
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced H2 recovery by coupling the water–gas shift reaction with in situ CO2 capture and mineralization using earth abundant Ca- and Mg-silicates and hydroxides†
Xun
Gao
a,
Divya
Prasad
a,
Mahadeo A.
Mahadik
a and
Greeshma
Gadikota
 *ab
*ab
aSchool of Civil and Environmental Engineering, Cornell University, Ithaca, NY 14853, USA
bSmith School of Chemical and Biological Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: gg464@cornell.edu; Tel: +1 607 255 4796
First published on 3rd December 2024
Abstract
Decarbonization of clean energy carriers such as H2 by coherent integration of multiphase chemical pathways with inherent carbon mineralization is a thermodynamically downhill pathway designed for a sustainable climate, energy, and environmental future. In this effort, a low-temperature water–gas shift reaction (WGSR) with Pt/Al2O3 catalysts is integrated with in situ carbon mineralization in a multiphase reaction environment. The hypothesis that Pt-based catalysts favor selective formation of H2 over CH4 has been investigated. H2 yields increased by 30.8%, 9.5%, 8.3%, and 1.7% in the presence of Ca(OH)2, Mg(OH)2, Mg2SiO4, and CaSiO3 relative to the blank experiment without the sorbent at constant experimental conditions of 250 °C and reaction time of 12 hours in the presence of Pt/Al2O3 catalyst with initial CO and N2 pressures of 8 bar and 12 bar, respectively. These studies unlock the feasibility of advancing single-step multiphase pathways for enhancing H2 yields with inherent CO2 capture and mineralization for a low carbon and sustainable energy and resource future.
1. Introduction
Demand for low-cost and abundant fossil energy resources has exacerbated greenhouse gas emissions (GHGs) leading to detrimental impacts on climate and the environment.1 To mitigate the detrimental impacts of GHG emissions and realize a carbon-neutral future, it is essential to develop novel chemical pathways to produce clean energy carriers such as H2 while capturing and storing CO2 emissions.2,3 As an alternative to conventional multi-step processes to capture, compress, and store CO2 emissions, intensified approaches that simultaneously inherently capture and store CO2 emissions in a single step via reactive separation are needed.4,5 The abundance of renewable energy resources such as biomethane serves as a viable feedstock for H2 production as an alternative to fossil-derived methane. H2 has emerged as a preferred energy carrier given the high energy density of H2 (122 MJ kg−1) compared to alternative fossil fuel-derived resources5 and the absence of CO2 emissions on combustion.6Current commercial H2 production involves energy-intensive hydrocarbon reforming, by which the non-renewable hydrocarbon fuel is reformed into carbon monoxide (CO) to release H2 over metal catalysts.7 As one of the primary reforming techniques, steam reforming (SR) is widely studied due to its relatively low operating temperature and high purity of the H2 product.8 The general reaction for steam reforming can be represented as follows:
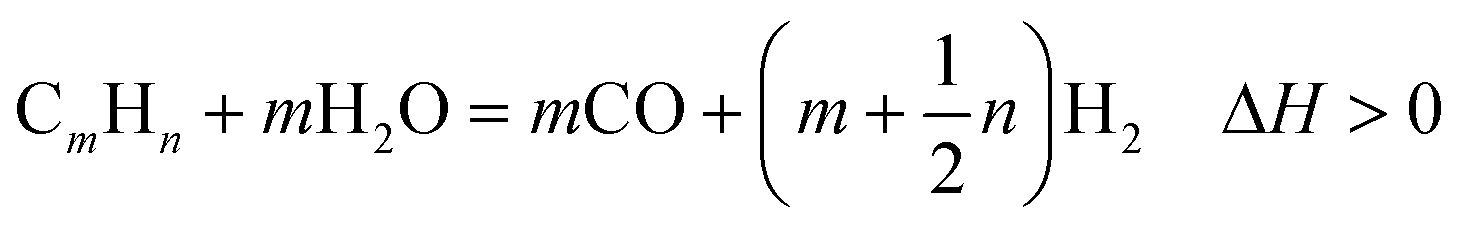 | (1) |
In this expression, when m = 1 and n = 4, the reforming reaction corresponds to steam methane reforming (SMR), which is the most extensively applied approach for industrial H2 production with thermal efficiencies of up to 85%.8 SMR is typically coupled with subsequent water–gas shift reaction (WGSR) and methanation to further enhance H2 yield and purity.9 As an effective supplement to SMR, the WGSR generates additional H2 while simultaneously reducing the concentrations of CO at intermediate temperatures (200–450 °C).10 The literature review by Wagner on catalytic advancements11 and fundamental insights on kinetic rates and challenges towards water gas shift are pioneering studies focused on optimizing this reaction.9,12 Thermodynamically, WGSR is favored at low temperatures because it is a reversible exothermic process using steam as the hydrogen source as eqn (2) indicates:
| CO + H2O → CO2 + H2 ΔH298K = −41.1 kJ mol−1 | (2) |
However, in the low-temperature regime, WGSR is challenged by sluggish kinetics and low CO conversions. The limiting kinetics of WGSR can be overcome by harnessing catalysts to enhance CO conversion kinetics and yields at low temperatures, for which various noble (Pt, Pd, Ru) and non-metal (Cu, Zn, Fe, Cr)-based supported catalysts have been reported in precedent literature.13–15 Especially, Pt-based materials have been reported to be promising metals for WGSR in the medium to low temperature (250–350 °C) regime due to their high stability in oxidizing environments.16
Even though steam methane reforming and WGSR are widely deployed for H2 production, challenges associated with producing high-purity H2 and achieving high selectivity remain.17 The competing co-presence of CO and CO2 along with H2 from gasification and WGSR at the effluent limits H2 selectivity and its yield. Consequently, the reactor effluent gases must be purified by the pressure swing adsorption process operating at high pressures (1 MPa) to produce a significant amount of high-purity H2.
This challenge necessitates the development of sustainable multiphase chemical pathways for selective and energy-efficient reactive separation of H2 from CO2 and CO. Harnessing alkaline materials such as Ca- and Mg-bearing hydroxides and silicates is a thermodynamically favorable pathway to enhance H2 yield. The proposed enhanced WGSR concept works based on the Le Chatelier's principle, in which the reversible gas phase WGSR when integrated with a slurry bearing Ca- and Mg-bearing hydroxides and silicates enhances H2 yield via in situ CO2 capture and mineralization.18 This integrated concept has the potential to significantly improve reaction efficiencies, minimize capital cost, and simplify the process configuration from the two-step conventional catalytic process to a single – step process. This approach also circumvents the thermodynamic limitations of conventional WGSR thereby enhancing the rate of forward reaction for H2 generation, driving the reaction towards the product side by shifting the equilibrium, and allowing high conversions of CO and steam to H2 and solid carbonates.
The approach of coupling carbon mineralization as a pathway to capture and crystallize CO2 emissions as solid carbonates is a significant departure from conventional carbon capture and storage (CCS) technologies in which CO2 is first captured using solvents, sorbents, and membranes and is then injected into geologic reservoirs for storage.19–23 The permanence and stability of solid Ca- and Mg-bearing carbonates24 motivate the coupling of carbon mineralization as a pathway to separate CO2 emissions and enhance H2 yields. Table 1 summarizes the state-of-the-art literature reports focused on the enhancement towards WGS performance and H2 yields.18,25–33
| Feedstock | Sorbents | Catalyst | Temperature [°C] | Pressure [bar] | Sorbents amount [g] | Catalyst amount [g] | H2 yields [%] | COx yields [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| CO2, CO, steam, H2, N2 | LDHs | Iron–chromium | 400 | 28 | 891 | 434 | 99.83 | 0.17% | 18 |
| CO, steam, Ar | LDHs | Cu/ZnO/Al2O3 | 400 | 1 | 48.45 | 48.45 | 99.99 | 0.01% | 25 |
| CO, steam, N2 | CaO/Al2O3 | Ni nanoparticle | 400 | NA | 0.25 | 0.0075 | 76–98 | NA | 26 |
| CO, steam, N2 | CaO/Al2O3 | Pt/Al2O3 | 350 | NA | NA | NA | High purity | NA | 27 |
| CO, steam, Ar | CaO/Ca12Al14O33 | Fe/Mn | 400–700 | 1 | 0.88 | 0.12 | 88–95 | 5–12 | 28 |
| CO, steam | CaO-based | Pd/Ni/Co | 425–550 | 1 | 20 | 1 | 98–99 | 0.35 | 29 |
| CO, steam, H2, He | CaO/NaOH | Pt/ZDC | 300–600 | 1–11 | NA | NA | 51–100 | 0–49 | 30 |
| CO, steam | AMS/MgO/CaCO3 | Cu/Ce0.6Zr0.4O2 | 275–375 | 12 | 2.625 | 2.625 | 86–99 | 1–14 | 31 |
| H2, CO, steam, N2 | Na–Mg based | Cu/ZnO/Al2O3 | 375 | 1 | 5 | 5 | High purity | NA | 32 |
| CO2, N2, H2 | CaO | MOx (Al, Ce, Ti, Zr) | 600–750 | NA | 0.2 | 0.1 | NA | NA | 33 |
| H2, CO, N2 | Mg/Ca hydroxide and silicate | Pt/Al2O3 | 250–300 | 20 | 3 | 3 | 72.67 | 27.33 | This work |
Carbon mineralization involves the reaction between silicate, hydroxide, and oxide minerals bearing divalent cations (e.g., Ca2+, Mg2+ ions) and CO2 to produce the respective carbonates. The negative free energy change of the reaction indicates the spontaneity of the process and the chemical stability of the carbonate products.34 Apart from the spontaneity of the reaction, carbon mineralization has several advantages. First, the natural abundance of starting materials guarantees easy accessibility and affordable prices. Besides, the alkaline minerals have a large CO2 storage capacity, providing an enormous CO2 storage potential worldwide. Additionally, the typical products from carbon mineralization, including silica, carbonates, and metal oxides, usually have multiple industrial applications after purification.
For carbon mineralization, the starting materials could be magnesium silicate minerals such as (Mg,Fe)2SiO4 (olivine), Mg2SiO4 (forsterite), MgO (periclase), CaO (lime), CaSiO3 (wollastonite), and Ca2+- and Fe2+-rich silicates (e.g., larnite). These minerals are known to spontaneously react with atmospheric CO2 to produce solid carbonates.35–37 These geo-inspired mineralization pathways motivated advances in engineered analogs to accelerate mineralization.34,38 Prior work by Gadikota and co-workers showed that olivine carbon mineralization extents as high as 85% are achieved at 185 °C, pCO2 of 139 atm, reaction time of 3 hours, and in 1.0 M NaCl and 0.64 M NaHCO3 solution with particle sizes of 30 μm or less.39–41 The observed high extents of carbon mineralization at the temperature conditions that are similar to that of the water gas shift reaction motivated the coupling of this mineralization pathway.
As an alternative to these hydrothermal routes, direct gas – solid mineralization routes42,43 are explored but challenged by slow kinetics when Mg- or Ca-bearing silicates are used. In this context, sorbent-enhanced water gas shift reactions (WGSR) in direct gas–solid modes were extensively studied.44–47 Compared with the widely used membrane separation approach to separate CO2 in WGSR, the sorbent-enhanced WGSR possesses multiple advantages, including more favorable and uniform kinetics, greater material durability, and larger CO2 capture capacity.30,48 Conventional sorbent-enhanced water–gas shift reactions occur in fluidized bed reactors with steam-saturated CO2 flow.32,49 The selected sorbents are typically metal oxides (e.g., CaO) or layered double hydroxide (LDH) (e.g., hydrotalcite) with high CO2 capture capacity.18,25,26 However, direct gas–solid carbonation is very kinetically limited and tends to be greatly enhanced with the participation of aqueous media, which dissolves the alkaline resource to release metal cations that capture CO2 to form carbonate species.43,50–52
While Ca-bearing oxides have been reported to be effective for enhanced water gas shift reaction in a direct gas–solid reaction mode,27,29,30 the direct use of earth abundant Mg- and Ca-bearing silicates in this mode is significantly challenged by mass transfer limitations and slow kinetics of reaction.39–41 The direct use of Mg- and Ca-bearing silicates circumvents the need to use additional reagents to produce more reactive Ca- and Mg-bearing hydroxides. To accelerate mass transfer and the kinetics of CO2 capture using earth-abundant Mg- and Ca-bearing silicates, aqueous routes which involve the dissolution of silicates to release Mg2+ or Ca2+ ions for capturing CO2 produced from the WGSR to produce the respective carbonates are investigated in this work.53 Prior studies have reported that the dissolution of silicate minerals facilitates more robust mass transfer and accelerates carbon mineralization.54–56 Prior studies have demonstrated enhanced H2 production using Mg(OH)2 for in situ CO2 capture and mineralization.57 However, the influence of directly using of Mg- and Ca-bearing silicates for the enhanced water gas shift reaction has not been reported to date. The conventional hypothesis is that silica precipitated as Mg- and Ca-bearing silicates limit mass transfer and thus lower reactivity with CO2 to produce the respective Ca- and Mg-bearing carbonates. However, this hypothesis has not been evaluated in the context of harnessing earth-abundant Ca- and Mg-bearing silicates to enhance the WGSR with in situ CO2 capture and mineralization, and is therefore, the focus of the proposed investigations.
Several favorable multiphase chemical interactions are necessary for this approach to be feasible. First, the reactivity of CO and steam in the presence of Pt/Al2O3 catalyst needs to be robust in producing CO2 and H2 as the products in sufficient quantities. Second, CO2 needs to be soluble in the aqueous phase to facilitate the forward WGSR for enhancing H2 production. Third, the dissolved CO2 needs to react with Mg- and Ca-bearing silicates to produce the respective carbonates. Fourth, the precipitated silica should not limit mass transfer and the subsequent carbon mineralization behavior. To unlock the full potential of enhanced H2 production coupled with carbon mineralization, it is essential to identify the rate limiting factor and advance strategies to overcome these limitations. To this end, the multiphase reaction pathways are assembled such that the gas phase catalytic WGSR is coupled with slurry phase carbon mineralization as shown in Fig. 1. The associated CO2 from WGSR reacts with water to produce carbonate species, which subsequently react with dissolved calcium or magnesium ions obtained from the sorbents for in situ carbon mineralization. To identify and address the factors limiting H2 production coupled with carbon mineralization, several key research questions are addressed: (i) what are the chemical mechanisms underlying enhanced H2 conversion with in situ carbon mineralization using Mg- and Ca-bearing hydroxides and silicates? (ii) What is the influence of the silicate vs. hydroxide reactivity on enhancing H2 production with carbon mineralization? (iii) In this multi-step reaction pathway, what is the rate limiting step and how can this step be accelerated?
