
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple redox model of low-T NO + CO adsorption onto Pd-CHA as effective passive NOx adsorbers†
Umberto
Iacobone
 a,
Andrea
Gjetja
a,
Nicola
Usberti
a,
Isabella
Nova
a,
Enrico
Tronconi
*a,
Djamela
Bounechada
b,
Roberta
Villamaina
b,
Maria Pia
Ruggeri
b,
Andrew P. E.
York
b,
Loredana
Mantarosie
b and
Jillian
Collier
b
a,
Andrea
Gjetja
a,
Nicola
Usberti
a,
Isabella
Nova
a,
Enrico
Tronconi
*a,
Djamela
Bounechada
b,
Roberta
Villamaina
b,
Maria Pia
Ruggeri
b,
Andrew P. E.
York
b,
Loredana
Mantarosie
b and
Jillian
Collier
b
aLaboratory of Catalysis and Catalytic Processes, Dipartimento di Energia, Politecnico di Milano - Via La Masa, 34, 20156 Milano, Italy. E-mail: enrico.tronconi@polimi.it; Tel: +39 02 23993264
bJohnson Matthey Technology Centre - Blounts Court Road, Sonning Common, Reading RG4 9NH, UK
First published on 18th November 2024
Abstract
Pd-exchanged chabazite (Pd-CHA) catalysts show NO adsorption and desorption features which comply well with the requirements for low-T passive NOx adsorber (PNA) applications. An earlier work based on transient adsorption tests investigated the NO storage pathway on Pd-CHA, a still debated topic in the literature. Such research highlighted a Pd-redox mechanism (Pd2+ ↔ Pd+) underlying the NO storage chemistry over these systems. CO and NO were capable of reducing Pd2+ at low temperatures, and the newly formed Pd+ acted as the main NO storage site. Increasing temperatures activated a Pd-oxidation process, which reduced the fraction of Pd+ sites, and consequently the NO storage, but was inhibited by H2O. Herein we challenge quantitatively such a scheme relying on transient kinetic analysis. We show that a simple redox kinetic model of NO + CO storage on Pd-CHA, based on the above, reproduces the main features of the species evolution and of the NO storage observed under variable operating conditions over Pd-CHA samples with two Pd-loadings, thus lending support to the proposed Pd-redox chemistry.
1. Introduction
The mitigation of NOx emissions from diesel-fuelled vehicles under cold-start conditions is still a prominent challenge in the environmental sector. The progressive enhancements of modern internal combustion engines have increased their fuel-efficiency, however lowering the exhaust gas temperatures. This aspect has worsened the deNOx performance of the current state-of-the-art aftertreatment technologies, such as lean NOx traps (LNT) and NH3-selective catalytic reduction (NH3-SCR) since their respective catalysts are characterized by light-off temperatures typically above 200 °C.1–4 Thus, extended warm-up phases are required by the catalytic converters to reach an appropriate thermal regime for pollutant abatement. This presents the cold-start period as the main source of NOx during vehicular operation.5 To cope with the increasing tightening of environmental regulations, a promising option is the adoption of systems such as passive NOx adsorbers (PNAs), upstream with respect to the main catalytic converter. These systems are designed to trap the low-temperature NOx (low-T, <200 °C), in particular NO, for later thermal release once the downstream reduction catalysts become active.6–12 Among several catalysts tested as potential PNA materials, Pd-exchanged small-pore zeolites (Pd-CHA) have emerged as the catalyst of choice. The remarkable capacity to adsorb low-T NOx, mainly as NO, an appropriate desorption temperature window, the resistance to sulphur and hydrocarbon poisoning, and the good hydrothermal stability make Pd-CHA an ideal material for these applications in comparison to other formulations (e.g., ceria/alumina-supported Pd/Pt, Pd-BEA).9,10,13–18 Nevertheless, despite extensive investigation, no consensus has been reached yet on the NOx adsorption/desorption pathways for such systems and the nature of the associated active sites.Several works have reported the existence of a variety of Pd sites for NO adsorption, in the form of clusters, nanoparticles, and metal-exchanged sites, supported by experimental and theoretical findings.6,7,9,13,14,19–23 Among these, isolated Pd cations are recently emerging as prominent and effective low temperature NO adsorption sites.9,14,20,24,25
Combined experimental evidence from kinetic studies, DFT computational work, XPS, EXAFS, DRIFT, and FT-IR spectroscopy suggests the role of Pd2+ and Pd+, acting as the NO storage centres, forming nitrosyl complexes, following both adsorption pathways and a Pd-redox chemistry where NO reduces Pd2+ to Pd+, releasing NO2.9,13,14,20,24–30 The identification of the primary storage site remains, however, controversial, as some suggest Pd+ (ref. 22, 23 and 26–28) while others support oxidized Pd species.13,31 Furthermore, it has been acknowledged that the nature of the Pd species can change also according to the catalyst features (silica–alumina ratio, Pd loading, zeolite framework), the pre-treatment, the reaction conditions, and the zeolite synthesis method.11,13,25,26,29,32,33 Similarly to NO, also CO has been proposed as a Pd reducing agent, enhancing the system storage due to its stronger interaction with Pd, and releasing CO2 as a result.26 A negative impact of water in the NO storage over different Pd-exchanged zeolites has been observed, supported by theoretical calculations, which demonstrated the lowering of the NO binding energies with Pd in its presence.9,13,14,31
Kinetic studies were conducted to elucidate the features of transient NO uptake and desorption over Pd-promoted zeolites, following complex reaction pathways. In the works of Ambast et al.27,34 and Yao et al.,21 PNA monolith materials were tested, and satisfactory kinetic descriptions were obtained considering reaction mechanisms with multiple-steps (9 and 10, respectively) for NO-only feed, with 9 additional steps in the presence of CO. Several adsorption phases, with the intermediate formation of surface complexes, and two-step Pd2+ reductions, releasing NO2 or CO2, were proposed. Ambast et al.27,34 accounted for Pd cations-only as active NO storage sites (i.e. Z-[Pd2+OH]+, Z2Pd2+, and ZPd+), while Yao et al.21 included also PdO as an additional adsorption centre. Furthermore, CO was proposed to further reduce Pd+ and PdO to Pd0 and metallic Pd respectively.21
In this context, in a previous study we reported NO adsorption chemistry over a Pd-CHA sample, relying on transient adsorption experiments at variable operative conditions.35 The simple integral analysis of the gas phase transients permitted the interrogation of the underlying NO storage mechanism, which clearly followed a Pd-redox chemistry. The majority of palladium existed initially as Pd2+, which was readily reduced to Pd+ by CO, or NO, acting as reducing agents. The study highlighted a more favourable NO storage under reducing conditions, i.e. on Pd+, while the activation of Pd-oxidation processes, converting Pd+ back to Pd2+, decreased the available NO adsorption sites thus leading to a decrement in the overall NO storage.23,35 Interestingly, the presence of water apparently inhibited Pd-oxidation, resulting in a positive effect on NO storage which was not observed previously in the literature. These findings were recently supported by an independent DFT investigation.23
With the intent to challenge quantitatively the NO adsorption chemistry proposed in previous work,35 a transient kinetic redox model has been herein developed and applied to describe the NO adsorption pathway. To strengthen the analysis, the model has been fitted to transient data covering three different gas phase compositions (effects of NO, CO, and H2O) over four adsorption temperatures in the low-T range (100–200 °C) and collected over two catalysts characterized by different Pd-loadings. Notably, the proposed kinetic model simplifies those already reported in the literature,21,27,34 relying on fewer reaction steps and fewer intermediate complexes, while further showcasing the central role of the Pd-redox chemistry in the NO storage mechanism.
