Removal of gaseous Hg0 by Cl-loaded carbonaceous material prepared from rice husk
Received
2nd September 2024
, Accepted 22nd October 2024
First published on 31st October 2024
Abstract
The main objective of this study was to investigate the removal of gaseous Hg0 from the residue obtained during the chlorination of rice husk char to recover silica. First, the chlorine content, chlorine form, pore size, and chlorination reaction of the chlorination residue of rice husk char were investigated. Subsequently, the adsorption performance of gaseous Hg0 and adsorption form of Hg0 were examined. Through the chlorination of rice husk char (RC) at 1000 °C for 10 min, silicon (Si) in the char could be separated and recovered, and carbonaceous material doped with 9.0% Cl could be prepared. The temperature-programmed desorption (TPD) analysis of the char before and after chlorination revealed that the oxygen-containing functional groups on the surface of char were part of the Cl adsorption sites. Additionally, the X-ray photoelectron spectroscopy (XPS) analysis showed that Cl was mainly present in the C–Cl bonds of the chlorination residue (RCC). The effect of temperature on the performance was small in the tested range (40–160 °C). The adsorption performance of the prepared RCC was superior to those of chlorine- and sulfur-loaded carbons, as reported in previous studies. The results of TPD analysis after the adsorption tests indicated that the adsorption form of Hg0 was mainly HgCl2.
1. Introduction
Mercury (Hg) is the most volatile and toxic element released into the atmosphere from both anthropogenic and natural sources. Hg emitted into the atmosphere is dispersed worldwide by prevailing winds. In contrast, inorganic Hg discharged into water is converted into organic Hg, which accumulates in biological organisms in highly toxic forms.1 Controlling Hg emissions is important and the Minamata Convention was adopted for the same purpose in 2013 at an international diplomatic conference.2 According to an investigation by the United Nations Environment Program (UNEP), approximately 25% of the Hg released into the atmosphere originates from coal combustion and industrial waste/sewage sludge treatment facilities.3 Therefore, the Hg emissions from these facilities in flue gas emissions must be controlled. In general, activated carbon is effective in removing Hg from flue gas,4–6 and the form of Hg adsorption on activated carbon fields has previously been elucidated.6–8 However, the effects of the impurities and functional groups in activated carbon on Hg removal remain unknown. Conversely, Hg removal by activated carbon loaded with calcium chloride (CaCl2) and sulfide has been investigated, and these chloride and sulfide loadings have been reported to be effective for Hg removal.9–17 Moreover, Hg removal using impregnated activated carbon has indicated that the adsorption performance of Hg is dominated by chemisorption rather than physical adsorption.8,14,17
We studied the selective recovery of silica from rice husk char (RC) by carbo-chlorination. We found that Cl atoms were loaded by the reaction of Cl2 gas with carbonaceous matter in the char.18,19 The amount of Cl loaded onto carbonaceous materials during chlorination is temperature-dependent, and Cl remains in the order of several percent after silica is separated from the char.18,19 Furthermore, the use of a chlorination residue (chlorinated rice husk char; RCC) for Hg ion adsorption has shown high performance in terms of the affinity between Cl atoms and Hg.18,19 Therefore, the RCC may be effective in removing Hg from flue gas.
Therefore, the main objective of this study was to investigate the removal of gaseous Hg0 from the residue obtained during the chlorination of RC to recover silica. First, the adsorption performance of gaseous Hg0 of the Cl-loaded carbonaceous material was evaluated using chlorinated RC. Subsequently, the adsorption of Hg0 onto the RCC was examined.
2. Experimental
2.1 Samples
Rice husk (RH) was used as the sample. Table 1 shows the elemental analysis and ash composition of RH. The sample was pyrolyzed in a gas-distribution fixed-bed reactor at 800 °C in high-purity N2 (99.999%) at a heating rate of 10 °C min−1 to prepare the RC. The ash composition of RC was as follows: Si, Ca, Mg, K, Na, P, Mn, Fe, and Al contents of 22, 0.3, 0.13, 0.73, 0.73, 0.20, 0.09, 0.05, and 0.02 wt% (dry), respectively (Table 1). The RC was ground and sieved through a 200-mesh and then used for the chlorination experiments.
Table 1 Composition of slag samples used in this study
| Code |
Elemental analysis, wt% (dry-ash-free; daf) |
Ash, wt% |
Ash composition, wt% (ash content basis) |
| C |
H |
N |
S + Oa |
Si |
Ca |
Mg |
K |
Na |
P |
Mn |
Fe |
Al |
|
Estimated by difference.
Not analyzed.
RCC prepared by chlorination at 1000 °C for 10 min.
|
| RH |
46.9 |
5.88 |
0.53 |
46.7 |
20.4 |
9.1 |
0.11 |
0.05 |
0.28 |
0.02 |
0.08 |
0.03 |
0.01 |
0.01 |
| RC |
89.1 |
1.96 |
0.51 |
8.4 |
49.5 |
21.6 |
0.31 |
0.13 |
0.73 |
0.73 |
0.20 |
0.09 |
0.05 |
0.02 |
| RCCc |
72.1 |
0.15 |
0.40 |
27.4 |
8.6 |
3.5 |
0.3 |
0.09 |
0.01 |
0.02 |
0.05 |
n.a.b |
— |
— |
2.2 Chlorination
A circulating fixed-bed reactor was used for the chlorination of RC.18,19 The reactor temperature was measured and controlled by vertically inserting a thermocouple from outside the electric reactor and bringing it in contact with the outside of the reaction tube. In the experiments, 1.0 g of the sample was placed in an alumina boat, inserted into the center of the reaction tube, and heated in Cl2 (99.99%) to a maximum temperature of 200–1000 °C at a rate of temperature increase of 20 °C min−1, with a holding time of 0–60 min. The chlorine gas and volatile products were completely absorbed by a trap containing a sodium hydroxide solution placed at the outlet of the reaction tube. After the target temperature or time was reached, the gas was switched to N2 (99.9995% purity), and the furnace was cooled to room temperature. The sample was then removed from the reactor and weighed to calculate the solid sample yield using eqn (1).where Y is the yield (%), and W0 and Wi are the sample weights (g) before and after chlorination, respectively. The ash content in the solid sample recovered after chlorination was determined by weighing the sample after combustion at 815 °C in accordance with JIS M8812. The volatilization rate of ash during chlorination was calculated using eqn (2).where REA is the release extent of ash (%), and A0 and Ai are the amounts of ash (g) in the sample before and after chlorination, respectively. Although the details are described below, in this study, the chlorination residue prepared at 1000 °C for 10 min is denoted as RCC and used for Hg0 adsorption. The RCC analyses values also shown in Table 1.
The ash in the prepared samples was dissolved in a mixed acid of HCl/HNO3/HF, according to previous reports.20 Measurement method of chlorine content in samples before and after chlorination has been described in section 2.6. The content of each element in the dissolved solution was determined by inductively coupled plasma optical emission spectrometry (ICP-OES). The volatility of each element during chlorination was calculated using eqn (3).
where REE is the release extent of the element (%), and
Ei and
E0 are the amounts of ash components in the ashing sample before and after chlorination, respectively.