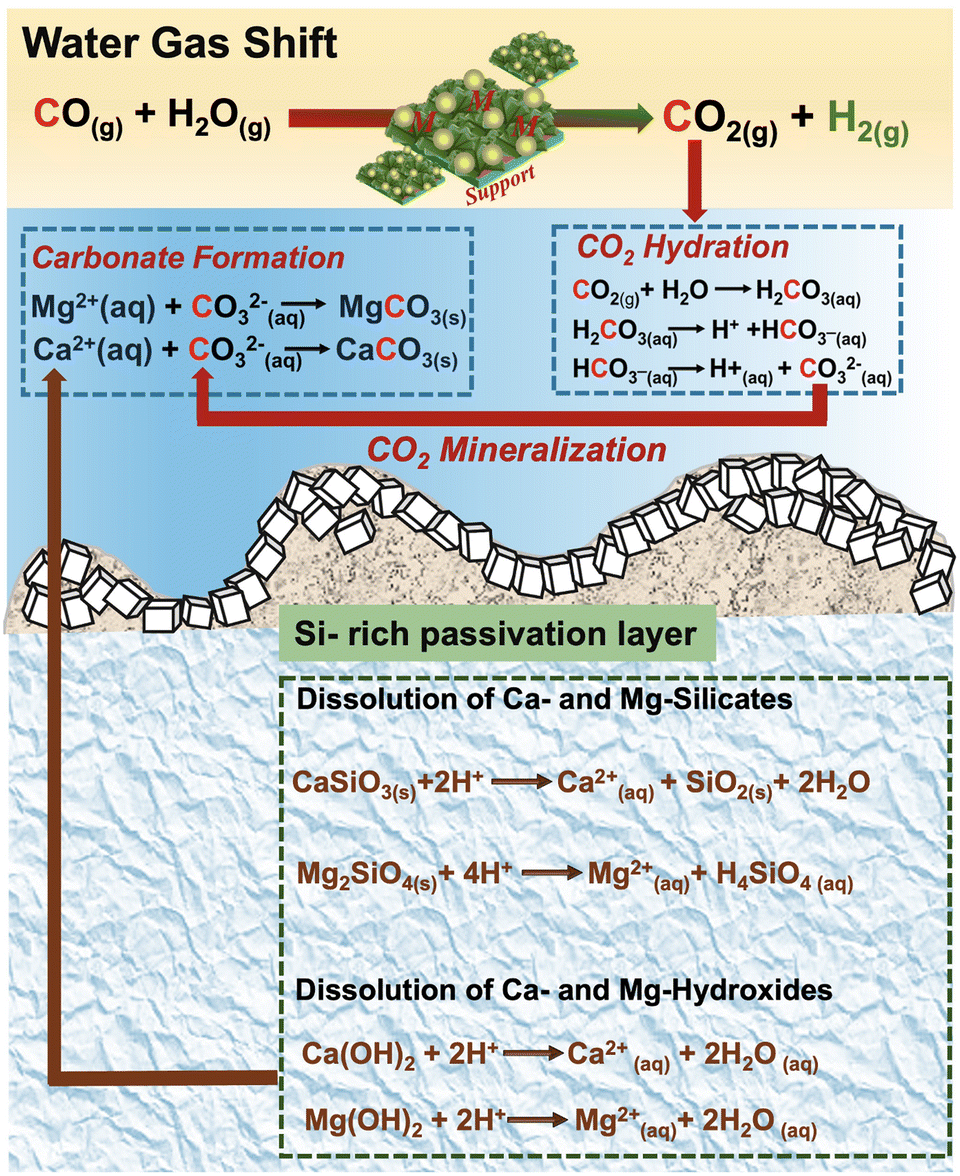 | ||
| Fig. 1 Schematic representation of the pathway to enhance H2 conversion by integrating the water gas shift reaction integrated with carbon mineralization of Ca- and Mg-bearing hydroxides and silicates. | ||
Addressing these questions unlock new insights into the mechanisms and the scientific feasibility of enhancing H2 conversion with in situ CO2 capture and mineralization using Mg- and Ca-bearing silicates and contrasting with that of the respective hydroxides.
2. Experimental methods
2.1 Materials and methods
All experiments to investigate enhancement in H2 conversion with the WGSR coupled with in situ CO2 capture and mineralization are conducted in the presence of platinum on alumina support (Pt/Al2O3) catalyst.27 The Pt/Al2O3 catalyst is selected due to its high activity, superior thermal stability, and high product specificity to H2 and CO2.58–60 Specifically, Pt/Al2O3 catalyst is sized as 3.2 mm pellets with a composition of 0.5 wt% Pt and is procured from Sigma Aldrich, USA. Mg-bearing hydroxide (brucite (Mg(OH)2)) and silicate (forsterite (Mg2SiO4)) used for in situ CO2 capture and mineralization are obtained from Fisher Chemical and Xi'An Function Material Group Co. Ltd, respectively. Ca-bearing hydroxide (portlandite (Ca(OH)2)) and silicate (wollastonite (CaSiO3)) also used for in situ CO2 capture and mineralization are acquired from Thermo Fischer Scientific Co., USA. These Mg- and Ca-bearing silicates and hydroxides are ball-milled in a bench-top mixer mill (8000 M Mixer/Mill® by SPEX® Sample Prep) to obtain raw materials with comparable mean particle sizes (5.57 μm to 10.38 μm). The particle sizes of the milled samples are determined using laser diffraction particle size analyzer (Anton Paar). Deionized water (18.2 MΩ cm, Millipore) is used throughout all the experiments as required. Ultra-high purity CO gas (99.999% purity) is supplied by Airgas Co., USA.2.2 Experimental setup for enhanced WGSR and carbon mineralization
Experiments to investigate enhanced H2 conversion with in situ CO2 capture and mineralization are conducted in a 50 mL stainless steel high-pressure stirred reactor (Micro Bench Top Reactor, Parr Instruments Co., USA) in a multiphase reaction environment. A schematic representation of the experimental setup is shown in Fig. 2. The reactor is a stirred cylindrical system with an inner diameter of 33 mm and a height of 57 mm. It features a 4-blade Rushton turbine stirrer with a 20 mm diameter, positioned 9 mm above the bottom along the central axis of the reactor. Given the stirring rate and the dimensions of the stirrer, efficient mixing and mass transfer in the aqueous phase is expected during the reaction. Simultaneously, CO and N2 are introduced through a gas inlet pipe (inner diameter of 3 mm) into the headspace for gas-phase reactions and to build internal pressure. Elevated internal pressures are preferred for these multiphase reactions, as the increased CO partial pressure shifts the equilibrium of the WGSR towards higher H2 and CO2 production, enhancing the subsequent dissolution and diffusion of CO2. Catalytic conversion of CO and steam occurs in the gas phase in the presence of Pt/Al2O3 catalyst. The surface area, pore volume, and average pore diameter of Pt/Al2O3 catalyst are 163.13 m2 g−1, 0.303 cc g−1, and 6.282 nm, respectively (see Table S1 in ESI†). Prior to CO gas injection, 3 g (0.5 wt%) of the commercial Pt/Al2O3 catalyst is placed in an aluminum basket and suspended in the headspace of the reactor ensuring the basket is not in contact with the sorbent slurry. CO flow rate from the cylinder is controlled by using a mass flow controller (MFC). CO2 capture and mineralization occurs in a slurry phase bearing 3 g of Mg- and Ca-bearing hydroxides and silicates, mixed with 10 mL of deionized water. The initial solid-to-liquid weight ratio is set at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, resulting in mole percentages of 8.48% for Mg(OH)2, 3.70% for Mg2SiO4, 6.80% for Ca(OH)2, and 4.45% for CaSiO3, respectively. The sorbent-to-catalyst ratio is maintained at a consistent weight ratio of 1:1 across all cases.
:10, resulting in mole percentages of 8.48% for Mg(OH)2, 3.70% for Mg2SiO4, 6.80% for Ca(OH)2, and 4.45% for CaSiO3, respectively. The sorbent-to-catalyst ratio is maintained at a consistent weight ratio of 1:1 across all cases.
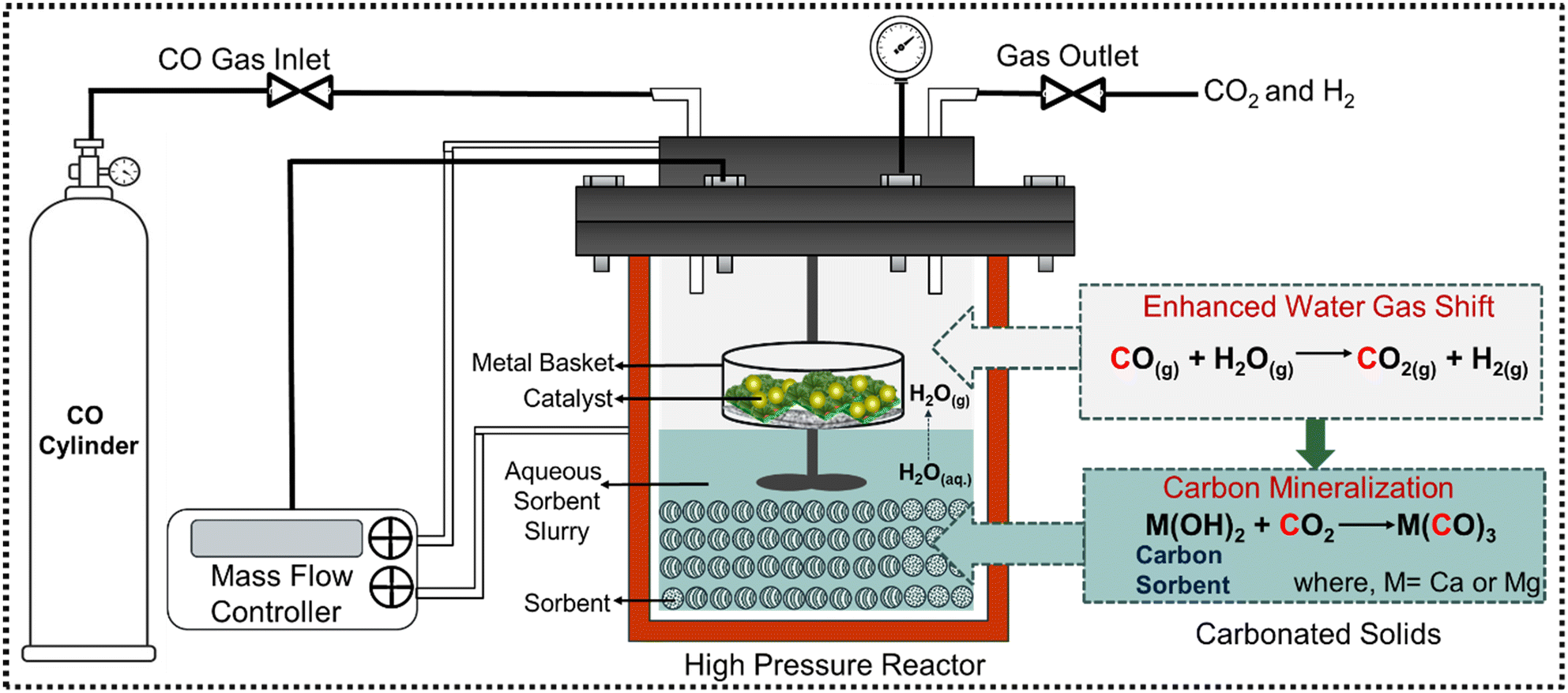 | ||
| Fig. 2 Schematic representation of the experimental setup for coupling the water gas shift reaction (WGSR) with in situ CO2 capture and mineralization for enhanced H2 conversion. | ||
The headspace of the reactor is purged with CO gas for 15 minutes to evacuate gaseous impurities before it is pressurized and sealed tightly. After reaching the desired pressure set point for CO at 20 bar, the heater is switched on to achieve a setpoint of 250 °C with a constant stirring rate of 300 rpm, signaling the start of the experiment. Steam is generated by the vaporization of water in the slurry. The corresponding partial pressure of steam is determined by the temperature in the steam table (Table S2.1 and 2.2†). The reactor outlet is sealed with heating tape to avoid the possibility of steam condensation and is connected directly to a gas chromatograph to analyze the concentrations of gaseous products. The pH of the slurry increased to 9.95 for Mg(OH)2, 8.69 for Mg2SiO4, 12.37 for Ca(OH)2, and 8.98 for CaSiO3, respectively. The experiments are performed at different reaction times ranging from 3 to 12 hours. After the completion of the reaction, the reactor is cooled down to ambient temperature. Following this, the outlet valve is slowly opened to purge the gaseous products in the micro-GC and the carbonate-bearing solid products are collected for further analysis and characterization. Additionally, enhancement in H2 yield resulting from CO2 capture and mineralization is investigated as a function of reaction time and the alkaline sorbent type.
Additional experiments are conducted to determine the exact moles of each gas in the reactor. The experimental set-up is the same as described previously. In this approach, 8 bar of CO is injected into the reactor with 12 bar of N2. N2 is used as a reference due to its inert nature and unchanged pressure over the course of the reaction. This approach enables the calculation of the moles for the other gaseous components based on the initial N2 content. The temperature is increased to a setpoint of 300 °C, and the reaction is performed for 12 hours, while keeping other reaction conditions unchanged, to investigate the influence of Mg- and Ca-bearing hydroxides and silicates on enhancing H2 conversions.