2. Materials and methods
2.1 Catalyst samples and experimental set-up
Two Pd-CHA catalysts in the form of powder, both supplied by Johnson Matthey, have been considered in this study. The reference material was characterized in a previous work by ICP-MS analysis, resulting in a Pd loading of 0.84% w/w and SiO2/Al2O3 (SAR) of 25.35 To study the impact of the Pd-loading on the PNA functionality, a second sample with Pd-content and SAR respectively of 0.50% w/w and 25 have been herein investigated, too. Throughout the manuscript, the two catalysts are labelled as 0.84% Pd and 0.50% Pd. A lean hydrothermal aging procedure (LHA) at 750 °C for 16 h in air with 10% H2O ensured the proper Pd particle redispersion forming exchanged ions, with negligible PdOx clusters, as shown by UV-vis and EXAFS analysis on both the 0.84% Pd and 0.5% Pd catalysts.35 This is also proven by the equimolar NO adsorption in respect to the Pd detected by ICP-MS (NO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd = 1:1) in NO + O2 wet tests at low temperature.35
:Pd = 1:1) in NO + O2 wet tests at low temperature.35
Two tubular quartz micro-flow reactors were prepared (ID ≈ 6 mm), one per catalyst, each loaded with 130 mg of powder mixture (40 mg of catalyst + cordierite diluent). A thermocouple inserted in the catalyst bed was used to measure the reactor temperature. The samples were tested in an experimental set-up where helium was used as gas mixture dilutant. The reactants (NO, CO, O2) were dosed with mass flow controllers (Brooks Instruments) and traced with Argon, while the water vapor feed concentration was regulated with an electrically heated saturator. A combination of a UV analyser (ABB LIMAS 11 HW) + IR analyser (ABB URAS 14) with a mass spectrometer (QGA Hiden Analytical) enabled to monitor the reactor outlet composition (NO, NO2, CO, CO2, O2, H2O, Ar). Additional information on the experimental set-up and protocols can be found in previous publications.35–37
2.2 Transient adsorption/desorption protocol
The transient adsorption/desorption procedure to evaluate the Pd-CHA functionality as PNA materials, with variable gas phase composition and adsorption temperature, was described previously.35A constant GHSV of 300000 cm3 h−1 gcat−1 (STP) has been adopted in the tests herein presented. Prior to each experiment, a high temperature treatment with 10% v/v of O2 at 500 °C was performed to remove previously adsorbed species and fully oxidize the palladium.35 A cooling phase in the same oxidizing atmosphere followed. If required, H2O was added to the feed when close to the test operative temperature. An initial 50 min isothermal adsorption phase was carried out in the presence of different feed gas mixtures (NO + O2 dry-gas, NO + O2 wet, CO + NO + O2 dry-gas; NO = 0–200 ppm, CO = 0–1500 ppm, O2 = 10% v/v, H2O = 0–5% v/v) and temperatures (100, 120, 150, 200 °C). Additionally, dedicated tests with the CO + NO + O2 feed mixture, where the NO step feed was delayed with respect to CO, were performed (called “CO + O2 + delayed NO” in the following). Initially applied on the 0.84% Pd sample, these protocols have been replicated on 0.50% Pd as well, limited to the adsorption temperature of 100 °C.35
It is well known that, at low temperatures, CO can reduce Pd to its metallic form, originating sintering phenomena: accordingly, we avoided to work at temperatures below 100 °C. In addition, all the tests, both with NO and with CO, have been run in the constant presence of 10% v/v of oxygen. In our experience, this further reduces the chances of fully reducing the palladium sites. To prove that no deactivation occurred throughout different tests in these conditions, Fig. SI.1† shows a sequence of NO + O2 temperature programmed surface reaction (TPSR) runs in a wet atmosphere: each run was executed after a CO + O2 test and no significant difference between them is apparent, indicating that the Pd sites were not considerably altered by repeated exposure to CO.
2.3 NO adsorption kinetic model
As discussed in the Introduction, the NO adsorption mechanism on Pd-CHA remains debated, with different pathways proposed in the literature.6,7,9,13,14,19–21 In the present work, we mainly rely on the mechanistic investigation of NO trapping performed previously over the 0.84% Pd.23,35 The goal of this work is to create a simple but chemically based kinetic mechanism which can accurately represent the behaviour of the PNA system in the low-temperature regime and use it to validate our mechanistic understanding with a minimum set of reaction steps. For this purpose, while a large variety of reactions has been considered in literature, several of them were ruled out in this work. An example of ruled-out reactions is the thermal desorption of NO or CO: in fact, these reactions are not relevant for the present kinetic analysis due to their very poor activity in the low-temperature regime, as addressed here.35 Consistently with these considerations, the assumed reaction steps are summarized in Table 1.| Reaction no. | Reaction step | Reaction rate expression [s−1] |
|---|---|---|
| (R1) | 2Pd2+OH + CO → 2Pd+ + CO2 + H2O |
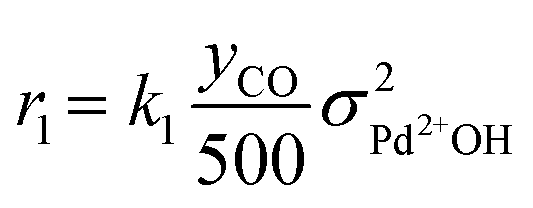
|
| (R2) | Pd+ + CO → Pd+CO |
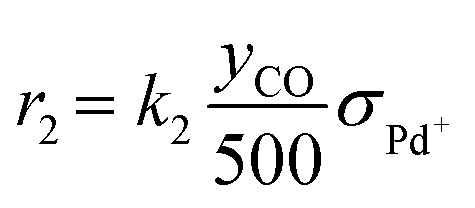
|
| (R3) | Pd+CO + NO → Pd+NO + CO |
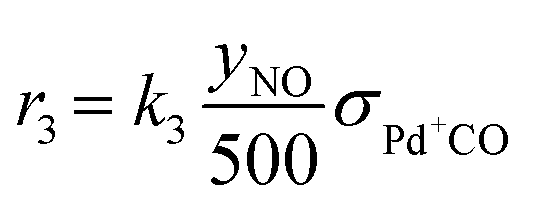
|
| (R4) | Pd2+OH + NO ⇌ Pd2+OH(NO) |

|
| r des4 = kdes4σPd2+OH(NO) | ||
| (R5) | 2Pd2+OH(NO) + NO → 2Pd+NO + NO2 + H2O |

|
| (R6) | 2Pd+NO + ½O2 + H2O → 2Pd2+OH + 2NO |

|
| (R7) | 2Pd+CO + ½O2 + H2O → 2Pd2+OH + 2CO |
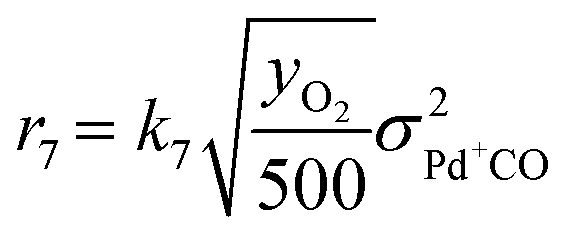
|
Analysis of flow reactor experiments led to the proposal of a Pd-redox NO adsorption chemistry, wherein Pd cycles between Pd2+ and Pd+, involving a strong link between the Pd reduction extent and the NO storage capacity.23,35 As discussed in the Introduction, the role of a redox chemistry in Pd-CHA systems has been proposed in several works. However, herein we regard it as the main pathway leading to stable NO adsorption.9,13,14,20,24–30 CO primarily, and NO to a lesser extent, act as Pd2+ reductants according to a dual-site catalytic chemistry producing CO2 or NO2 respectively ((R1) and (R5)).14,23,26,27,34,38
The key role of ZPd2+OH in such a mechanism has already been reported, even though the Pd2+ population may include also other species, like the doubly ligated Z2Pd2+.13,14,23,31 In fact, it is generally accepted that Z2Pd2+ species are not prone to be reduced, contrary to the redox-active ZPd2+OH.27 In our tests we were able to fully reduce the Pd catalyst with dry CO + O2 already at 100 °C, producing CO2.35 Thus, even if our oxidized Pd-CHA samples may not contain exclusively ZPd2+OH in their pristine state, all the Pd2+ cations need eventually to be converted to such species in order to be reduced and then adsorb CO. Considering that H2O molecules are available on the zeolite, a possible explanation is provided by the hydrolysis reaction Z2Pd2+ + H2O ⇌ ZPd2+OH + ZH+, whose equilibrium is shifted towards the monodentate ZPd2+OH form by its consecutive reactivity with CO (a so-called scavenging reaction). This is similar to the Z2Cu2+ hydrolysis documented over Cu-CHA catalysts for NH3-SCR.39
In a recent FTIR in operando study of CO adsorption on Pd-CHA, Lietti and co-workers40 have reported that, at room temperature and under dry-gas conditions, CO was adsorbed onto Z2Pd2+ sites. When water was added to the gas feed, though, a complete shift of the IR bands towards the region of Pd(I) carbonyls proved the hydrolysis of the Z2Pd2+ sites to ZPd2+OH, which are readily reducible and enabled adsorption of CO in a stable form as Pd+–CO. This picture is fully consistent with our CO storage mechanism, steps (R1) and (R2) in Table 1, and supports the assumption of ZPd2+OH as the dominant oxidized Pd species, as well as the precursor of the primary storage site for NO and CO, i.e., Pd+, in our simple redox model.
While the evidence for the conversion of Z2Pd2+ sites to ZPd2+OH under wet conditions (i.e., with H2O in the feed stream) experiments are solid,40 this is less clear for our experiments under dry-gas conditions (i.e., with no H2O added to the feed stream). Indeed, in our protocols the catalysts are pre-heated to 500 °C also to dehydrate the powder samples. However, we speculate that this pretreatment did not remove all the moist in the small-pores zeolite cages of our Pd-CHA systems, so that the residual H2O could still promote the hydrolysis step. Accordingly, we retain the assumption of the Pd2+ population consisting primarily of ZPd2+OH for dry-gas conditions, too.
