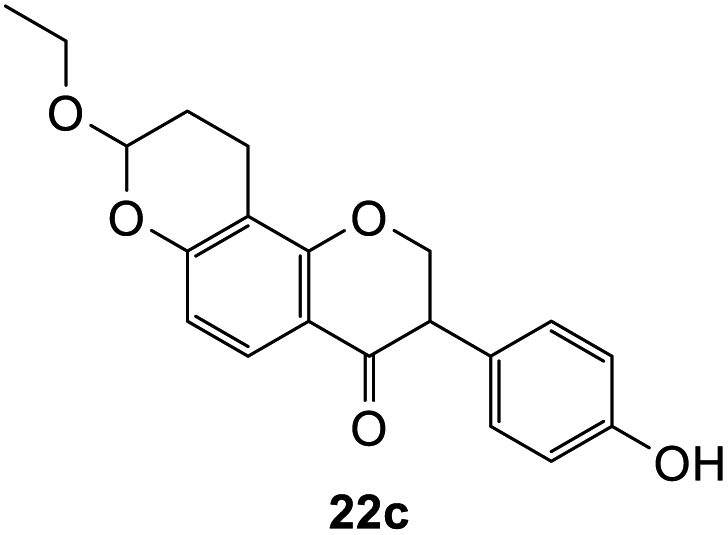 |
10 | 8 | 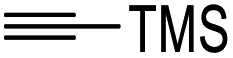 |
48 h | 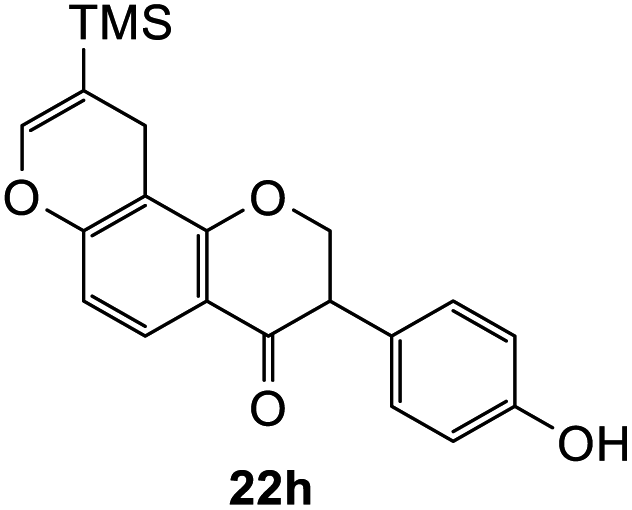 |
0 |
| 4 | 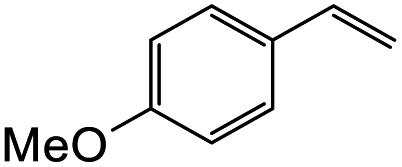 |
48 h | 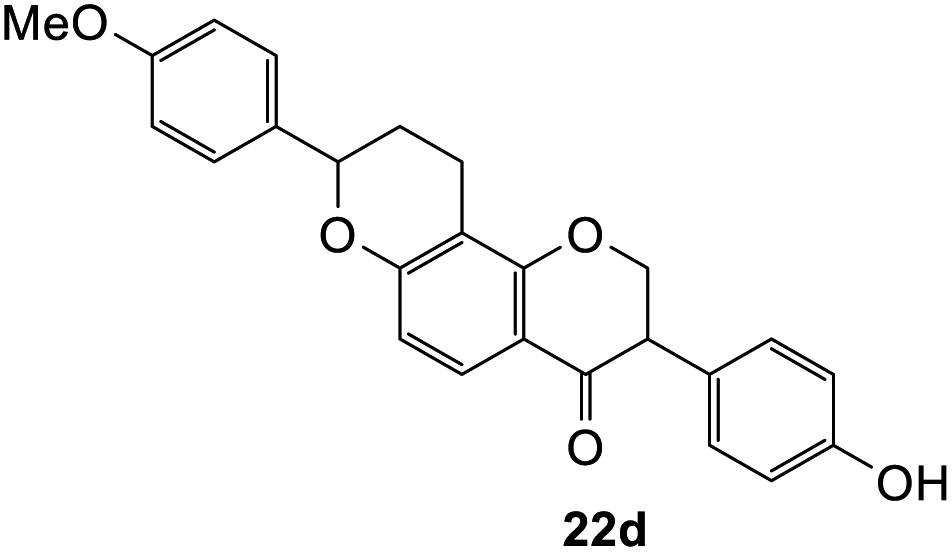 |
33 | 9 |  |
48 h |  |
0 |
| 5 | 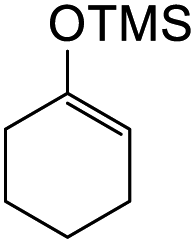 |
48 h | 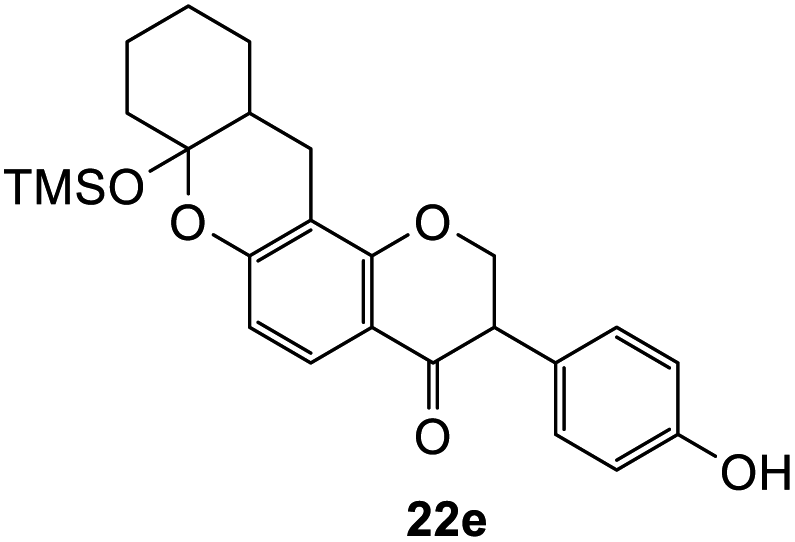 |
27 |
To further expand the substrate scope, silane-containing dienophiles were explored. Using the same conditions as depicted in Scheme 7, Mannich base 11 was treated with (cyclohex-1-en-1-yloxy)trimethylsilane (Table 4, entry 5), providing the fused-ring system containing trimethylsiloxyl group 22e. However, with (cyclopent-1-en-1-yloxy)trimethylsilane (Table 4, entry 6), the cycloadduct 22f obtained was suggested to undergo hydrolysis giving a hydroxyl group instead of the trimethylsiloxyl group. The use of allyltrimethylsilane, ethynyltrimethylsilane and phenylacetylene as dienophiles failed to generate the desired cycloadducts 22g, 22h, and 22i respectively (Table 4, entries 7–9) and only the starting Mannich base 11 was isolated from the reaction mixtures. The lack of nucleophilicity of these dienophiles again contributed to such observations, where the Diels–Alder reactions could not be facilitated.
We also investigated the reactivity of o-QM intermediates 21 derived from Mannich base 11 towards the Michael addition using various nucleophiles, providing diarylmethane products 23a–d (Scheme 8). A key feature in the 1H NMR spectra of the Michael adducts is the signal from the methylene bridge connecting C8 of the isoflavanone core with the nucleophilic moiety, which appeared as a singlet at around δH 3.70 ppm.
 | ||
| Scheme 8 Michael addition of in situ generated o-QM 21 derived from Mannich base 11. | ||
The reaction between o-QM 20 and 3,5-dimethoxyphenol led to Michael adduct 23a, where the 3,5-dimethoxyphenol reacted at the position between the two methoxy groups (Table 5, entry 1). Different regioselectivity was observed when the Mannich base 11 was treated with dimethoxyaniline (Table 5, entry 2). The 1H NMR spectrum of the product suggests that the dimethoxyaniline moiety is unsymmetrical. However, another possible regioisomer, where the methylene bridge attached to the more sterically hindered position of the dimethoxyaniline moiety, was not observed. When thiophenol was used as the nucleophile, a thioether linkage was formed instead of the methylene bridge (Table 5, entry 3). As confirmed by the 1H NMR spectrum, the methylene protons of the thioether linkage resonated at a more downfield region of δH 4.24 ppm compared with a typical methylene bridge (ca. δH 3.70 ppm). The thiol-Michael addition reaction is hypothesized to be catalyzed by dimethylamine, which was produced as a by-product during the o-QM formation. The amine deprotonates the thiol to generate the thiolate anion, a stronger nucleophile than the π electrons in the benzene ring that attacks the o-QM 20. However, the use of 4-chlorothiophenol generated the corresponding Michael adduct in lower yield compared to thiophenol (Table 5, entry 4). This was attributed to the presence of the electron-withdrawing chlorine substituent in the nucleophile, further enforcing the importance of using electron-rich nucleophiles for such reactions.
| Entry | Nucleophile | Product | Yield [%] | Entry | Nucleophile | Product | Yield [%] |
|---|---|---|---|---|---|---|---|
| 1 | 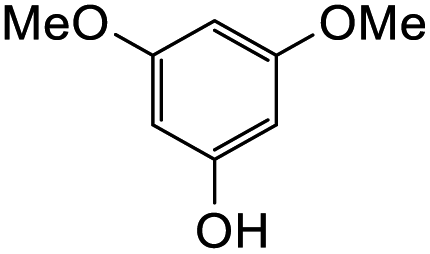 |
 |
8 | 4 | 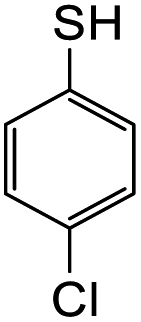 |
 |
16 |
| 2 |  |
 |
17 | 5 | 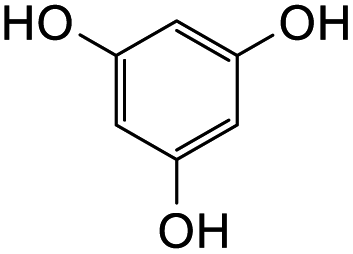 |
 |
0 |
| 3 | 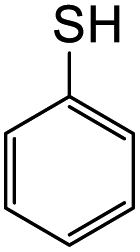 |
 |
41 | 6 | 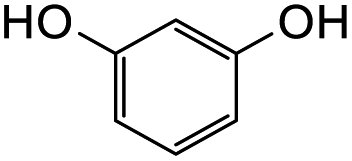 |
 |
0 |
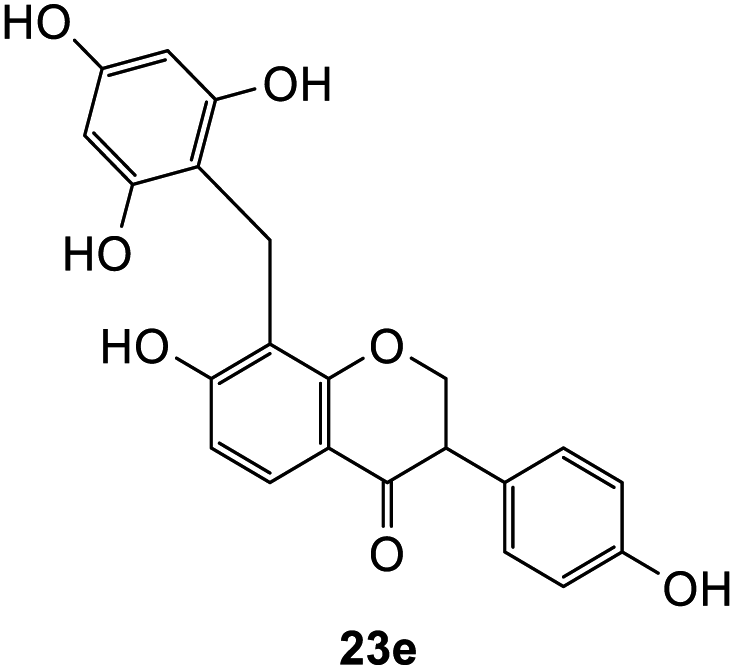
Further attempts using phloroglucinol and resorcinol had failed to generate the desired products 23e–f respectively (Table 5, entries 5 and 6) as they are non-activated as compared to dimethoxyphenol and dimethoxyaniline even after prolonged heating or drying the reagent before use. In the two cases, dihydrodaidzein 7 was isolated instead. This indicates that electron-rich nucleophiles are required for the Michael addition reaction to the o-QM intermediate. The [4 + 2] cycloaddition and Michael addition reaction proceeded with electron-rich alkenes and nucleophiles, which led to low overall yields for most o-QMs adducts. A possible reason for the low yields could be due to self-dimerization between the o-QM intermediate, leading to reduced efficiency of this reaction.47 Overall, these methods pave the way for accessing fused-ring and diarylmethane products from flavanones and isoflavanones that are not synthesized in nature, which might show interesting biological activity.
