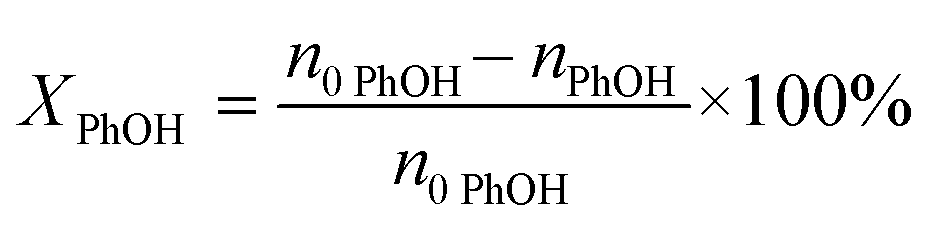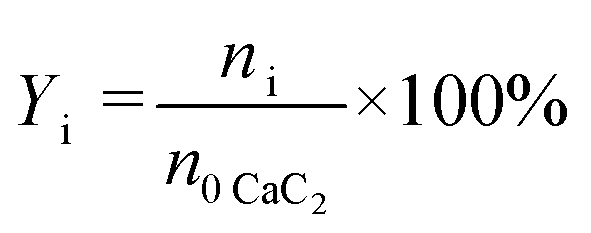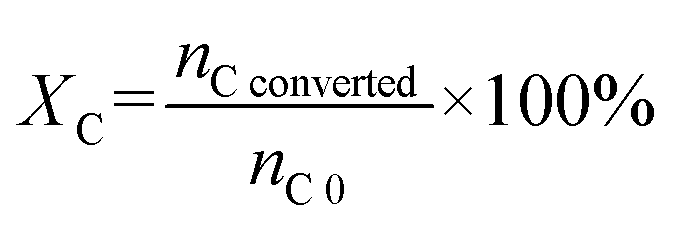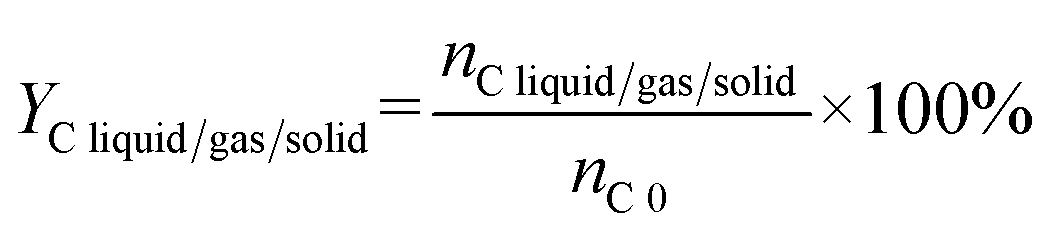Synthesis of ethylphenols and xanthenes via reaction of calcium carbide and phenol: experimental and theoretical studies†
Received
17th August 2024
, Accepted 28th October 2024
First published on 8th November 2024
Abstract
Calcium carbide (CaC2) is a platform chemical for various organic synthesis, and monomeric phenol (PhOH) is expected to be produced via biomass conversion in the near future. This work explores their downstream product during reaction at 300–400 °C without additional solvent and catalyst. The reaction matrix was investigated by density functional theory (DFT) calculation and characterization of the solid product. Results indicate that in addition to ethylphenols, xanthenes are unexpectedly formed with a yield of 26.0% at 350 °C. DFT calculation indicates that PhOH is firstly alkylated by CaC2 to form vinylphenol or dehydrated intermolecularly to form diphenyl ether. Xanthenes are then formed through two pathways: dehydration of vinylphenol with PhOH and then cyclization; alkylation and cyclization of diphenyl ether with CaC2-derived acetylene. Ethylphenols are formed through hydrogenation of vinylphenol where PhOH provides hydrogen. Vinylphenol hydrogenation for ethylphenols exhibits a competitive advantage over vinylphenol dehydration for xanthenes. X-ray diffraction (XRD) of the solid product indicates that CaC2 is converted to calcium phenoxide. Isomolecular electrostatic potential maps suggest that calcium phenoxide exerts a catalytic effect on the alkylation and dehydration reactions. This work provides a novel protocol for xanthene synthesis and an in situ efficient utilization method of the acetenyl group.
1. Introduction
Calcium carbide (CaC2) is widely recognized as a versatile platform chemical for organic synthesis and is highly regarded for its environmentally friendly and sustainable nature.1,2 Direct reactions of CaC2 with organic compounds have been widely explored in recent years, including vinylation of alcohol/phenol (PhOH),3 addition reaction with aldehyde/ketone,4 alkyne propylation of tertiary amine,5 Favorskii reaction with aromatic aldehyde,6 and exchange, substitution, and cross-coupling reactions.7 In these reactions, CaC2 initially reacts with trace amounts of water to produce acetylene and calcium hydroxide; subsequently, the in situ generated acetylene further reacts with various organic compounds, wherein CaC2 primarily serves as a stable and reliable source of acetylene. Nevertheless, Our recent work indicated that reaction of CaC2 with acetone forms methylbenzene and methylnaphthalene8 where CaC2 not only catalyzes acetone condensation by its superior nucleophilicity but also participates in aromatization reaction via calcium acetylide. Furthermore, Reaction of CaC2 with ethanol indicated that the solid product calcium ethoxide could catalyze ethanol condensation to n-butanol.9 These findings suggest that CaC2 can serve as a safer alternative in all acetylene-involved reactions,10 while its derivatives exhibit catalytic potential in specific reactions.
With the rapid development in biomass conversion to monomeric PhOH,11 PhOH has the potential to emerge as another platform chemical in the near future. Zhang et al. investigated the reaction between CaC2 and PhOH at 140 °C using Cs2CO3 as the catalyst and DMSO with 7 vol% water as the solvent.3 They observed that the main product is phenyl vinyl ether for 16 h. Ananikov et al. studied the catalytic effect of F− on the reaction between CaC2 and dimethylphenol at 130 °C and the main product is also phenyl vinyl ether.12 In both cases, these reactions can be categorized as vinylation reactions involving PhOH and CaC2 (termed O-alkylation). It was reported that PhOH can also be alkylated by acetylene to form vinylphenol and diaryl compounds over acidic catalysts or zeolites over 250 °C (ref. 13–15) (termed C-alkylation). Given that CaC2 has both an alkynyl moiety and good catalytic activity, we tentatively explored its reactivity towards PhOH, leading to a surprising formation of xanthenes alongside alkylation products such as 2- and 4-ethylphenol. It was reported that the market size of xanthenes exceeded $1.06 trillion in 2021.16 Some are naturally occurring compounds isolated from herbs and fungi. Reaction of PhOH with specific structural aldehydes or acetals was also reported to synthesize xanthenes, but the xanthenes are all with substituents. The total yield is between 20% and 40% over nano ZnO and MgO.17,18
Since the reaction of PhOH and CaC2 is a novel and simple protocol for xanthene synthesis, the effect of reaction conditions on the product yield is elaborated in this work. The reaction matrix and pathways are also investigated by controlled experiments, density functional theory (DFT) calculation and detailed characterization of the solid product using XRD, infrared spectroscopy, elemental analysis and ESR.
2. Materials and methods
2.1 Raw materials
CaC2 with a purity of 91% was ground and sieved to 80–100 mesh in a glovebox before being used for experiments. PhOH, calcium hydroxide, dihydrophenanthrene (DHP) and tetrahydronaphthalene (THN) were commercial reagents and utilized as received.
2.2 Reaction experiments
The reaction of PhOH with CaC2 was carried out in a high-throughput reaction furnace housing 20 parallel glass tube reactors, each with dimensions of 2.4 mm × 100 mm, which has been detailed in our previous work.19 CaC2 (2.5 ± 0.05 mg) and a certain amount of PhOH were loaded into the tube reactors inside a glovebox. The reactors were purged with Ar to remove air, fused by a blast burner and then inserted into the furnace preheated to an assigned temperature. The reactants' temperature could be elevated to the desired level within 2 min without overshooting. After a certain reaction time, the reactors were taken out and immersed in an ice-water bath for rapid cooling. Nine parallel experiments were performed at each reaction condition to facilitate subsequent product analysis. The detailed implementation operations can be found in Fig. S1 and S2.†
2.3 Analysis of products
The cool reactor was put into a latex tube filled with 0.1 MPa N2 and broken manually to release the gas product. The gas product was quantified by a gas chromatograph (Agilent 7820B) with an external standard method. Inorganic gases were analyzed using a thermal conductivity detector (TCD) and tandem columns of Porapak Q (6 ft × 1/8 in × 2.0 mm) and a 5A molecular sieve (6 ft × 1/8 in × 2.0 mm) with N2 as the carrier gas. The temperatures of the inlet, column and detector were maintained at 200 °C, 75 °C, and 250 °C, respectively. Organic gases were analyzed using a flame ionization detector (FID) and a GsBP-PLOT Al2O3 “S” capillary column (50 m × 0.53 mm × 10 μm) with N2 (30 mL min−1) as the carrier gas. The temperatures of the inlet, column, and detector were set at 200 °C, 80 °C, and 250 °C, respectively.
The broken reactor was immersed in 1.5 mL of tetrahydrofuran (THF) for 12 h, and then the solution was filtered to isolate the liquid product. Qualitative analysis of the liquid product was performed using a GC-MS instrument (Agilent 7890B-5977A) equipped with an HP-5MS fused silica capillary column (30 m × 0.25 mm × 0.25 μm). He was employed as the carrier gas at a flow rate of 1.0 mL min−1. The injector temperature was set at 250 °C, and the column temperature program was 40 °C for 5 min, 3 °C min−1 to 180 °C, 180 °C for 5 min and 20 °C min−1 to 280 °C. The mass spectrometer was operated in electron ionization mode at 70 eV with a scan range of 30 to 350 m/z. Quantitative analysis of the liquid product was performed on a GC instrument (Agilent 7890B) with an FID detector and n-tetradecane as the internal standard under the same conditions as mentioned above.
The other reactors from the parallel experiments were broken and subjected to vacuum drying at 80 °C for 72 h to obtain the solid product. X-ray diffraction (XRD) analysis of the solid product was conducted using a D8FOCUS powder diffractometer with Cu Kα radiation (λ = 1.5432 Å), operating at 40 kV and 40 mA. The scanning speed was set at 10 min−1 and the scan range was 3° to 90°. The total carbon content in the solid product was determined using an elemental analyzer (vario EL CUBE).