The proposal of two ZPd2+OH species involved in (R1) and (R5) originated from a quantitative analysis of the reacting system: throughout the different NO adsorption tests, a maximum 1:2 molar ratio between CO2, or NO2, release and the overall Pd content was noted under both wet and dry-gas conditions, consistent with the involvement of an O-donor species and with the two-electron exchange requirements of (R1) and (R5), where the Pd2+ reduction to Pd+ needs one electron transfer per Pd. Notably, the results from the CO oxidation tests indicate that two Pd2+OH cations are always available in close proximity, both under dry-gas and wet conditions.35 This suggests that the Pd sites are mobile, possibly because H2O-solvated. While this is reasonable for wet experiments,40 in the case of dry-gas experiments we have again to assume, similar to the previous considerations on the Z2Pd2+ hydrolysis, that residual H2O in the zeolite can confer enough mobility to the Pd sites to enable the CO oxidation activity we have observed also in dry-gas conditions.
The detailed analysis of the Pd-speciation is outside the scope of this work: it must be mentioned, however, that the same considerations above may be consistent also with the involvement of Pd dimers (e.g., [Pd–O–Pd]2+).13,14,23,31 In fact, for the CO to CO2 oxidation reaction (R1), similar conclusions were drawn by several studies conducted previously using CO as a probe molecule to assess the presence of binuclear Cu2+ species, originating from the coupling of mobile Cu2+OH ions, over Cu-CHA catalysts for NH3-SCR applications.36,39,41,42
Once reduced, Pd+ acts as the main NO adsorption site,23,26–28 forming Pd+NO; adsorption of CO is also possible (R2), however when NO is present CO is displaced from Pd+, and the competitive NO adsorption is favoured (R3).29,35 In addition to this primary NO storage mechanism, a weak reversible NO storage on the oxidized Pd2+ is also accounted for by reaction (R4), mainly active when CO is not present.23,26 The palladium utilization factor (i.e., adsorbed NO/Pd molar ratio) was shown to be a decreasing function of temperature.35 Indeed, an increasing temperature progressively activates Pd+ oxidation: Pd+ is converted back to Pd2+OH, thus leading to a decrease in the number of available NO and CO adsorption sites. This process is herein accounted for by (R6) and (R7) in Table 1.21 Globally, five Pd-species are included in the considered adsorption scheme: two oxidized palladium species – Pd2+OH and Pd2+OH(NO) – and three reduced species – Pd+, Pd+CO and Pd+NO.
Remarkably, the adsorption of NO onto Brønsted Acid Sites (BAS) has been disregarded. Indeed, previous work on the proton form of the CHA zeolite demonstrated that, in dry-gas conditions, only minor amounts of NO were weakly adsorbed, which were readily desorbed isothermally in helium, while under wet conditions NO adsorption was not observed at all.35
The kinetic analysis of the transient NO adsorption steps for the 0.50% Pd and 0.84% Pd samples relies on the abovementioned reaction mechanism. Table 1, 3rd column, lists the turnover rate expressions of the k-reactions (rk) adopted for the reaction steps ((R1) to (R7)), where kk are apparent rate constants. These equations assume first-order reaction kinetics in the gaseous species (NO, CO), with the NO and CO concentrations normalized by a reference feed concentration (500 ppm). Conversely, a second-order dependence on the oxidized and reduced Pd fractions, respectively, has been postulated for the two-electron exchange Pd reduction and re-oxidation steps ((R1) and (R5)–(R7)). While alternative first-order dependences would result in a similar goodness of fit, the adopted second-order assumption is in line with the recent literature on kinetic modelling of PNA systems, see e.g. the dual-site steps in the models published by Olsson and coworkers21 and by Harold and coworkers.27
To estimate the associated rate parameters, the transient gas phase species evolution was fitted adopting a transient integral plug-flow model of the test reactor, modelled as a cascade of 20 isothermal, isobaric continuous stirred tank reactors (CSTR). A similar approach has been successfully utilized in the kinetic analysis of transient NH3-SCR experiments over Cu-CHA catalysts.37,43,44 The mass balance equations for the gaseous species and for the Pd sites in each CSTR are reported in Tables 2 and 3, respectively.
| Equation no. | Gaseous species | Gas phase mass balance [−] |
|---|---|---|
| (1) | NO |

|
| (2) | CO |

|
| (3) | NO2 |
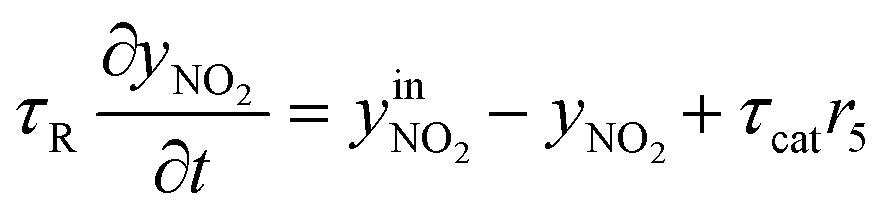
|
| (4) | CO2 |

|
| Equation no. | Pd-species | Pd-species mass balance [molPdj molPdtot−1 s−1] |
|---|---|---|
| (5) | Pd2+OH |

|
| (6) | Pd+NO |

|
| (7) | Pd+ |
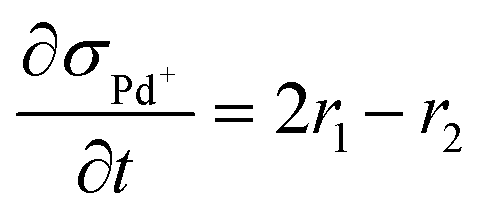
|
| (8) | Pd+CO |

|
| (9) | Pd2+OH(NO) | σ Pd2+OH(NO) = 1 − σPd2+OH − σPd+NO − σPd+ − σPd+CO |
For each i-gas species, yin and y are respectively the inlet and outlet molar fractions in each CSTR, τR is the residence time in each CSTR and τcat is the contact time, evaluated as nPd/F [molPd mol−1 s−1], where nPd is the Pd loading in each CSTR and F is the molar feed flow. σj is the molar fraction of a Pd j-species in each CSTR, as nj/nPd [molPdj molPd−1] and nPd is the respective molar content.
A code written in FORTRAN language has been used for numerical integration of the PDE system in time for each CSTR reactor. For this purpose, we used the SLSODE solver for ordinary differential equations (ODEs), originally proposed for initial-value problems with stiff and non-stiff systems of first order ODEs by Hindmarsh,45 and successfully implemented in earlier works.37,43
To validate the NO adsorption chemistry proposed in this work, a simple kinetic modelling approach has been implemented. Individual portions of the low-T redox model were fitted independently to dedicated sets of experimental data. Thus, k1 and k2 were estimated by adapting the model to the CO and CO2 transients of the CO + O2 + delayed NO experiment (first part, in absence of NO). Subsequently, k3 was calibrated to account for the NO transients and the CO release with the delayed NO feed. The CO + O2 + delayed NO test was also used to extract an estimate of k7 from the steady-state production of CO2 both in the first and in the second part of the run. Additionally, the NO + O2 tests were used to estimate k4–k6 independently, fitting the NO and NO2 profiles for k4, the NO2 peak for k5 and the steady-state production of NO2 for k6 together with the fit of the NO + O2 TPSR. Then, the co-feed of CO and NO, the water effect in the presence of only NO, the adsorption temperature effect, and the Pd-loading impact have all been predictively simulated just relying on the previously estimated rate parameters.
The goal of studying the PNA kinetics in absence and in presence of water is to elucidate the effect of H2O on the different reaction steps, primarily to validate quantitatively our mechanistic understanding, rather than deriving empirical kinetic dependences on H2O, which is out of scope and would require a dedicated set of experiments at different H2O feed concentration.
3. Experimental results and kinetic modelling
The transient NO and/or CO adsorption and desorption tests are performed following the experimental protocols described in Materials and methods section, varying the gas phase feed composition, the Pd content, and the adsorption temperature. While the experimental results over the 0.84% Pd sample have been reported in our previous work,35 herein we focus on the kinetic modelling of the isothermal adsorption step. The complete list of the estimated apparent rate constants is provided in Tables SI.1 and SI.2 of the ESI.†3.1 Gas mixture composition effect
We start by discussing the kinetic analysis of the NO adsorption step with different feed gas mixtures at 100 °C over the 0.84% Pd sample. Fig. 1 shows the experimental data (dots) in the presence of a CO + O2 + delayed NO feed mixture. | ||
| Fig. 1 CO + O2 + delayed NO feed mixture: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic fit (solid lines) over 0.84% Pd sample. T = 100 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 0–200 ppm, CO = 1500 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
Upon feeding CO, a CO2 production is readily observed, characterized by an initial peak and a monotonic decreasing trend until reaching an almost null steady state value.35 The involvement of oxygen in the system chemistry can be ruled out, and the CO2 formation is thus ascribed to the Pd2+OH reduction by CO according to the dual site CO oxidation step (R1) in Table 1. Concurrently, CO is weakly adsorbed on the reduced Pd+ and the Pd+CO could be oxidized back by oxygen according to reactions (R2) and (R7), respectively. The bonding of NO with Pd+ sites is however favoured with respect to CO, resulting in a displacement of CO upon NO feed, as described by reaction (R3).29,35 The quantitative analysis of such experiments has been reported earlier; it highlighted: i) the complete reduction of Pd2+ to Pd+ by CO, ii) an equimolar displacement between the desorbed CO and the stored NO, iii) a complete Pd utilization, as confirmed by the NO/Pd molar ratio in Fig. 2.35 Consistent with the proposed reaction scheme, no release of NO2 is observed during the NO feed, since the NO2 formation step (R5) requires the presence of Pd2+OH, which was already reduced by CO during the initial step feed. Thus, no further reactions are considered here.
 | ||
| Fig. 2 Comparison between the experimental NO storage efficiency (red bars) and the model estimate (blue bars), with different gas phase compositions, on the 0.84% Pd sample at 100 °C. | ||
Such an experimental protocol enables to estimate the apparent rate constants of (R1), (R2), and (R7) (k1, k2 and k7) independently from (R3) (k3). The result of the kinetic fit is shown in Fig. 1 (continuous lines), where an accurate description of all the gas species transients is apparent. Indeed, both the CO and CO2 features are faithfully reproduced during the CO-only feed. The same fit quality is observed for the displacement phenomena caused by the NO step feed. Furthermore, the model successfully predicts the integral NO storage, as plotted in Fig. 2. The Pd utilization factor (NO/Pd) is evaluated extrapolating the model to very long times, corresponding to fully developed steady-state profiles.
The kinetic analysis of the NO + O2 system, in the absence of CO, enable effective estimation of the apparent rate constants of reactions (R4) to (R6) (k4 to k6), independently from the previously described CO-related phenomena. Fig. 3 displays the experimental data, in dry-gas (A) and wet (B) conditions.
 | ||
| Fig. 3 NO + O2 step feed on the 0.84% Pd sample, under dry-gas (A) and wet (B) conditions: nominal inlet concentrations (dashed lines), experimental results (dots), kinetic fit (solid lines, A) and kinetic simulation (solid lines, B). T = 100 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 0 ppm, O2 = 10% v/v, H2O = 0–5% v/v. | ||
In close correspondence to the previous CO-step feed, a release of NO2 is detected once NO is stepwise introduced in the feed stream. However, unlike CO2, the rate of release of NO2 is slow, despite the fast NO adsorption. Thus, a reversible adsorption on the oxidized Pd2+OH is proposed (R4), forming the Pd2+OH(NO) species.23,26,38 Subsequently, NO interacts with such complexes being oxidized to NO2 (R5), thus reducing two Pd species similarly to the CO pathway (R1). In the absence of water (Fig. 3A), an incomplete Pd oxidation is observed. Since NO is a weaker Pd reductant than CO, this experiment is likely affected by Pd-reoxidation phenomena (R6) even at low temperatures, resulting in an incomplete NO storage efficiency (NO/Pd = 0.84, as shown in Fig. 2).35 Conversely, when cofeeding water (Fig. 3B), complete NO storage is recovered (NO/Pd = 1.00). This positive effect is rationalized by an inhibition effect of H2O on the Pd-oxidation reaction (R6), which otherwise hinders the NO reduction extent of Pd2+.35
The kinetic model successfully fits the transient species evolutions in the case of the NO + O2 mixture, with and without water added to the feed, as shown in Fig. 3A and B (solid lines). The apparent rate constants (k4 to k6), are estimated from the first case only, while the water effect has been simulated by simply deactivating the Pd-oxidation reaction (R6). However, the very few ppm of NO2 in the NO + O2 dry-gas experiment do not enable a reliable estimation of the rate constant for (R6). Thus, k6 has been estimated by fitting the TPSR run in the NO + O2 dry-gas atmosphere shown in Fig. 4 (brown dots). This test follows the one shown in Fig. 3A, and shows how NOx are desorbed when the temperature is raised up to 500 °C with a ramp of 15 °C min−1. To fit the TPSR profile, an Arrhenius form of the rate constant is needed  , where the two parameters to be fitted are k0 and EA. The brown solid line Fig. 4 shows the model fit of the TPSR curve, which is in close agreement with the experimental trace. The model is also able with good accuracy to follow the NO and NO2 dynamics over temperature during the T-ramp, as in Fig. SI.2.†
, where the two parameters to be fitted are k0 and EA. The brown solid line Fig. 4 shows the model fit of the TPSR curve, which is in close agreement with the experimental trace. The model is also able with good accuracy to follow the NO and NO2 dynamics over temperature during the T-ramp, as in Fig. SI.2.†
 | ||
| Fig. 4 Temperature programmed surface reaction (TPSR) test in a NO + O2 dry-gas atmosphere on the 0.84% Pd-CHA sample: NO feed concentration (dashed line), experimental NOx trace (dots), kinetic fit (solid lines). T = 100–500 °C at 15 °C min−1, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Feed: NO = 200 ppm, CO = 0 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
Finally, once we combine both k4 and k5 estimated from the adsorption test with k6 estimated from the desorption step, the predicted NO storage capacity, albeit with some minor over/under-estimations, closely matches the experimental one for both the NO + O2 dry-gas and wet tests (Fig. 2), so that the simple approach in the simulation of the H2O impact (turning off the (R6)) appears to be effective.
Lastly, the case with the co-feed of NO and CO is considered, with the experimental species evolutions shown in Fig. 5. The system features resemble closely those observed in the delayed feed case (Fig. 1), namely CO2 production followed by CO displacement in correspondence to the NO adsorption, with similar considerations related to the quantitative analysis.35
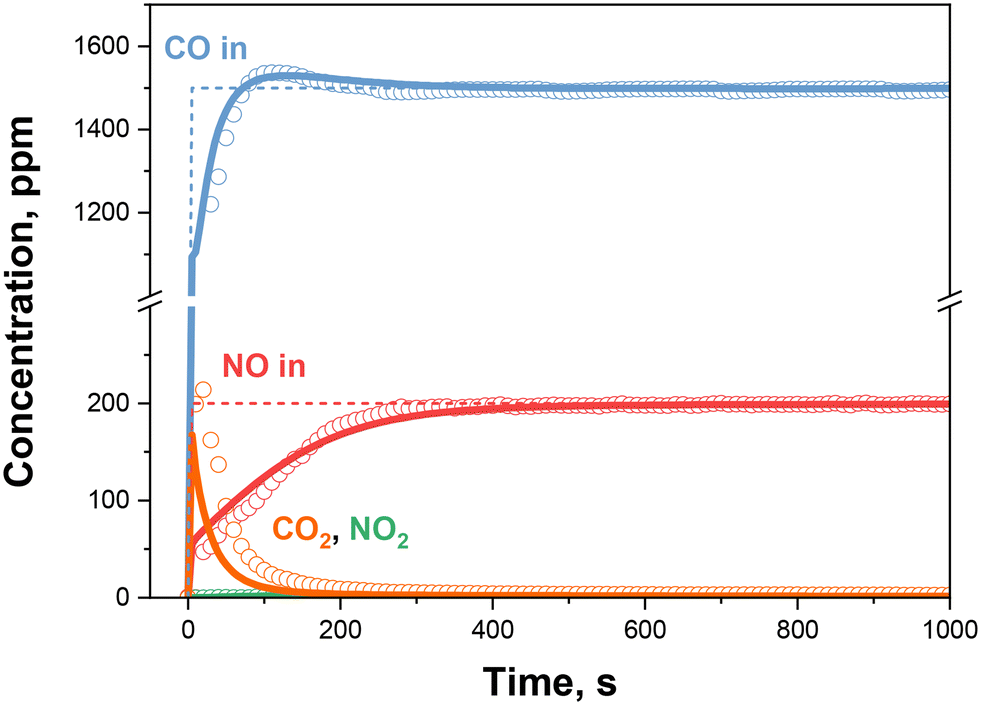 | ||
| Fig. 5 CO + NO + O2 step feed: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic simulations (solid lines) over the 0.84% Pd sample. T = 100 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 1500 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
The solid lines in Fig. 5 represent the model simulation based on the apparent rate constants previously estimated from the CO + O2 + delayed NO (k1, k2, k3 and k7) and NO + O2 (k4, k5 and k6) runs. A satisfactory prediction of the evolution of both species and of the NO integral storage (as shown in Fig. 2) is apparent, validating the model based on a simple Pd-redox scheme where CO acts as the main reducing species, according to a dual-site mechanism over Pd2+OH, and where Pd+ is the main adsorption site. Furthermore, the negligible NO2 production expected according to the considered kinetic model, once CO is present, is also found in agreement with the experimental results.
3.2 Pd-loading effect
A duplicate set of experiments with the NO + O2 dry-gas, NO + O2 wet and CO + NO + O2 dry-gas feed mixtures has been carried out at 100 °C over the 0.50% Pd sample. The results are shown here for the first time, in combination with the kinetic analysis of the adsorption step.Fig. 6 illustrates the experiment for the NO + O2 mixture, in the absence (Fig. 6A) and presence (Fig. 6B) of 5% v/v H2O in the feed stream. Both experiments show the same features detected over the higher Pd-loading sample (0.84% Pd), namely an instantaneous NO adsorption upon its injection, concurrent with slow NO2 production. Thus, the system could be described following the same reaction scheme discussed for the higher loading sample ((R4) to (R6)). Remarkably, both tests show an NO storage efficiency close to one, see Fig. 7 (0.97 and 0.95 for the dry-gas and wet mixture, respectively), in contrast to the 0.84% Pd sample where a promotion effect is observed with water.35 This may be rationalized by the limited NO capacity to reduce Pd2+, as earlier discussed. Accordingly, a lower loading may enable the reactant to completely reduce to Pd+ the overall palladium content even without water, while at a higher loading the effect of Pd-oxidation may become more relevant, preventing the complete reduction. Indeed, in the NO + O2 tests, the NO storage efficiency is determined by the balance between the reductive NO adsorption steps ((R4) + (R5)), and the oxidative NO desorption step (R6), which compete with different rates at the different temperatures. This closed redox loop determines the fraction of Pd+NO, and, thus, the amount of adsorbed NO.
 | ||
| Fig. 6 NO + O2 mixture: nominal feed concentrations (dashed lines), experimental results (dots) and kinetic simulations (solid lines) over the 0.50% Pd sample, under dry-gas (A) and wet (B) conditions. T = 100 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 0 ppm, O2 = 10% v/v, H2O = 0–5% v/v. | ||
 | ||
| Fig. 7 Comparison between the experimental (red bars) and the kinetic model (purple bars) NO storage efficiencies with different feed gas phase compositions, over the over 0.50% Pd sample, at 100 °C. | ||
It is important to assess the effect of the Pd loading on the rates of reactions (R4)–(R6) which govern the NO storage. The rate equation for the reversible adsorption of NO on Pd2+OH (R4) is first order in the reactive palladium species. Thus, its kinetic constants (kads4 and kdes4) remain unaltered when changing the Pd loading. Instead, (R5) and (R6) (the reduction and oxidation of Pd in presence of NO) are second order in their respective palladium species (as shown in Table 1), and their rate constants are proportional to the Pd loading. Thus, to extrapolate the kinetic constant to a different loading from the previously assessed estimates for the 0.84% Pd sample, they must be proportionally adapted as follows (n is the number of moles in the sample):
 | (10) |
Similarly, in the simultaneous presence of NO and CO, the 0.50% Pd sample resembles its higher loading counterpart (Fig. 8): a carbon dioxide production upon CO feed (R1), a CO displacement in correspondence to the NO adsorption ((R2) and (R3)), a negligible NO2 production and the complete Pd utilization are observed, as reported in Fig. 7. The kinetic simulation, now accounting for all the reactions and using the adjusted rate constants for (R1)–(R7) is shown in Fig. 8 as solid lines. The model simulation adheres to the experimental CO profile, however, somewhat underestimating the initial CO2 peak and predicting a slower NO adsorption rate.
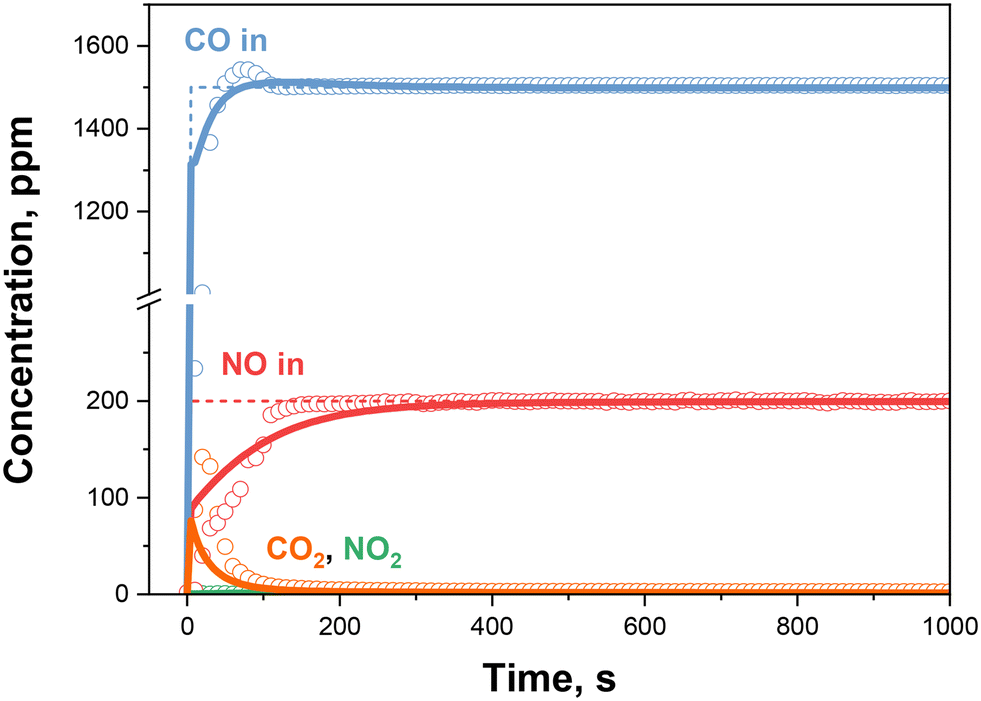 | ||
| Fig. 8 CO + NO + O2 mixture: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic simulation (solid lines) over 0.50% Pd sample. T = 100 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 1500 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
Such a simplified approach in the description of the Pd-loading effect has been shown to be effective, capturing the main system features, and with a predicted NO storage efficiency close to the experimental one (0.97, Fig. 7). Furthermore, in the case of the 100 °C systems it confirms the adequacy of the Pd-redox chemistry and of the kinetic modelling methodology herein adopted, albeit further improvements may be required to enhance its accuracy.
3.3 Adsorption temperature effect
The kinetic analysis of the adsorption temperature effect (120 to 200 °C) for all the gas feed mixtures (NO + O2 dry-gas, NO + O2 wet, CO + O2 + delayed NO, CO + NO + O2) is examined over the 0.84% Pd sample in the following sections. | ||
| Fig. 9 NO + O2 dry-gas mixture: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic fit (solid lines) over 0.84% Pd sample, at 120 °C (A), 150 °C (B) and 200 °C (C). T = 120/150/200 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 0 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
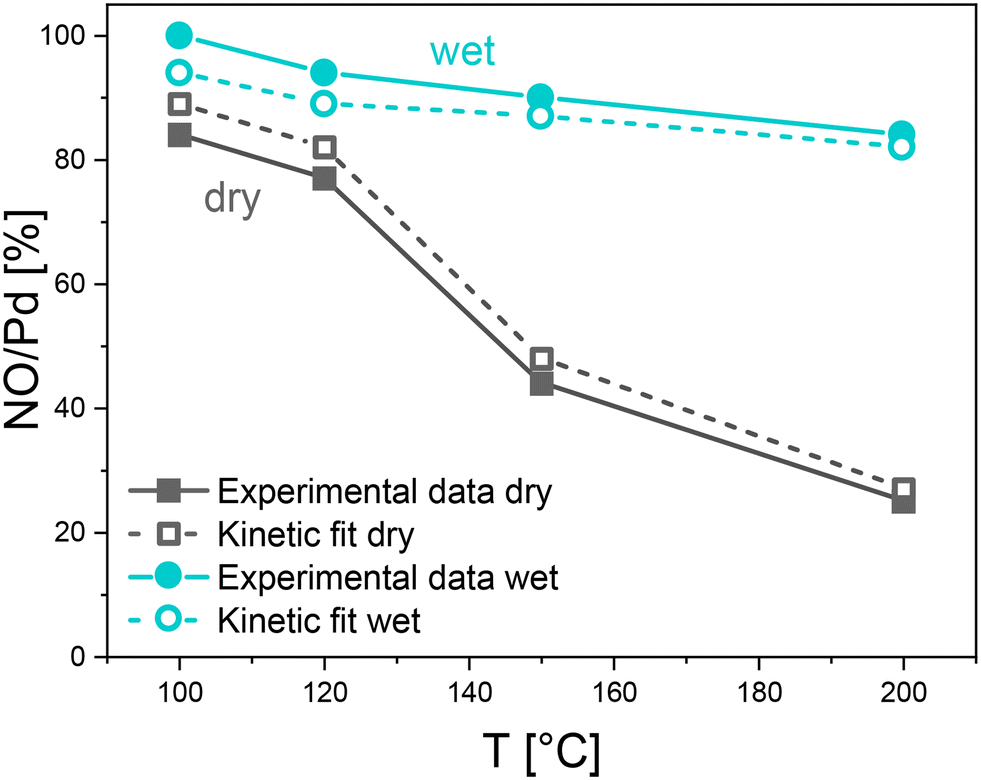 | ||
| Fig. 10 Comparison between the experimental NO storage (full symbols) and the kinetic model evaluation (empty symbols), for the NO + O2 mixture both in dry-gas (grey squares + lines) and wet (water blue circles + lines), over the 0.84% Pd sample, at different adsorption temperatures. | ||
The transient species concentration trends predicted by the kinetic model adhere to the experimental data, accounting for an increasing adsorption rate and a progressive change in the NO and NO2 steady states. The monotonic decrease of the integral NO storage is also correctly traced, as documented in Fig. 10 (grey empty symbols), however with a slight overestimation with respect to the experimental data. The rate constants (k4 to k6) have been accordingly fitted at different temperatures ((R6) together with the TPSR fit, as seen at 100 °C), and the estimates are illustrated in terms of Arrhenius plots in Fig. SI.3.† The highest activation energy is estimated for (R6) (EA = 69.3 kJ mol−1), coherently with the proposed temperature effect where Pd-oxidation becomes progressively more relevant at higher temperatures. Nevertheless, non-negligible energy barriers have been estimated also for the other reactions.
