
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of an exfoliated kaolinite–poly(urea–formaldehyde) nanocomposite
Hervé Barye Tatang*a,
Jacques Richard Macheb,
Cyrill Joël Ngally Sabouangc,
Angelina Razafitianamaharavod,
Renaud Gleyd,
Sakeo Konga and
Jean Aimé Mbey *a
*a
aLaboratory of Applied Inorganic Chemistry, Department of Inorganic Chemistry, University of Yaoundé I, P.O. Box 812, Yaoundé, Cameroon. E-mail: mbey25@yahoo.fr; jean-aime.mbey@facsciences-uy1.cm; barye.tatang@facsciences-uy1.cm
bDepartments of Chemistry, Higher Teacher Training College, University of Bamenda, P.O. Box 39, Bambili, Cameroon
cSchools of Geology and Mining Engineering, University of Ngaoundere, P.O. Box 115, Meiganga, Cameroon
dUniversité de Lorraine, CNRS, LIEC, F-54000 Nancy, France
First published on 29th January 2025
Abstract
In this study, kaolinite–poly(urea–formaldehyde) was successfully prepared through the polymerization of urea intercalated within the kaolinite structure. Polymerization was carried out under ambient conditions by immersing kaolinite–urea in formaldehyde. Evidence of urea intercalation and polymerization was obtained from FTIR, XRD, and thermal analysis (TG–DSC). The XRD pattern of the kaolinite–poly(urea–formaldehyde) composite shows that polymerization induces exfoliation of the kaolinite layers, leading to the formation of a nanocomposite. Textural analysis through nitrogen adsorption on raw kaolinite and kaolinite–urea demonstrates that polymerization mainly occurs within the interlayer. Water resistance tests show that poly(urea–formaldehyde) within the nanocomposite is less sensitive to decomposition when immersed in water compared to pristine poly(urea–formaldehyde). Additionally, a self-healing effect was observed for poly(urea–formaldehyde) in the nanocomposite, indicating that fragments from the decomposition of the intercalated polymer when immersed in water remain trapped within the composite matrix owing to interactions with kaolinite mineral layers. Modeling of kaolinite–poly(urea–formaldehyde) interactions allows for the proposal of a mechanism for the interlayer polymerization of intercalated urea.
1. Introduction
Formaldehyde-based resins have found extensive applications in various sectors, including adhesives,1,2 the wood industry,3–5 agriculture6,7 and encapsulation.2,8–12 However, the release of formaldehyde by these polymers is still challenging, given that formaldehyde is classified as a Group 1 carcinogen by the International Agency for Research on Cancer (IARC).13–15 Hence, formaldehyde's high reactivity, volatility, and toxicity demand a robust mitigation strategy. To address this issue, researchers have focused on synthetic pathways that reduce formaldehyde content14,16–19 or incorporate highly reactive molecules to capture formaldehyde.3,10,20–22 Although effective in reducing emissions, these methods often result in materials with compromised water resistance and mechanical properties. Recent advancements have explored the incorporation of mineral fillers, specifically kaolinite, to enhance both formaldehyde mitigation and overall resin performance.4,12,18,20,21,23–25 These studies demonstrate a successful reduction in formaldehyde emissions alongside an improvement in the mechanical properties of the resulting resins.Chen et al.24 investigated the incorporation of reactive kaolinite into poly(urea–formaldehyde) and reported notable results, including low formaldehyde emissions, good thermal stability, and improved water resistance. However, the strong internal cohesion of kaolinite hinders its homogeneous dispersion within the polymer matrix, thereby limiting interactions between the mineral and the polymer. Kaolinite is a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 phyllosilicate comprising alternating siliceous tetrahedral sites (SiO2) and aluminous octahedral sites (Al(OH)3) with interlayer space. Within this space, neighboring octahedral and tetrahedral sites are strongly bound through hydrogen bonds and dipole interactions arising from the layer's asymmetry,26,27 making the structure poorly dispersible. It can therefore be postulated that increased kaolinite dispersion may result in improved properties of kaolinite–polymer composites. Studies on kaolinite structural organization show that the weakening of interlayer bonding is a key process for improving dispersion, which is achievable through a convenient intercalation–deintercalation process.28,29
:1 phyllosilicate comprising alternating siliceous tetrahedral sites (SiO2) and aluminous octahedral sites (Al(OH)3) with interlayer space. Within this space, neighboring octahedral and tetrahedral sites are strongly bound through hydrogen bonds and dipole interactions arising from the layer's asymmetry,26,27 making the structure poorly dispersible. It can therefore be postulated that increased kaolinite dispersion may result in improved properties of kaolinite–polymer composites. Studies on kaolinite structural organization show that the weakening of interlayer bonding is a key process for improving dispersion, which is achievable through a convenient intercalation–deintercalation process.28,29
The cohesive nature of kaolinite can be weakened by intercalating small polar molecules into its interlayer spaces.28 Among the directly intercalated molecules, dimethyl sulfoxide, formamide, and urea are largely reported in the literature.30–32 The case of urea is of interest for the preparation of a kaolinite–poly(urea–formaldehyde) composite. The intercalation of urea into kaolinite is favored by its numerous interaction sites.31–34 The presence of amino and carbonyl groups in urea enables its retention within the interlayer space by establishing hydrogen bonds respectively with the hydrogen atoms of octahedral hydroxyls and the basal oxygen atoms of the tetrahedral sites.28 Additionally, the nitrogen atom in the amino group also forms hydrogen bonds with the hydrogen atoms of hydroxyls in the octahedral sites.34,35 This retention opens up the possibility of a urea–formaldehyde polymerization process within the kaolinite interlayer space.
The polymerization mechanism of urea–formaldehyde resins is well-documented in the literature.3,14,36–38 The process involves an initial methylolation (hydroxymethylation) of urea, followed by a polycondensation reaction. Recent studies by Wibowo et al.14 identify four stages in the formation of this resin, depending on the synthesis process. The first stage, corresponding to the addition reaction phase, results in short-chain polymers such as mono-, bis- and tris(hydroxyethyl)urea, which exhibit good crystallinity. The second and third stages, related to the condensation phase, lead to an amorphous polymer characterized by numerous cross-links through oxymethylene. Finally, the fourth stage results in a long-chain polymer that also possesses good crystallinity due to the linear structure, which is formed due to the formation of a methylene bridge.
Furthermore, previous studies have reported the exfoliation/delamination of kaolinite through in situ polymerization processes.27,39,40 It is reported that for exfoliation to occur, the departure of the pre-intercalated molecule and the insertion of the monomer must happen simultaneously. This result clearly indicates that a prior intercalation process is necessary to allow the insertion of the monomer into the interlayer space of the mineral. Furthermore, it is well established that the classic in situ polymerization process is generally initiated by a reaction damper.27,39,41 The formation of kaolinite-based nanocomposites through polymerization at room temperature ((28 ± 1)) °C) of molecules that can be directly intercalated in its interstitial space may be of interest as the displacement stage of the intercalated species is suppressed. Such a process is of interest for an efficient and rapid synthesis of kaolinite-based nanocomposites, which reduce the use of chemicals and the need for intermediate complexes.
Mechanisms of urea intercalation into kaolinite, as well as the polymerization of urea and formaldehyde to form poly(urea–formaldehyde) (UF), are well-established. However, the interlayer polymerization of intercalated urea with formaldehyde is not reported, whereas the formation of the kaolinite–urea complex suggest that one could induce interlayer polymerization for the preparation of the kaolinite–poly(urea–formaldehyde) composite. In particular, it is expected that an exfoliated kaolinite–poly(urea–formaldehyde) nanocomposite will be formed. Additionally, the exfoliation of kaolinite layers will probably influence the polymer properties, which could be tuned for improved or new applications.
Hence, the proposed aim of this study is to elaborate the kaolinite–poly(urea–formaldehyde) (KUF) composite through interlayer polymerization at room temperature of urea directly intercalated in the kaolinite interlayer. The mechanism of this polymerization is proposed through the modeling of the urea–kaolinite and urea–formaldehyde interactions. The structural characteristics (exfoliation/delamination, microstructural changes) and some physico-chemical properties (water resistance, thermal stability) of this novel material were evaluated using Fourier-transform Infrared Spectroscopy (FTIR), X-ray diffraction (XRD), and thermogravimetric analysis coupled with differential scanning calorimetry (TGA-DSC). Prior to interlayer polymerization, textural analyses of the intercalated precursor (kaolinite–urea) was done through nitrogen adsorption in order to access the reactive surface and estimate surface vs. internal polymerization.
2. Materials and methods
The kaolinite sample (K) used in this study was sourced from the locality of Mayouom in Cameroon. It was wet sieved at 45 μm, dried at ambient temperature ((28 ± 1) °C), then ground and sieved over a 160 μm mesh, and stored in a closed glass jar. Its chemical composition and crystallinity, reported in previous studies,28,42 indicate a high content (76.4%) in kaolinite with good structural organization (Hinckley index: 0.74; P0: 1.24; Slope Ratio: 0.92). The chemical composition of major elements (in oxide percentages) in the clay is as follows: % SiO2: 44.28; % Al2O3: 34.21; % Fe2O3: 1.97; % K2O: 1.23; % MgO: 0.24; % CaO: 0.04; % MnO: 0.01; % TiO2: 3.39; % P2O5: 0.26 and LOI: 13.7%. Analytical grade urea (purity: 98%) and formaldehyde (40%) were utilized for the intercalation and polymerization processes.The intercalation of urea into kaolinite was conducted in an aqueous suspension, following a modified approach by Makó et al.33 Typically, a mixture of kaolin with a half-saturated aqueous urea solution at a mass/volume ratio of 1:3 was prepared at ambient temperature ((28 ± 1) °C). The suspension was then stirred in a closed jar for 15 minutes and left to stand for 14 days to ensure optimal intercalation under soft condition. The resulting suspension was filtered, dried, ground, and labeled as KU.
For the interlayered polymerization process, approximatively 20 g of the cake obtained after filtration from the intercalation process was collected and dispersed in 60 mL of formaldehyde at an approximate mass/volume ratio of 1:3 at room temperature. The mixture was agitated daily for 5 minutes in a closed glass jar for 14 days, filtered, dried, ground and labeled as KUF.
The synthesis of UF resin was adapted from Park et al. and Ferra et al.1,43 52 mL of formaldehyde was placed in a reactor kettle, heated to 60 °C, and the pH was adjusted to 8 with sodium hydroxide. Subsequently, 20 g of urea was added, and the mixture was heated to 90 °C for 45 minutes. The pH was then adjusted to 5 with acetic acid for condensation. A second addition of urea was done sequentially with 7 g in order to control the cross-linking while ensuring a urea/formaldehyde ratio greater than 1, which limited the formation of oxymethylene bridges by the condensation of formaldehyde molecules.1,43 After this stage, a control was prepared to verify the effective formation of the polymer through the adsorption of a small amount of solution using a dropper and then dropped into a beaker filled with water until a white mist appeared. The appearance of this mist indicates that the polymer was successfully synthesized and the reaction was stopped by adding a sodium hydroxide solution (6 M) until the pH reached 9. After 7 minutes, the heating was stopped and the resin, without washing, was cooled to room temperature, air-dried and labeled as UF.