2.4 Formula description
The PhOH conversion (XPhOH) was calculated using eqn (1), and the yields of products (Yi) were calculated using eqn (2). Considering the excess PhOH in the reaction, both the liquid and gas product yields were calculated based on the amount of CaC2.| | 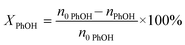 | (1) |
| | 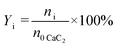 | (2) |
where n0 represents the initial amount of the reactants, and ni represents the amount of product i.
Carbon conversion (XC) and carbon yield of liquid/gas/solid phase (YC liquid/gas/solid) in carbon balance analysis followed eqn (3) and (4), respectively.
| | 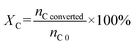 | (3) |
| | 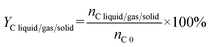 | (4) |
where
nC converted represents the total carbon in converted PhOH and CaC
2,
nC 0 represents the total carbon in initial PhOH and CaC
2,
nC liquid/gas/solid represents the total carbon in liquid/gas/solid products.
2.5 DFT calculation
DFT calculation was performed using Gaussian 09 software to simulate the reaction routes. The transition state structure was determined and optimized at the B3LYP/6-311g(d, p) level. Whether the transition state structure connected the reactant and product was verified by the intrinsic reaction coordinate (IRC) method. Single-point energy calculation was carried out at the M06-2X/def2-TZVP level and corrected using Shermo software.20 Through optimizing the configurations of reactant, transition state and products, the potential energy curves of the relevant reactions were computed to discern the priority of different reaction mechanisms and rates, thus facilitating the derivation of a reasonable reaction path.
3. Results and discussion
3.1 Liquid product characterization and carbon balance analysis
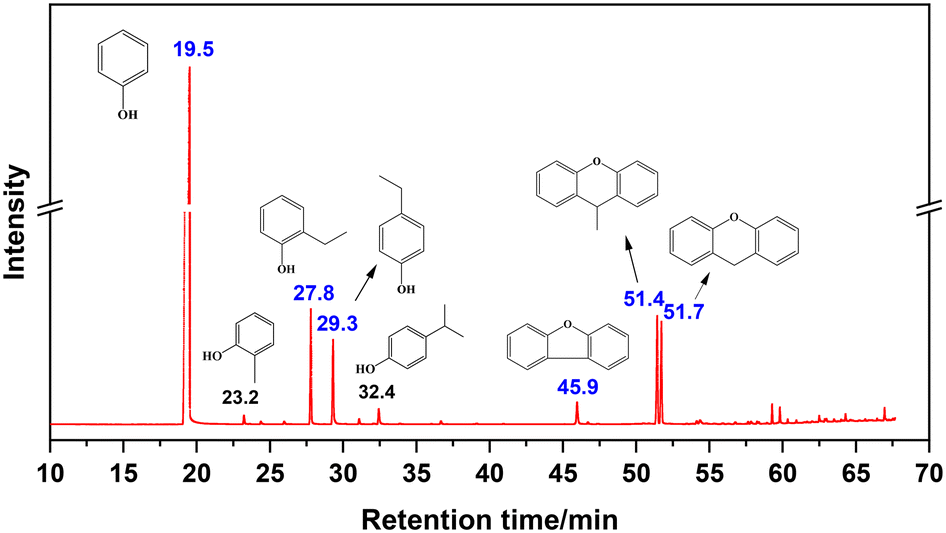
Fig. 1 shows the total ion flow chromatogram of the liquid product obtained from the reaction conducted at 350 °C for 2 h, with its mass spectral information detailed in Fig. S3.† The chromatogram reveals a retention time of 19.5 min for unreacted PhOH, followed by five main peaks, descending in retention time (marked in blue), representing 2-ethylphenol, 4-ethylphenol, dibenzofuran, 9-methylxanthene, and xanthene, in turn. Minor quantities of 2-methylphenol and 4-isopropylphenol were also detected (marked in black). Notably, the product composition significantly differs from that in the vinylation reaction of PhOH with CaC2 in the presence of strong alkalis and trace water, which yields phenyl vinyl ether as the main product.3
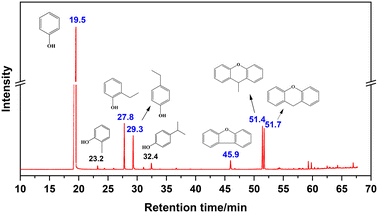 |
| | Fig. 1 Total ion chromatogram of liquid products (main products are marked in blue, by-products are marked in black). | |
Table 1 lists the carbon balance results obtained under various experimental conditions, indicating the carbon balance rate of approximately 90% ± 3% (total carbon yield of the three-phase product divided by carbon conversion). For enhanced data accuracy, the results for each reaction condition represent the average of three parallel experiments.
Table 1 Carbon balance analysis under different reaction conditions
|
T/°C |
t/h |
X
C/% |
Y
C liquid/% |
Y
C gas/% |
Y
C solid/% |
Carbon balance/% |
The mass of CaC2 is 2.5 mg (0.036 mmol) and the molar ratio of PhOH/CaC2 is 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. :1. |
| 350 |
0.5 |
36.0 |
4.5 |
2.2 |
25.8 |
90.3 |
| 350 |
2 |
43.2 |
9.8 |
0.6 |
27.5 |
87.8 |
| 350 |
6 |
51.8 |
12.2 |
0.1 |
35.9 |
93.1 |
| 300 |
2 |
34.7 |
5.3 |
1.3 |
24.6 |
89.9 |
| 400 |
2 |
54.2 |
10.2 |
0.4 |
38.8 |
91.2 |
3.2 Effect of reaction conditions on the liquid product yield
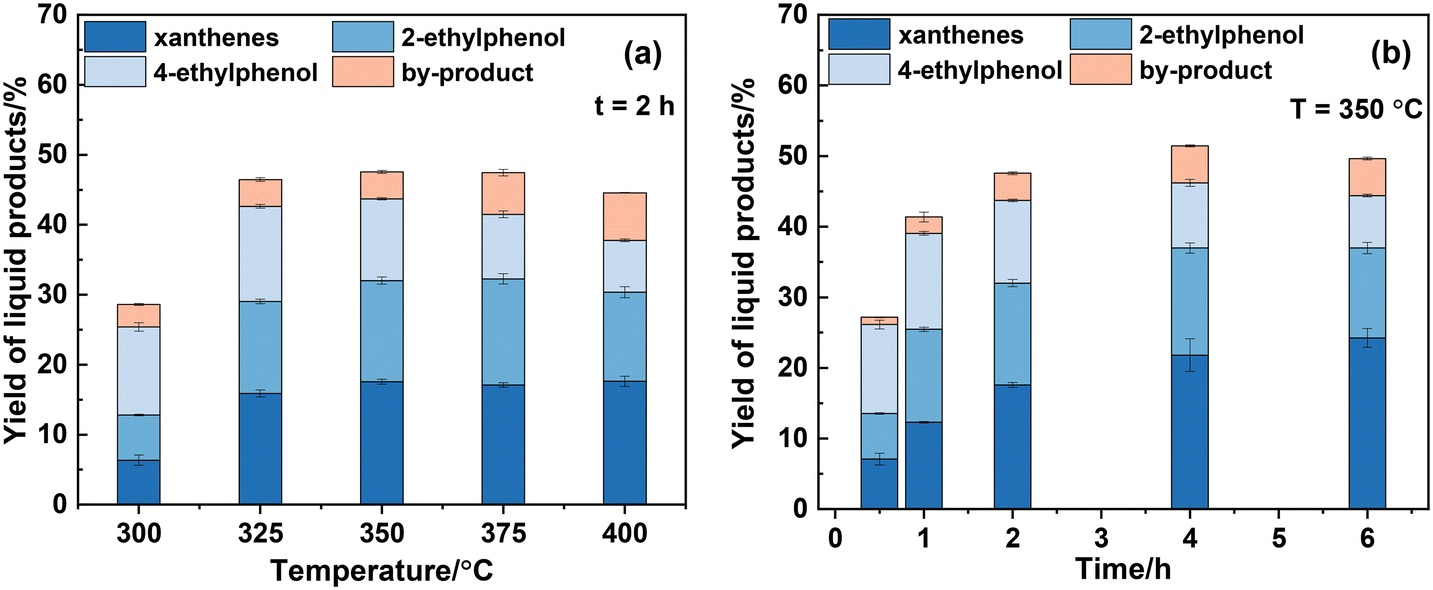
Fig. 2 shows the influence of reaction temperature and time on the yield of liquid products where xanthenes refer to 9-methylxanthene and xanthene collectively. It should be pointed out that due to the synthesis process of dibenzofuran not involving the utilization of CaC2, dibenzofuran is consequently excluded from the yield calculation of the liquid product. Additionally, minor amounts of acetylene were detected when the solid product obtained at 300 °C for 0.5 h was reacted with water, but not the solid products from other conditions. This indicated that CaC2 is fully converted under other reaction conditions with 100% conversion. As shown in Fig. 2(a), the total yield of both ethylphenols increases to a maximum value of 26% at 350 °C and then decreases with further increase in temperature. In contrast, the yield of xanthenes increases consistently, reaching its maximum of about 18% at 400 °C. The yield of other alkylphenols, mainly methylphenol and isopropylphenol, also increases consistently with temperature.
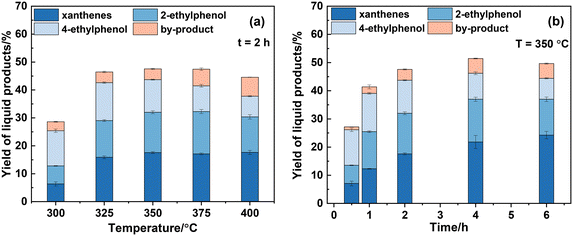 |
| | Fig. 2 Effect of reaction temperature (a) and time (b) on the yield of liquid products. The mass of CaC2 is 2.5 mg (0.036 mmol) and the molar ratio of PhOH/CaC2 is 8:1. | |
From the viewpoint of xanthenes and ethylphenols, 350 °C was selected to investigate the effect of reaction time on the product yield, as shown in Fig. 2(b). It is evident that the yields of 2-ethylphenol, 4-ethylphenol, and xanthenes at 0.5 h are 7.3%, 8.8%, and 8.1%, respectively. As the reaction time is prolonged, the yield of xanthenes gradually increases to 24.2% at 6 h. The total yield of ethylphenols peaks at 27.6% at 4 h, then slightly decreases to 25.4% at 6 h which is accompanied by an increase in the yield of other alkylphenols. This suggests the cleavage of the ethyl side chain of ethylphenols at high temperature and prolonged reaction time.