To assess the anti-proliferative properties, several cycloadditions adduct were tested in vitro against MDA-MB-231 triple-negative breast cancer cell lines using Alamar Blue assay. As shown in Table 6, the introduction of fused-ring system on 6-hydroxyflavanone 5 caused lower anti-proliferative activities compared to the parental compound. On the other hand, cycloadducts derived from 7-hydroxyflavanone 6 displayed slightly improved anti-proliferative activity against MDA-MB-231 breast cancer cell lines, though it was very weak. For cycloadducts synthesized from dihydrodaidzein 7, only compound 22d bearing 8-(p-methoxyphenyl) exhibited improved anti-proliferative activity compared to dihydrodaidzein 7 with the IC50 value of 35.1 ± 0.7 μM. These findings pave the way for incorporating other dienophiles, which might lead to enhanced biological activity. Additional studies are also needed to further assess the anti-proliferative activity of Michael adducts.
| Compound | IC50/μM |
|---|---|
| 5 | 42.9 ± 1.7 |
| 13a | >100 |
| 13b | >100 |
| 13c | >100 |
| 6 | >100 |
| 15a | 66.5 ± 2.8 |
| 15b | 43.2 ± 1.8 |
| 15c | 88.6 ± 1.5 |
| 7 | >100 |
| 22a | >100 (but ∼100) |
| 22b | >100 |
| 22c | >100 |
| 22d | 35.1 ± 0.7 |
Experimental
General procedures
All reactions requiring anhydrous conditions were performed in oven dried glassware under a nitrogen atmosphere unless otherwise stated. All the reagents were purchased from commercial sources and used without further purification.Reactions were monitored using thin layer chromatography (TLC) using aluminum plates coated with silica gel GF254. Compounds were detected by short and long wave-length ultraviolet light. Gravity column chromatography was carried out using 40–63 micron silica gel. Preparative reversed phase HPLC was performed using a PDA detector (254 nm) and C18 column (150 mm x 10 nm) on a gradient elution of 2–100% over 56 min with a flow rate of 5 mL min−1. 0.1% Formic acid/MilliQ water was used as eluent A and 0.1% formic acid/acetonitrile was used as eluent B. 1H NMR spectral data are reported as follows: chemical shifts are in parts per million (ppm) and internally referenced relative to the solvent nuclei (δ); multiplicity; observed coupling constant (J) in hertz (Hz); proton count; as signment. Multiplicities are assigned as singlet (s), doublet (d), doublet of doublets (dd), doublet of triplets (dt), triplet, (t), doublet of doublet of doublets (ddd), triplet of doublets (td), multiplet (m), and broad singlet (br s) where appropriate. 13C NMR spectra were recorded in the designated solvents, and chemical shifts are reported in ppm internally referenced relative to the solvent nuclei. Melting points were measured using a Mel-Temp melting point apparatus and are uncorrected. Infrared spectra were recorded using an attenuated total reflection FTIR Spectrometer. High-resolution mass spectra were measured at 70 eV using a quadrupole analyzer and are reported with ion mass/charge (m/z) ratios as values in atomic mass units. All procedure involving use of a pressure tube at high temperature were performed following safety guidelines from the manufacturer.
Synthesis
:5) as the eluent to afford the corresponding products. Further purification was performed using preparative reversed phase HPLC when required.
5-((Dimethylamino)methyl)-6-hydroxy-2-phenylchroman-4-one (9). To a solution of 5 (0.75 g, 3.13 mmol, 1 eq.) in 1,4-dioxane (30 mL) was added bis(N,N-dimethylamino)methane (0.85 mL, 6.25 mmol, 2.0 eq.). The mixture was heated to reflux for 24 h. After completion of reaction, the solvent was evaporated and the resulting crude product was chromatographed over silica gel (230–400 mesh) using DCM
:MeOH (95:5) as eluent to afford 9 as an orange solid (0.71 g, 76%). M.p. 109–111 °C; IR (neat): υmax 3356, 2920, 1674, 1460, 1274, 1004, 903, 825, 759, 698 cm−1; UV (MeOH): λmax 348 nm (ε 56259 cm−1 M−1), 308 (9456), 230 (51382); 1H NMR (400 MHz, CDCl3): δ 7.37–7.48 (m, 5H, H2′, H3′, H4′, H5′, H6′), 7.02 (d, J = 8.8 Hz, 1H, H7), 6.92 (d, J = 8.8 Hz, 1H, H8), 5.38 (dd, J = 2.8, 13.7 Hz, 1H, H2), 4.37 (d, J = 3.3 Hz, 2H, N–CH2), 3.06 (dd, J = 13.7, 16.9 Hz, 1H, H3α), 2.84 (d, J = 2.8, 16.9 Hz, 1H, H3β), 2.38 (s, 6H, 2 × CH3); 13C NMR (100 MHz, CDCl3): δ 194.7 (C4), 156.2 (C8a), 154.5 (C6), 139.3 (C1′), 129.0 (C3′, C5′), 128.8 (C4′), 126.3 (C2′, C6′), 125.0 (C7), 121.5 (C4a), 119.5 (C5), 118.4 (C8), 79.0 (C2), 59.2 (CH2–N), 46.8 (C3), 44.5 (2 × CH3). HRMS (+ESI): (C18H19NO3) calcd m/z 298.1438 [M + H]+, found m/z 298.1433.
2-Phenyl-2,3,10,11,11a,12-hexahydro-9H-dipyrano[2,3-b:3′,2′-f]chromen-1(7aH)-one (13a). Flavanone 13a was synthesized following general procedure 1 using the Mannich base 9 (207 mg, 0.70 mmol, 1 eq.) and 3,4-dihydro-2H-pyran (1.47 mL, 16.11 mmol, 23.0 eq.). The mixture was heated at 155 °C for 23 h. The product was obtained as a white solid (28 mg, 12%). M.p. 154–155 °C; IR (neat): υmax 2928, 1672, 1600, 1466, 1282, 1200, 1136, 1087, 999, 935, 831, 755, 699 cm−1; UV (MeOH): λmax 348 nm (ε 47
430 cm−1 M−1), 308 (5719), 225 (17156); 1H NMR (400 MHz, acetone-d6): δ 7.56–7.58 (m, 2H, H2′, H6′), 7.42–7.46 (m, 2H, H3′, H5′), 7.36–7.40 (m, 1H, H4′), 7.05 (d, J = 8.7 Hz, 1H, H11), 6.91 (d, J = 8.7 Hz, 1H, H12), 5.51 (ddd, J = 3.1, 8.5, 13.2 Hz, 1H, H2), 5.25 (dd, J = 2.5, 12.8 Hz, 1H, H9a), 3.90–3.95 (m, 1H, H8α), 3.63–3.67 (m, 1H, H8β), 3.19–3.26 (m, 2H, H5), 3.09 (dd, J = 13.2, 16.4 Hz, 1H, H3α), 2.81 (dd, J = 3.1, 16.4 Hz, 1H, H3β), 2.13–2.17 (m, 1H, H5a), 1.61–1.70 (m, 2H, H7), 1.57–1.60 (m, 2H, H6); 13C NMR‡ (100 MHz, acetone-d6): δ 193.9 (193.8) (C4), 158.3 (158.2) (C12a), 148.5 (148.3) (C10a), 140.6 (140.5) (C1′), 129.5 (C3′, C5′), 129.3 (129.2) (C4′), 127.2 (C2′, C6′), 125.2 (125.1) (C11), 122.4 (122.3) (C4a′), 119.7 (119.5) (C4a), 118.1 (118.0) (C12), 96.9 (96.8) (C9a), 79.9 (79.8) (C2), 63.4 (62.9) (C8), 46.7 (46.6) (C3), 32.1 (C5a), 28.8 (29.4) (C5), 25.2 (25.4) (C6), 24.1 (24.4) (C7). HRMS (+ESI): (C21H20O4) calcd m/z 359.1254 [M + Na]+, found m/z 359.1250.
7-Ethoxy-2-phenyl-2,3,9,10-tetrahydropyrano[3,2-f]chromen-1(8H)-one (13b). Flavanone 13b was synthesized following general procedure 1 using the Mannich base 9 (80 mg, 0.27 mmol, 1 eq.) and ethyl vinyl ether (0.52 mL, 5.43 mmol, 20.1 eq.). The mixture was heated at 155 °C for 23 h. The product was obtained as a yellow sticky solid (26 mg, 30%). IR (DCM): υmax 2920, 1679, 1467, 1318, 1267, 1200, 1062, 1010, 964, 823, 699 cm−1. UV (MeOH): λmax 343 nm (ε 11
743 cm−1 M−1), 308 (2206), 225 (2984); 1H NMR (400 MHz, acetone-d6): δ 7.55–7.59 (m, 2H, H2′, H6′), 7.38–7.47 (m, 3H, H3′, H4′, H5′), 7.04 (d, J = 8.9 Hz, 1H, H9), 6.89 (d, J = 8.9 Hz, 1H, H10), 5.49 (dd, J = 3.2, 13.1 Hz, 1H, H2), 5.25 (t, J = 2.6 Hz, 1H, H7), 3.77–3.83 (m, 1H, H1′′α), 3.60–3.66, (m, 1H, H1′′β), 3.25–3.36 (m, 1H, H5α), 3.11–3.17 (m, 1H, H5β), 3.09 (dd, J = 13.1, 16.1 Hz, 1H, H3α), 2.78 (dd, J = 2.9,16.1 Hz, 1H, H3β), 1.95–2.02 (m, 1H, H6α), 1.77–1.89 (m, 1H, H6β), 1.10–1.16 (m, 3H, 2′′-CH3); 13C NMR‡ (100 MHz, acetone-d6): δ 193.8 (193.9) (C4), 158.0 (158.2) (C10a), 147.4 (147.5) (C8a), 140.7 (C1′), 129.5 (C3′, C5′), 129.3 (C4′), 127.2 (C2′, C6′), 125.8 (125.7) (C9), 124.8 (125.0) (C4a′), 119.3 (119.6) (C4a), 117.8 (117.8) (C10), 97.2 (97.1) (C7), 79.8 (79.7) (C2), 64.2 (C1′′), 46.7 (46.8) (C3), 26.9 (27.1) (C6), 20.8 (21.0) (C5), 15.6 (C2′′). HRMS (+ESI): (C20H20O4) calcd m/z 347.1254 [M + Na]+, found m/z 347.1252.
2-Phenyl-2,3,9,10,10a,11-hexahydrofuro[2,3-b]pyrano[3,2-f]chromen-1(7aH)-one (13c). Flavanone 13c was synthesized following general procedure 1 using the Mannich base 9 (100 mg, 0.34 mmol, 1 eq.) and 2,3-dihydrofuran (0.51 mL, 6.75 mmol, 19.8 eq.). The mixture was heated at 155 °C for 17 h. The product was obtained as colourless needle-like crystals (23 mg, 21%). M.p. 148–149 °C; IR (neat): υmax 2950, 1665, 1455, 1312, 1268, 1202, 1066, 909, 829, 754, 692 cm−1. UV (THF): λmax 348 nm (ε 2514 cm−1 M−1), 239 (4352); 1H NMR (400 MHz, acetone-d6): δ 7.56–7.59 (m, 2H, H2′, H6′), 7.42–7.47 (m, 2H, H3′, H5′), 7.36–7.40 (m, 1H, H4′), 7.03 (dd, J = 2.2, 8.9 Hz, 1H, H10), 6.91 (dd, J = 3.4, 8.9 Hz, 1H, H11), 5.54 (dd, J = 2.3, 13.6 Hz, 1H, H2), 5.52 (dd, J = 2.0, 15.9 Hz, 1H, H8a), 3.81–3.93 (m, 2H, H7), 3.67–3.76 (m, 1H, H5α), 3.14–3.20 (m, 1H, H5β), 3.07 (dd, J = 13.1, 17.3 Hz, 1H, H3α), 2.80 (dd, J = 2.7, 16.3 Hz, 1H, H3β), 2.72–2.76 (m, 1H, H5a), 2.07–2.12 (m, 1H, H6α), 1.49–1.61 (m, 1H, H6β). 13C NMR‡ (100 MHz, acetone-d6): δ 194.1 (193.9) (C4), 158.3 (158.2) (C11a), 148.8 (C9a), 140.5 (140.4) (C1′), 129.4 (C3′, C5′), 129.2 (C4′), 127.1 (C2′, C6′), 125.6 (125.5) (C10), 124.3 (124.1) (C4a′), 119.9 (119.5) (C4a), 117.9 (117.8) (C11), 102.0 (101.8) (C8a), 79.8 (79.7) (C2), 68.3 (68.2) (C7), 46.7 (46.6) (C3), 38.1 (38.0) (C5a), 29.4 (C6), 25.2 (24.9) (C5). HRMS (+ESI): (C20H18O4) calcd m/z 323.1278 [M + H]+, found m/z 323.1278.
2,7-Diphenyl-2,3,9,10-tetrahydropyrano[3,2-f]chromen-1(8H)-one (13d). Flavanone 13d was synthesized following general procedure 1 using the Mannich base 9 (150 mg, 0.50 mmol, 1 eq.) and styrene (1.16 mL, 10.12 mmol, 20.2 eq.). The mixture was heated at 155 °C for 44 h. The product was obtained as a white solid (45 mg, 25%). M.p. 159–160 °C; IR (neat): υmax 2918, 1677, 1600, 1468, 1325, 1268, 1199, 1162, 1007, 939, 835, 750, 695 cm−1. UV (MeOH): λmax 342 nm (ε 5845 cm−1 M−1), 307 (1283), 231 (642); 1H NMR (400 MHz, acetone-d6): δ 7.56–7.59 (m, 2H, H2′, H6′), 7.46–7.49 (m, 2H, H2′′, H6′′), 7.43–7.46 (m, 2H, H3′, H5′), 7.38–7.42 (m, 3H, ArH), 7.30–7.35 (m, 1H, ArH), 7.12 (d, J = 8.9 Hz, 1H, H9), 6.93 (d, J = 8.9 Hz, 1H, H10), 5.54 (dd, J = 3.1, 13.1 Hz, 1H, H2), 5.06 (t, J = 2.4 Hz, 1H, H7), 3.18–3.38 (m, 2H, H5), 3.13 (dd, J = 13.2, 16.2 Hz, 1H, H3α), 2.79 (dd, J = 2.8, 16.2 Hz, 1H, H3β), 2.22–2.30 (m, 1H, H6α), 1.91–2.03 (m, 1H, H6β); 13C NMR‡ (100 MHz, acetone-d6): δ 193.9 (193.7) (C4), 158.2 (157.9) (C10a), 150.7 (150.6) (C8a), 142.9 (142.8) (C1′′), 140.6 (C1′), 129.5 (C3′, C5′), 129.3 (C3′′, C5′′), 129.2 (C4′), 128.6 (128.5) (C4′′), 127.2 (C2′, C6′), 126.9 (126.8) (C2′′, C6′′), 125.7 (C9), 124.1 (124.0) (C4a′), 119.8 (119.5) (C4a), 118.0 (117.9) (C10), 79.8 (C2), 77.8 (77.6) (C7), 46.8 (46.6) (C3), 30.6 (30.5) (C6), 25.9 (25.5) (C5). HRMS (+ESI): (C24H20O3) calcd m/z 379.1305 [M + Na]+, found m/z 379.1303.