The water resistance of both dried pristine UF and KUF was evaluated by immersing 1 g of each material in 20 mL of distilled water for 5, 10, 20 minutes, and 1 hour and in 40 mL during 3 hours with continuous agitation at 200 rpm, followed by drying to a constant weight. The remaining UF quantity after immersion was determined using eqn (1) for pristine UF and eqn (2), which utilizes FTIR spectra for UF within the composite.
 | (1) |
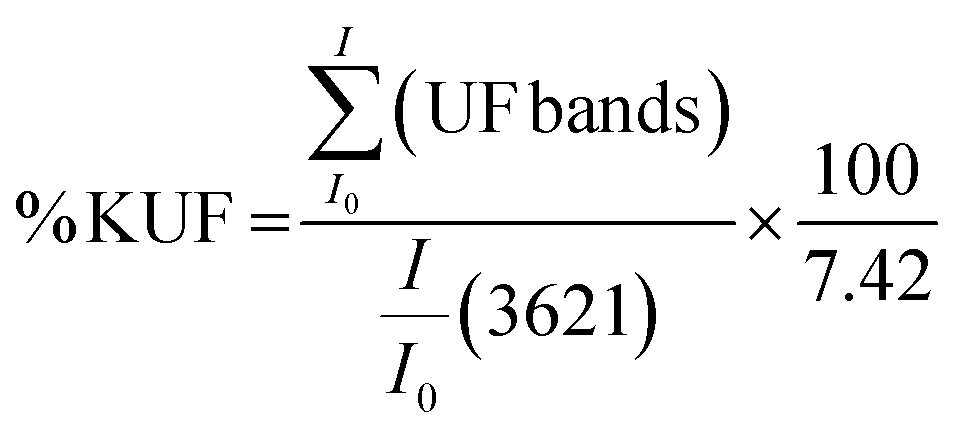 | (2) |
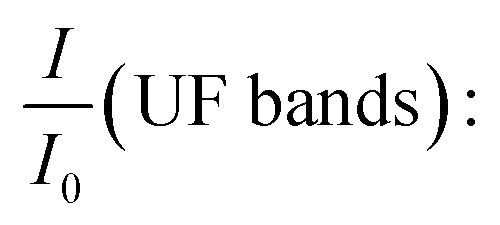 the relative intensities of bands at 1653, 1544, 1388 and 1270 cm−1 of UF in the composite;
the relative intensities of bands at 1653, 1544, 1388 and 1270 cm−1 of UF in the composite; 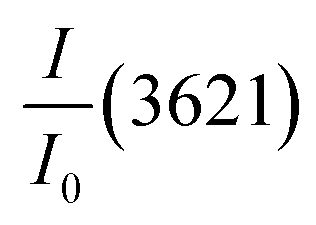 : the relative intensity inner hydroxyls band of kaolinite at 3621 cm−1 determined using the baseline method from the FTIR spectra and 7.42 is the ratio value of
: the relative intensity inner hydroxyls band of kaolinite at 3621 cm−1 determined using the baseline method from the FTIR spectra and 7.42 is the ratio value of 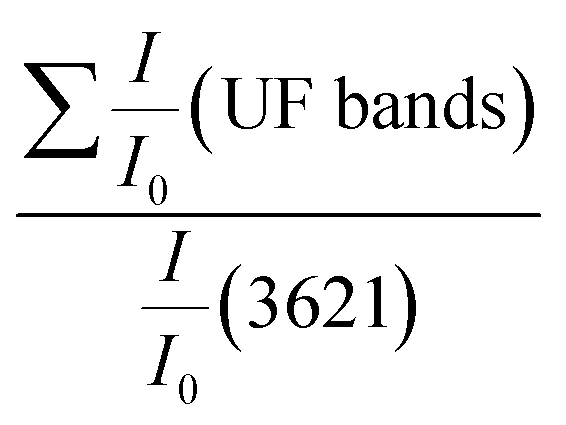 in the composite before immersion. The inner hydroxyls band of kaolinite at 3621 cm−1 is used as a reference, given that its intensity is not affected by the modification of H-bonding in the kaolinite due to exfoliation/delamination.
in the composite before immersion. The inner hydroxyls band of kaolinite at 3621 cm−1 is used as a reference, given that its intensity is not affected by the modification of H-bonding in the kaolinite due to exfoliation/delamination.
2.1. Nitrogen adsorption at 77 K
Nitrogen adsorption–desorption isotherms at 77 K were obtained using a self-built setup, where pressures were monitored with Baratron-type pressure sensors provided by Edwards. The samples were outgassed at 110 °C under vacuum before adsorption using high-purity nitrogen (N55 > 99.9995%). The specific surface area (SSA) was calculated utilizing the Brunauer–Emmett–Teller (BET) equation, assuming a cross-sectional area of 16.3 Å2 for nitrogen. The study estimated the error in SSA determination at ±1 m2 g−1. Mesoporous structure analysis was conducted using a modified Kelvin equation for slit-shaped pores on the desorption branch between 0.4 ≤ P/P0 ≤ 0.98 following the method of Delon and Dellys,44 with nitrogen-adsorbed layer thickness (t) calculated using the Jura & Harkins45,46 equation.2.2. Fourier transform infrared (FTIR) spectroscopy
Fourier transform infrared (FTIR) spectroscopy analysis was performed in the diffuse reflectance mode using a BRUKER Alpha P Fourier transformed spectrometer in the range from 400 to 4000 cm−1. Each spectrum was obtained as an accumulation of 40 scans.The interlayer changes were monitored by evaluating the interlayer cohesion or hydrogen bond extension in the interlayer space using the P0 test (eqn (3)).28,47,48
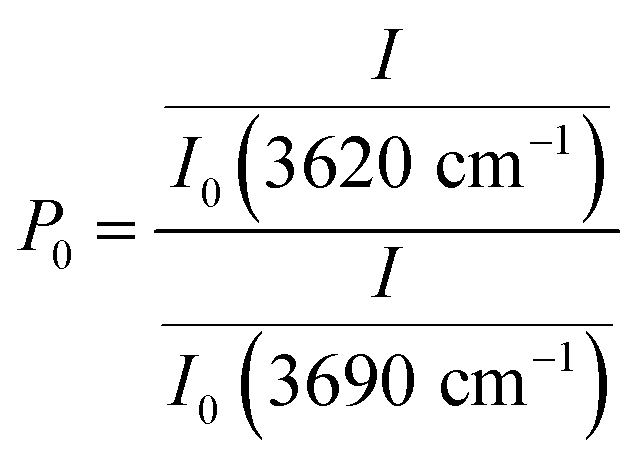 | (3) |
2.3. X-ray diffraction
The X-ray diffraction patterns of the samples were obtained from the University of Liege using a Bruker AXS D8 advance diffractometer equipped with Cu Kα radiation (λ = 1.5406 Å), operating under a voltage of 40 kV and an intensity of 40 mA. The diffraction patterns were recorded at a step size of 0.02° for a step time of 2 seconds over the 2 theta range from 5° to 70°. The <2 μm fraction for oriented slides was obtained through the dispersion of clay in water from which drops of the supernatant were deposited on a glass slide. After air drying, the oriented samples were subjected to glycolation and thereafter to firing at 550 °C. The XRD patterns of the oriented, glycolated, and fired samples were recorded at the University of Lorraine using a Bruker D8 advance diffractometer equipped with a 1D LynxEye linear detector. The diffraction patterns were recorded using Cu-Kα radiation (λ = 1.5406 Å) operating under a voltage of 40 kV and a current of intensity of 40 mA. Scanning was performed at a step rate of 0.035°/3 s from 5° to 35° (2θ).The structural changes were evaluated by determining the coherent scattering domain size (D) using the full width at half maximum (height) (β) of the reflection associated with the d001 and d002 basal spacing (with a preference for d002) and Scherrer's equation (eqn (4)).49 For this calculation, the level of disorder in the direction associated with the reflection used was assumed to be the same. The number of layers per crystallite (NL) was obtained by dividing the preferential coherent scattering domain thickness by the main basal spacing associated with the 001 reflection of the mineral.
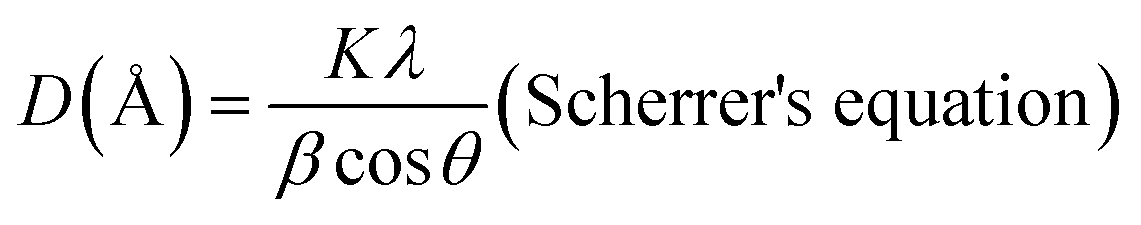 | (4) |
| K = 0.89 (constant), λ = 1.5406 (Å) |
The intercalation level of urea in kaolinites was assessed by calculating the intercalation ratio.26 This parameter was obtained using the intercalation reflection intensity and the residual kaolinite basal reflection intensity in the treated kaolinite.26,30 For this calculation, the degree of orientation of the intercalated and non-intercalated particles was assumed to be the same.