2.3 Adsorption of gaseous Hg0
The adsorption performances of RC and RCC for gaseous Hg0 were evaluated using a fixed-bed reactor with a gas distribution system (Fig. 1). The apparatus comprised a gaseous Hg0 generator, adsorption bed, and mercury analyzer. A 0.1-g sample was introduced onto quartz wool packed in a stainless tube reactor and connected to the reaction system. The Hg generator was temperature controlled to adjust the Hg concentration to 100 ppb of Hg0 in the gas supplied to the adsorbent. To prevent Hg from adhering to the piping, the piping was heated to more than 100 °C with a ribbon heater before and after the reaction. In the experiment, the reactor was heated to 60 or 140 °C in N2 gas, 100 ppb of Hg0/N2 was circulated over the sample, and the Hg concentration at the reactor outlet was analyzed online at intervals of 1 s using a portable mercury measuring device (EMP-2) equipped with a mercury gas pretreatment device (Nippon Instruments Corp., WLE-9). The amount of adsorbed mercury was calculated using eqn (4). The spatial velocity was 1.2 × 105 h−1.where MA is the mercury adsorption rate, and C0 and Ci are the Hg concentrations at the reactor outlet before the start of the reaction and at an arbitrary time, respectively. In this study, the breakthrough point was defined as Ci/C0 = 0.1. In this study, test temperatures of 60 and 140 °C were selected as the temperatures near the flue-gas desulfurization (FGD) equipment and bag filter, respectively. Cl utilization was calculated by eqn (5).| | | Cl utilization, % = Removal amount of gaseous-Hg0(mg g−1)/Cl amount in RCC (mg g−1) × 100 | (5) |
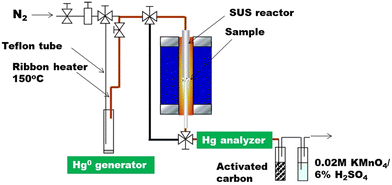 |
| | Fig. 1 Diagram of the Hg0 adsorption apparatus. | |
2.4 Characterization
In the X-ray photoelectron spectroscopy (XPS) analysis, the powdered sample was held on an indium platelet, and Mg-Kα (10 kV × 10 mA) was used as the source of radiation under vacuum with pressures ranging from 4 × 10−7 to 7 × 10−7 Pa. The background of the observed Cl 2p and C 1s XPS spectra was removed using the Shirley method, and the binding energy was corrected using that of In 3d5/2 (444.9 eV) of In2O3.21 The oxygen-containing functional groups of the samples before and after chlorination were examined using temperature-programmed desorption (TPD).22–25 In the experiments, samples were first kept in a fixed-bed reactor with gas distribution and then heated in He at a heating rate of 10 °C min−1 up to a temperature of 950 °C. The CO and CO2 production profiles were separated based on previous reports and the amount of each oxygen-containing functional group was evaluated.22–25 The pore properties of the samples were calculated by the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods after measuring the N2 adsorption/desorption isotherms by the N2 adsorption method. The chlorine contents of the samples before and after chlorination were determined by ion chromatography (DIONEX DX120) using an automatic combustion device (Yanaco). The form of Hg present in the sample after the adsorption of gaseous Hg0 was examined using TPD. After the adsorption experiment, high-purity N2 gas was distributed in the reactor to reduce the Hg concentration in the reactor outlet gas to below 1 ppb. Subsequently, the reactor was heated to 600 °C at a heating rate of 10 °C min−1, and the Hg0 generated in the process was analyzed online at intervals of 1 s with the aforementioned mercury analyzer.
3. Results and discussion
3.1 Changes in the release extent of ash-composed elements and pore structure with chlorination of RC
Table 2 presents the yield, ash volatility, volatility of ash constituents, Cl content, and pore properties after the chlorination of RC. The yield increased up to a temperature of 500 °C and then decreased with increasing temperature. Conversely, ash volatility increased with increasing temperature and remained almost constant at 1000 °C. The volatilized ash components showed that more than 70% of the Si, K, Na, and P originally present in the RC were released, whereas the volatilization rates of Ca and Mg were low. Table 1 presents the ash compositions of RC and RCC. The chlorination treatment of RC reduced Si, K, Na, P, and some Mg. The main ash components in the RCC were the remaining Si and Ca. Thermodynamic equilibrium calculations showed that SiCl4 (g) was a volatile form of Si, suggesting that this method can be used to recover Si from rice husk. The Cl content showed a maximum value (22%) at 600–700 °C and subsequently decreased with increasing temperature, but still remained 9% at 1000 °C for 10 min. According to previous reports, Cl2 dissociates on the carbonaceous surface and reacts with carbon,26 and the increase in Cl up to a temperature of 700 °C may be ascribed to the adsorption of Cl2-derived Cl on the RC surface. However, the decrease in Cl content above a temperature of 800 °C may be attributed to the desorption of Cl adsorbed on the carbonaceous material and chloride volatilization of the ash constituents. According to our previous study, when demineralized RC with an ash content of 1.0% was chlorinated in the same manner, the amount of Cl increased in the same temperature range as the RC and subsequently decreased with increasing temperature. Therefore, this indicates that Cl is adsorbed onto the carbonaceous materials in the RC.
Table 2 Summary of yields of the chlorinated residue, removal extent of ash, and pore structure
| Temp.,°C/holding time at 1000 °C, min |
Y
, wt% |
REAb, % |
REEc, % |
Cl, % |
Pore structure |
| Si |
Ca |
Mg |
K |
Na |
P |
S
BET
m2 g−1 |
Volumee m3 g−1 |
D(v)f |
|
Yield.
Volatile extent of ash.
Volatile extent of element.
Specific surface area calculated by the Brunauer–Emmett–Teller (BET) method.
Total pore volume calculated at Pi/P0 = 0.9.
Average pore diameter calculated by the Barrett–Joyner–Halenda (BJH) method.
|
| 400 |
109 |
5 |
5 |
0 |
0 |
0 |
0 |
0 |
12 |
<10 |
0.01 |
4.1 |
| 500 |
111 |
9 |
10 |
0 |
0 |
1 |
1 |
5 |
18 |
10 |
0.02 |
3.8 |
| 600 |
106 |
19 |
25 |
0 |
0 |
1 |
17 |
14 |
22 |
40 |
0.03 |
3.8 |
| 700 |
83 |
45 |
50 |
0 |
0 |
1 |
36 |
43 |
22 |
155 |
0.10 |
3.7 |
| 800 |
70 |
57 |
62 |
0 |
0 |
3 |
45 |
53 |
18 |
195 |
0.19 |
3.7 |
| 900 |
63 |
63 |
68 |
0 |
3 |
17 |
57 |
60 |
15 |
245 |
0.19 |
3.6 |
| 1000 |
52 |
75 |
75 |
0 |
10 |
49 |
60 |
67 |
11 |
315 |
0.18 |
3.6 |
| 10 |
44 |
80 |
84 |
4 |
28 |
99 |
97 |
75 |
9 |
335 |
0.18 |
3.5 |
| 20 |
45 |
80 |
83 |
8 |
29 |
100 |
98 |
75 |
9 |
330 |
0.18 |
3.5 |
Subsequently, the pore properties were examined. The surface area increased rapidly above a temperature of 700 °C, reaching 355 m2 g−1 after 10 min of holding at 1000 °C. A similar increase was observed in the pore volume. Comparing the changes in Si volatility and pore properties, the temperature at which the former increased coincided with that at which the pore properties developed. In particular, the development of pores was caused by the volatilization of Si in RC. On examining the pore size distribution, a profile with a peak at approximately 4 nm was observed at all temperatures, as indicated by the average pore size D(v) presented in Table 2, and the peak intensity increased with increasing temperature. This indicates that Si exists in RC with a size of approximately 4 nm. Based on the above results, RC chlorinated at 1000 °C for 10 min was used as RCC in this study.