7-(4-Methoxyphenyl)-2-phenyl-2,3,9,10-tetrahydropyrano[3,2-f]chromen-1(8H)-one (13e). Flavanone 13e was synthesized following general procedure 1 using the Mannich base 9 (90 mg, 0.30 mmol, 1 eq.) and p-methoxy styrene (0.81 mL, 6.05 mmol, 20.2 eq.). The mixture was heated at 155 °C for 24 h. The product was obtained as a yellow solid (42 mg, 36%). M.p. 163–164 °C; IR (neat): υmax 2920, 1670, 1613, 1513, 1455, 1378, 1245, 1172, 1024, 940, 820, 768; UV (MeOH): λmax 348 nm (ε 3207 cm−1 M−1), 263 (8850), 230 (32
887), 201 (33157); 1H NMR (400 MHz, acetone-d6): δ 7.57–7.59 (m, 2H, H2′, H6′), 7.43–7.46 (m, 2H, H3′, H5′), 7.38–7.41 (m, 3H, H4′, H2′′, H6′′), 7.09 (dd, J = 3.2, 8.9 Hz, 1H, H9), 6.95 (dd, J = 2.1, 8.9 Hz, 2H, H3′′, H5′′), 6.92 (dd, J = 2.6, 8.9 Hz, 1H, H10), 5.54 (dd, J = 3.0, 13.1 Hz, 1H, H2), 5.00 (t, J = 2.5 Hz, 1H, H7), 3.81 (s, 3H, –OCH3), 3.32–3.39 (m, 1H, H5α), 3.18–3.28 (m, 1H, H5β), 3.13 (dd, J = 13.1, 16.1 Hz, 1H, H3α), 2.79 (dd, J = 3.0, 16.1 Hz, 1H, H3β), 2.20–2.26 (m, 1H, H6α), 1.93–2.02 (m, 1H, H6β); 13C NMR‡ (100 MHz, acetone-d6): δ 193.9 (193.7) (C4), 160.4 (160.3) (C4′′), 158.1 (157.9) (C10a), 150.9 (150.8) (C8a), 140.7 (140.6) (C1′), 134.8 (134.8) (C1′′), 129.5 (C3′, C5′), 129.3 (129.2) (C4′), 128.3 (128.2) (C2′′, C6′′), 127.2 (127.2) (C2′, C6′), 125.7 (125.7) (C9), 124.1 (124.0) (C4a′), 119.9 (119.6) (C4a), 118.0 (117.9) (C10), 114.6 (C3′′, C5′′), 79.8 (79.8) (C2), 77.6 (77.4) (C7), 55.6 (OMe), 46.8 (46.6) (C3), 26.1 (25.6) (C6), 23.4 (C5). HRMS (+ESI): (C25H22O4) calcd m/z 409.1410 [M + Na]+, found m/z 409.1406.
7-Methyl-2,7-diphenyl-2,3,9,10-tetrahydropyrano[3,2-f]chromen-1(8H)-one (13f). Flavanone 13f was synthesized following general procedure 1 using the Mannich base 9 (60 mg, 0.20 mmol, 1 eq.) and α-methyl styrene (0.53 mL, 4.08 mmol, 20.4 eq.). The mixture was heated at 155 °C for 23 h. The product was obtained as a yellow oil (9 mg, 12%). IR (DCM): υmax 2922, 1679, 1466, 1372, 1271, 1161, 1069, 937, 823, 763, 699 cm−1; UV (THF): λmax 353 nm (ε 7891 cm−1 M−1); 1H NMR (400 MHz, acetone-d6): δ 7.53–7.56 (m, 2H, H2′, H6′), 7.41–7.45 (m, 4H, ArH), 7.28–7.39 (m, 4H, ArH), 7.22–7.24 (m, 1H, ArH), 7.20–7.21 (m, 1H, ArH), 6.93 (dd, J = 3.0, 9.0 Hz, 1H, H10), 5.46 (dd, J = 3.1, 13.5 Hz, 1H, H2), 3.23–3.29 (m, 1H, H5α), 3.02 (dd, J = 13.2, 16.1 Hz, 1H, H3α), 2.72 (dd, J = 3.1, 16.1 Hz, 1H, H3β), 2.68–2.72 (m, 1H, H5β), 2.52–2.60 (m, 2H, H6), 1.59 (s, 3H, CH3); 13C NMR‡ (100 MHz, acetone-d6): δ 193.6 (193.8) (C4), 157.9 (157.7) (C10a), 149.4 (149.5) (C8a), 146.3 (146.2) (C1′′), 140.6 (C1′), 129.4 (C3′, C5′), 129.2 (C3′′, C5′′), 129.2 (C4′), 127.6 (C4′′), 127.1 (C2′, C6′), 125.9 (C9), 125.8 (C2′′, C6′′), 123.8 (C4a′), 119.4 (119.2) (C4a), 118.0 (117.9) (C10), 79.8 (79.6) (C2), 78.4 (78.2) (C7), 46.6 (46.5) (C3), 32.8 (32.6) (C6), 31.1 (CH3), 23.4 (23.3) (C5); HRMS (+ESI): (C25H22O3) calcd m/z 393.1461 [M + Na]+, found m/z 393.1460.
7-Methyl-2-phenyl-7-(3-(prop-1-en-2-yl)phenyl)-2,3,9,10-tetrahydropyrano[3,2-f]chromen-1(8H)-one (13g). Flavanone 13g was synthesized following general procedure 1 using the Mannich base 9 (60 mg, 0.20 mmol, 1 eq.) and 1,3-diisopropenylbenzene (0.69 mL, 4.03 mmol, 20.1 eq.). The mixture was heated at 155 °C for 14 h. The product was obtained as a yellow oil (17 mg, 21%). IR (DCM): υmax 2970, 1675, 1597, 1461, 1371, 1327, 1266, 1159, 1075, 933, 891, 799 cm−1; UV (MeOH) λmax 348 nm (ε 17
816 cm−1 M−1), 308 (2997), 230 (2381); 1H NMR (300 MHz, acetone-d6): δ 7.58–7.59 (m, 1H, ArH), 7.52–7.56 (m, 2H, H2′, H6′), 7.40–7.45 (m, 2H, ArH), 7.34–7.39 (m, 3H, ArH), 7.28–7.32 (m, 1H, ArH), 7.24 (dd, J = 2.0, 9.0 Hz, 1H, H9), 6.93 (dd, J = 2.2, 9.0 Hz, 1H, H10), 5.45 (dd, J = 2.8, 13.2 Hz, 1H, H2), 5.34–5.36 (m, 1H, =CH2), 5.05–5.07 (m, 1H, =CH2), 3.22–3.33 (m, 1H, H5α), 3.02 (dd, J = 13.1, 16.2 Hz, H3α), 2.69 (dd, J = 3.0, 16.2 Hz, 1H, H3β), 2.55–2.69 (m, 3H, H5, H6), 2.09–2.11 (m, 3H, CH3), 1.61 (s, 3H, CH3); 13C NMR‡ (100 MHz, acetone-d6): δ 192.8 (C4), 157.1 (156.8) (C10a), 148.6 (C8a), 145.3 (C1′′), 143.3 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 141.1 (C3′′), 139.7 (C1′), 128.5 (2 × ArCH), 128.4 (2 × ArCH), 126.2 (2 × ArCH), 125.0 (ArCH), 124.3 (ArCH), 123.9 (ArCH), 123.0 (ArC), 122.1 (ArCH), 118.6 (C4a), 117.2 (C10), 112.0 (CH2), 78.9 (78.7) (C2), 77.6 (77.4) (C7), 45.7 (C3), 31.6 (31.8) (C6), 30.3 (29.9) (CH3), 22.5 (22.3) (C5), 21.0 (CH3); HRMS (+ESI): (C28H26O3) calcd m/z 433.1774 [M + Na]+, found m/z 433.1771.
CH2), 141.1 (C3′′), 139.7 (C1′), 128.5 (2 × ArCH), 128.4 (2 × ArCH), 126.2 (2 × ArCH), 125.0 (ArCH), 124.3 (ArCH), 123.9 (ArCH), 123.0 (ArC), 122.1 (ArCH), 118.6 (C4a), 117.2 (C10), 112.0 (CH2), 78.9 (78.7) (C2), 77.6 (77.4) (C7), 45.7 (C3), 31.6 (31.8) (C6), 30.3 (29.9) (CH3), 22.5 (22.3) (C5), 21.0 (CH3); HRMS (+ESI): (C28H26O3) calcd m/z 433.1774 [M + Na]+, found m/z 433.1771.
8,8-Dimethyl-2-phenyl-2,3,8,9,10,12-hexahydropyrano[3,2-a]xanthene-1,11-dione (13h). Flavanone 13h was synthesized following general procedure 1 using the Mannich base 9 (32 mg, 0.11 mmol, 1 eq.) and 3-(dimethylamino)-5,5-dimethylcyclohex-2-en-1-one (18 mg, 0.11 mmol, 1 eq.). The mixture was heated at 155 °C for 21 h. The product was obtained as a pale-yellow solid (32 mg, 79%). M.p. 238–239 °C; IR (neat): υmax 2955, 1683, 1634, 1597, 1466, 1389, 1308, 1221, 1146, 1018, 832, 758 cm−1; UV (THF) λmax 348 nm (ε 22
167 cm−1 M−1), 287 (95482), 255 (80729), 236 (83275); 1H NMR (400 MHz, acetone-d6): δ 7.57–7.59 (d, J = 8.1 Hz, 2H, H2′, H6′), 7.43–7.47 (m, 2H, H3′, H5′), 7.38–7.41 (m, 1H, H4′), 7.24 (d, J = 8.9 Hz, 1H, H11), 7.01 (d, J = 8.9 Hz, 1H, H12), 5.59 (dd, J = 2.7, 13.0 Hz, 1H, H2), 3.87 (d, J = 6.6 Hz, 2H, H5), 3.14 (dd, J = 13.1, 16.4 Hz, 1H, H3α), 2.86 (dd, J = 2.7, 16.4 Hz, H3β), 2.45 (s, 2H, H9), 2.28 (s, 2H, H7), 1.12 (s, 6H, 8-CH3); 13C NMR (100 MHz, acetone-d6): δ 196.7 (C6), 193.4 (C4), 164.3 (C9a), 160.2 (C12a), 145.4 (C10a), 140.2 (C1′), 129.5 (C3′, C5′), 129.4 (C4′), 127.2 (C2′, C6′), 124.8 (C11), 123.6 (C4a′), 119.2 (C4a), 118.5 (C12), 109.0 (C5a), 79.8 (C2), 51.1 (C7), 46.3 (C3), 41.4 (C9), 32.6 (C8), 28.4 (2 × CH3), 22.4 (C5); HRMS (+ESI): (C24H22O4) calcd m/z 397.1410 [M + Na]+, found m/z 397.1408.
8-((Dimethylamino)methyl)-7-hydroxy-2-phenylchroman-4-one (10). To a solution of 6 (1.24 g, 5.18 mmol, 1 eq.) in 1,4-dioxane (50 mL), was added bis(N,N-dimethylamino)methane (1.41 mL, 10.34 mmol, 2.0 eq.). The mixture was heated at reflux for 22 h. After completion of reaction, the solvent was evaporated. The resulting crude product was chromatographed over silica gel using DCM
:MeOH (95:5) as eluent to give 10 as a yellow solid (0.96 g, 62%). M.p. 144–145 °C; IR (neat): υmax 3011, 2956, 1654, 1583, 1440, 1361, 1310, 1210, 1103, 1050, 999, 819, 751, 698 cm−1; UV (THF) λmax 305 nm (ε 16622 cm−1 M−1), 279 (29794); 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 8.7 Hz, 1H, H5), 7.44–7.46 (m, 3H, H3′, H4′, H5′), 7.36–7.43 (m, 2H, H2, H6′), 6.53 (d, J = 8.7 Hz, 1H, H6), 5.44 (dd, J = 3, 13 Hz, 1H, H2), 3.81 (s, 2H, CH2), 2.98 (dd, J = 13.0, 16.8 Hz, 1H, H3α), 2.82 (dd, J = 3.0, 16.8 Hz, 1H, H3β), 2.38 (s, 1H, 2 × CH3); 13C NMR (100 MHz, CDCl3): δ 190.6 (C4), 166.9 (C8a), 160.5 (C7), 139.4 (C1′), 129.0 (C3′, C5′), 128.8 (C5), 128.2 (C4′), 126.2 (C2′, C6′), 113.6 (C4a), 111.8 (C6), 108.0 (C8), 79.9 (C2), 55.5 (CH2–N), 44.6 (2 × CH3), 44.5 (C3); HRMS (+ESI): (C18H19NO3) calcd m/z 298.1438 [M + Na]+, found m/z 298.1433.
2-Phenyl-7-((tetrahydro-2H-pyran-2-yl)oxy)chroman-4-one (15a). Flavanone 15a was synthesized following general procedure 1 using the Mannich base 10 (100 mg, 0.34 mmol, 1 eq.) and 3,4-dihydro-2H-pyran (0.61 mL, 6.73 mmol, 19.8 eq.). The mixture was heated at 155 °C for 43 h. The product was obtained as a white solid (20 mg, 12%). M.p. 102–103 °C; IR (neat): υmax 2921, 2851, 1683, 1601, 1437, 1325, 1252, 1138, 1089, 937, 898 cm−1; UV (THF) λmax 306 nm (ε 26
440 cm−1 M−1), 278 (55101), 239 (46825), 232 (42251), 217 (86923); 1H NMR (400 MHz, acetone-d6): δ 7.67 (dd, J = 2.2, 8.7 Hz, 1H, H5), 7.62 (d, J = 8.7 Hz, 2H, H2′, H6′), 7.46–7.50 (m, 2H, H3′, H5′), 7.39–7.43 (m, 1H, H4′), 6.57 (dd, J = 2.1, 8.7 Hz, H6), 5.64 (dd, J = 3.1, 12.9 Hz, 1H, H2), 5.39 (dd, J = 2.7, 4.7 Hz, 1H, H7a), 3.90–3.97 (m, 1H, H9α), 3.67–3.72 (m, 1H, H9, H9β), 3.05 (dd, J = 12.8, 16.9 Hz, 1H, H3α), 2.84 (dd, J = 3.1, 16.9 Hz, 1H, H3β), 2.78–2.80 (m, 1H, H12α), 2.64–2.71 (m, 1H, H12β), 2.19–2.26 (m, 1H, H11a), 1.53–1.75 (m, 4H, H10, H11); 13C NMR‡ (100 MHz, acetone-d6): δ 189.0 (189.0) (C4), 160.0 (160.0) (C12a′), 158.7 (158.6) (C6a), 139.2 (139.1) (C1′), 128.1 (128.1) (C3′, C5′), 127.8 (C4′), 125.7 (ArCH), 125.6 (ArCH), 124.9 (124.9) (ArCH), 114.1 (114.0) (C4a), 110.0 (110.0) (C6), 108.1 (108.0) (C12a), 96.4 (96.3) (C7a), 79.1 (79.0) (C2), 61.8 (61.5) (C9), 43.2 (43.0) (C3), 30.1 (30.0) (C11a), 23.4 (23.3) (C11), 22.8 (22.6) (C10), 22.3 (22.1) (C12). HRMS (+ESI): (C21H20O4) calcd m/z 359.1254 [M + Na]+, found m/z 359.1252.The earlier fractions were confirmed as the corresponding ring-opened product 16a.