Table 2 shows the influence of the molar ratio of PhOH/CaC2 on the product composition. Taking the reaction at 350 °C for 2 h as an example (entries 1–5), it is observed that as the molar ratio of PhOH/CaC2 increases from 1:1 to 16:1, the yield of each product exhibits an upward trend, with the total product yield increasing from 1.8% to 59.4%. Specifically, the yield of xanthenes increases from 0 to 21.2%, and the total yield of ethylphenols reaches 34.3%, while the yield of other products is 3.9% at 16:1. The total product yield of 69.4% is achieved at a PhOH/CaC2 molar ratio of 16:1 at 350 °C for 6 h (entry 6), where the yields of 2-ethylphenol, 4-ethylphenol and xanthenes are 21.2%, 17.1% and 26.0%, respectively. In addition, alkylation of PhOH with hydrocarbons/alcohols mainly generates alkyl-substituted phenols as shown in Table 3, with total yields between 50% and 76.5%. In contrast, our study demonstrates that the alkylation of PhOH with CaC2 produces not only ethylphenols but also the high-value compound xanthenes. The total yield of these products is 64.3% without use of any catalyst, which is comparable to the yields reported in the literature.21–24
Table 2 Effect of PhOH dosage on the yield of liquid products
| Entry |
PhOH/CaC2 |
Time (h) |
X
PhOH (%) |
Y
i (%) |
| Xanthenes |
2-Ethylphenol |
4-Ethylphenol |
Others |
Total |
| The mass of CaC2 is 2.5 mg (0.036 mmol) and the temperature is 350 °C; (I) PhOH/CaC2/DHP = 8:1:2 (molar ratio); (II) PhOH/CaC2/THN = 8:1:2 (molar ratio); (III) PhOH/CaC2/diphenyl ether = 8:1:1 (molar ratio). |
| 1 |
1:1 |
2 |
60.3 |
ND |
0.7 |
1.1 |
ND |
1.8 |
| 2 |
2:1 |
2 |
54.8 |
ND |
2.2 |
4.8 |
ND |
7.0 |
| 3 |
4:1 |
2 |
49.9 |
8.3 |
5.6 |
9.2 |
1.7 |
24.8 |
| 4 |
8:1 |
2 |
42.8 |
17.6 |
14.4 |
11.7 |
3.8 |
47.5 |
| 5 |
16:1 |
2 |
24.9 |
21.2 |
17.6 |
16.7 |
3.9 |
59.4 |
| 6 |
16:1 |
6 |
29.0 |
26.0 |
21.2 |
17.1 |
5.1 |
69.4 |
| 7I |
8:1 |
2 |
32.1 |
5.6 |
13.1 |
26.3 |
8.5 |
53.5 |
| 8II |
8:1 |
2 |
29.6 |
8.8 |
16.5 |
22.6 |
6.8 |
54.7 |
| 9III |
8:1 |
2 |
39.9 |
20.7 |
13.2 |
10.8 |
5.8 |
50.5 |
Table 3 Results of alkylation of phenols with hydrocarbons
| Entry |
Reactant |
Reaction conditions |
Products (yield) |
Ref. |
|
Vertical packed bed reactor; online detection.
|
| 1 |
PhOH and propylene |
350 °C, zeolites |
2/4-Isopropylphenol (1.9–9.5%)a |
21
|
| 2 |
PhOH and cyclohexene |
160 °C, 4 h |
2/4-Cyclohexylphenol (57.4%) |
22
|
| HBEA-150 |
2,4-Dicyclohexylphenol (18.9%) |
| 3 |
PhOH and cyclohexene |
160 °C, 4 h |
2/4-Cyclohexylphenol (32.3%) |
22
|
| HY-80 |
2,4-Dicyclohexylphenol (31.4%) |
| 2,6-Dicyclohexylphenol (12.8%) |
| 4 |
PhOH and methanol |
350 °C, 8 h |
Anisole (∼30%) |
23
|
| 10% ZrO2–30% WO3–SiO2 |
o-Cresol (∼20%) |
| 5 |
PhOH and methanol |
450 °C, 2 h |
Polyalkylated compounds (72%) |
24
|
| Beta-2 catalyst |
| 6 |
PhOH and CaC2 |
350 °C, 6 h |
2/4-Ethylphenol (38.3%) |
Our work |
| No catalyst |
Xanthenes (26.0%) |
3.3 Transformation of CaC2
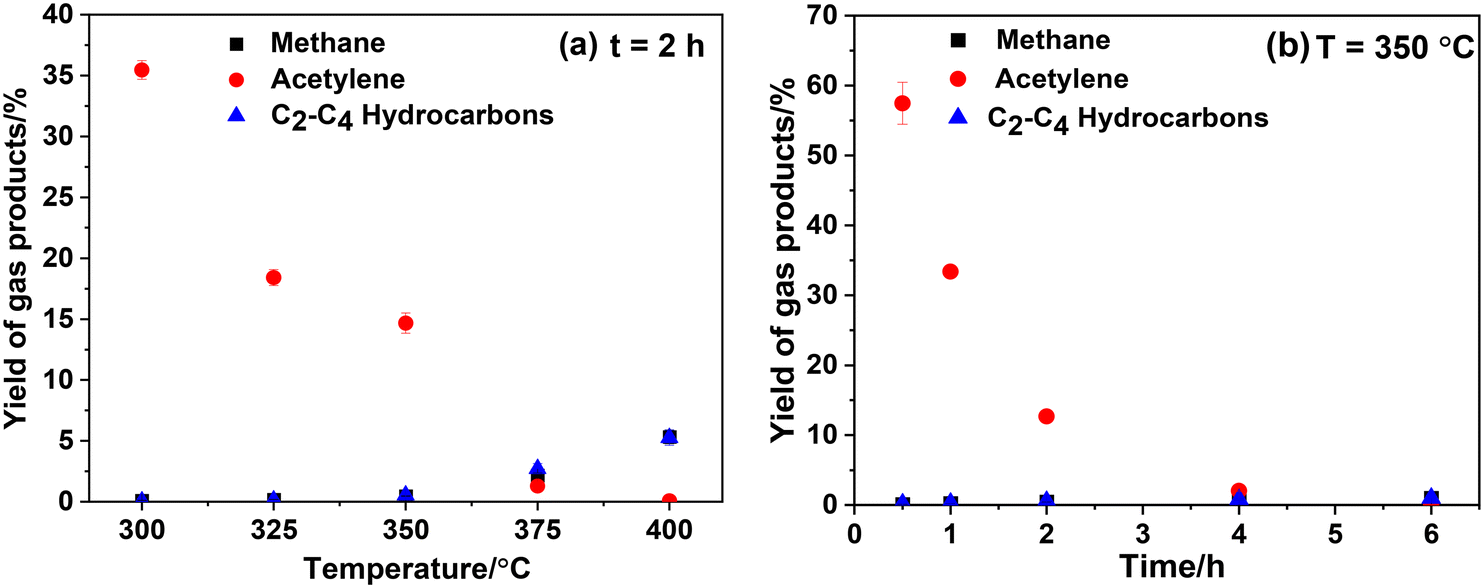
Both ethylphenols and xanthenes contain aliphatic carbon atoms. Given that the reaction temperature is insufficient to break the benzene rings, the alkyne group in CaC2 may participate in the formation of ethylphenols and xanthenes. To disclose the role of the alkyne group in CaC2 during the reaction and to trace the transformation of CaC2, the composition of gas products and the morphological characteristics of solid products were examined. Fig. 3 shows the composition of gas products at various temperatures and times, including acetylene, methane, C2–C4 alkanes and olefins (collectively referred to as C2–C4 hydrocarbons), with acetylene being the predominant product. As shown in Fig. 3(a), with increasing reaction temperature, the yield of acetylene gradually decreases to 0. However, the yields of methane and C2–C4 hydrocarbons significantly increase from 350 °C to 400 °C, reaching a yield of 5.2% for both at 400 °C. This phenomenon suggests that side-chain cleavage of alkylphenol products starts at 350 °C and becomes severe at 400 °C which is consistent with the results of Fig. 2. Fig. 3(b) shows a gradual decrease in acetylene yield from 57.5% for 0.5 h to 0 for 6 h at 350 °C, indicating that the acetylene was gradually converted to liquid or solid products.