3.2 Cl form in RCC
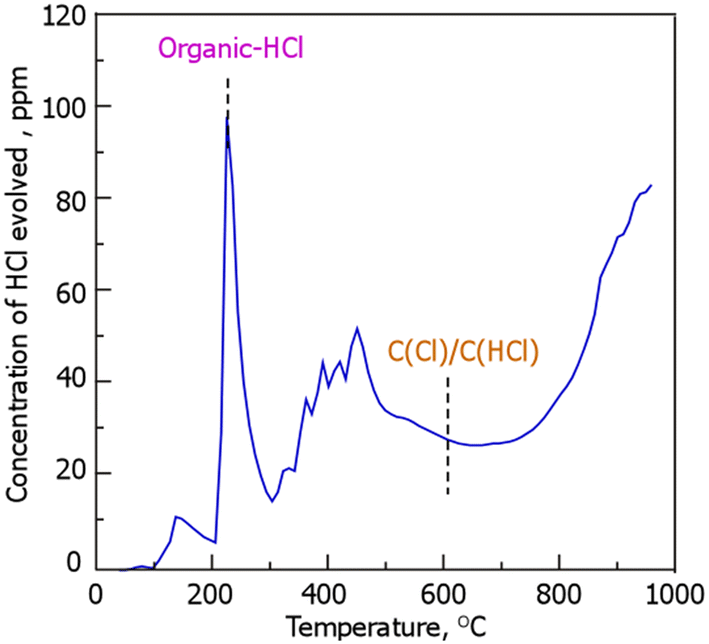
Fig. 2 shows the results of HCl desorption behavior during TPD to examine the presence of Cl in RCC; HCl desorption was also observed above a temperature of 700 °C, suggesting that the desorption below 200 °C was ascribed to the volatilization of physically adsorbed HCl. Conversely, desorption above 200 °C is assumed to be attributed to the cleavage of the chemical bonds of the Cl species in RCC. In accordance with previous reports, organic-HCl and C(Cl)/C(HCl), wherein Cl or HCl is bound to the carbon structure, show HCl desorption peaks at temperatures around 250 and 600 °C, respectively.27–30 Although the source of the Cl species being desorbed as HCl above 300–500 and 700 °C is currently unknown, it is clear that Cl is at least present in RCC in the organic-HCl, and carbonaceous materials combined with the Cl and HCl (C(Cl)/C(HCl)) forms. According to previous reports on Cl release during the thermal treatment of biomass, Cl species are classified into those present below 400 °C and those that volatilize at 700–900 °C;31 the former represents organic Cl species, such as methyl chloride (organo-chlorine (C–Cl)),32 and the latter is mainly formed owing to the sublimation of metal chlorides.33 The HCl-TPD profile in Fig. 2 showed that 17% of the Cl (1.5%-Cl) of the total Cl content (9%-Cl) in the RCC was evolved as HCl gas. In other words, most of the Cl (83%) is present in RCC in a thermally stable form. As mentioned above, in the HCl-TPD profile in Fig. 2, the C(Cl)/C(HCl) peak was assigned based on previous reports. However, this peak alone is not considered to represent the total amount of C(Cl) present. In other words, it is possible that there may be other C–Cl bonds that are very thermally stable. As presented in Table 1, the RCC used in this study contained low amounts of metal species, lesser than the amounts of Si and Ca. Conversely, Ca, which has the second highest content after Si, is considered to form CaCl2, and the melting point of this species is 770 °C. Therefore, the Cl derived from CaCl2 may react with carbonaceous materials and be released as HCl. The details of this process will be the subject of future studies.
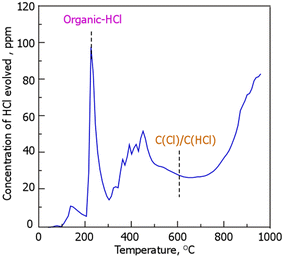 |
| | Fig. 2 HCl evolution profile in RCC during temperature-programmed desorption. (RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). | |
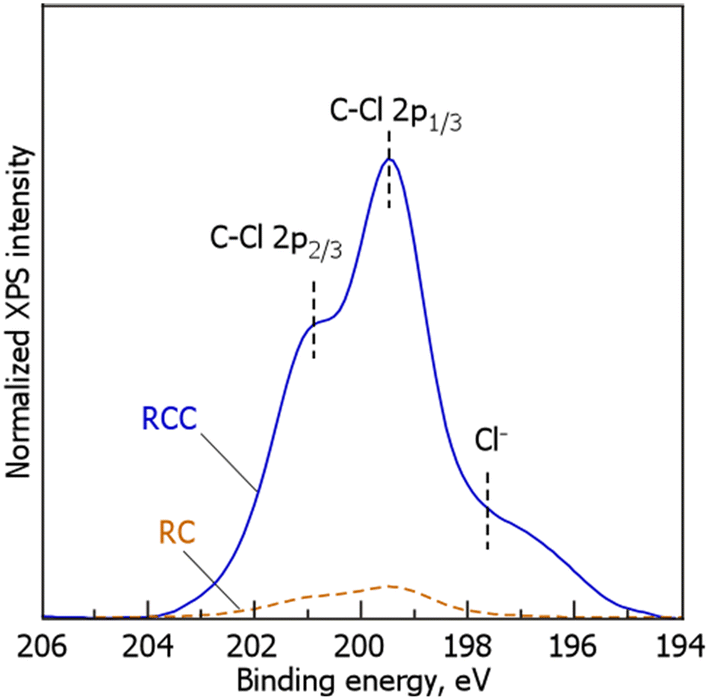
Fig. 3 shows the Cl 2p XPS spectrum of RCC during TPD. For RC, weak main and shoulder peaks were observed at 199 and 201 eV, respectively. In contrast, in RCC, in addition to the main and shoulder peaks at 199 and 201 eV, respectively, similar to that in RC, a weak shoulder peak was observed at 197–198 eV. In accordance with previous reports,34–39 the peaks at 199 and 201 eV were attributed to the C–Cl 2p1/3 and C–Cl 2p2/3 bonds, respectively. Although the detailed chemical form of Cl is unknown, the Cl may possibly exist in a chemically bonded state with the carbonaceous material during the chlorination of RC. According to previous reports, hydrocarbon species and Cl2 follow eqn (6)–(8).40,41
| | | Substitution reaction: C–H + Cl2 (g) → C–Cl + HCl (g) | (6) |
| | Addition reaction of the unconjugated double bond: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C + Cl2 (g) → ClC–CCl C + Cl2 (g) → ClC–CCl | (7) |
| | | Dehydrogenation reaction: HC–CH + Cl2 (g) → CC + 2HCl (g) | (8) |
Notably, in the pyrolysis of coal, the decomposition of OH and CHO occurs at 300–550 °C, the decomposition of –CH
3, –C
nH
m, and –CH
2– and the dehydrogenation of –CH
2– occur at 450–750 °C; above 750 °C, a dehydrogenation reaction occurs around the aromatic condensation ring. In this study, char was prepared by the pyrolysis of biomass (rice husk) at 800 °C, which has weaker bonds between each structure than coal, and C–H and HC–CH do not likely exist as substitution and dehydrogenation reaction sites with Cl
2 as expressed in
eqn (6)–(8). Therefore, Cl
2 is mainly incorporated into the carbon structure as ClC–CCl through the addition reaction of the non-conjugated double bond (
eqn (7)). This is supported by the observation of HCl being derived from C(Cl)/C(HCl) at around 600 °C in the TPD profile of HCl shown in
Fig. 2 and the presence of the C–Cl bond in the XPS analysis shown in
Fig. 3. From these results, we concluded that most of the doped Cl exists in a thermally stable C–Cl bonded state.