2.3 Product analysis and characterization
After the reaction, the compositions of effluent gases are analyzed by micro gas chromatography (GC) with the calibration data shown in Fig. S1.† The changes in the composition and structure of the Mg- and Ca-bearing hydroxides and silicates are investigated using infrared (IR) spectroscopy collected in an attenuated total reflection (ATR) mode using an attenuated total reflection-Fourier transform infrared spectrometer (ATR-FTIR, Nicolet™ iS50, Waltham, MA), and X-ray diffractometry (XRD, Bruker D8 Advance ECO powder diffractometer, Bruker), respectively. Furthermore, the morphology and particle sizes are determined using the field emission scanning electron microscope (FESEM, LEO 1550 FESEM, Bruker) and particle size analyzer (Anton Paar). The surface area and the pore size distributions of the sorbents before and after the experiments were determined by N2 adsorption–desorption isotherms using the Brunauer–Emmett–Teller technique (BET) (Quantachrome Autosorb iQ Analyzer, Boynton Beach, FL) with the BJH model. Prior to measuring the adsorption–desorption isotherms, the samples were outgassed at 140 °C for 12 hours. A ±5% BET instrument error was accounted for all the analysed samples. The concentration of formate in the aqueous phase was measured using a 500 MHz 1H NMR spectrometer (Bruker AVIII 500) using a solvent suppression method. The calibration line for formate concentration and its corresponding slope, determined using DMSO as internal standards, are shown in Fig. S2.†3. Results and discussion
3.1 Reaction pathways involved in coupled WGSR with in situ CO2 capture and mineralization for enhanced H2 conversion
The multiphase reactions involved in coupling the WGSR with in situ CO2 capture and mineralization for enhanced H2 conversion include: (i) low-temperature conversion of CO and steam over Pt/Al2O3 catalyst to produce CO2 and H2; (ii) dissolution of Mg- and Ca-bearing hydroxides and silicates to release Mg2+ and Ca2+ ions, respectively and promote alkalinity; (iii) dissolution of the produced gaseous CO2 in the alkaline aqueous phase; (iv) reactivity of the hydrated CO2 with Mg2+ and Ca2+ ions to mineralize into Mg and Ca-carbonates; and (v) generation of high-purity H2 due to equilibrium shift due to CO2 capture and mineralization.The motivation for using Pt/Al2O3 catalyst stems from the need to direct the formation of H2 as opposed to CH4.61 Prior analogous studies showed that Pt or Ni/Al2O3 catalysts are effective in reforming aqueous biomass oxygenates such as ethylene glycol and glycerol to produce H2 and CO2 preferentially over CH4.62 Therefore, in this study, Pt/Al2O3 catalysts are used to lower the activation energy barrier, accelerate reaction kinetics, suppress the formation of CH4, and promote the formation of H2 and CO2. Furthermore, the in situ capture and conversion of gaseous CO2 in the presence of alkaline Mg- and Ca-bearing hydroxides and silicates to produce the respective carbonates favors the forward equilibrium shift towards more CO conversion and H2. Dissolution of Mg – or Ca – bearing hydroxides and silicates releases Mg2+ and Ca2+ cations which capture and solubilize CO2 to produce the respective solid carbonates. For example, in this work, the pH of the alkaline slurry is observed to be in the range of 8.69–12.37 for the Mg- and Ca-bearing hydroxides and silicates. This pH is ideal to capture and solubilize CO2 from the gas phase to produce the respective (bi)carbonate species.
To probe the mechanisms underlying enhanced H2 conversion by coupling the WGSR with in situ CO2 capture and mineralization, the first set of experiments are conducted by injecting CO gas. Steam required for the WGSR is provided by the slurry. The ratio of CO to steam is calculated and quantified from the steam table (Table S2.1 and 2.2, ESI†). In this approach, the effluent gas compositions are measured using micro gas chromatography (GC), and the molar percentage of each phase is normalized by the total amount of H2, CO, and CO2. This approach provides insights into the relative abundance of the gas phase compositions. In this scenario, the relative gas phase composition of H2 can be elevated if CO2 is absorbed by the liquid phase, even if CO conversion is unchanged. Therefore, CO conversion is determined for quantifying the enhancement in WGSR using the following expression:
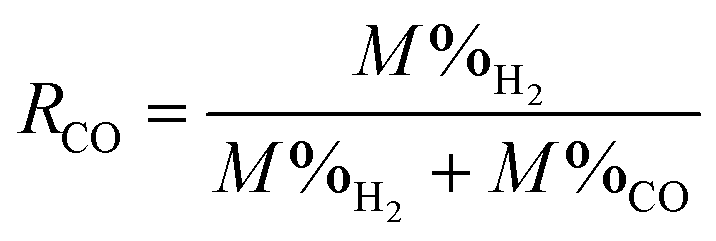 | (3) |
Additionally, the exact number of moles of each gas is necessary to determine the theoretical maximum yield of H2 and gaseous CO2 compositions. To this end, additional experiments are conducted with a moderate amount of N2 in the system as the baseline. In this approach, the absolute number of moles of N2 is constant throughout the experiment.
This approach enables the determination of the exact moles of H2, CO, and CO2 by the van der Waals equation (as shown below) from the normalized mole percentages after the multi-step reactions.
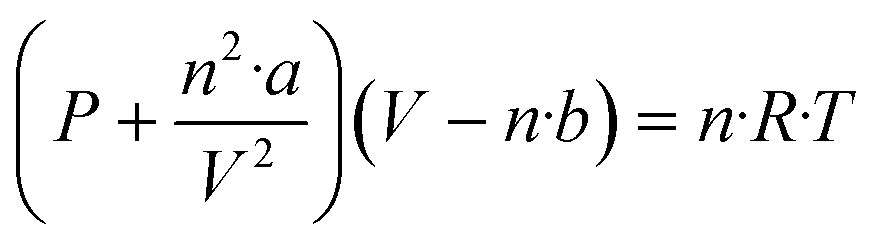 | (4) |
3.2 Thermodynamic considerations for enhanced H2 conversion by coupling the WGSR with in situ CO2 capture and mineralization
The reaction conditions for enhanced H2 conversion by coupling the WGSR with in situ CO2 capture and mineralization is equilibrium-limited and dictated by the thermodynamics for WGSR to form CO2 and H2 and CO2 capture and mineralization to form Mg- and Ca-carbonates.48 The detailed reactions associated with coupling the WGSR with in situ CO2 capture and mineralization are below:| Mg(OH)2(s) + CO(g) + H2O(g) → MgCO3(s) + H2(g) + H2O(l) | (5) |
| Mg2SiO4(s) + 2CO(g) + 2H2O(g) → 2MgCO3(s) + SiO2(s) + 2H2(g) | (6) |
| Ca(OH)2(s) + CO(g) + H2O(g) → CaCO3(s) + H2(g) + H2O(l) | (7) |
| CaSiO3(s) + CO(g) + H2O(g) → CaCO3(s) + SiO2(s) + H2(g) | (8) |
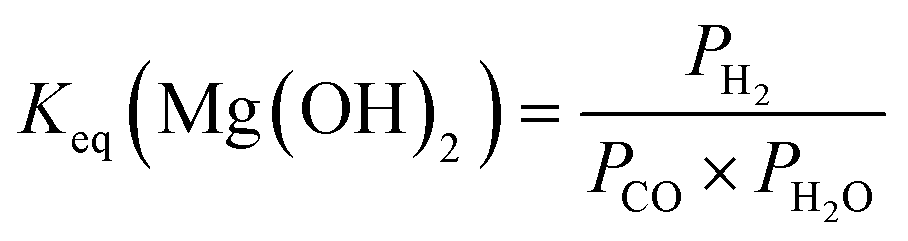 | (9) |
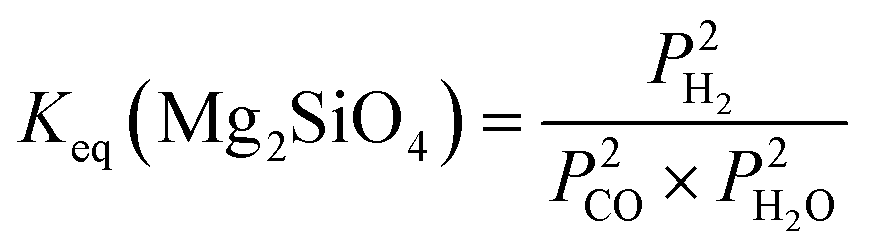 | (10) |
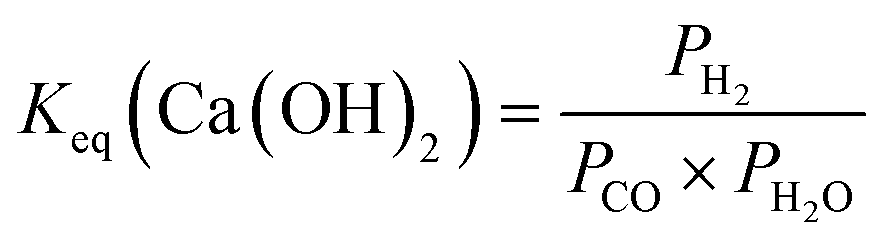 | (11) |
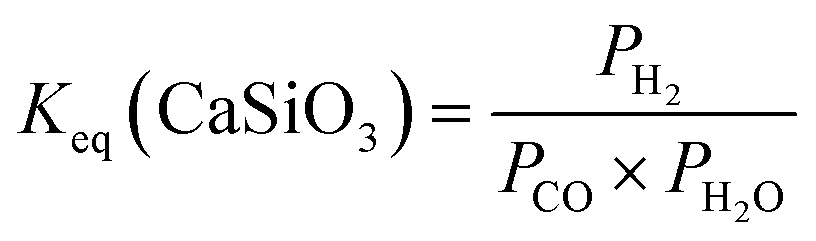 | (12) |
 | (13) |
:1 are used to determine theoretical CO conversions using Mg(OH)2, Mg2SiO4, Ca(OH)2, and CaSiO3 as alkaline sources (Fig. 3).
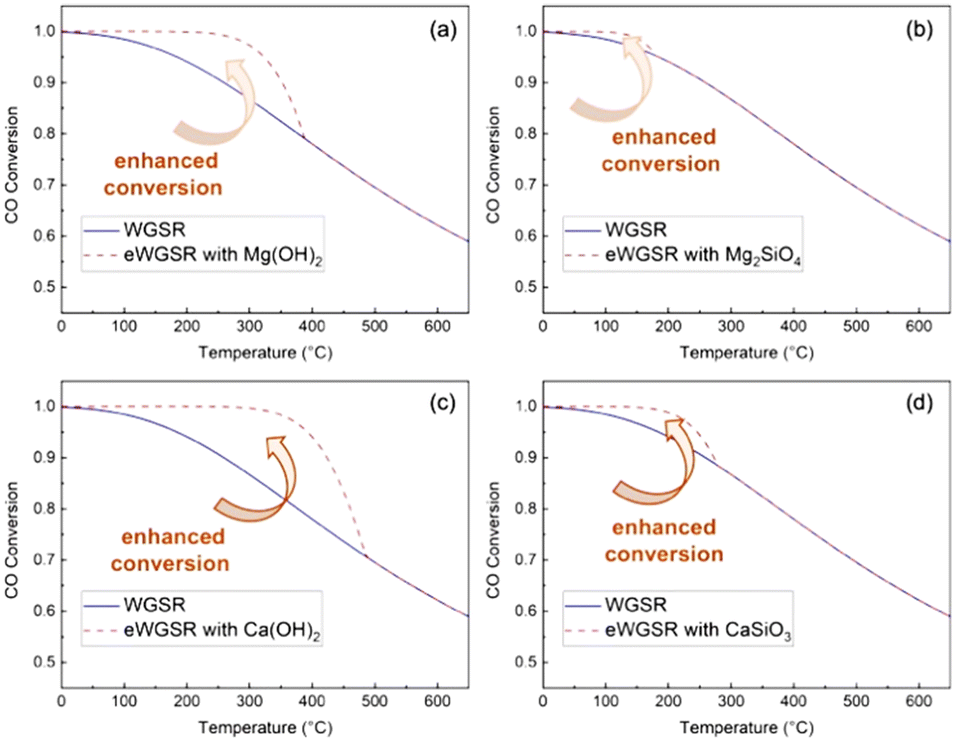 | ||
| Fig. 3 Enhancement in WGSR as determined from CO conversions based on thermodynamic evaluations in gas–solid reaction mode using (a) Mg(OH)2, (b) Mg2SiO4, (c) Ca(OH)2, and (d) CaSiO3 for the in situ CO2 capture. The initial steam-to-CO ratio is 1:1 with adequate sorbents for carbon mineralization. Results for Mg(OH)2 agree with prior studies.64 | ||
Notably, the WGSR and enhanced WGSR (or eWGSR) conversions define the theoretical percentage of CO converted to CO2 at the equilibrium. CO2 conversions are calculated from the partial pressure of H2, CO, and steam. The theoretical CO conversion of Mg(OH)2 confirms prior reported calculations.64 CO conversions at equilibrium decrease with increasing temperature due to the exothermic nature of the reactions (Fig. 3). Compared with conventional WGSR, a significant enhancement in CO conversion is noted at temperatures below 350 °C, indicating that Mg- and Ca-bearing hydroxides and silicates are effective in aiding CO (and H2) conversion by capturing CO2. It is interesting to note that at temperatures below 350 °C, Mg(OH)2 and Ca(OH)2 enhance CO conversion at 250 °C and 300 °C due to in situ capture of gaseous CO2.
In contrast, the enhancement in CO conversion realized using Mg- and Ca-bearing silicates is achieved below 200 °C. Based on these thermodynamic analyses, it is evident that CO conversion can be enhanced using Ca- and Mg-bearing silicates in gas–solid reaction modes. To overcome the mass transfer limitations associated with gas–solid reactions, multiphase gas–liquid–solid reactions are proposed to accelerate the kinetics of WGSR when coupled with aqueous CO2 capture and mineralization pathways at temperatures below 300 °C. These experimental conditions are a significant departure from conventional sorbent-enhanced WGSR operating in a gas–solid mode to capture CO2.