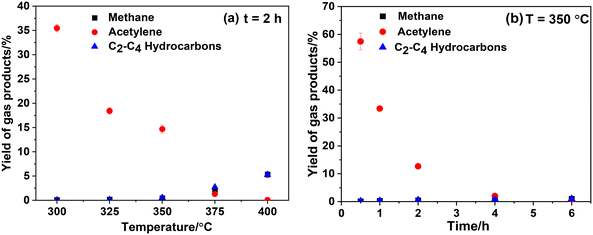 |
| | Fig. 3 Effect of reaction temperature (a) and time (b) on the yield of gas products. The mass of CaC2 is 2.5 mg (0.036 mmol) and the molar ratio of PhOH/CaC2 is 8:1. | |
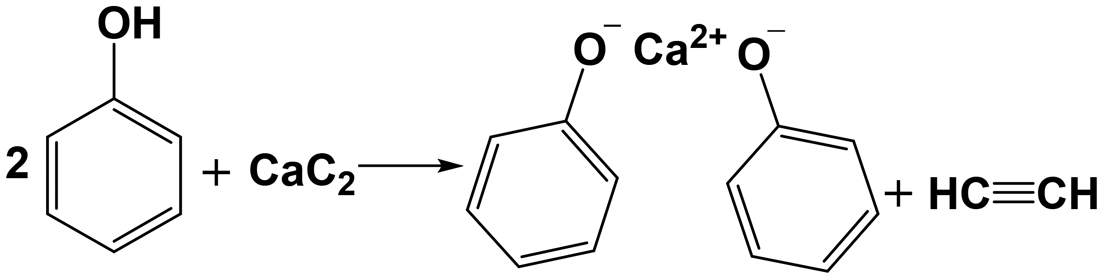
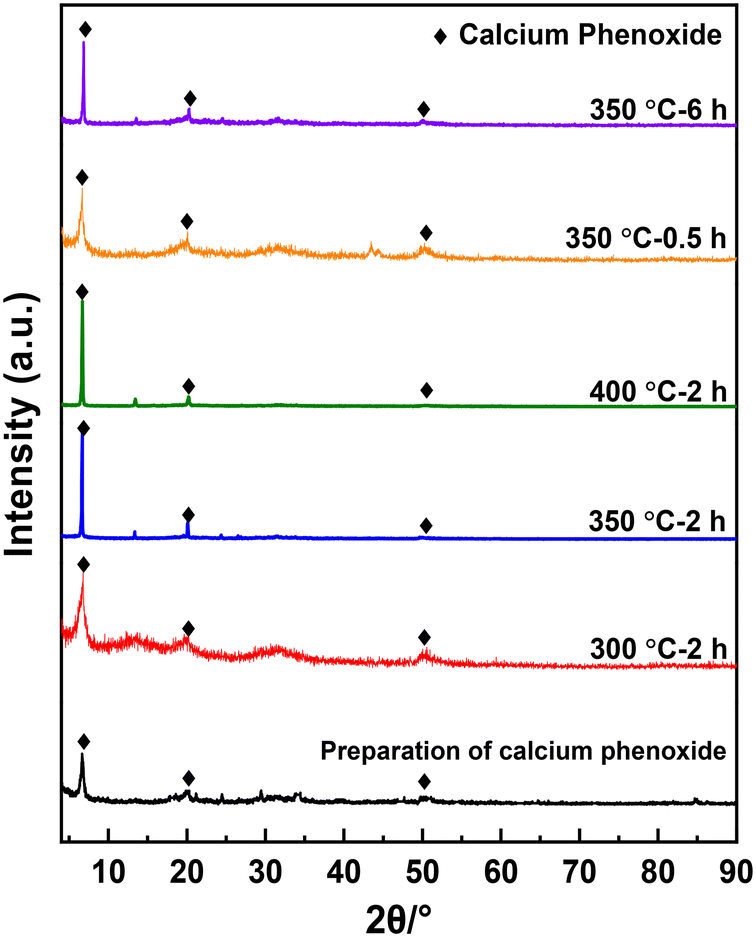
Fig. 4 presents the XRD spectra of the solid products, all of which exhibit a strong peak at 2θ = 10.7° and two faint peaks at 2θ = 19.8° and 49.8°. These peaks are not cataloged in the XRD spectra database but likely to be the diffraction peaks of calcium phenoxide formed through Re. 1. Formation of acetylene shown in Fig. 3 may support this reaction. To verify the attribution of the three peaks, calcium phenoxide was synthesized through the reaction between calcium hydroxide and PhOH by referencing the literature25 and subsequently subjected to XRD analysis (Fig. S4†). The location of the strongest peak, 2θ = 10.7°, is the same as that of the solid products, which confirms the formation of calcium phenoxide.
| | 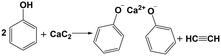 | (Re. 1) |
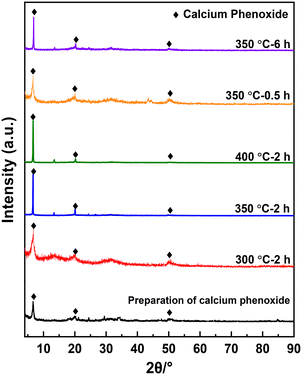 |
| | Fig. 4 XRD of the solid products obtained under different reaction conditions. | |
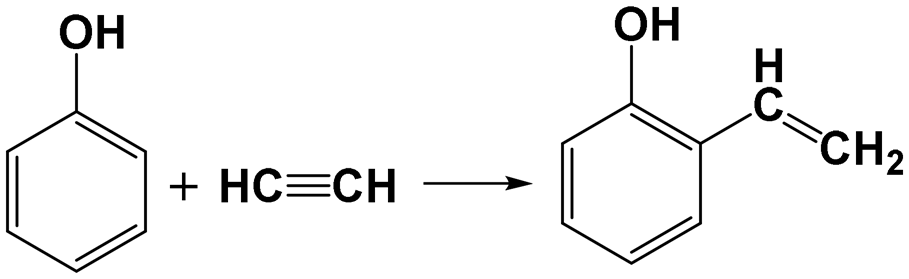
3.4 Reaction pathway analysis
It was reported that PhOH can undergo alkylation with acetylene via Friedel–Crafts reaction, resulting in the formation of 2-vinylphenol (see Re. 2) and minor quantities of diaryl compounds, utilizing sulfuric acid as the catalyst.15 Vinylphenol may form a tricyclic structure akin to xanthene through cyclization and rearrangement with aldehydes26 or alcohols.27 The hydrogenation of vinylphenol will yield ethylphenol. This information indicates that vinylphenol could serve as a potential intermediate of the liquid products. However, vinylphenol is not observed in all the liquid products, and therefore DFT calculation is conducted to evaluate the validity of this hypothesis.| | 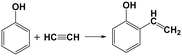 | (Re. 2) |
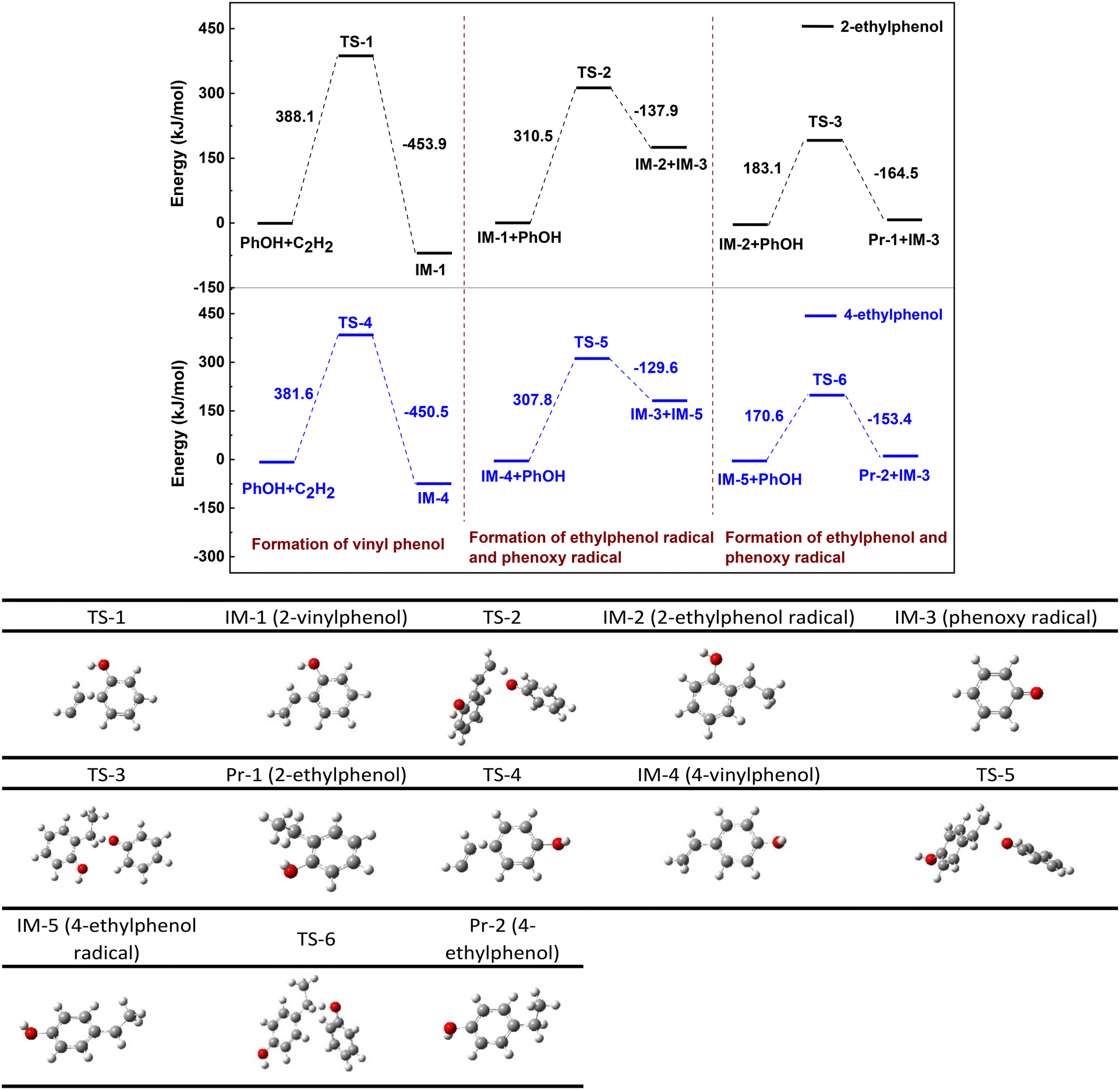
3.4.1 Formation of ethylphenols.
Fig. 5 shows the energy barrier diagram of each step during reaction of PhOH with acetylene to form 2- and 4-ethylphenols. The bond length parameters for each transition state are detailed in Fig. S5† and the accuracy of the transition state structure at each reaction stage is confirmed by IRC curves, as depicted in Fig. S6.† The black line in Fig. 5 represents the formation route of 2-ethylphenol (Pr-1), while the blue one corresponds to that of 4-ethylphenol (Pr-2). Taking the black line as an example, the transition state, intermediate and hydrogenation route are discussed below.