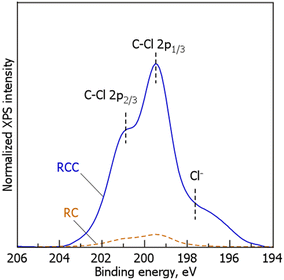 |
| | Fig. 3 Cl 2p XPS spectra of RC and RCC. (RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). | |
3.3 Evolution profiles of CO and CO2 in RC and RCC during TPD
According to previous reports, Cl2 and carbonaceous matter are reported to undergo substitution reactions with oxygen-containing functional groups, as expressed in eqn (9)–(10).40| | | 2COOH + Cl2 → 2COCl + H2O + 0.5O2 | (9) |
| | | 2COH + Cl2 → 2CCl + H2O + 0.5O2 | (10) |
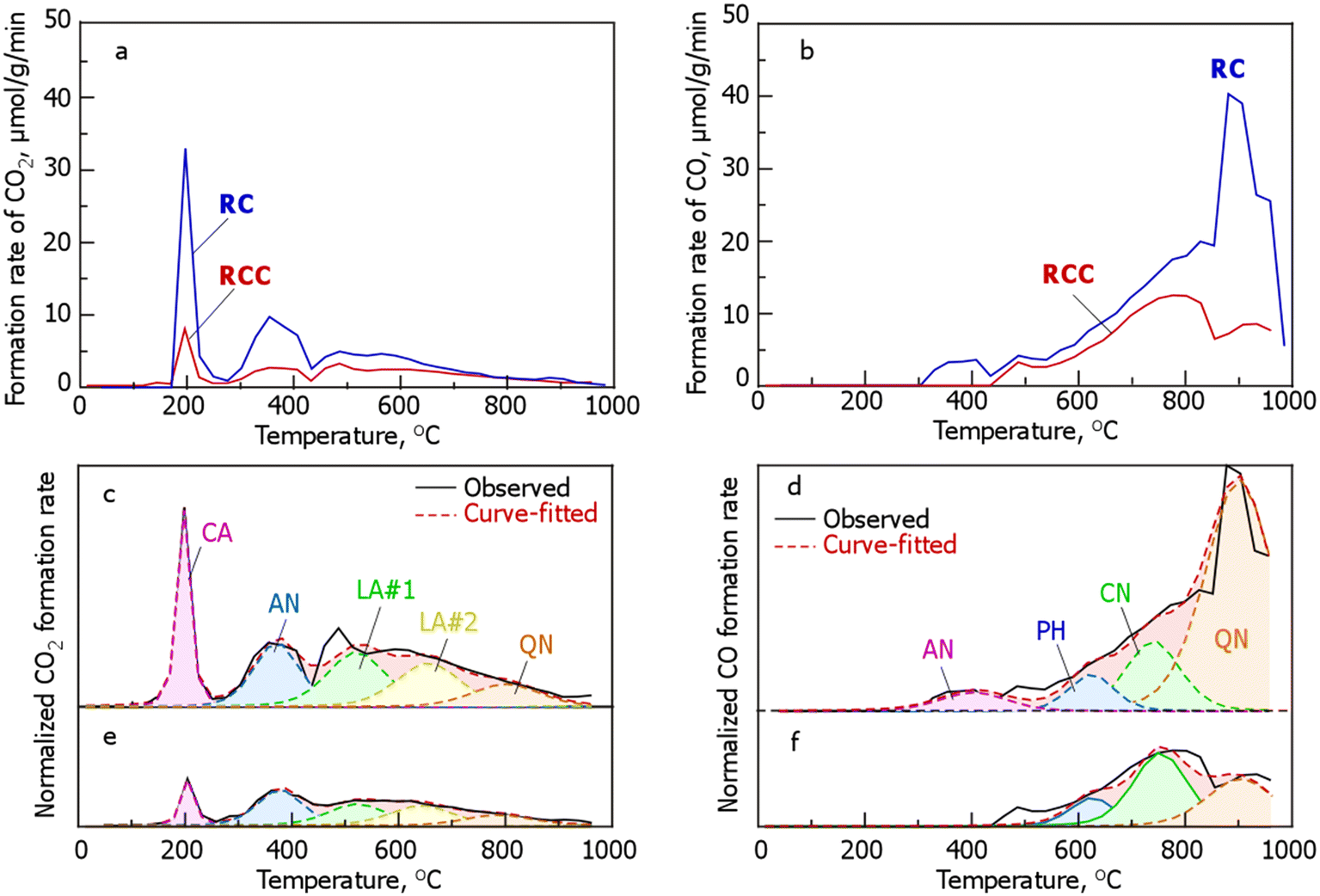
To further investigate the incorporation of Cl into RCC, the changes in the oxygen-containing functional groups based on the desorption behavior of CO and CO2 during the TPD of RC and RCC are shown in Fig. 4. The profile also showed a large peak around 200 °C and weak peaks at 300–450 and 450–550 °C. CO desorption occurs above 300 °C, with weak peaks at 350–400 and 500 °C, and the amount of desorption increases with increasing temperature, with a main peak at around 900 °C. The shape of the CO2 desorption profile of RCC is similar to that of RC; however, the intensity of each peak (the amount produced) is smaller. The desorption profile showed weak and broad peaks around 500, 750, and 900 °C. Compared with RC, in RCC, CO production at 300–400 °C and the production peak at 800–900 °C disappeared, and the desorption amounts of CO and CO2 were smaller than those of RC.