2.4. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)
Thermal analyses were conducted using a combined TGA–DSC device SETARAM at the University of Liege. Forty milligrams (40 mg) of each sample was placed in an alumina crucible and heated at a rate of 10 °C min−1 from room temperature to 800 °C.3. Results and discussion
3.1. Texture, mineralogy and thermal behavior of raw and urea-intercalated kaolinite
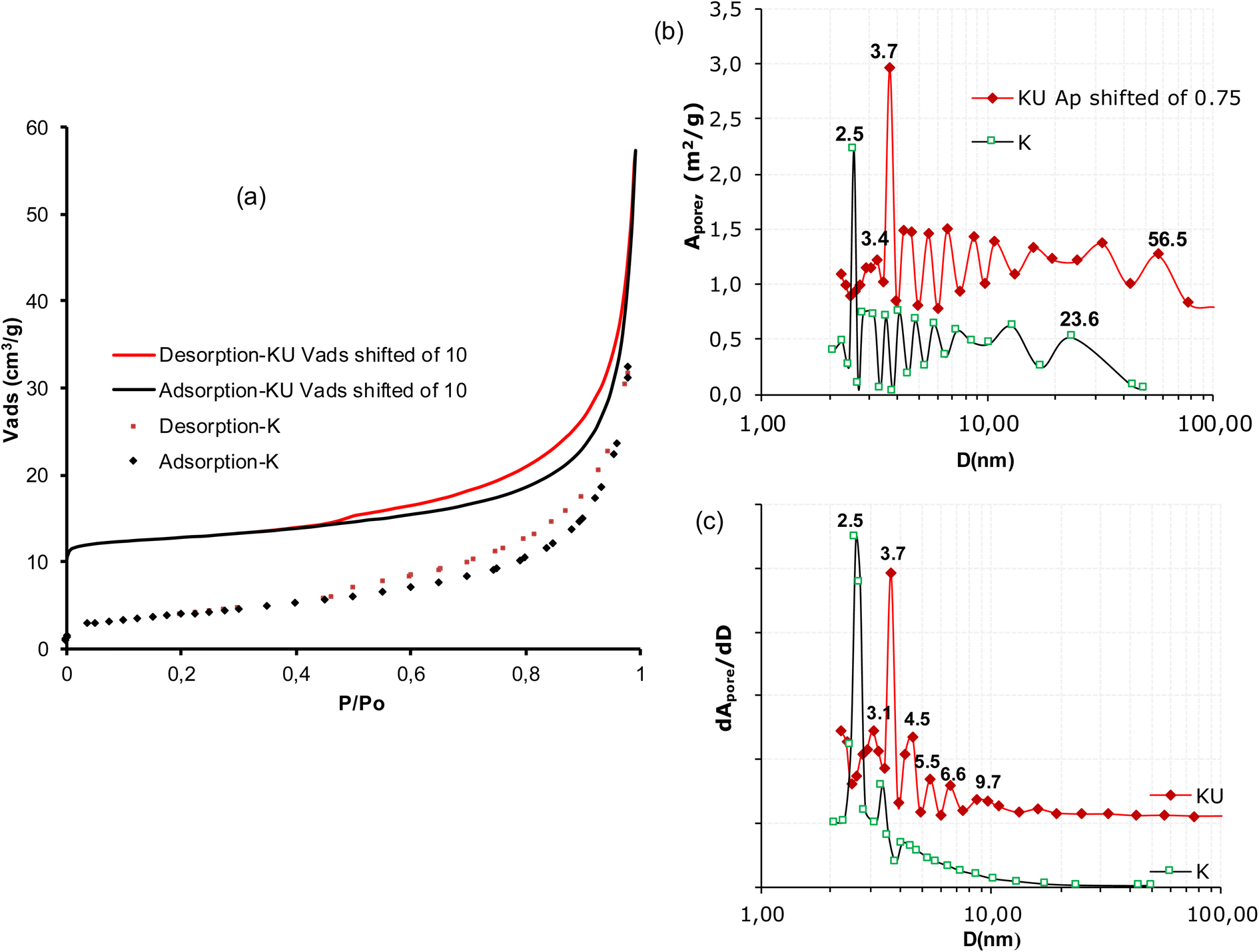 | ||
| Fig. 1 (a) Nitrogen adsorption–desorption isotherms of raw (K) and urea intercalated kaolinite (KU) at 77 K; (b) spectra of specific areas of mean pore openings; (c) distribution spectra of mean pore openings. | ||
| Samples | K | KU | KUF | UF | |||
|---|---|---|---|---|---|---|---|
| a d: Basal distance; D: crystallite sizes; NL: number of layers per crystallite; SBET: BET specific area; ∑Apore: Cumulative pore area and IR: Intercalation Ratio. | |||||||
| Reflection | 001 | 002 | 001 | 002 | 001 | 002 | 4 |
| d (Å) | 7.14 | 3.58 | 10.7 | 3.58 | >44.1 | 3.59 | |
| D (Å) | 190 | 174 | 229 | 209 | / | 210 | 69 |
| NL | 24 | 20 | <5 | / | |||
| Test P0 | 1.24 | 0.74 | 0.97 | / | |||
| SBET | 14.3 | 10.1 | / | / | |||
| ∑APore | 12.46 | 10.02 | / | / | |||
| IR (%) | / | 86 | / | / | |||
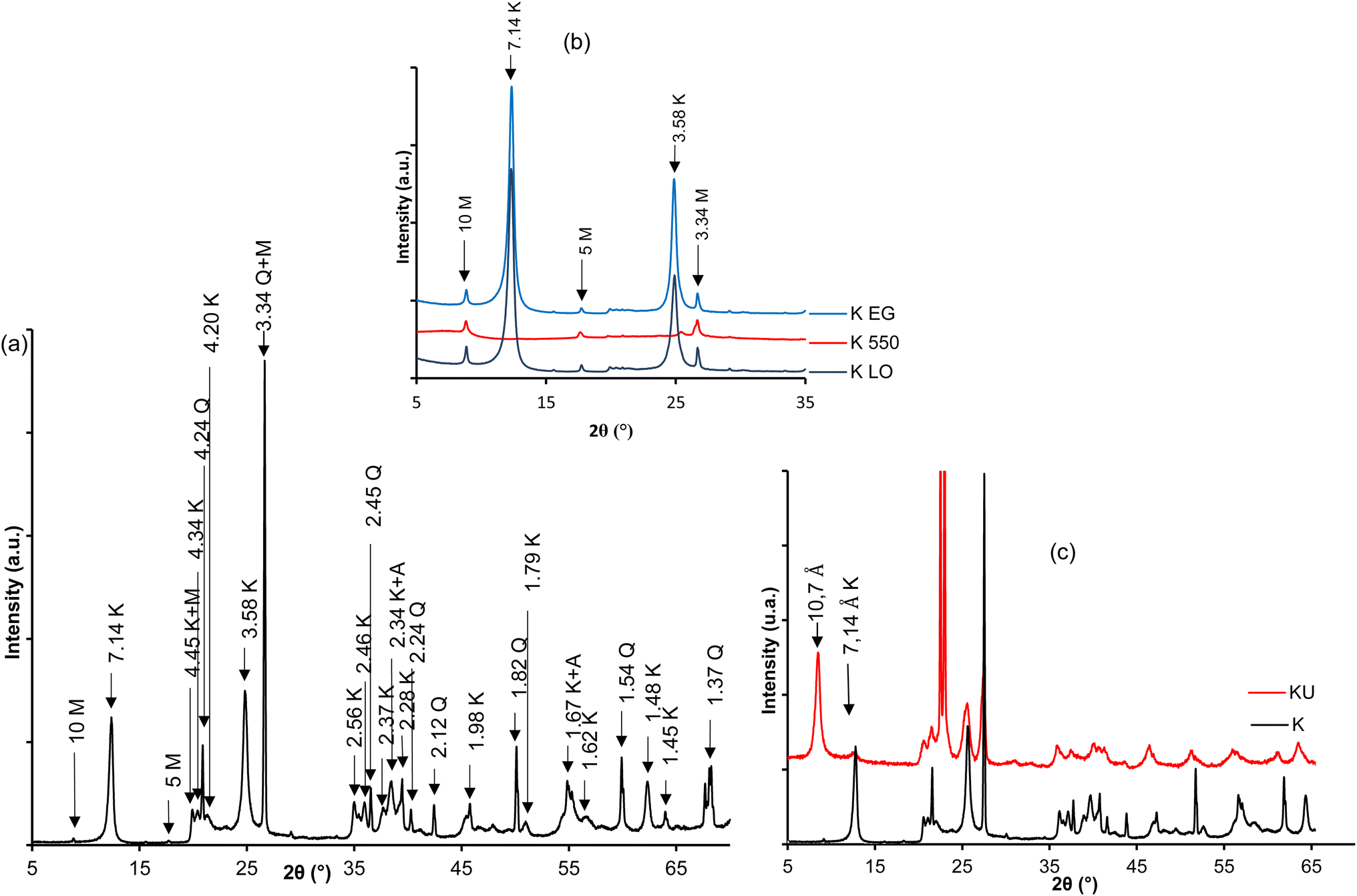 | ||
| Fig. 2 XRD patterns of raw and urea-intercalated kaolinite: (a) bulk sample; (b) oriented, heated at 550 °C and glycolated, and (c) urea-intercalated (KU). | ||
After the intercalation of urea, the reflection of kaolinite shifts to 10.7 Å (Fig. 2c), characteristic of the monolayer insertion of urea molecules in the interlayer space of kaolinite.34 The evaluated intercalation ratio indicates a value of 86%, reflecting a good insertion of urea relative to the low defects on the kaolinite sheets used.28 The low intensities of reflections characteristic of excess urea are due to the appropriate choice of the saturation ratio of the urea aqueous solution.
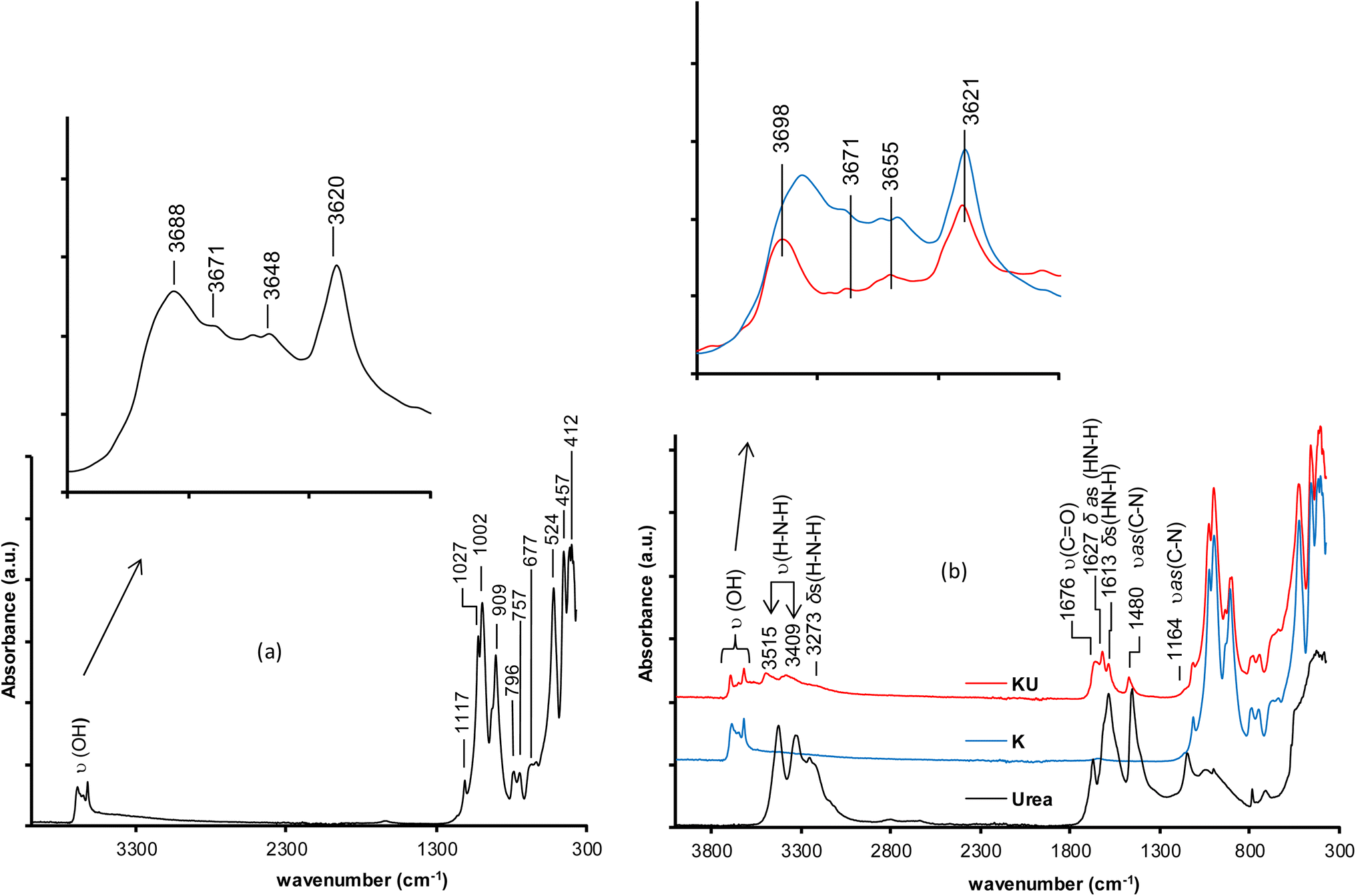 | ||
| Fig. 3 FTIR spectra of (a) raw kaolinite; (b) urea, kaolinite (K) and urea-intercalated kaolinite (KU). ν: Stretching vibrations; υ as: asymmetric stretching vibrations; δ: bending vibrations; δas: asymmetric bending vibrations; δs: symmetric bending vibrations. | ||
The intercalation of urea into kaolinite is highlighted by the appearance of new bands and the displacement of characteristic vibration bands of kaolinite (Fig. 3b). These phenomena indicate the interactions between kaolinite and urea. The disturbances observed on the characteristic vibration bands of kaolinite hydroxyls and the loss in intensity of the stretching vibration bands of C–N bonds at 1480 cm−1 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1676 cm−1 from the urea spectrum indicate the formation of hydrogen bonds between the heteroatoms (nitrogen and eventually oxygen atoms) of urea and the hydrogen atoms (AlO–H) of the octahedral site of kaolinite. The appearance of a bending vibration band at 1627 cm−1 attributable to new N–H bonds reflects the hydrogen bonds formed between urea hydrogen atoms and oxygen atoms (Si–O) of the tetrahedral site of kaolinite.35 The characteristic bands of the stretching vibration of interlayer urea H–N–H bonds appear at 3515 and 3409 cm−1, and the bending vibration is related to the bands at 3273 and 1613 cm−1. At 1164 cm−1, the stretching vibration band of the C–N bond was observed.