3.3 Enhancement in WGSR using Mg(OH)2 and Mg2SiO4
To enhance H2 conversion with in situ CO2 capture, it is essential to use a suitable catalyst that will convert CO and steam to produce H2 and CO2 and an alkaline source to capture CO2.65 Prior studies have reported enhancement in H2 conversion via sorbent – enhanced WGSR using Ca-bearing sorbents.46 Enhancement in H2 yields were also reported by coupling the WGSR with slurry phase CO2 capture and mineralization using Mg(OH)2.66 However, Mg(OH)2 needs to be directly mined or extracted from earth – abundant Mg-silicates. Eliminating this additional mining or extraction step and directly using earth – abundant Mg-silicate minerals for enhancing H2 conversion while capturing CO2 is less explored but highly transformative. To address this knowledge gap, experiments are conducted to contrast the influence of Mg(OH)2vs. Mg2SiO4 on enhancing H2 yield with in situ CO2 capture. The abundance of olivine (Mg,Fe)2SiO4 bearing forsterite (Mg2SiO4) in the natural environment motivates the direct use of these materials for CO2 capture.39 To investigate the enhancement in H2 yield with CO2 capture, the gaseous products at the effluent are analyzed. Following this, the structures and morphologies of the carbonate-bearing products are characterized separately.Initially, a blank experiment is performed by considering only the WGSR over Pt/Al2O3 catalyst, to investigate its catalytic activity in the low-temperature regime. Fig. 4(a) represents the gas mole percentages when feeding only 20 bar CO at 250 °C for 9 hours. Fig. 4(b) shows the conversion of CO for the blank experiment and the cases with Mg(OH)2 and Mg2SiO4. In the absence of the alkaline resource, CO conversion of 72.4% is noted (Fig. 4(b)). Gas analyses did not indicate any CH4 formation demonstrating that the Pt/Al2O3 catalyst is effective in converting CO and steam to CO2 and H2. The observed higher selectivity towards H2 using low-temperature Pt/Al2O3 catalyst is consistent with previous literature on WGSR.27 Compared with the blank experiment without any alkaline source, WGSR reactions in the presence of a slurry bearing Mg(OH)2 or Mg2SiO4 enhanced CO conversion and H2 yield and lowered the concentration of gaseous CO2 due to in situ CO2 capture. In contrast to the blank experiment without the alkaline source, CO conversions in the presence of Mg(OH)2 and Mg2SiO4 at 250 °C are 81% and 79.7%, respectively, as seen in Fig. 4(b). The molar compositions of H2 and CO2 in the gas phase are 50.8% and 30.3% for H2 and CO2 for the blank experiment. In contrast, H2 concentrations increase by 18% and 9.5%, and CO2 concentrations decrease by 15.9% and 6.1% in the presence of Mg(OH)2 and Mg2SiO4 compared to the blank experiment (Fig. 4(a)).
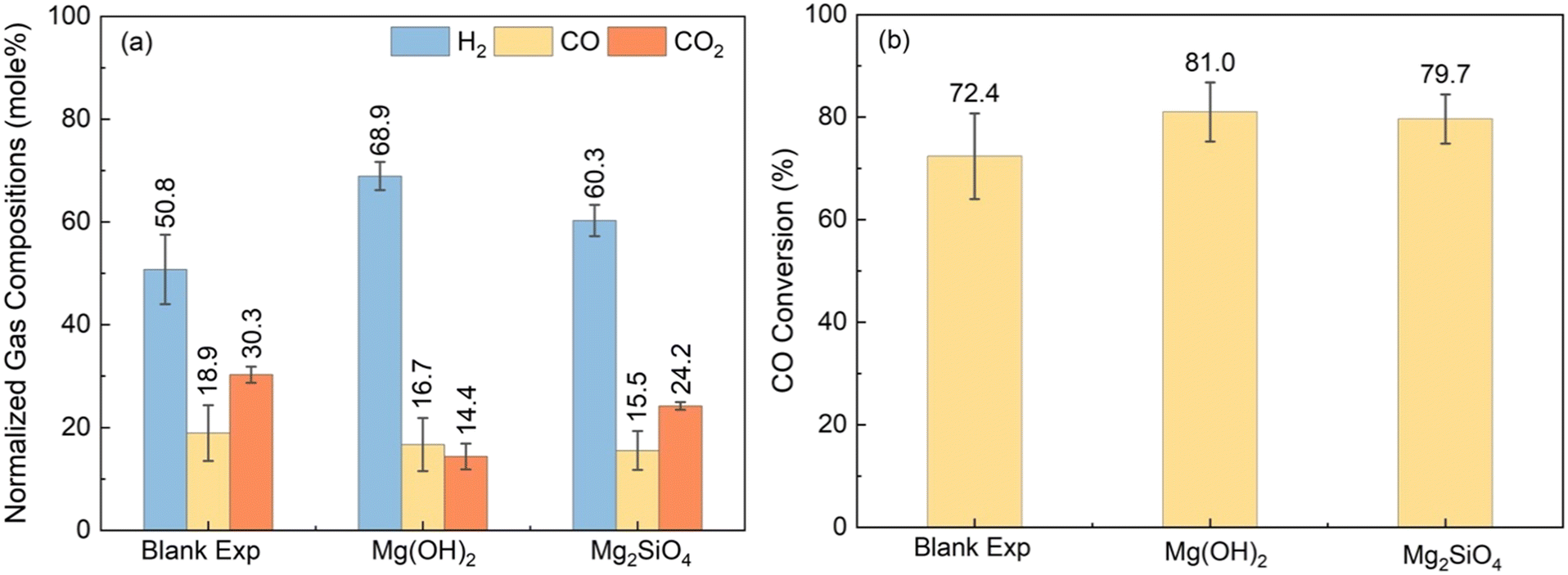 | ||
| Fig. 4 Contrasting the influence of Mg(OH)2 and Mg2SiO4 on the water gas shift reaction (WGSR) as determined from H2, CO, and CO2 compositions in (a) and CO conversions in (b). Experiments are conducted at 250 °C, 20 atm for 9 h. The blank experiment is conducted over 3 g of Pt/Al2O3 catalyst without alkaline source. The Mg(OH)2 and Mg2SiO4 cases are conducted over 3 g of Pt/Al2O3 catalyst with a sorbent-to-liquid weight ratio of 3:10, leading to mole percentages of 8.48% for Mg(OH)2 and 3.70% for Mg2SiO4, respectively. | ||
The relative composition of H2 in the gas phase is 8.6% higher when Mg(OH)2 is used for CO2 capture as opposed to Mg2SiO4. CO2 concentrations are nearly 10% lower when Mg(OH)2 is used as opposed to Mg2SiO4. Higher H2 concentrations and lower CO2 concentrations in the presence of Mg(OH)2versus Mg2SiO4 are attributed to the faster dissolution kinetics of Mg(OH)2 relative to Mg2SiO4.67 The enhanced release of Mg2+ ions favors CO2 capture and the forward WGSR reaction which results in enhanced H2 conversions and CO consumption (Fig. 4(a) and (b)).
3.4 Enhancement in H2 conversion with in situ CO2 capture as a function of time using Mg(OH)2 and Mg2SiO4 materials
One of the key uncertainties in enhancing H2 conversions with in situ CO2 capture is the influence of the reactivity of the alkaline resources in hydrothermal environments. In this context, even though it is known that Mg(OH)2 is more soluble and has faster kinetics of dissolution compared to Mg2SiO4, the kinetics associated with accelerating WSGR are not well understood. Several factors could be competing in this integrated and enhanced WGSR approach.First, the gas phase conversion of CO and steam to H2 and CO2 can be limiting. Second, the solubility of CO2 and reactivity of Ca- and Mg-bearing silicates and hydroxide to capture CO2 can be slow. Based on the changes in the compositions of the gas phase over time, the factors limiting the conversion of CO and H2O to H2 with in situ CO2 capture can be determined. To this end, experiments are conducted with Mg(OH)2 and Mg2SiO4 at 250 °C, 20 atm as a function of reaction time. The normalized gas phase compositions for reactions conducted at 3, 6, 9, and 12 hours are shown in Fig. 5(a) and (b) in the presence of Mg(OH)2 and Mg2SiO4, respectively.
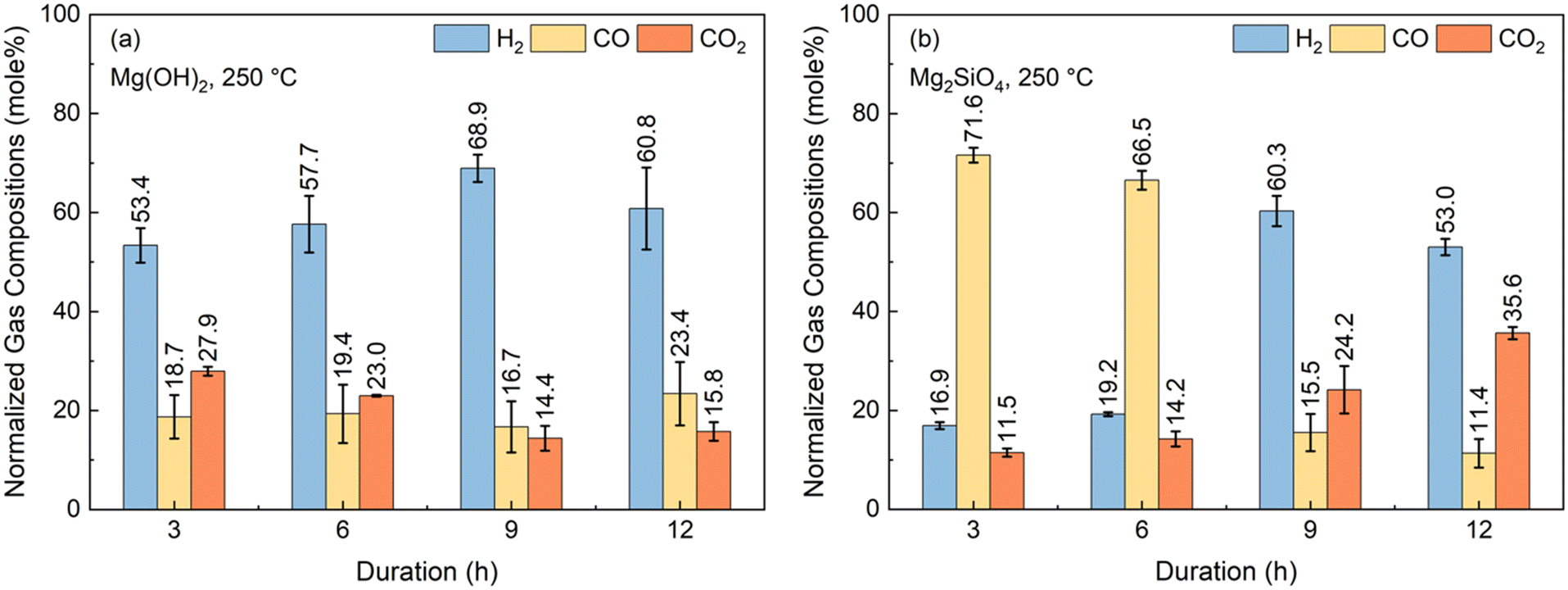 | ||
| Fig. 5 Determination of time dependence on the concentrations of H2, CO, and CO2 when the WGSR is coupled with in situ CO2 capture and mineralization using (a) Mg(OH)2 and (b) Mg2SiO4. The Mg(OH)2 and Mg2SiO4 cases are conducted at 250 °C and 20 atm over 3 g of Pt/Al2O3 catalyst with a sorbent-to-liquid weight ratio of 3:10, leading to mole percentages of 8.48% for Mg(OH)2 and 3.70% for Mg2SiO4, respectively. | ||
In the first three hours of the reaction with Mg(OH)2 as the alkaline source, H2 composition is 53.4% while that of CO and CO2 are 18.7 and 27.9%, respectively. An increase in H2 composition to 68.9% and a decrease in CO and CO2 compositions to 16.7% and 14.4%, respectively are noted in the 9-hour cases. These results indicate that Mg(OH)2 is effectively enhancing CO conversions and H2 yields with inherent CO2 capture. Therefore, the WGSR is not the limiting factor in H2 conversion in the first nine hours of the reaction. However, increasing the reaction time from 9 hours to 12 hours, lowered H2 compositions from 68.9% to 60.9%. CO and CO2 compositions increased from 16.7 to 23.4% and 14.4 to 15.8%, respectively, when the reaction time is increased from 9 hours to 12 hours. These results suggest that CO2 capture is the likely limiting factor as the reaction time increases from 9 to 12 hours. This observation can be attributed to the fact that at increased reaction times, the availability of Mg2+ ions for CO2 capture is likely decreasing over time. Slower dissolution rates of Mg(OH)2 over time due to mass transfer arising from the formation of Mg-carbonates68 or due to the saturation of the solution with Mg2+ ions are attributed to the decrease in H2 and increase CO2 compositions at 12 hours.
In contrast, Mg2SiO4 for in situ CO2 capture yielded significantly different results compared to the use of Mg(OH)2 (Fig. 5(b)). H2 compositions are 16.9% and 19.2% respectively, at 3 hours and 6 hours of reaction time, while CO compositions are 71.6% and 66.5% in the presence of Mg2SiO4. These results indicate that the WGSR is the limiting step in the first 6 hours of reaction in the presence of Mg2SiO4. This is likely due to the slower dissolution behavior of Mg2SiO4 to release Mg2+ ions compared to that of Mg(OH)2. As a result, in situ CO2 capture and subsequent enhancement in H2 yields is limited in the presence of Mg2SiO4 in the first six hours of the reaction. At 9 hours of reaction time, H2 composition is 60.3%. CO composition of 66.5% at 6 hours decrease to 15.5% at 9 hours of reaction time (Fig. 5(b)). These results indicate that while the kinetics of WGSR are slower in the first 6 hours of the reaction in the presence of Mg2SiO4 compared to in the presence of Mg(OH)2, comparable compositions are achieved when the reaction time is 9 hours.