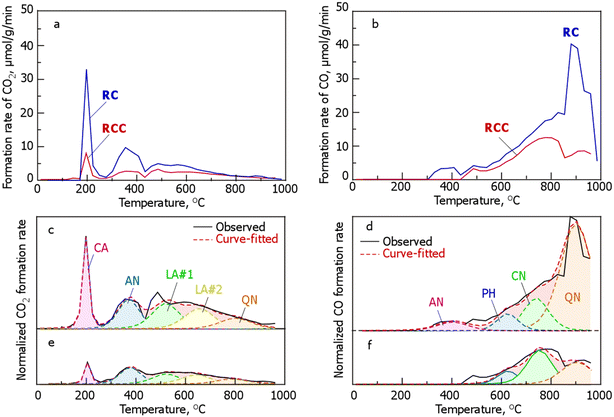 |
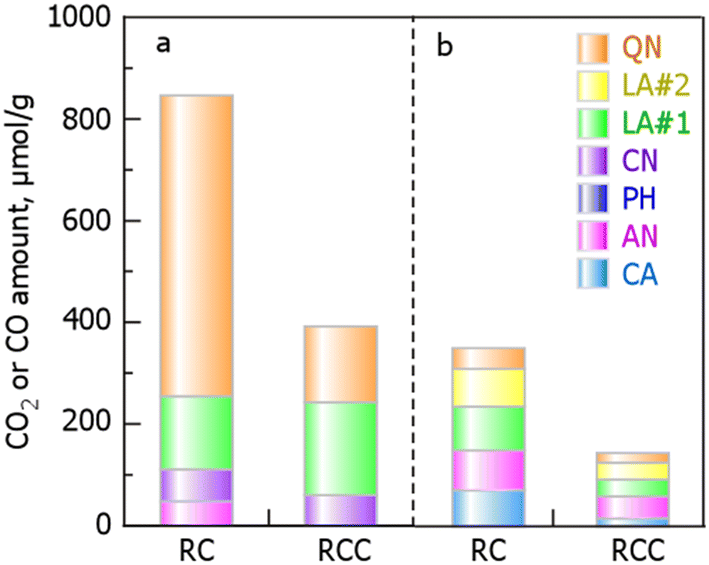
| | Fig. 4 Evolution profiles of CO (a) or CO2 (b) from RC and RCC during TPD and curve fitting of the formation rates of (c and e) CO2 and (d and f) CO during the TPD of (c and d) RC and (e and f) RCC. The abbreviations in the CO2 evolution profile are defined as follows: CA, carboxylic anhydrides; AN, anhydrides; LA#1, lactones at lower temperature; LA#2, lactones at higher temperature; QN, quinones as a result of the Boudouard reaction. Those in the CO evolution profile are defined as follows: AN, anhydrides; PH, phenols; CN, carbonyls; QN, quinone. (RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). | |
Notably, in TPD, the peaks of the generation rate of CO and CO2 originate from oxygen-containing functional groups in carbonaceous materials. The production rate profiles of CO2 in RC (Fig. 5a) were derived from carboxyls (CA, –COOH), anhydrides (AN, OC –O–CO), lactones at lower temperatures (LA#1, O–CO), lactones at higher temperatures (LA#2, O–CO), and quinone (QN, as a result of the Boudouard reaction). The production rate profiles of CO in RC (Fig. 4) were derived from AN (OC–O–CO), phenols (PH, –OH), carbonyls (CN, –CO–), and QN (OC–CO). The production rate profile of CO2 in RCC (Fig. 4c) was classified into five functional group species, similar to that of RC, with the following order: CA (11%) < QN (13%) < LA#1 (23%)  LA#2 (23%). In the formation rate profile of CO in RCC, the peak attributed to AN decomposition, which was observed in RC, disappeared and was composed of PH (15%), CN (47%), and QN (38%) (Fig. 4).
LA#2 (23%). In the formation rate profile of CO in RCC, the peak attributed to AN decomposition, which was observed in RC, disappeared and was composed of PH (15%), CN (47%), and QN (38%) (Fig. 4).
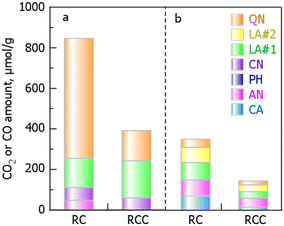 |
| | Fig. 5 Changes in the amounts of oxygen-containing functional groups in the sample before and after chlorination. The amounts of oxygen-containing functional groups derived from (a) CO and (b) CO2. (RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). | |
The oxygen-containing functional groups obtained from the curve-fitting results are presented in Table 3 and shown in Fig. 5. The amounts of CO and CO2 produced by RCC were less than half those of RC, indicating that the amount of oxygen-containing functional groups changed after the chlorination of RC. A comparison of the amount of each oxygen-containing functional group indicates that Cl2 reacts with AN (OC–O–CO), QN (OC–CO), and LA (O–CO), in addition to CA and CN expressed in eqn (9) and 10, and is incorporated into the carbon structure. We concluded that the functional groups that easily react with Cl2 are CA, AN, LA, and QN, depending on the degree of decrease in the amount of each oxygen-containing functional group in RC and RCC.
Table 3 Summaries of the oxygen-containing functional groups in RC and RCC
| Sample |
Amount of CO2 evolved, μmol g−1 |
Amount of CO evolved, μmol g−1 |
| CAa |
ANb |
LA#1c |
LA#2d |
QNe |
ANb |
PHf |
CNg |
QNh |
|
Carboxylic anhydrides.
Anhydrides.
Lactones at lower temperature.
Lactones at higher temperature.
Quinones as a result of the Boudouard reaction.
Phenols.
Carbonyls.
Quinone.(RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). |
| RC |
71 |
78 |
86 |
74 |
41 |
48 |
63 |
144 |
591 |
| RCC |
16 |
43 |
33 |
33 |
19 |
0 |
60 |
183 |
149 |
3.4 Breakthrough curves of the adsorption of gaseous Hg0 on RC and RCC
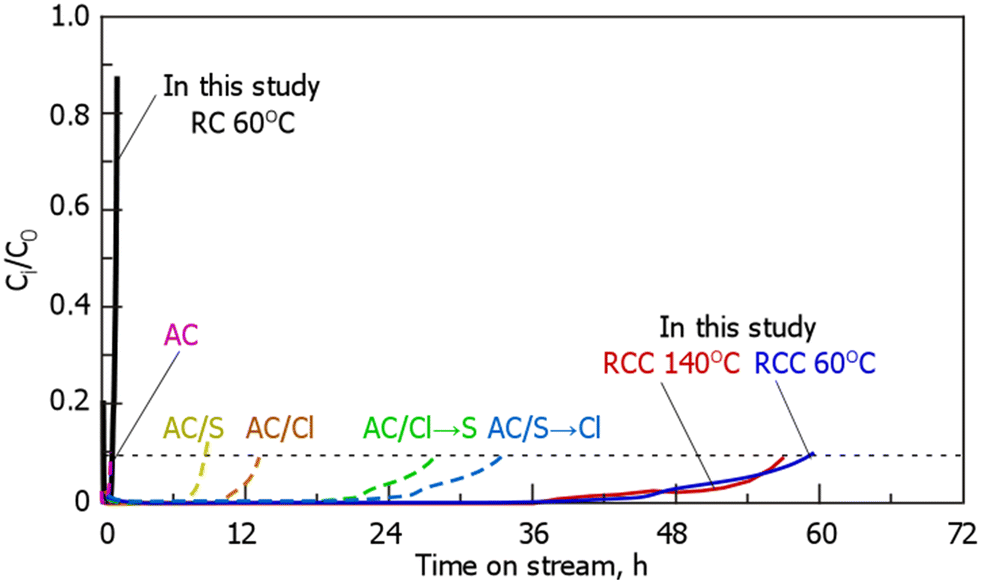
Fig. 6 shows the breakthrough curves of gaseous Hg0 for RC and RCC. During treatment with RC at 60 °C, Hg0 adsorption reached breakthrough immediately after the start of the experiment, whereas during treatment with RCC at 140 °C, Hg0 was not detected until 36 h. After 36 h, Hg0 was detected at the reactor outlet and reached breakthrough (Ci/C0 = 0.1) at 57–60 h. Comparison between the breakthrough curves of RC and RCC during the treatment at 60 °C indicated that the latter showed a better Hg0 adsorption performance than the former. The Hg0 adsorption breakthrough curves of the Cl- and S-loaded carbonaceous materials reported in a previous study are shown in Fig. 6 and presented in Table 4.14 The experimental conditions in the previous study14 are almost the same as those in our study in terms of the Hg concentration and gas flow rate, but the weight of the adsorbent is different; thus, the combined results are weight-corrected (2 g → 0.1 g). This adsorbent outperformed the adsorption performance of carbonaceous materials loaded with S, Cl, or both, as reported previously.14 Assuming that Hg0 is only adsorbed on Cl sites in the sample, the Cl utilization at the RCC breakthrough point is 3–4%, which is larger than that (0.7–2%) reported previously.14 Notably, the Cl content of AC-Cl was 11%, which was approximately 1.46-fold higher than that of RCC, but the Hg adsorption was smaller than that of RCC. Although it is not possible to compare the dispersibility of chlorine in AC-Cl and RCC used in previous reports, the dispersibility of the Cl species was considered to be small in AC-Cl. RCC as prepared by treating RC with Cl2 gas, and we assumed that a high Hg removal rate was achieved owing to the presence of fine Cl on the carbonaceous surface. In addition, the difference of Cl chemical in RCC and reported AC-Cl also may affect of Hg0 removal performance.
