
Transesterification or polymerization? Reaction mechanism and kinetics of 2-(diethylamino)ethyl methacrylate with methanol and the competitive effect on free-radical polymerization†
Judith
Cabello-Romero
a,
Román
Torres-Lubián
a,
Javier Francisco
Enríquez-Medrano
a,
Adrián
Ochoa-Terán
b,
Jesús
Jara-Cortés
 c and
Iván
Zapata-González
*d
c and
Iván
Zapata-González
*d
aDepartment of Macromolecular and Nanomaterial Chemistry, Centro de Investigación en Química Aplicada, Saltillo, Coahuila 25294, Mexico
bCentro de Graduados e Investigación en Química, Instituto Tecnológico de Tijuana, Baja California 22000, Mexico
cUniversidad Académica de Ciencias Básicas e Ingeniería, Universidad Autónoma de Nayarit, Nayarit 63155, Mexico
dDepartment of Polymerization Processes, Centro de Investigación en Química Aplicada, Blvd. Enrique Reyna Hermosillo No. 140, Saltillo, Coahuila C.P. 25294, Mexico. E-mail: ivan.zapata@ciqa.edu.mx
First published on 21st October 2024
Abstract
Transesterification of 2-(diethylamino)ethyl methacrylate (DEAEMA) with methanol leads to the formation of methyl methacrylate (MMA) and 2-(diethylamino)ethanol; this alcoholysis reaction is studied by Density Functional Theory (DFT) calculations and in situ1H-NMR measurements. The transesterification mechanism involves the cooperative effect of methanol. Second-order transesterification kinetics and Arrhenius parameters (A and Ea) are reported. Furthermore, the competition between transesterification and (co)polymerization between DEAEMA and the MMA transesterification product, using 2,2-azobis (2-methylpropionitrile) (AIBN) as an initiator at 70 °C, has been analysed. In experiments with a DEAEMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol molar ratio of 1:46 the copolymerization results in a large proportion of the MMA copolymer composition (FMMA) of 60 mol%; with an equimolar ratio the transesterification is avoided and the FMMA is only 2 mol%. FMMA can also be tuned by modification of the DEAEMA:AIBN molar ratio. Therefore, this work provides guidelines for the synthesis of well-defined poly(DEAEMA) and poly(DEAEMA-co-MMA) in primary alcohols.
:methanol molar ratio of 1:46 the copolymerization results in a large proportion of the MMA copolymer composition (FMMA) of 60 mol%; with an equimolar ratio the transesterification is avoided and the FMMA is only 2 mol%. FMMA can also be tuned by modification of the DEAEMA:AIBN molar ratio. Therefore, this work provides guidelines for the synthesis of well-defined poly(DEAEMA) and poly(DEAEMA-co-MMA) in primary alcohols.
Introduction
For decades, the family of ternary amino-containing methacrylates (TAMAs) has been very used in the synthesis of multi-stimuli-responsive (co)polymers,1–3 with 2-(diethylamino)ethyl methacrylate (DEAEMA) and 2-(dimethylamino)ethyl methacrylate (DMAEMA) monomers being the most commonly used due to their commercial availability. The protonation–deprotonation of N in the amino functionality has been demonstrated to occur directly as a result of an applied gradient of pH in the environment for TAMA-based (co)polymers. As a result, electrostatic repulsions occur between amino groups and the solvent, leading to phase transition or conformational changes.2,4 Furthermore, the (dialkylamino) ethyl groups produce temperature sensitivity that generates a low critical solution temperature (LCST) at around 40–50 °C.4,5 Since properties such as LCST,6 particle size7 and ζ-potential,2 as well as biocompatibility8 are influenced by the copolymer composition, it is essential to accurately quantify the DEAEMA content in the polymer chains.Although poly(DEAEMA) and its copolymers have significant relevance in several fields of application and a great number of studies have been reported in the last decades,9–11 the kinetic basis of TAMA polymerization reported hitherto is scarce. Generally, TAMA-based (co)polymers have been synthesized by free radical polymerization in bulk and homogeneous phases using aqueous, organic, and inorganic solvents or a mixture of them. However, since DEAEMA and DMAEMA are weak bases with pKa of 8.8 (ref. 12) and 8.4 (ref. 13) at 25 °C, respectively, and some solvents can act as nucleophiles, e.g. protic solvents (alcohols and water), a solvolysis reaction can be carried out.
The most studied solvolysis of TAMAs has been the hydrolysis in H2O. Firstly, van de Wetering et al.13 analysed the impact of hydrolysis on three TAMAs and the results showed that half of the initial DMAEMA lost the amino functionality at 17 h with pH 7.4 and 37 °C. This was ascribed to an interaction between the protonated dimethylamino group and the ester carbonyl, which makes the ester more susceptible to nucleophilic attack of a hydroxyl ion and leads to an efficient hydrolysis. At alkaline pH (>pKa), no differences in stability between the compounds were found. The results of Zheng et al.14 indicated the dependency of DMAEMA hydrolysis on pH and temperature, where its rate increases with more basic media, high temperatures, and also with bulky alkyl substituents attached to the N atom. The estimated Arrhenius parameters for a pseudo first-order kinetic constant were the frequency factor of ln(A) = 18.25 s−1 and activation energy of Ea = 72.2 ± 3 kJ mol−1 for a temperature interval of 25 to 60 °C in inherent pH.15 More recently, Ajogbege et al.16 investigated the influence of pH and temperature on the Arrhenius parameters for the hydrolysis of DEAEMA in D2O using in situ1H-NMR spectroscopy measurements, where the protonated state of DMAEMA provided more stability. These findings resulted in values of pseudo first-order parameters for ln(A) of 10 and 17.4 s−1 and Ea, of 55.4 and 69.6 kJ mol−1 as the pH increased from 8 to 10.1, respectively. During the DMAEMA homopolymerization conducted with the 2,2′-azobis(2-methyl-propionamidine) dihydrochloride initiator in D2O at 50 and 60 °C, the formation of deuterated methacrylic acid (MAA-d1) and a hydroxyalkylamine produced by hydrolysis significantly affected the kinetic behaviour, molecular weight and composition of the obtained poly(DMAEMA-co-MAA-d1). A copolymer composition of MAA-d1 (FMMA-d1) greater than 30 mol% was formed using a solution pH of 10.1. However, hydrolysis was completely avoided by adjusting the pH of the solution to 1.0 and 4.0, due to the fully ionized DMAEMA. Indeed, TAMAs are very susceptible to hydrolysis under specific pH conditions, but the resulting TAMA polymers are stable to solvolysis and modification of their compositions is less significant.13,17 In contrast, polymers based on tertiary amino-containing acrylates are very susceptible to solvolysis.18
Although the hydrolysis of TAMAs, especially DMAEMA, and its influence on polymerization have recently been addressed, the transesterification of TAMAs (alcoholysis) has not been deeply analysed to the best of our knowledge. Generally, primary alcohols, such as methanol (CH3OH), act as nucleophiles and attack esters, resulting in a new alcohol and a new ester compound. Due to the fact that ester transesterification with alcohol is carried out in a very slow transformation, since alcohols are poor nucleophiles and ester contains basic leaving groups, it is common practice to increase its rate by adding an acid catalyst,19 a conjugate base of the reacting alcohol, or a metal catalyst.20–24 However, according to Bories-Azeau and Armes,25 DMAEMA is susceptible to transesterification with primary alcohols without the presence of a catalyst, leading to the formation of methyl methacrylate (MMA) and 2-(dimethylamino)alcohol, Scheme 1. Although this finding has been reported for more than 20 years, no studies have been conducted to explain it and propose a mechanism for the transesterification of the TAMA family. The relative propensity of TAMAs toward alcoholysis has been reported as DEAEMA > DMAEMA > 2-(diisopropylamino)ethyl methacrylate, suggesting a base-catalyzed transesterification.25,26 DMAEMA polymerization using CH3OH at ambient temperature carried out by Bories-Azeau and Armes resulted in an FMMA of 26 mol%, therefore, the MMA formed in situ was able to integrate with the DMAEMA monomer and generate poly(DMAEMA-co-MMA). In the same study, a mixture of water/CH3OH was used as a solvent to increase the polymerization rate and avoid the formation of poly(DMAEMA-co-MMA), but no details on the pH of the experiments were provided. Additionally, a high selectivity of TAMA-based polymers (conditions for no TAMA transesterification) was observed when a secondary alcohol (such as 2-propanol) was used as a solvent. In fact, this finding was explored in RAFT polymerization using tert-butanol to avoid transesterification, first by Arredondo et al.27 conducting DMAEMA experiments and later by Quiñonez Angulo28 for copolymerization of poly(ethylene glycol)methyl ether methacrylate (PEGMA) and either DEAEMA or DMAEMA.
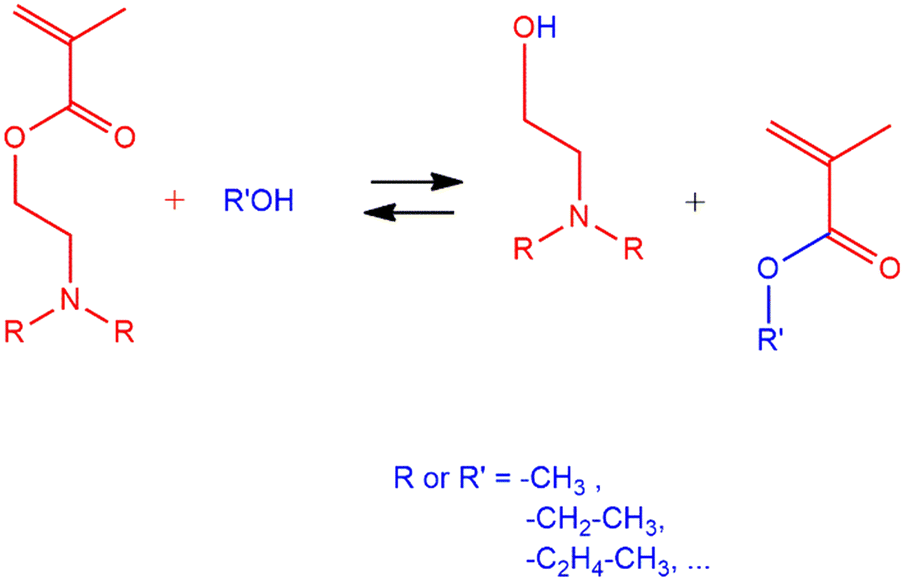 | ||
| Scheme 1 Transesterification of DEAEMA with primary alcohol. | ||
Although the products of the TAMA transesterification have been observed using only primary alcohols at low or medium temperatures,25–28 the same reaction has not produced another methacrylate monomer in large amounts during processes involving an alkyl methacrylate with primary alcohols, indicating that N in the amino group must play a key role in the process.