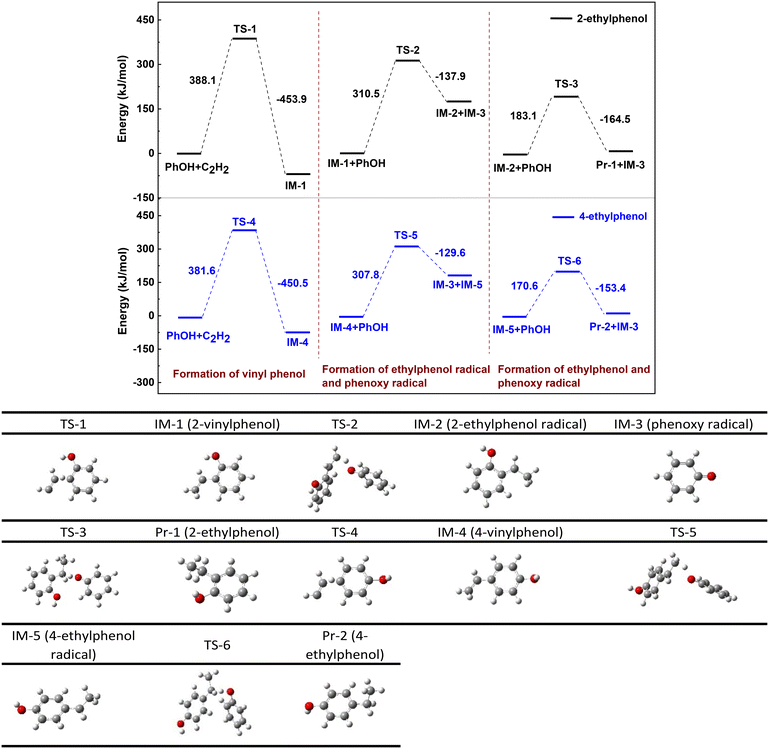 |
| | Fig. 5 Energy barrier diagram of PhOH reaction with acetylene to form ethylphenols. The black line is the formation route of 2-ethylphenol and the blue one is that of 4-ethylphenol. | |
The first column of DFT calculation indicates that bending of the C–H bond and elongation of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond within the acetylene molecular structure allow the ortho H of the hydroxyl of PhOH approaching acetylene to form 2-vinylphenol (IM-1), an intermediate of the reaction, with an energy barrier of 388.1 kJ mol−1. The conversion from IM-1 to Pr-1 necessitates hydrogenation, where the hydrogen must derive from PhOH since none of the hydrogen atom is in CaC2. Both hydroxyl groups and hydrogens located at ortho and para positions on PhOH serve as potential hydrogen sources. DFT calculation indicates that the O–H bond in the hydroxyl group has the lowest dissociation energy, being 301.6 kJ mol−1 (Table 4), while the dissociation energy of Car–H is as high as 392–397 kJ mol−1. Consequently, cleavage of the O–H bond should be the primary source for IM-1 hydrogenation. If this hypothesis holds true, cleavage of the O–H bond of PhOH is accompanied by the formation of phenoxy radical. It is known that radicals are predominantly unstable and easily undergo polymerization. Elemental analysis of the solid product indicates that its carbon content is about 24.6–38.8% (Table S1†), significantly exceeding the theoretical carbon content of calcium phenoxide (approximately 24%), suggesting transformation of carbon into the solid product at higher temperature or prolonged time. Infrared spectroscopy results confirm both hydroxyl groups and ether bonds in these solid products (Fig. S7†), while electron spin resonance (ESR) analysis reveals a free radical concentration of approximately 9.67 × 1016 spins per g (Fig. S8†). These phenomena confirm the polymerization of phenoxy radicals, as observed over 2,6-dimethylphenol.28
C bond within the acetylene molecular structure allow the ortho H of the hydroxyl of PhOH approaching acetylene to form 2-vinylphenol (IM-1), an intermediate of the reaction, with an energy barrier of 388.1 kJ mol−1. The conversion from IM-1 to Pr-1 necessitates hydrogenation, where the hydrogen must derive from PhOH since none of the hydrogen atom is in CaC2. Both hydroxyl groups and hydrogens located at ortho and para positions on PhOH serve as potential hydrogen sources. DFT calculation indicates that the O–H bond in the hydroxyl group has the lowest dissociation energy, being 301.6 kJ mol−1 (Table 4), while the dissociation energy of Car–H is as high as 392–397 kJ mol−1. Consequently, cleavage of the O–H bond should be the primary source for IM-1 hydrogenation. If this hypothesis holds true, cleavage of the O–H bond of PhOH is accompanied by the formation of phenoxy radical. It is known that radicals are predominantly unstable and easily undergo polymerization. Elemental analysis of the solid product indicates that its carbon content is about 24.6–38.8% (Table S1†), significantly exceeding the theoretical carbon content of calcium phenoxide (approximately 24%), suggesting transformation of carbon into the solid product at higher temperature or prolonged time. Infrared spectroscopy results confirm both hydroxyl groups and ether bonds in these solid products (Fig. S7†), while electron spin resonance (ESR) analysis reveals a free radical concentration of approximately 9.67 × 1016 spins per g (Fig. S8†). These phenomena confirm the polymerization of phenoxy radicals, as observed over 2,6-dimethylphenol.28
Table 4 Bond dissociation energy of PhOH
| The molecular structure of PhOH |
Entry |
Bond-breaking position |
Bond dissociation energy ΔG (kJ mol−1) |
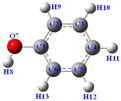
|
1 |
O7–H8 |
301.6 |
| 2 |
C2–H9 |
397.3 |
| 3 |
C4–H11 |
392.2 |
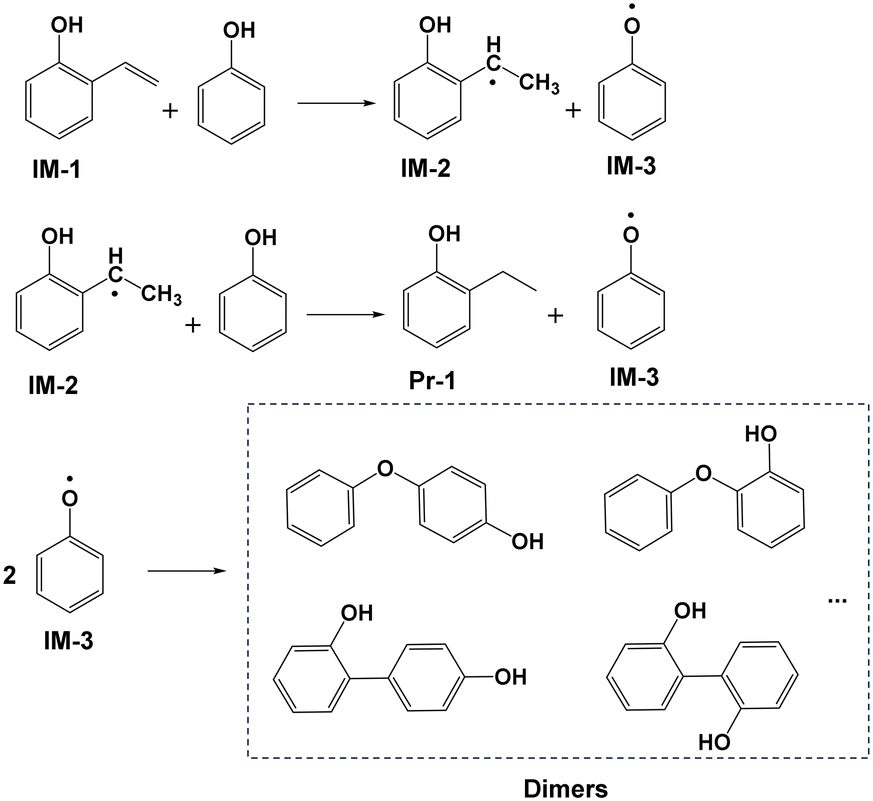
The results of DFT calculations regarding the hydrogenation of IM-1 by PhOH are presented in columns 2 and 3 of Fig. 5. Specifically, IM-1 approaches the H of the O–H bond in PhOH, which leads to extension of the O–H bond and ultimately resulting in the combination of H with the vinyl group in IM-1 to form 2-ethylphenol radical (IM-2). This process has a reaction free energy of 310.5 kJ mol−1 and is accompanied by the formation of phenoxy radical (IM-3). Subsequently, IM-2 continues to combine with H in the hydroxyl group of another PhOH molecule in a similar reaction as before, resulting in the products Pr-1 and IM-3. The energy barrier of this reaction process is 183.1 kJ mol−1. The three energy barriers reveal that formation of IM-1 (388.1 kJ mol−1) is more energetically demanding than its subsequent hydrogenation to Pr-1 (310.5, 183.1 kJ mol−1) and thus establishing it as the rate-limiting step. Easy conversion of vinylphenol is consistent with little detectable vinylphenol in solution. For clarity, Fig. 6 shows the reaction route from IM-1 to Pr-1. Following this step, two molecules of IM-3 will combine to form the dimer. According to reports in the literature,29 dimers may exist in multiple combination forms, and then these dimers will polymerize again to form complex solid polymers.
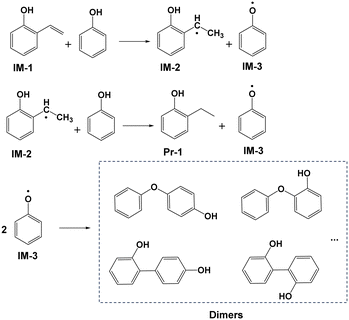 |
| | Fig. 6 Possible pathway for hydrogenation of 2-vinylphenol (IM-1) to form 2-ethylphenol (Pr-1). | |
Formation of 4-ethylphenol follows a similar route to that of 2-ethylphenol but with lower energy barriers, 381.6, 307.8 and 170.6 kJ mol−1, respectively. This information indicates that the synthesis of 4-ethylphenol is more favorable than that of 2-ethylphenol, which is consistent with the experimental results at 300 °C for 2 h and 350 °C for 0.5 h (Fig. 2). It is noted that the inconsistency at higher temperatures for a long time is due to instability (decomposition) of 4-ethylphenol.30
Since hydrogen donation of PhOH is important for ethylphenol formation, the common hydrogen-donating solvents, dihydrophenanthrene (DHP) and tetrahydronaphthalene (THN), were tested for the reaction of PhOH and CaC2. The results presented in Table 2 (entries 7 and 8) indicate a reduction in PhOH conversion (from 42.8% to 32.1% and 29.6%, respectively), alongside an increase in the total ethylphenol yield (from 26.1% to 39.4% and 39.1%). This finding further confirms that PhOH serves as the hydrogen source in the absence of alternative hydrogen sources and more vinylphenols are hydrogenated in DHP and THN than in PhOH.