(E)-1-(6-Hydroxy-3,4,4a,10a-tetrahydro-2H,5H-pyrano[2,3-b]chromen-7-yl)-3-phenylprop-2-en-1-one (16a). Yellow needle-like crystals (14 mg, 8%). M.p. 174–175 °C; IR (neat): υmax 2936, 1621, 1570, 1487, 1420, 1356, 1299, 1228, 1109, 1038, 975, 923, 853, 751 cm−1; UV (THF) λmax 331 nm (ε 42
284 cm−1 M−1), 212 (27012); 1H NMR (400 MHz, acetone-d6): δ 13.92 (s, 1H, OH), 8.09 (d, J = 8.9 Hz, 1H, H10′), 7.99 (d, J = 15.5 Hz, 1H, Hβ), 7.89 (d, J = 15.5 Hz, 1H, Hα), 7.86–7.88 (m, 2H, H2, H6), 7.47–7.48 (m, 3H, H3, H4, H5), 6.45 (d, J = 8.9 Hz, 1H, H9′), 5.42 (d, J = 2.4 Hz, 1H, H7′a), 3.88–3.96 (m, 1H, H6′α), 3.66–3.75 (m, 1H, H6′β), 2.76–2.78 (m, 1H, H3′α), 2.66 (dd, J = 4.2, 17.3 Hz, 1H, H3′β), 2.20–2.28 (m, 1H, H3′a), 1.53–1.75 (m, 4H, H4′, H5′); 13C NMR (100 MHz, acetone-d6): δ 193.1 (CO), 165.0 (C2′), 160.9 (C8′a), 145.0 (Cα), 135.9 (C1), 131.5 (C4), 130.5 (C10′), 129.8 (C3, C5), 129.7 (C2, C6), 121.7 (Cβ), 114.4 (C1′), 109.1 (C2′a), 109.0 (C9′), 98.0 (C7′a), 62.8 (C6′), 31.6 (C3′a), 24.6 (C4′), 24.4 (C5′), 23.5 (C3′). HRMS (+ESI): (C21H20O4) calcd m/z 359.1254 [M + Na]+, found m/z 359.1256.
2-Phenyl-2,3,7a,9,10,10a-hexahydro-4H,11H-furo[2,3-b]pyrano[2,3-f]chromen-4-one (15b). Flavanone 15b was synthesized following general procedure 1 using the Mannich base 10 (100 mg, 0.34 mmol, 1 eq.) and 2,3-dihydrofuran (0.51 mL, 6.73 mmol, 19.8 eq.). The mixture was heated at 155 °C for 43 h. The product obtained (13 mg, 12%) after column chromatography was further purified by preparative HPLC using a 5 μM column (150 × 22 mm ID) at a flow rate of 5 mL min−1 with a gradient elution from 2% to 100% B (solvent A: 0.1% formic acid/MilliQ water) and solvent B (0.1% formic acid/acetonitrile) over 56 min to afford the expected product 15b as a yellow solid (6 mg, 6%). M.p. 121 °C; IR (neat): υmax 2921, 1680, 1601, 1439, 1321, 1104, 1068, 939, 891 cm−1; 1H NMR (400 MHz, acetone-d6): δ 7.65 (dd, J = 2.1, 8.7 Hz, 1H, H5), 7.60 (d, J = 8.9 Hz, 2H, H2′, H6′), 7.44–7.48 (m, 2H, H3′, H5′), 7.37–7.41 (m, 1H, H4′), 6.50 (dd, J = 2.3, 8.7 Hz, 1H, H6), 5.64 (dd, J = 3.0, 14.2 Hz, 1H, H2), 5.61 (d, J = 4.2 Hz, 1H, H7a), 4.00–4.06 (m, 1H, H9α), 3.91–3.98 (m, 1H, H9β), 3.03 (dd, J = 12.6, 16.7 Hz, 1H, H3α), 2.90–2.95 (m, 2H, H11), 2.81 (dd, J = 3.1, 16.7 Hz, 1H, H3β), 2.72–2.80 (m, 1H, H10a), 2.10–2.18 (m, 1H, H10α), 1.56–1.68 (m, 1H, H10β); 13C NMR‡ (100 MHz, acetone-d6): δ 190.4 (190.3) (C4), 161.5 (161.5) (C11a′), 160.3 (C6a), 140.6 (140.5) (C1′), 129.5 (C3′, C5′), 129.2 (C4′), 127.1 (C2′, C6′), 126.5 (C5), 115.6 (115.5) (C4a), 111.7 (C6), 108.8 (108.7) (C11a), 101.9 (101.8) (C7a), 80.6 (80.4) (C2), 68.6 (68.6) (C9), 44.6 (44.5) (C3), 37.2 (37.1) (C10a), 28.4 (28.3) (C10), 20.2 (20.1) (C11). HRMS (+ESI): (C20H18O4) calcd m/z 345.1097 [M + Na]+, found m/z 345.1094.
The earlier fractions were confirmed as the corresponding ring-opened product 16b.
(E)-1-(5-Hydroxy-2,3,3a,9a-tetrahydro-4H-furo[2,3-b]chromen-6-yl)-3-phenylprop-2-en-1-one (16b). Yellow solid (6 mg, 5%). M.p. 153 °C; IR (neat): υmax 2894, 1609, 1590, 1488, 1419, 1357, 1219, 1111, 1062, 932, 852, 768 cm−1; UV (THF) λmax 330 nm (ε 48
257 cm−1 M−1), 237 (15312), 213 (24016); 1H NMR (400 MHz, acetone-d6): δ 13.93 (s, 1H, OH), 8.09 (d, J = 8.7 Hz, 1H, H9′), 7.99 (d, J = 15.4 Hz, 1H, Hβ), 7.90 (d, J = 15.4 Hz, 1H, Hα), 7.87–7.88 (m, 2H, H2, H6), 7.47–7.48 (m, 3H, H3, H4, H5), 6.39 (d, J = 8.7 Hz, 1H, H8′), 5.64 (d, J = 4.1 Hz, 1H, H6′a), 4.02–4.05 (m, 1H, H5′α), 3.96–4.00 (m, 1H, H5′β), 2.87–2.99 (m, 2H, H3′), 2.11–2.18 (m, 1H, H3′a), 1.56–1.67 (m, 2H, H4′); 13C NMR (100 MHz, acetone-d6): δ 193.1 (CO), 165.1 (C2′), 161.0 (C7′a), 145.0 (Cα), 135.9 (C1), 131.5 (C4), 130.7 (C9′), 129.8 (C3, C5), 129.7 (C2, C6), 121.6 (Cβ), 114.3 (C1′), 109.2 (C8′), 108.2 (C2′a), 102.1 (C6′a), 68.7 (C5′), 37.2 (C3′a), 28.2 (C4′), 19.6 (C3′); HRMS (+ESI): (C20H18O4) calcd m/z 323.1278 [M + H]+, found m/z 323.1275.
8-Ethoxy-2-phenyl-2,3,9,10-tetrahydro-4H,8H-pyrano[2,3-f]chromen-4-one (15c). Flavanone 15c was synthesized following general procedure 1 using the Mannich base 10 (100 mg, 0.34 mmol, 1 eq.) and ethyl vinyl ether (0.64 mL, 6.73 mmol, 19.8 eq.). The mixture was heated at 155 °C for 6 h. The product was obtained as a yellow oil (8 mg, 7%). IR (DCM): υmax 2922, 1681, 1601, 1438, 1325, 1243, 1168, 1119, 1048, 948, 869 cm−1; UV (THF) λmax 307 nm (ε 13
948 cm−1 M−1), 276 (33089); 1H NMR (400 MHz, acetone-d6): δ 7.64–7.66 (m, 1H, H5), 7.59–7.63 (m, 2H, H2′, H6′), 7.44–7.48 (m, 2H, H3′, H5′), 7.38–7.42 (m, 1H, H4′), 6.55 (dd, J = 4.0, 8.8 Hz, 1H, H6), 5.62 (dd, J = 3.2, 12.9 Hz, 1H, H2), 5.35–5.38 (m, 1H, H8), 3.80–3.89 (m, 1H, H1′′α), 3.64–3.72 (m, 1H, H1′′β), 3.04 (dd, J = 3.4, 16.6 Hz, 1H, H3α), 2.79–2.80 (m, 1H, H3β), 2.59–2.76 (m, 2H, H10), 2.01–2.03 (m, 1H, H9α), 1.82–1.96 (m, 1H, H9β), 1.15 ppm (t, J = 7.20 Hz, 3H, CH3); 13C NMR‡ (100 MHz, acetone-d6): δ 190.6 (190.5) (C4), 161.0 (160.9) (C10a′), 159.3 (159.2) (C6a), 140.6 (140.6) (C1′), 129.5 (C3′, C5′), 129.2 (129.1) (C4′), 127.1 (C2′, C6′), 126.1 (C5), 115.4 (115.5) (C4a), 112.1 (C6), 111.9 (111.8) (C10a), 98.3 (98.1) (C8), 80.6 (80.3) (C2), 64.6 (64.5) (C1′′), 44.6 (44.5) (C3), 26.3 (26.1) (C9), 15.5 (15.4) (C10), 15.3 (CH3). HRMS (+ESI): (C20H20O4) calcd m/z 347.1254 [M + Na]+, found m/z 347.1252.The earlier fractions were confirmed as the corresponding ring-opened product 16c.
(E)-1-(2-Ethoxy-5-hydroxychroman-6-yl)-3-phenylprop-2-en-1-one (16c). Yellow oil (6 mg, 5%); IR (DCM): υmax 2930, 1617, 1605, 1489, 1420, 1360, 1228, 1107, 1050, 947, 867, 773 cm−1; UV (THF) λmax 327 nm (ε 16
770 cm−1 M−1), 240 (4087); 1H NMR (400 MHz, acetone-d6): δ 13.88 (s, 1H, OH), 8.08 (d, J = 8.9 Hz, 1H, H8′), 7.99 (d, J = 15.5 Hz, 1H, Hβ), 7.89 (d, J = 15.5 Hz, 1H, Hα), 7.86–7.89 (m, 2H, H2, H6), 7.47–7.48 (m, 3H, H3, H4, H5), 6.45 (d, J = 8.9 Hz, 1H, H7′), 5.38 (t, J = 3.1 Hz, 1H, H5′), 3.84–3.88 (m, 1H, H1′′α), 3.65–3.71 (m, 1H, H1′′β), 2.67–2.76 (m, 2H, H3′), 2.60–2.66 (m, 1H, H4′α), 1.87–1.96 (m, 1H, H4′β), 1.15 (t, J = 3.0 Hz, 3H, CH3); 13C NMR (100 MHz, acetone-d6): δ 193.2 (CO), 164.4 (C2′), 160.0 (C6′a), 144.9 (Cα), 135.9 (C1), 131.5 (C4), 130.2 (C8′), 129.8 (C3, C5), 129.7 (C2′, C6′), 121.6 (Cβ), 114.3 (C1′), 111.6 (C2′a), 109.6 (C7′), 98.5 (C5′), 64.6 (C1′′), 26.3 (C4′), 15.4 (C3′), 15.0 (CH3); HRMS (+ESI): (C20H20O4) calcd m/z 347.1254 [M + Na]+, found m/z 347.1253.
8-(4-Methoxyphenyl)-2-phenyl-2,3,9,10-tetrahydro-4H,8H-pyrano[2,3-f]chromen-4-one (15d). Flavanone 15d was synthesized following general procedure 1 using the Mannich base 10 (150 mg, 0.50 mmol, 1 eq.) and p-methoxystyrene (0.66 mL, 4.96 mmol, 9.9 eq.). The mixture was heated at 155 °C for 6 h. The product was obtained as a pale-yellow solid (14 mg, 7%). M.p. 173 °C; IR (neat): υmax 2935, 1673, 1583, 1511, 1437, 1327, 1242, 1167, 1105, 1067, 1030, 914, 825, 818, 742; UV (MeOH) λmax 282 nm (ε 26
897 cm−1 M−1), 222 (57040), 202 (59822); 1H NMR (400 MHz, acetone-d6): δ 7.66 (dd, J = 3.3; 8.8 Hz, 1H, H5), 7.59–7.62 (m, 2H, H2′, H6′), 7.43–7.48 (m, 2H, H3′, H5′), 7.37–7.41 (m, 3H, H4′, H2′′, H6′′), 6.96 (d, J = 8.6 Hz, 2H, H3′′, H5′′), 6.58 (d, J = 8.8 Hz, 1H, H6), 5.63 (dd, J = 3.3, 13.9 Hz, 1H, H8), 5.13 (dd, J = 2.1, 19.4 Hz, 1H, H2), 3.81 (s, 3H, OMe), 2.98–3.08 (m, 1H, H3α), 2.72–2.81 (m, 3H, H3β, H10), 2.21–2.27 (m, 1H, H9α), 1.96–2.21 (m, 1H, H9β); 13C NMR‡ (100 MHz, acetone-d6): δ 190.5 (190.4) (C4), 162.4 (162.2) (C4′′), 161.4 (161.3) (C10a′), 160.5 (160.4) (C6a), 140.7 (140.6) (C1′), 134.0 (C1′′), 129.5 (C3′, C5′), 129.2 (129.1) (C4′), 128.3 (128.2) (C2′′, C6′′), 127.1 (127.0) (C2′, C6′), 126.2 (126.1) (C5), 115.2 (115.1) (C4a), 114.6 (C3′′, C5′′), 112.0 (C6), 111.3 (111.3) (C10a), 80.5 (80.4) (C8), 78.7 (78.5) (C2), 55.6 (OMe), 44.5 (C3), 29.5 (C9), 20.1 (19.9) (C10). HRMS (+ESI): (C25H22O4) calcd m/z 387.1591 [M + Na]+, found m/z 387.1590.The earlier fractions were confirmed as the corresponding ring-opened product 16d.