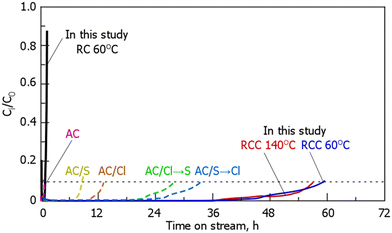 |
| | Fig. 6 Breakthrough curves of gaseous-Hg0 on RC and RCC. (The results of AC/S, AC/Cl, AC/Cl → S, AC/S → Cl are obtained from a previous study14). (RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). | |
Table 4 Summaries of the breakthrough curves of gaseous-Hg0 and their comparison
| Sample |
In this study |
Ref. 14 |
| RC |
RCC |
AC |
AC/Cla |
AC/Sb |
AC/Cl → Sc |
AC/S → Cld |
|
CaCl2-loaded AC.
K2S-loaded AC.
K2S-loaded AC/Cl (K2S loaded after the preparation of AC/Cl).
CaCl2-loaded AC/S (CaCl2 loaded after the preparation of AC/S).
The amount at breakthrough point.
Not analyzed. (RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min). |
| Temperature, °C |
60 °C |
60 °C |
140 °C |
140 °C |
| Breakthrough time, h |
0.39 |
56 |
56 |
0.15 |
13 |
8 |
28 |
32 |
| Adsorbed-Hg, mg g−1e |
0.07 |
8.7 |
11 |
0.05 |
4.1 |
2.5 |
9.2 |
10.3 |
| S or Cl content, % |
0.1 |
9.0 |
9.0 |
n.a.f |
11 |
8.3 |
Cl,7.2; S,5.5 |
S,1.0; Cl,23 |
3.5 Adsorption form of gaseous Hg0 on RCC
Although XPS analysis of spent RCC was performed, Hg analysis was not possible due to the overlap of the Si and Hg spectral ranges. Therefore, adsorption form of gaseous Hg0 in RCC was investigated by TPD method. Fig. 7 shows the desorption behavior of Hg when RCC was subjected to TPD after rupture to examine the adsorption behavior of gaseous Hg0 on RCC. The desorption of Hg from the RCC was observed at a temperature above 150 °C. According to previous reports, the adsorption of Hg0 onto Cl species existing as C–Cl on C occurs according to eqn (11)–(15).42–45| | | Hg0 (ad) + C–Cl (ad) → C–Hg–Cl (ad) | (12) |
| | | 2Hg0 (ad) + 2C–Cl (ad) → Hg2Cl2 (ad) | (13) |
| | | Hg0 (ad) + 2C–Cl (ad) → HgCl2 (ad) | (14) |
| | | Hg2Cl2 (ad) + 2C–Cl (ad) → 2HgCl2 (ad) | (15) |
Therefore, in this study, Hg was adsorbed as Hg2Cl2 and HgCl2. Therefore, based on previous studies of the desorption behavior of Hg observed during the TPD mass spectrometry (MS) with carbon and Hg compounds,46 curve fitting was conducted to examine the form of Hg adsorption. The results showed that 89% of the Hg was adsorbed as HgCl2, and the remaining 11% was in the form of HgO. However, the amount of Hg adsorbed did not match that desorbed from the TPD (approximately 2/3 is not balanced). In previous reports, wherein Cl- and S-impregnated carbon were compared, the adsorption stability of elemental Hg was found to be lower.47,48 Therefore, some Hg may have been desorbed during gas displacement in the reactor before the TPD experiment. Conversely, Hg may also be present in a thermally stable form owing to strong interaction with Cl, and the details of this phenomenon require further investigation.
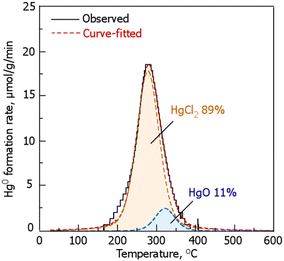 |
| | Fig. 7 The evolution profile of Hg0 in RCC during TPD. RCC preparation conditions: chlorination temperature, 1000 °C; holding time, 10 min. | |
According to previous reports on the effect of oxygen-containing functional groups on the adsorption of gaseous-Hg0 on carbonaceous materials,47,48 it has been reported that carboxyl groups, carbonyl groups, and ester groups are the sites for Hg0 adsorption. Thus, the presence of HgO in RCC may be due to the reaction of oxygen-containing functional groups with gaseous-Hg0. However, the amount of these oxygen-containing functional groups in RCC is small compared to that in RC, and the effect is small considering the fact that RC quickly reaches saturation as shown in Fig. 6. According the report that examined gaseous-Hg0 removal by calcium chloride loaded activated carbon,49–51 albeit under air atmosphere, it has been reported that the oxidation reaction of gaseous-Hg0 occurs by calcium chloride to produce oxy-mercury chloride (HgCl2·nHgO). As shown in Table 1, the chlorinated residue, RCC, contains Ca, and its form is assumed to be a chloride species. Therefore, it is possible that various chlorine species contained in RCC affect gaseous-Hg0 adsorption (e.g., as HgO in addition to HgCl2), and a detailed mechanism is a future issue to be elucidated. In addition, it is possible that Cl species other than C–Cl affect Hg0 adsorption. However, it was difficult to clearly identify Cl species other than C–Cl in this study. Therefore, the identification of Cl species other than C–Cl and their effects on Hg0 adsorption are also issues for future investigation.
4. Conclusions
In this study, we investigated the removal of gaseous Hg0 using the residue obtained during the chlorination of RC to recover silica. The following conclusions were drawn.
Through chlorination of RC at 1000 °C for 10 min, Si in the char could be separated and recovered, and a carbonaceous material doped with 9.0% Cl could be prepared.
TPD analysis of the char before and after chlorination revealed that the oxygen-containing functional groups on the surface of char were part of the Cl adsorption site.
Based on the XPS analysis, Cl was mainly present on RCC through C–Cl bonding.
The removal performances of gaseous Hg0 by RC and RCC were tested. The former reached breakthrough soon after the start of the test, whereas the removal performance of RCC was maintained up to 56 h, and the effect of temperature on the performance was small in the tested range (40–140 °C).
The adsorption performance of the prepared RCC was superior to those of chlorine- and sulfur-loaded carbons, as reported in previous studies.
The results of the TPD analysis after the adsorption tests revealed that the adsorbed form of Hg0 was mainly HgCl2.
Data availability
The data that support the findings of this study are available from the corresponding author, [T.S.], upon reasonable request.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was supported in part by a Grant-in-Aid for Challenging Research (Exploratory) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, Iron and Steel Institute of Japan (ISIJ), and Steel Foundation for Environmental Protection Technology (SEPT).
References
- W. Fitzgerald, D. Engstrom, R. Mason and E. Nater, The case for atmospheric mercury contamination in remote areas, Environ. Sci. Technol., 1998, 32(1), 1–7 CrossRef CAS.
-
UNEP, Minamata convention on mercury, https://mercuryconvention.org/2013 (Accessed 25.7.2016).
-
UNEP, Global Mercury Assessment 2013, United Nations Pubns, 2015 Search PubMed.
- S. V. Krishnan, B. K. Gullet and W. Jozewicz, Sorption of elemental mercury by activated carbons, Environ. Sci. Technol., 1994, 28(8), 1506–1508 CrossRef CAS PubMed.
- J. A. Korpiel and R. D. Vidic, Effect of sulfur impregnation method on activated carbon uptake of gas-phase mercury, Environ. Sci. Technol., 1997, 31(8), 2319–2325 CrossRef CAS.
- M. Takaoka, T. Yamamoto, N. Takeda, K. Oshita, T. Tanaka and T. Uruga, Determination of chemical states of mercury on activated carbon using XANES, AIP Conf. Proc., 2007, 882, 283 CrossRef CAS.
- F. E. Huggins, N. Yap, G. P. Huffman and C. L. Senior, XAFS characterization of mercury captured from combustion gases on sorbents at low temperatures, Fuel Process. Technol., 2003, 82(2–3), 167–196 CrossRef CAS.
-
K. Nishimura, M. Takaoka and N. Takeda, Effect of chemical properties of carbon on de novo synthesis of PCDD/PCDF, in Proceedings of the Annual Conference of the Japan Society of Waste Management Experts, 2004, pp. 901–903, (in Japanese) Search PubMed.
- M. Takaoka, N. Takeda and T. Fujiwara, Experimental studies on the removal mechanism of mercury vapor by synthetic fly ash, Taiki Kankyo Gakkaishi, 2000, 3(1), 51–62 Search PubMed , (in Japanese).
- M. Takaoka, N. Takeda, Y. Shimaoka and T. Fujiwara, Removal of mercury in flue gas by the reaction with sulfide compounds, Toxicol. Environ. Chem., 2000, 73(1–2), 1–16 Search PubMed.
- W. Liu, R. D. Vidic and T. D. Brown, Optimization of sulfur impregnation protocol for fixed-bed application of activated carbon-based sorbents for gas-phase mercury removal, Environ. Sci. Technol., 1998, 32(4), 531–538 CrossRef CAS.
- J. A. Korpiel and R. D. Vidic, Effect of sulfur impregnation method on activated carbon uptake of gas-phase mercury, Environ. Sci. Technol., 1997, 31(8), 2319–2326 CrossRef CAS.
- H. C. Hsi, M. J. Rood, R. A. Massoud, S. Chen and R. Chang, Effects of sulfur impregnation temperature on the properties and mercury adsorption capacities of activated carbon fibers (ACFs), Environ. Sci. Technol., 2001, 35(13), 2785–2791 CrossRef CAS PubMed.
- A. Sano, M. Takaoka and K. Shiota, Vapor-phase elemental mercury adsorption by activated carbon co-impregnated with sulfur and chlorine, Chem. Eng. J., 2017, 315, 598–607 CrossRef CAS.
- A. E. Morris, W. D. Kirk and Q. C. Jia, Roles of sulfuric acid in elemental mercury removal by activated carbon and sulfur-impregnated activated carbon, Environ. Sci. Technol., 2012, 46, 7905–7912 CrossRef PubMed.
- I. R. Ie, C. H. Hung, Y. S. Jen, C. S. Yuan and W. H. Chen, Adsorption of vapor-phase elemental mercury (Hg0) and mercury chloride (HgCl2) with innovative composite activated carbons impregnated with Na2S and S0 in different sequences, Chem. Eng. J., 2013, 229(1), 469–476 CrossRef CAS.
- X. Li, Y. J. Lee and S. Heald, XAFS characterization of mercury captured on cupric chloride-impregnated sorbents, Fuel, 2012, 93, 618–624 CrossRef CAS.
- Y. Mochizuki, J. Bud, J. Liu, M. Takahashi and N. Tsubouchi, Mercury (II) ion adsorption performance of Cl-loaded carbonaceous material prepared by chlorination of pyrolyzed rice husk char, J. Clean. Prod., 2021, 305, 127176 CrossRef CAS.
- Y. Mochizuki, J. Bud, J. Liu, M. Takahashi and N. Tsubouchi, Production of Silicone tetrachloride from rice husk by chlorination and performance of mercury adsorption from aqueous solution of the chlorinated residue, ACS Omega, 2020, 5, 29110–29120 CrossRef CAS.
- N. Tsubouchi, Y. Mochizuki, Y. Ono, K. Uebo, T. Takanohashi and N. Sakimoto, Sulfur and Nitrogen Distributions during Coal Carbonization and the Influences of These Elements on Coal Fluidity and Coke Strength, ISIJ Int., 2014, 54, 2439–2445 CrossRef CAS.
- N. Tsubouchi, N. Ohtaka and Y. Ohtsuka, Reactions of Hydrogen Chloride with Carbonaceous Materials and the Formation of Surface Chlorine Species, Energy Fuels, 2016, 30(3), 2320–2327 CrossRef CAS.
- N. Tsubouchi, Y. Ohtsuka, H. Hashimoto, T. Yamada and H. Hashimoto, Chemical characterization of unburned carbon in coal fly ashes by use of TPD/TPO and LRS methods, Environ. Sci. Technol., 2015, 49, 5189–5194 CrossRef CAS PubMed.
- N. Tsubouchi, S. Ohtsuka, Y. Nakazato and Y. Ohtsuka, Formation of hydrogen chloride during temperature-programmed pyrolysis of coals with different ranks, Energy Fuels, 2005, 19, 554–560 CrossRef CAS.
- N. Li, J. Zhu, X. Ma, Q. Zha and C. Song, Tailoring of surface oxygen-containing functional groups and their effect on adsorptive denitrogenation of liquid hydrocarbons over activated carbon, AIChE J., 2013, 59, 1236–1244 CrossRef CAS.
- M. Almarri, X. Ma and C. Song, Role of surface oxygen-containing functional groups in liquid-phase adsorption of nitrogen compounds on carbon-based adsorbents, Energy Fuels, 2009, 23, 3940–3947 CrossRef CAS.
- V. T. Amorebieta and A. J. Colussi, Direct study of the catalytic decomposition of chlorine and chloromethanes over carbon films, Int. J. Chem. Kinet., 1985, 17, 849–858 CrossRef CAS.
- N. Tsubouchi, E. Kasai, K. Kawamoto, H. Noda, Y. Nakazato and Y. Ohtsuka, Functional forms of carbon and chlorines in dust samples formed in the sintering process of iron ores, Tetsu-to-Hagane, 2005, 91, 751–756 CrossRef CAS.
- N. Tsubouchi, T. Saito, N. Ohtaka and Y. Ohtsuka, Evolution of hydrogen chloride and change in the chlorine functionality during pyrolysis of Argonne premium coal samples, Energy Fuels, 2013, 27, 87–96 CrossRef CAS.
- N. Tsubochi, H. Hashimoto, N. Ohtake and Y. Ohtsuka, Chemical characterization of dust particles recovered from bag filters of electric arc furnaces for steelmaking: Some factors influencing the formation of hexachlorobenzene, J. Hazard. Mater., 2010, 193, 116–124 CrossRef.
- N. Tsubouchi, Y. Mochizuki, Y. Wang and Y. Ohtsuka, Fate of the chlorine in coal in the heating process, ISIJ Int., 2018, 58, 227–235 CrossRef CAS.
- H. Chen, X. Chen, Z. Qiao and H. Liu, Release and transformation behavior of Cl during pyrolysis of torrefied rice straw, Fuel, 2016, 183, 145–154 CrossRef CAS.
- S. B. Saleh, J. P. Flensborg, T. K. Shoulaifar, Z. Sarossy, B. B. Hansen, H. Egsgaard, N. DeMartini, P. A. Jensen, P. Glarborg and K. Dam-Johansen, Energy Fuels, 2014, 28, 3738–3746 CrossRef CAS.
- P. A. Jensen, F. J. Frandsen, K. Dam-Johansen and B. Sander, Experimental investigation of transformation and release to gas phase of potassium and chlorine during straw pyrolysis, Energy Fuels, 2000, 14, 1280–1285 CrossRef CAS.
- M. Takeda, A. Ueda, H. Hashimoto, T. Yamada, N. Suzuki, M. Sato, N. Tsubouchi, Y. Makazato and Y. Ohtsuka, Fate of the chlorine and fluorine in a sub-bituminous coal during pyrolysis and gasification, Fuel, 2006, 85, 235–242 CrossRef CAS.
- N. Tsubouchi, Y. Mochizuki, Y. Shinohara, A. Kawashima and Y. Ohtsuka, Formation of surface chlorine species by low temperature reaction of HCl with metal-loaded carbon, Fuel, 2019, 246, 51–59 CrossRef CAS.
- N. Tsubouchi, T. Saito, N. Ohtaka, Y. Nakazato and Y. Ohtsuka, Chlorine release during fixed-bed gasification of coal chars with carbon dioxide, Energy Fuels, 2013, 27, 5076–5082 CrossRef CAS.
- N. Tsubouchi, N. Ohtaka and Y. Ohtsuka, Reactions of hydrogen chloride with carbonaceous materials and the formation of surface chlorine species, Energy Fuels, 2016, 30, 2320–2327 CrossRef CAS.
- A. Pérez-Cadenas, F. Maldonado-Hódar and C. Moreno-Castilla, On the nature of surface acid sites of chlorinated activated carbons, Carbon, 2003, 41, 473–478 CrossRef.
- Y. Xu, X. Zeng, G. Luo, B. Zhang, P. Xu, M. Xu and H. Yao, Chlorine-Char composite synthesized by co-pyrolysis of biomass wastes and polyvinyl chloride for elemental mercury removal, Fuel, 2016, 183, 73–79 CrossRef CAS.
- J. González, M. del C. Ruiz, A. Bohe and D. Pasquevich, Oxidation of carbons in the presence of chlorine, Carbon, 1999, 37, 1979–1988 CrossRef.
-
Principles of extractive metallurgy, ed. F. Habashi, Gordon and Breach Science Publisher, New York, 1980, vol. II, pp. 172–174 Search PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitass and J. J. M. Orfao, Modification of the surface chemistry of activated carbons, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- Y. Xu, X. Zeng, B. Zhang, X. Zhu, M. Zhou, R. Zou, P. Sun, G. Luo and H. Yao, Experiment and Kinetic Study of Elemental Mercury Adsorption over a Novel Chlorinated Sorbent Derived from Coal and Waste Polyvinyl Chloride, Energy Fuels, 2016, 30, 10635–10642 CrossRef CAS.
- T. Wang, J. Wu, Y. Zhang, J. Liu, Z. Sui, H. Zhang, W.-Y. Chen, P. Norris and W.-P. Pan, Increasing the chlorine active sites in the mesopores of biochar for improved mercury adsorption, Fuel, 2018, 229, 60–67 CrossRef CAS.
- G. Li, S. Wang, Q. Wu, F. Wang and B. Shen, Mercury sorption study of halides modified bio-chars derived from cotton straw, Chem. Eng. J., 2016, 302, 305–313 CrossRef CAS.
- S. Wu, R. Katayama, W. Yang, M. A. Uddin and E. Sasaoka, Study on speciation of HgCl2 supported over activated carbon in simulated flue gases by temperature-programmed decomposition desorption mass spectrometry, Fuel, 2018, 219, 69–75 CrossRef CAS.
- H. Zhang, T. Wang, Z. Sui, Y. Zhang, B. Sun and W.-P. Pana, Enhanced mercury removal by transplanting sulfur-containing functional groups to biochar through plasma, Fuel, 2019, 253, 703–712 CrossRef CAS.
- R. D. Vidic and D. P. Siler, Vapor-phase elemental mercury adsorption by activated carbon impregnated with chloride and chelating agents, Carbon, 2001, 39, 3–14 CrossRef CAS.
- P. Sun, B. Zhang, X. Zeng, G. Luo, X. Li, H. Yao and C. Zheng, Deep study on effects of activated carbon's oxygen functional groups for elemental mercury adsorption using temperature programmed desorption method, Fuel, 2017, 200, 100–106 CrossRef CAS.
- S. Guo and G. Guo, Enhancement of gaseous mercury (Hg0) adsorption for the modified activated carbons by surface acid oxygen function groups, J. Groundwater Sci. Eng., 2018, 6, 104–114 Search PubMed.
- M. Takaoka, N. Takeda and T. Fujiwara, Experimental Studies on the Removal Mechanism of Mercury Vapor, by Synthetic Fly Ash, Taiki Kankyo Gakkaishi, 2000, 35, 51–62 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Momone
Yoshizawa
,
Javzandogole
Bud
and
Yuuki
Mochizuki
*,
Momone
Yoshizawa
,
Javzandogole
Bud
and
Yuuki
Mochizuki

 LA#2 (23%). In the formation rate profile of CO in RCC, the peak attributed to AN decomposition, which was observed in RC, disappeared and was composed of PH (15%), CN (47%), and QN (38%) (Fig. 4).
LA#2 (23%). In the formation rate profile of CO in RCC, the peak attributed to AN decomposition, which was observed in RC, disappeared and was composed of PH (15%), CN (47%), and QN (38%) (Fig. 4).