O at 1676 cm−1 from the urea spectrum indicate the formation of hydrogen bonds between the heteroatoms (nitrogen and eventually oxygen atoms) of urea and the hydrogen atoms (AlO–H) of the octahedral site of kaolinite. The appearance of a bending vibration band at 1627 cm−1 attributable to new N–H bonds reflects the hydrogen bonds formed between urea hydrogen atoms and oxygen atoms (Si–O) of the tetrahedral site of kaolinite.35 The characteristic bands of the stretching vibration of interlayer urea H–N–H bonds appear at 3515 and 3409 cm−1, and the bending vibration is related to the bands at 3273 and 1613 cm−1. At 1164 cm−1, the stretching vibration band of the C–N bond was observed.
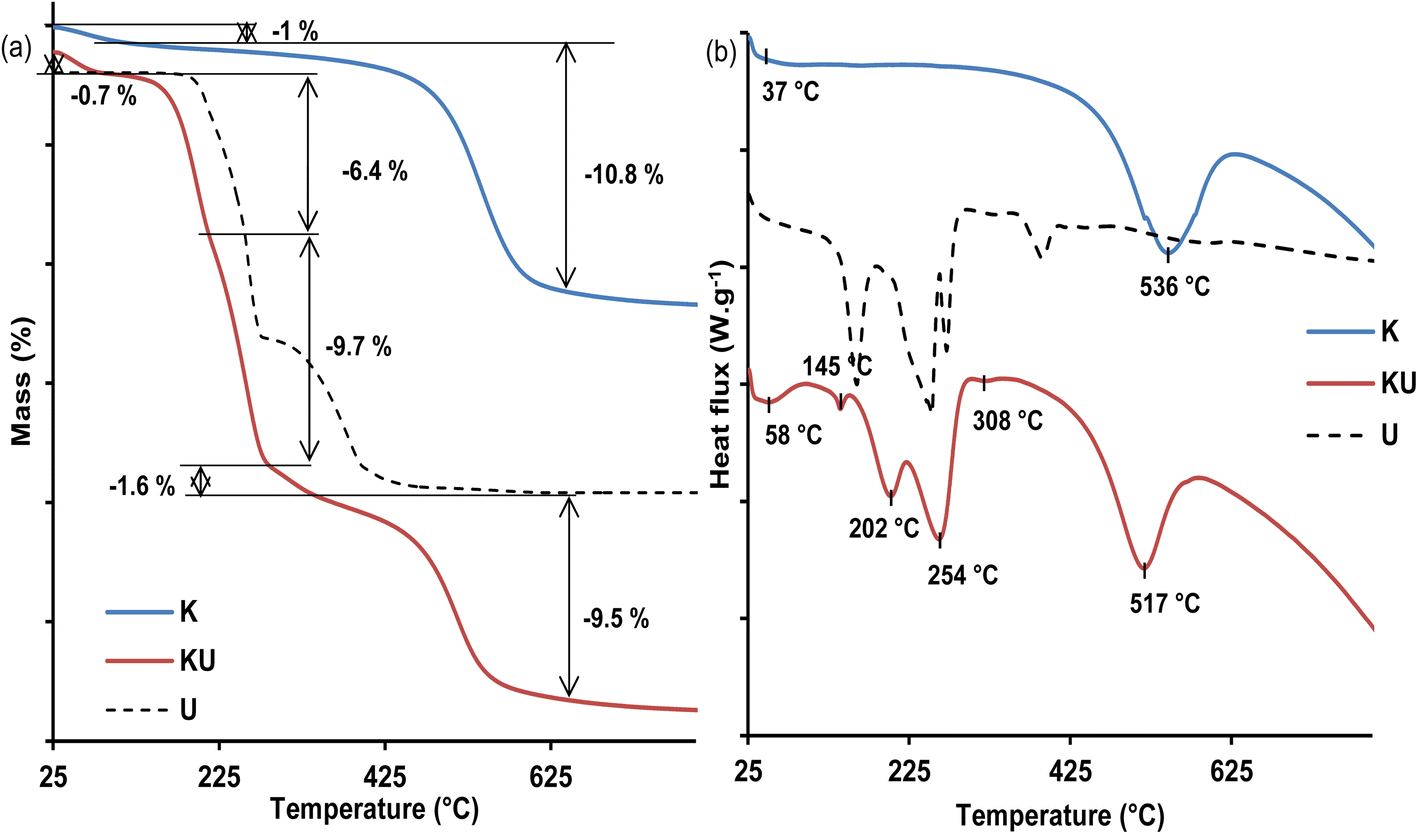 | ||
| Fig. 4 Thermal analyses of urea (U), raw kaolinite (K) and urea-intercalated kaolinite (KU): (a) TGA; (b) DSC. | ||
The thermogram of the urea-intercalated sample (KU) exhibits four additional endothermic events on its DSC curve (Fig. 4b). First, an event at 145 °C is not associated with any mass loss on TGA (Fig. 4a) and is attributed to the melting of non-intercalated urea.33,52 The second endothermic event is located at 202 °C and is associated in the TGA curve (Fig. 4a) to a mass loss of about 6.4%, related to the beginning of decomposition of urea, which continues at the third DSC phenomenon at 254 °C with an additional mass loss of about 9.7%. The starting at 202 °C is dominated by the non-intercalated urea, while at 254 °C, it is mostly dominated by the intercalated urea. The decomposition products are made of biuret, cyanuric acid, ammelide, and melamine,34,52 and their evaporation is indicated by the fourth endothermic event at about 308 °C, resulting in a residual mass loss of about 1.6% in the TGA curve (Fig. 4a). The presence of urea within the interlayer space of kaolinite was further confirmed by the decreased dehydroxylation temperature of kaolinite to 517 °C, with an associated TGA mass loss estimated at 9.5%. In comparison to dehydroxylation loss of 10.8% in the raw kaolinite, it was suggested that kaolinite sheet dehydroxylation starts earlier and is associated with intercalated urea decomposition within the KU complex.