(E)-1-(5-Hydroxy-2-(4-methoxyphenyl)chroman-6-yl)-3-phenylprop-2-en-1-one (16d). Yellow solid (15 mg, 8%); M.p. 179–180 °C; IR (neat): υmax 2932, 1584, 1498, 1359, 1219, 1111, 1027, 975, 856, 810, 768 cm−1; UV (MeOH) λmax 337 nm (ε 27
206 cm−1 M−1), 224 (30336), 202 (45292); 1H NMR (400 MHz, acetone-d6): δ 13.99 (s, 1H, OH), 8.09 (d, J = 8.9 Hz, 1H, H8′), 8.01 (d, J = 15.4 Hz, 1H, Hβ), 7.90 (d, J = 15.4 Hz, 1H, Hα), 7.87–7.89 (m, 2H, H2, H6), 7.47–7.48 (m, 3H, H3, H4, H5), 7.41 (d, J = 8.5 Hz, 2H, H2′′, H6′′), 6.97 (d, J = 8.9 Hz, 2H, H3′′, H5′′), 6.47 (d, J = 8.9 Hz, 1H, H7′), 5.15 (dd, J = 2.3, 10.3 Hz, 1H, H5′), 3.82 (s, 3H, OCH3), 2.82–2.83 (m, 1H, H3′α), 2.70–2.79 (m, 1H, H3′β), 2.25–2.30 (m, 1H, H4′α), 2.06–2.09 (m, 1H, H4′β); 13C NMR (100 MHz, acetone-d6): δ 193.1 (CO), 165.0 (C2′), 163.0 (C4′′), 160.5 (C6′a), 144.9 (Cα), 135.9 (C1), 134.0 (C1′′), 131.5 (C4), 130.2 (C8′), 129.8 (C3, C5), 129.7 (C2, C6), 128.4 (C2′′, C6′′), 121.7 (Cβ), 114.7 (C3′′, C5′′), 114.0 (C1′), 111.1 (C2′a), 109.5 (C7′), 78.9 (C5′), 55.6 (OCH3), 29.3 (C4′), 19.6 (C3′); HRMS (+ESI): (C25H22O4) calcd m/z 409.1410 [M + Na]+, found m/z 409.1415.
8-Methyl-2,8-diphenyl-2,3,9,10-tetrahydro-4H,8H-pyrano[2,3-f]chromen-4-one (15e). Flavanone 15e was synthesized following general procedure 1 using the Mannich base 10 (100 mg, 0.34 mmol, 1 eq.) and α-methyl styrene (0.87, 6.73 mmol, 19.8 eq.). The mixture was heated at 155 °C for 16 h. The product obtained (12 mg, 10%) after column chromatography was further purified by preparative HPLC using a 5 μM column (150 × 22 mm ID) at a flow rate of 5 mL min−1 with a gradient elution from 2% to 100% B (solvent A: 0.1% formic acid/MilliQ water) and solvent B (0.1% formic acid/acetonitrile) over 56 min to afford the expected product as a white solid (5 mg, 4%). M.p. 127–128 °C; IR (neat): υmax 2969, 1670, 1580, 1434, 1329, 1258, 1193, 1099, 902, 817, 757 cm−1; UV (THF) λmax 305 nm (ε 13959 cm−1 M−1), 279 (35
526), 240 (39045); 1H NMR (400 MHz, acetone-d6): δ 7.66–7.70 (dd, J = 3.8, 8.6 Hz, 1H, H5), 7.52–7.56 (m, 2H, H2′, H6′), 7.37–7.44 (m, 5H, ArH), 7.31–7.36 (m, 4H, ArH), 7.23–7.27 (m, 2H, ArH), 6.72 (dd, J = 5.1, 8.7 Hz, 1H, H6), 5.54 (dd, J = 2.9, 13.0 Hz, 1H, H2), 2.98 (dd, J = 12.7, 16.7 Hz, 1H, H3α), 2.78–2.79 (m, 1H, H3β), 2.69–2.75 (m, 1H, H10α), 2.52–2.63 (m, 1H, H10β), 2.07–2.28 (m, 2H, H9), 1.64 (d, J = 2.2 Hz, 3H, CH3). 13C NMR‡ (100 MHz, acetone-d6): δ 190.5 (190.3) (C4), 161.3 (161.2) (ArC), 161.1 (161.1) (ArC), 145.9 (145.7) (C1′′), 140.5 (140.6) (C1′), 129.4 (129.4) (2 × ArCH), 129.4 (129.3) (2 × ArCH), 129.2 (ArCH), 127.8 (ArCH), 127.0 (2 × ArCH), 126.3 (126.2) (ArCH), 125.7 (125.6) (2 × ArCH), 115.1 (115.0) (C4a), 112.2 (112.1) (C6), 111.1 (111.0) (C10a), 80.6 (C2), 80.3 (80.2) (C8), 44.6 (44.4) (C3), 32.2 (32.0) (C9), 30.9 (CH3), 17.7 (C10). HRMS (+ESI): (C25H22O3) calcd m/z 371.1642 [M + H]+, found m/z 371.1640.The earlier fractions were confirmed as the corresponding ring-opened product 16e.
(E)-1-(5-Hydroxy-2-methyl-2-phenylchroman-6-yl)-3-phenylprop-2-en-1-one (16e). Yellow oil (9 mg, 7%). IR (DCM): υmax 2929, 1615, 1602, 1488, 1361, 1275, 1223, 1113, 1033, 763 cm−1; UV (THF) λmax 333 nm (ε 17
522 cm−1 M−1), 241 (4668); 1H NMR (400 MHz, acetone-d6): δ 13.88 (s, 1H, OH), 8.11 (d, J = 9.2 Hz, 1H, H8′), 7.99 (d, J = 15.5 Hz, 1H, Hβ), 7.88 (d, J = 15.5 Hz, 1H, Hα), 7.86–7.88 (m, 2H, H2, H6), 7.46–7.48 (m, 3H, H3, H4, H5), 7.43 (d, J = 7.9 Hz, 2H, H2′′, H6′′), 7.31–7.35 (m, 2H, ArH),7.22–7.25 (m, 1H, ArH), 6.62 (d, J = 9.2 Hz, 1H, H7′), 2.70–2.79 (m, 1H, H3′α), 2.54–2.60 (m, 1H, H3′β), 2.11–2.25 (m, 2H, H4′), 1.66 (s, 3H, CH3); 13C NMR (100 MHz, acetone-d6): δ 193.0 (CO), 164.8 (C2′), 161.8 (C6′a), 145.8 (C1′′), 144.8 (Cα), 135.9 (C1), 131.5 (C4), 130.4 (C8′), 129.8 (C3, C5), 129.7 (C2, C6), 129.3 (C3′′, C5′′), 127.8 (C4′′), 125.6 (C2′′, C6′′), 121.7 (Cβ), 114.0 (C1′), 110.9 (C2′a), 109.7 (C7′), 80.6 (C5′), 32.3 (C4′), 30.4 (CH3), 17.2 (C3′); HRMS (+ESI): (C25H22O4) calcd m/z 371.1642 [M + H]+, found m/z 371.1644.
(E)-1-(2-Bromo-8-hydroxy-9H-benzo[e][1,2,4]triazolo[5,1-b][1,3]oxazin-7-yl)-3-phenylprop-2-en-1-one (18). To a mixture of the Mannich base 10 (150 mg, 0.50 mmol, 1 eq.) and K2CO3 (209 mg, 1.51 mmol, 3.0 eq.) in DMA (10 mL), 3,5-dibromo-1,2,4-triazole (115 mg, 0.51 mmol, 1.0 eq.) was added. The reaction mixture was heated to 150 °C in a pressure tube for 4 h. After completion of reaction, the reaction mixture was cooled to room temperature and poured into water (50 mL). The precipitated yellow solid was filtered, washed with water, and dried to give 18 as a yellow solid (117 mg, 58%). M.p. 219–221 °C; IR (neat): υmax 1638, 1624, 1545, 1532, 1354, 1291, 1209, 1103, 984, 823, 770 cm−1; UV (THF) λmax 331 nm (ε 36
636 cm−1 M−1); 1H NMR (400 MHz, DMSO-d6): δ 13.82 (br s, 1H, OH), 8.51 (d, J = 8.6 Hz, 1H, H10′), 8.09 (d, J = 15.5 Hz, 1H, Hα), 7.94–7.96 (m, 2H, H2, H6), 7.92 (d, J = 15.5 Hz, 1H, Hβ), 7.49–7.50 (m, 3H, H3, H4, H5), 7.01 (d, J = 8.6 Hz, 1H, H9′), 5.17 (s, 2H, CH2–N); 13C NMR (100 MHz, DMSO-d6): δ 192.8 (CO), 160.8 (C2′), 152.6 (C8′a), 152.4 (C7′a), 145.9 (Cβ), 136.7 (C6′), 134.2 (C1), 132.0 (C10′), 131.3 (C4), 129.4 (C2, C6), 129.0 (C3, C5), 120.5 (Cα), 116.3 (C1′), 107.8 (C9′), 104.8 (C2′a), 42.6 (C3′). HRMS (+ESI): (C18H12BrN3O3) calcd for 79Br m/z 419.9954 [M + Na]+, found m/z 419.9947; calcd for 81Br m/z 421.9934 [M + Na]+, found m/z 421.9926.
9-Bromo-2-phenyl-2,3-dihydro-4H,12H-chromeno[8,7-e][1,2,4]triazolo[5,1-b][1,3]oxazin-4-one (19). To a solution of 18 (30 mg, 0.075 mmol, 1 eq.) in MeOH (2 mL) was added concentrated HCl (0.13 mL, 1.58 mmol, 21 eq.). The reaction was heated to reflux for 72 h using a reflux setup. The reaction mixture was cooled to room temperature and poured into ice-water. The precipitated yellow solid (24 mg) was subjected to preparative reverse-phase HPLC using a 5 μM column (150 × 22 mm ID) at a flow rate of 5 mL min−1 with a gradient elution from 2% to 100% B (solvent A: 0.1% formic acid/H2O) and solvent B (0.1% formic acid/ACN) over 56 min to afford 19 as a pale-yellow solid (3 mg, 10%) and the starting material 18 (8 mg, 27%). M.p. 236 °C; IR (neat): υmax 2919, 1699, 1606, 1532, 1521, 1442, 1270, 1232, 1094, 1060, 982, 886, 799 cm−1; UV (THF) λmax 313 nm (ε 15
610 cm−1 M−1), 269 (20628); 1H NMR (400 MHz, DMSO-d6): δ 7.85 (d, J = 8.8 Hz, 1H, H5), 7.56–7.58 (m, 2H, H2′, H6′), 7.40–7.48 (m, 3H, H3′, H4′, H5′), 7.07 (d, J = 8.8 Hz, 1H, H6), 5.84 (dd, J = 3.1, 12.4 Hz, 1H, H2), 5.21 (s, 2H, CH2–N), 3.26 (dd, J = 12.4, 16.9 Hz, 1H, H3α), 2.98 (dd, J = 3.1, 16.9 Hz, 1H, H3β); 13C NMR (100 MHz, DMSO-d6): δ 189.8 (C4), 158.3 (C12b), 152.7 (C6a), 152.2 (C7a), 138.3 (C9), 136.7 (C1′), 128.7 (C3′, C5′), 128.6 (C4′), 127.3 (C5), 126.3 (C2′, C6′), 117.5 (C4a), 110.4 (C6), 105.4 (C12a), 79.7 (C2), 42.9 (CH2–N), 42.7 (C3); HRMS (+ESI): (C18H12BrN3O3) calcd for 79Br m/z 419.9954 [M + Na]+, found m/z 419.9952; calcd for 81Br m/z 421.9934 [M + Na]+, found m/z 421.9929.
:EtOAc (95:5) as the eluent to afford the corresponding products. Further purification was performed using preparative reversed phase HPLC when required.
8-((Dimethylamino)methyl)-7-hydroxy-3-(4-hydroxyphenyl)chroman-4-one (11). To a solution of dihydrodaidzein 7 (1.20 g, 4.68 mmol, 1 eq.) in ethanol (60 mL) was added bis(N,N-dimethylamino)methane (1.28 mL, 9.37 mmol, 2.0 eq.). The reaction mixture was heated to reflux for 1 h. The product precipitated from the reaction mixture and was collected by filtration to give 11 as a white fine solid (0.92 g, 61%). M.p. 274–275 °C; IR (neat): υmax 3280, 2962, 1651, 1582, 1439, 1384, 1111, 946, 805, 745 cm−1. UV (MeOH): λmax 329 nm (ε 15
863 cm−1 M−1), 260 (6509), 221 (19014); 1H NMR (300 MHz, DMSO-d6): δ 7.59 (d, J = 8.7 Hz, 1H, H5), 7.03 (d, J = 8.7 Hz, 2H, H2′, H6′), 6.69 (d, J = 8.7 Hz, 2H, H3′, H5′), 6.41 (d, J = 8.7 Hz, 1H, H6), 4.56 (d, J = 6.6 Hz, 2H, H2), 3.87 (t, J = 6.6 Hz, 1H, H3), 3.74 (s, 2H, CH2–N), 2.32 (s, 6H, N(CH3)2); 13C NMR (75 MHz, DMSO-d6): δ 190.5 (C4), 166.8 (C7), 160.4 (C8a), 156.5 (C4′), 129.7 (C2′, C6′), 127.6 (C5), 126.3 (C1′), 115.2 (C3′, C5′), 112.4 (C8), 111.1 (C6), 107.7 (C4a), 71.4 (C2), 53.8 (CH2–N), 50.0 (C3), 43.7 (2 × CH3). HRMS (+ESI): (C18H19NO4) calcd m/z 314.1387 [M + H]+, found m/z 314.1382.