As in the case of Mg2SiO4, a decrease in H2 compositions and increase in CO2 compositions is observed when the reaction time is 12 hours. H2 compositions decrease from 60.3% to 53.0% while CO2 compositions increase from 24.2% to 35.6% at 12 hours of reaction. The relative decrease in H2 compositions and increase in CO2 compositions at 12 hours is attributed to the mass transfer limitations associated with the dissolution of Mg- silicate and Mg-hydroxide. Therefore, these studies indicate that WGSR can be the initial slow step in the presence of Mg2SiO4 as opposed to Mg(OH)2 during the first 6 hours of reaction. Non-monotonic changes in the concentrations of H2, CO, and CO2 observed as the reaction time increases from 9 hours to 12 hours are likely due to limitations from the slow dissolution of Mg(OH)2 and Mg2SiO4 to release Mg2+ ions for CO2 capture.
3.5 Influence of Mg- and Ca-bearing hydroxides and silicates on enhanced H2 conversion with in situ CO2 capture
Contrasting the influence of Mg- and Ca-bearing silicates and hydroxides on enhanced H2 conversion coupled with in situ CO2 capture can unlock new opportunities for the utilization of a wide range of alkaline feedstocks. Ca-bearing hydroxides and silicates are abundant in fly ash and slags.69 Mg-bearing hydroxides and silicates are abundant in ultramafic and mafic mine tailings.70 To this end, experiments are conducted at 300 °C in the presence of 8 bar of CO and 12 bar of N2 in the gas phase and Mg(OH)2, Ca(OH)2, Mg2SiO4, or CaSiO3 to capture CO2 in a slurry phase over a reaction time of 12 hours, as noted in section 3.1. Pt/Al2O3 catalyst is used as a low-temperature WGSR catalyst. N2 gas is used as a reference due to its inert nature and constant quantity before and after the reaction which enables the determination of the moles of gas products formed. The influence of Mg(OH)2, Ca(OH)2, Mg2SiO4, or CaSiO3 on compositions of H2, CO, and CO2 are shown in Fig. 6.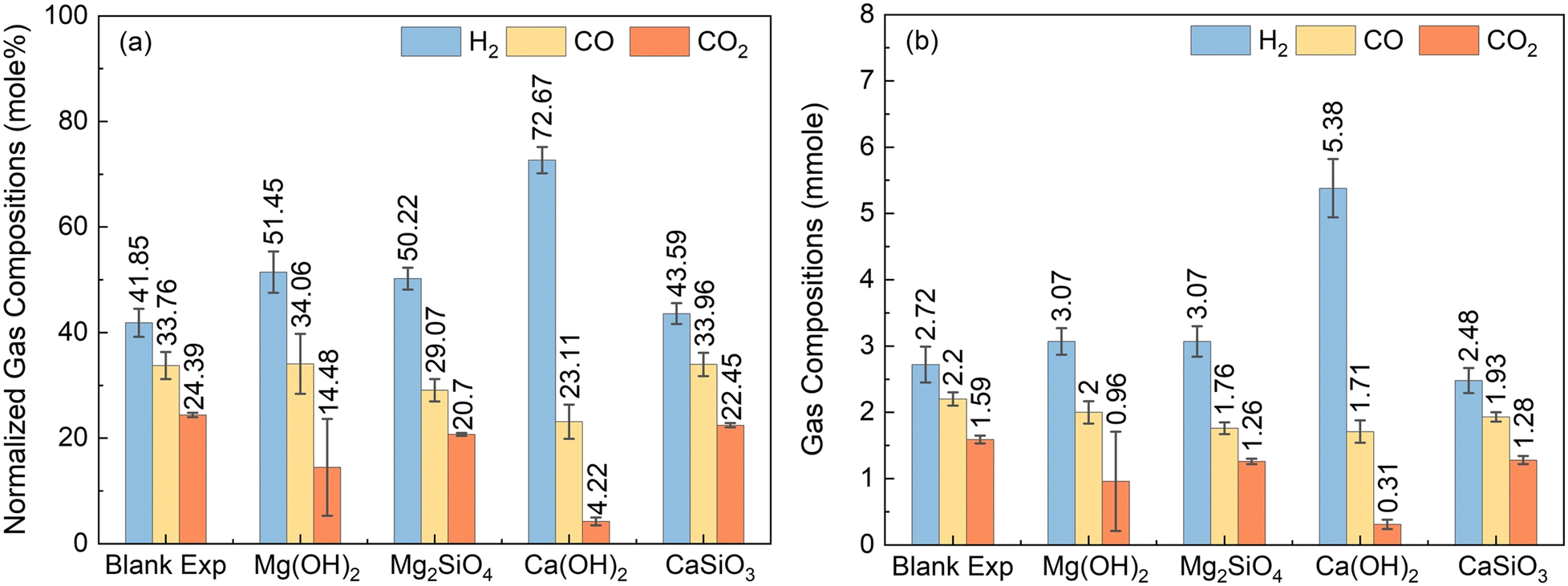 | ||
| Fig. 6 Contrasting the influence of alkaline resources such as Mg(OH)2, Mg2SiO4, Ca(OH)2, and CaSiO3 on enhancing H2 yield with in situ CO2 capture and mineralization where (a) represents normalized gas mole percentages and (b) represents exact gas moles. Experiments are conducted at 300 °C, 20 atm for 12 h. The blank experiment is conducted over 3 g of Pt/Al2O3 catalyst without any alkaline source. The test experiments are conducted over 3 g of Pt/Al2O3 catalyst with a sorbent-to-liquid weight ratio of 3:10, leading to mole percentages of 8.48% for Mg(OH)2, 3.70% for Mg2SiO4, 6.80% for Ca(OH)2, and 4.45% for CaSiO3, respectively. | ||
The influence of WGSR alone in the absence of Ca- and Mg-bearing hydroxide and silicate determined at the same experimental conditions as the others showed that H2, CO, and CO2 account for 41.85%, 33.76%, and 24.39%, respectively (Fig. 6(a)). Relative to this blank experiment, the highest enhancement in H2 conversion and CO2 suppression was observed in the presence of Ca(OH)2. In this best case scenario with Ca(OH)2, H2 yield is 72.67% which is 30.82% higher compared to the blank experiment. CO2 yields are suppressed by 20.17% to 4.22% relative to the blank experiment indicating that Ca(OH)2 in the slurry is highly effective in capturing CO2 emissions. Significant enhancement in WGSR is also evident from the changes in CO compositions which are lowered by 10.65% compared to the blank experiment. In comparison to Ca(OH)2, the H2 yields are ∼50–51% in the presence of Mg(OH)2 and Mg2SiO4 which are lower. Interestingly, CO compositions are 5% lower when Mg2SiO4 is used compared to Mg(OH)2, indicating WGSR is enhanced in the presence of Mg2SiO4. However, the higher solubility of Mg(OH)2 compared to Mg2SiO4 enhances CO2 capture lowering CO2 compositions by 6.22% when Mg(OH)2 is used related to Mg2SiO4.67
The reactivity of CaSiO3 in enhancing H2 yield and suppressing CO2 emissions is lower compared to the other alkaline sources. Compared to the blank experiment, the composition of H2 is 1.74% higher, CO is similar, and CO2 is ∼2% lower. While these results indicate that CaSiO3 enhances H2 conversion and enables CO2 capture, substantial changes relative to the blank experiment are not observed. The composition data in mmoles are reported in Fig. 6(b) for use in subsequent modeling efforts. The results reported in Fig. 6 indicate that the effectiveness of the alkaline sources in enhancing H2 yields while capturing CO2 emissions follows this order: CaSiO3 < Mg2SiO4 < Mg(OH) 2< Ca(OH)2. As hypothesized, the reactivity of Ca- and Mg-bearing hydroxides in enhancing H2 yield and suppressing CO2 emissions is higher compared to that of the corresponding silicates. The higher solubility and faster dissolution rates of Ca- and Mg-bearing hydroxides compared to the corresponding silicates facilities enhanced CO2 capture and H2 conversion.67
3.6 Mechanistic insights underlying enhancement in H2 conversion coupled with in situ CO2 capture and mineralization
The key reasons for the differences in the reactivity of Ca- and Mg-bearing silicates vs. hydroxides are the slower kinetics of dissolution of silicates and the formation of a silica passivation layer which is known to limit mass transfer.71,72Fig. 7 represents the multiphase chemical reaction mechanisms associated with carbon mineralization by harnessing hydroxides versus silicates. While the CO2 hydration reactions to produce bicarbonate and carbonate species remain unchanged, the leaching of the metal into the solution results in the formation of a silica-rich layer in the case of silicates unlike in hydroxides. It is interesting to note that despite the slower kinetics of Mg-silicate dissolution and the formation of a silica layer that can limit mass transfer, comparable gas phase compositions of H2, CO, and CO2 are noted with Mg(OH)2 and Mg2SiO4 (Fig. 6). However, the kinetics associated with enhancing H2 conversion with in situ CO2 capture and mineralization can differ significantly as noted in Fig. 5. The slow kinetics of WGSR observed up to 6 hours in the presence of Mg2SiO4 are attributed to limited availability of Mg2+ ions to capture CO2 and facilitate the forward reaction. However, beyond 6 hours of reaction time, the observed enhancement in H2 yields and corresponding decrease in CO2 emissions is attributed to the availability of sufficient Mg2+ ions to capture CO2 and aid the forward WGSR to produce H2.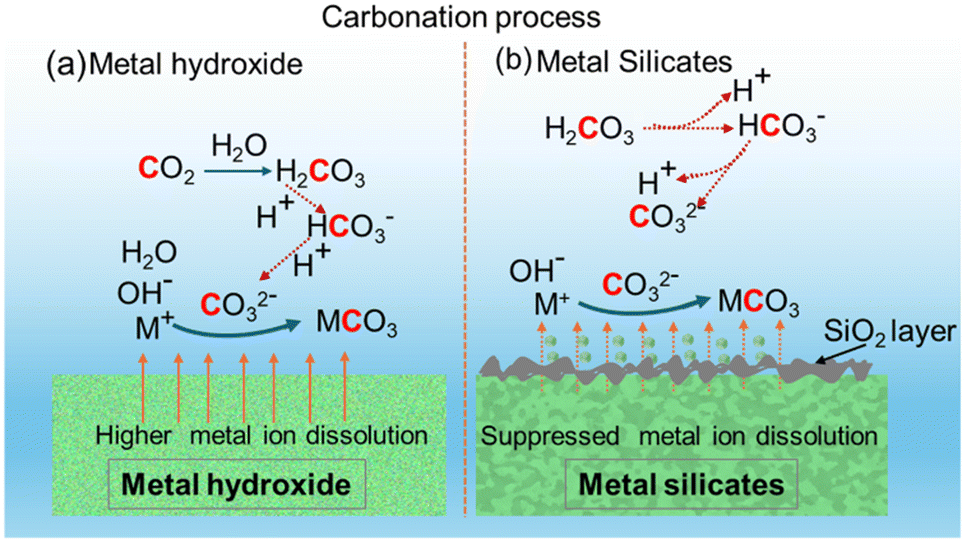 | ||
| Fig. 7 Schematic diagram of surface reactions, dissolution, and mineralization process over (a) metal hydroxides and (b) metal silicates sorbents. | ||
Prior studies have shown that temperatures above 100 °C and high partial pressures of CO2 aid the carbon mineralization of olivine.39,73 The solubility of CO2, and dissolution of Mg2SiO4 to release Mg2+ ions for capturing CO2 and producing carbonates at the experimental conditions used in this study are analogous to those reported in prior studies. Therefore, despite the slow initial kinetics of Mg2SiO4, the release of Mg2+ ions over time results in comparable H2, CO, and CO2 compositions as when Mg(OH)2 is used. It is also interesting to note that Mg2SiO4 is more effective in enhancing WGSR compared to CaSiO3. Prior studies reported that CaSiO3 is reactive at temperatures as low as 110 °C for carbon mineralization, unlike Mg2SiO4.74 Possible reasons for the lower-than-expected reactivity of CaSiO3 are the formation of a silica passivation layer that is thicker and limits mass transfer at the experimental conditions reported in this study. To uncover if CO2 uptake by the alkaline slurry results in the formation of solid carbonates, detailed structural and morphological analyses are investigated and reported in the following section.