3.2. Characterization of pristine poly(urea–formaldehyde) (UF) and kaolinite–UF intercalated resin: bonding evolution, crystallinity change and thermal analysis
O stretching vibration band of urea to lower wavenumbers at 1629 cm−1 reflects a change in the structure of amide I, likely due to hydrogen substitution in the amino groups by methylene (N-methylolation). The decrease in the intensity of the C–N elongation vibration band at 1441 cm−1 from urea in favor of strong bands at 1548 and 1251 cm−1 characterizing new C–N vibrations in UF indicates the restructuring of amide II (N-methylolation). The intense vibration band of C–O–C and possibly CH2–N–CH2 bonds at 1135 cm−1 and C–C–O bonds at 1031 cm−1 suggest the significant presence of methylene and oxymethylene (ether) linkages. These results well correspond to the stage #114 of the obtained resin, including a great presence of the methylene ether linkage. Various polymerization mechanisms of UF and probable structures of the obtained polymers are clearly outlined in the literature.14,36,38
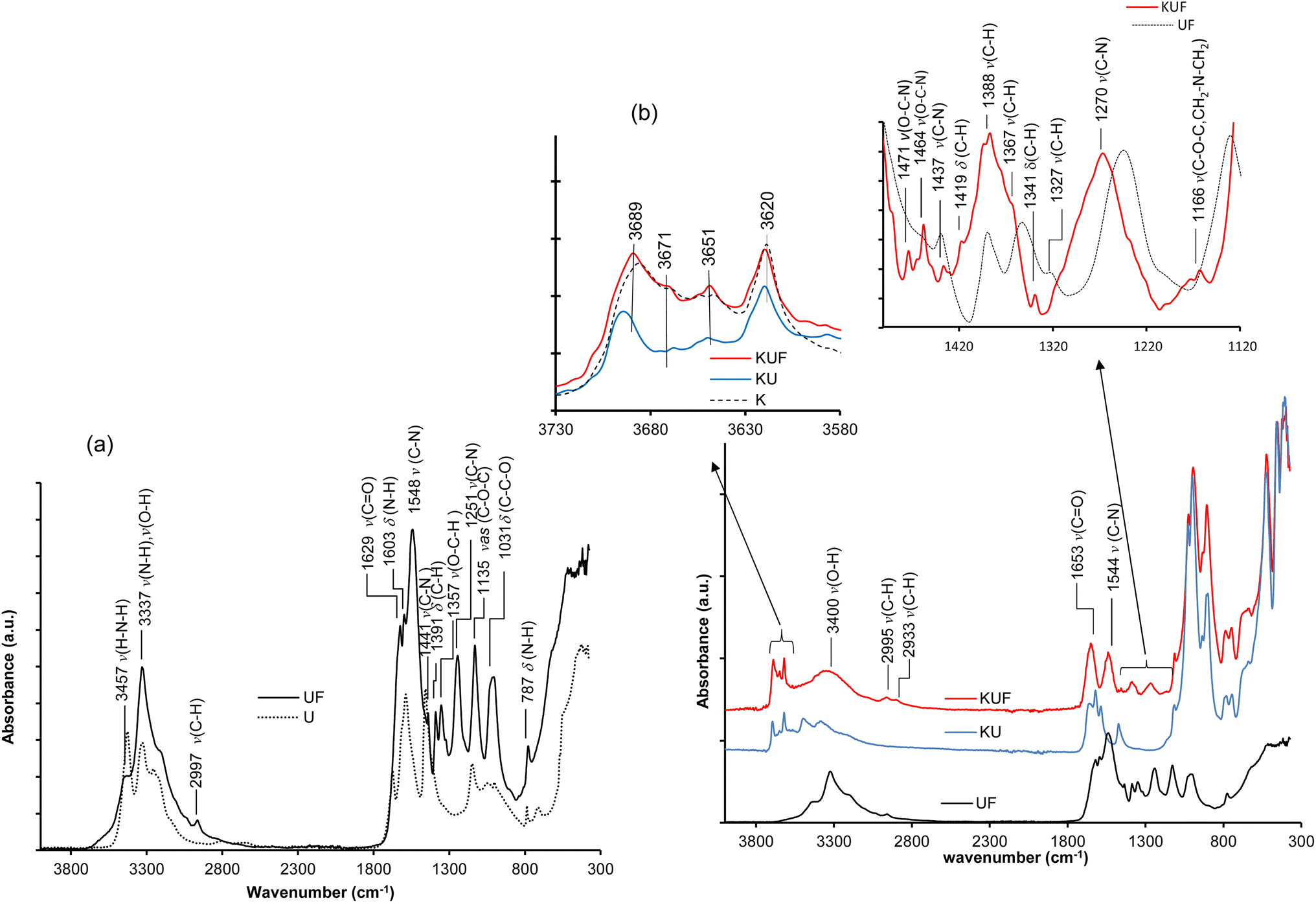 | ||
| Fig. 5 FTIR spectra of (a) free poly(urea–formaldehyde) (UF) and urea (U); (b) urea-intercalated kaolinite (KU), and intercalated kaolinite–poly(urea–formaldehyde) (KUF). v: Stretching vibrations; vas: asymmetric stretching vibrations; δ: bending vibrations. | ||
In kaolinite–UF intercalated resin (KUF), in addition to the characteristic bands of kaolinite bonds, the characteristic of UF bonds with higher wavenumbers are observed in the FTIR spectrum in Fig. 5b. This difference in the vibration wavenumbers of UF within the kaolinite compared to the pristine UF clearly characterizes an in situ position within the interlayer space of kaolinite. Furthermore, the restructuring of the hydroxyl vibration bands of kaolinite back to their initial states indicates that the hydrogen bonds previously formed with urea have been broken. However, the changes observed in the intensities and wavenumbers of these hydroxyl bond bands are due to the new hydrogen bonds they establish with the water molecules formed during polymerization (see section 3.3.3). At about 3400 cm−1, the observed rounded vibration band characterizes the bonds of water formed during polymerization. The vibrational bands of methylene (–CH2) bonds characterizing CH2–N and N–CH2–N groups were observed at about 2995, 1419, 1388, and 1367 cm−1. Those characterizing CH2–O and O–CH2–O groups were observed at about 2633 and 1341 cm−1, respectively. Additionally, the bending vibration band of -C-H in the O–CH2–N group was observed at about 1471 and 1464 cm−1. At 1653 cm−1, the carbonyl (CO) vibration band manifests with a high intensity, indicating its freedom (unbound) in the interlayer space. The intensification of the new C–N bands at 1544 and 1270 cm−1 characterizes the numerous N-methylene cross-links formed during condensation. The absence of characteristic –N–H vibration bands indicates complete amino group methylation during the process. The very low intensity of CH2–O bands indicates the low presence of methylene ether linkages.
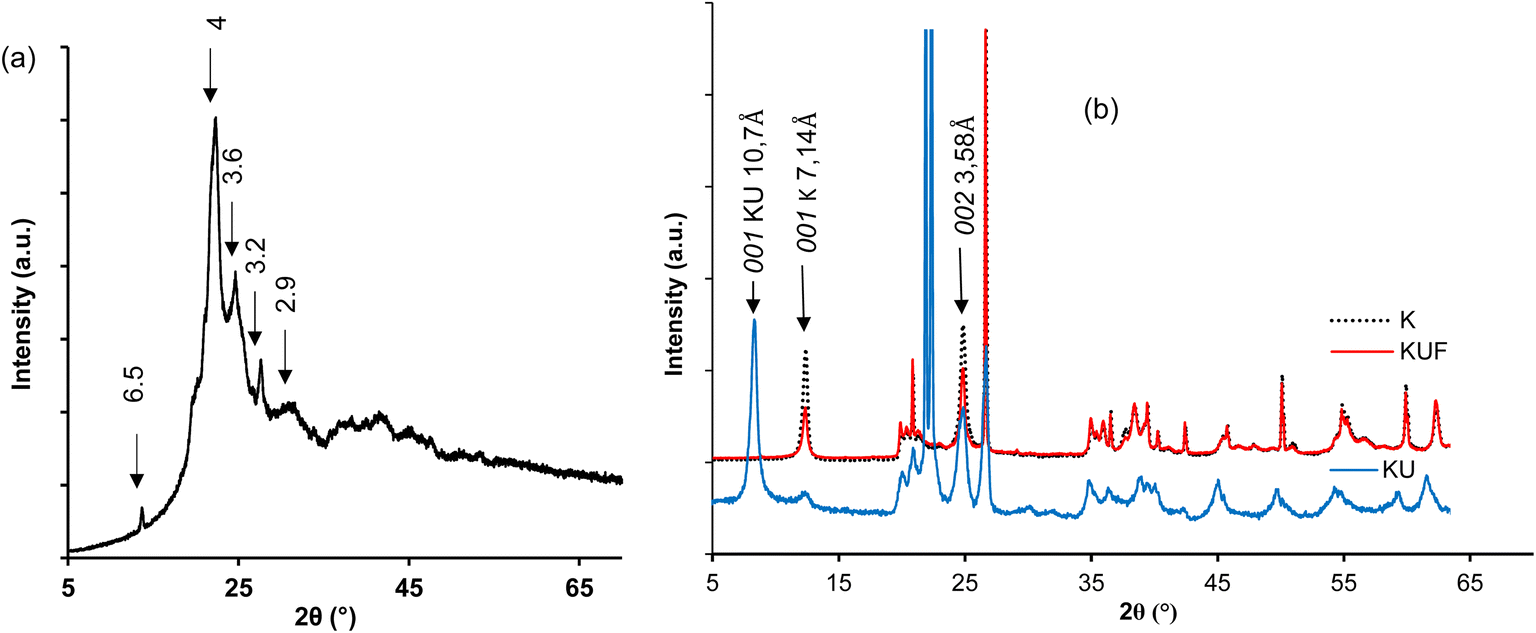 | ||
| Fig. 6 XRD patterns of (a) pristine poly(urea–formaldehyde) (UF); (b) kaolinite (K), urea-intercalated kaolinite (KU) and intercalated kaolinite–poly(urea–formaldehyde) (KUF). | ||
The X-ray diffraction pattern of KUF (Fig. 6b) reveals the complete disappearance of the main reflection of urea-intercalated kaolinite at 10.7 Å. Additionally, the residual main kaolinite reflection at 7.16 Å was low as compared to the one in the raw kaolinite. It was then concluded that polymerization induces the exfoliation of the kaolinite. Consequently, the polymer chains formed effectively dispersed the mineral layers, which lost their periodicity, imparting to the new material a dimension along the  axis that is undetectable by XRD analysis. The residual d001 of kaolinite was probably associated with the part of kaolinite that was not intercalated by urea and the deintercalation domain occurring from the displacement of some urea out of the kaolinite interlayer in the formaldehyde solution. The characteristic reflections of UF are non-observable due to the dominance of the mineral reflection peak. A total absence of excess urea characteristic reflections was also noted, indicating a complete polymerization of urea both in the interlayer and at the surface of the clay sheets.
axis that is undetectable by XRD analysis. The residual d001 of kaolinite was probably associated with the part of kaolinite that was not intercalated by urea and the deintercalation domain occurring from the displacement of some urea out of the kaolinite interlayer in the formaldehyde solution. The characteristic reflections of UF are non-observable due to the dominance of the mineral reflection peak. A total absence of excess urea characteristic reflections was also noted, indicating a complete polymerization of urea both in the interlayer and at the surface of the clay sheets.