2-Hydroxy-1-(5-hydroxy-2,3,3a,9a-tetrahydro-4H-furo[2,3-b]chromen-6-yl)-2-(4-hydroxyphenyl)propan-1-one (20). To a suspension of Mannich base 11 (121 mg, 0.38 mmol, 1 eq.) in DMF (10 mL) was added 2,3-dihydrofuran (0.58 mL, 7.71 mmol, 20.3 eq.). The reaction mixture was heated at 150–155 °C in a pressure tube for 28 h. After completion of reaction, the solvent was evaporated in vacuo, and the resulting crude was added methanol. The precipitated solid was filtered, washed with methanol, and air-dried to give product 20 as an off-white solid (39 mg, 28%). M.p. 252–253 °C; IR (neat): υmax 3350, 2927, 1686, 1592, 1435, 1281, 1214, 1048, 935, 830 cm-1; UV (THF): λmax 317 nm (ε 8018 cm−1 M−1), 269 (11
083), 239 (9408); 1H NMR (400 MHz, DMSO-d6): δ 9.55 (s, 1H, OH), 7.38 (d, J = 8.6 Hz, 1H, H4), 7.22 (d, J = 8.8 Hz, 2H, H2′, H6′), 6.76 (d, J = 8.8 Hz, 2H, H3′, H5′), 6.59 (d, J = 8.6 Hz, 1H, H5), 5.71 (d, J = 4.1 Hz, 1H, H6a), 3.99–4.04 (m, 1H, H8α), 3.90–3.96 (m, 1H, H8β), 2.88–3.02 (m, 2H, H10), 2.69–2.77 (m, 1H, H9a), 2.10–2.18 (m, 1H,H9α), 1.69 (s, 3H, CH3), 1.50–1.61 (m, 1H, H9β); 13C NMR (100 MHz, DMSO-d6): δ 199.1 (C1), 171.0 (C11), 161.5 (C5a), 157.6 (C4′), 128.5 (C1′), 126.0 (C2′, C6′), 123.4 (C4), 115.4 (C3′, C5′), 112.7 (C5), 111.5 (C1a), 103.9 (C10a), 101.1 (C6a), 90.4 (C2), 67.7 (C8), 35.4 (C9a), 27.1 (C9), 23.3 (C3), 18.3 (C10). HRMS (+ESI): (C20H20O6) calcd m/z 357.1333 [M + H]+, found m/z 357.1328.
3-(4-Hydroxyphenyl)-2,3,7a,9,10,10a-hexahydro-4H,11H-furo[2,3-b]pyrano[2,3-f]chromen-4-one (22a). Isoflavanone 22a was synthesized following general procedure 2 using the Mannich base 11 (121 mg, 0.39 mmol, 1 eq.) and 2,3-dihydrofuran (0.58 mL, 7.67 mmol, 19.7 eq.). The product was obtained as a white solid (23 mg, 18%). M.p. 230–231 °C; IR (neat): υmax 3265, 2953, 1679, 1587, 1515, 1438, 1357, 1228, 1166, 1096, 1025, 936, 888 cm−1; UV (THF): λmax 307 nm (ε 6429 cm−1 M−1), 283 (8425), 269 (12
282), 240 (9846); 1H NMR (400 MHz, DMSO-d6): δ 9.36 (s, 1H, OH), 7.58 (d, J = 8.7 Hz, 1H, H5), 7.04 (d, J = 8.7 Hz, 2H, H2′, H6′), 6.71 (d, J = 8.7 Hz, 2H, H3′, H5′), 6.51 (d, J = 8.7 Hz, 1H, H6), 5.63 (d, J = 4.2 Hz, 1H, H7a), 4.62–4.66 (m, 2H, H2), 3.94–4.01 (m, 2H, H9), 3.86–3.91 (m, 1H, H3), 2.82 (d, J = 4.1 Hz, 2H, H11), 2.64–2.72 (m, 1H, H10a), 2.04–2.11 (m, 1H, H10α), 1.43–1.54 (m, 1H, H10β); 13C NMR‡ (100 MHz, DMSO-d6): δ 191.1 (190.9) (C4), 160.5 (160.4) (C1a), 158.8 (158.7) (C6a), 156.6 (C4′), 129.8 (129.6) (C2′, C6′), 126.0 (C1′), 125.9 (C5), 115.2 (C3′, C5′), 114.4 (114.3) (C4a), 110.8 (110.6) (C6), 107.3 (C11a), 100.6 (C7a), 71.6 (71.5) (C2), 67.7 (C9), 50.1 (49.9) (C3), 35.6 (C10a), 27.1 (CH2), 19.0 (CH2). HRMS (+ESI): (C20H18O5) calcd m/z 361.1046 [M + Na]+, found m/z 361.1045.
3-(4-Hydroxyphenyl)-2,3,7a,10,11,11a-hexahydro-4H,9H,12H-dipyrano[2,3-b:2′,3′-f]chromen-4-one (22b). Isoflavanone 22b was synthesized following general procedure 2 using the Mannich base 11 (150 mg, 0.48 mmol, 1 eq.) and 3,4-dihydro-2H-pyran (0.91 mL (97%), 9.64 mmol, 20.1 eq.). The product was obtained as a white solid (22 mg, 13%). M.p. 223–224 °C; IR (neat): υmax 3380, 2947, 1675, 1603, 1516, 1435, 1356, 1230, 1038, 989 cm−1; UV (THF): λmax 307 nm (ε 6061 cm−1 M−1), 282 (8528), 270 (12
157), 239 (8880); 1H NMR (400 MHz, DMSO-d6): δ 9.36 (s, 1H, OH), 7.59 (d, J = 8.2 Hz, 1H, H5), 7.03 (d, J = 8.6 Hz, 2H, H2′, H6′), 6.71 (d, J = 8.6 Hz, 2H, H3′, H5′), 6.57 (d, J = 8.6 Hz, 1H, H6), 5.39 (d, J = 2.9 Hz, 1H, H7a), 4.61–4.65 (m, 2H, H2), 3.91–3.97 (m, 1H, H3), 3.80–3.86 (m, 1H, H9α), 3.64–3.66 (m, 1H, H9β), 2.73 (dd, J = 2.4, 6.2 Hz, 1H, H12α), 2.52–2.56 (m, 1H, H12β), 2.15–2.16 (m, 1H, H11a), 1.44–1.66 (m, 4H, 10, 11-CH2); 13C NMR‡ (100 MHz, DMSO-d6): δ 191.1 (191.0) (C4), 160.3 (160.2) (C1a), 158.6 (158.5) (C6a), 156.6 (C4′), 129.8 (129.6) (C2′, C6′), 126.0 (C1′), 125.9 (C5), 115.3 (C3′, C5′), 114.4 (114.3) (C4a), 110.5 (C6), 108.2 (108.1) (C12a), 96.4 (C7a), 71.6 (71.5) (C2), 59.8 (C8), 50.1 (49.9) (C3), 29.9 (29.8) (C11a), 23.7 (23.6) (CH2), 22.8 (22.7) (CH2), 22.3 (22.2) (CH2). HRMS (+ESI): (C21H20O5) calcd m/z 353.1384 [M + H]+, found m/z 353.1385.
8-Ethoxy-3-(4-hydroxyphenyl)-2,3,9,10-tetrahydro-4H,8H-pyrano[2,3-f]chromen-4-one (22c). Isoflavanone 22c was synthesized following general procedure 2 using the Mannich base 11 (150 mg, 0.48 mmol, 1 eq.) and ethyl vinyl ether (0.92 mL, 9.61 mmol, 20.0 eq.). The product obtained (32 mg, 20%) after column chromatography was further purified by preparative HPLC using a 5 μM column (150 × 22 mm ID) at a flow rate of 5 mL min−1 with an isocratic elution of 50% solvent B (solvent A: 0.1% formic acid/H2O) and solvent B (0.1% formic acid/ACN) over 56 min to afford the expected product as a white solid (10 mg, 10%). M.p. 241–242 °C; IR (neat): υmax 3316, 2930, 1667, 1583, 1517, 1436, 1354, 1227, 1094, 921 cm−1; UV (THF): λmax 307 nm (ε 6705 cm−1 M−1), 281 (9394), 268 (14
875), 239 (9394); 1H NMR (400 MHz, DMSO-d6): δ 9.41 (br s, 1H, OH), 7.59 (d, J = 8.8 Hz, 1H, H5), 7.04 (d, J = 8.7 Hz, 2H, H2′, H6′), 6.71 (d, J = 8.7 Hz, 2H, H3′, H5′), 6.55 (d, J = 8.8 Hz, 1H, H6), 5.38 (t, J = 3.6 Hz, 1H, H8), 4.59–4.69 (m, 2H, H2), 3.91–3.97 (m, 1H, H3), 3.73–3.81 (m, 1H, H1′′α), 3.61–3.69 (m, 1H, H1′′β), 2.62–2.68 (m, 1H, H10α), 2.53–2.57 (m, 1H, H10β), 1.96–2.02 (m, 1H, H9α), 1.83–1.89 (m, 1H, H9β), 1.12 (t, J = 7.1 Hz, 3H, H2′′). 13C NMR‡ (100 MHz, DMSO-d6): δ 191.2 (C4), 159.8 (C1a), 157.9 (C6a), 156.6 (C4′), 129.7 (129.6) (C2′, C6′), 125.9 (125.8) (C1′), 125.7 (C5), 115.2 (C3′, C5′), 114.2 (C4a), 111.1 (C6), 110.3 (110.2) (C9a), 97.0 (96.9) (C8), 71.5 (71.4) (C2), 63.6 (63.5) (C1′′), 50.0 (C3), 25.0 (24.9) (C9), 15.0 (CH3), 14.3 (14.2) (C10). HRMS (+ESI): (C20H20O5) calcd m/z 341.1384 [M + H]+, found m/z 341.1382.
3-(4-Hydroxyphenyl)-8-(4-methoxyphenyl)-2,3,9,10-tetrahydro-4H,8H-pyrano[2,3-f]chromen-4-one (22d). Isoflavanone 22d was synthesized following general procedure 2 using the Mannich base 11 (120 mg, 0.38 mmol, 1 eq.) and p-methoxystyrene (1.02 mL, 7.66 mmol, 20.2 eq.). The product was obtained as a white solid (51 mg, 33%). M.p. 263–264 °C; IR (neat): υmax 3285, 2918, 1657, 1580, 1511, 1435, 1357, 1175, 1029 cm−1; UV (THF): λmax 288 nm (ε 13
321 cm−1 M−1), 270 (15011), 240 (12758); 1H NMR (400 MHz, DMSO-d6): δ 9.37 (s, 1H, OH), 7.59 (d, J = 8.6 Hz, 1H, H5), 7.36 (d, J = 8.6 Hz, 2H, H2′′, H6′′), 7.05 (d, J = 8.7 Hz, 2H, H2′, H6′), 6.96 (d, J = 8.6 Hz, 2H, H3′′, H5′′), 6.71 (d, J = 8.5 Hz, 2H, H3′, H5′), 6.57 (d, J = 8.7 Hz, 1H, H6), 5.10–5.14 (m, 1H, H8), 4.62–4.71 (m, 2H, H2), 3.89–3.98 (m, 1H, H3), 3.76 (s, 3H, OCH3), 2.69–2.74 (m, 2H, H10), 2.16–2.19 (m, 1H, H9α), 1.94–2.05 (m, 1H, H9β); 13C NMR‡ (100 MHz, DMSO-d6): δ 191.1 (191.0) (C4), 160.9 (C4′′), 160.3 (160.2) (C1a), 159.0 (C6a), 156.6 (C4′), 132.6 (ArC), 129.7 (129.6) (C2′, C6′), 127.6 (127.5) (C2′′, C6′′), 126.1 (C1′), 126.0 (C1′′), 125.7 (C5), 115.2 (115.3) (C3′, C5′), 113.8 (C3′′, C5′′), 109.9 (111.0) (C6), 77.2 (77.1) (C8), 71.5 (C2), 55.1 (OCH3), 50.0 (49.9) (C3), 27.7 (27.8) (C9), 18.8 (18.7) (C10). HRMS (+ESI): (C24H22O7) calcd m/z 403.1574 [M + H]+, found m/z 403.1541.
3-(4-Hydroxyphenyl)-7a-((trimethylsilyl)oxy)-2,3,7a,8,10,11,11a,12-octahydro-4H,9H-pyrano[2,3-a]xanthen-4-one (22e). Isoflavanone 22e was synthesized following general procedure 2 using the Mannich base 11 (152 mg, 0.48 mmol, 1 eq.) and (cyclohex-1-en-1-yloxy)trimethylsilane (0.94 mL, 4.83 mmol, 10.1 eq.). The product was obtained as an off-white solid (57 mg, 27%). M.p. 197–198 °C; IR (neat): υmax 3350, 2934, 1657, 1584, 1516, 1435, 1362, 1245, 1174, 1093, 913 cm−1; UV (THF): λmax 268 nm (ε 14
649 cm−1 M−1), 238 (9386); 1H NMR (400 MHz, acetone-d6): δ 8.31 (s, 1H, OH), 7.69 (d, J = 8.9 Hz, 1H, H5), 7.13 (d, J = 8.5 Hz, 2H, H2′, H6′), 6.79 (d, J = 8.5 Hz, 2H, H3′, H5′), 6.53 (d, J = 8.9 Hz, 1H, H6), 4.63–4.70 (m, 2H, H2), 3.84–3.88 (dd, J = 5.4, 7.3 Hz, 1H, H3), 2.43–2.48 (m, 2H, H8), 2.19–2.27 (m, 1H, H12α), 1.97–2.02 (m, 1H, H11a), 1.67–1.73 (m, 1H, H11α), 1.65–1.67 (m, 1H, H12β), 1.62–1.63 (m, 1H, H10α), 1.53–1.57 (m, 1H, H9α), 1.49–1.52 (m, 1H, H11β), 1.33–1.42 (m, 1H, H10β), 1.06–1.15 (m, 1H, H9β), 0.02 (s, 9H, 3 × CH3); 13C NMR‡ (100 MHz, acetone-d6): δ 191.9 (191.5) (C4), 161.8 (161.7) (C1a), 158.8 (158.7) (C6a), 157.6 (C4′), 130.8 (130.6) (C2′, C6′), 128.1 (127.8) (C1′), 126.7 (126.6) (C5), 116.2 (C3′, C5′), 115.8 (115.7) (C12a), 112.1 (112.0) (C6), 110.0 (109.9) (C4a), 100.4 (100.4) (C7a), 72.9 (73.0) (C2), 51.9 (51.7) (C3), 39.5 (C12), 38.6 (38.5) (C11a), 29.6 (C9), 25.5 (C10), 24.4 (C11), 22.6 (22.5) (C8), 1.83 (3 × CH3). HRMS (+ESI): (C25H30O5Si) calcd m/z 461.1755 [M + Na]+, found m/z 461.1749.
7a-hydroxy-3-(4-hydroxyphenyl)-2,3,7a,8,9,10,10a,11-octahydro-4H-cyclopenta[b]pyrano[2,3-f]chromen-4-one (22f). Isoflavanone 22f was synthesized following general procedure 2 using the Mannich base 11 (120 mg, 0.38 mmol, 1 eq.) and (cyclopent-1-en-1-yloxy)trimethylsilane (0.68 mL, 7.66 mmol, 20.2 eq.). The product was obtained as a light-yellow sticky solid (34 mg, 25%). IR (DCM): υmax 3287, 2962, 1715, 1586, 1512, 1438, 1362, 1263, 1093 cm−1; UV (THF): λmax 288 nm (ε 169
500 cm−1 M−1), 269 (179366), 241 (187471); 1H NMR (400 MHz, acetone-d6): δ 7.64 (d, J = 8.7 Hz, 1H, H5), 7.13 (d, J = 8.5 Hz, 2H, H2′, H6′), 6.79 (d, J = 8.5 Hz, 2H, H3′, H5′), 6.62 (d, J = 8.7 Hz, 1H, H6), 4.63–4.66 (m, 2H, H2), 3.81–3.86 (m, 1H, H3), 2.98–3.04 (m, 1H, H11α), 2.61–2.68 (m, 1H, H11β), 2.44–2.52 (m, 1H, H10a), 2.20–2.27 (m, 1H, H10α), 2.08–2.18 (m, 1H, H10β), 1.98–2.02 (m, 1H, H9α), 1.70–1.80 (m, 2H, H8), 1.60–1.73 (m, 1H, H9β); 13CNMR‡ (100 MHz, acetone-d6): δ 191.6 (C4), 162.5 (C11a′), 162.2 (162.1) (C6a), 157.5 (C4′), 130.5 (130.5) (C2′, C6′), 128.0 (128.0) (C1′), 127.4 (C5), 116.1 (C3′, C5′), 115.4 (C7a), 115.0 (C11a, C4a), 110.7 (110.6) (C6), 72.6 (72.5) (C2), 51.6 (51.5) (C3), 49.4 (C10a), 38.1 (C10), 30.0 (C9), 23.2 (C11), 21.1 (C8). HRMS (+ESI): (C21H20O5) calcd m/z 375.1203 [M + Na]+, found m/z 375.1199.
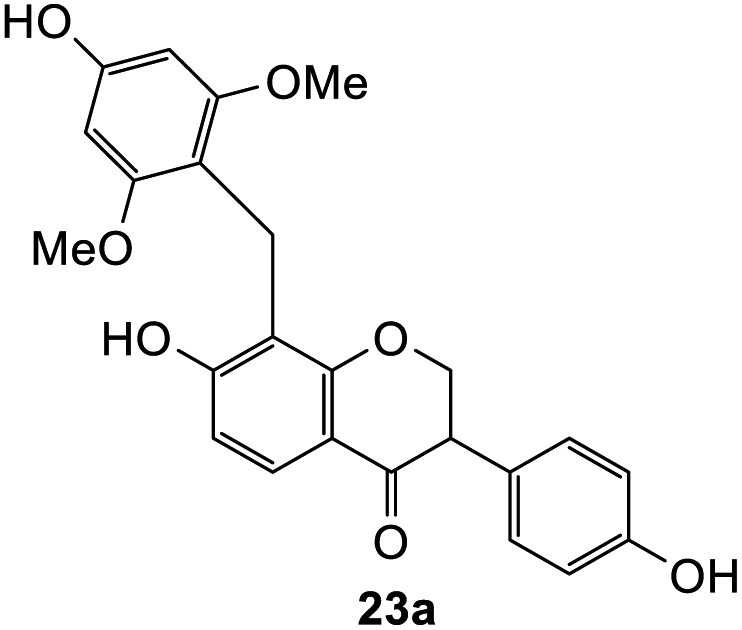
7-Hydroxy-8-(4-hydroxy-2,6-dimethoxybenzyl)-3-(4-hydroxyphenyl)chroman-4-one (23a). Isoflavanone 23a was synthesized following general procedure 2 using the Mannich base 11 (120 mg, 0.38 mmol, 1 eq.) and 3,5-dimethoxyphenol (60 mg, 0.38 mmol, 1 eq.). The product was obtained as an orange sticky solid (13 mg, 8%). IR (DCM): υmax 3157, 2926, 1590, 1512, 1437, 1241, 1195, 1109 cm−1; UV (THF): λmax 288 nm (ε 9505 cm−1 M−1), 269 (11
701), 241 (18629); 1H NMR (400 MHz, DMSO-d6): δ 9.87 (s, 1H, OH-7), 9.31 (s, 1H, OH-4′), 9.17 (s, 1H, OH-4′′), 7.45 (d, J = 8.5 Hz, 1H, H5), 7.00 (d, J = 8.5 Hz, 2H, H2′, H6′), 6.67 (d, J = 8.5 Hz, 2H, H3′, H5′), 6.45 (d, J = 8.5 Hz, 1H, H6), 6.00 (s, 2H, H3′′, H5′′), 4.48–4.53 (m, 2H, H2), 3.76–3.78 (m, 1H, H3), 3.72 (s, 2H, CH2), 3.59 (s, 6H, 2 × OCH3); 13C NMR (100 MHz, DMSO-d6): δ 191.2 (CO), 162.1 (C7), 161.2 (C8a), 158.7 (C2′′, C6′′), 156.7 (C4′′), 156.4 (C4′), 129.7 (C2′, C6′), 126.7 (C1′), 125.3 (C5), 115.3 (C8), 115.2 (C3′, C5′), 113.2 (C4a), 109.8 (C6), 107.1 (C1′′), 92.2 (C3′′, C5′′), 71.2 (C2), 55.5 (2 × OCH3), 50.0 (C3), 16.4 (CH2). HRMS ESI (C24H22O7) calcd m/z 423.1438 [M + H]+, found m/z 423.1438.
8-(2-Amino-4,6-dimethoxybenzyl)-7-hydroxy-3-(4-hydroxyphenyl)chroman-4-one (23b). Isoflavanone 23b was synthesized following general procedure 2 using the Mannich base 11 (120 mg, 0.38 mmol, 1 eq.) and 3,5-dimethoxyaniline (59 mg, 0.38 mmol, 1 eq.). The product was obtained as a brown sticky solid (28 mg, 17%). IR (DCM): υmax 3368, 3236, 2929, 1590, 1513, 1436, 1241, 1203, 1090, 1023, 998 cm−1; UV (THF): λmax 288 nm (ε 8429 cm−1 M−1), 268 (10
494), 241 (16310); 1H NMR (400 MHz, DMSO-d6): δ 9.32 (s, 1H, OH), 7.50 (d, J = 8.7 Hz, 1H, H5), 6.99 (d, J = 8.4 Hz, 2H, H2′, H6′), 6.65 (d, J = 8.4 Hz, 2H, H3′, H5′), 6.52 (d, J = 8.7 Hz, 1H, H6), 5.87 (d, J = 2.2 Hz, 1H, H3′′), 5.86 (d, J = 2.2 Hz, 1H, H5′′), 4.55 (d, J = 6.2 Hz, 2H, H2), 3.80 (t, J = 6.2 Hz, 1H, H3), 3.70 (s, 3H, OMe), 3.67 (s, 2H, CH2), 3.64 (s, 3H, OMe); 13C NMR (100 MHz, DMSO-d6): δ 191.1 (C4), 161.7 (C7), 161.1 (C8a), 158.9 (C4′′), 158.7 (C6′′), 156.4 (C4′), 147.3 (C2′′), 129.5 (C2′, C6′), 126.4 (C1′), 125.8 (C5), 115.2 (C3′, C5′), 114.1 (C4a), 113.5 (C4a), 110.0 (C6), 104.5 (C1′′), 93.2 (C3′′), 88.1 (C5′′), 71.2 (C2), 55.5 (OCH3), 54.6 (OCH3), 49.9 (C3), 16.7 (CH2). HRMS (+ESI): (C24H23NO6) calcd m/z 422.1598 [M + H]+, found m/z 422.1597.
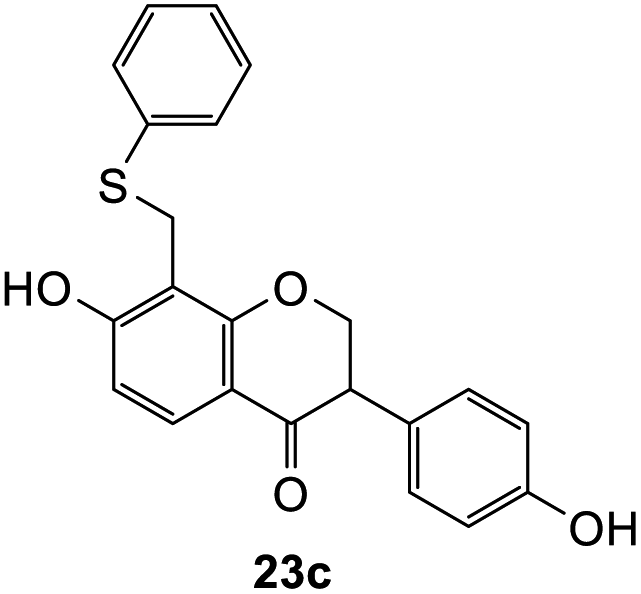
7-Hydroxy-3-(4-hydroxyphenyl)-8-((phenylthio)methyl)chroman-4-one (23c). Isoflavanone 23c was synthesized following general procedure 2 using the Mannich base 11 (150 mg, 0.48 mmol, 1 eq.) and thiophenol (0.05 mL, 0.49 mmol, 1.0 eq.). The product was obtained as colourless crystals (75 mg, 41%). M.p. 159–160 °C; IR (neat): υmax 3300, 2974, 1648, 1589, 1515, 1439, 1373, 1215, 1092, 1045 cm−1; UV (THF): λmax 288 nm (ε 9612 cm−1 M−1), 265 (15
138), 243 (18960); 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 8.5 Hz, 1H, H5), 7.41–7.44 (m, 2H, H2′′, H6′′), 7.27–7.32 (m, 2H, H3′′, H5′′), 7.17–7.23 (m, 1H, H4′′), 7.12 (d, J = 8.5 Hz, 2H, H2′, H6′), 6.79 (d, J = 8.7 Hz, 2H, H3′, H5′), 6.65 (d, J = 8.7 Hz, 1H, H6), 4.50–4.53 (m, 2H, H2), 4.24 (s, 2H, CH2–S), 3.80 (t, J = 6.9 Hz, 1H, H3); 13C NMR (100 MHz, acetone-d6): δ 191.3 (C4), 162.6 (C7), 162.1 (C8a), 157.6 (C4′), 138.4 (C1′′), 131.1 (C2′′, C6′′), 130.7 (C2′, C6′), 129.6 (C3′′, C5′′), 128.5 (C5), 128.0 (C1′), 127.0 (C4′′), 116.2 (C3′, C5′), 115.4 (C4a), 112.9 (C8), 110.7 (C6), 72.8 (C2), 51.6 (C3), 27.5 (S–CH2). HRMS (+ESI): (C22H18O4S) calcd m/z 401.0818 [M + Na]+, found m/z 401.0819.
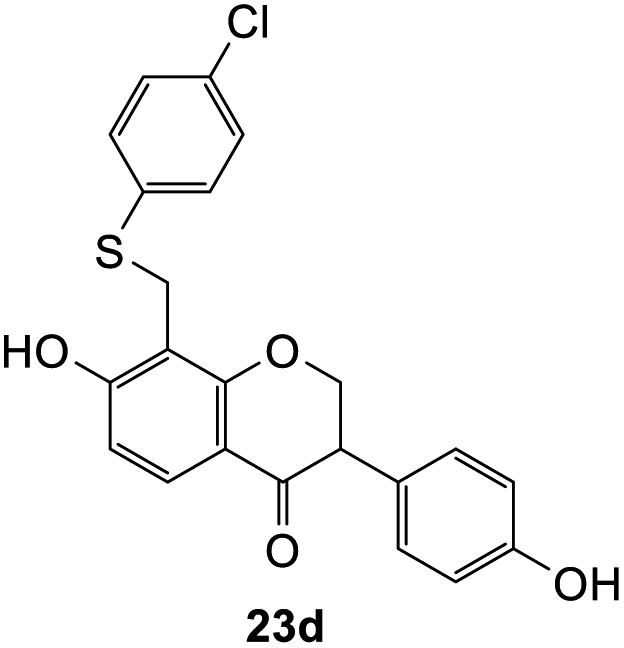
8-(((4-Chlorophenyl)thio)methyl)-7-hydroxy-3-(4-hydroxyphenyl)chroman-4-one (23d). Isoflavanone 23d was synthesized following general procedure 2 using the Mannich base 11 (121 mg, 0.39 mmol, 1 eq.) and p-chlorothiophenol (56 mg, 0.39 mmol, 1.0 eq.). The product obtained (40 mg, 25%) after column chromatography was further purified by preparative HPLC using a 5 μM column (150 × 22 mm ID) at a flow rate of 5 mL min−1 with a gradient elution of 2–100% solvent B (solvent A: 0.1% formic acid/H2O) and solvent B (0.1% formic acid/ACN) over 56 min to afford the expected product as a white solid (26 mg, 16%). M.p. 199–200 °C; IR (neat): υmax 3101, 2925, 1672, 1590, 1513, 1440, 1377, 1277, 1092, 1002 cm−1; UV (THF): λmax 288 nm (ε 15
607 cm−1 M−1), 264 (25598), 242 (28406); 1H NMR (400 MHz, DMSO-d6): δ 9.39 (br s, 1H, OH), 7.57 (d, J = 8.7 Hz, 1H, H5), 7.38 (d, J = 8.9 Hz, 2H, H2′′, H6′′), 7.34 (d, J = 8.9 Hz, 2H, H3′′, H5′′), 7.01 (d, J = 8.7 Hz, 2H, H2′, H6′), 6.69 (d, J = 8.7 Hz, 2H, H3′, H5′), 6.54 (d, J = 8.7 Hz, 1H, H6), 4.47–4.49 (m, 2H, H2), 4.11 (s, 2H, CH2–S), 3.79–3.82 (m, 1H, H3); 13CNMR (100 MHz, DMSO-d6): δ 190.7 (C4), 162.1 (C7), 160.7 (C8a), 156.5 (C4′), 136.1 (C1′′), 130.7 (C2′′, C6′′), 130.6 (C4′′), 129.6 (C2′, C6′), 128.7 (C3′′, C5′′), 127.5 (C5), 126.1 (C1′), 115.2 (C3′, C5′), 113.5 (C4a), 110.7 (C6) 109.8 (C8), 71.4 (C2), 50.2 (C3), 26.2 (S–CH2). HRMS (+ESI): (C22H17ClO4S) calcd m/z 435.0428 [M + Na]+, found m/z 435.0429.