On the other hand, poly(TAMA-co-MMA) presents exceptional characteristics and has been widely used in several fields, e.g. against the adsorption of bovine serum albumin,29 free-standing hydrogels,30 antimicrobial agents,31 drug delivery nanodevices,32etc. The use of alcohols for the synthesis of methacrylate copolymers is also common, due to the enhanced propagation rate with regard to bulk or organic solvents.33
Moreover, tandem transesterification and copolymerization should have a complex kinetic mechanism with a penultimate effect and solvent-dependent reactivity ratios.34
Even though knowledge of the reaction conditions to achieve the homopolymerization of DEAEMA, namely the avoidance of the transesterification during the reaction, is very important for the development of some smart materials, this route of synthesis can provide an easy way for the copolymerization of poly(DEAEMA-co-MMA), where a specific copolymer composition is desirable. This focus has been examined in the present work; we first propose a mechanism of DEAEMA transesterification based on Density Functional Theory (DFT) calculations. With this part of the information, we subsequently investigated the transesterification rate of DEAEMA using methanol-d4 (CD3OD) as a unique solvent at three levels of initial weight DEAEMA ratio (ωM0) and four temperatures. The rate coefficients were then estimated to satisfy the complete kinetic picture. Finally, we analysed the competition between the transesterification and the (co)polymerization processes, and they were characterized in terms of their monomer conversions, degree of alcoholysis, remaining monomer composition, and copolymer composition. In both experimental parts, in situ1H-NMR measurements were used as the principal experimental tool to avoid unnecessary sample manipulations.
Experimental
Materials
2-(Diethylamino)ethyl methacrylate (DEAEMA, 99%) and methanol-d4 (CD3OD, 99% atoms D) were used in the transesterification reactions, in which DEAEMA was purified by stirring in the presence of inhibitor remover beads (for hydroquinone and monomethyl ether hydroquinone). In polymerization reactions, in addition to the aforementioned reagents, chloroform-d (CDCl3, 99.8% of the D atom), chloroform (CHCl3 ACS reagent, 99.8%), methanol (CH3OH, ACS reagent, 99.8%), 2,2-azobis (2-methylpropionitrile) (AIBN, 99%) and 1,3,5-trioxane (99%) were added to recipes, in which AIBN was recrystallized twice from ethanol before use. All other reagents were used as received and purchased from Sigma-Aldrich.Transesterification
Transesterification reactions were performed in quick pressure valve NMR tubes of 5 mm diameter, by heating and acquiring 1H-NMR spectra along time on a Bruker Avance III HD 400N 400 MHz spectrometer with a 5 mm multinuclear BBI-decoupling probe with a Z grad. The spectrometer is equipped with a SampleXpress 5 mm tube autosampler and a BCU II flow unit to control the temperature of the probe. The 1H-NMR spectrum acquisition was conducted with 1H 30° pulse direct excitation, a total of 8 scans, 65 K complex data points, a spectral width of 3874 Hz, a recovery delay of 1 s and an acquisition time of 8.4 s. No apodization function was used prior to the Fourier transformation. The chemical shifts (ppm) were relative to the remaining non-deuterated CH3OH signals (from CD3OD) which were used as the internal reference.Homogeneous solutions of DEAEMA and CD3OD were prepared in glass vials, and initial weight DEAEMA ratios of ωM0 = 10, 30 and 60 wt%. Aliquots of the prepared solutions were transferred to NMR tubes resulting in a sample solution height of approximately 5.5 cm (∼0.50 g). The prepared solutions were degassed by three freeze–pump–thaw cycles prior to heating. Subsequently, each NMR tube was sealed and inserted into the NMR instrument, which was programmed to heat up and perform automatic collection of 1H-NMR spectra, every so often from 17 to 24 hours. The reaction temperatures were 26.8, 43.5, 60 and 70 °C.
As an example, for the ωM0 = 10 wt% (1:46 (DEAEMA:D3OD) molar ratio) experiment at 70 °C: in a glass vial 0.049 g (0.26 mmol) of DEAEMA were weighed, followed by 0.44 g (12.27 mmol) of CD3OD, and the capped vial was vortexed for 5 minutes. A fraction of the resulting homogeneous solution was transferred to the NMR tube and then degassed. The sealed NMR tube was placed on the instrument and thermally equilibrated at 26.8 °C, then a first 1H-NMR spectrum was acquired and with this, after the corresponding signals were integrated, the real DEAEMA:CD3OD molar ratio was checked. After that, the probe was heated to 26.8 °C (to avoid loss of quality of the shimming sample, the auto-shimming mode was activated in the Topspin 3.6.2 software), and as soon as the target temperature was reached the first spectrum was acquired. Then, the spooler mode in the Topspin software was used to program the acquisition of the remaining spectra as a function of time, for 24 hours. Finally, the signals corresponding to the transesterification products in the 1H-NMR spectra were integrated using the Bruker Topspin software.
13C (100.5 MHz) and two-dimensional (COSY, HSQC) NMR spectra were obtained at room temperature with a 400 MHz Bruker Avance III HD 400 N spectrometer (with a 5 mm multinuclear BBI-decoupling probe with a Z grad).
Free radical (co)polymerization
Similar to the procedure described above, homogeneous solutions of DEAEMA as a monomer, CH3OH as an alcohol source, AIBN as an initiator, 1,3,5-trioxane as an internal reference (for species quantification), and CDCl3 as a deuterated solvent were prepared in glass vials. Aliquots of the prepared solutions were transferred to the NMR tubes resulting in a sample solution height of approximately 5.50 cm (∼0.50 mL). The prepared solutions were degassed by three freeze–pump–thaw cycles before heating. Subsequently, each NMR tube was sealed and inserted into the NMR instrument, which was programmed to heat up and perform automatic collection of 1H-NMR spectra. Different monomer:alcohol source molar ratios were evaluated at 70 °C, for 24 hours of reaction.
As an example, for an experiment with the 1:8 (DEAEMA:CH3OH) molar ratio: in a glass vial 7.78 × 10−3 g (4.74 × 10−2 mmol) of AIBN were weighed, followed by 0.034 g (0.38 mmol) of 1,3,5-trioxane, 0.81 g (4.39 mmol) of DEAEMA, 1.17 g (36.40 mmol) of CH3OH and 1.49 g (12.39 mmol) of CDCl3. The capped vial was vortexed for 5 minutes, then a fraction of the resulting homogeneous solution was transferred to the NMR tube and then degassed. The sealed NMR tube was placed in the NMR equipment to carry out the copolymerization reaction, following the same procedure described for the case of transesterifications.
Theoretical calculations
To rationalize the mechanism of the DEAEMA transesterification reaction with CH3OH, electronic structure calculations using DFT were performed to obtain the relevant intermediaries and transition states involved in the process. Calculations were carried out using the B3LYP functional (unrestricted Kohn–Sham approach) and the set of def2TZVP bases, including dispersion corrections (D3BJ) and implicitly considering the effect of the solvent CH3OH by using the polarizable continuum model.35–39 Since several parts of the potential energy surface of the DEAEMA–CH3OH adduct are relatively flat, the accuracy for the evaluation of the two-electron integrals was increased (Acc2E = 14) and, a finer grid (SuperFineGrid) was used for the evaluation of the exchange-correlation numerical integrations in the DFT part. In addition, a stricter criterion (opt = tight) was considered for the force limits and the step size to determine the convergence in the geometry optimization. The nature of the stationary points was verified by evaluating the vibrational frequencies. To check the connectivity between the stationary points, intrinsic coordinate reaction calculations were performed. All of the above was performed using the Gaussian 16 program.40Results and discussion
Transesterification
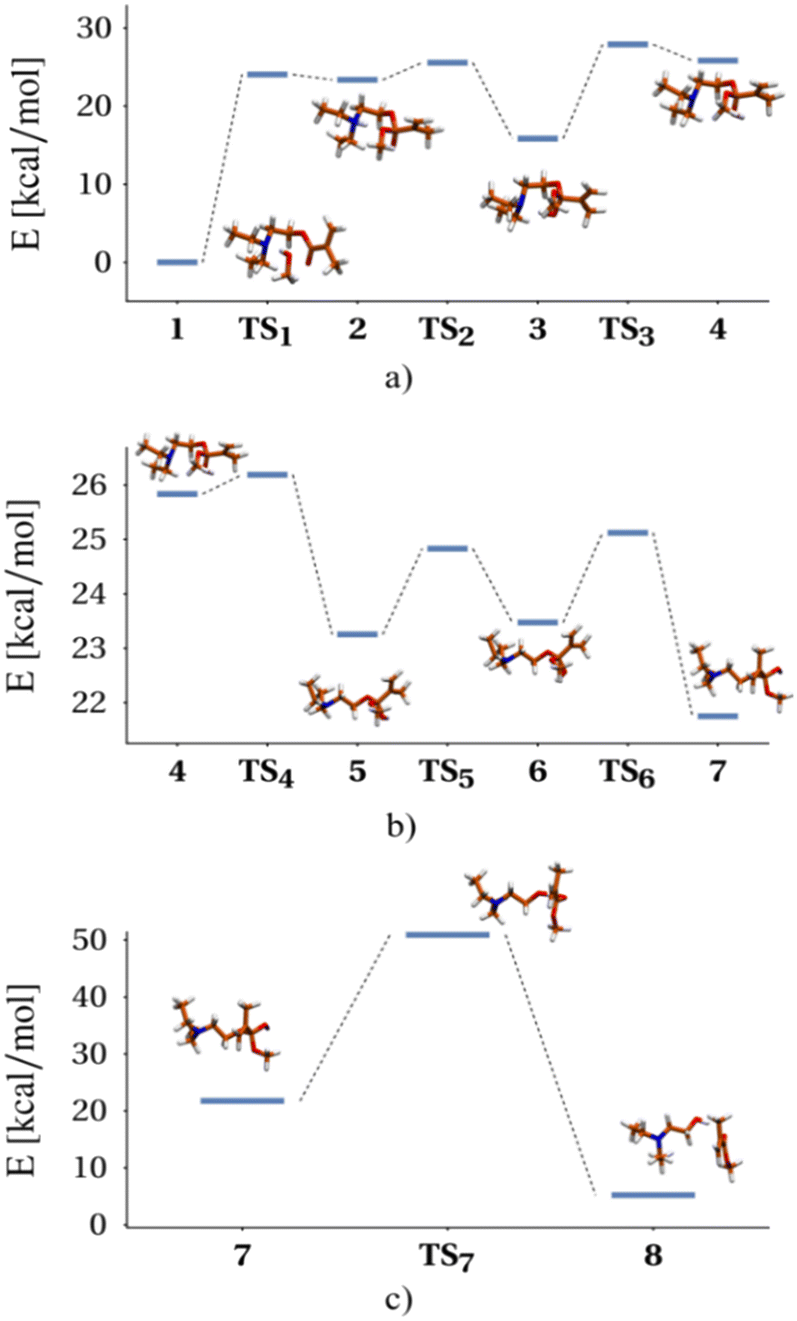 | ||
| Fig. 1 Reaction scheme for the transesterification process of DEAEMA with a CH3OH molecule, obtained from electronic structure calculations at the UB3LYP-PCM(methanol)/def2TZVP level of theory: a) methanol attack to carbonyl, b) adduct conformational changes, c) ester C–O bond cleavage. Bold numbers denote minima (reactants, intermediaries and products) on the potential energy surface, while TSi denotes the i-th transition state. Energies are referenced to 1 (see Fig. S1 and S2 in the ESI† for a detailed version of the diagram). | ||
In stage I (Fig. 1a), the limiting step involves the formation of the DEAEMA–methanol adduct, where, to access the corresponding transition state (TS1) from an initial DEAEMA–CH3OH intermolecular complex, it is necessary to overcome an energy barrier of 24.05 kcal mol−1 (100.62 kJ mol−1). Regarding changes in molecular geometries, by going from 1 to 2 the methanol molecule transfers the OH proton to the amine, while the CH3O moiety forms an O–C bond with the carbonyl of the ester. Then, passing through TS2, the amine proton migrates to the oxygen of the carbonyl group, and subsequently, a rotation of this hydrogen around the C–O bond takes place in the opposite direction of the amine (E3 − ETS3 = 12.07 kcal mol−1), allowing the recovery of the conformational flexibility around the central part of the molecule. This paves the way for the structural changes to be performed in the following stage, because it is known that the tetrahedral addition intermediate is unstable and N,N-diethylethanol is released in order to recover the carbonyl group and form the methyl methacrylate.