3.7 Structural and morphological analyses of the alkaline sources
To determine if the captured CO2 only dissolves in the aqueous phase or reacts to produce solid carbonates, the changes in the structural and morphological features of the alkaline resources are determined. The crystalline features in unreacted and reacted Ca- and Mg-bearing silicates and hydroxides using X-ray diffraction (XRD) measurements (Fig. 8). As shown in Fig. 8(a), the unreacted Mg(OH)2 exhibits the typical diffractogram of hexagonal Mg(OH)2 brucite structure with characteristic XRD peaks at 33.2°, 38.1°, 50.6°, 58.7°, 68.3°, and 72.1° (see peaks in black). These peaks are well in accordance with the reported literature (PDF 44-1482).75 The diffractogram of the reacted solid (peak in red) revealed the existence of magnesite (MgCO3) peaks along with suppression of Mg(OH)2 intensities. This co-existence indicates the dissolution of Mg(OH)2 and the capture and conversion of CO2 to produce MgCO3.73 To investigate if MgCO3 is formed starting from Mg2SiO4 precursors, XRD data of the unreacted and reacted materials are collected and shown in Fig. 8(b). The X-ray diffraction pattern corresponding to the monoclinic structure of unreacted Mg2SiO4 is shown in Fig. 8(b).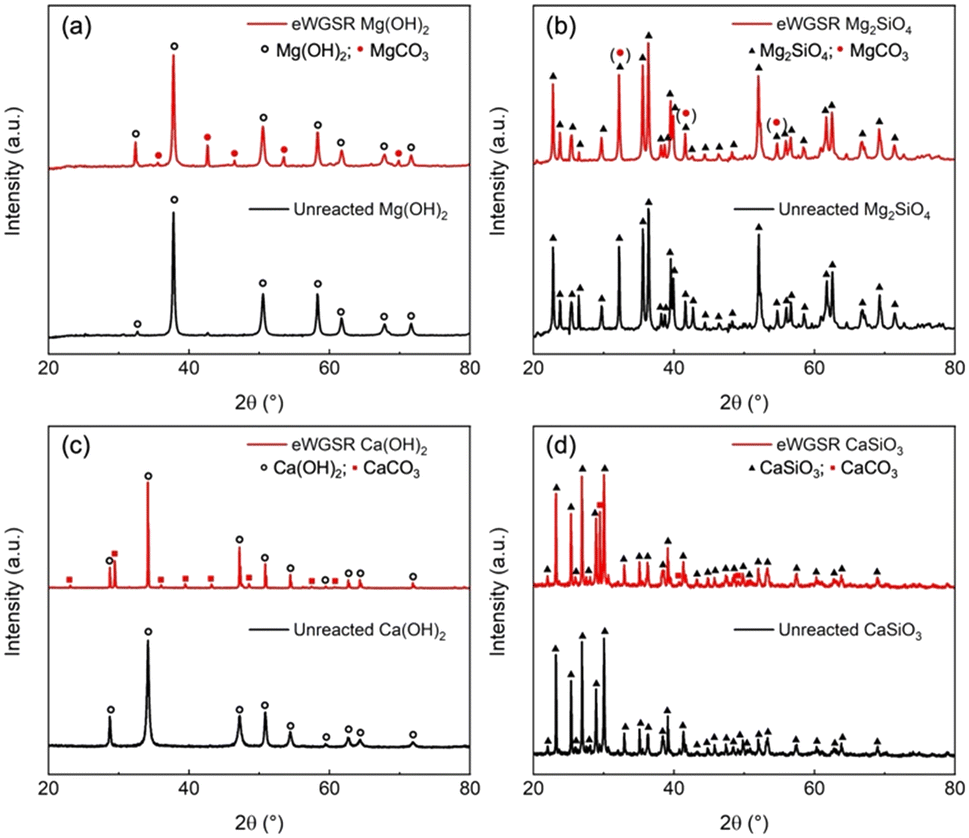 | ||
| Fig. 8 Determination of the crystalline phases before and after reaction starting with (a) Mg(OH)2, (b) Mg2SiO4, (c) Ca(OH)2, and (d) CaSiO3 using X-ray diffraction (XRD) analyses. | ||
The reacted Mg2SiO4 product exhibits a very similar scattering pattern as the unreacted material, which can results from overlapping between (104) peak of the generated magnesite and (130) peak of the residual Mg2SiO4.76,77 To further distinguish the different phases and prove the formation of carbonate, additional characterization is required. Interestingly, the absence of the nesquehonite (MgCO3·3H2O) or hydromagnesite (Mg5(CO3)4(OH)2·4H2O) phases in our case is due to the higher stability of anhydrous magnesite (MgCO3) at elevated temperatures and pressures.39 Additionally, the formation of Mg-carbonate can be influenced by numerous other factors, including the reaction time, ionic strength or the addition of reagents including salts.39 Thus, in this study, the formation of anhydrous magnesite (MgCO3) can be attributed to the application of high temperatures of 250–300 °C with elevated CO2 partial pressures in the range of 10–20 atm for reaction durations of 9–12 hours.
The structural arrangement characterization of the Ca cases shows a similar trend with Mg cases, in which the anhydrous calcite (CaCO3) is the unique carbonate product due to the favorable reaction conditions. Fig. 8(c) illustrates the XRD peaks of unreacted Ca(OH)2 at 28.7°, 34.1°, 47.2°, 50.6°, 54.3°, 62.3°, and 64.3° corresponds to the (100), (101), (102) (110), (111), (201) and (112) planes, respectively. Several studies have reported similar patterns of Ca(OH)2.78,79 After the enhanced WGSR, the reacted product shows the presence of both Ca(OH)2 and CaCO3 phases (JCPDS 84-1276 and 85-1108, respectively). The occurrence of peaks at 23.1°, 29.5°, 31.6°, 36.1°, 39.5°, 43.3°, 47.2°, 47.6°, 48.6°, 57.5°, 61.1°, and 64.5° corresponds to (012), (104), (006), (110), (114), (202), (024), (018), (116), (112) (119), and (300) planes, respectively. These XRD patterns corresponded to the rhombohedral crystal structure of CaCO3.80 Furthermore, Fig. 8(d) shows the diffraction peaks of unreacted sorbents can be indexed as the CaSiO3.81 For the reacted CaSiO3, the XRD peaks resemble the co-occurrence of CaSiO3 and CaCO3 phases, which is a low-temperature polymorph of calcium carbonate (JCPDS 00-005-0586).82
Furthermore, the morphologies of the reacted Mg(OH)2, Ca(OH)2, Mg2SiO4, and CaSiO3 materials are determined using FE-SEM to identify carbonate-bearing phases. Fig. 9(b) shows the cube-like structure of MgCO3 with the appearance of residual Mg(OH)2 after carbon mineralization, confirming the carbonate formation after the enhanced WGSR. Similarly, the cube-shaped MgCO3 particles emerged in the case of reacted Mg2SiO4 materials (Fig. 9(d)).
 | ||
| Fig. 9 Morphology of (a) unreacted Mg(OH)2; (b) reacted Mg(OH)2; (c) unreacted Mg2SiO4; (d) reacted Mg2SiO4; (e) unreacted Ca(OH)2; (f) reacted Ca(OH)2; (g) unreacted CaSiO3; (h) reacted CaSiO3 determined by scanning electron microscope measurements. | ||
Interestingly, the granular particles covered on cubic MgCO3 particles can be attributed to the residual Mg2SiO4 and associated SiO2 layer resulting from silicate dissolution. Similar phenomena are also observed in the reacted Ca(OH)2 and CaSiO3 materials (Fig. 9(f) and (h)). Additionally, the large hexagonal particles indicate the significant dissolution and recrystallization of initial Ca(OH)2 sorbents (Fig. 9(f)), and the elongated particles correspond to the residual CaSiO3 materials (Fig. 9(g) and (h)). Interestingly, the XRD analysis does not show the clear existence of crystalline phases rich in silica, indicating the potential presence of an amorphous silica-rich layer or glassy SiO2 coating that may cause the suppression in metal dissolution in CaSiO3, Mg2SiO4, responsible for the low extents of carbon mineralization.
As a further clarification of the sorbents' morphological evolution, the surface area and the pore distribution determined by BET analysis are listed in Table 2. The decreased surface area in Mg(OH)2, Mg2SiO4, and Ca(OH)2 cases indicates the formation of larger carbonate particles like MgCO3 and CaCO3 (Fig. 9(a)–(f)). Notably, the significant surface area reduction in the Ca(OH)2 case aligns with its more effective CO2 mineralization and the corresponding greater carbonate formation. In contrast, due to the larger particle size and limited pore volume in the unreacted CaSiO3 compared with the generated CaCO3, the small increase in the surface area and pore volume after reaction are attributed to the dominance of dissolution over carbon mineralization (Fig. 9(g) and (h)). Furthermore, the pore size distributions determined using the BJH model are illustrated in Fig. S4.† The unreacted hydroxide sorbents exhibit relatively narrow pore distributions, centered at approximately 4.01 nm for Mg(OH)2 and 3.72 nm for Ca(OH)2. Post-reaction, significant pore structure rearrangements are observed, leading to reductions in both pore volume and diameter, particularly for Ca(OH)2. Additionally, the formation of MgCO3 results in more well-ordered pore and particle size distributions compared to the Mg2SiO4 precursor. Conversely, significant changes in the pore size distribution of CaSiO3 before and after reaction are not observed due to the relatively low extent of carbon mineralization.
| Unreacted alkaline sources | Reacted alkaline sources | |||||
|---|---|---|---|---|---|---|
| Surface area (m2 g−1) | Pore volume (cm3 g−1) | Mean pore diameter (nm) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Mean pore diameter (nm) | |
| Mg(OH)2 | 9.78 | 0.047 | 4.01 | 6.48 | 0.05 | 3.93 |
| Mg2SiO4 | 17.41 | 0.050 | 2.81 | 14.93 | 0.08 | 3.93 |
| Ca(OH)2 | 24.29 | 0.107 | 3.72 | 4.81 | 0.01 | 1.50 |
| CaSiO3 | 5.00 | 0.020 | 2.66 | 6.20 | 0.02 | 3.94 |
Attenuated total reflection Fourier transform infrared (ATR-FTIR) analysis is also performed on unreacted sorbents and reacted products to confirm carbonate formation after the enhanced WGSR. As shown in Fig. 10(a) and (c), unreacted hydroxide sorbents exhibited only one peak between 3600–3700 cm−1, indicating the purity of starting materials (Mg(OH)2, Ca(OH)2).83 On the other hand, the occurrence of new peaks around 1400–1500 cm−1, 850–890 cm−1, and 700–750 cm−1 corresponds to C–O asymmetric stretching vibrations, the C–O bending vibration, and the in-plane bending vibration of O–C–O, respectively, in the carbonated products formed after the enhanced WGSR.84,85 Furthermore, peaks shown in ATR-FTIR of unreacted Mg2SiO4 sorbents around 600–650 cm−1 are consistent with SiO4 bending and stretching modes, respectively, while the peak around 1000 cm−1 represents asymmetric stretching modes of Si–O–Si.86,87 As shown in Fig. 10(b), despite its very low intensity, the peak at 1100 cm−1 could be interpreted as a symmetric C–O stretching vibration band indicating the carbon mineralization of Mg2SiO4.84 Moreover, in the case of CaSiO3 materials, the presence of peaks approximately 600–700 cm−1, 900 cm−1 corresponds to the symmetrical stretching of O–Si–O bonds and Si–O–Ca bonds, while the peaks between 1000–1100 cm−1 correspond to a symmetrical stretching of Si–O–Si bonds, respectively.88 As shown in Fig. 10(d), the typical peaks from silicate remained constant after the enhanced WGSR with a little shoulder around 1400 cm−1, indicating the asymmetric stretching mode of C–O, proving the carbon mineralization of CaSiO3 material.85
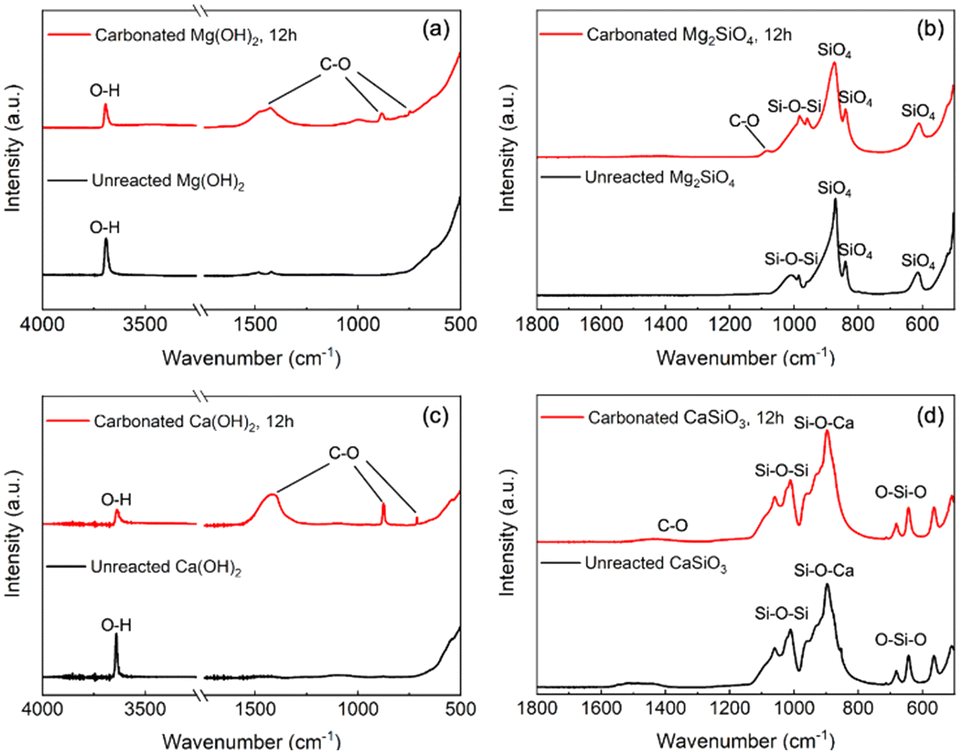 | ||
| Fig. 10 Identification of different functional groups using attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy measurements in unreacted and reacted (a) Mg(OH)2; (b) Mg2SiO4; (c) Ca(OH)2; (d) CaSiO3 materials. | ||
Further, to investigate the effect of eWGSR on the metal hydroxides and metal silicates, XPS analyses are conducted. The XPS spectra were calibrated based on the carbon C–C peak (284.8 eV).89 Fig. S5(a1) and (a2)† shows the O 1s deconvolution result for unreacted and reacted Mg(OH)2 materials. The high-resolution XPS spectrum of unreacted Mg (OH)2 exhibit a peak at binding energy of 531.7 eV which suggests the existence of hydroxyl (OH) species. Furthermore, the reacted Mg(OH)2 materials showed slight changes in O 1s spectrum (Fig. S5(a2)†). Binding energy (BE) peaks are observed at 531.8 eV and 533.34 eV which correspond to hydroxyl (OH) and carbonate (CO32−) species, respectively.90,91 Similar trends are observed within the unreacted and reacted Ca(OH)2 materials. The hydroxyl (OH) peak at 531.9 eV exclusively appears in the unreacted Ca(OH)2 while the hydroxyl peak at 531.8 eV and carbonate peak at 532.9 co-exists in the reacted Ca(OH)2 material (Fig. S5(b1) and (b2)†). To further determine the influence of enhanced WGSR on metal silicates (CaSiO3 and Mg2SiO4), the valance state of the oxygen in the unreacted and reacted silicates are studied using XPS analyses. Fig. S6† shows the carbon (C1s) spectra from unreacted and reacted Mg2SiO4, CaSiO3 materials consisting of two main components arising from C–C (∼284.4 eV) and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (carboxyl, ∼288.6 eV).92,93 Compared to unreacted counterparts, the reacted materials exhibit higher intensity signal of C 1s indicating the presence of carbonate (CO32−) species. The C 1s peak values observed between 289.5 eV and 289.7 eV are well-matched with the carbonate (CO32−) species in the reported literature.94,95
C (carboxyl, ∼288.6 eV).92,93 Compared to unreacted counterparts, the reacted materials exhibit higher intensity signal of C 1s indicating the presence of carbonate (CO32−) species. The C 1s peak values observed between 289.5 eV and 289.7 eV are well-matched with the carbonate (CO32−) species in the reported literature.94,95
Insights into the changes in the structure of silicon are obtained from XPS analyses. Fig. S7(a1)† shows that O 1s peak at 531.35 eV corresponds to the SiO4 tetrahedra in the unreacted Mg2SiO4.96 Additionally, the O1s peak tail at a higher binding energy region around 533.6 eV could result from minor amorphous SiO2 content.97 On the other hand, the reacted Mg2SiO4 materials ((Fig. S7(a2)†) show deconvoluted peaks at 531.1, 532.3, and 533.5 eV which corresponds to the residual Mg2SiO4, the generated carbonate and the amorphous SiO2 resulting from silicate dissolution.98–101 On the contrary, Fig. S7(b1)† shows that unreacted CaSiO3 exhibits a SiO2 peak at 533.5 eV other than the typical SiO3 peak at 532 eV, indicating the presence of an amorphous phase in the unreacted sorbent.97,102 After the enhanced WGSR, the O1s peaks of the reacted CaSiO3 shift towards a high binding energy range. More specifically, the existence of deconvoluted O1s peaks at 531.4 eV, 532.9 eV, and 533.3 eV correspond to the residual SiO3, generated carbonate and SiO2, which confirms carbonate formation and the dissolution of calcium silicate material.