3.4.2 Formation of xanthenes.
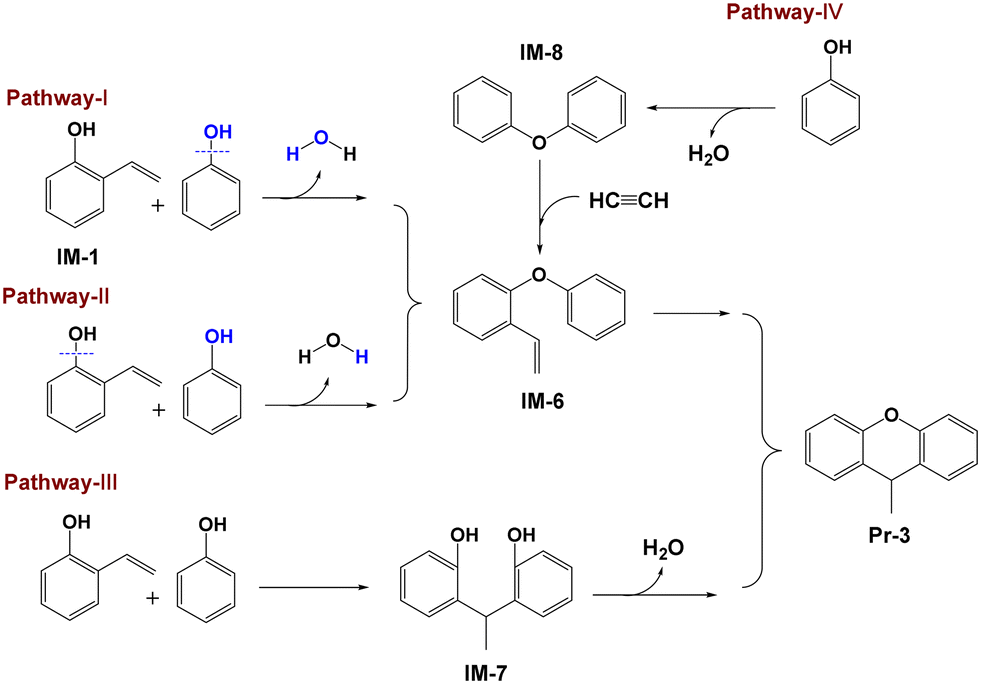
2-Vinylphenol (IM-1) may also serve as an intermediate in the formation of xanthene because IM-1 has the capability to form a 6-membered heterocyclic ring. If this is indeed the case, xanthene formation will compete with 2-ethylphenol formation for IM-1. Addition of DHP and THN obviously decreases the yield of xanthenes, from 17.6% to 5.6% and 8.8%, respectively, confirming the competition reactions. The reduction in xanthene yield subsequent to the addition of DHP or THN might be attributed to the decreased formation of phenoxyl radicals. However, elemental balance analysis indicates that the reaction between phenoxyl radicals and IM-1 does not lead to xanthene formation. Thus, it is plausible that xanthene is exclusively formed through the reaction between PhOH and IM-1. Reaction of IM-1 and PhOH to form 9-methylxanthene (Pr-3) involves dehydration and cyclization reactions with three possible pathways as indicated in Fig. 7.
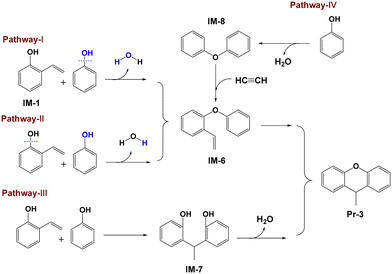 |
| | Fig. 7 Four possible formation routes of 9-methylxanthene. | |
One pathway involves the intermolecular dehydration of IM-1 with PhOH to form 1-phenoxy-2-vinylbenzene (IM-6) and then cyclization of IM-6 to yield Pr-3, as indicated by pathway I (Fig. 7). This pathway involves the reaction of PhOH's hydroxyl group with the hydrogen in the hydroxyl group of IM-1 to produce H2O. The energy barrier of for pathway I is 403.5 kJ mol−1 (Fig. 8). Another pathway is similar to pathway I except for the intermolecular dehydration step indicated by pathway II (Fig. 7), involving the combination of the hydroxyl group of IM-1 and the hydrogen in the hydroxyl group of PhOH to form H2O. The energy barrier of for pathway II is 391.1 kJ mol−1 (Fig. 8). Comparing pathways I and II, it can be concluded that pathway II is kinetically favored. Therefore, during the dehydration reaction between PhOH and IM-1, it predominantly follows pathway II where IM-1 donates its hydroxyl group and PhOH provides its hydrogen atom to form water. This conclusion is supported by bond dissociation energies (Table S4†) of IM-1 and PhOH as well. The subsequent cyclization of IM-6 into Pr-3 involves twisting of the C–H bond and elongation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond within the vinyl group of IM-6, ultimately cyclizing with the adjacent benzene ring to form Pr-3, which has an energy barrier of 379.0 kJ mol−1 (Fig. 8).
C bond within the vinyl group of IM-6, ultimately cyclizing with the adjacent benzene ring to form Pr-3, which has an energy barrier of 379.0 kJ mol−1 (Fig. 8).
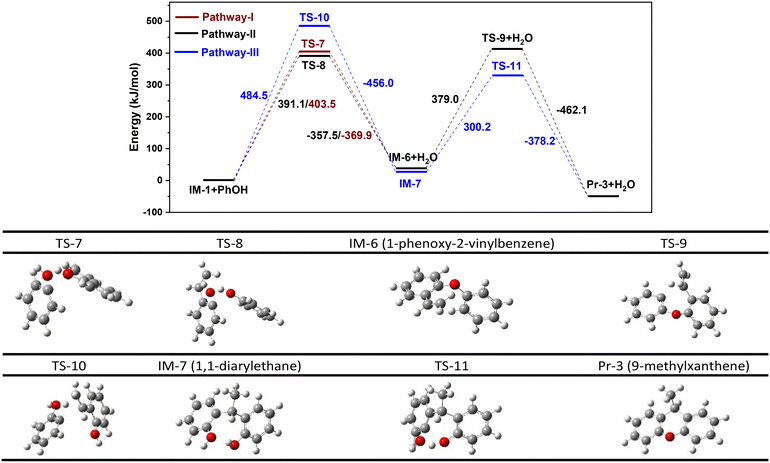 |
| | Fig. 8 Energy barrier diagram of vinylphenol reaction with PhOH to form 9-methylxanthene. Pathways I and II: dehydration is followed by a cyclization reaction (I: dehydroxylation of PhOH and dehydrogenation of 2-vinylphenol; II: dehydrogenation of PhOH and dehydroxylation of 2-vinylphenol). Pathway III: cyclization is followed by a dehydration reaction. | |
The last pathway involves a reaction between the vinyl group of IM-1 and the ortho hydrogen of PhOH, resulting in the formation of 1,1-diarylethane (IM-7), which subsequently undergoes an intramolecular dehydration reaction to form Pr-3 (pathway III in Fig. 7). Pathway III begins similarly, with twisting of the C–H bond and elongation of the CC bond within the vinyl group of IM-1 with an energy barrier of 484.5 kJ mol−1 (Fig. 8). This value is significantly higher than that for the first step of pathway II, suggesting that pathway III is less likely to occur during the actual reaction process, although the following step of pathway III has a lower energy barrier of 300.2 kJ mol−1. Therefore, it can be concluded that pathway II represents the predominant mechanism for xanthene formation.
It is noted that the energy barrier for the hydrogenation of IM-1 to Pr-1 (310.5 kJ mol−1) is lower than that for the formation of Pr-3 (391.1 kJ mol−1), but the yield of xanthenes consistently exceeds that of 2-ethylphenol. This discrepancy suggests potential alternative routes for xanthene formation that do not involve IM-1 as an intermediate. Given the strong dehydrating property of CaC2, an additional synthetic pathway is proposed (pathway IV in Fig. 7). Two PhOH molecules undergo a dehydration reaction to form diphenyl ether,31,32 which subsequently undergoes alkylation with acetylene to form IM-6 as reported in the literature.33 Conversion of IM-6 to Pr-3 is analogous to that observed in pathway II. The presence of dibenzofuran in the product (Fig. 1) may originate either from the pyrolysis reaction of diphenyl ether34 or from the intramolecular dehydration reaction of the dimer 2,2′-dihydroxybiphenyl (Fig. 6).35 However, as CaC2 is not implicated in either of these pathways, and given that the dibenzofuran yield based on PhOH does not exceed 1%, the specific generation mechanism of dibenzofuran will not be expounded upon in detail.
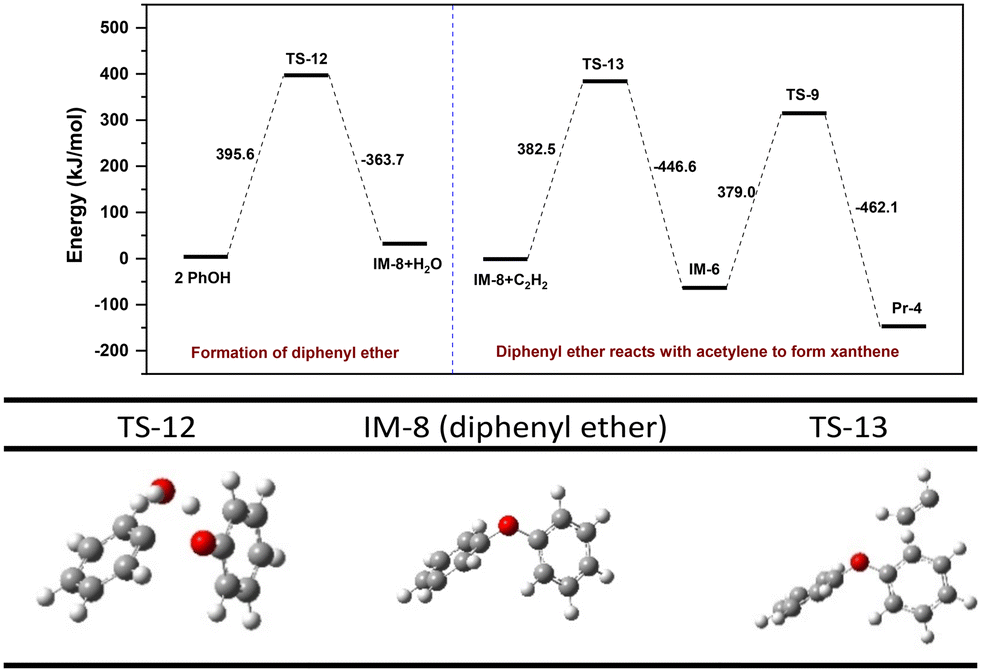
DFT calculation of pathway IV is shown in Fig. 9. Intermolecular dehydration of two PhOH to form diphenyl ether (IM-8) presents an energy barrier of 395.6 kJ mol−1. Due to the high symmetry of IM-8, the reaction barriers for alkylation at both benzene ring with acetylene are theoretically identical. However, to facilitate subsequent cyclization into a 6-membered heterocycle, the alkylation reaction should occur at the ortho position of the ether bond of IM-8. The free energy barrier for the alkylation and subsequent cyclization steps are 382.5 kJ mol−1 and 379.0 kJ mol−1, respectively. This suggests that formation of IM-8 is the rate-limiting step. The preceding discussion indicates that the reaction energy barriers for the rate-limiting step of pathways II and IV are very close, 391.1 vs. 395.6 kJ mol−1, indicating viability of both pathways in the actual process. A control experiment (entry 9 in Table 2) demonstrates that addition of diphenyl ether increases the yields of xanthenes by 3.1% along with a small increase in dibenzofuran, indicating that pathway IV is not the primary synthesis route of xanthenes.
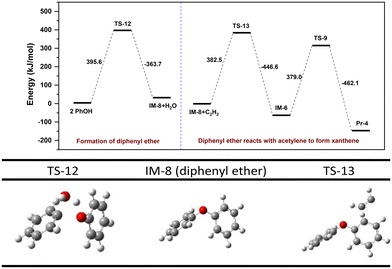 |
| | Fig. 9 Energy barrier diagram of an alternative reaction pathway (pathway IV) to 9-methylxanthene. | |
Nevertheless, the presence of pathway IV well explains the experimental observations in Table 2. When the molar ratio of PhOH to CaC2 is relatively low (entries 1 and 2), xanthenes predominantly form through pathway II. In this pathway, 2-ethylphenol competes with xanthenes and exhibits a competitive advantage by yielding higher quantities than xanthenes. With increasing molar ratios of PhOH to CaC2 (entries 3–5), the contribution of xanthenes synthesized via pathway IV becomes more significant, resulting in yields that surpass those of 2-ethylphenol. Addition of a hydrogen-donating solvent, DHP or THN, inhibits the formation pathway of xanthenes via 2-vinylphenol, while the diphenyl ether pathway is less affected. This leads to a significant decrease in the yield of xanthenes but not to 0 (Table 2, entries 7 and 8, and Table S2†). Since the free energy barriers for the formation of intermediates vinylphenol and diphenyl ether are 388.1/381.6 and 395.6 kJ mol−1, respectively, this suggests that PhOH preferentially reacts with acetylene to form vinylphenols, which is supported by the total yield of ethylphenols being significantly higher than that of xanthenes.
3.5 Utilization efficiency of PhOH and CaC2
Based on the preceding analysis, PhOH may convert to calcium phenoxide, ethylphenols and xanthenes. The formation of calcium phenoxide consumes two PhOH molecules, whereas xanthenes or ethylphenols require two or three PhOH molecules, respectively. Therefore, to facilitate the formation of ethylphenols and xanthenes, the molar ratio of PhOH/CaC2 should exceed 2. To evaluate the effective utilization of PhOH, the selectivity of liquid products (including ethylphenols and xanthenes) and calcium phenoxide was calculated and the results are shown in Table 5. It indicates that PhOH is also consumed by an unidentified product that is likely to be present in the solid phase, as the gas and liquid products have been accurately detected. The selectivity of the unidentified product is from 8% to 17%, indicating an effective utilization of PhOH between 83% and 92%. Additionally, the alkynyl groups in CaC2 are predominantly converted to ethylphenols and xanthenes along with minor amounts of acetylene, and each molecule of these products formed consumes one alkynyl group. According to Table 2, under optimal experimental conditions, the effective utilization of alkynyl groups approximates 69.4%, suggesting that the unidentified product also consumes alkynyl groups. The selectivity of the undefined product is equal to 100% minus the sum of the selectivity of the liquid products and acetylene. These findings are detailed in Table 5, where it is observed that the PhOH consumption by the unidentified product is roughly equal to the CaC2 consumption at low temperatures or over a short time (see the data in parentheses). Considering the tendency of unsaturated bonds in vinylphenol to polymerize, it is plausible that the unidentified product is polyvinylphenol. Moreover, FT-IR analysis of the solid product confirms the presence of aromatic compounds with alkyl side chains (Fig. S7†). A potential direction to optimize the reaction between CaC2 and PhOH is to inhibit polymerization of vinyl products.
Table 5 Conversion pathways of phenol under different reaction conditions
| Temp. (°C) |
t (h) |
X
PhOH (%) |
S
PhOH i (%) |
S
CaC2 unidentified product (%) |
| Liquid product |
Calcium phenoxide |
Unidentifieda |
| The mass of CaC2 is 2.5 mg (0.036 mmol) and the molar ratio of PhOH/CaC2 is 8:1. Subtraction. The molar ratio of PhOH/CaC2 is 16:1. Number in parentheses is the amount of PhOH or CaC2 consumed by the undefined product. |
| 300 |
2 |
35.4 |
25.4 |
64.4 |
10.2 (0.0104)c |
29.9 (0.0108) |
| 325 |
2 |
39.4 |
34.0 |
58.1 |
7.9 (0.0089) |
33.1 (0.0119) |
| 350 |
2 |
42.8 |
35.1 |
53.3 |
11.6 (0.0144) |
36.6 (0.0132) |
| 375 |
2 |
47.4 |
31.9 |
50.8 |
17.3 (0.0236) |
48.1 (0.0173) |
| 350 |
0.5 |
34.7 |
21.6 |
70.6 |
7.8 (0.0078) |
19.8 (0.0071) |
| 350 |
1 |
38.7 |
27.9 |
62.3 |
9.8 (0.0109) |
27.2 (0.0098) |
| 350 |
4 |
45.8 |
36.7 |
50.6 |
12.7 (0.0168) |
43.4 (0.0156) |
| 350b |
2 |
24.9 |
41.8 |
49.8 |
8.4 (0.0121) |
28.9 (0.0104) |
3.6 Summary of reaction pathways
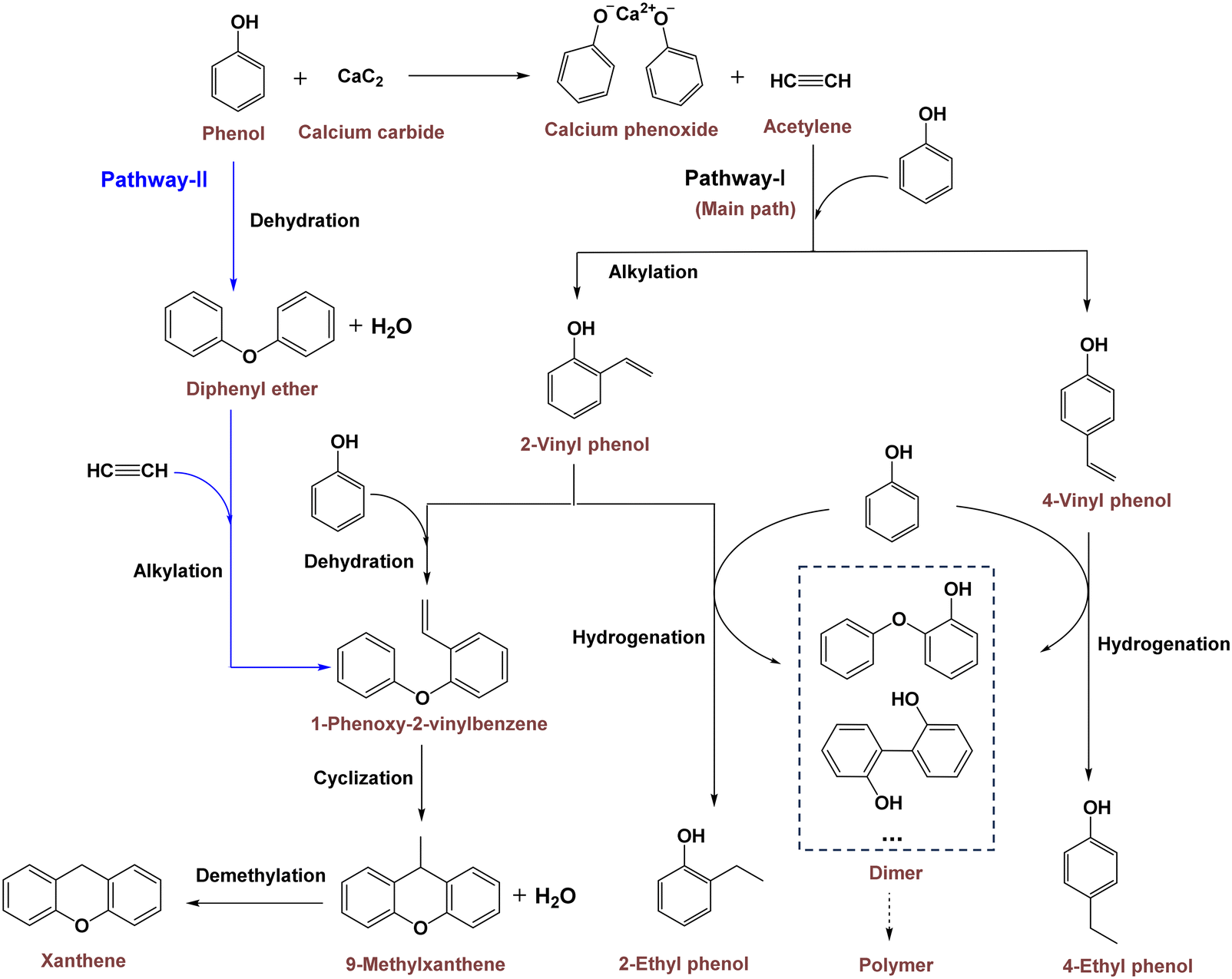
The reaction network for synthesizing xanthenes and ethylphenols from PhOH and CaC2 is summarized in Scheme 1. Initially, CaC2 and PhOH rapidly convert into acetylene and calcium phenoxide. Subsequently, alkylation of PhOH with acetylene serves as the primary reaction, leading to the formation of the intermediate 2- or 4-vinylphenol as shown in pathway I. 4-Vinylphenol converts to 4-ethylphenol through hydrogenation by PhOH. Following hydrogen donation, the phenoxy moiety of PhOH undergoes polymerization to generate a complex polymer in the solid. Conversely, 2-vinylphenol can also yield 2-ethylphenol or react with another PhOH molecule to form 1-phenoxy-2-vinylbenzene. This compound subsequently cyclizes to yield 9-methylxanthene, which decomposes into xanthene at high temperature (Table S3†). Moreover, 1-phenoxy-2-vinylbenzene can also be synthesized through alkylation of diphenyl ether, an intermolecular dehydration product of PhOH, with acetylene, as shown in pathway II.