Cell viability assays
The cell viability assay was conducted following literature method.48 MDA-MB-231 cells were seeded at 3000 cells per well in 96-well plates to ensure sustained exponential growth for 4 days. Cells were treated 24 h after seeding with 1 to 500 μM of compounds. After 72 h drug incubation, the treatment-media was replaced with 10% Alamar Blue in fresh media and the cells were incubated for another 6 h. The metabolic activity was detected by spectrophotometric analysis by assessing the absorbance of Alamar blue. Cell proliferation was determined and expressed as a percentage of untreated control cells. The determination of IC50 values was done using GraphPad Prism 6.Conclusions
In conclusion, o-QM intermediates derived from 6-hydroxyflavanone, 7-hydroxyflavanone, and dihydrodaidzein had been successfully utilized to generate fused-ring systems and diarylmethane products. The resultant yields of the analogues varied depending on the reactivity of the dienophiles or nucleophiles. With o-QMs derived from 6-hydroxyflavanone, the cycloadducts were formed in relatively higher yield when electron-rich enaminone was used as a dienophile. Reactions involving o-QMs derived from 7-hydroxyflavanone generated a mixture of both ring-closed and ring-opened products in comparatively lower yields. A thioether bridge was formed instead of a methylene bridge when sulfur-containing nucleophiles were used in the Michael addition reaction of Mannich base derived from dihydrodaidzein. A rationale has been postulated, in which the thiolate anion behaves as a stronger nucleophile than the π electrons in the benzene ring. Several cycloadducts exhibited slightly improved anti-proliferative activity against MDA-MB-231 breast cancer cell lines as compared to their corresponding parent compounds, although the activity was low. Overall, the reactions involving o-QM intermediates remain a useful method to generate a wide range of cycloaddition adducts and diarylmethane products.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: N. K. and D. B., methodology: V. A. N., software N/A, validation: V. A. N., formal analysis: V. A. N., investigation: V. A. N., resources: N. K. and D. B., data curation, writing – original draft: V. A. N., writing – review & editing: V. F., D. S. W., N. K. and D. B., visualization: V. A. N. and V. F. supervision: N. K., D. B. and D. S. W., project administration: N. K. and D. B., funding acquisition: N. K. and D. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NMR and BMSF facility, University of New South Wales. V. A. N gratefully acknowledges the Indonesia Endowment Fund for Education Agency (LPDP) for a postgraduate scholarship.Notes and references
- M.-H. Pan, C.-S. Lai and C.-T. Ho, Anti-inflammatory activity of natural dietary flavonoids, Food Funct., 2010, 1, 15 RSC.
- A. Sharma, H. Singh Tuli and A. K. Sharma, Chemistry and Synthetic Overview of Flavonoids, in Current Aspects of Flavonoids: Their Role in Cancer Treatment, ed. H. Singh Tuli, Springer Singapore, Singapore, 2019, pp. 23–38 Search PubMed.
- A. M. Pereira, H. Cidade and M. E. Tiritan, Stereoselective Synthesis of Flavonoids: A Brief Overview, Molecules, 2023, 28, 426 CrossRef CAS PubMed.
- A. Bianco, C. Cavarischia and A. Farina, et al., A new synthesis of flavonoids via Heck reaction, Tetrahedron Lett., 2003, 44, 9107–9109 CrossRef CAS.
- K. Tanaka and T. Sugino, Efficient conversion of 2′-hydroxychalcones into flavanones and flavanols in a water suspension medium, Green Chem., 2001, 3, 133–134 RSC.
- C. M. Brennan, I. Hunt and T. C. Jarvis, et al., Stereoelectronic effects in ring closure reactions: the 2′-hydroxychalcone – flavanone equilibrium, and related systems, Can. J. Chem., 1990, 68, 1780–1785 CrossRef CAS.
- R. Dixon, Genistein, Phytochemistry, 2002, 60, 205–211 CrossRef CAS PubMed.
- M. Furusawa, Y. Ido and T. Tanaka, et al., Novel, complex flavonoids from Mallotus philippensis (Kamala tree), Helv. Chim. Acta, 2005, 88, 1048–1058 CrossRef CAS.
- L. G. Bahrin, P. G. Jones and H. Hopf, Tricyclic flavonoids with 1, 3-dithiolium substructure, Beilstein J. Org. Chem., 2012, 8, 1999–2003 CrossRef CAS PubMed.
- Y. Luan, H. Sun and S. E. Schaus, Iron-Catalyzed Rearrangements and Cycloaddition Reactions of 2 H -Chromenes, Org. Lett., 2011, 13, 6480–6483 CrossRef CAS PubMed.
- N. Majumdar, K. A. Korthals and W. D. Wulff, Simultaneous Synthesis of Both Rings of Chromenes via a Benzannulation/o-Quinone Methide Formation/Electrocyclization Cascade, J. Am. Chem. Soc., 2012, 134, 1357–1362 CrossRef CAS PubMed.
- J. H. George, M. D. Hesse and J. E. Baldwin, et al., Biomimetic Synthesis of Polycyclic Polyprenylated Acylphloroglucinol Natural Products Isolated from Hypericum papuanum, Org. Lett., 2010, 12, 3532–3535 CrossRef CAS PubMed.
- K. K. Hong, G. E. Ball and D. S. Black, et al., The mosaic of rottlerin, J. Org. Chem., 2015, 80, 10668–10674 CrossRef CAS PubMed.
- K. Nakatani, N. Higashida and I. Saito, Highly efficient photochemical generation of o-Quinone methide from Mannich bases of phenol derivatives, Tetrahedron Lett., 1997, 38, 5005–5008 CrossRef CAS.
- Đ. Škalamera, C. Bohne and S. Landgraf, et al., Photodeamination Reaction Mechanism in Aminomethyl p -Cresol Derivatives: Different Reactivity of Amines and Ammonium Salts, J. Org. Chem., 2015, 80, 10817–10828 CrossRef PubMed.
- S. Bilgiç, O. Bilgiç and B. Büyükkıdan, et al., Synthesis of Chromans from the Reaction of O-quinone Methide Precursor with Substituted Styrenes, J. Chem. Res., 2007, 2007, 76–79 CrossRef.
- M. S. Frasinyuk, G. P. Mrug and S. P. Bondarenko, et al., Antineoplastic Isoflavonoids Derived from Intermediate ortho-Quinone Methides Generated from Mannich Bases, ChemMedChem, 2016, 11, 600–611 CrossRef CAS PubMed.
- P. Mallepaka, J. R. Yerrabelly and H. Yerrabelly, Synthesis and anti-cancer activity of diverse oxa-carbocycle fused isoflavones using the olefin metathesis, Arkivoc, 2023, 8, 202312130 Search PubMed.
- D. Wang, L. Hou and L. Wu, et al., Synthesis and Anti-tumor Activities of Novel Oxazinyl Isoflavonoids, Chem. Pharm. Bull., 2012, 60, 513–520 CrossRef CAS PubMed.
- M. M. Hann, A. R. Leach and G. Harper, Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS PubMed.
- F. Lovering, Escape from Flatland 2: complexity and promiscuity, Med. Chem. Commun., 2013, 4, 515 RSC.
- T. J. Ritchie and S. J. F. Macdonald, The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design?, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS PubMed.
- E. E. Allen, C. Zhu and J. S. Panek, et al., Multicomponent condensation reactions via ortho-quinone methides, Org. Lett., 2017, 19, 1878–1881 CrossRef CAS PubMed.
- A. V. Lukashenko, V. A. Osyanin and D. V. Osipov, et al., Reaction of Push–Pull Enaminoketones and in Situ Generated ortho-Quinone Methides: Synthesis of 3-Acyl-4 H-chromenes and 2-Acyl-1 H-benzo [f] chromenes as Precursors for Hydroxybenzylated Heterocycles, J. Org. Chem., 2017, 82, 1517–1528 CrossRef CAS PubMed.
- D. Zhou, K. Mao and J. Zhang, et al., Organocatalytic annulation of aldehydes and o-quinone methides: A facile access to dihydrocoumarins, Tetrahedron Lett., 2016, 57, 5649–5652 CrossRef CAS.
- H. Jian, K. Liu and W.-H. Wang, et al., Construction of 9-functionalized xanthenes via Diels-Alder reaction of stable ortho-quinone methides and arynes, Tetrahedron Lett., 2017, 58, 1137–1141 CrossRef CAS.
- T. P. Pathak and M. S. Sigman, Applications of ortho-Quinone Methide Intermediates in Catalysis and Asymmetric Synthesis, J. Org. Chem., 2011, 76, 9210–9215 CrossRef CAS PubMed.
- L. Caruana, M. Fochi and L. Bernardi, The emergence of quinone methides in asymmetric organocatalysis, Molecules, 2015, 20, 11733–11764 CrossRef CAS PubMed.
- V. A. Osyanin, D. V. Osipov and M. R. Demidov, et al., Potassium trinitromethanide as a 1, 1-ambiphilic synthon equivalent: access to 2-nitroarenofurans, J. Org. Chem., 2014, 79, 1192–1198 CrossRef CAS PubMed.
- R. M. Jones, C. Selenski and T. R. Pettus, Rapid syntheses of benzopyrans from o-OBOC salicylaldehydes and salicyl alcohols: a three-component reaction, J. Org. Chem., 2002, 67, 6911–6915 CrossRef CAS PubMed.
- J.-J. Zhao, S.-B. Sun and S.-H. He, et al., Catalytic Asymmetric Inverse-Electron-Demand Oxa-Diels–Alder Reaction of In Situ Generated ortho-Quinone Methides with 3-Methyl-2-Vinylindoles, Angew. Chem., Int. Ed., 2015, 54, 5460–5464 CrossRef CAS PubMed.
- N. J. Willis and C. D. Bray, ortho-Quinone Methides in Natural Product Synthesis, Chem.–Eur. J., 2012, 18, 9160–9173 CrossRef CAS PubMed.
- Y. Chen, S. L. Cass and S. K. Kutty, et al., Synthesis, biological evaluation and structure–activity relationship studies of isoflavene based Mannich bases with potent anti-cancer activity, Bioorg. Med. Chem. Lett., 2015, 25, 5377–5383 CrossRef CAS PubMed.
- Y. L. Garazd, T. N. Panteleimonova and M. M. Garazd, et al., Modified Coumarins. 11. Synthesis and Biological Activity of Mannich Bases of Substituted 1, 3-Dihydrocyclopenta [c] chromen-4-ones, Chem. Nat. Compd., 2003, 39, 330–336 CrossRef CAS.
- K. Stefanska, H. Jedrzejewska and M. Wierzbicki, et al., The Inverse Demand Oxa-Diels–Alder Reaction of Resorcinarenes: An Experimental and Theoretical Analysis of Regioselectivity and Diastereoselectivity, J. Org. Chem., 2016, 81, 6018–6025 CrossRef CAS PubMed.
- H. Sugimoto, S. Nakamura and T. Ohwada, Retro-Diels−Alder Reaction of 4 H -1,2-Benzoxazines to Generate o -Quinone Methides: Involvement of Highly Polarized Transition States, J. Org. Chem., 2007, 72, 10088–10095 CrossRef CAS PubMed.
- A.-C. Knall and C. Slugovc, Inverse electron demand Diels–Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- V. A. Osyanin, A. V. Lukashenko and D. V. Osipov, et al., Synthesis of 2-nitro-1H-benzo [f] chromenes, Chem. Heterocycl. Compd., 2015, 50, 1528–1533 CrossRef CAS.
- N. Chopin, H. Gerard and I. Chataigner, et al., Benzofurans as efficient dienophiles in normal electron demand [4+ 2] cycloadditions, J. Org. Chem., 2009, 74, 1237–1246 CrossRef CAS PubMed.
- C.-H. Chen, P. D. Rao and C.-C. Liao, Furans Act as Dienophiles in Facile Diels− Alder Reactions with Masked o-Benzoquinones, J. Am. Chem. Soc., 1998, 120, 13254–13255 CrossRef CAS.
- C. Liu, M. Shen and B. Lai, et al., Condition-Determined Multicomponent Reactions of 1,3-Dicarbonyl Compounds and Formaldehyde, ACS Comb. Sci., 2014, 16, 652–660 CrossRef CAS PubMed.
- J. Yang, J.-N. Tan and Y. Gu, Lactic acid as an invaluable bio-based solvent for organic reactions, Green Chem., 2012, 14, 3304 RSC.
- V. Ramesh, S. Shanmugam and N. S. Devi, Er(OTf)3 -Catalyzed Multicomponent Synthesis of 3,4-Dihydro-2 H -pyran via Hetero-Diels-Alder Reaction under Ambient Temperature, ChemistrySelect, 2018, 3, 3652–3658 CrossRef CAS.
- A. Genest, D. Portinha and E. Fleury, et al., The aza-Michael reaction as an alternative strategy to generate advanced silicon-based (macro) molecules and materials, Prog. Polym. Sci., 2017, 72, 61–110 CrossRef CAS.
- D. Osipov, V. Osyanin and L. Voskressensky, et al., Reactions of o-Quinone Methides with Halogenated 1H-Azoles: Access to Benzo[e]azolo[1,3]oxazines, Synthesis, 2017, 49, 2286–2296 CrossRef CAS.
- A. K. Salakka, T. H. Jokela and K. Wähälä, Multiple hydride reduction pathways in isoflavonoids, Beilstein J. Org. Chem., 2006, 2, 16 Search PubMed.
- M. S. Singh, A. Nagaraju and N. Anand, et al., ortho-Quinone methide (o-QM): a highly reactive, ephemeral and versatile intermediate in organic synthesis, RSC Adv., 2014, 4, 55924–55959 RSC.
- E. M. H. Yee, M. B. Brandl and D. S. Black, et al., Synthesis of isoflavene-thiosemicarbazone hybrids and evaluation of their anti-tumor activity, Bioorg. Med. Chem. Lett., 2017, 27, 2454–2458 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08704f |
| ‡ Additional peaks are due to diastereomers. |
| This journal is © The Royal Society of Chemistry 2025 |