Further, Fig. S7(c1) and (d1)† depicts the existence of Si2p broad peak at the binding energy of 102.7 eV and 103.2 eV for unreacted Mg2SiO4 and CaSiO3, respectively.103 It's worth noting that the SiO2 peaks around 104 eV appear in both the silicates, corresponding to the results from O1s peak.104 However, after carbon mineralization, Si 2p deconvoluted spectra shows the diminished SiO4 or SiO3 peaks with intensified SiO2 peaks for both silicates, indicating the dissolution of silicate and the generation of Si-rich passivation layer.105,106 Additionally, the relatively high amorphous SiO2 component in the unreacted CaSiO3 compared to Mg2SiO4 likely suppresses the dissolution of CaSiO3, thus resulting in lowering H2 yields compared to Mg2SiO4 materials.
To quantitatively evaluate the carbonate phases, the carbon/metal ratios are calculated before and after the enhanced WGSR based on the atom% from XPS (Fig. S8†). The detected carbon phase in the unreacted carbonate-free sorbents can be attributed to the adventitious carbon on samples' surface due to the carbon contamination during air exposure.107 However, neglecting the impact from adventitious carbon, the carbon/metal atomic ratio after the enhanced WGSR increased to 0.58 with Mg(OH)2, 0.78 with Ca(OH)2, 0.06 with Mg2SiO4, and 0.1 with CaSiO3, respectively. In terms of the carbonate fraction, the increased carbon/metal ratio of Mg2SiO4 requires doubling to compare with other sources due to its doubled stoichiometric coefficient of metal ion, which results in a 0.12 increase in Mg2SiO4 carbon/metal atomic ratio after the enhanced WGSR. In this case, the varying trend in carbon/metal atomic ratio increases perfectly aligns with the enhanced WGSR: Ca(OH)2 > Mg(OH)2 > Mg2SiO4 > CaSiO3, indicating the strong positive correlation between the enhanced H2 yields and carbonate formation.
3.8 Insights on the mechanisms of the enhanced WGSR and in situ CO2 capture and carbon mineralization
Given the multiphase reaction described in Fig. 1, the key reaction mechanisms involved in the enhanced WGSR can be delineated as CO oxidation and CO2 capture. To be more specific, the CO oxidation proceeds via two potential approaches simultaneously: (i) the gaseous-phase WGSR with the assistance of noble-metal (Pt/Al2O3) catalyst (eqn (1)) and (ii) the aqueous-phase CO reaction to form formate followed by subsequent formate decomposition (eqn (14) and (15)).108| CO(aq) + OH−(aq) → HCOO−(aq) | (14) |
| HCOO−(aq) + H2O(aq) → CO2(g) + H2(g) + OH−(aq) | (15) |
On the other hand, the generated CO2 from both CO oxidation mechanisms further hydrated to form bicarbonate and carbonate species and released protons into the aqueous phase (eqn (16)).110 Simultaneously, the dissolution of the alkaline resources released the metal cations (M2+), which could be enhanced by the increasing proton level. Consequently, the metal cations combine with carbonate species (CO32−, HCO3−) precipitated out when the solubility limit is reached (eqn (17)). In our case, the carbonate products tend to be in anhydrous form due to their favored chemical stability at the operating temperature (250–300 °C) in this multiphase reaction environment,111 which is also confirmed by the characterization of the solid products.
| CO2(aq) + H2O(l) → HCO−3(aq) + H+(aq) → CO2−3(aq) + 2H+(aq) | (16) |
| M2+(aq) + CO2−3(aq) → MCO3(s) | (17) |
4. Conclusions
In this study, the feasibility of the enhanced WGSR integrated with in situ thermodynamically downhill CO2 capture and mineralization is investigated in the presence of Mg- and Ca-hydroxides and silicates to co-produce H2 and carbonate-bearing materials. Calibrated tuning of the multiphase chemical interactions is unlocked by deriving the thermodynamical models of the multiphase reactions, where the theoretical prediction of the CO conversion is mutually corroborated with the experimental results. For example, 30.82% more H2 yields with 20.17% less CO2 emission achieved with Ca(OH)2 compared with independent WGS reaction without in situ CO2 capture and mineralization. Thus, these studies proved the hypothesis that the in situ capture and mineralization of gaseous CO2 depletes CO2 gas compositions and favors the forward reaction to produce H2. The higher solubility and dissolution rates of Ca- and Mg-hydroxides favors enhanced H2 yields. Comparable compositions of H2 are achieved with Mg(OH)2 and Mg2SiO4 when reacted for 12 hours at 300C despite initial slower kinetics of CO conversion with Mg2SiO4 in the first 6 hours of the reaction.Among the studied alkaline resources for enhanced WGSR, Ca(OH)2 exhibited unbeatable CO2 capture over the other sorbents (CO2 compositions: Ca(OH)2 (4.22%) < Mg(OH)2 (14.48%) < Mg2SiO4 (20.7%) < CaSiO3 (22.45%) < blank experiment (24.39%) respectively), when feeding a mixture of CO and N2 at 300 °C and 20 bar. Additionally, the temporal evolution in H2 yields and CO2 capture revealed that the kinetic limitation of the overall reaction was dependent on the sorbent dissolution behavior, especially for the silicate sorbents. The amorphous Si-rich layer generated from silicate dissolution and mineralization passivated the alkaline resources and restricted the rate of enhanced WGSR, particularly in the case of CaSiO3. The dissolution of Mg- and Ca-bearing hydroxides and silicates, formation of the silica passivation layer, and anhydrous metal carbonate products in the reacted alkaline resources are confirmed from structural and morphological analyses. Detailed characterization and analyses confirmed that anhydrous carbonates (MgCO3, CaCO3) are the dominant carbonate – bearing products without any associated hydrated carbonate formations. Quantitative analyses of the reacted products showed that the carbonate content corresponded directly to the trend in H2 yields. Evidence of comparable H2 yields with Mg2SiO4 and Mg(OH)2 indicates the feasibility of directly using silicates for enhanced WGSR as opposed to extracting Mg(OH)2 from Mg2SiO4 resources, thus enabling more material and energy efficient pathways for producing H2 with inherent CO2 capture and storage in the form on durable Mg- and Ca-bearing carbonates. This approach can also unlock new opportunities in harnessing renewable biomethane for co-producing H2 with inherent CO2 capture and mineralization to produce carbonates for use in construction materials by harnessing energy from renewable sources, thus enabling carbon-negative pathways for a sustainable energy and resource future.
Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Author contributions
Greeshma Gadikota – supervision, conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, validation, visualization, writing – original draft, writing – review & editing. Xun Gao – conceptualization, data curation, formal analysis, investigation, methodology, resources, software, validation, visualization, writing – original draft, writing – review & editing; Divya Prasad – investigation, validation, visualization, writing – review & editing; Mahadeo A. Mahadik – investigation, validation, visualization, writing – review & editing.Conflicts of interest
Greeshma Gadikota is the co-founder of Carbon To Stone which is commercializing technologies for industrial decarbonization and carbon management. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
All the analytical lab work has been performed at the School of Civil and Environmental Engineering, Cornell University. The authors acknowledge the use of shared facilities at the Cornell Center for Materials Research (CCMR). The DOE CAREER Award supports G. G. and X. G.'s contributions through the Office of Science: DE-SC0020263. The authors also gratefully acknowledge the support of Ivan Kuzmenko at APS and Peilong Lu at Cornell University for assisting in this effort.References
- R. E. H. Sims, H. H. Rogner and K. Gregory, Energy Policy, 2003, 31, 1315–1326 CrossRef.
- International Energy Agency (IEA), Global Energy Review: CO2 Emissions in 2021, IEA, 2022 Search PubMed.
- D. Prasad, K. N. Patil, N. K. Chaudhari, H. Kim, B. M. Nagaraja and A. H. Jadhav, Catal. Rev.: Sci. Eng., 2022, 64, 356–443 CrossRef CAS.
- M. A. Rosen and S. Koohi-Fayegh, Energy, Ecol. Environ., 2016, 1, 10–29 CrossRef.
- M. A. Pellow, C. J. M. Emmott, C. J. Barnhart and S. M. Benson, Energy Environ. Sci., 2015, 8, 1938–1952 RSC.
- I. K. Kapdan and F. Kargi, Enzyme Microb. Technol., 2006, 38, 569–582 CrossRef CAS.
- P. Nikolaidis and A. Poullikkas, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CrossRef CAS.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- M. I. Temkin, Adv. Catal., 1979, 28, 173–287 CAS.
- C. Rhodes, G. J. Hutchings and A. M. Ward, Catal. Today, 1995, 23, 43–58 CrossRef CAS.
- C. Ratnasamy and J. P. Wagner, Catal. Rev.:Sci. Eng., 2009, 51, 325–440 CrossRef CAS.
- H. Bohlbro, J. Catal., 1964, 3, 207–215 CrossRef CAS.
- W. Xu, R. Si, S. D. Senanayake, J. Llorca, H. Idriss, D. Stacchiola, J. C. Hanson and J. A. Rodriguez, J. Catal., 2012, 291, 117–126 CrossRef CAS.
- P. Mierczynski, W. Maniukiewicz and T. P. Maniecki, Cent. Eur. J. Chem., 2013, 11, 912–919 CAS.
- D. W. Jeong, W. J. Jang, J. O. Shim, W. B. Han, H. S. Roh, U. H. Jung and W. L. Yoon, Renewable Energy, 2014, 65, 102–107 CAS.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y. W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589, 396–401 CAS.
- L. Chen, Z. Qi, S. Zhang, J. Su and G. A. Somorjai, Catalysts, 2020, 10, 858 CrossRef CAS.
- E. R. Van Selow, P. D. Cobden, P. A. Verbraeken, J. R. Hufton and R. W. Van Den Brink, Ind. Eng. Chem. Res., 2009, 48, 4184–4193 CrossRef CAS.
- J. Gibbins and H. Chalmers, Energy Policy, 2008, 36, 4317–4322 CrossRef.
- I. Sreedhar, T. Nahar, A. Venugopal and B. Srinivas, Renewable Sustainable Energy Rev., 2017, 76, 1080–1107 CrossRef CAS.
- R. Ben-Mansour, M. A. Habib, O. E. Bamidele, M. Basha, N. A. A. Qasem, A. Peedikakkal, T. Laoui and M. Ali, Appl. Energy, 2016, 161, 225–255 CrossRef CAS.
- R. Khalilpour, K. Mumford, H. Zhai, A. Abbas, G. Stevens and E. S. Rubin, J. Cleaner Prod., 2015, 103, 286–300 CrossRef CAS.
- C. A. Scholes, M. T. Ho, D. E. Wiley, G. W. Stevens and S. E. Kentish, Int. J. Greenhouse Gas Control, 2013, 17, 341–348 CAS.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- H. M. Jang, K. B. Lee, H. S. Caram and S. Sircar, Chem. Eng. Sci., 2012, 73, 431–438 CAS.
- S. M. Kim, A. Armutlulu, A. M. Kierzkowska, D. Hosseini, F. Donat and C. Müller, Sustainable Energy Fuels, 2020, 4, 713–729 CAS.
- M. S. Duyar, R. J. Farrauto, M. J. Castaldi and T. M. Yegulalp, Ind. Eng. Chem. Res., 2014, 53, 1064–1072 CAS.
- C. Zhang, Y. Li, Z. He, J. Zhao and D. Wang, Appl. Catal., B, 2022, 314, 121474 CAS.
- T. Noor, M. V. Gil and D. Chen, Appl. Catal., B, 2014, 150–151, 585–595 CrossRef CAS.
- R. W. Stevens, A. Shamsi, S. Carpenter and R. Siriwardane, Fuel, 2010, 89, 1280–1286 CrossRef CAS.
- Y. Hu, H. Cui, Z. Cheng and Z. Zhou, Chem. Eng. J., 2019, 377, 119823 CrossRef CAS.
- C. H. Lee and K. B. Lee, Appl. Energy, 2017, 205, 316–322 CrossRef CAS.
- S. Sun, C. Zhang, S. Chen, X. Zhao, Y. Wang, S. Xu and C. Wu, R. Soc. Open Sci., 2023, 10, 230067 CrossRef CAS.