 |
| | Scheme 1 Possible route of PhOH reaction with CaC2 to form ethylphenols and xanthenes. | |
The alkylation of PhOH by acetylene to form the intermediate vinylphenol is a critical step in the reaction. Conventionally, the alkylation of PhOH by olefins or alkynes is believed to necessitate acid catalysis. Despite the alkaline nature of CaC2 and calcium phenoxide, recent studies indicated that the Ca2+ ion in calcium alkoxide acts as an active Lewis acid site.19,36 Calcium phenoxide exhibits structural similarities with calcium alkoxides and displays a positively charged region near the Ca2+ ion, as shown in the isomolecular electrostatic potential maps (Fig. S9†). This charged region makes the Ca2+ ion more electrophilic, enabling it to serve as a Lewis acid catalytic site.37 Meanwhile, calcium ion-catalyzed dehydroxylation of phenolic compounds has been reported in related literature.38 Thus, calcium phenoxide potentially catalyzes alkylation, cyclization and dehydration reactions. This discovery prompts a need for further research to fully understand the catalytic processes and mechanisms. Additionally, residual calcium phenoxide can react with acid to regenerate PhOH, thereby participating in the reaction cycle and enhancing atom utilization (Table S5†).
4. Conclusions
It is observed that ethylphenols and xanthenes can be synthesized by the reaction of CaC2 and PhOH in the absence of catalyst and solvent above 350 °C. At the molar ratio of PhOH/CaC2 of 16, the yields of ethylphenols and xanthenes reach 38.3% and 26.0%, respectively, at 350 °C for 6 h. Formation of ethylphenols begins with conversion of CaC2 to calcium phenoxide and acetylene. PhOH is alkylated by acetylene to form 2- or 4-vinylphenol, which is subsequently hydrogenated by PhOH to form 2- or 4-ethylphenol; meanwhile, accompanying phenoxy radicals tend to polymerize into solid polymers. Formation of xanthenes follows two pathways with similar energy barriers. One involves the intermolecular dehydration of 2-vinylphenol with PhOH to form 1-phenoxy-2-vinylbenzene, and the other involves dehydration of PhOH to form diphenyl ether and alkylation of diphenyl ether by acetylene to form 1-phenoxy-2-vinylbenzene. 1-Phenoxy-2-vinylbenzene then undergoes cyclization to form 9-methylxanthene, and 9-methylxanthene converts to xanthene through demethylation. The formation of both 2-ethylphenol and xanthene represents competitive reactions when using 2-vinylphenol as the intermediate and the energy barrier of the rate-limiting step is 310.5 kJ mol−1 for 2-ethylphenol and 391.1 kJ mol−1 for xanthenes. Additionally, the solid product, calcium phenoxide, may function as a Lewis acid catalyst, specifically with the Ca ion as the catalytic site to catalyze alkylation, cyclization and dehydration reactions.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Xin Liu: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft. Yuxin Yan: supervision, visualization, writing – review & editing. Zhenyu Liu: supervision, methodology. Qingya Liu: project administration, funding acquisition, supervision, resources, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (22178020).
Notes and references
- A. Li, H. Song, X. Xu, H. Meng, Y. Lu and C. Li, ACS Sustainable Chem. Eng., 2018, 6, 9560–9565 CrossRef CAS.
- K. S. Rodygin, Y. A. Vikenteva and V. P. Ananikov, ChemSusChem, 2019, 12, 1483–1516 CrossRef CAS.
- S. P. Teong, D. Yu, Y. N. Sum and Y. Zhang, Green Chem., 2016, 18, 3499–3502 RSC.
- Y. Li, H. Meng, Y. Lu and C. Li, Ind. Eng. Chem. Res., 2016, 55, 5257–5262 CrossRef CAS.
- M. S. Ledovskaya and V. V. Voronin, Tetrahedron, 2023, 133720 CrossRef CAS.
- Z. Li, L. He, R. Fu, G. Song, W. Song, D. Xie and J. Yang, Tetrahedron, 2016, 72, 4321–4328 CrossRef CAS.
- K. S. Rodygin, M. S. Ledovskaya, V. V. Voronin, K. A. Lotsman and V. P. Ananikov, Eur. J. Org. Chem., 2021, 2021, 43–52 CrossRef CAS.
- D. Wang, Z. Liu and Q. Liu, Ind. Eng. Chem. Res., 2019, 58, 6226–6234 CrossRef CAS.
- D. Wang, Z. Liu and Q. Liu, RSC Adv., 2019, 9, 18941–18948 RSC.
- K. S. Rodygin, G. Werner, F. A. Kucherov and V. P. Ananikov, Chem. – Asian J., 2016, 11, 965–976 CrossRef CAS PubMed.
- P. Asawaworarit, P. Daorattanachai, W. Laosiripojana, C. Sakdaronnarong, A. Shotipruk and N. Laosiripojana, Chem. Eng. J., 2019, 356, 461–471 CrossRef CAS.
- G. Werner, K. S. Rodygin, A. A. Kostin, E. G. Gordeev, A. S. Kashin and V. P. Ananikov, Green Chem., 2017, 19, 3032–3041 RSC.
- B. Wang, C. W. Lee, T.-X. Cai and S.-E. Park, Catal. Lett., 2001, 76, 219–224 CrossRef CAS.
- M. Yamaguchi, M. Arisawa, K. Omata, K. Kabuto, M. Hirama and T. Uchimaru, J. Org. Chem., 1998, 63, 7298–7305 CrossRef CAS.
- M. Yamaguchi, Pure Appl. Chem., 1998, 70, 1091–1096 CrossRef CAS.
-
Mordor Intelligence, Xanthene Derivatives Market Size & Share Analysis-Growth Trends & Forecasts (2024-2029), https://www.mordorintelligence.com/industry-reports/xanthene-derivatives-market, 2024. (accessed 20 February 2024).
- A. S. Burange, K. G. Gadam, P. S. Tugaonkar, S. D. Thakur, R. K. Soni, R. R. Khan, M. S. Tai and C. S. Gopinath, Environ. Chem. Lett., 2021, 19, 3283–3314 CrossRef CAS.
- S. S. Kahandal, A. S. Burange, S. R. Kale, P. Prinsen, R. Luque and R. V. Jayaram, Catal. Commun., 2017, 97, 138–145 CrossRef CAS.
- X. Liu, F. Yuan, Y. Pan, S. Shi, N. Li, Z. Liu and Q. Liu, Chem. Eng. J., 2023, 475, 146250 CrossRef CAS.
- T. Lu and Q. Chen, Comput. Theor. Chem., 2021, 1200, 113249 CrossRef CAS.
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, Appl. Catal., A, 2013, 459, 114–120 CrossRef CAS.
- Y. Liu, G. Cheng, E. Baráth, H. Shi and J. A. Lercher, Appl. Catal., B, 2021, 281, 119424 CrossRef CAS.
- K. H. Bhadra and G. D. Yadav, Appl. Catal., A, 2018, 562, 67–78 CrossRef CAS.
- M. Bregolato, V. Bolis, C. Busco, P. Ugliengo, S. Bordiga, F. Cavani, N. Ballarini, L. Maselli, S. Passeri and I. Rossetti, J. Catal., 2007, 245, 285–300 CrossRef CAS.
- H. Stanley and T. Lundy, Oral Surg., Oral Med., Oral Pathol., 1972, 34, 818–827 CrossRef CAS.
- J. Jang and D. Y. Kim, Asian J. Org. Chem., 2022, 11, e202200052 CrossRef CAS.
- Y.-P. Han, X.-R. Song, Y.-F. Qiu, X.-S. Li, H.-R. Zhang, X.-Y. Zhu, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2016, 18, 3866–3869 CrossRef CAS PubMed.
- J. Gao, S. H. Zhong and R. A. Zingaro, J. Mol. Catal. A: Chem., 2004, 207, 15–20 CrossRef CAS.
- H. Higashimura, K. Fujisawa, M. Kubota and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1955–1962 CrossRef CAS.
- Y. Liao, M. d'Halluin, E. Makshina, D. Verboekend and B. F. Sels, Appl. Catal., B, 2018, 234, 117–129 CrossRef CAS.
- H. E. Ungnade, Chem. Rev., 1946, 38, 405–446 CrossRef CAS PubMed.
- A. de Klerk and R. J. Nel, Ind. Eng. Chem. Res., 2007, 46, 7066–7072 CrossRef CAS.
- P. A. Jelzowa, M. M. Koton, K. Minejewa and K. Ssurnina, Chem. Zentralbl., 1963, 134, 20821–20822 Search PubMed.
- T. Ishida, R. Tsunoda, Z. Zhang, A. Hamasaki, T. Honma, H. Ohashi, T. Yokoyama and M. Tokunaga, Appl. Catal., B, 2014, 150, 523–531 CrossRef.
- F. P. Petrocelli and M. T. Klein, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 635–641 CrossRef CAS.
- D. Wang, Z. Liu and Q. Liu, J. Phys. Chem. C, 2019, 123, 22932–22940 CrossRef CAS.
- G. Li, J. H. Stenlid, M. S. Ahlquist and T. Brinck, J. Phys. Chem. C, 2020, 124, 14696–14705 CrossRef CAS.
- W. Wang, R. Lemaire, A. Bensakhria and D. Luart, J. Anal. Appl. Pyrolysis, 2022, 163, 105479 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Qingya
Liu
and
Qingya
Liu