 | ||
| Fig. 11 NO + O2 wet mixture: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic fit (solid lines) over the 0.84% Pd sample, at 120 °C (A), 150 °C (B) and 200 °C (C). T = 120/150/200 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 0 ppm, O2 = 10% v/v, H2O = 5% v/v. | ||
As to the 100 °C test, the water effect with variable adsorption temperature is simulated relying on the kinetic constants previously assessed in the absence of water; only the rate constants for (R5) and (R6) (kwet5 and kwet6) are fitted to account for the water effect on the Pd-reduction/oxidation reactions. The Arrhenius plot is shown in Fig. SI.4.† An accurate match with the data is found at all temperatures for the NO and NO2 trends (Fig. 11) and for the integral NO storage (Fig. 10).
 | ||
| Fig. 12 CO + O2 + delayed NO mixture: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic simulation (solid lines) over 0.84% Pd sample at 120 °C (A) and 150 °C (B). T = 120/150 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: CO = 1500 ppm, NO = 200 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
The experimental results at different temperatures are compared in Fig. SI.5.† The transient profiles of CO and CO2 are very similar at 100, 120, and 150 °C, indicating almost negligible activation energy for both the CO-mediated reduction of palladium (R1) and CO adsorption (R2). Indeed, by simulating the CO + O2 tests with k1 and k2 estimated at 100 °C, the CO and CO2 transients are well reproduced (Fig. 12, solid lines), as was the case for the test at 100 °C. For the CO release due to NO insertion (R3), the same conclusions are obtained comparing the experimental results at different temperatures, as shown in Fig. SI.6.† Indeed, the NO consumption profile does not show substantial differences at increasing temperatures. The same could be said for the CO release profile, apart from the initial and final steady states of CO due to (R7) being activated with temperature. Empirically, this means that the rate constant k3 is not changing with temperature. The substitution of CO with NO seems then to be a non-activated reaction, at least up to 150 °C, suggesting that likely activated reactions (such as CO desorption) are not part of this competitive mechanism.
The predicted NO storage efficiency at 120 and 150 °C is consistent with the experimental trend but slightly underestimated, as shown in Fig. 13. Based on all the previous estimates of the rate constants, the steady-state production of CO2 enables the k7 fit at different temperatures, providing a model which can accurately predict the CO2 value in the sole presence of CO and O2. Furthermore, in the presence of both CO and NO, the predicted CO2 steady-state value is again close to the experimental value, indicating the correct estimation.
 | ||
| Fig. 13 Comparison between the experimental NO storage efficiency (full symbols) and the kinetic model prediction (empty symbols), for the CO + O2 + delayed NO mixture, over the 0.84% Pd sample, at different adsorption temperatures. | ||
When plotting the estimates of the rate constants in Arrhenius form (Fig. SI.3†), it is interesting to note that the activation energies of (R6) dry-gas and (R7) are comparable (EA equal to 69.3 and 65.9 kJ mol−1, respectively), possibly indicating similarity of the mechanisms of Pd+ reoxidation, independently of the adsorbed species (Pd+X, where X = CO or NO).
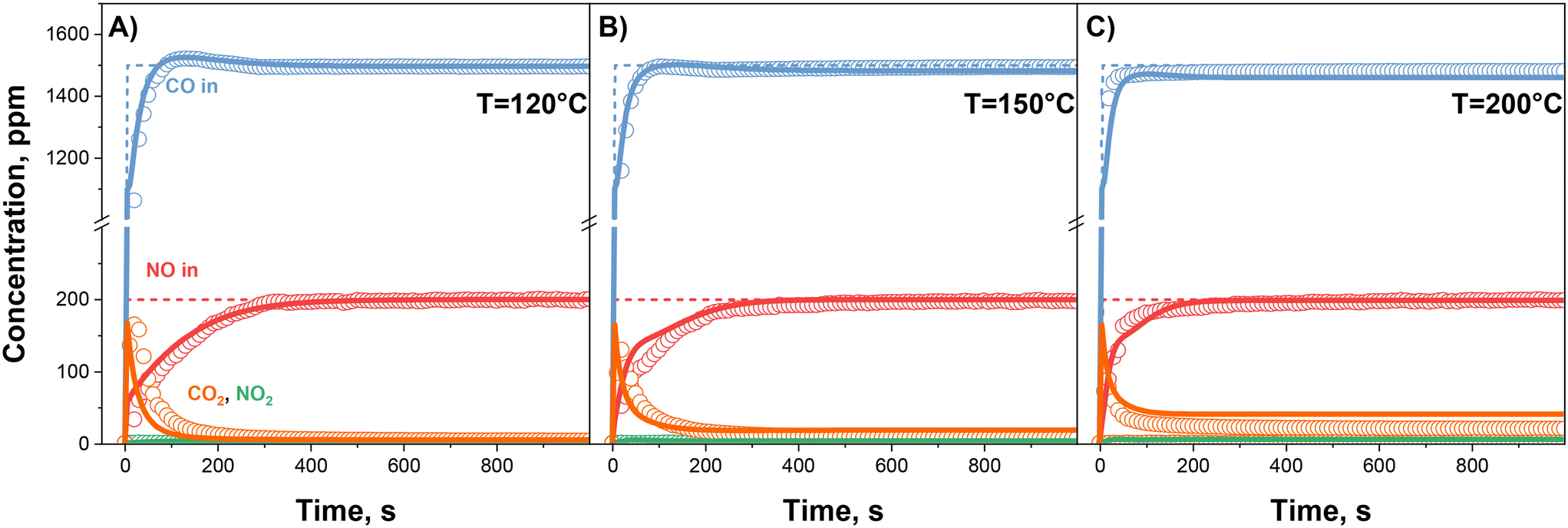 | ||
| Fig. 14 CO + NO + O2 mixture: nominal inlet concentrations (dashed lines), experimental results (dots) and kinetic simulation (solid lines) over 0.84% Pd sample at 120 °C (A), 150 °C (B) and 200 °C (C). T = 120/150/200 °C, Wcat = 40 mg, GHSV = 300000 cm3 h−1 gcat−1 (STP). Gas feed: NO = 200 ppm, CO = 1500 ppm, O2 = 10% v/v, H2O = 0% v/v. | ||
 | ||
| Fig. 15 Comparison between the experimental NO storage efficiency (full symbols) and the kinetic model predictions (empty symbols), for the CO + NO + O2 mixture, over the 0.84% Pd sample, at different adsorption temperatures. | ||
An effective simulation of the gas phase trends and of the integral NO storage is achieved at all temperatures (Fig. 14), with minor deviations from the experimental features, by superimposing the rate constants assessed at 100 °C for the CO-related reactions ((R1) to (R3)) and the temperature dependent estimates for the remaining ones ((R4) to (R7)).
As a last step, a simple sensitivity analysis of all the kinetic parameters has been run by incrementing (3×) and decrementing (1/3×) the rate constants and comparing the related model simulations to the experimental data. Results are displayed in Fig. SI.7 of the ESI:† they indicate that the model predictions are sensitive to all the rate parameter estimates, that we can therefore regard as sufficiently robust.
4. Conclusions
This study has focused on the kinetic modelling of low-temperature NO adsorption transients over two Pd-CHA systems for PNA applications, characterized by different metal loadings (0.84% Pd and 0.50% Pd), considering different operating conditions. The goal was to gain kinetic and fundamental insight into the still debated NO adsorption chemistry.A Pd-redox chemistry (Pd2+ ↔ Pd+) previously developed has been herein considered, where CO and NO act as reducing elements for Pd2+ (assumed to be in the form of Pd2+OH), while the Pd re-oxidation is ascribed to O2, and Pd+ is proposed as the main NO adsorption site. A simple reacting scheme has been implemented in a transient micro-kinetic model and fitted to experiments on the two samples, with variable gas phase compositions and adsorption temperatures in the 100–200 °C range. The adopted modelling methodology focused firstly to independently assess the rate constants of two sets of reactions ((R1) to (R3) – invariant with T, (R4) to (R7) – T dependent), relying on dedicated experimental protocols (NO + O2 dry-gas/wet, CO + O2 + delayed NO). Then we proceeded to simulate the Pd-loading effect (0.50% Pd) on the CO + NO + O2 systems.