In stage II (Fig. 1b), a series of conformational changes occur, involving small energy barriers of less than 2 kcal mol−1, where the system adopts the reactive conformation necessary to facilitate the breaking of the OC bond. Finally, in stage III (Fig. 1c), the system must be supplied with 29.15 kcal mol−1 (121.96 kJ mol−1) to pass through TS7, in order to carry out the cleavage of the C–O bond, as well as the migration of hydrogen, to give rise to the observed products of the reaction. A recreation of the reaction mechanism relating the reagents, transition states, intermediates and products is seen in the file Transesterification_reaction_movie.
Other reaction routes exist, but the energy changes involved are larger, so they do not represent major kinetic competition to the proposed mechanism. As an example, the direct deprotonation of CH3OH by the amine involves an initial unfavourable energy change of 47.43 kcal mol−1. Besides the presence of the Michael addition product was not identified in the NMR experiments, so this other possibility was ruled out.
However, the energy barriers obtained in the proposed mechanism differ by a factor of 2, from the estimated value of the activation energy of the kinetic analysis using the experimental data (vide infra). This difference remains relatively unchanged if, instead of using electronic energies, suitable Gibbs free energies obtained from gas-phase thermochemical calculations are used. We attribute the difference to the inadequate description of the solvent by explicitly considering only one molecule of CH3OH. It is well known that the calculated energy barriers for proton migration reactions, in hydrogen-bonded solvents, decrease when more than one solvent molecule is explicitly considered, due to a cooperative effect.41–44 Thus, as an additional possibility, transition states analogous to TS1 and TS7 were obtained, but considering two methanol molecules in the calculations.
Fig. S3 in the ESI† shows the structures obtained for these transition states. In the analogous case of TS7, when considering the two solvent molecules, the barrier decreases by 6.23 kcal mol−1 with respect to that observed in Fig. 1. Moreover, in this situation, the conformational changes of stage II are not necessary, since because of the increased flexibility induced by the additional solvent molecule, the reaction can take place right after the amine transfers the proton to the carbonyl. On the other hand, taking into consideration a CH3OH molecule bonded to the carbonyl in the TS1 analogue, the barrier decreases by 5.13 kcal mol−1. In this case, the electrophilic character of the carbonyl increases by the interaction with the molecule of solvent and the other methanol molecule may react with the carbonyl initiating the transesterification process. These examples show that the consideration of an additional solvent molecule moves the observed energy barriers in the right direction, with reference to the experimental values. Importantly, the values of these barriers are very similar to those reported for transesterification reactions in analogous systems using an acid catalyst.45 A more general mechanism considering TS1 and TS7 transition states from the theoretical analysis is provided in Scheme 2.