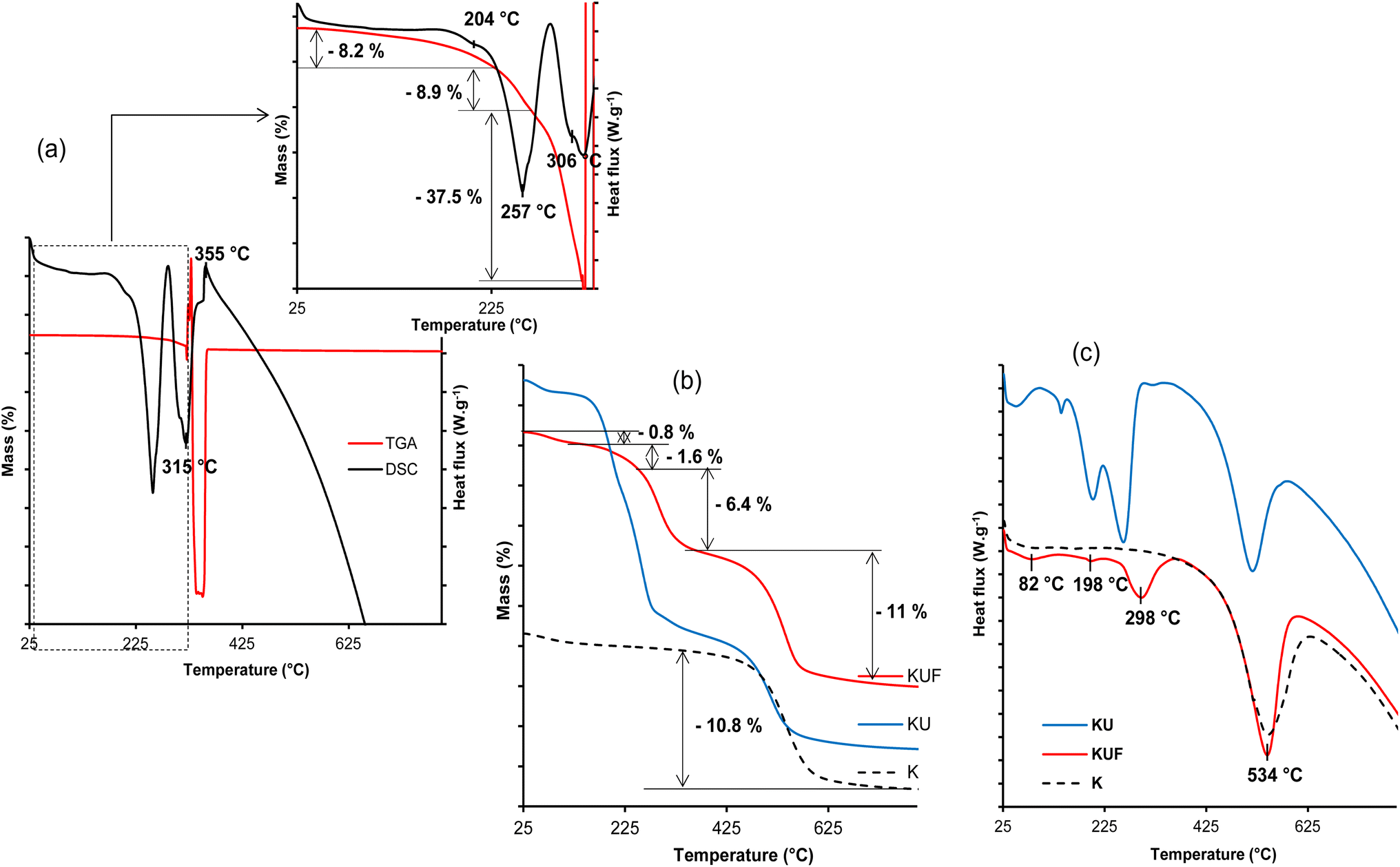 | ||
| Fig. 7 (a) TGA–DSC of pristine poly(urea–formaldehyde); (b) TGA and (c) DSC of kaolinite (K), urea-intercalated kaolinite (KU) and intercalated kaolinite–poly(urea–formaldehyde) (KUF). | ||
Thermal analysis of KUF reveals four (04) events. At 82 °C, a 0.8% mass loss occurs, attributed to the removal of hydration water and potentially excess formaldehyde. The evaporation of interstitial water formed during the condensation process occurs at about 198 °C, accounting for 1.6% of the material. Unlike the pristine poly(urea–formaldehyde), the UF within the kaolinite decomposes in a single step at 298 °C (Fig. 7c) to form low reactive products such as carbon dioxide, water and nitrogen with a 6.4% mass loss (Fig. 7b), indicating its stabilization by interactions with the clay sheets. This suggestion is supported by the higher decomposition temperature compared to the pristine polymer. The UF interactions with the kaolinite layers are also revealed by the dehydroxylation of the clay, which occur at 534 °C in the KUF, whereas in the KU, it occurred at 517 °C. This relatively higher dehydroxylation temperature of kaolinite layers after the formation of the poly(urea–formaldehyde) polymer within the interlayer is associated with an increase in the extent of hydroxyl groups associated with the bonding extension caused by water molecules, produced during the polycondensation step. The urea–formaldehyde polymer contributes to hydroxyl interactions extension, which induce increased cohesion of the kaolinite layer, resulting in a shift (increase) in the dehydroxylation temperature. This hydroxyl interaction extension is supported by the P0 test (Table 1), from which the hydroxyl interaction extension is more prominent in KUF than in KU, justifying a higher dehydroxylation temperature for kaolinite in KUF. The mass loss of 11% associated with this dehydroxylation is coherent with that of the raw kaolinite (10.8%) (Fig. 7b).
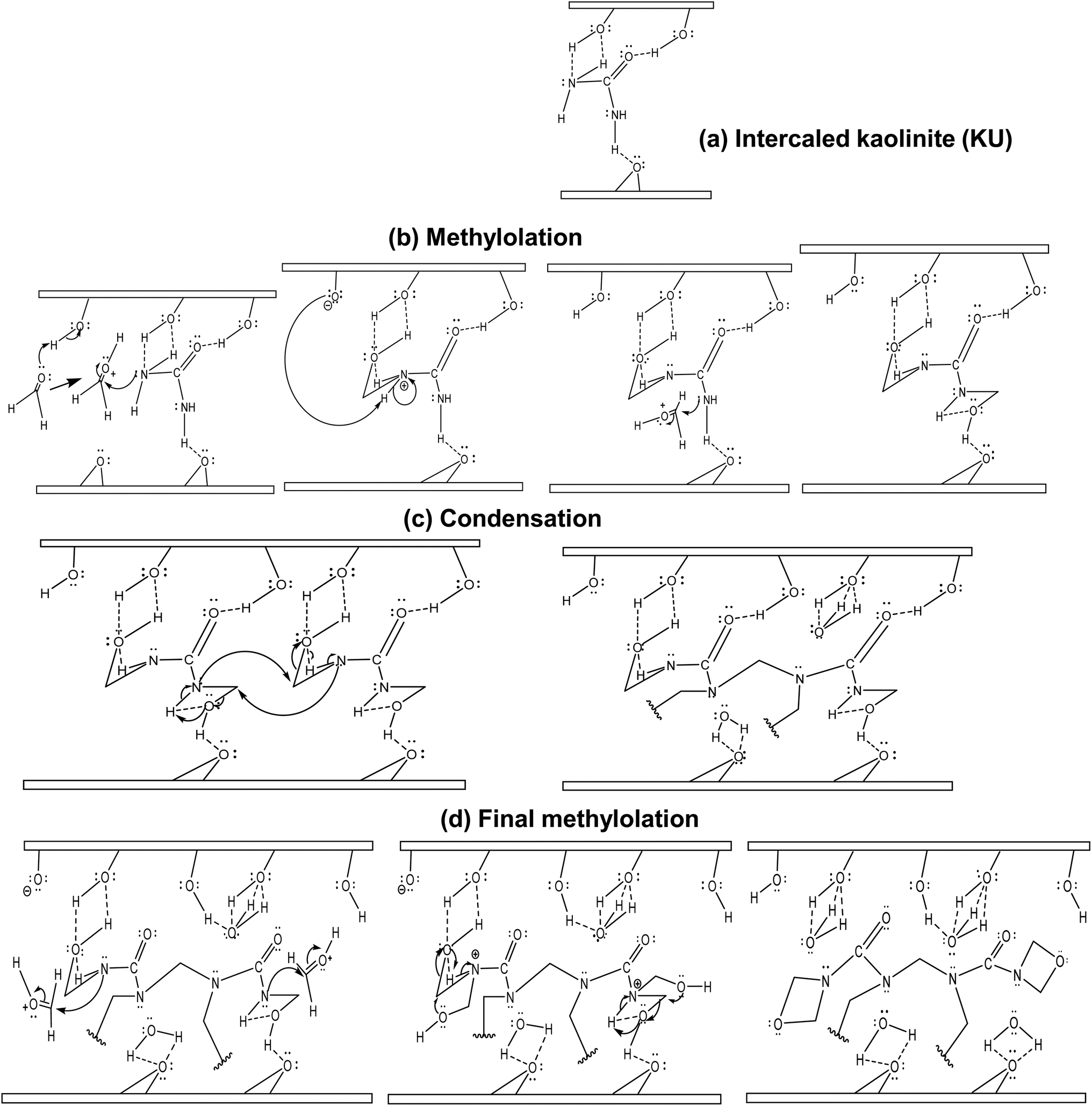 | ||
| Fig. 8 Proposed mechanism for the in situ polymerization of urea–formaldehyde in kaolinite interstitial space: (a) urea-intercalated kaolinite; (b) first methylolation; (c) condensation; (d) final methylolation. | ||
The in situ polymerization process would start with a classical methylolation: firstly, the activation of the carbonyl group of formaldehyde molecules by the surface hydroxyl protons within the interlayer space, followed by the formation of –NH–CH2 bonds by the attack of the carbonyl electrophilic site (Fig. 8b). For condensation, the formed methylols react with adjacent urea–formaldehyde monomer nitrogen atoms, releasing interlayer water molecules that would form stronger hydrogen bonds (Fig. 8c) with octahedral and tetrahedral sites. A final methylation occurs with the formation of terminal 1,3-oxazetidine cycles (Fig. 8d), releasing more interstitial water molecules. This indicates that UF within the interlayer space is only bound to kaolinite by low van der Waals' interactions. Furthermore, monitoring interstitial cohesion using the P0 test indicates values of 1.24 for raw kaolinite, 0.74 for intercalated kaolinite and 0.97 for the nanocomposite (Table 1). This suggests that the strong cohesion in raw kaolinite has been disrupted by urea intercalation and that a more symmetrical repartition of internal and external AlO–H is observed after polymerization. This symmetrical repartition indicates the exfoliated structure of the layer after polymerization. Monitoring the d-spacing using the Bragg equation reveals an increase to 3.56 Å after urea intercalation. Subsequently, a loss of periodicity, indicating an expansion of the interlayer space beyond the detectable range of XRD (>44.1 Å), occurs after polymerization (Table 1) relative to the raw kaolinite. These structural changes may account for the increase in the pore openings shown in the pore spectra (see section 3.1.1), which account for progressive exfoliation/delamination following the different stages of the process. Additionally, evaluating the number of layers per crystallite (NL) provides an initial value of 24 for the raw mineral, then 20 for the urea-intercalated kaolinite, and less than 5 for the clay–polymer composite, indicating an exfoliated structure for the kaolinite in the obtained nanocomposite.