A satisfactory match of the model predictions with both the observed gas phase concentration transients and the NO integral storage was found at 100 °C, thus supporting the underlying chemistry. CO, or NO, reduces Pd2+OH according to a dual-site mechanism, releasing CO2, or NO2, and so enabling the NO adsorption onto Pd+. However, while CO reacts readily with Pd2+OH, NO is less active, requiring a pre-adsorption step, forming Pd2+OH(NO). Notably, the fitted rate constants allowed to predict the effect of changing the Pd loading, at 100 °C, with different gas phase reactants.
Concerning the adsorption temperature effect, up to 200 °C an effective description of the NO + O2 dry-gas system was achieved by considering the progressive activation of a Pd-oxidation process (R6): as Pd+ is re-oxidized, fewer NO adsorption sites remain available, which agrees closely with the observed drop of the NO storage. In the presence of water (NO + O2 wet), the Pd+ reoxidation process is, however, inhibited. In addition, water promotes a higher NO2 production peak. Thus, including account of such effects in the related steps of the model – i.e., promoting reaction (R5) and inhibiting (R6) – enabled to describe successfully the NO storage at variable temperatures.
Finally, the CO + O2 + delayed NO tests allowed to assess that reactions (R1)–(R3) are temperature independent, since the related transient profiles do not change significantly with T, when below 200 °C. The steady state production of CO2 allowed to tune the T-dependence of the Pd+CO oxidation (R7), which was found in close similarity to the T-dependence of the Pd+NO oxidation, suggesting that there is just one Pd+ oxidation step. Lastly, accurate simulations of the CO + NO + O2 system were obtained by using the rate constants fitted on the independent NO + O2 and CO + O2 + delayed NO test runs.
The methodology based on the independent estimation of the rate constants employed in this work has therefore confirmed the validity of the proposed simple low-temperature Pd-redox chemistry for Pd-CHA PNA systems in a broad range of conditions.
Data availability
The data supporting this article are included in the main text and the ESI.† Additional data are not available.Author contributions
Umberto Iacobone: investigation, validation, formal analysis, visualization, data curation, writing – original draft. Andrea Gjetja: investigation, validation, formal analysis, visualization, data curation, writing – original draft. Nicola Usberti: formal analysis, data curation, writing – review & editing. Isabella Nova: conceptualization, resources, supervision, writing – review & editing. Enrico Tronconi: conceptualization, resources, supervision, project administration, funding acquisition, writing – review & editing. Djamela Bounechada: conceptualization, writing – review & editing. Roberta Villamaina: investigation, conceptualization, writing – review & editing. Maria Pia Ruggeri: conceptualization, resources. Andrew P. E. York: conceptualization, writing – review & editing. Loredana Mantarosie: conceptualization. Jillian Collier: conceptualization, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by Johnson Matthey.References
- C. K. Lambert, Perspective on SCR NO: X control for diesel vehicles, React. Chem. Eng., 2019, 4, 969–974, 10.1039/c8re00284c.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Recent advances in automotive catalysis for NOx emission control by small-pore microporous materials, Chem. Soc. Rev., 2015, 44, 7371–7405, 10.1039/c5cs00108k.
- I. Nova and E. Tronconi, Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, 2014, DOI:10.1007/978-1-4899-8071-7.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, The ammonia selective catalytic reduction activity of copper-exchanged small-pore zeolites, Appl. Catal., B, 2011, 102, 441–448, DOI:10.1016/j.apcatb.2010.12.022.
- H. Y. Chen, S. Mulla, E. Weigert, K. Camm, T. Ballinger, J. Cox and P. Blakeman, Cold Start Concept (CSC™): A Novel Catalyst for Cold Start Emission Control, SAE Int. J. Fuels Lubr., 2013, 6, 372–381, DOI:10.4271/2013-01-0535.
- H. Zhao, A. J. Hill, L. Ma, A. Bhat, G. Jing and J. W. Schwank, Progress and future challenges in passive NO adsorption over Pd/zeolite catalysts, Catal. Sci. Technol., 2021, 11, 5986–6000, 10.1039/d1cy01084k.
- Y. Gu and W. S. Epling, Passive NOx adsorber: An overview of catalyst performance and reaction chemistry, Appl. Catal., A, 2019, 570, 1–14, DOI:10.1016/j.apcata.2018.10.036.
- J. Lee, J. R. Theis and E. A. Kyriakidou, Vehicle emissions trapping materials: Successes, challenges, and the path forward, Appl. Catal., B, 2019, 243, 397–414, DOI:10.1016/j.apcatb.2018.10.069.
- H. Y. Chen, J. E. Collier, D. Liu, L. Mantarosie, D. Durán-Martín, V. Novák, R. R. Rajaram and D. Thompsett, Low Temperature NO Storage of Zeolite Supported Pd for Low Temperature Diesel Engine Emission Control, Catal. Lett., 2016, 146, 1706–1711, DOI:10.1007/s10562-016-1794-6.
- J. R. Theis and C. K. Lambert, An assessment of low temperature NOx adsorbers for cold-start NOx control on diesel engines, Catal. Today, 2015, 258, 367–377, DOI:10.1016/j.cattod.2015.01.031.
- O. Mihai, L. Trandafilović, T. Wentworth, F. F. Torres and L. Olsson, The Effect of Si/Al Ratio for Pd/BEA and Pd/SSZ-13 Used as Passive NOx Adsorbers, Top. Catal., 2018, 61, 2007–2020, DOI:10.1007/s11244-018-1017-z.
- A. Porta, T. Pellegrinelli, L. Castoldi, R. Matarrese, S. Morandi, S. Dzwigaj and L. Lietti, Low Temperature NOx Adsorption Study on Pd-Promoted Zeolites, Top. Catal., 2018, 61, 2021–2034, DOI:10.1007/s11244-018-1045-8.
- K. Khivantsev, F. Gao, L. Kovarik, Y. Wang and J. Szanyi, Molecular Level Understanding of How Oxygen and Carbon Monoxide Improve NOx Storage in Palladium/SSZ-13 Passive NOx Adsorbers: The Role of NO+ and Pd(II)(CO)(NO) Species, J. Phys. Chem. C, 2018, 122, 10820–10827, DOI:10.1021/acs.jpcc.8b01007.
- Y. Zheng, L. Kovarik, M. H. Engelhard, Y. Wang, Y. Wang, F. Gao and J. Szanyi, Low-Temperature Pd/Zeolite Passive NOx Adsorbers: Structure, Performance, and Adsorption Chemistry, J. Phys. Chem. C, 2017, 121, 15793–15803, DOI:10.1021/acs.jpcc.7b04312.
- Y. Ji, S. Bai and M. Crocker, Al2O3-based passive NOx adsorbers for low temperature applications, Appl. Catal., B, 2015, 170–171, 283–292, DOI:10.1016/j.apcatb.2015.01.025.
- D. Yao, P. H. Ho, R. F. Ilmasani, J. C. Wurzenberger, T. Glatz, D. Creaser and L. Olsson, Enhanced CO resistance of Pd/SSZ-13 for passive NOx adsorption, Chem. Eng. J., 2023, 460, 141681, DOI:10.1016/j.cej.2023.141681.
- D. Yao, P. H. Ho, J. C. Wurzenberger, T. Glatz, W. Di, R. F. Ilmasani, D. Creaser and L. Olsson, Deciphering SO2 poisoning mechanisms for passive NOx adsorption: A kinetic modeling approach and development of a high-resistance catalyst, Chem. Eng. J., 2024, 487, 150406, DOI:10.1016/j.cej.2024.150406.
- L. Castoldi, S. Morandi, P. Ticali, R. Matarrese and L. Lietti, An Assessment of Zeolite Framework Effect for Low-Temperature NOX Adsorbers, Catalysts, 2023, 13, 962, DOI:10.3390/catal13060962.
- A. Wang, K. Lindgren, M. Di, D. Bernin, P. A. Carlsson, M. Thuvander and L. Olsson, Insight into hydrothermal aging effect on Pd sites over Pd/LTA and Pd/SSZ-13 as PNA and CO oxidation monolith catalysts, Appl. Catal., B, 2020, 278, 119315, DOI:10.1016/j.apcatb.2020.119315.
- J. Lee, Y. S. Ryou, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Investigation of the active sites and optimum Pd/Al of Pd/ZSM–5 passive NO adsorbers for the cold-start application: Evidence of isolated-Pd species obtained after a high-temperature thermal treatment, Appl. Catal., B, 2018, 226, 71–82, DOI:10.1016/j.apcatb.2017.12.031.
- D. Yao, R. Feizie Ilmasani, J. C. Wurzenberger, T. Glatz, J. Han, A. Wang, D. Creaser and L. Olsson, Kinetic modeling of CO assisted passive NOx adsorption on Pd/SSZ-13, Chem. Eng. J., 2022, 428, 132459, DOI:10.1016/j.cej.2021.132459.
- M. Kaushik, T. S. Khan, M. A. Haider and D. Bhatia, Effect of H2O and CO on the NO Operating Cycle in Pd/CHA Passive NOx Adsorbers, J. Phys. Chem. C, 2024, 128, 2814–2827, DOI:10.1021/acs.jpcc.3c04999.
- M. Kaushik, G. Shrivastav, T. S. Khan, M. A. Haider and D. Bhatia, The Operating Cycle of NO Adsorption and Desorption in Pd-Chabazite for Passive NO xAdsorbers, Langmuir, 2021, 37, 13799–13809, DOI:10.1021/acs.langmuir.1c01383.
- K. Khivantsev, N. R. Jaegers, L. Kovarik, J. C. Hanson, F. Tao, Y. Tang, X. Zhang, I. Z. Koleva, H. A. Aleksandrov, G. N. Vayssilov, Y. Wang, F. Gao and J. Szanyi, Achieving Atomic Dispersion of Highly Loaded Transition Metals in Small-Pore Zeolite SSZ-13: High-Capacity and High-Efficiency Low-Temperature CO and Passive NOx Adsorbers, Angew. Chem., Int. Ed., 2018, 57, 16672–16677, DOI:10.1002/anie.201809343.
- Y. S. Ryou, J. Lee, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Activation of Pd/SSZ-13 catalyst by hydrothermal aging treatment in passive NO adsorption performance at low temperature for cold start application, Appl. Catal., B, 2017, 212, 140–149, DOI:10.1016/j.apcatb.2017.04.077.
- A. Gupta, S. B. Kang and M. P. Harold, NOx uptake and release on Pd/SSZ-13: Impact Of Feed composition and temperature, Catal. Today, 2021, 360, 411–425, DOI:10.1016/j.cattod.2020.01.018.
- M. Ambast, A. Gupta, B. M. M. Rahman, L. C. Grabow and M. P. Harold, NOx adsorption with CO and C2H4 on Pd/SSZ-13: Experiments and modeling, Appl. Catal., B, 2021, 286, 119871, DOI:10.1016/j.apcatb.2020.119871.
- P. Kim, J. Van Der Mynsbrugge, M. Head-Gordon and A. T. Bell, Experimental and Theoretical Studies of Pd Cation Reduction and Oxidation during NO Adsorption on and Desorption from Pd/H-CHA, J. Phys. Chem. C, 2022, 126, 18744–18753, DOI:10.1021/acs.jpcc.2c06357.
- L. Castoldi, R. Matarrese, S. Morandi, P. Ticali and L. Lietti, Low-temperature Pd/FER NOx adsorbers: Operando FT-IR spectroscopy and performance analysis, Catal. Today, 2021, 360, 317–325, DOI:10.1016/j.cattod.2020.02.019.
- C. Descorme, P. Gélin, M. Primet and C. Lécuyer, Infrared study of nitrogen monoxide adsorption on palladium ion-exchanged ZSM-5 catalysts, Catal. Lett., 1996, 41, 133–138, DOI:10.1007/BF00811479.
- K. Mandal, Y. Gu, K. S. Westendorff, S. Li, J. A. Pihl, L. C. Grabow, W. S. Epling and C. Paolucci, Condition-Dependent Pd Speciation and NO Adsorption in Pd/Zeolites, ACS Catal., 2020, 10, 12801–12818, DOI:10.1021/acscatal.0c03585.
- D. Mei, F. Gao, J. Szanyi and Y. Wang, Mechanistic insight into the passive NOx adsorption in the highly dispersed Pd/HBEA zeolite, Appl. Catal., A, 2019, 569, 181–189, DOI:10.1016/j.apcata.2018.10.037.
- K. Khivantsev, N. R. Jaegers, L. Kovarik, S. Prodinger, M. A. Derewinski, Y. Wang, F. Gao and J. Szanyi, Palladium/Beta zeolite passive NOx adsorbers (PNA): Clarification of PNA chemistry and the effects of CO and zeolite crystallite size on PNA performance, Appl. Catal., A, 2019, 569, 141–148, DOI:10.1016/j.apcata.2018.10.021.
- M. Ambast, S. A. Malamis and M. P. Harold, Coupled uptake and conversion of C12H26 and NO on Pd/SSZ-13: Experiments and modeling, Chem. Eng. J., 2021, 423, 129958, DOI:10.1016/j.cej.2021.129958.
- R. Villamaina, U. Iacobone, I. Nova, E. Tronconi, M. P. Ruggeri, L. Mantarosie, J. Collier and D. Thompsett, Mechanistic insight in NO trapping on Pd/Chabazite systems for the low-temperature NOx removal from Diesel exhausts, Appl. Catal., B, 2021, 284, 119724, DOI:10.1016/j.apcatb.2020.119724.
- U. Iacobone, I. Nova, E. Tronconi, R. Villamaina, M. P. Ruggeri, J. Collier and D. Thompsett, Appraising Multinuclear Cu2+ Structure Formation in Cu-CHA SCR Catalysts via Low-T Dry CO Oxidation with Modulated NH3 Solvation, ChemistryOpen, 2022, 11, e202200186, DOI:10.1002/open.202200186.
- N. Usberti, F. Gramigni, N. D. Nasello, U. Iacobone, T. Selleri, W. Hu, S. Liu, X. Gao, I. Nova and E. Tronconi, An experimental and modelling study of the reactivity of adsorbed NH3 in the low temperature NH3-SCR reduction half-cycle over a Cu-CHA catalyst, Appl. Catal., B, 2020, 279, 119397, DOI:10.1016/j.apcatb.2020.119397.
- R. Matarrese, L. Castoldi, S. Morandi, P. Ticali, M. C. Valsania and L. Lietti, NO adsorption/desorption pathways over Pd/SSZ-13 low temperature NOx adsorbers investigated by operando FT-IR spectroscopy and microreactor study, Appl. Catal., B, 2023, 331, 122723, DOI:10.1016/j.apcatb.2023.122723.
- U. Iacobone, I. Nova, E. Tronconi, R. Villamaina, M. P. Ruggeri, J. Collier and D. Thompsett, Transient CO Oxidation as a Versatile Technique to Investigate Cu2+ Titration, Speciation and Sites Hydrolysis on Cu–CHA Catalysts: The Cu Loading Effect, Top. Catal., 2023, 66, 761–770, DOI:10.1007/s11244-023-01813-8.
- S. Morandi, L. Castoldi, R. Matarrese and L. Lietti, Novel insights on the Pd speciation in Pd/SSZ-13 and on the role of H2O in the Pd reduction by CO, Spectrochim. Acta, Part A, 2025, 324, 124945, DOI:10.1016/j.saa.2024.124945.
- R. Villamaina, U. Iacobone, I. Nova, M. P. Ruggeri, J. Collier, D. Thompsett and E. Tronconi, Low-T CO Oxidation over Cu−CHA Catalysts in Presence of NH 3: Probing the Mobility of Cu II Ions and the Role of Multinuclear Cu II Species, ChemCatChem, 2020, 12, 3843–3848, DOI:10.1002/cctc.202000991.
- U. Iacobone, W. S. Epling, I. Nova and E. Tronconi, Transient kinetic analysis of CO oxidation over a Cu-SSZ-13 catalyst proves complete conversion of ZCu2+(OH)- cations to binuclear Cu2+ species, Chem. Eng. Sci., 2024, 292, 119970, DOI:10.1016/j.ces.2024.119970.
- F. Gramigni, N. D. Nasello, N. Usberti, U. Iacobone, T. Selleri, W. Hu, S. Liu, X. Gao, I. Nova and E. Tronconi, Transient Kinetic Analysis of Low-Temperature NH3-SCR over Cu-CHA Catalysts Reveals a Quadratic Dependence of Cu Reduction Rates on CuII, ACS Catal., 2021, 11, 4821–4831, DOI:10.1021/acscatal.0c05362.
- N. D. Nasello, U. Iacobone, N. Usberti, A. Gjetja, I. Nova, E. Tronconi, R. Villamaina, M. P. Ruggeri, D. Bounechada, A. P. E. York and J. Collier, Investigation of Low-Temperature OHC and RHC in NH3–SCR over Cu-CHA Catalysts: Effects of H2O and SAR, ACS Catal., 2024, 14, 4265–4276, DOI:10.1021/acscatal.4c00118.
- A. Hindmarsh, {ODEPACK}. A Collection of ODE System Solvers, IMACS Transactions on Scientific Computation, 1983, vol. 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00324a |
| This journal is © The Royal Society of Chemistry 2025 |