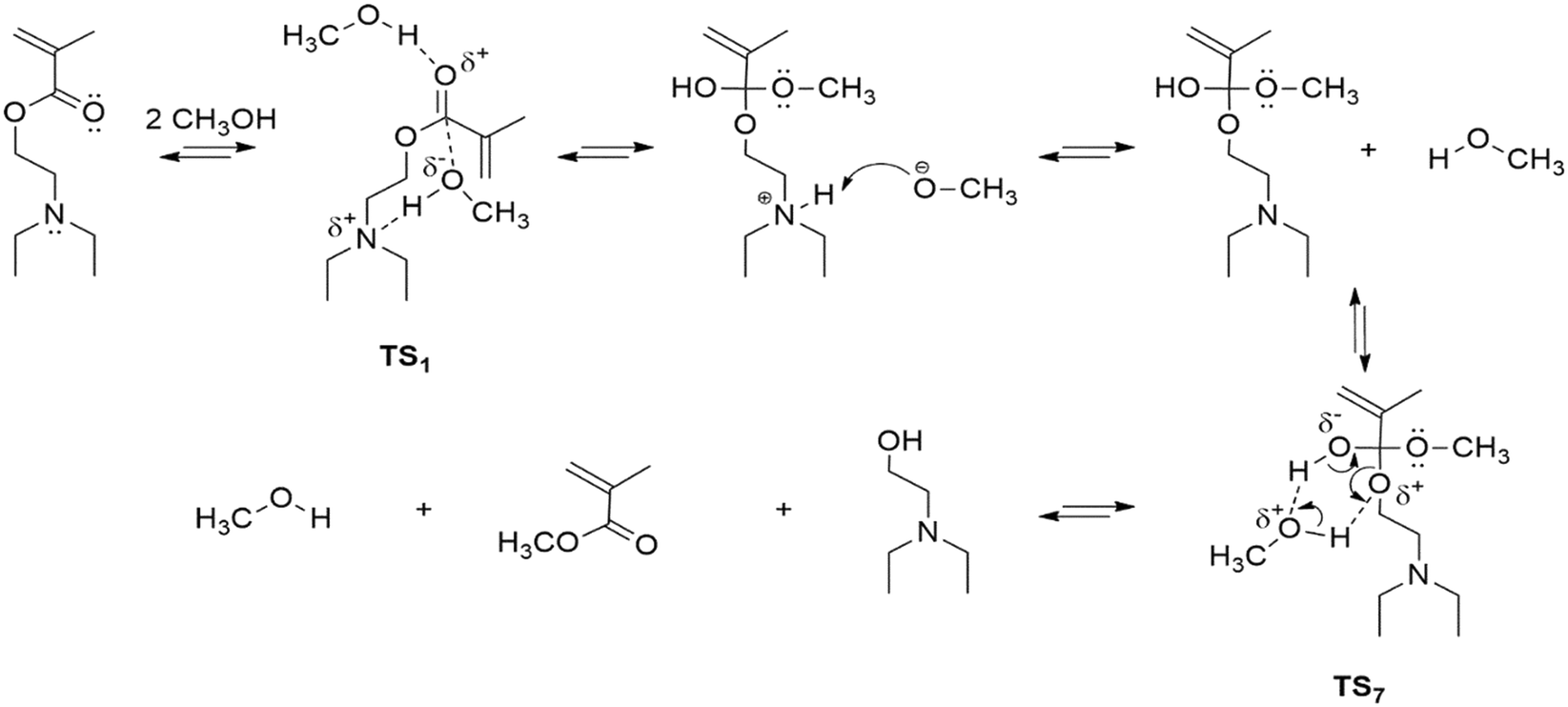 | ||
| Scheme 2 Plausible mechanism for transesterification of DEAEMA with CH3OH. | ||
On the other hand, Fig. 2 shows the signal assignations of the 1H-NMR spectra at 70 °C during 17 h of reaction with the DEAEMA monomer presenting the following peaks (DOCD3, 400 MHz): (1a) 6.10 ppm (1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), (1b) 5.61 ppm (1H, CHC), (5) 4.26 ppm (2H, CH2–O), (6) 2.79 ppm (2H, CH2–N), (7) 2.62 ppm (4H, (CH2)2–N), (3) 1.95 ppm (3H, CH3–C), and (8) 1.07 ppm (6H, 2(CH3–C)). Transesterification products have peaks and multiplicities at different chemical shifts (δ) than those of DEAEMA, MMA-d3: (1a*) 6.07 ppm (1H, CHC), (1b*) 5.59 ppm (1H, CHC), (9*), 3.64 ppm (3H, CH3–C) (for MMA no deuterated, CH3OH), (3*) 1.95 ppm (CH3, CH3–C); and hydroxyalkylamine: (OH): 4.33 ppm (1H, HO–C), (5*) 3.62 ppm (2H, CH2–O), (6* and 7*) 2.61 ppm (2H + 4H; CH2–N, (CH2)2–N), and (8*) 1.07 ppm (6H, 2(CH3–C)). Additionally, the peak at 4.35 ppm corresponds to water contained in the methanol.46 The 1H-NMR spectra of DEAEMA and MMA monomers in CD3Cl are shown in Fig. S4 and S5 in the ESI.† HSQC NMR spectroscopy was used to confirm the assignment of the signals, as shown in Fig. S6 in the ESI.†
C), (1b) 5.61 ppm (1H, CHC), (5) 4.26 ppm (2H, CH2–O), (6) 2.79 ppm (2H, CH2–N), (7) 2.62 ppm (4H, (CH2)2–N), (3) 1.95 ppm (3H, CH3–C), and (8) 1.07 ppm (6H, 2(CH3–C)). Transesterification products have peaks and multiplicities at different chemical shifts (δ) than those of DEAEMA, MMA-d3: (1a*) 6.07 ppm (1H, CHC), (1b*) 5.59 ppm (1H, CHC), (9*), 3.64 ppm (3H, CH3–C) (for MMA no deuterated, CH3OH), (3*) 1.95 ppm (CH3, CH3–C); and hydroxyalkylamine: (OH): 4.33 ppm (1H, HO–C), (5*) 3.62 ppm (2H, CH2–O), (6* and 7*) 2.61 ppm (2H + 4H; CH2–N, (CH2)2–N), and (8*) 1.07 ppm (6H, 2(CH3–C)). Additionally, the peak at 4.35 ppm corresponds to water contained in the methanol.46 The 1H-NMR spectra of DEAEMA and MMA monomers in CD3Cl are shown in Fig. S4 and S5 in the ESI.† HSQC NMR spectroscopy was used to confirm the assignment of the signals, as shown in Fig. S6 in the ESI.†
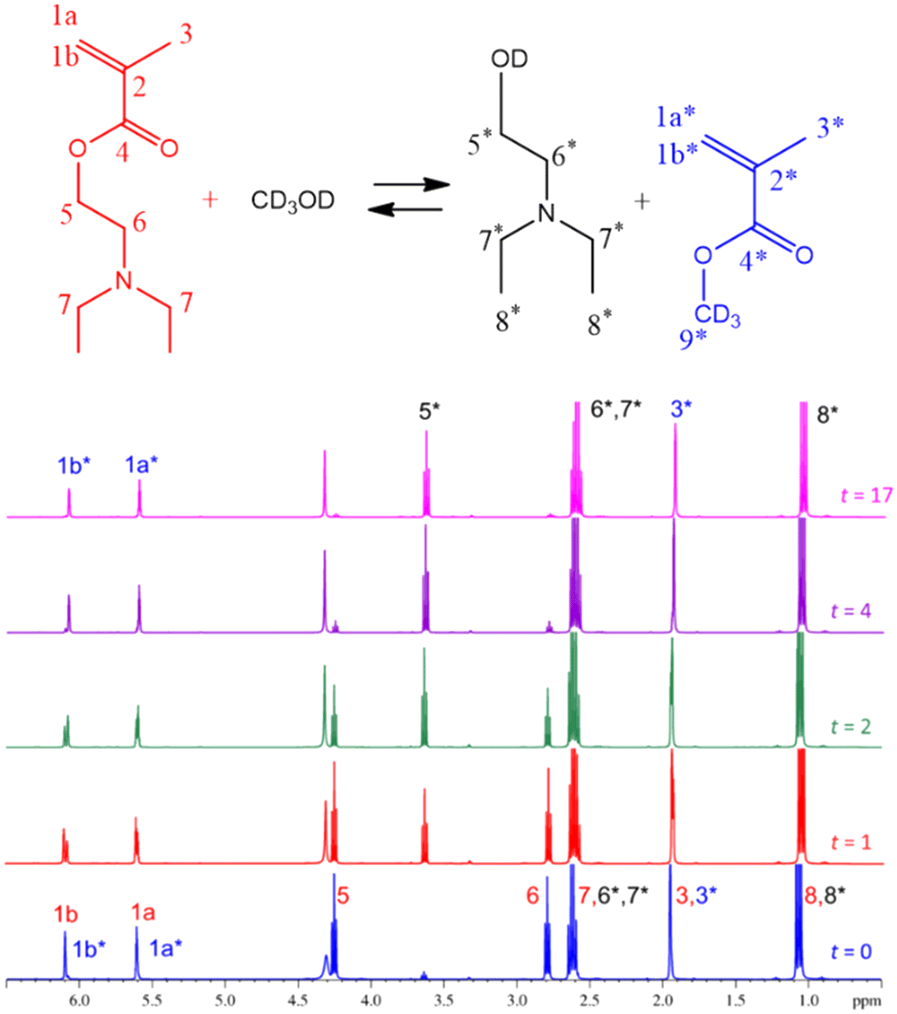 | ||
| Fig. 2 Scheme of transesterification between DEAEMA and CD3OD, and stacked 1H-NMR spectra at t = 0, 1, 2, 4, and 17 h for transesterification between DEAEMA and CD3OD with an initial ratio ωM0 = 10 wt% at 70 °C. | ||
:1 molar ratio between the products on the right of the scheme shown in Fig. 2. Da (mol%) is estimated with the following eqn (1): | (1) |
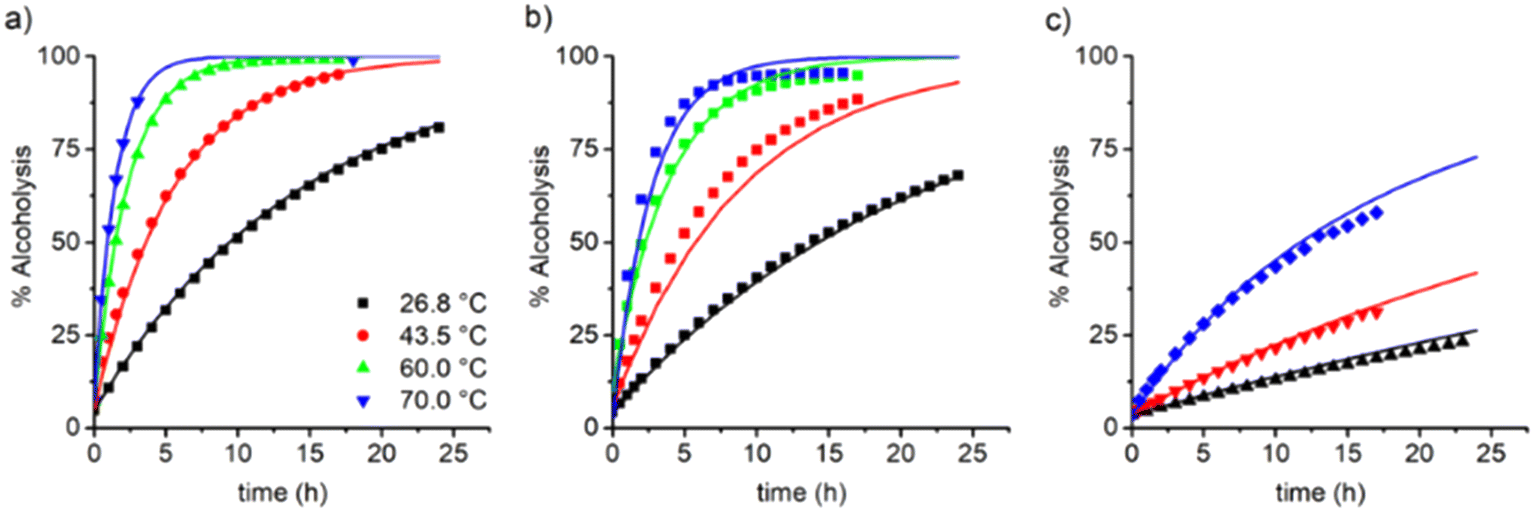 | ||
| Fig. 3 Evolution of the degree of alcoholysis (Da, mol%) in time and variation of the reaction temperature (labels). The value of ωM0 was varying at: a) 10, b) 30, and c) 60 wt%. Symbols are the experimental data obtained via1H-NRM in situ measurements and lines represent a model prediction with a second-order kinetic equation and using eqn (2)–(6). | ||
 | (2) |
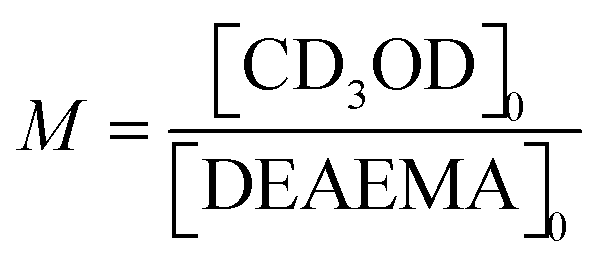 . Furthermore, when t = t1/2 the value of
. Furthermore, when t = t1/2 the value of 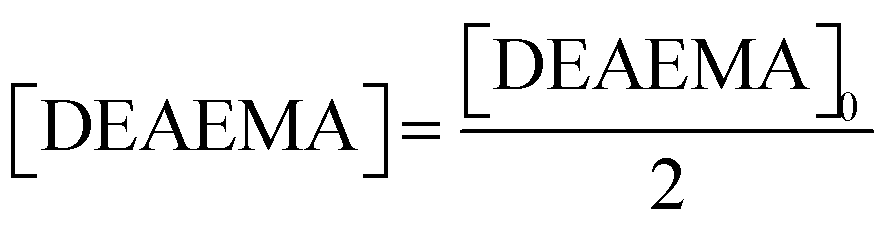 .
.  and the half-life for the second-order reactions is calculated using the following eqn (3).
and the half-life for the second-order reactions is calculated using the following eqn (3).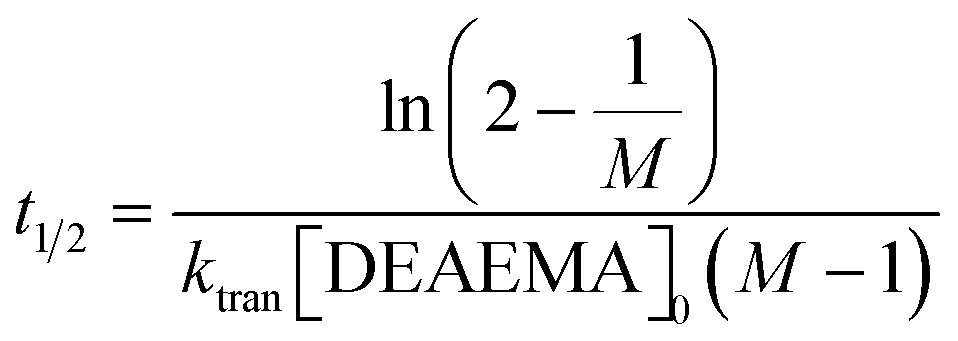 | (3) |
| ω M0 (wt%) | T (°C) | k tran (L mol−1 s−1) | R 2 | t 1/2 (h) |
|---|---|---|---|---|
| 10 | 26.8 | 8.66 × 10−7 | 1 | 10.04 |
| 43.5 | 2.29 × 10−6 | 0.99 | 3.82 | |
| 60.0 | 5.12 × 10−6 | 0.99 | 1.70 | |
| 70.0 | 8.93 × 10−6 | 0.99 | 0.97 | |
| 30 | 26.8 | 7.08 × 10−7 | 0.99 | 15.83 |
| 43.5 | 1.97 × 10−6 | 0.99 | 5.68 | |
| 60.0 | 3.75 × 10−6 | 0.99 | 2.97 | |
| 70.0 | 6.23 × 10−6 | 0.99 | 1.79 | |
| 60 | 26.8 | 2.97 × 10−7 | 0.99 | 70.11 |
| 43.5 | 6.19 × 10−7 | 0.99 | 34.87 | |
| 70.0 | 1.63 × 10−6 | 0.99 | 13.17 |
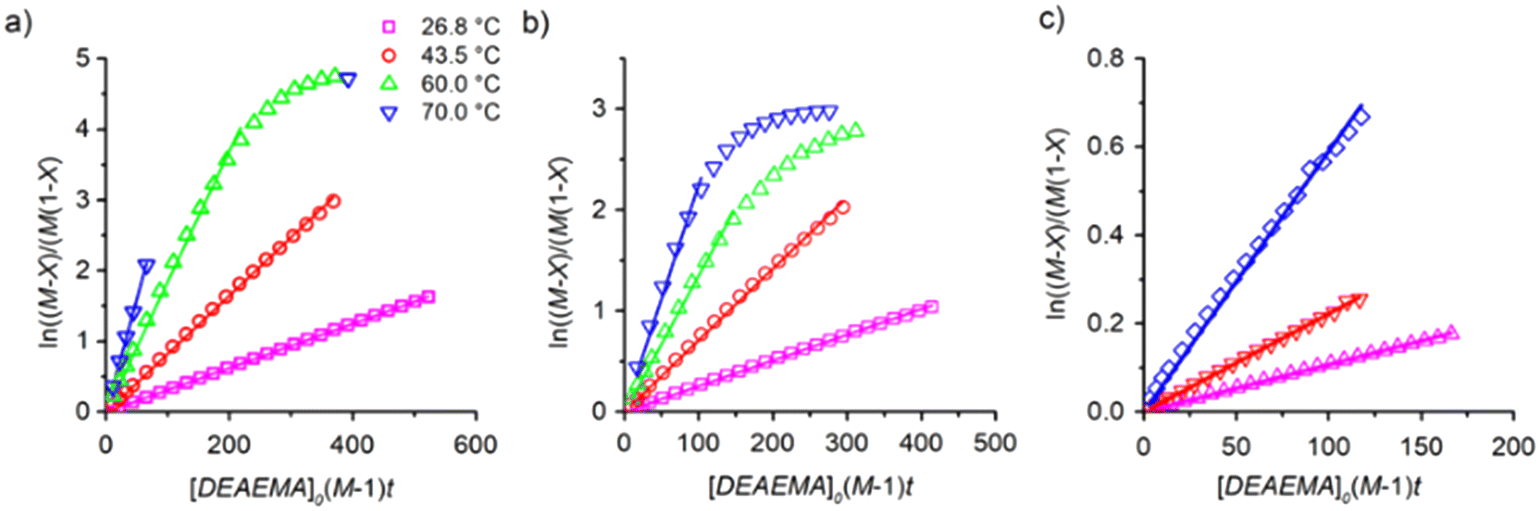 | ||
| Fig. 4 Second-order kinetic plots for transesterification analysed with eqn (3), varying the temperature (labels) and ωM0: a) 10 wt%, b) 30 wt% and c) 60 wt%. Symbols are the experimental data and lines are regressions with the estimated values shown in Table 1. | ||
The dependency of temperature on the DEAEMA kinetic parameters was investigated at four levels, from a low temperature (26.8 °C) to a typical temperature (70 °C) for free radical polymerization. The higher temperature was limited for the boiling point of CD3OD (65.4 °C),48 but in a closed system with a vacuum, its temperature can be increased to 70 °C without any change in the solvent phase. The lower limit was approximated to the standard ambient temperature (25 °C). As the temperature increases, the transesterification is significantly faster, as shown in Fig. 3 for all the values of ωM0, and ln(ktran) is also linearly increased as a function of T−1, Fig. 5. Surprisingly, the plot shows an important shift in both the slopes of the linear regressions, which is proportional to the activation energy (Ea), also in the intercept that is related to the frequency factor (A) in the Arrhenius expression, eqn (4):
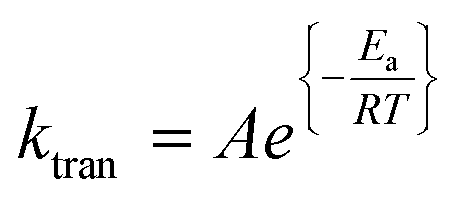 | (4) |
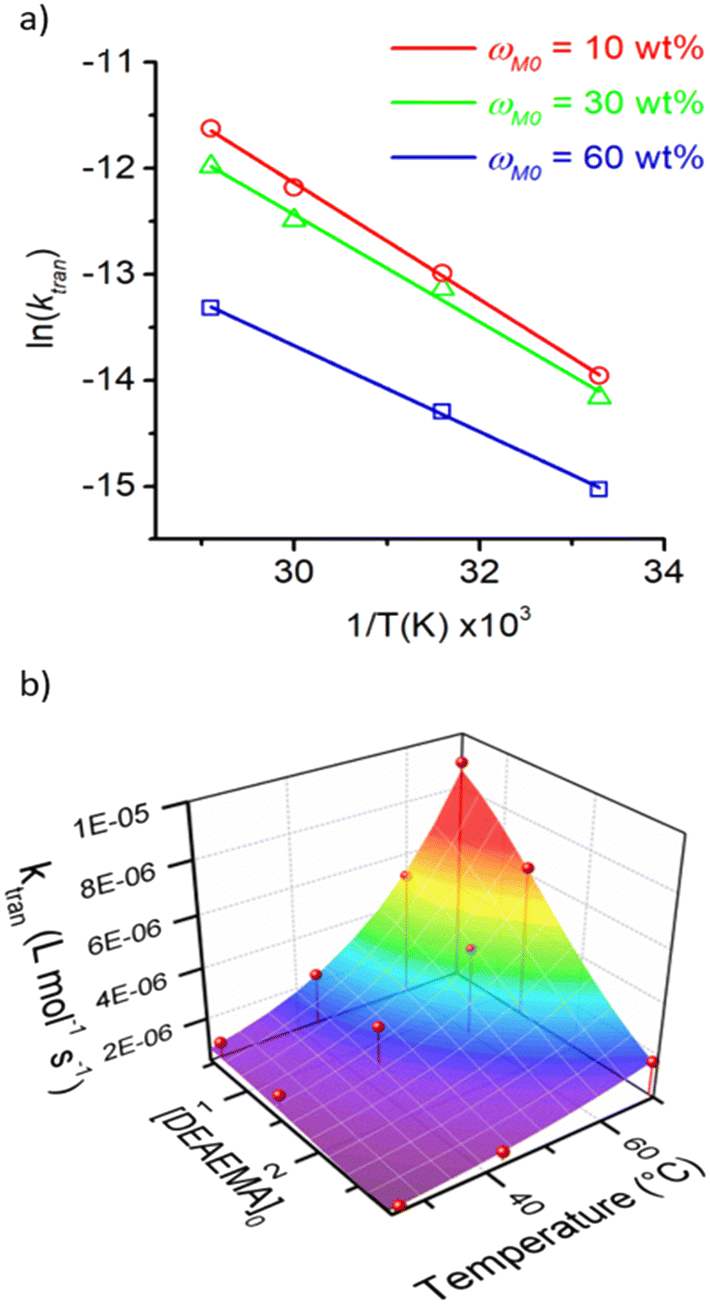 | ||
| Fig. 5 Kinetic analysis of the transesterification rate coefficient in the temperature interval from 26.8 to 70 °C and ωM0 from 10 to 60 wt%. a) Arrhenius plots and b) 3D plot of the evolution of ktran as a function of [DEAEMA]0 and temperature. Symbols denote the experimental data and lines the regressions. The 3D surface has been calculated according to eqn (4)–(6). | ||
| ω M0 (wt%) | M (−) | ln(A) (L mol−1 s−1) | E A (kJ mol−1) | R 2 |
|---|---|---|---|---|
| 10 | 49.93 | 4.35 (±0.37) | 45.65 (±0.99) | 0.998 |
| 30 | 13.42 | 2.76 (±0.79) | 42.09 (±2.14) | 0.992 |
| 60 | 3.46 | −1.48 (±0.17) | 33.77 (±0.43) | 0.999 |
Empirical expressions have been derived to capture the influence of ωM0 on the frequency factor, eqn (5), and the activation energy, eqn (6), using polynomial regressions for both kinetic parameters, as shown in Fig. S10 in the ESI.†
| ln(A) (L mol−1 s−1) = 4.77 − 0.03ωM0 − 1.24 × 10−3ωM02 | (5) |
| EA (kJ mol−1) = 46.84 − 0.10ωM0 − 1.99 × 10−3ωM02 | (6) |
Copolymerization of DEAEMA and MMA
To understand the effect of the transesterification reaction on the free radical polymerization, a set of experiments was conducted in NMR tubes containing CD3OD, DEAEMA, and AIBN as reagents at 70 °C. It is proposed that due to transesterification during the reaction, MMA-d3 formed in situ can react with the remaining DEAEMA monomer and produce poly(DEAEMA-co-MMA-d3), resulting in a concurrent tandem polymerization and transesterification, Scheme 3. The copolymer composition depends on the concentrations of DEAEMA and CD3OD, as well as the temperature, as demonstrated below. | ||
| Scheme 3 Copolymerization of DEAMA and MMA-d3. | ||
 | ||
| Fig. 6 Partial 13C-NMR (100 MHz, r.t., δ in ppm) spectra of copolymers for the molar ratios of DEAEMA:CD3OD (or CH3OH) of: a) 1:0, pure poly(DEAEMA), b) 1:4, c) 1:8, d) 1:1 and e) 1:46. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :methanol on the kinetics, the remaining monomer, and copolymer composition.
First, a highly diluted experiment analogous to the previous one with a DEAEMA:CD3OD = 1:46 molar ratio (ωM0 = 10 wt%), but with the addition of the AIBN initiator (DEAEMA:AIBN = 92:1 molar ratio), is performed. Fig. 7 shows the 1H-NMR spectra of the mixture of DEAEMA, CD3OD, and AIBN at t = 0, and the generation of sub-products and polymers at t = 0, 1, 3, 6 and 24 h of reaction. Although a rigorous methodology was performed to avoid transesterification before acquisition of the first spectrum (t = 0), the signal at 3.65 ppm (5*) indicates the formation of 2-(diethylamino)ethanol and the signals at 5.60 and 6.07 ppm confirm the generation of MMA-d3. As shown in the stacked spectra, the amount of remaining MMA-d3 is almost equal to DEAEMA in the mixture at 1 h, and after 6 h the signal 5 corresponding to the DEAEMA monomer has vanished. Additionally, a wide signal at 4.1 ppm (e) is observed after 15 min, corresponding to the –OCH2– of the DEAEMA units incorporated in the copolymer.
:methanol on the kinetics, the remaining monomer, and copolymer composition.
First, a highly diluted experiment analogous to the previous one with a DEAEMA:CD3OD = 1:46 molar ratio (ωM0 = 10 wt%), but with the addition of the AIBN initiator (DEAEMA:AIBN = 92:1 molar ratio), is performed. Fig. 7 shows the 1H-NMR spectra of the mixture of DEAEMA, CD3OD, and AIBN at t = 0, and the generation of sub-products and polymers at t = 0, 1, 3, 6 and 24 h of reaction. Although a rigorous methodology was performed to avoid transesterification before acquisition of the first spectrum (t = 0), the signal at 3.65 ppm (5*) indicates the formation of 2-(diethylamino)ethanol and the signals at 5.60 and 6.07 ppm confirm the generation of MMA-d3. As shown in the stacked spectra, the amount of remaining MMA-d3 is almost equal to DEAEMA in the mixture at 1 h, and after 6 h the signal 5 corresponding to the DEAEMA monomer has vanished. Additionally, a wide signal at 4.1 ppm (e) is observed after 15 min, corresponding to the –OCH2– of the DEAEMA units incorporated in the copolymer.
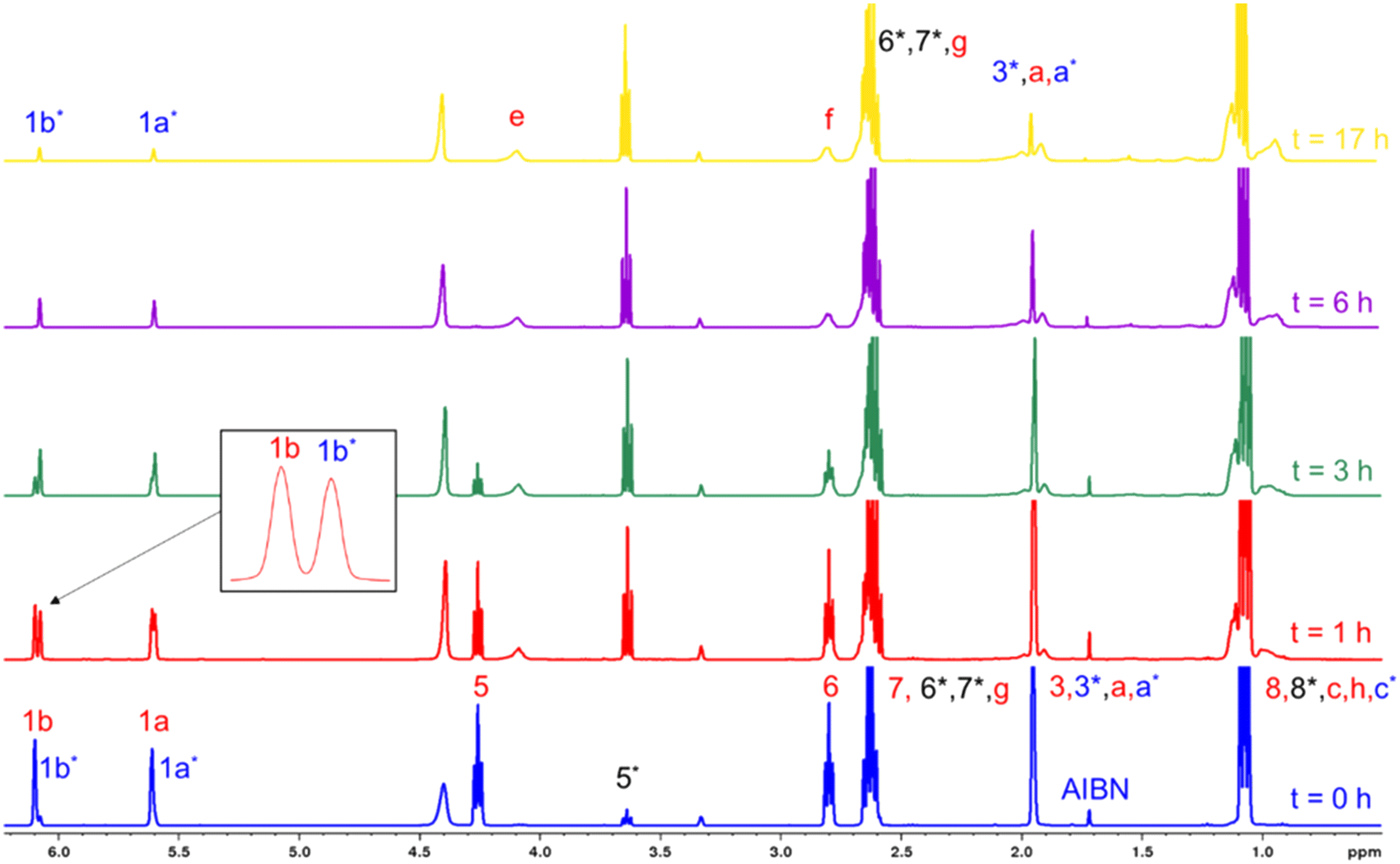 | ||
| Fig. 7 Stacked 1H-NMR spectra (400 MHz, r.t., δ in ppm) at t = 0, 1, 3, 6, and 24 h for copolymerization of DEAEMA and MMA-d3 with an initial ratio ωM0 = 10 wt% and DEAEMA:AIBN = 92:1 molar ratio in CD3OD at 70 °C. The signal assignments correspond to Scheme 3. | ||
The fraction of MMA formed during the process (Da) is estimated with the equimolar formation of 2-(diethylamino)ethanol. DEAEMA conversion (XDEAEMA) is the ratio between the moles of DEAEMA in the copolymer and the initial moles of DEAEMA. The remaining monomer composition (fDEAEMA and fMMA-d3) and the copolymer composition (FDEAEMA and FMMA-d3) are also estimated. All ratios are described in section S.8 in the ESI.†
In Fig. 8a, the profiles of XDEAEMA, Da, and the percentage of remaining DEAEMA (DEAEMA) are illustrated as a function of time. Analysing the data, only a low proportion (26.7 mol%) of [DEAEMA]0 is polymerized and stopped prematurely at 5 h. Since the transesterification rate is relatively higher than the polymerization rate, a large amount of DEAEMA is transformed into MMA-d3 as indicated by the Da value of 73.3 mol%. While MMA-d3 is only generated before 5 h of reaction (Xtot = 60 mol%), it is consumed continuously throughout the process. Under these conditions, it is clear that the final mixture is composed of poly(DEAEMA-co-MMA-d3) and poly(MMA-d3), the former is generated in the first stage when DEAEMA and MMA-d3 monomers are present in the medium and the latter is generated when the DEAEMA monomer has been totally exhausted and then the MMA-d3 monomer only remains in the reaction mixture, Fig. 8a and b. The copolymer is enriched with DEAEMA for Xtot lower than 60 mol%, but this is constantly decreasing as MMA-d3 is generated and DEAEMA is exhausted, at 60% an equimolar composition of DEAEMA:MMA (50:50 mol%) is reached, and above that conversion, the MMA-d3 copolymer composition presents a high value of FMMA = 63 mol%. In fact, a white solution was observed after a day of experiment; the precipitated mass was separated and characterized by 13C-NMR and 1H-NMR, resulting in the proportion of poly(MMA-d3) of 95 mol%, Fig. S11–S13 in the ESI.† Therefore, the final material only contains a low proportion of amino functionality, which will dramatically affect its thermo- and pH-sensitivity properties.
 | ||
| Fig. 8 Copolymerization/transesterification of DEAEMA in a mixture of methanol/CDCl3 at 60 °C to form poly(DEAEMA-co-MAA) with DEAEMA:AIBN molar ratio of 92:1 and DEAEMA:methanol of: a)–c) 1:46, d)–f) 1:8, g)–i) 1:4, and j)–l) equimolar, 1:1. a), d), g) and j) consumption profiles of: DEAEMA (DEAEMA), DEAEMA incorporated into (co)polymer (XDEAEMA), and degree of transesterification (Da), b), e), h) and k) remaining monomer composition (mol%), c), f), i) and l) copolymer composition (mol%). CD3OD was only used for the experiment with a 1:50 (DEAEMA:alcohol) molar ratio and for the other experiments CH3OH was used. | ||
On the other hand, different studies have used solvent mixtures to increase the miscibility of some components of the TAMA-containing polymer system, such as protic solvents (alcohols, water, etc.) and non-protic organic solvents.53–55 Additionally, the combination of solvents is used in the actual study to increase the DEAEMA concentration regarding the alcohol, avoiding high viscosity of the medium and the problems with the acquisition of the spectra.
Here, a solvent mixture of CH3OH/CD3Cl was used and the kinetic behaviour of transesterification and copolymerization was followed by using 1H-NMR in situ measurements. Chloroform acts as an inert solvent in the reaction since it does not present any inter- and/or intramolecular interaction. However, the amount of CH3OH gives rise to the transesterification of DEAEMA and the MMA subproduct (no deuterated). According to the previous results of this work, the transesterification rate should suffer a decrease when the DEAEMA content increases, Fig. 3; hence, 1:8 (ωM0 = 41 wt%), 1:4 (ωM0 = 58 wt%), and 1:1 (ωM0 = 84.6 wt%) DEAEMA:CH3OH molar ratios were analysed.
In the next two experiments, the amount of DEAEMA is increased in the molar ratios of DEAEMA:CH3OH at 1:8 and 1:4 to explore the kinetic behaviour under moderate dissolution conditions using concurrent tandem transesterification and polymerization, and these reaction conditions are commonly used in TAMA (co)polymerization.
An excess of alcohol in 1:8 (DEAEMA:CH3OH) molar ratio was first studied. In this case, DEAEMA is consumed after 10 h (see DEAEMA) by a copolymerization pathway (XDEAEMA) faster than transesterification (Da), Fig. 8d. Therefore, an increase in the DEAEMA content results in a significant impact on the transesterification, reducing its rate and promoting the copolymerization between DEAEMA and the MMA formed in situ. The estimated values of XMMA are higher than those of the previous experiment with only CD3OD as a solvent; after 17 h the DEAEMA and MMA monomers were consumed totally consumed (Xtot > 97%). As shown in Fig. 8e, both monomers are present throughout the reaction, giving rise to a copolymer in solution without the possible existence of homopolymer chains; below Xtot = 75% the remaining monomer mixture is reached by DEAEMA and above it a higher level of MMA is observed. A system of competitive reactions is obtained under these conditions, because both kinetic steps contribute to the growth of the chains throughout the process, resulting in a random copolymer with a final composition of 67 and 33 mol% DAEAMA and MMA, respectively, Fig. 8f. A value of FDEAEMA = 62 mol% was estimated in the sample after purification of the monomers. Additionally, experiments in Schleck tubes were carried out at different reaction times of 0.5, 4 and 7 h. After purification, the samples were analysed by Differential Scanning Calorimetry (DSC). The glass transition temperatures (Tg) were −18.5, −11, and −6.6 at 0.5, 4, and 7 h, respectively, Fig. S14 in the ESI.† As a result, the copolymer composition is enriched with MMA during the tandem process, which is consistent with the in situ1H-NMR analysis.