3.3. Water resistance of the UF-intercalated kaolinite compared to pristine UF
Upon immersion of pristine UF and the nanocomposite in water, variations are observed in terms of masses and adsorption bands from FTIR, as depicted in Fig. 9. The percentages of the remaining UF after immersion were obtained using eqn (1) and (2) for the pristine UF and the composite, respectively. The remaining mass of the UF after immersion in water shows a decrease in the amount of UF with increasing immersion time (Fig. 9c). Using the FTIR bands for the UF within the composite also shows a decrease in the UF up to twenty minutes of immersion; thereafter, the UF seems to recombine within the kaolinite layers. This decrease in both UF and the KUF of the polymer is attributed to its degradation through the cleavage of ether linkages and the opening of 1,3-oxazetidine rings. The actual rate of decrease seems to be higher for pristine UF due to its direct exposure to water molecules, while when associated with kaolinite, the mineral layer seems to play a barrier role that limits the degradation of UF in the composite material. The decrease observed in the composite is further confirmed by the intensity of O–H stretching associated with the free water molecule bands at 3400 cm−1 (Fig. 9a). This significant decrease in intensity is believed to result from the opening of the 1,3-oxazetidine ring, leading to the formation of methylol groups, thus enabling the modification of H-bonding in the kaolinite interlayer due to the involvement of methylol groups. This suggestion is confirmed by the changes in the vibrational bands of the surface hydroxyls in the kaolinite interlayers (Fig. 9b).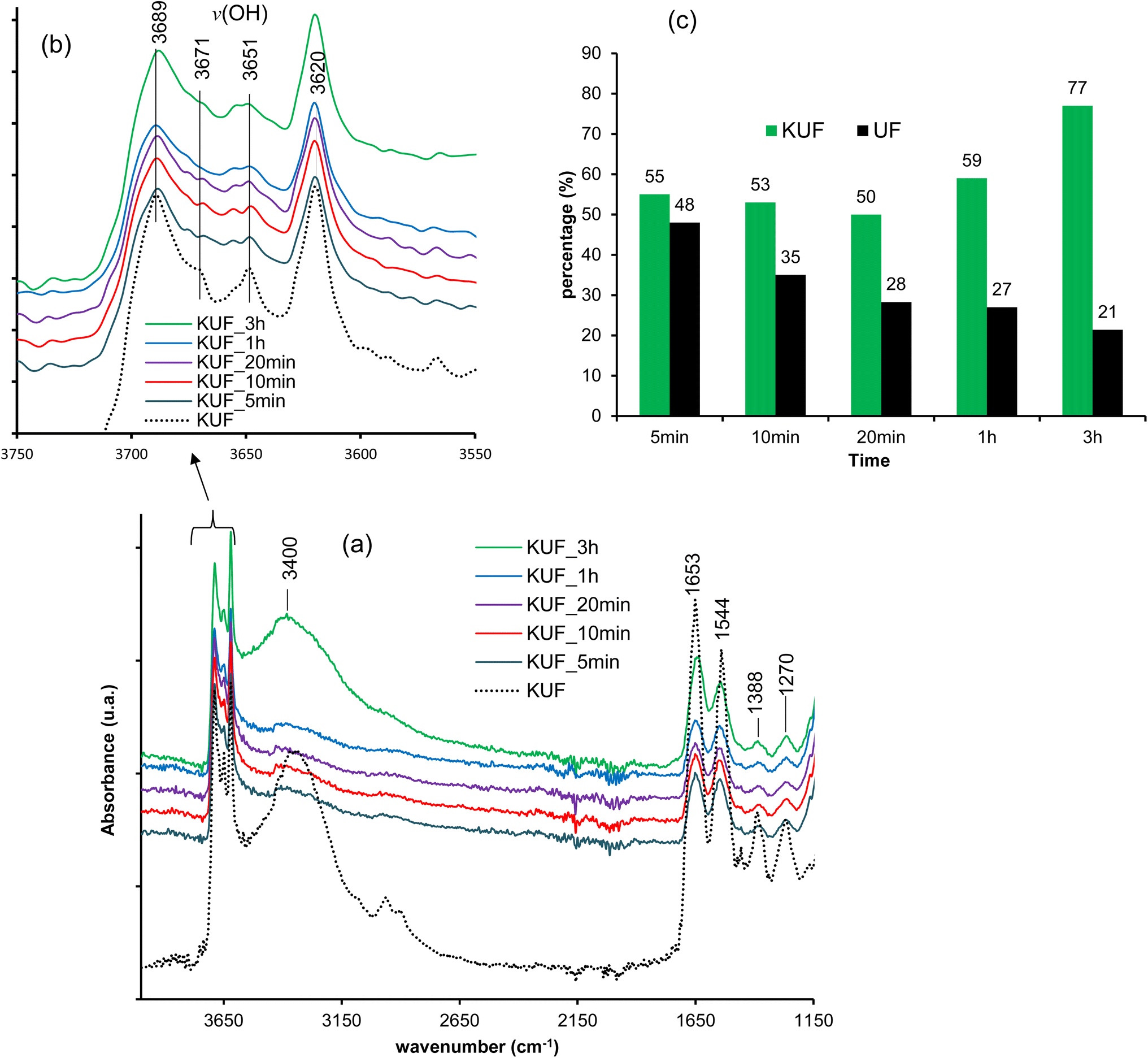 | ||
| Fig. 9 Characterization of UF-intercalated kaolinite and pristine UF after various immersion times in water: (a) FTIR spectra; (b) hydroxyl vibrational bands domain and (c) percentages of UF in the composite and percentages of pristine UF after different immersion times. | ||
The re-increase in the amount of UF in the composite, as observed from 1 hour of immersion, suggests that the degradation induced by water leads to fragments that remain trapped by the mineral layers and, these fragments recombine to regenerate the polymer, indicating the self-healing capacity of the nanocomposite. The same observation is made after 3 hours of immersion. This regeneration of the UF within the KUF from 1 hour and beyond is also supported by the increase in the stretching band of O–H from the free water molecule at 3400 cm−1 from 1 hour, which is more obviously visible after 3 hours. It is also evident from the kaolinite interlayer O–H stretching, which is almost similar after 3 hours to those of the pristine KUF (Fig. 9b).
4. Conclusion
In this study, the kaolinite–poly(urea–formaldehyde) (KUF) composite was synthesized for the first time through the interlayer polymerization of urea-intercalated within the kaolinite. The evidence of this polymerization was accessed using XRD, FTIR, thermal analysis and the water resistance tests. The polymerization mechanism of urea–formaldehyde within the kaolinite interlayer was also discussed.The analysis of the kaolinite structural changes, from urea intercalation to polymerization, initially indicated the appearance of characteristic vibrational bands of intercalated urea molecules and the hydrogen bonds they formed within the kaolinite, as observed via FTIR spectroscopy. This was further confirmed by X-ray diffraction, thermal analyses (TGA/DSC), and nitrogen adsorption, showing shifts in the main reflection of kaolinite from 7.14 Å to 10.7 Å, an increase in the decomposition temperature of the intercalated urea, while the kaolinite dehydroxylation temperature decreases. B.E.T analysis from nitrogen adsorption reveal a widening of the average pore openings. The urea intercalation ratio was found to be 86% from the XRD patterns. Subsequently, results from FTIR and thermal analyses indicated the appearance of bands and thermal events characteristic of crosslinked poly(urea–formaldehyde) in the composite material. This presence within the kaolinite interlayer space was confirmed by the disappearance of the reflection of urea-intercalated kaolinite and the enhancement of UF thermal behavior. The kaolinite in the obtained material was mostly exfoliated, as indicated by the low intensity of the characteristic reflection of kaolinite at 7.14 Å. This domain was associated with urea displacement from the kaolinite–urea complex when immersed in formaldehyde. The exfoliated structure was also confirmed by the evaluation of the number of layers per crystallite. As the characteristic peak of urea was not observed on the kaolinite–poly(urea–formaldehyde) composite, it was concluded, regarding the intercalation ratio, that polymerization largely occurs within the interlayer space.
The analysis of these results led to a proposed polymerization mechanism of urea–formaldehyde in kaolinite, which consists of a hydroxymethylation step of urea by formaldehyde, followed by a polycondensation step, predominantly forming methylene linkages and a final methylolation step, leading to the formation of 1,3-oxazetidine cycles. In the first step, the stabilization of the reaction is enhanced by H-bonding with the mineral in the second stage; the released water molecules from polycondensation contribute to enhanced mineral–organic H-bonding. The released water from the second to the third step generated van der Waals interactions that ensure bridging between the formed polymer chains and the mineral surface, indicating that poly(urea–formaldehyde) in the interlayer space interacts with the kaolinite surface and improve the extension of hydroxyl bonding. Such a configuration is of interest in controlled release or adsorption processes.
The water resistance test on the obtained composite shows a moderate decomposition of poly(urea–formaldehyde) and the retention of the decomposition fragments by the mineral layers, which allow the self-healing behavior of the composite after 1 hour of immersion. The synthesized poly(urea–formaldehyde) within the kaolinite interlayer space is more water-resistant than the conventionally synthesized urea–formaldehyde resins.
Data availability
All the data are included in the manuscript and source files are available upon request to the corresponding author.Author contributions
Tatang Hervé Barye: conceptualization, methodology, validation, investigation, data curation, formal analysis, visualization, writing – original draft, writing – review & editing. Mbey Jean Aimé: conceptualization, methodology, validation, resources, investigation, data curation, formal analysis, visualization, project administration, supervision, writing – review & editing. MACHE Jacques Richard: methodology, resources, investigation, visualization, writing – review & editing. Ngally Sabouang C. J.: methodology, resources, visualization, writing – review & editing. Angelina Razafitianamaharavo: investigation, resources, data curation Renaud GLEY: investigation, resources. KONG Sakeo: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially carried out at the Pôle de Compétences Physico-Chimie et Environnement, LIEC Laboratory UMR 7360 CNRS-Université de Lorraine. The Cameroonian Minister of Higher Education is acknowledged for the special research allowance to the research staff in the state Universities. The authors are thankful to Dr Isabelle BIHANNIC, for access facilitation to the Pôle de Compétences Physico-Chimie et Environnement. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- B.-D. Park, E.-C. Kang and J. Y. Park, Differential Scanning Calorimetry of Urea–Formaldehyde Adhesive Resins, Synthesized under Different pH Conditions, J. Appl. Polym. Sci., 2006, 100, 422–427 CrossRef CAS.
- M. B. Kashani, A. Salimi, M. J. Zohuriaan-Mehr and A. Hanifpour, Preparation of poly (urea-formaldehyde) microcapsules for use in capsular adhesive, J. Polym. Res., 2019, 26, 270 CrossRef.
- R. Ghafari, K. DoostHosseini, A. Abdulkhani and S. A. Mirshokraie, Replacing formaldehyde by furfural in urea formaldehyde resin: effect on formaldehyde emission and physical–mechanical properties of particleboards, Eur. J. Wood Wood Prod., 2016, 74, 609–616 CrossRef CAS.
- E. Roumeli, E. Papadopoulou, E. Pavlidou, G. Vourlias, D. Bikiaris, K. M. Paraskevopoulos and K. Chrissafis, Synthesis, characterization and thermal analysis of urea–formaldehyde/nanoSiO2 resins, Thermochim. Acta, 2012, 527, 33–39 CrossRef CAS.
- T. Zorba, E. Papadopoulou, A. Hatjiissaak, K. M. Paraskevopoulos and K. Chrissafis, Urea-formaldehyde Resins Characterization by Thermal Analysis and FTIR method, J. Therm. Anal. Calorim., 2008, 92, 29–33 CrossRef CAS.
- Y. Guo, Y. Shi, Q. Cui, X. Zai, S. Zhang, H. Lu and G. Feng, Synthesis of Urea-Formaldehyde Fertilizers and Analysis of Factors Affecting These Processes, processes, 2023, 11, 3251 CrossRef CAS.
- P. Zhang, P. Huang, H. Sun, J. Ma and B. Li, The structure of agricultural microplastics (PT, PU and UF) and their sorption capacities for PAHs and PHE derivates under various salinity and oxidation treatments, Environ. Pollut., 2020, 257, 113525 CrossRef CAS PubMed.
- J. Kothari and J. O. Iroh, Self-Healing Poly(urea formaldehyde) Microcapsules: Synthesis and Characterization, polymers, 2023, 15, 1668 CrossRef CAS PubMed.
- G. Li, Y. Feng, P. Gao and X. Li, Preparation of Mono-Dispersed Polyurea-Urea Formaldehyde Double Layered Microcapsules, Polym. Bull., 2008, 60, 725–731 CrossRef CAS.
- J. Li, S. Wang, H. Liu, N. Liu and L. You, Preparation and Application of Poly(melamine-ureaformaldehyde) Microcapsules Filled with Sulfur, Polym.-Plast. Technol. Eng., 2011, 50, 689–697 CrossRef CAS.
- J. Li, S. Yang, Y. Muhammad, M. Sahibzada, Z. Zhu, T. Liu and S. Liao, Fabrication and application of polyurea formaldehyde-bioasphalt microcapsules as a secondary modifier for the preparation of high selfhealing rate SBS modified asphalt, Constr. Build. Mater., 2020, 246, 118452 CrossRef CAS.