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce and D. H. Sharp, Energy, 1995, 20, 1153–1170 CrossRef CAS.
- D. E. Giammar, R. G. Bruant and C. A. Peters, Chem. Geol., 2005, 217, 257–276 CrossRef CAS.
- P. B. Kelemen, R. Aines, E. Bennett, S. M. Benson, E. Carter, J. A. Coggon, J. C. De Obeso, O. Evans, G. Gadikota, G. M. Dipple, M. Godard, M. Harris, J. A. Higgins, K. T. M. Johnson, F. Kourim, R. Lafay, S. Lambart, C. E. Manning, J. M. Matter, K. Michibayashi, T. Morishita, J. Noël, K. Okazaki, P. Renforth, B. Robinson, H. Savage, R. Skarbek, M. W. Spiegelman, E. Takazawa, D. Teagle, J. L. Urai and J. Wilcox, in Energy Procedia, Elsevier Ltd, 2018, vol. 146, pp. 92–102 Search PubMed.
- B. P. McGrail, H. T. Schaef, A. M. Ho, Y. J. Chien, J. J. Dooley and C. L. Davidson, J. Geophys. Res.:Solid Earth, 2006, 111, 1–13 Search PubMed.
- K. S. Lackner, Annual Review of Energy and the Environment, 2002, 27, 193–232 CrossRef.
- G. Gadikota, J. Matter, P. Kelemen and A. H. A. Park, Phys. Chem. Chem. Phys., 2014, 16, 4679–4693 RSC.
- G. Gadikota, J. Matter, P. Kelemen, P. V. Brady and A. H. A. Park, Fuel, 2020, 277, 117900 CrossRef CAS.
- G. Gadikota, E. J. Swanson, H. Zhao and A. H. A. Park, Ind. Eng. Chem. Res., 2014, 53, 6664–6676 CrossRef CAS.
- R. Baciocchi, G. Costa, A. Polettini, R. Pomi and V. Prigiobbe, Energy Procedia, 2009, 1, 4851–4858 CrossRef CAS.
- R. Zevenhoven, S. Teir and S. Eloneva, Energy, 2008, 33, 362–370 CrossRef CAS.
- C. R. Müller, R. Pacciani, C. D. Bohn, S. A. Scott and J. S. Dennis, Ind. Eng. Chem. Res., 2009, 48, 10284–10291 CrossRef.
- E. R. van Selow, P. D. Cobden, A. D. Wright, R. W. van den Brink and D. Jansen, Energy Procedia, 2011, 4, 1090–1095 CrossRef CAS.
- Y. Liu, Z. Li, L. Xu and N. Cai, Ind. Eng. Chem. Res., 2012, 51, 11989–11997 CrossRef CAS.
- J. Boon, K. Coenen, E. van Dijk, P. Cobden, F. Gallucci and M. van Sint Annaland, Adv. Chem. Eng., 2017, 51, 3–96 Search PubMed.
- D. Iruretagoyena, K. Hellgardt and D. Chadwick, Int. J. Hydrogen Energy, 2018, 43, 4211–4222 CrossRef CAS.
- T. J. Stadler, P. Barbig, J. Kiehl, R. Schulz, T. Klövekorn and P. Pfeifer, Energies, 2021, 14, 355 CrossRef CAS.
- J. Fagerlund, J. Highfield and R. Zevenhoven, RSC Adv., 2012, 2, 10380–10393 RSC.
- L. Zhao, L. Sang, C. Jun, J. Ji and H. H. Teng, Environ. Sci. Technol., 2010, 44, 406–411 CrossRef CAS.
- A. I. Fernández, J. M. Chimenos, M. Segarra, M. A. Fernández and F. Espiell, Hydrometallurgy, 1999, 53, 155–167 CrossRef.
- W. K. O'Connor, D. C. Dahlin, G. E. Rush, S. J. Gerdemann, L. R. Penner and R. P. Nilsen, Aqueous Mineral Carbonation: Mineral Availability, Pretreatment, Reaction Parametrics, and Process Studies, DOE/ARC-TR-04-002, Albany Research Center, Albany, 2005 Search PubMed.
- A. I. Fernández, J. M. Chimenos, M. Segarra, M. A. Fernández and F. Espiell, Hydrometallurgy, 1999, 53, 155–167 CrossRef.
- J. S. Loring, C. J. Thompson, C. Zhang, Z. Wang, H. T. Schaef and K. M. Rosso, J. Phys. Chem. A, 2012, 116, 4768–4777 CrossRef CAS PubMed.
- W. J. J. Huijgen, G. J. Witkamp and R. N. J. Comans, Environ. Sci. Technol., 2005, 39, 9676–9682 CrossRef CAS PubMed.
- G. Gadikota, Nat. Rev. Chem., 2020, 4, 78–89 CrossRef CAS PubMed.
- M. Bilal and S. D. Jackson, Appl. Catal., A, 2017, 529, 98–107 CrossRef CAS.
- R. Y. Chein, Y. H. Lin, Y. C. Chen, Y. P. Chyou and J. N. Chung, Int. J. Hydrogen Energy, 2014, 39, 18854–18862 CrossRef CAS.
- I. Kocemba, I. Śmiechowicz, M. Jędrzejczyk, J. Rogowski and J. M. Rynkowski, Catalysts, 2021, 11, 1475 CrossRef CAS.
- E. Baraj, K. Ciahotný and T. Hlinčík, Fuel, 2021, 288, 119817 Search PubMed.
- P. Ochonma, C. Noe, S. Mohammed, A. Mamidala and G. Gadikota, React. Chem. Eng., 2023, 8, 1943–1959 RSC.
- B. R. Smith, J. M. Loganathan and M. Shekhar Shantha, Int. J. Chem. React. Eng., 2010, 8, 1–32 Search PubMed.
- K. J. Fricker, Magnesium Hydroxide Sorbents for Combined Carbon Dioxide Capture and Storage In Energy Conversion Systems, PhD Thesis, Columbia University, 2014 Search PubMed.
- D. P. Harrison, Ind. Eng. Chem. Res., 2008, 47, 6486–6501 CrossRef CAS.
- E. Abbasi, A. Hassanzadeh, S. Zarghami, H. Arastoopour and J. Abbasian, Fuel, 2014, 137, 260–268 CrossRef CAS.
- J. L. Palandri and Y. K. Kharaka, Open-File Rep. – U. S. Geol. Surv., 2004, 1068, 1–64 Search PubMed.
- B. Li, L. Wei, H. Yang, X. Wang and H. Chen, Energy, 2014, 68, 248–254 CrossRef CAS.
- Z. Giergiczny, Cem. Concr. Res., 2019, 124, 105826 CrossRef CAS.
- A. Polat, P. W. U. Appel and B. J. Fryer, Gondwana Res., 2011, 20, 255–283 CrossRef CAS.
- S. V. Golubev, O. S. Pokrovsky and J. Schott, Chem. Geol., 2005, 217, 227–238 CrossRef CAS.
- G. Rim, A. K. Marchese, P. Stallworth, S. G. Greenbaum and A.-H. A. Park, Chem. Eng. J., 2020, 396, 125204 CrossRef CAS.
- S. Katre, P. Ochonma, H. Asgar, A. M. Nair, K. Ravi and G. Gadikota, Phys. Chem. Chem. Phys., 2024, 26, 9264–9283 RSC.
- S. J. Gerdemann, W. K. O'Connor, D. C. Dahlin, L. R. Penner and H. Rush, Environ. Sci. Technol., 2007, 41, 2587–2593 CrossRef CAS PubMed.
- R. Kurosawa, M. Takeuchi and J. Ryu, RSC Adv., 2021, 11, 24292–24311 RSC.
- W. Liang, Y. Yin, L. Wang, L. Chen and H. Li, J. Alloys Compd., 2017, 702, 346–351 CrossRef CAS.
- Z. F. Cheng, X. Hu, Y. Li and Z. Y. Ling, J. Am. Ceram. Soc., 2016, 99, 2688–2692 CrossRef CAS.
- Y. H. Lee, H. Eom, S. M. Lee and S. S. Kim, RSC Adv., 2021, 11, 8306–8313 RSC.
- H. Zheng, Y. He, Y. Zhu, L. Liu and X. Cui, RSC Adv., 2021, 11, 12476–12483 RSC.
- K. S. P. Karunadasa, C. H. Manoratne, H. M. T. G. A. Pitawala and R. M. G. Rajapakse, J. Phys. Chem. Solids, 2019, 134, 21–28 CrossRef CAS.
- L. Ernawati, R. A. Wahyuono, A. D. Laksono, A. Ningrum, K. Handayani and A. Sabrina, IOP Conf. Ser.:Mater. Sci. Eng., 2021, 1053, 012001 CAS.
- R. Lakshmi, V. Velmurugan and S. Sasikumar, Combust. Sci. Technol., 2013, 185, 1777–1785 CrossRef CAS.
- T. N. Ramesh and A. Taj, Int. J. Sci. Res., 2013, 1, 58 Search PubMed.
- C. H. Park, J. H. Lee, E. Jang, K. B. Lee and J. H. Kim, Chem. Eng. J., 2017, 307, 503–512 CrossRef CAS.
- A. Z. Noah, M. A. El Semary, A. M. Youssef and M. A. El-Safty, Egypt. J. Pet., 2017, 26, 33–40 CrossRef.
- S. H. Tamin, S. B. R. S. Adnan, M. H. Jaafar and N. S. Mohamed, Ionics, 2018, 24, 2665–2671 CrossRef CAS.
- A. Saberi, B. Alinejad, Z. Negahdari, F. Kazemi and A. Almasi, Mater. Res. Bull., 2007, 42, 666–673 CrossRef CAS.
- M. V. Reddy and M. Pathak, J. Inorg. Organomet. Polym. Mater., 2018, 28, 2187–2195 CrossRef CAS.
- K. H. Tan, A. Iqbal, F. Adam, N. H. H. Abu Bakar, M. N. Ahmad, R. M. Yusop and H. Pauzi, RSC Adv., 2019, 9, 38760–38771 RSC.
- J. P. Holgado, G. Munuera, J. P. Espinos and A. R. Gonzalez-Elipe, Appl. Surf. Sci., 2000, 158, 164–171 CrossRef CAS.
- P.-C. Huang, A. C. Vilando, T.-H. Ha and M.-C. Lu, Sustainable Environ. Res., 2024, 34, 11 CrossRef CAS.
- X. Y. Peng, X. X. Liu, D. Diamond and K. T. Lau, Carbon, 2011, 49, 3488–3496 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed.
- C. M. Chen, J. Q. Huang, Q. Zhang, W. Z. Gong, Q. H. Yang, M. Z. Wang and Y. G. Yang, Carbon, 2012, 50, 659–667 CrossRef CAS.
- A. Dufourny, C. Julcour, J. Esvan, L. Cassayre, P. Laniesse and F. Bourgeois, Front. Clim., 2022, 4, 946735 CrossRef.
- B. Han, Y. Yang, J. Li, H. Deng and C. Yang, Int. J. Electrochem. Sci., 2018, 13, 9166–9182 CrossRef CAS.
- M. T. Nichols, W. Li, D. Pei, G. A. Antonelli, Q. Lin, S. Banna, Y. Nishi and J. L. Shohet, J. Appl. Phys., 2014, 115, 094105 CrossRef.
- M. J. Guittet, J. P. Crocombette and M. Gautier-Soyer, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 125117 CrossRef.
- A. A. Khassin, T. M. Yurieva, M. P. Demeshkina, G. N. Kustova, I. Sh. Itenberg, V. V. Kaichev, L. M. Plyasova, V. F. Anufrienko, I. Yu. Molina, T. V. Larina, N. A. Baronskaya and V. N. Parmon, Phys. Chem. Chem. Phys., 2003, 5, 4025–4031 RSC.
- H. Xu, Q. Wang, H. Xiao, X. Li, X. Su, M. Tang, L. Chen and S. Li, RSC Adv., 2019, 9, 1319–1326 RSC.
- J. Esvan, G. Berger, S. Fabre, E. Bêche, Y. Thébault, A. Pages and C. Charvillat, Geochim. Cosmochim. Acta, 2022, 335, 124–136 CrossRef CAS.
- L. Liu, J. Liu, L. Zhao, Z. Yang, C. Lv, J. Xue and A. Tang, Environ. Sci. Pollut. Res., 2019, 26, 8721–8736 CrossRef CAS PubMed.
- X. Gao, H. Asgar, I. Kuzmenko and G. Gadikota, Microporous Mesoporous Mater., 2021, 327, 111381 CrossRef CAS.
- A. Kaur, P. Chahal and T. Hogan, IEEE Electron Device Lett., 2016, 37, 142–145 CAS.
- D. A. Shirley, High-Resolution X-Ray Photoemission Spectrum of the Valence Bands of Gold, Phys. Rev. B: Solid State, 1972, 5, 4709 CrossRef.
- T. V. Larina, L. S. Dovlitova, V. V. Kaichev, V. V. Malakhov, T. S. Glazneva, E. A. Paukshtis and B. S. Bal'Zhinimaev, RSC Adv., 2015, 5, 79898–79905 RSC.
- G. Greczynski and L. Hultman, ChemPhysChem, 2017, 18, 1507–1512 CrossRef CAS.
- D. C. Elliott, R. T. Hallen and L. J. Sealock, Ind. Eng. Chem. Process Des. Dev., 1983, 22, 431–435 CAS.
- W. A. R. Slegeir, R. S. Sapienza, R. Rayford and L. Lam, Organometallics, 1982, 1, 1728–1730 CrossRef CAS.
- A. H. G. Cents, D. W. F. Brilman and G. F. Versteeg, Chem. Eng. Sci., 2005, 60, 5830–5835 CrossRef CAS.
- M. Hänchen, V. Prigiobbe, R. Baciocchi and M. Mazzotti, Chem. Eng. Sci., 2008, 63, 1012–1028 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00480a |
| This journal is © The Royal Society of Chemistry 2025 |