In the next experiment, the concentration of the CH3OH was decreased and the profiles of XDEAEMA, XMMA, Da, and DEAEMA as a function of time for a 1:4 (DEAEMA:CH3OH) molar ratio are plotted in Fig. 8g, the remaining monomer composition fDEAEMA and fMMA in Fig. 8h, and the copolymer composition FDEAEMA and FMMA in Fig. 8i. Fast consumption of the DEAEMA monomer is observed, depleting in 10 h. Only 10% of the initial concentration of DEAEMA is consumed by transesterification to produce MMA and 90% by polymerization, thus the process is governed by a high copolymerization rate. It is assumed that the copolymerization runs under starved MMA conditions, in which the MMA is consumed as soon as it is generated. Therefore, the remaining monomer mixture is predominantly composed of DEAEMA during the reaction, Fig. 8h. The resulting copolymer is enriched with 91% DEAEMA and only 9% MMA as shown in Fig. 8i, which is very different in terms of the composition of 67% DEAEMA and 33% MMA obtained in the experiment conducted with a 1:4 (DEAEMA:CH3OH) molar ratio.
Finally, an equimolar ratio of DEAEMA:CH3OH was employed with the objective of completely avoiding transesterification with DEAEMA. In fact, this goal was achieved because it is supported by the very low resulting values of Da during the 17 h of reaction, Fig. 8j. From the total initial DEAEMA, an amount of XDEAEMA = 98 mol% is polymerized and 2 mol% is lost through transesterification, leading to a homopolymerization of DEAEMA. Additionally, the consumption of DEAEMA is slow under these conditions, since it is exhausted over the course of 10 h compared to the time of 5 h for that experiment with a molar ratio of 1:8 (DEAEMA:CH3OH), Fig. 8aversusFig. 8j. Both the remaining monomer mixture (Fig. 8k) and the copolymer compositions (Fig. 8l) are enriched with poly(DEAEMA) in the course of the 17 h reaction, producing DEAEMA chains with a high FDEAEMA of 98 mol%, which was confirmed after sample purification (FDEAEMA = 99%), see Fig. S15–S17 in the ESI.†
:AIBN decreased from 92:1 to 200:1 in the experiment with ωM0 = 10 wt% at 70 °C (only CD3OD as solvent), maintaining constant the ratio M; the comparison between both experiments is illustrated in Fig. 9. A slower copolymerization rate is reflected in the lower conversion values of Xtot, XDEAEMA and XMMA-d3 shown in Fig. 9a and b for a molar ratio 200:1; e.g., the final value of Xtot of 83% for the 92:1 molar ratio is twice that for the 200:1 ratio (39%). The profile of Xtot for the experiment with the lower amount of initiator reflects a strong change of slope at 4 h due to the exhaustion of DEAEMA. The lower consumption of the DEAEMA monomer by polymerization leads to a higher availability of the monomer for its transesterification with CD3OD and increases the degree of alcoholysis Da to 90 mol%, Fig. 9a. The analogous profile of Da estimated for the transesterification experiments illustrated in Fig. 3a is compared with that estimated in the copolymerization for the molar ratio DEAEMA:AIBN = 200:1, resulting in an overlap of both curves. Therefore, the process has high selectivity to transesterification and MMA-d3 formation under these reaction conditions.
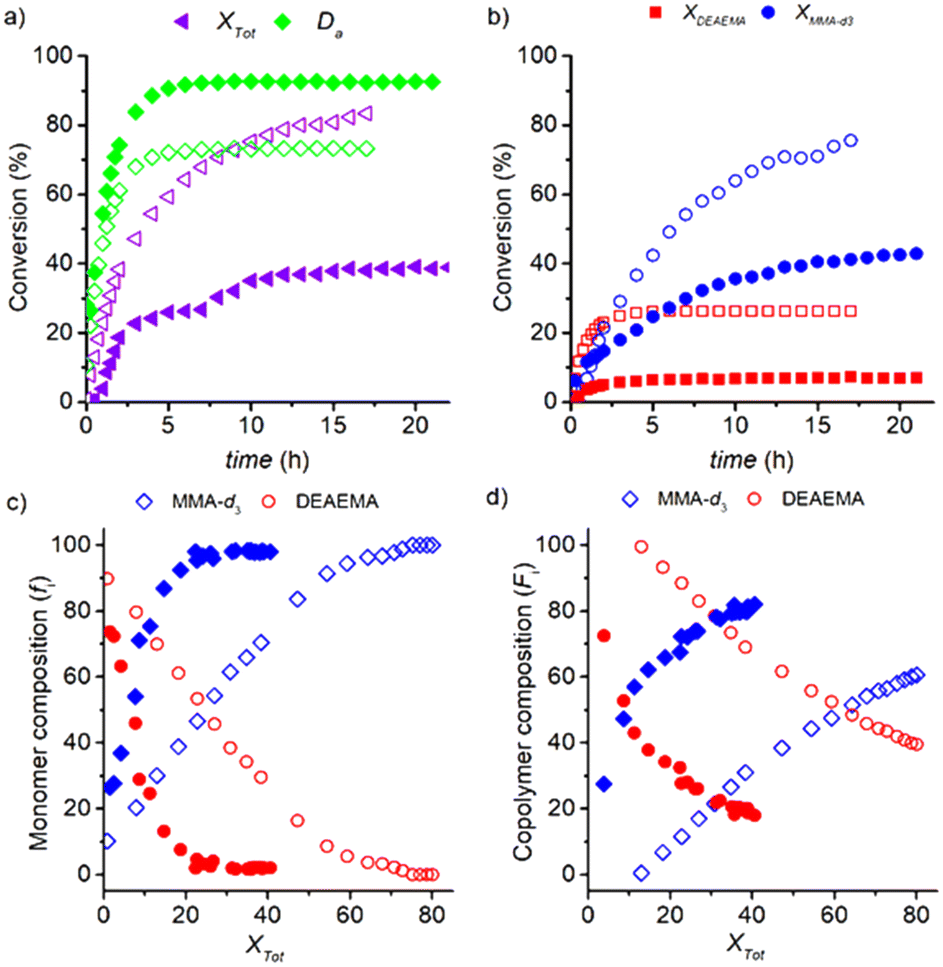 | ||
| Fig. 9 Copolymerization/transesterification of DEAEMA in CD3OD at 60 °C to form poly(DEAEMA-co-MAA-d3) with 92:1 (DEAEMA:AIBN) molar ratio (open symbols) or 200:1 (closed symbols) both with 1:50 (DEAEMA:CD3OD) molar ratio, a) degree of transesterification (Da) and total conversion (Xtot). b) Consumption profiles of DEAEMA incorporated into (co)polymer (XDEAEMA) and MMA incorporated into (co)polymer (XMMA). c) Remaining monomer composition, fi (mol%). d) Cumulative copolymer composition, Fi (mol%). | ||
Decreasing the concentration level of the initiator results in a change in the remaining monomer mixture from a DEAEMA-enriched solution to an MMA-enriched solution in a short period, but only 40% of Xtot can be achieved in 24 h, Fig. 9a and c. In the first stage of the reaction, the copolymer chains are formed with a high DEAEMA content, but since MMA is generated by a fast transesterification, the chains are enriched with MMA until they reach 80% of the accumulated composition at 22 h, Fig. 9d. It is estimated that 37 mol% of the total mass of macromolecules are formed by homopolymer chains of MMA-d3, which are generated above Xtot = 20 mol%. In fact, a white precipitate was observed after 24 h of reaction, Fig. S18 in the ESI.†
These results indicate that batch DEAEMA polymerizations in CH3OH (and possibly other primary alcohols) under mild temperature and low levels of initial concentration of the initiator undergo a high selectivity for transesterification and in situ formation of the MMA monomer, leading to a copolymer with strong composition drift (even homopolymer chains) and loss of amino functionality in the final material. Such conditions could be easily achieved in reversible-deactivation radical polymerizations (RDRP),56–58 specifically during processes with significantly long preequilibrium periods. However, gradient and alternated copolymers could be prepared via concurrent tandem RDRP and transesterification such as in previous studies,59 but without the use of metal catalysts.
Conclusions
The mechanism of transesterification of DEAEMA with methanol to produce MMA and aminoethanol has been elucidated via a DFT approach; the postulated mechanism for an equimolar ratio of reagents comprises three principal stages: i) a limiting step of the amine protonation while the CH3O moiety forms an O–C bond with the carbonyl of the ester; then the adduct suffers structural chances to recover the conformational flexibility around the central part of the molecule; ii) this step is principally governed for conformational changes of the adduct, and iii) a cleavage of the C–O bond in the adduct, as well as the migration of hydrogen, giving rise to the observed products of the reaction. It is important to note that a cooperative effect of the solvent molecules that participate in the hydrogen bonding between DEAEMA and methanol is plausible, which leads to a reduction in the energetic barriers of the transition states and regeneration of the methanol molecules in the products.In situ NMR spectroscopy and a second-order reaction were used in the kinetic studies of the transesterification of DEAEMA with a primary alcohol. Surprisingly, the experiment with ωM0 = 10 wt% and T = 26.8 °C resulted in an almost complete transesterification process (Da = 80%) after only one day. However, increasing the content of DEAEMA (higher ωM0), the transesterification degree is lowered, which is consistent with the hypothesis of the cooperative effect of solvent molecules, because Ea decreases as the value of ωM0 increases. A second order expression is presented to describe the transesterification rate coefficient with Arrhenius parameters depending on ωM0, showing a good fit with the experimental data over a wide temperature interval.
Additionally, the operating conditions of the transesterification of DEAEMA that give rise to MMA generation, both monomers in the presence of a thermal initiator and competitive (co)polymerization, were also studied. The kinetic complexity of this system increases as a result of the in situ formation of MMA during the reaction, the initiator decomposition rate, homopolymerization, and copolymerization processes. The incorporation of MMA into the propagating chains was validated via NMR analysis. For highly diluted DEAEMA experiments (46:1 (DEAEMA:CH3OH) molar ratio), a fast transesterification rate was observed, leading to a mixture with MMA composition as high as 67 mol%, with most of them forming homopolymer chains due to fast DEAEMA exhaustion in the first stage of reaction. However, if an equimolar ratio of DEAEMA:CH3OH is analysed, the transesterification is practically avoided, giving rise to a DEAEMA copolymer composition of 98 mol%; therefore, the process is mainly governed by DEAEMA homopropagation, preserving the amino functionality. Furthermore, the amount of MMA transesterification product, as well as the copolymer composition, can also be regulated by modifying the concentration of initiator. These results provide guidelines for the synthesis of well-defined DEAEMA copolymers in primary alcoholic media used in stimuli-responsive materials in which avoiding transesterification is desirable. However, transesterification offers a new route of synthesis for the solution free radical copolymerization of TAMAs and alkyl or functionalized methacrylates, and efforts are being made to model the kinetics and implement RDRP techniques.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, I. Z. G.; methodology: J. C. R., J. F. E. M., and R. T. L.; validation, J. C. R., R. T. L.; investigation: all the authors; NMR and laboratory resources, J. F. E. M. and R. T. L.; DFT calculations and discussions, J. J. C. and A. O. T.; writing—original draft preparation, I. Z. G., writing—review and editing, I. Z. G., A. O. T. and R. T. L.; visualization, I. Z. G.; project administration: I. Z. G.; funding acquisition: I. Z. G. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ivan Zapata-González thanks the CIQA for financial support through the program of Internal projects, number 6754.Notes and references
- J. Ramos, J. Forcada and R. Hidalgo-Alvarez, Chem. Rev., 2014, 114(1), 367–428, DOI:10.1021/cr3002643.
- A. González-Urías, I. Zapata-González, A. Licea-Claverie, A. F. Licea-Navarro, J. Bernaldez-Sarabia and K. Cervantes-Luevano, Colloids Surf., B, 2019, 182, 110365, DOI:10.1016/j.colsurfb.2019.110365.
- Q. R. Huang, W. Volksen, E. Huang, M. Toney, C. W. Frank and R. D. Miller, Chem. Mater., 2002, 14(9), 3676–3685, DOI:10.1021/cm020014z.
- S. H. Cho, M. S. Jhon, S. H. Yuk and H. B. Lee, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 595–598, DOI:10.1002/(SICI)1099-0488(199703)35:4<595::AID-POLB7>3.0.CO;2-P.
- M. Okubo, H. Ahmad and T. Suzuki, Colloid Polym. Sci., 1998, 276, 470–475, DOI:10.1007/s003960050268.
- M. P. J. Miclotte, S. B. Lawrenson, S. Varlas, B. Rashid, E. Chapman and R. K. O'Reilly, ACS Polym. Au, 2021, 1(1), 47–58, DOI:10.1021/acsami.2c15024.
- A. Pikabea, J. Ramos and J. Forcada, Part. Part. Syst. Charact., 2013, 31, 101–109, DOI:10.1002/ppsc.201300265.
- D. S. Spencer, A. B. Shodeinde, D. W. Beckman, B. C. Luu, H. R. Hodges and N. A. Peppas, J. Controlled Release, 2021, 332, 608–619, DOI:10.1016/j.jconrel.2021.03.004.
- V. D. Lechuga-Islas, M. Trejo-Maldonado, I. Anufriev, I. Nischang, İ. Terzioğlu, J. Ulbrich, R. Guerrero-Santos, L. E. Elizalde-Herrera, U. S. Schubert and C. Guerrero-Sánchez, Macromol. Biosci., 2023, 23, 2200262, DOI:10.1002/mabi.202200262.
- V. D. Lechuga-Islas, G. Festag, M. Rosales-Guzman, O. E. Vega-Becerra, R. Guerrero-Santos, U. S. Schubert and C. Guerrero-Sanchez, Eur. Polym. J., 2020, 124, 109457, DOI:10.1016/j.eurpolymj.2019.10.
- R. Yañez-Macias, I. Alvarez-Moises, I. Perevyazko, A. Lezov, R. Guerrero-Santos, U. S. Schubert and C. Guerrero-Sanchez, Macromol. Chem. Phys., 2017, 218(10), 1700065, DOI:10.1002/macp.201700065.
- A. Darabi, P. G. Jessop and M. F. Cunningham, Chem. Soc. Rev., 2016, 45, 4391, 10.1039/C5CS00873E.
- P. van de Wetering, N. J. Zuidam, M. J. Van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31(23), 8063–8068, DOI:10.1021/ma980689g.
- P. Zheng, X. Su, C. Fei, X. Shi, H. Yin and Y. Feng, J. Polym. Sci., Part B: Polym. Phys., 2018, 56(12), 914–923, DOI:10.1002/polb.24606.
- The estimated values using linear regression and the temperature and ln(k) values obtained from Table 1 of ref. 11 are A = 3.10 × 107 s−1 and 69.51 ± 3 kJ mol−1.
- O. Ajogbeje, I. Lacík and R. A. Hutchinson, Polym. Chem., 2023, 14(21), 2624–2639, 10.1039/D3PY00350G.
- N. P. Truong, Z. M. J. Burges, N. A. McMillan and M. J. Monteiro, Biomacromolecules, 2011, 12(5), 1876–1882, DOI:10.1021/bm200219e.
- P. Cotanda, D. B. Wright, M. Tyler and R. K. A. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(16), 3333–3338, DOI:10.1002/pola.26730.
- P. Y. Bruice, Organic Chemistry, Person Education Inc, Prentice Hall, 5th edn, 2007, p. 744 Search PubMed.
- J. Otera, Transesterification, Chem. Rev., 1993, 93, 1449–1470, DOI:10.1021/cr00020a004.
- J. Otera and J. Nishikido, Esterification: Methods, Reactions, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2009, DOI:10.1002/9783527627622.
- G. A. Grasa, R. Singh and S. P. Nolan, Synthesis, 2004, 7, 971–985, DOI:10.1055/s-2004-822323.
- H. E. Hoydonckx, D. E. De Vos, S. A. Chavan and P. A. Jacobs, Top. Catal., 2004, 27, 83–96, DOI:10.1023/B:TOCA.0000013543.96438.1a.
- J. Q. Ng, H. Arima, T. Mochizuki, K. Toh, K. Matsui, M. Ratanasak, J. Y. Hasegawa, M. Hatano and K. Ishihara, ACS Catal., 2021, 11(1), 199–207, DOI:10.1021/acscatal.0c04217.
- X. Bories-Azeau and S. P. Armes, Macromolecules, 2002, 35(27), 10241–10243, DOI:10.1021/ma021388g.
- P. Quiñonez-Angulo, R. A. Hutchinson, Á. Licea-Claveríe, E. Saldívar-Guerra and I. Zapata-González, Polym. Chem., 2021, 12(37), 5289–5302, 10.1039/d1py00750e.
- J. Arredondo, P. Champagne and M. F. Cunningham, Polym. Chem., 2019, 10(15), 1938–1946, 10.1039/C8PY01803K.
- P. Quiñonez Angulo, Synthesis, Kinetic Study and Applications of Smart Copolymers with Amino-Containing Groups and PEGMAs, Ph.D. Thesis, Tecnológico de Tijuana/TecNM, B.C., México, 2022 Search PubMed.
- S. Mushtaq, M. A. Abbas, H. Nasir, A. Mahmood, M. Iqbal, H. A. Janjua and N. M. Ahmad, Sci. Rep., 2023, 13, 4572, DOI:10.1038/s41598-023-27515-5.
- J. Y. Zheng, M. J. Tan, P. Thoniyot and X. J. Loh, RSC Adv., 2015, 5(76), 62314–62318, 10.1039/C5RA12816A.
- L. M. Thoma, B. R. Boles and K. Kuroda, Biomacromolecules, 2014, 15(8), 2933–2943, DOI:10.1021/bm500557d.
- Q. Chen, W. Lin, H. Wang, J. Wang and L. Zhang, RSC Adv., 2016, 6(72), 68018–68027, 10.1039/c6ra10757e.
- S. Beuermann, Macromolecules, 2004, 37(3), 1037–1041, DOI:10.1021/ma0354647.
- S. K. Fierens, P. H. M. Van Steenberge, M. F. Reyniers, D. R. D'hooge and G. B. Marin, React. Chem. Eng., 2018, 3(2), 128–145 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648–5652, DOI:10.1063/1.464913.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98(45), 11623–11627, DOI:10.1021/j100096a001.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/B508541A.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094, DOI:10.1021/cr9904009.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- S. Y. Park and D. J. Jang, J. Am. Chem. Soc., 2010, 132(1), 297–302, DOI:10.1021/ja907491u.
- S. Morpurgo, M. Bossa and G. Morpurgo, Adv. Quantum Chem., 2000, 36, 169–183, DOI:10.1016/S0065-3276(08)60483-9.
- Y. Zhao and T. Gennett, Phys. Rev. Lett., 2014, 112, 076101, DOI:10.1103/PhysRevLett.112.076101.
- S. Banerjee, A. Pabbathi, M. C. Sekhar and A. Samanta, J. Phys. Chem. A, 2011, 115(33), 9217–9225, DOI:10.1021/jp206232b.
- P. L. Silva, C. M. Silva, L. Guimarães and J. R. Pliego, Theor. Chem. Acc., 2015, 134, 1591, DOI:10.1007/s00214-014-1591-5.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29(9), 2176–2179, DOI:10.1021/om100106e.
- O. Levespiel, The chemical Reactor Omnibook, OSU Book Stores, Inc., Corvallis, Oregon, 1989, p. 2.3 Search PubMed.
- Sigma Aldrich, https://www.sigmaaldrich.com/MX/en/product/aldrich/343803, acceded on July 4th, 2024.
- G. Aksnes and P. Froyen, Acta Chem. Scand., 1966, 20(6), 1451–1455, DOI:10.3891/acta.chem.scand.20-1451.
- M. Přádný and S. Ševčík, Makromol. Chem., 1985, 186(1), 111–121, DOI:10.1002/macp.1985.021860111.
- P. A. Mirau, Practical guide to understanding the NMR of polymers, John Wiley & Sons, 2005 Search PubMed.
- G. Nguyen, M. Matlengiewicz and D. Nicole, Analusis, 1999, 27(10), 847–853, DOI:10.1051/analusis:1999152.
- M. Baowei, L. H. Gan, Y. Y. Gan, X. Li, P. Ravi and K. C. Tam, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5161–5169, DOI:10.1002/pola.20228.
- B. Mao, L. H. Gan, Y. Y. Gan, X. Li, P. Ravi and K. C. Tam, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(20), 5161–5169, DOI:10.1002/pola.20228.
- E. Tang, K. Du, X. Feng, M. Yuan, S. Liu and D. Zhao, Eur. Polym. J., 2015, 66, 228–235, DOI:10.1016/j.eurpolymj.2015.01.041.
- M. Manguian, M. Save and B. Charleux, Macromol. Rapid Commun., 2006, 27(6), 399–404, DOI:10.1002/marc.200500807.
- L. Zhu, S. Powell and S. G. Boyes, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(8), 1010–1022, DOI:10.1002/pola.27529.
- E. Tang, K. Du, X. Feng, M. Yuan, S. Liu and D. Zhao, Eur. Polym. J., 2015, 66, 228–235, DOI:10.1016/j.eurpolymj.2015.01.041.
- K. Nakatani, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 13600–13601, DOI:10.1021/ja9058348.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00406j |
| This journal is © The Royal Society of Chemistry 2025 |