- C. Li, M. Wang, Z. Liu, Y. Xu, C. Zhou and L. Wang, Kaolinite-armoured polyurea microcapsules fabricated on Pickering emulsion: controllable encapsulation and release performance of a lipophilic compound, Clay Miner., 2021, 56, 46–54 CrossRef CAS.
- International Agency for Research on Cancer (IARC), Formaldehyde, 2-butoxyethanol, and 1-tert-butoxy-2-propanol, IARC monographs on the evaluation of carcinogenic risks to humans, IARC, Lyon, France, 2006, vol. 88 Search PubMed.
- E. S. Wibowo, B.-D. Park and V. Causin, Hydrogen-Bond-Induced Crystallization in Low-Molar-Ratio Urea−Formaldehyde Resins during Synthesis, Ind. Eng. Chem. Res., 2020, 59, 13095–13104 CrossRef CAS.
- A. Pizzi and K. Mittal, Handbook of Adhesive Technology, Revised and Expanded, CRC Press, 2003 Search PubMed.
- M. Chiavarini, R. Bigatto and N. Conti, Synthesis of Urea-Formaldehyde Resins: NMR Studies on Reaction Mechanisms, Die Angewandte Makromolekulare Chemie, 1978, 70, 49–58 CrossRef CAS.
- G. E. Myers, How mole ratio of UF resin affects formaldehyde emission and other properties: A literature critique, For. Prod. J., 1984, 34, 35–41 CAS.
- E. S. Wibowo and B.-D. Park, Cure kinetics of low-molar-ratio urea-formaldehyde resins reinforced with modified nanoclay using different kinetic analysis methods, Thermochim. Acta, 2020, 686, 178552 CrossRef CAS.
- Z. Wu, X. Xi, L. Yu, L. Su, H. Lei, G. Du and Z. Yin, An eco-friendly urea-formaldehyde resin: preparation structure and properties, Wood Res., 2018, 63, 45–56 CAS.
- Y. Ming, J. Hu, J. Xing, M. Wu and J. Qu, Preparation of polyurea/melamine formaldehyde double-layered self-healing microcapsules and investigation on core fraction, J. Microencapsulation, 2016, 33, 307–314 CrossRef CAS PubMed.
- B. Pang, M.-K. Li, S. Yang, T.-Q. Yuan, G.-B. Du and R.-C. Sun, Eco-Friendly Phenol−Urea−Formaldehyde Co-condensed Resin Adhesives Accelerated by Resorcinol for Plywood Manufacturing, ACS Omega, 2018, 3, 8521–8528 CrossRef CAS PubMed.
- H. Yuan, G. Li, L. Yang, X. Yan and D. Yang, Development of melamine–formaldehyde resin microcapsules with low formaldehyde emission suited for seed treatment, Colloids Surf., B, 2015, 128, 149–154 CrossRef CAS PubMed.
- H. Lei, G. Du, A. Pizzi and A. Celzard, Influence of Nanoclay on Urea-Formaldehyde Resins for Wood Adhesives and Its Model, J. Appl. Polym. Sci., 2008, 109, 2442–2451 CrossRef CAS.
- S. Chen, X. Lu, T. Wang and Z. Zhang, Preparation and characterization of urea-formaldehyde resin/reactivekaolinite composites, Particuology, 2016, 24, 203–209 CrossRef CAS.
- J. Song, S. Chen, X. Yi, X. Zhao, J. Zhang, X. Liu and B. Liu, Preparation and Properties of the Urea-Formaldehyde Res-In/Reactive Halloysite Nanocomposites Adhesive with Low-Formaldehyde Emission and Good Water Resistance, polymers, 2021, 13, 2224 CrossRef CAS PubMed.
- J. A. Mbey, F. Thomas, C. J. N. Sabouang, Liboum and D. Njopwouo, An insight on the weakening of the interlayer bonds in a Cameroonian kaolinite through DMSO intercalation, Appl. Clay Sci., 2013, 83–84, 327–335 CrossRef CAS.
- C. Detellier and S. Letaief, Kaolinite–Polymer Nanocomposites in Developments in Clay Science, Elsevier, 2013, vol. 5, pp. 707–719 Search PubMed.
- H. B. Tatang, J. A. Mbey, C. J. N. Sabouang, J. R. Mache, R. Gley and S. Kong, Kaolinites structural defects related to urea and dimethyl sulfoxide intercalation, Appl. Clay Sci., 2024, 255, 107415 CrossRef CAS.
- J. A. Mbey, S. Hoppe and F. Thomas, Cassava starch–kaolinite composite film. Effect of clay content and clay modification on film properties, Carbohydr. Polym., 2012, 88, 213–222 CrossRef CAS.
- J. A. Mbey, J. M. Siéwé, C. J. N. Sabouang, A. Razafitianamaharavo, S. Kong and F. Thomas, DMSO Intercalation in Selected Kaolinites: Influence of the Crystallinity, chemengineering, 2020, 4, 66 CrossRef CAS.
- M.-S. Y. Elhadj and F. X. Perrin, Influencing parameters of mechanochemical intercalation of kaolinite with urea, Appl. Clay Sci., 2021, 213, 106250 CrossRef CAS.
- É. Makó, A. Kovács and T. Kristóf, Influencing parameters of direct homogenization intercalation of kaolinite with urea, dimethyl sulfoxide, formamide, and N-methylformamide, Appl. Clay Sci., 2019, 182, 105287 CrossRef.
- É. Makó, J. Kristóf, E. Horváth and V. Vágvölgyi, Kaolinite–urea complexes obtained by mechanochemical and aqueous suspension techniques—A comparative study, J. Colloid Interface Sci., 2009, 330, 367–373 CrossRef PubMed.
- S. Zhang, Q. Liu, F. Gao, X. Li, C. Liu, H. Li, S. A. Boyd, C. T. Johnston and B. J. Teppen, Mechanism Associated with Kaolinite Intercalation with Urea: Combination of Infrared Spectroscopy and Molecular Dynamics Simulation Studies, J. Phys. Chem. C, 2017, 121, 402–409 CrossRef CAS PubMed.
- R. L. Frost, J. Kristof, L. Rintoul and J. T. Kloprogge, Raman spectroscopy of urea and urea-intercalated kaolinites at 77 K, Spectrochim. Acta, Part A, 2000, 56, 1681–1691 CrossRef PubMed.
- B. Arab and A. Shokuhfar, Molecular dynamics simulation of cross-linked urea-formaldehyde polymers for self-healing nanocomposites: prediction of mechanical properties and glass transition temperature, J. Mol. Model., 2013, 19, 5053–5062 CrossRef CAS PubMed.
- C. Gonçalves, J. Pereira, N. Paiva, J. Ferra, J. Martins, F. Magalhães, A. Barros-Timmons and L. Carvalho, Impact of the Synthesis Procedure on Urea-Formaldehyde Resins Prepared by Alkaline−Acid Process, Ind. Eng. Chem. Res., 2019, 58, 5665–5676 CrossRef.
- Q.-N. Sun, C.-Y. Hse and T. F. Shupe, Effect of Different Catalysts on Urea–Formaldehyde Resin Synthesis, J. Appl. Polym. Sci., 2014, 131, 40644 CrossRef.
- J. C. M. Neto, N. R. do Nascimento, R. H. Bello, L. A. de Verçosa, J. E. Neto, J. C. M. da Costa and F. R. V. Diaz, Kaolinite Review: Intercalation and Production of Polymer Nanocomposites, Eng. Sci., 2022, 17, 28–44 Search PubMed.
- J. C. M. Neto, S. P. R. Kimura, M. G. Adeodato, J. E. Neto, N. R. do Nascimento and L. M. F. Lona, Intercalation and Exfoliation Mechanism of Kaolinite During the Emulsion Polymerization, Chem. Eng. Trans., 2017, 57, 1453–1458 Search PubMed.
- S. Letaief, J. Leclercq, Y. Liu and C. Detellier, Single Kaolinite Nanometer Layers Prepared by an In Situ Polymerization-Exfoliation Process in the Presence of Ionic Liquids, Langmuir, 2011, 27, 15248–15254 CrossRef CAS PubMed.
- J. A. Mbey, F. Thomas, A. Razafitianamaharavo, C. Caillet and F. Villiéras, A comparative study of some kaolinites surface properties, Appl. Clay Sci., 2019, 172, 135–145 CrossRef CAS.
- J. M. Ferra, P. C. Mena, J. Martins, A. M. Mendes, M. R. N. Costa, F. D. Magalhães and L. H. Carvalho, Optimization of the Synthesis of Urea-Formaldehyde Resins using Response Surface Methodology, J. Adhes. Sci. Technol., 2010, 24, 1454–1471 CrossRef.
- P. Didier, D. Perret, Y. Tardy and D. Nahon, Equilibres entre kaolinites ferrifères, goethltes alumineuses et hématites alumineuses dans les systèmes cuirassés. Rôle de l’activité de l’eau et de la taille des pores/Equilibria between Fekaolinites, Al-goethites and Al-hematltes in ferricretes. Part of water activity and pore size, Sci. Geol., Bull., 1985, 38, 383–397 CrossRef CAS.
- G. Jura and W. D. Harkins, Surfaces of Solids. XI. Determination of the Decrease (T) of Free Surface Energy of a Solid by an Adsorbed Film, J. Am. Chem. Soc., 1944, 66, 1356–1362 CrossRef CAS.
- I. Siminiceanu, I. Lazau, Z. Ecsedi, L. Lupa and C. Burciag, Textural Characterization of a New Iron-Based Ammonia Synthesis Catalyst, Chem. Bull. ‘POLITEHNICA’ Univ. (Timişoara), 2008, 53(67), 38–44 Search PubMed.
- J.-M. Cases, L. Olivier, Y. Jacques and D. jean-François, Etudes des propriétés cristallochimiques, morphologiques, superficielles de kaolinites désordonnées, Bull. Mineral., 1982, 105, 5 Search PubMed.
- T. W. Parker, A classification of kaolinites by infrared spectroscopy, Clay Miner., 1969, 8, 135 CrossRef CAS.
- A. L. Patterson, The Scherrer Formula for X-Ray Particle Size Determination, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- J. C. P. Broeckhoff and W. P. Van Beek, Scanning studies on capillary condensation and evaporation of nitrogen, J. Chem. Soc., Faraday Trans., 1979, 1(75), 42–55 RSC.
- P. G. Rouxhet, N. Samudacheata and H. J. O. Anton, Attribution of the OH Stretching Bands of Kaolinite, Clay Miner., 1977, 12, 171 CrossRef CAS.
- Q. Liu, S. Zhang, H. Cheng, D. Wang, X. Li, X. Hou and R. L. Frost, Thermal behavior of kaolinite–urea intercalation complex and molecular dynamics simulation for urea molecule orientation, J. Therm. Anal. Calorim., 2014, 117, 189–196 CrossRef CAS.
- M. A. Arshad, A. Maaroufi, G. Pinto, S. EL-Barkany and A. Elidrissi, Morphology, thermal stability and thermal degradation kinetics of cellulose-modified urea–formaldehyde resin, Bull. Mater. Sci., 2016, 39, 1609–1618 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |