
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessment of fine-tuned large language models for real-world chemistry and material science applications†
Joren
Van Herck‡
 a,
María Victoria
Gil‡
ab,
Kevin Maik
Jablonka‡
acd,
Alex
Abrudan
e,
Andy S.
Anker
fg,
Mehrdad
Asgari
h,
Ben
Blaiszik
ij,
Antonio
Buffo
k,
Leander
Choudhury
l,
Clemence
Corminboeuf
m,
Hilal
Daglar
n,
Amir Mohammad
Elahi
a,
Ian T.
Foster
ij,
Susana
Garcia
o,
Matthew
Garvin
o,
Guillaume
Godin
p,
Lydia L.
Good
eq,
Jianan
Gu
r,
Noémie
Xiao Hu
a,
Xin
Jin
a,
Tanja
Junkers
s,
Seda
Keskin
n,
Tuomas P. J.
Knowles
et,
Ruben
Laplaza
m,
Michele
Lessona
k,
Sauradeep
Majumdar
a,
Hossein
Mashhadimoslem
u,
Ruaraidh D.
McIntosh
v,
Seyed Mohamad
Moosavi
w,
Beatriz
Mouriño
a,
Francesca
Nerli
x,
Covadonga
Pevida
b,
Neda
Poudineh
o,
Mahyar
Rajabi-Kochi
w,
Kadi L.
Saar
e,
Fahimeh
Hooriabad Saboor
y,
Morteza
Sagharichiha
z,
K. J.
Schmidt
i,
Jiale
Shi
aaab,
Elena
Simone
k,
Dennis
Svatunek
ac,
Marco
Taddei
x,
Igor
Tetko
pad,
Domonkos
Tolnai
r,
Sahar
Vahdatifar
z,
Jonathan
Whitmer
abae,
D. C. Florian
Wieland
r,
Regine
Willumeit-Römer
r,
Andreas
Züttel
af and
Berend
Smit
*a
a,
María Victoria
Gil‡
ab,
Kevin Maik
Jablonka‡
acd,
Alex
Abrudan
e,
Andy S.
Anker
fg,
Mehrdad
Asgari
h,
Ben
Blaiszik
ij,
Antonio
Buffo
k,
Leander
Choudhury
l,
Clemence
Corminboeuf
m,
Hilal
Daglar
n,
Amir Mohammad
Elahi
a,
Ian T.
Foster
ij,
Susana
Garcia
o,
Matthew
Garvin
o,
Guillaume
Godin
p,
Lydia L.
Good
eq,
Jianan
Gu
r,
Noémie
Xiao Hu
a,
Xin
Jin
a,
Tanja
Junkers
s,
Seda
Keskin
n,
Tuomas P. J.
Knowles
et,
Ruben
Laplaza
m,
Michele
Lessona
k,
Sauradeep
Majumdar
a,
Hossein
Mashhadimoslem
u,
Ruaraidh D.
McIntosh
v,
Seyed Mohamad
Moosavi
w,
Beatriz
Mouriño
a,
Francesca
Nerli
x,
Covadonga
Pevida
b,
Neda
Poudineh
o,
Mahyar
Rajabi-Kochi
w,
Kadi L.
Saar
e,
Fahimeh
Hooriabad Saboor
y,
Morteza
Sagharichiha
z,
K. J.
Schmidt
i,
Jiale
Shi
aaab,
Elena
Simone
k,
Dennis
Svatunek
ac,
Marco
Taddei
x,
Igor
Tetko
pad,
Domonkos
Tolnai
r,
Sahar
Vahdatifar
z,
Jonathan
Whitmer
abae,
D. C. Florian
Wieland
r,
Regine
Willumeit-Römer
r,
Andreas
Züttel
af and
Berend
Smit
*a
aLaboratory of Molecular Simulation (LSMO), Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), Rue de l’Industrie 17, CH-1951 Sion, Switzerland. E-mail: Berend.Smit@epfl.ch
bInstituto de Ciencia y TecnologÍa del Carbono (INCAR), CSIC, Francisco Pintado Fe 26, 33011 Oviedo, Spain
cLaboratory of Organic and Tecnolog'ıa Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstrasse 10, 07743 Jena, Germany
dHelmholtz Institute for Polymers in Energy Applications Jena (HIPOLE Jena), Lessingstrasse 12-14, 07743 Jena, Germany
eYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
fDepartment of Energy Conversion and Storage, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark
gDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK
hDepartment of Chemical Engineering & Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK
iDepartment of Computer Science, University of Chicago, Chicago, IL 60637, USA
jData Science and Learning Division, Argonne National Laboratory, Lemont, IL 60439, USA
kDepartment of Applied Science and Technology (DISAT), Politecnico di Torino, 10129 Turino, Italy
lLaboratory of Catalysis and Organic Synthesis (LCSO), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
mLaboratory for Computational Molecular Design (LCMD), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
nDepartment of Chemical and Biological Engineering, Koç University, Rumelifeneri Yolu, Sariyer, 34450 Istanbul, Turkey
oThe Research Centre for Carbon Solutions (RCCS), School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
pBIGCHEM GmbH, Valerystraße 49, 85716 Unterschleißheim, Germany
qLaboratory of Chemical Physics, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, USA
rInstitute of Metallic Biomaterials, Helmholtz Zentrum Hereon, Geesthacht, Germany
sPolymer Reaction Design Group, School of Chemistry, Monash University, Clayton, VIC 3800, Australia
tCavendish Laboratory, Department of Physics, University of Cambridge, Cambridge CB3 0HE, UK
uDepartment of Chemical Engineering, University of Waterloo, Waterloo, N2L3G1, Canada
vInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
wChemical Engineering & Applied Chemistry, University of Toronto, Toronto, Ontario M5S 3E5, Canada
xDipartimento di Chimica e Chimica Industriale, Unità di Ricerca INSTM, Università di Pisa, Via Giuseppe Moruzzi 13, 56124 Pisa, Italy
yChemical Engineering Department, University of Mohaghegh Ardabili, P. O. Box 179, Ardabil, Iran
zDepartment of Chemical Engineering, College of Engineering, University of Tehran, Tehran, Iran
aaDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
abDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA
acInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9, 1060, Vienna, Austria
adInstitute of Structural Biology, Molecular Targets and Therapeutics Center, Helmholtz Munich - Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH), Ingolstädter Landstraße 1, 85764 Neuherberg, Germany
aeDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA
afLaboratory of Materials for Renewable Energy (LMER), Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), Rue de l'Industrie 17, CH-1951 Sion, Switzerland
First published on 22nd November 2024
Abstract
The current generation of large language models (LLMs) has limited chemical knowledge. Recently, it has been shown that these LLMs can learn and predict chemical properties through fine-tuning. Using natural language to train machine learning models opens doors to a wider chemical audience, as field-specific featurization techniques can be omitted. In this work, we explore the potential and limitations of this approach. We studied the performance of fine-tuning three open-source LLMs (GPT-J-6B, Llama-3.1-8B, and Mistral-7B) for a range of different chemical questions. We benchmark their performances against “traditional” machine learning models and find that, in most cases, the fine-tuning approach is superior for a simple classification problem. Depending on the size of the dataset and the type of questions, we also successfully address more sophisticated problems. The most important conclusions of this work are that, for all datasets considered, their conversion into an LLM fine-tuning training set is straightforward and that fine-tuning with even relatively small datasets leads to predictive models. These results suggest that the systematic use of LLMs to guide experiments and simulations will be a powerful technique in any research study, significantly reducing unnecessary experiments or computations.
1 Introduction
The traditional machine-learning workflow starts with the painstaking process of harvesting the literature for the relevant data. Some help can be obtained from text harvesting programs.1 These data can be used to find correlations in the properties or synthesis of molecules or materials or correlations in any other relevant chemical question. For this, it is crucial to describe the system with features fed into a model. Ultimately, the trained model allows us to make predictions from the features of unknown materials. These models typically improve when more data becomes available.In chemistry and material science, however, the amount of experimental data is often, if not always, a bottleneck. Therefore, it is essential to have some leverage. One way of doing this is by expanding a dataset with computer simulations.2 Alternatively, we can leverage knowledge of the system. For example, suppose we want to predict the pressure of a gas at a given density and temperature; we can focus our machine learning (ML) on predicting the deviations from the ideal gas law.3 Another option is to introduce descriptors with proper inductive biases that capture our understanding of the underlying systems.4
Another way of leveraging knowledge is through transfer learning. Imagine that one has a lot of data on some particular properties of a class of materials but, as is typical in many practical applications, not enough data for the property of interest. The idea of transfer learning is that we can train a model on the properties for which we have a lot of data and subsequently fine-tune this model for the property of interest.5 The mechanism of this fine-tuning or transfer learning is that one only retrains a small part of, for example, a transformer model (or of an added layer) and hence leverages all the pre-trained information locked in the model's part that remains unchanged. This fine-tuned model can then be used to make predictions for these properties.
In this context, a remarkable recent discovery is that one can also use fine-tuned large language models (LLMs) to answer chemistry and material science questions for which the base LLM would not know the answer.6,7 LLMs are pre-trained (without supervision) on web-scale data. Their training is to predict the next likely character (or word) to complete a sentence. For example, if we use GPT-3 (e.g., via ChatGPT) to ask a specific chemical question, say, if the high-entropy alloy Tb0.5Y0.5 is a single phase, it will reproduce the knowledge is has. GPT-3 would not know the answer (GPT-4 knows more chemistry8). Hence, it will likely not get an answer to such chemical questions. However, we can fine-tune an LLM with experimental data of high-entropy alloys, of which we know whether it is a single phase or not. This gives us a new model that only aims to predict whether a particular high entropy alloy is a single phase.
In addition, the fact that these LLMs use natural language as input instead of a descriptor is one of its most attractive features; it creates a convenient way for researchers to interact with data and tools. Numerous successful chemical applications exploiting this power of LLMs exist today, ranging from tools that summarize literature to the deployment of “chatbots” for experimental instrumentation.9
These general-purpose LLMs are important because they do not require pre-training, can be used for any chemical question, and do not require knowledge of machine learning. In our previous work, we measured the potential of LLMs in solving chemical problems against conventional machine learning specifically developed and optimized for that problem.7 We showed that LLM models fine-tuned on classification, regression, and inverse design problems can be competitive with current state-of-the-art machine learning models. For this, we searched for chemical problems with a known ML solution and validated our approach against it.
In this work, we want to go a step further and attempt to address relevant chemical questions from a more practical point of view. This implies that most of the data have not been curated or selected previously for machine learning studies but are the data that researchers have at hand. The case studies we present are guided by the questions these researchers have.
With this data, we performed simple “experiments”. First, we asked whether fine-tuned LLMs show any signs of learning. To address this question, we split the dataset in half and used a simple classification to test if the model would classify the data correctly on a holdout set that the model has not seen in training. Accurate binary classifications can be particularly useful in experimental scenarios where precise numerical values are not necessary, simplifying and facilitating the decision-making process in routine research. Such classifiers can already be of great practical interest, as they mimic daily “yes” or “no” questions of researchers, e.g., “Can we synthesize this molecule?” or “Will property X of this molecule be high or low?”. Having access to accurate predictions of the answer of these questions has the potential to facilitate chemical workflows, reducing computational or experimental resources. Technically, this first step involved the fine-tuning of a model using a standard setting without any optimization. For this step, we used open-source models, which we tuned using parameter-efficient fine-tuning techniques.10–12 The models showed some learning for almost all problems. The extent of learning depends on the dataset and the complexity of the question. Inevitably, the performance of such models will not be optimal for every study; therefore, we optimized the models by performing basic hyperparameter optimization in those cases.
In the following sections, we outline the methodology and then summarize the main conclusions of each case study. The corresponding section of the ESI† provides a detailed account of each study. The main aim of these summaries is to illustrate the range of chemical questions that can be addressed. The discussion section summarizes the lessons we have learned from these case studies.
2 Methods
Researchers from different disciplines presented 22 datasets. We used these datasets in case studies to better understand the potential and limitations of fine-tuning LLMs. For these case studies, we used three open-source LLMs: GPT-J-6B,13 Llama-3.1-8M,14 and Mistral-7B.15 The LLMs used are smaller models than, for example, GPT-3 (and GPT-4). Our previous results show that GPT-3 typically performs better, i.e., it requires less training data to get similar performance.7 However, this increase in accuracy does not compensate for the fact that with open-source models, anybody can reproduce our results.To present the results in a structured manner, we have organized the case studies into three categories: Materials and properties, Reactions and synthesis, and Systems and applications.
Each case study was approached similarly. The first step requires converting the dataset into a set of questions and answers that we can use for fine-tuning. We obtained some general knowledge of the systems' scientific background for this. This background is given in more detail in the ESI† and summarized at the beginning of each paragraph describing the case studies. In the ESI,† one can find more details on how the dataset was obtained.
The first test we carried out was a standard test to determine if our fine-tuned LLM learned anything. This test was a simple classification problem in which we split the dataset into two equally populated categories. Depending on the case study, these categories were high/low, good/bad, optimal/non-optimal, etc. This simple classification allows for a simple benchmark: random guessing. The minimum criterion the LLM should outperform on the test set is to do better than random guessing. This random guess corresponds to the situation where we have zero knowledge of the system. We will refer to this experiment as the “base case”. Hence, any model that does better might be of practical use. In addition, we also compared the performance of LLMs with that of two “traditional” ML models, i.e., random forest (RF) and XGBoost. Even before training and using these models, the potential advantages of LLMs became apparent here. As these traditional models require numeric inputs, an additional step was often necessary to convert the features of the received datasets.
If our base case model outperformed the benchmark, the next step is to make the LLM more useful. In most practical applications, one has more data on poor-performing materials than optimal materials. However, for fine-tuning LLMs, one also needs a reasonable number of materials above the performance threshold, distinguishing poor from top-performing materials. This may require more data than we have available for a specific case study and may require us to optimize the model further. This part will be specific to each case study.
We follow the same fine-tuning method as in our previous work (the reader is referred to the original work7 for details on the fine-tuning) except that we now used GPT-J-6B, Llama-3.1-8B, and Mistral-7B. In this procedure, the chemical context is formatted in a single representation as a question (Table 1). The binary class is given as a numeric value, i.e., 0 or 1, representing the respective chemical property.
| Representation | Completion | Real |
|---|---|---|
| What is 〈Property〉 of 〈Materia〉? | 0 or 1 | Low or high |
The first iteration used default fine-tuning hyperparameter values (see ESI Note 2†). This allowed us to gain some insights into whether such an approach can be used as a black box without expertise in using LLMs or if some tweaking is needed to get sufficiently accurate results. After analyzing the first result, in some case studies, increasing the number of epochs, i.e., the times the model sees the training data, significantly increased the model's performance. This gives us insights into the fine-tuning procedure. This second step typically requires some more experience with these LLMs.
3 Results and discussion – case studies
3.1 Materials and properties
Following a bottom-up approach, most chemical applications start from fundamental research of the structure–property relationships. Therefore, it is no surprise that a thorough understanding of the structures at hand is necessary before proceeding to field-specific applications. However, the chemical space is vast and complex, and finding optimal solutions often requires extensive screening and analysis. Alternatively, chemical properties can be predicted from the structural features of a molecule.16 We demonstrate how our approach can help predict chemical properties in various case studies and more effectively guide computational and experimental research.The remarkable aspect of these results is that we have a hypothetical model polymer for which simulations compute the free energies. Yet, the LLM can correlate a sequence of 20 (arbitrarily chosen) characters of the type “A” and “B” to the free energy, suggesting no potential data leakage.
As the melting point of many chemicals is reported, we first studied how well ChatGPT (OpenAI's GPT-3.5) can classify the melting point as high or low. Using the front-end interface, we prompted “What is the melting point of 〈name of molecule〉?” and saw that only 50% of the time it predicted this correctly, which is no better than random guessing. In contrast, models trained on the IUPAC name reached an accuracy of 66%. Interestingly, our fine-tuned models trained on the SMILES of the molecules could predict the melting point with an accuracy of 69% (GPT-J). The fine-tuned model proves to compete with traditional ML models.
As the dynamic viscosity value of many chemicals is also reported, we evaluated (via ChatGPT) how well OpenAI's GPT-3.5 model can classify the viscosity as high or low. Our prompt was “What is the dynamic viscosity of 〈name of molecule〉?” Our results showed that viscosity was not better predicted than random guessing, with an accuracy of 55% when the chemical name was provided as input to the model.
In contrast, for a median split balanced dataset, with a training set size of 80 examples and 30 fine-tuning epochs, the fine-tuned LLM model GPT-J reached an accuracy of 80% for binary classification (which is comparable to traditional ML models). We also trained a model to predict whether a chemical had a dynamic viscosity in the top 28% of the values in the dataset. After reducing the dataset size to obtain a balanced dataset, we also obtained a predictive accuracy of 80% (GPT-J) using a training set of 50 data points by increasing the number of fine-tuning epochs to 140.
We found that the fine-tuned models can deal with incomplete and multivariable inputs, reaching accuracies of 94% (Mistral, comparable to traditional ML models) to predict the classification of the material either belonging to the class high or low amount of second phases. Interestingly, we acquired similar accuracies when only using the production route to represent the model irrespective of the individual process parameters. Despite the small dataset, the LLM is able to catch the material science properties and classify them accordingly.
This is an interesting case for an LLM model, as a protein represented by a string, like RRGDGRRRG…GGGRGQGGRGR, can be inputted directly in the prompt (see ESI Note 3.6†). We obtained accuracies reaching 95% (GPT-J) for models that distinguish proteins on their phase separation propensity, which is similar to the accuracy obtained by Saar et al..26 We want to stress that no extra data manipulation was needed. The protein sequence as received was used as input for the prompt, again demonstrating the versatility of LLMs.
In addition, we carried out some experiments in which we changed the original sequences (e.g., making them shorter or creating randomized sequences of the same letter/amino acid composition). The most interesting observation was that a model trained on randomized sequences of the same letters resulted in a relatively small drop in accuracy from 95% to 86%, which shows that a significant part (but not all) of the predictive ability can be obtained from the protein's sequence composition without any positional information of sequence order. Interestingly, the addition of positional information was also not found to increase the performance in predicting the apparent and shear modulus of materials.27
This is also an interesting case for an LLM model since the scattering pattern consists of a very long series of numbers represented as a string. With our approach, we obtained an accuracy of 97% (Mistral) to predict the structure type of nanoparticles from scattering patterns simulated from 7 highly unbalanced structure types with between 5 and 100 atoms (30 epochs, 1800 data points). We found that, for complex input variables, where the information is embedded along long sequences, using a relatively large training set size, the fine-tuned model can predict an unbalanced dataset with 7 classes. However, if the number of training data points is very low (200), the fine-tuned model is not even predictive on a balanced dataset.
For the prediction of the number of atoms in the nanomaterial, we obtained accuracies of 98% (Mistral) and 93% (Llama) for datasets with 4 and 10 balanced classes, respectively.
These results are comparable to those obtained with traditional ML models for the 4-class dataset but notably superior to those obtained with ML models for the more challenging 10-class classification task. However, from a practical point of view, given the interest in predicting the number of atoms with very high accuracy, we also developed a regression model. The LLM regression models predicted the number of atoms with an R2 of 99% (Llama and Mistral) and a maximum absolute error (MAE) below one atom (for comparison, R2 of random forest and XGBoost was 93% and 94%, respectively, while MAE was 5.1 and 4.7, respectively), i.e., LLMs showed an excellent performance.
The various notations of 211 TAGs provided an interesting test case to examine the influence of the representation on the predictions. We compared the IUPAC name, InChI code, Omega notation, and SMILES of the TAGs. Interestingly, with similar accuracies of 92% (GPT-J and Llama), we see excellent performance, slightly higher than that obtained with “traditional” ML models (86–88%).
Fig. 1 shows an overview of the results of the case studies on Materials and properties.
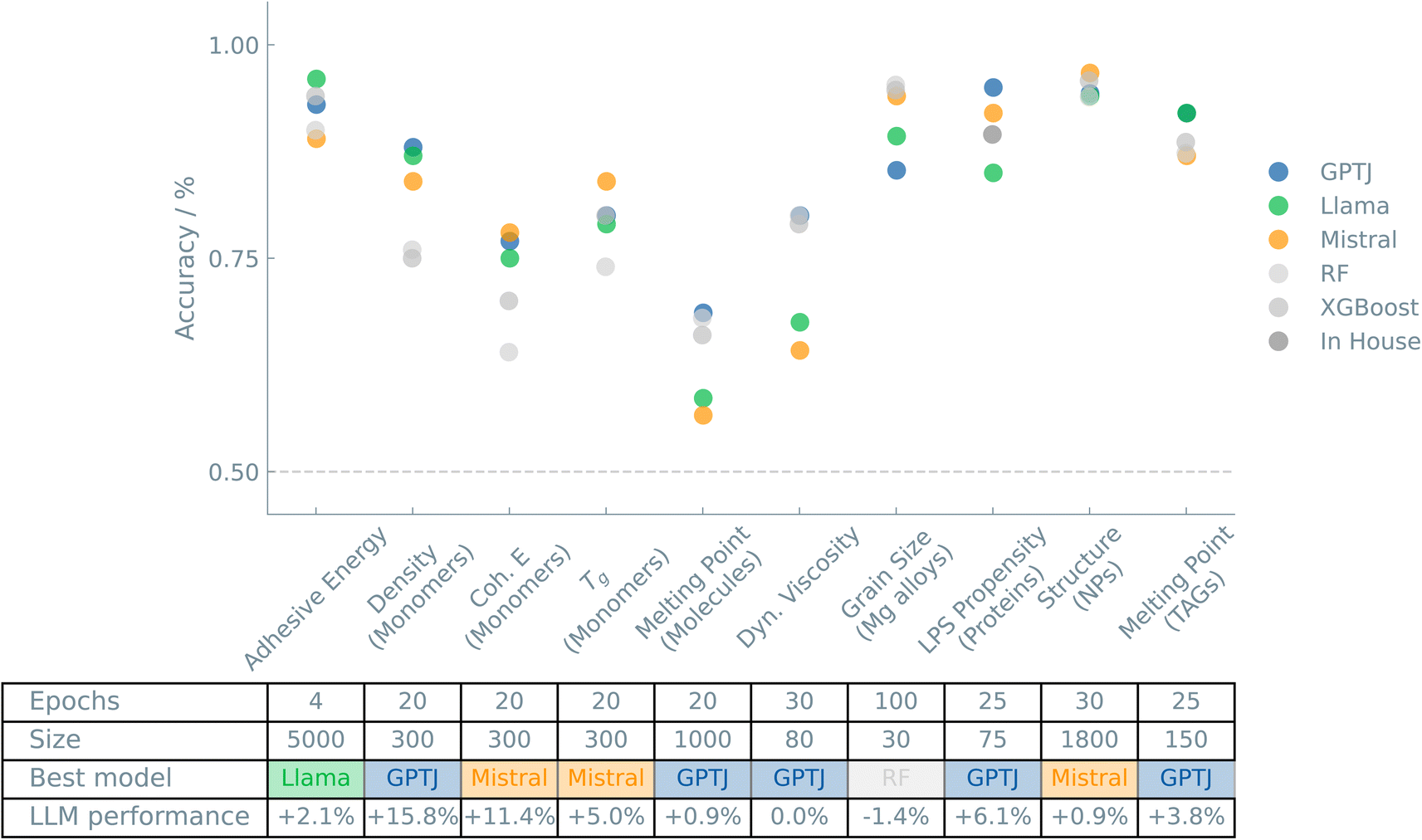 | ||
| Fig. 1 Overview of the “Materials and properties” case studies. The accuracies of binary classifiers are plotted for the “Materials and properties” case studies. Three different LLMs (GPT-J, Llama, and Mistral) and two traditional ML models (random forest (RF) and XGBoost) are compared. The dashed line indicates the zero-rule baseline of 50% accuracy. The table summarizes the number of epochs (for fine-tuning the LLMs), training set size, the best-performing model, and the relative difference between the best LLM and the best “traditional” model. | ||
3.2 Reactions and synthesis
Going beyond chemical properties, we explored the potential of our methodology in predicting reaction outcomes. Predicting reaction outcomes is a field in which conventional theory has made little progress. The complexity and specificity of chemical reactions make developing a general theoretical framework extremely challenging.30 Practically, indicating the success rate or yield could prioritize the synthesis of top candidates or omit protocols that lead to certain failures, thereby saving resources, time, and money.This case study is a great example of how machine learning, specifically LLMs, can significantly impact expensive computational studies. This model can be used as a first screening to filter out poor-performing structures. More expensive calculations can then be used for a more detailed analysis of well-performing structures.
While excellent in performance, the scientific relevance of evaluating these systems based on a single threshold is limited. Rather, a small range of continuous descriptor values is often considered ‘good.’ All values above and below this range are then considered ‘poor.’ From the dataset of catalysts, only 3.8% was labeled a ‘good’ catalyst. As a result, we were forced to reduce our training set significantly to get a balanced dataset. Nevertheless, even with a training size of 100 data points, the model was able to classify 79% (GPT-J) of the test data correctly.
An interesting future strategy would be to use an LLM model combined with more expensive quantum calculations. Initially, one would aim for a band of ‘good’ structures that is broader than one would like from a catalysis point of view but more balanced to get a more accurate model. Then, if we get more ‘good’ materials, we will retrain the model with a narrower band.
Not only are high-throughput kinetic screenings an excellent way to gain in-depth insights into reaction mechanisms, but they also produce datasets that can be used to train ML models and guide further research and development. Here, only two reaction parameters were varied. Combining screenings in a multi-parameter landscape with predictive models could accelerate polymer synthesis optimizations.
In this study, we predicted various photocatalytic properties of MOFs, thereby assessing whether a given material has the right band alignments for water splitting and absorbs visible light. We used the elemental composition of the MOF's linker and metal node to represent the material. The fine-tuned LLMs could successfully predict the various properties with accuracies higher than 90%.
This quantity issue is reflected in the provided dataset, which has only 25 different reaction conditions. Taking a yield of 20% as the success threshold could create a fairly balanced dataset. After training for just ten epochs on a training set size of 20 examples, the fine-tuned models could not recognize the prompt/completion structure and thus failed to output a meaningful prediction. Using a training set size of 20 and increasing the number of epochs to 50 leads to the expected binary responses, i.e., 0 or 1, with an average accuracy of 89% (GPT-J).
Fig. 2 shows an overview of the results of the case studies on Reactions and synthesis.
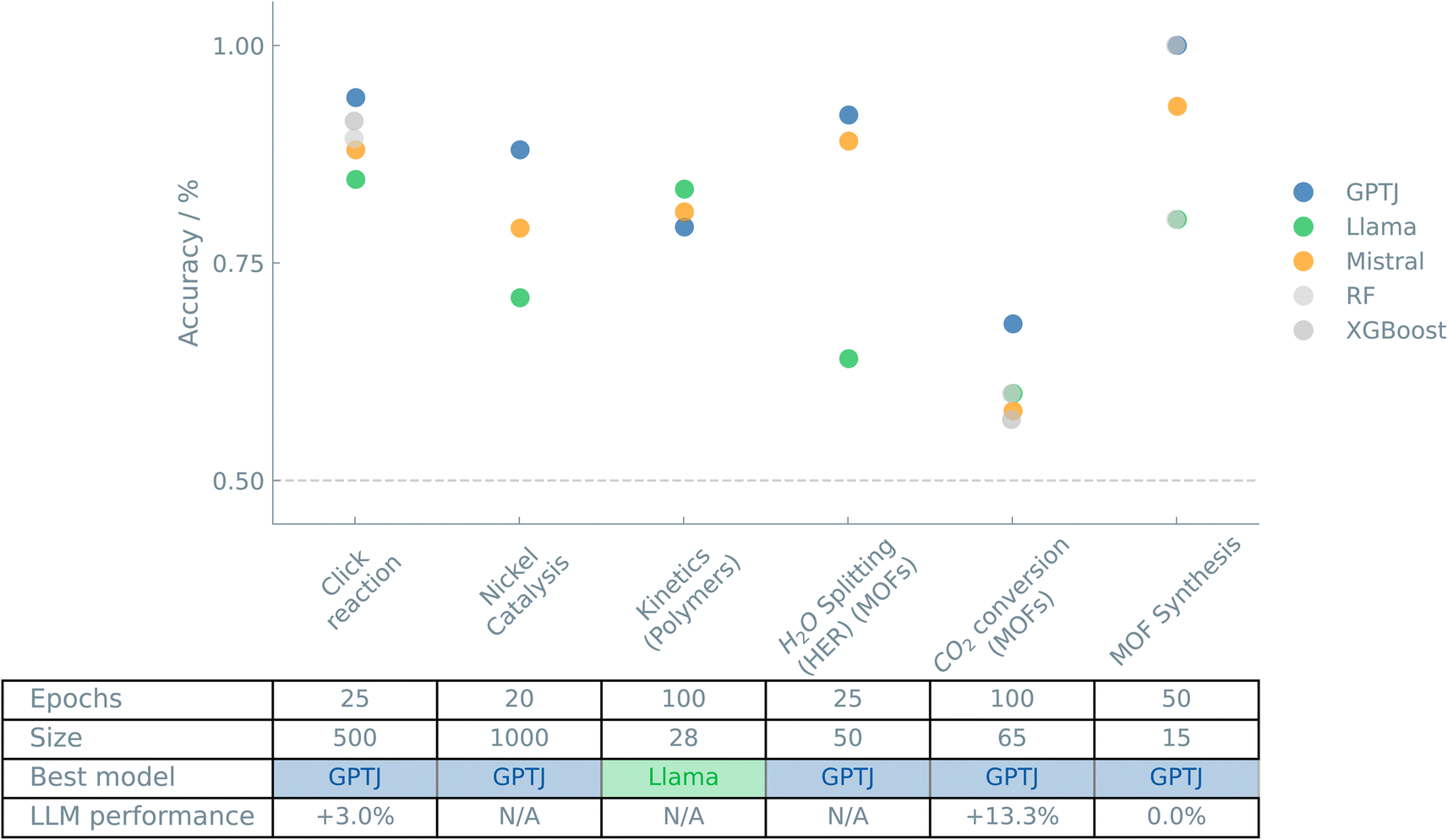 | ||
| Fig. 2 Overview of the “Reactions and synthesis” case studies. The accuracies of binary classifiers are plotted for the “Reactions and synthesis” case studies. Three different LLMs (GPT-J, Llama, and Mistral) and two traditional ML models (random forest (RF) and XGBoost) are compared. The dashed line indicates the zero-rule baseline of 50% accuracy. The table summarizes the number of epochs (for fine-tuning the LLMs), training set size, the best-performing model, and the relative difference between the best LLM and the best “traditional” model. | ||
3.3 Systems and applications
Here, we explore the potential of LLMs to predict the outcomes of different systems and applications.32 Knowing the effect of processing parameters on process performance can help to optimize a system and increase its efficiency. Gaining knowledge of the influence of the experimental conditions on the results can allow us to tailor the process according to our specific objectives.Some MOFs (e.g., ZIFs) have different isomers with the same chemical building blocks. Therefore, it is interesting to investigate whether adding further details on the structure in the prompt will improve learning. Apart from the MOFid and the uptake and diffusion values, Daglar and Keskin33 included 20 additional simulated features of the MOF structures, all of which are numeric and grouped based on their chemical and physical relevance. We used a combined feature vector (per group) to create a prompt for predicting the binary class, i.e., above or below the median, for helium diffusion. In the first experiment, we included the largest cavity diameter, pore limiting diameter, and the pore size ratio, i.e., group A. Secondly, a prompt with the density, pore volume, porosity, and surface area, i.e., group B, was created. For these “single group” experiments, we obtained an accuracy of 68% and 62%, respectively, for groups A and B. Only when we combined groups A and B, thus creating a prompt with seven features, we obtained a performance improvement (73% accuracy). Adding eight extra features related to the elemental composition increased the accuracy to 77%. We tested various models for predicting helium diffusion and saw very comparable results among the three tested LLMs.
The heat of formation of a metal hydride is often used as an indicator for their potential hydrogen storage use. Theoretically, this value is related to the equilibrium pressure. We, therefore, started our experiment by validating if fine-tuned LLMs could capture this relation. The ML-HydPARK dataset created by Witman et al.36 contains 430 metal hydrides with their respective heat of formation and equilibrium pressure (see ESI Note 5.2†). In these initial experiments, we used the median heat of formation as the threshold for the binary classification. We fine-tuned an LLM that could answer the question “Is the heat of formation, and thus its potential for hydrogen storage, of a metal hydride with an equilibrium pressure of 〈value〉 high or low?”. Such models indeed predicted the heat of formation from a material's equilibrium pressure with an accuracy of 76% (GPT-J).
In an alternative approach, we hypothesized that the metal in the material is an indicator of success. We substituted the equilibrium pressure with the elemental formula of the material and repeated the training. Instead of a numeric feature, we now describe the material with a simple textual string, fully exploiting the potential of LLMs. These binary classification models performed significantly better, with an accuracy of 85% (GPT-J). A possible explanation for this increase in performance might be rooted in the augmented information present in the chemical composition of the material. When we combined both the pressure and the chemical formula in the feature vector, we saw a slightly higher accuracy than with the model trained on only pressure values (reaching an accuracy value of 86% with the Llama model), suggesting that the additional chemical information had extra predictive power.
From a practical point of view, a realistic threshold for defining promising materials would be useful. As suggested by the literature, this range (heat of formation values between −40 kJ mol−1 to −20 kJ mol−1) created a slightly unbalanced dataset. Nevertheless, acceptable performances were still achieved with accuracies of 75% (GPT-J).
The dataset contains data on 33 biomass precursors and ten activating agents. We fine-tuned LLMs to predict the BET surface area and CO2 adsorption capacity from the biomass precursor, activation conditions, adsorbent textural properties, and adsorption conditions. An interesting aspect of this example is that, unlike in conventional machine learning models that require conversion to numerical values, the biomass precursor's name and the activating agent's chemical formula were entered as textual strings into the model.
By taking the median as the threshold to classify adsorbent materials, an accuracy of 90% (Mistral, comparable to traditional ML models) was obtained for the prediction of the CO2 adsorption capacity from the precursor activation conditions, activated carbon textural properties, and adsorption conditions, with non-optimized hyperparameters. Since the dataset is smaller (training set of 65 data points), we had to increase the number of finetuning epochs from 30 to 140 to predict the BET surface area from the precursor activation conditions with an accuracy of 76% (Llama). We also found that models trained without the precursor name performed slightly worse than models trained on the full feature vector, indicating that the model could also learn some trends associated with the biomass name. For these tasks, LLMs performance is comparable to that of “traditional” ML models.
Under a more practical classification, we also evaluated a threshold value that would allow us to predict which materials are really ‘good,’ which forced us to reduce the training set to obtain a balanced dataset. Under these conditions, CO2 adsorption capacity was predicted with an accuracy of 82% (GPT-J) by increasing the number of fine-tuning epochs to 100. Likewise, using a smaller dataset, BET surface area was predicted with 75% (GPT-J) accuracy by increasing the number of fine-tuning epochs to 200.
The dataset was split to obtain a balanced binary classification problem. The relatively small size of the dataset forced us to use a maximum training set size of 25 example prompts. The first model with non-optimized hyperparameters showed no predictive power. By increasing the number of epochs from 30 to 100, we obtained an accuracy of 87% (Mistral) for the specific heat transfer surface and 100% for GOR (Mistral), which are values comparable to “traditional” ML models.
We also trained binary classification models using unbalanced datasets to simulate a more realistic case for finding top-performing conditions. The model did not perform better than the random guessing baseline of 80% to predict the specific heat transfer surface, given an accuracy of 80% (GPT-J), but showed acceptable performance in predicting GOR, with 93% accuracy (GPT-J).
A binary classification model trained on 45 example prompts (100 epochs) could predict whether a given sensor was in the top half performing conditions with 89% accuracy (GPT-J, obtaining similar results with other LLMs and “traditional” ML models).
With a rather small dataset of 19 data points, this case study tested the limits on the size of the training set. With slightly optimized hyperparameters by increasing the number of epochs to 120, a predictive accuracy of 71% (GPT-J) was achieved for a binary classification model that was able to predict whether a sensor was stable, i.e., had a response loss between days 5 and 15 of less than 12%.
Fig. 3 shows an overview of the results of the case studies on Systems and applications.
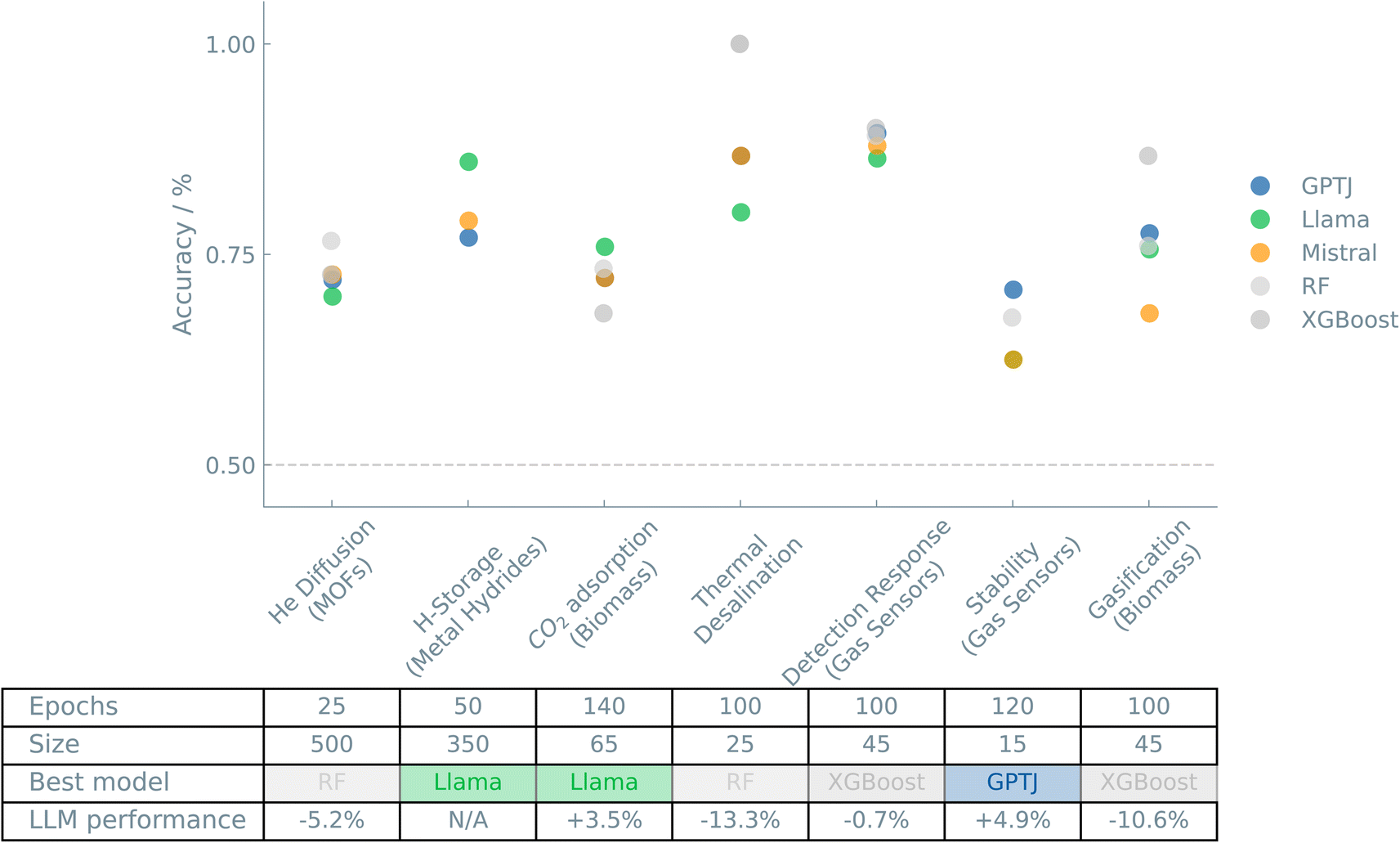 | ||
| Fig. 3 Overview of the “Systems and applications” case studies. The accuracies of binary classifiers are plotted for the “Systems and applications” case studies. Three different LLMs (GPT-J, Llama, and Mistral) and two traditional ML models (random forest (RF) and XGBoost) are compared. The dashed line indicates the zero-rule baseline of 50% accuracy. The table summarizes the number of epochs (for fine-tuning the LLMs), training set size, the best-performing model, and the relative difference between the best LLM and the best “traditional” model. | ||
4 Discussion
In chemistry, we mostly have to deal with a limited amount of data. Hence, it is essential to use leverage in any machine-learning approach applied to chemistry. In an LLM, one leverages the linguistic nuances, patterns, and knowledge captured in correlations harvested from large quantities of internet text. But it is not only knowledge, as is illustrated with the example with the hypothetical polymer notation; in this case the LLMs acts as a flexible probabilistic (n-gram-like) language model. This new source of data, combined with using natural language to interface with the model, makes this approach, potentially, more powerful than machine learning models trained only on conventional data sources.In this work, we tried to obtain some insights into the performance of such LLMs by looking at 22 case studies describing many different systems, ranging from predicting simple thermodynamic properties to device performance. The obvious question is whether it works. In this section, we try to answer these questions in parts.
We must remember that the original corpus of text used to train these models was not specifically curated for chemical questions. It is remarkable that we can create specific solutions for a range of chemical subfields, spanning from a molecular level to reaction kinetics to high-end applications.
In most of these case studies, the LLMs demonstrated their ability to predict basic structure–property relationships. Various cases concerning reactions showcased that LLMs can predict reaction outcomes and yield determination, thereby facilitating reaction optimizations, scoping studies, or catalyst designs. In our applied chemistry cases, the versatility of our approach was further underscored by predicting system parameters, thereby assisting the optimization of real-life chemical processes.
Our results also make clear that the LLM approach works best with a reasonably balanced dataset. However, in practice, one is often interested in the (small) subset of top-performing materials, and we observe that the training set quickly becomes too unbalanced to make sufficiently accurate predictions. The solution to this problem is to start with a model trained on a less narrow window. We typically observe that depending on the size of the dataset, this approach is better than random guessing. So, for problems that are too complex or for which we do not have any intuition, we already gain. If we then collect more data in the region of interest, we can narrow the window, making the model increasingly useful.
Our study provided useful insights into more specific issues related to featurization, feature importance, size of the dataset, and model used.
4.1 Featurization
Can we translate the prediction of the properties of a material or a chemical reaction into a set of simple questions and answers that can be used to fine-tune an LLM? Conventional machine learning requires featurization, i.e., converting the chemical system into a feature vector that quantifies the similarity between systems one wants to compare. Especially in experimental datasets, feature extraction is not as obvious as it might seem. Indeed, many tools and molecular fingerprints exist, but these often pose a burden for non-experts aiming to integrate machine learning into their workflows.As the case studies show, translating a chemical question into a prompt for fine-tuning is straightforward. The primary challenge is choosing how to represent a material or chemical. One can try one of a number of different representations or even use a combination of such representations (see Fig. 4). Standardized notations like SMILES can be exploited to represent chemical structures in LLMs. The readability of SMILES strings makes them convenient for researchers and chemical toolkits to interpret. We show that text-based descriptors like SMILES (see ‘Melting point of molecules’ study), MOFid (see ‘Gas uptake and diffusion of MOFs’ study), or even non-standardized strings (see ‘Adhesion energy of polymers’ and ‘Structure of nanoparticles’ studies) perform well in connecting structural information with physical/chemical properties or reaction outcomes. However, as Alampara et al.40 pointed out, adding structural information does not always give better results.
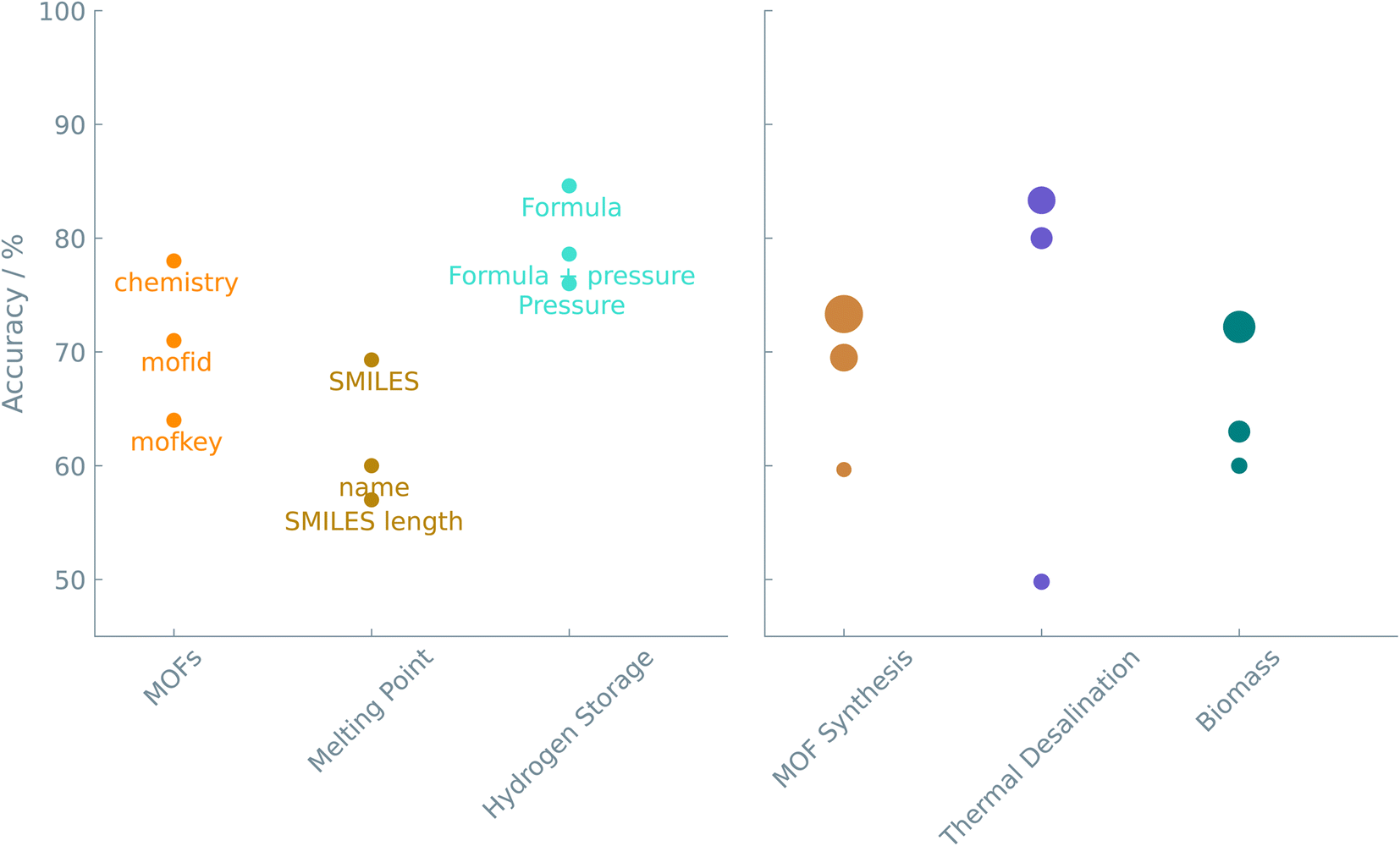 | ||
| Fig. 4 Accuracy of the different representations. The color indicates the particular case study. The same color coding is used in Fig. 5 to compare between all case studies. In the left figure, the annotations are the representations used in the prompt. In the right figure, the size of the circles is related to the number of epochs used. In these examples, the results shown were obtained from fine-tuning the GPT-J model. | ||
Machine learning approaches become even more powerful when dealing with multiple variables. Thus, we extended our prompts with additional data to allow for multi-variable predictions. For instance, in the ‘Hydrogen Storage Capacity of Metal Hydrides’ study, we combined molecular information and equilibrium pressure in one prompt to predict a material's heat of formation. Interestingly, we noticed that this longer prompt outperformed the model that was only trained on the pressure data. Similar trends are also seen in the ‘Gas Uptake and Diffusion of MOFs’ and ‘Photocatalytic Water Splitting Activity of MOFs’ studies. This methodology becomes particularly interesting for predicting the experimental success of a synthetic reaction. Reported reaction protocols are generally described as a combination of textual (e.g., reagents, solvent system) and numeric (e.g., reaction time, temperature) data. In the ‘CO2 Adsorption of Biomass-derived Adsorbents’ study, the dataset consisted of 8 variables used to fine-tune the models. Again, models trained without one of the textual variables performed slightly worse, highlighting the synergy between text-based data and LLMs.
4.2 Feature importance
Feature importance is often an interesting analysis of a trained ML model to know which features carry the most weight in the model. Synthetic chemists also do this daily by asking, for example, “Which parameter do I most likely need to vary to get the desired result?”. Combining natural language and machine learning, i.e., using LLMs, could facilitate this process and create an understandable approach to optimizing chemical systems. By iteratively removing one particular feature in the multi-variable prompt and assessing the accuracy of the resulting fine-tuned model, the influence of the respective parameter on the final objective can be evaluated. We used this approach in the ‘Gas Uptake and Diffusion of MOFs’ dataset. We saw high accuracies when structures were represented as 20 individual features. By removing descriptors, we noticed a drop in accuracies, hinting at the importance of the omitted descriptors on the final predictions.4.3 Size of the dataset
Data quantity, as well as quality, plays an important role in fine-tuning LLMs. We explored datasets ranging from as few as 20 to as many as 5000 entries. Fig. 5 summarises the accuracies we obtained for the different datasets. We see that our approach works well for datasets in the low-data regime. A consistent trend over all experiments suggests that models trained on larger datasets have excellent predictive performance. We typically get accuracies above 80%, allowing us to make balanced training sets if one wants to identify a (much smaller) sub-set of interesting materials.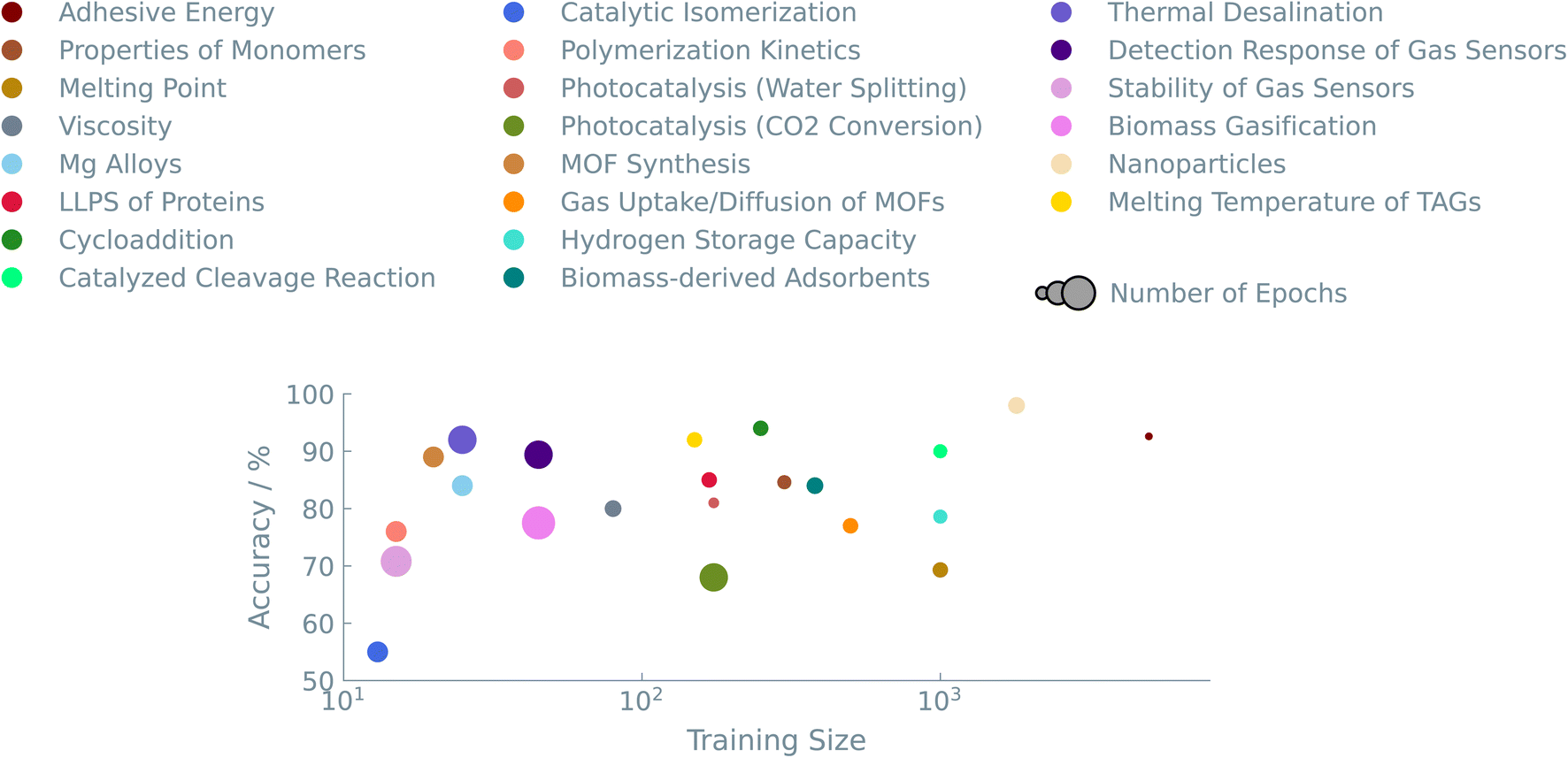 | ||
| Fig. 5 Accuracy as a function of the size of the dataset. The color indicates the case study, and the size of the circles is related to the number of epochs used. The represented results were obtained from fine-tuning the GPT-J model. | ||
On the other end of the plot, i.e., really small datasets (<20 data points), the LLMs initially have difficulties predicting any meaningful output. For experiments with small datasets, we slightly optimized the hyperparameters to increase the performance of the models. By increasing the number of times that the models see the training data, i.e., the number of epochs during training, the performance of these models came close to large dataset models in terms of accuracy (see Fig. 4 (right)).
4.4 Model selection
A last discussion point is dedicated to model selection. As with all machine learning applications, the particular model can significantly impact the predictive outcome. This is also the case in LLMs. The pool of LLMs is increasing at a fast pace. The community is growing, and prominent players in the AI landscape are now creating their own base models. Each of these models is trained on a different corpus of text, each with different parameters. In this work, we selected three models to fine-tune and compare their predictive performance from chemical datasets. In addition, the results were benchmarked against two commonly used “traditional” ML models, i.e., random forest and XGBoost. In Fig. 6, the case studies are plotted with increasing accuracy for each model. Interestingly, there were no significant differences among the different LLMs. We see that, in general, LLMs can compete with traditional ML models. In the majority of the case studies (>80%), the best LLM outperformed traditional ML models. The different types of case studies made it clear that superior non-LLMs are often found in ‘Systems and Applications’ datasets (Fig. 3). One possible explanation could lie in the greater specificity of the problem available data during pre-training of the LLMs. The higher performance of LLMs fine-tuned on SMILES notations, a more prevalent and intuitive representation, supports this hypothesis.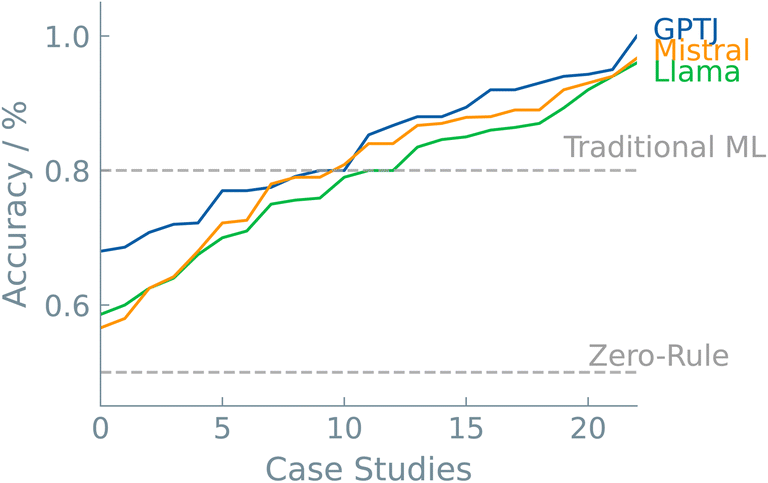 | ||
| Fig. 6 LLM comparison over case studies. The accuracies of all the studied case studies are plotted in increasing order for all three LLM models. Dashed lines show the zero-rule baseline of 50% and the average accuracy of the “traditional” ML models, i.e., 80%. | ||
5 Conclusions
For most, if not all, of these 22 case studies, the LLMs performed (much) better than random guessing and generally better than “traditional” ML models. This is a remarkable result, given that the LLMs were not pre-trained on the bulk of scientific literature. In addition, our effort in making a predictive model using an LLM is modest.We focused on binary classifications that provide a simple ‘yes’ or ‘no’ answer. We see such tasks as a first step; if the LLM did not outperform random guessing, we would conclude that there has been no learning. If we can sufficiently accurately predict such a simple classification, one can proceed to develop a regression model as the next step. Yet, even binary classifications can be useful, especially in experimental settings where a continuous value is often unnecessary to streamline decision-making. An accurate binary classifier can already facilitate various aspects of today's research. For example, ML-based screenings of a particular chemical system can significantly reduce computational resources or experimental work, e.g., “Is it worth doing this experiment?”.
Even a modest accuracy can be helpful if the alternative is random guessing or complex field-specific ML models. Moreover, we also show that these models improve significantly if more data is collected. In this context, we must mention the importance of balanced datasets. In most practical cases, there are many more failures than successful experiments. Hence, in our training, we had to reduce the training set to have a reasonably balanced dataset. If we were to use literature data, we would have the opposite problem. In most, if not all, studies, only successful results are published. Machine learning, like human learning, learns even more from its failures.41 Thus, if we want to take full advantage of the tools explored here, we need to rethink how data are reported.42
In addition to the remarkable performances of the trained models, we also want to stress that natural language in ML models facilitates various aspects of the case studies. By obviating the need to featurize the chemical system, this use of textual descriptors of molecules points to an attractive alternative interface to chemical knowledge suitable for non-experts. Moreover, we noticed that natural language greatly improves scientific interpretation, effective discussions, and communication between different research fields.
Data availability
The datasets and Jupyter Notebooks used in this work are available at https://github.com/JorenBE/GPT-Challenge.Author contributions
The author contributions are provided in the ESI.†Conflicts of interest
K. M. J. and A. S. A. have been contractors for OpenAI (as part of the red teaming network). K. L. S. is a consultant for Transition Bio.Acknowledgements
The research of J. V. H., and B. S. is supported by the Swiss Science Foundation through a Project Funding (214872) and Advanced Grant (216165). M. V. G. and C. P. gratefully acknowledge financial support from the Spanish Agencia Estatal de Investigación (AEI) through Grants TED2021-131693B-I00 (M. V. G. and C. P.) and CNS2022-135474 (M. V. G.), funded by MICIU/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. M. V. G. acknowledges support from the Spanish National Research Council (CSIC) through Programme for internationalization i-LINK 2023 (Project ILINK23047). M. V. G. acknowledges the access granted by the Galician Supercomputing Center (CESGA) to the FinisTerrae III supercomputer, funded by the Spanish Ministry of Science and Innovation, the Galician Government, and the European Regional Development Fund (ERDF), and the access granted by the CSIC to the Drago supercomputer. Parts of the work of K. M. J. were supported by the Carl Zeiss Foundation. S. G., M. G., N. P., B. S., and J. V. H. are partly supported by the USorb-DAC Project through a grant from The Grantham Foundation for the Protection of the Environment to RMI's climate tech accelerator program, Third Derivative. The work of A. S. A. is supported by Novo Nordisk Foundation grant NNF23OC0081359. S. M. M. and M. R. K. work is partly supported by grant number DSI-CGY3R1P16 from the Data Sciences Institute at the University of Toronto. M. A. expresses gratitude to the European Research Council (ERC) for evaluating the project with the reference number 101106377 titled “CLARIFIER” and accepting it for funding under the HORIZON TMA MSCA Postdoctoral Fellowships – European Fellowships. Furthermore, M. A. acknowledges the funding by UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee (EP/Y023447/1; organization reference:101106377). L. L. G., A. A., and T. P. J. K. gratefully acknowledge funding from the European Research Council under the European Union's Horizon 2020 research and innovation program through the ERC grant DiProPhys (agreement ID 101001615) (L. L. G., A. A., T. P. J. K.). The National Institutes of Health Oxford-Cambridge Scholars Program (L. L. G.), the Cambridge Trust's Cambridge International Scholarship (L. L. G.), the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (L. L. G.). The European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013; T. P. J. K.) and the Frances and Augustus Newman Foundation (T. P. J. K.). K. L. S. acknowledges funding from the Schmidt Science Fellowship in partnership with the Rhodes Trust and from St. John's College Research Fellowship programme. F. N. and M. T. thank the Italian MUR for provision of funding through the PRIN 2020 Project doMino (ref 2020P9KBKZ). The research of N. X. H. was supported by the NCCR MARVEL, a National Centre of Competence in Research, funded by the Swiss National Science Foundation (grant number 205602). B. M. acknowledges the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 945363. E. S. acknowledges the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 949229, CryForm).References
- O. Kononova, T. J. He, H. Y. Huo, A. Trewartha, E. A. Olivetti and G. Ceder, Opportunities and challenges of text mining in materials research, Iscience, 2021, 24(3), 102155, DOI:10.1016/j.isci.2021.102155
.
- H. Han and S. Choi, Transfer Learning from Simulation to Experimental Data: NMR Chemical Shift Predictions, J. Phys. Chem. Lett., 2021, 12, 3662–3668, DOI:10.1021/acs.jpclett.1c00578
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach, J. Chem. Theory Comput., 2015, 11, 2087–2096, DOI:10.1021/acs.jctc.5b00099
- J. T. Margraf, Science-Driven Atomistic Machine Learning, Angew. Chem., Int. Ed., 2023, 62(26), e202219170, DOI:10.1002/anie.202219170
-
J. Howard and S. Ruder, Universal Language Model Fine-tuning for Text Classification, 2018 Search PubMed
- C. M. Castro Nascimento and A. S. Pimentel, Do Large Language Models Understand Chemistry? A Conversation with ChatGPT, J. Chem. Inf. Model., 2023, 63, 1649–1655, DOI:10.1021/acs.jcim.3c00285
- K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero and B. Smit, Leveraging large language models for predictive chemistry, Nat. Mach. Intell., 2024, 6(2), 161–169, DOI:10.1038/s42256-023-00788-1
-
A. Mirza, et al., Are large language models superhuman chemists?, 2024 Search PubMed
- K. M. Jablonka,
et al., 14 examples of how LLMs can transform materials science and chemistry: a reflection on a large language model hackathon, Digital Discovery, 2023, 2, 1233–1250, 10.1039/D3DD00113J
- T. Dettmers, M. Lewis, Y. Belkada and L. Zettlemoyer, GPT3.int8(): 8-bit Matrix Multiplication for Transformers at Scale, Adv. Neural Inf. Process. Syst., 2022, 2198 Search PubMed
-
T. Dettmers, M. Lewis, S. Shleifer and L. Zettlemoyer, 8-bit Optimizers via Block-wise Quantization, The Tenth International Conference on Learning Representations, ICLR, 2022 Search PubMed
-
E. J. Hu, Y. Shen, P. Wallis, Z. Allen-Zhu, Y. Li, S. Wang and W. Chen, LoRA: LowRank Adaptation of Large Language Models, International Conference On Learning Representations, 2021 Search PubMed
-
B. Wang and A. Komatsuzaki, GPT-J-6B: A 6 Billion Parameter Autoregressive Language Model, 2021, https://github.com/kingoflolz/mesh-transformer-jax Search PubMed
- A. Dubey et al., The Llama 3 Herd of Models, arXiv, 2024, preprint, arXiv:2407.21783, DOI:10.48550/arXiv.2407.21783.
- A. Q. Jiang; A. Sablayrolles; A. Mensch; C. Bamford; D. S. Chaplot; D. d. l. Casas; F. Bressand; G. Lengyel; G. Lample; L. Saulnier, et al., Mistral 7B, arXiv, 2023, preprint, arXiv:2310.06825, DOI:10.48550/arXiv.2310.06825.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, Chemprop: A Machine Learning Package for Chemical Property Prediction, J. Chem. Inf. Model., 2024, 64, 9–17, DOI:10.1021/acs.jcim.3c01250
- J. Shi, M. J. Quevillon, P. H. A. Valença and J. K. Whitmer, Predicting Adhesive Free Energies of Polymer–Surface Interactions with Machine Learning, ACS Appl. Mater. Interfaces, 2022, 32, 37161–37169 CrossRef PubMed
- L. Schneider, M. Schwarting, J. Mysona, H. Liang, M. Han, P. M. Rauscher, J. M. Ting, S. Venkatram, R. B. Ross, K. J. Schmidt, B. Blaiszik, I. Foster and J. J. de Pablo,
In silico active learning for small molecule properties, Mol. Syst. Des. Eng., 2022, 7, 1611–1621, 10.1039/d2me00137c
-
H. Dieringa and K. U. Kainer, in Springer Handbook of Materials Data, ed. Warlimont, H. and Martienssen, W., Springer International Publishing, Cham, 2018, pp 151–159 Search PubMed
- M. Wolff, T. Ebel and M. Dahms, Sintering of Magnesium, Adv. Eng. Mater., 2010, 12, 829–836, DOI:10.1002/adem.201000038
- M. Wolff, J. G. Schaper, M. R. Suckert, M. Dahms, F. Feyerabend, T. Ebel, R. WillumeitRömer and T. Klassen, Metal Injection Molding (MIM) of Magnesium and Its Alloys, Metals, 2016, 6, 118 CrossRef
- M. Wolff, J. G. Schaper, M. Dahms, T. Ebel, K. U. Kainer and T. Klassen, Magnesium powder injection moulding for biomedical application, Powder Metall., 2014, 57, 331–340, DOI:10.1179/1743290114Y.0000000111
-
Fundamentals of Magnesium Alloy Metallurgy, ed. Pekguleryuz, M. O., Kainer, K. U. and Arslan Kaya, A., Woodhead Publishing Series in Metals and Surface Engineering; Woodhead Publishing, 2013, p iv, DOI:10.1016/B978-0-85709-0881.50012-4
-
Recrystallization and Related Annealing Phenomena, ed. Humphreys, F., and Hatherly, M., Elsevier, Oxford, 2nd edn, 2004, pp 527–540, DOI:10.1016/B978-008044164-1/50021-9
- S. Boeynaems, S. Alberti, N. L. Fawzi, T. Mittag, M. Polymenidou, F. Rousseau, J. Schymkowitz, J. Shorter, B. Wolozin, L. Van Den Bosch, P. Tompa and M. Fuxreiter, Protein Phase Separation: A New Phase in Cell Biology, Trends Cell Biol., 2018, 28, 420–435, DOI:10.1016/j.tcb.2018.02.004
- K. L. Saar, A. S. Morgunov, R. Qi, W. E. Arter, G. Krainer, A. A. Lee and T. P. Knowles, Learning the molecular grammar of protein condensates from sequence determinants and embeddings, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2019053118 CrossRef CAS PubMed
- N. Alampara, S. Miret and K. M. Jablonka, MatText: Do Language Models Need More than Text & Scale for Materials Modeling?, arXiv, 2024, preprint, arXiv:2406.17295, DOI:10.48550/arXiv.2406.17295.
- A. S. Anker, E. T. S. Kjær, E. B. Dam, S. J. L. Billinge, K. M. O. Jensen and R. Selvan, Characterising the Atomic Structure of Mono-Metallic Nanoparticles from X-Ray Scattering Data Using Conditional Generative Models, ChemRxiv, 2020, preprint, DOI:10.26434/chemrxiv.12662222.v1.
- E. T. S. Kjær, A. S. Anker, M. N. Weng, S. J. L. Billinge, R. Selvan and K. M. O. Jensen, DeepStruc: towards structure solution from pair distribution function data using deep generative models, Digital Discovery, 2023, 2, 69–80, 10.1039/d2dd00086e
- Z. Tu, T. Stuyver and C. W. Coley, Predictive chemistry: machine learning for reaction deployment, reaction development, and reaction discovery, Chem. Sci., 2023, 14, 226–244, 10.1039/D2SC05089G
- M. Cordova, M. D. Wodrich, B. Meyer, B. Sawatlon and C. Corminboeuf, Data-Driven Advancement of Homogeneous Nickel Catalyst Activity for Aryl Ether Cleavage, ACS Catal., 2020, 10, 7021–7031, DOI:10.1021/acscatal.0c00774
- M. R. Dobbelaere, P. P. Plehiers, R. Van de Vijver, C. V. Stevens and K. M. Van Geem, Machine Learning in Chemical Engineering: Strengths, Weaknesses, Opportunities, and Threats, Engineering, 2021, 7, 1201–1211, DOI:10.1016/j.eng.2021.03.019
- H. Daglar and S. Keskin, Combining Machine Learning and Molecular Simulations to Unlock Gas Separation Potentials of MOF Membranes and MOF/Polymer MMMs, ACS Appl. Mater. Interfaces, 2022, 14, 32134–32148 CrossRef CAS
- B. Bucior, A. Rosen, M. Haranczyk, Z. Yao, M. Ziebel, O. Farha, J. Hupp, J. Siepmann, A. Aspuru-Guzik and R. Snurr, Identification Schemes for Metal–Organic Frameworks To Enable Rapid Search and Cheminformatics Analysis, Cryst. Growth Des., 2019, 19(11), 6682–6697, DOI:10.1021/acs.cgd.9b01050
- N. Klopčič, I. Grimmer, F. Winkler, M. Sartory and A. Trattner, A review on metal hydride materials for hydrogen storage, J. Energy Storage, 2023, 72, 108456, DOI:10.1016/j.est.2023.108456
- M. Witman, S. Ling, D. M. Grant, G. S. Walker, S. Agarwal, V. Stavila and M. D. Allendorf, Extracting an Empirical Intermetallic Hydride Design Principle from Limited Data via Interpretable Machine Learning, J. Phys. Chem. Lett., 2019, 11, 40–47, DOI:10.1021/acs.jpclett.9b02971
- H. Mashhadimoslem, M. Vafaeinia, M. Safarzadeh, A. Ghaemi, F. Fathalian and A. Maleki, Development of Predictive Models for Activated Carbon Synthesis from Different Biomass for CO2 Adsorption Using Artificial Neural Net-works, Ind. Eng. Chem. Prod. Res. Dev., 2021, 60, 13950–13966, DOI:10.1021/acs.iecr.1c02754
- N. Darre and G. Toor, Desalination of Water: a Review, Curr. Pollut. Rep., 2018, 4, 1–8, DOI:10.1007/s40726-018-0085-9
- M. V. Gil, K. M. Jablonka, S. Garcia, C. Pevida and B. Smit, Biomass to energy: a machine learning model for optimum gasification pathways, Digital Discovery, 2023, 2, 929–940, 10.1039/d3dd00079f
- N. Alampara, S. Miret, K. M. Jablonka, MatText: Do Language Models Need More than Text & Scale for Materials Modeling?, arXiv, 2024, preprint, arXiv:2406.17295, DOI:10.48550/arXiv.2406.17295.
- S. M. Moosavi, A. Chidambaram, L. Talirz, M. Haranczyk, K. C. Stylianou and B. Smit, Capturing chemical intuition in synthesis of metal-organic frameworks, Nat. Commun., 2019, 10, 539, DOI:10.1038/s41467-019-08483-9
- K. M. Jablonka, L. Patiny and B. Smit, Making the collective knowledge of chemistry open and machine actionable, Nat. Chem., 2022, 14, 365–376, DOI:10.1038/s41557-022-00910-7
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04401k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |