
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective, regioselective, and positionally selective fluorogenic stapling of unprotected peptides for cellular uptake and direct cell imaging†
Naysilla L.
Dayanara
 ,
Juliette
Froelich
,
Pascale
Roome
and
David M.
Perrin
*
,
Juliette
Froelich
,
Pascale
Roome
and
David M.
Perrin
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T1Z1, Canada. E-mail: dperrin@chem.ubc.ca
First published on 5th November 2024
Abstract
Peptide stapling reactions represent powerful methods for structuring native α-helices to improve their bioactivity in targeting protein–protein interactions (PPIs). In light of a growing need for regio- and positionally selective stapling methods involving natural amino acid residues in their unprotected states, we report a rapid, mild, and highly chemoselective three-component stapling reation using a class of molecular linchpins based on 2-arylketobenzaldehydes (ArKBCHOs) that create a fluorescent staple, hereafter referred to as a Fluorescent Isoindole Crosslink (FlICk). This methodology offers positional selectivity favouring i, i + 4 helical staples comprising a lysine and cysteine, in the presence of competing nucleophiles on unprotected peptides. In our efforts to further validate this chemistry, we have successfully shown in vitro cytotoxicity of a FlICk-ed peptide (IC50 = 5.10 ± 1.27 μM), equipotent to an olefin-stapled congener. In harnessing the innate fluorescence of the thiol-isoindole, we report new blue-green fluorophores, which arise as a consequence of stapling, with appreciable quantum yields that enable direct cellular imaging in the assessment of cell permeability, thus bridging therapeutic potential with cytological probe development.
Introduction
Peptides are valuable drug leads for recognizing receptors implicated in cancer, inflammation and immune checkpoint therapy, and for disrupting protein–protein interactions (PPIs). While linear peptides recognize undruggable targets, their lack of defined structure, particularly in short peptides, accounts for low affinity and poor humoral stability. One common chemical approach to address these deficiencies is peptide stapling, which pre-organizes peptide secondary structure, thereby enhancing affinity, rigidity, stability, and even membrane permeability. In the past decade, numerous reports have disclosed new stapling methods1–41 including, but not limited to Cu-catalyzed alkyne coupling,1–3 alkyne-amination,4 triazole formation,5–8 Grubbs Ru-catalyzed cross-metathesis,9–14 Pd-catalyzed arylations of cysteine-thiols15 and amines,16 thio-etherifications,17–30 thioimidation,31,32 Lys–Lys stapling,33 triazine tethering,34 cysteine alkylation,35,36 iodonium-mediated thiol alkynylation,37 indole-arylations,25 oxadiazole synthesis,38 Petasis-borono–Mannich reactions,39 and isoindole synthesis.40,41 As stapling typically occurs late stage, it is widely appreciated that exceptional chemoselectivity is often required. This is readily achievable with non-canonical amino acid residues.2,3,10,13,25,42,43 With over 150 peptides in clinical trials as of 2023, new stapling methods represent platform technologies that are expected to improve peptide druggability and pharmacological performance as seen in recent examples,44,45 and as reviewed.46–60 The appearance of numerous recent reviews on stapling methods signifies a collective interest in this topic. Of further interest are fluorogenic stapling reactions, where emergent fluorescence signals reaction progress and generates a fluorescent peptide that allows for direct visualization of its association with biological targets,40,41,61,62 thereby obviating the need to append a fluorophore that may eventually alter target affinity. For example, Waser and coworkers61 and Zhu and coworkers,63 disclosed metal-catalyzed tryptophan crosslinking with a non-canonical amino acid reaction partner (Fig. 1a).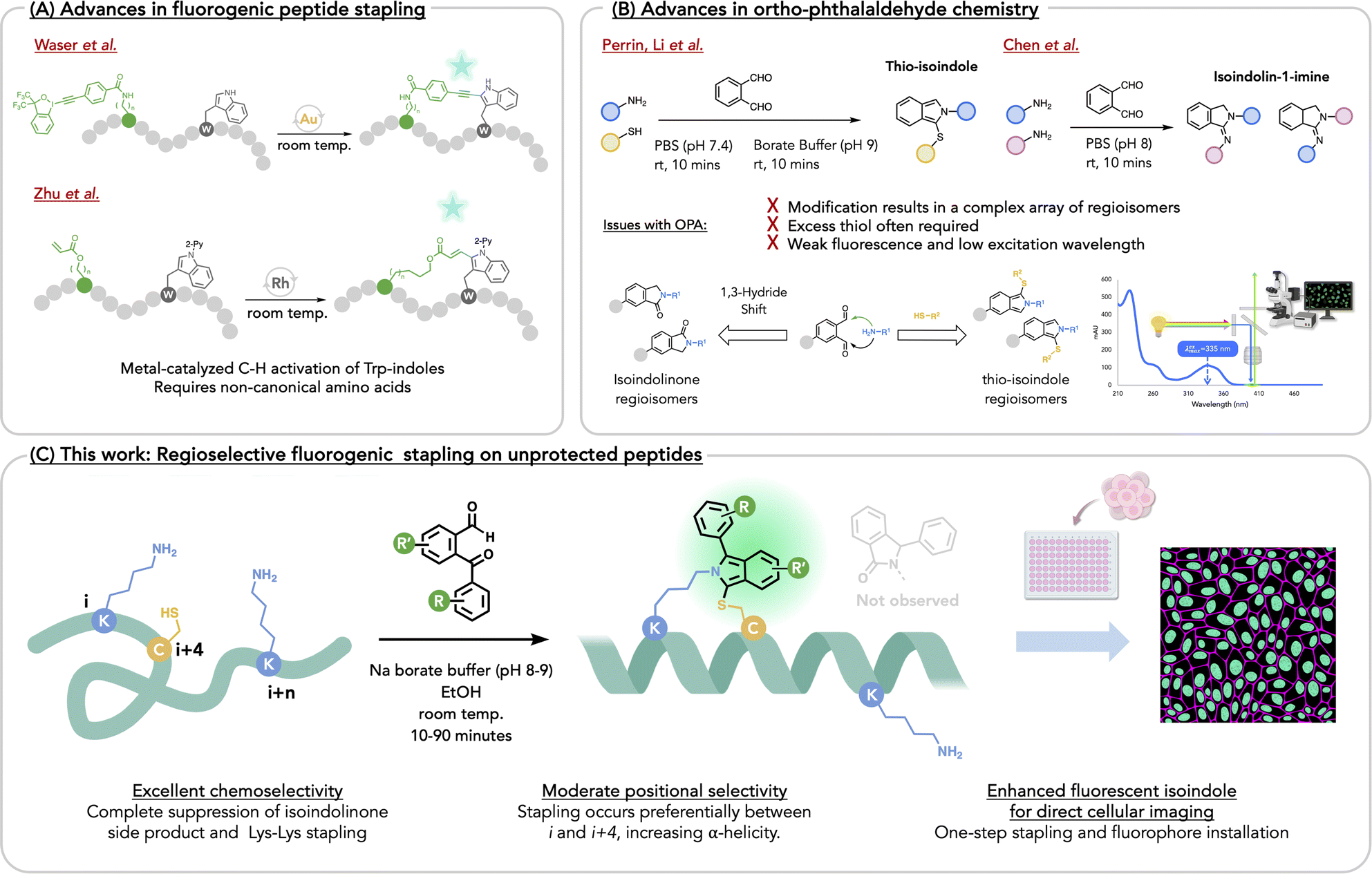 | ||
| Fig. 1 (A) Recent fluorogenic peptide stapling, utilizing metal-catalyzed C–H activation of Trp with non-canonical amino acid reaction partner. (B) Three-component and two-component ortho-phthalaldehyde chemistry suffers from complex regioisomer formation and isoindolinone side-product resulting in undesired N-capped residues. (C) This work: regioselective and positional selective isoindole α-helix stapling with 2-arylketobenzaldehydes (ArKBCHOs) with improved fluorescence and chemoselectivity, allowing for direct cellular imaging. | ||
In line with these elegant metal-mediated approaches, there is a growing interest in developing metal-free fluorogenic stapling reactions involving unprotected, natural amino acids. Such may result in staples whose lengths compare to well-known olefinic ones used to reinforce α-helices. Recently, Li's group41 and ours,40 examined fluorogenic stapling by exploiting a three-component reaction of Lys, Cys, and ortho-phthalaldehyde (OPA), resulting in a thio-isoindole (Fig. 1b). In 2019, Li and coworkers stapled small peptides at Lys–Cys with OPA and explored post-stapling reactions of the isoindole staple with electrophiles that allowed for enhanced stability and added functionality. Our group independently investigated the same stapling methodology and, in turn, probed its innate fluorescence, hereafter referring to the reaction as Fluorescent Isoindole Crosslinking (FlICking). Others extended this reaction to other heteroaryl-1,2-dialdehydes and acetylbenzaldehydes, with applications in peptide display and DNA-encoded technologies.64
More recently, Li and coworkers demonstrated the use of guanidine to suppress competing amine-only reactions that yield an isoindolinone or an isoindolin-1-imine, that arises from addition of one or two lysines, which also has been expertly investigated by Chen and coworkers.65,66 (Fig. 1b). Initially, we had designed a series of substituted OPAs that gave fluorescent isoindoles with appreciably higher quantum yields (ϕ = 31%), while the nitro-OPAs gave visibly orange, although non-fluorescent isoindoles. Yet, in all of these examples, the amine nucleophile adds indiscriminately to either aldehyde, giving a near-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of difficult-to-separate isoindole regioisomers, thereby limiting several important applications e.g. bioconjugation, drug development, which would benefit from single regioisomers.
:1 ratio of difficult-to-separate isoindole regioisomers, thereby limiting several important applications e.g. bioconjugation, drug development, which would benefit from single regioisomers.
Given the intractable regioisomerism that arises when using modified OPAs for isoindole stapling, we appreciated that replacing one of the aldehydes with a ketone would afford a simple solution to this problem.67,68 Following early reports on the use of 2-phenylketobenzaldehyde and benzoyl-quinoline-carboxaldehyde as analytical reagents for quantifying primary amines,67,69–71 we appreciated that ketone arylation would extend π-conjugation of the FlICk, thereby increasing the quantum yield and red-shifting the excitation λmax for the added advantage of greater compatibility with cell biology applications. With a solution to the regioisomer problem in view and scant evidence in the literature as to the use of 2-arylketobenzaldehydes (ArKBCHOs) as chemoselective peptide stapling agents, we sought an expansion of this chemical space for photophysical modulation and chemical functionalization that would provide many new stapling reagents for further applications (vide infra). Herein we report a robust methodology for the synthesis of a large family of ArKBCHOs as potential fluorogenic stapling reagents. With 35 ArKBCHOs prepared, including heteroaryl congeners synthesized, converted to corresponding isoindoles, and characterized with respect to their photophysical properties, we identified a number of stapling agents that afford bright, blue-green, fluorescent isoindole staples, while others provide visibly yellow-to-red isoindole chromophores.
Unexpectedly, ArKBCHOs appear to show an near-exclusive chemoselectivity for the three-component reaction with Lys–Cys, with suppression of the isoindolinone by-product. In the context of peptide stapling, select exemplars show good-to-excellent positional selectivity for late-stage stapling of unprotected peptides in the presence of a competing side-chain amine or thiol nucleophile. Such allows the construction of stapled α-helical peptides that show potent cytotoxicity. We further demonstrate the utility of this chemistry by using FlICk-stapled α-helices for direct fluorescent cell imaging to qualitatively assess cellular uptake (Fig. 1c).
Results and discussion
Using a Pb-mediated rearrangement,72–75 we constructed an array of substituted ArKBCHOs 6a–6ai (also Scheme S4†), from a number of modularly assembled substituted benzylidene hydrazides 5a–5ai. This synthetic route proved to be both simple and robust, lending itself to considerable functional group tolerance and enabling the rapid assembly of a large array of ArKBCHOs, including several unknown compositions, from simple and readily accessible starting materials.Commercially available carboxylic acids, 1a–1o (Scheme S1†), were converted to their corresponding hydrazides 2a–2o, which were then refluxed with salicylaldehyde derivatives 4a–4g (Scheme S2†) to afford benzylidene hydrazides 5a–5ai (Scheme S3†). Reaction with Pb(OAc)4 yielded 2-arylketobenzaldehydes (ArKBCHOs) 6a–6ai in moderate to high yields (Scheme S4†). In our initial choice of substituents, compounds 6b–6ab were designed to assess whether the nature of substituents, i.e., electron-withdrawing and electron-donating groups (EWG, EDG, respectively) and their position around the aromatic ring would perturb fluorescence.
We also explored “push–pull” pairings of EDGs and EWGs (6i, 6o–6u) for extending the π-conjugation in order to red-shift the excitation wavelength. As well, we synthesized 6af–6ai to demonstrate reaction compatibility with electron-deficient and electron-rich heterocycles, e.g., pyridyl and indoyl moieties. Notably, this reaction manifold proved compatible with several substituents, including azide, alkyne, nitrile, bromine, and iodine, which were chosen for their potential to afford additional functionalization. To fully characterize the photophysical properties of this family of isoindoles, we reacted 6a–6ai with model substrates: N-acetyl-cysteine and L-Arg-OH. A limited screening of reaction conditions was undertaken to optimize the conversion of isoindoles owing to the different solubility and reactivity of ArKBCHOs relative to OPA.
To test this, we reacted 6aa under a 1:1:1 stoichiometric ratio to optimize the conversion to isoindole 7aa (Table 1). Hence, ArKBCHO 6aa was dissolved at 50 mM in DMSO, which enhanced the solubility of 6aa in the aqueous reaction buffer. To determine the conversion to the desired isoindole from a 1:1:1 mixture of starting materials, we established a baseline run where all three reagents were added into stirring Na-borate buffer (pH 9) and a sample of this reaction was analysed by HPLC (<2 minutes following mixing of all reaction components). Rightly, this showed 0% conversion and thus provided a baseline chromatogram against which percent conversion to 7aa could be referenced (Table 1, entry 1). Following a standard protocol for intermolecular isoindole formation, the thiol was added in excess, and the reaction was stirred for 30 minutes.
|
|
|||
|---|---|---|---|
| Entry | NAc-Cys | Co-solvent:solvent |
% conv. to 7aad |
| a Baseline run was obtained by injecting the crude reaction mixture immediately after all reagents were added, amounting to <2 minutes of reaction time. b Prepared as 50 mM solution in H2O. c Prepared as 50 mM solution in H2O (0.1% formic acid). d % conversion was determined by HPLC peak integration of 7aa relative to peaks observed in the baseline run (entry 1). | |||
| 1a | 1b eq. | Na borate buffer pH 9 (aq.) | 0% |
| 2 | 3b eq. | Na borate buffer pH 9 (aq.) | 0% |
| 3 | 1b eq. | CH3CN:Na borate buffer pH 9 (aq.) |
0% |
| 4 | 1c eq. | DMSO:Na borate buffer pH 9 (aq.) |
39% |
| 5 | 1c eq. | EtOH:Na borate buffer pH 9 (aq.) |
66% |
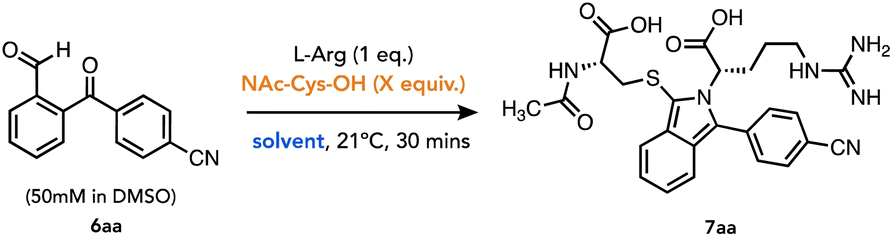
Unfortunately, in this initial set of conditions, we did not observe the emergence of a peak corresponding to an isoindole (Table 1, entry 2). Whereas DMSO enhanced the solubility of 6aa, the reaction proved to be slow, reaching an apparent maximal conversion of ca. 39% in 30 minutes while also becoming viscous and ultimately turbid, giving a fine, yellow precipitate, owing to the poor solubility of the isoindole product. These initial observations led us to add co-solvents e.g. CH3CN, DMSO, EtOH (Table 1, entries 3, 4, 5, respectively), in the hopes of dissolving the precipitate. Ultimately, EtOH improved the conversion from 39% to 66% without the noted turbidity (Table 1, entry 5). To our excitement, this reaction progressed under 1:1:1 stoichiometric ratio under these conditions.
To confirm the successful formation of 7aa, we completed characterization by NMR spectroscopy with key correlations depicted in Fig. 2 (for full spectral characterization, discussion, and peak assignments, see Fig. S1–S7 and Table S2 in ESI†). With optimized conditions established, we embarked on synthesizing and characterizing isoindoles 7a–7ai (Fig. 3). Isoindoles generated from this array generally show λex-max values ca. 360 nm, with varying quantum yields. Halogen situation has little effect on λex-max whilst heavier halogens (e.g. Br and I) attenuate fluorescence. Notably, fluoro-isoindole 7m exhibited blue fluorescence (ϕF = 45%) with its hypsochromic excitation λex-max of ca. 350 nm, making it sub-optimal for biological imaging applications. Interestingly, the introduction of electron-donating groups on the isoindole core imparted bathochromic shifts and significantly affected emission profiles. For example, methoxy-substituted isoindoles, 7g, 7i–l, and 7v, showed excitation maxima ca. 360 nm. Of this series, 7v (MeO-substituted), showed a high quantum yield (ϕF = 46%) with blue fluorescence. Replacement of the methoxy group with a dimethylamino-group (7w) red-shifted the emission further to give a bright green fluorophore (λex-max 494 nm, ϕF = 46%). With these results, we selected 6w as a choice fluorogenic stapling agent owing to its good quantum yield, red-shifted λex-max, and emission wavelength comparable to that of commercially available green fluorophores.76,77
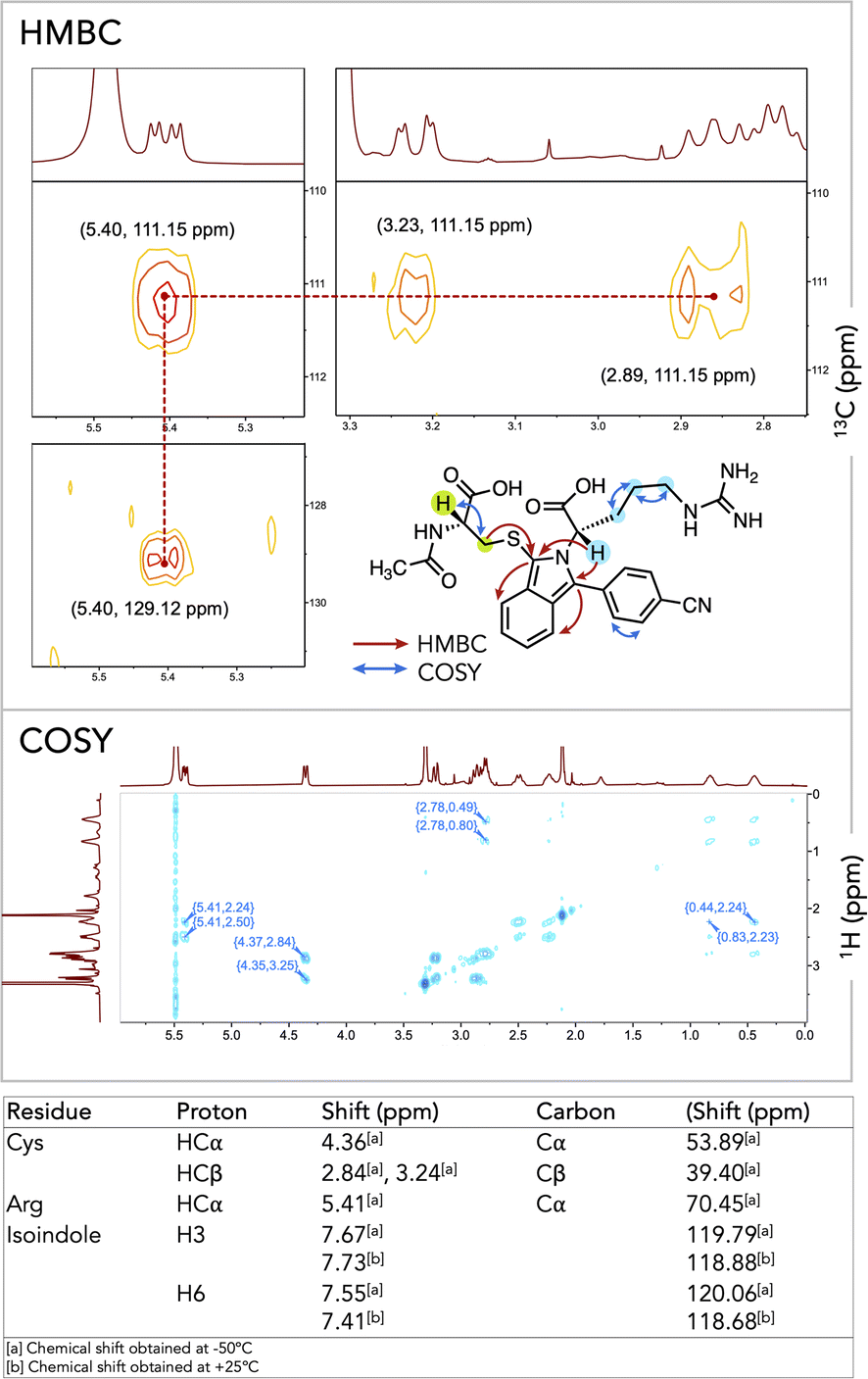 | ||
| Fig. 2 HMBC signals showing key correlation in thio-isoindole 7aa and COSY spectrum showing aliphatic proton correlation of Cys and Arg. | ||
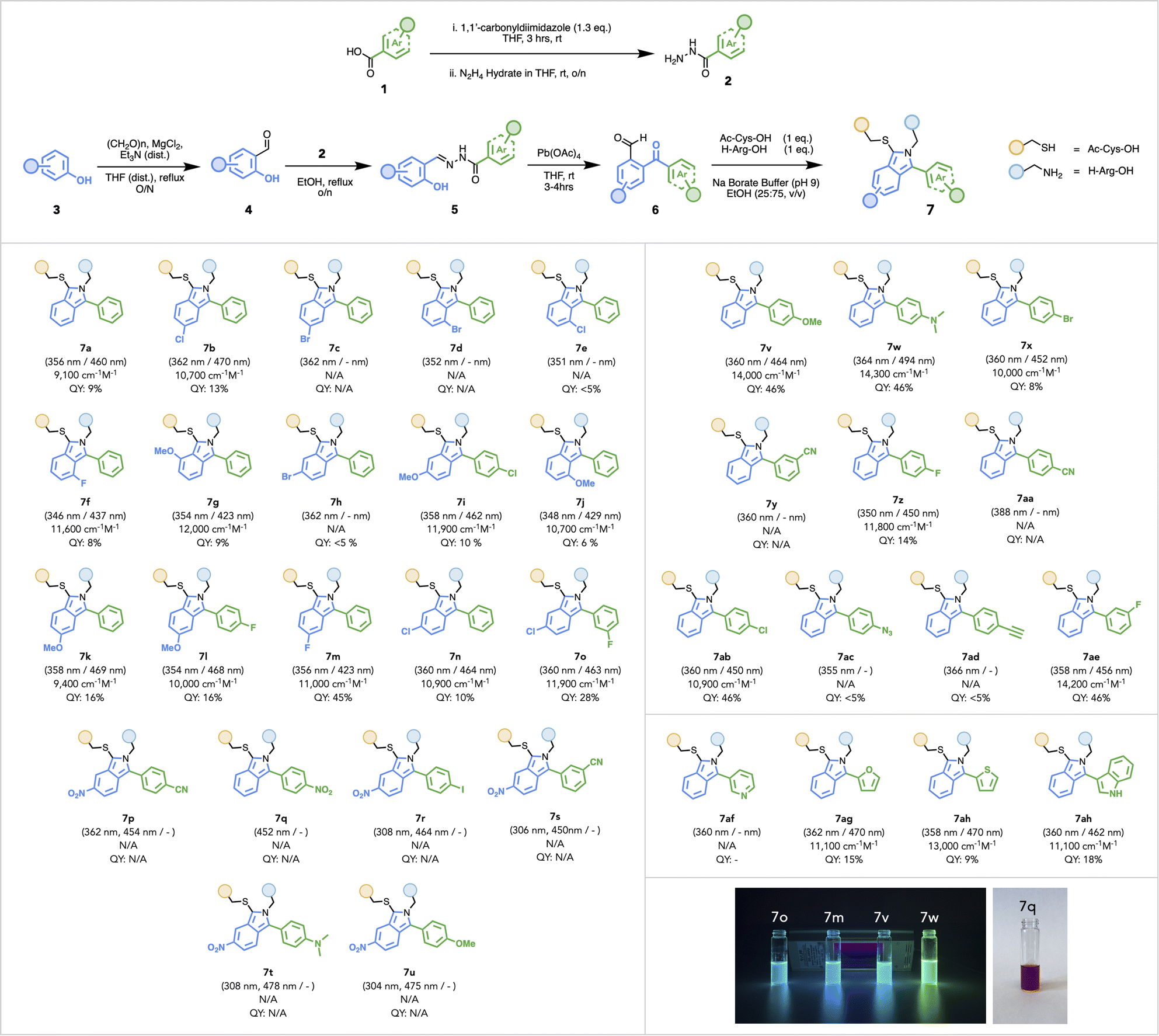 | ||
| Fig. 3 Synthesis of an array of regioselective thio-isoindoles 7a–7ai. Extinction coefficient was measured at respective λex-max N/A for compounds that did not show any fluorescence observed under 365 nm hand-held UV lamp, or where QY values were found to be <5%. | ||
The conventional three-component reaction with ortho-phthalaldehyde demands the addition of amine and thiol, for which excess thiol is typically added to suppress a competing two-component, amine-only reaction.10 Revisiting the intermolecular 1:1:1 ratio in converting 6aa to isoindole 7aa, we tested the chemoselectivity of 6w in a similar manner. Studies also showed that introducing the thiol in solution ahead of the amine does not alter the reaction equilibrium as it progresses towards the thiol-isoindole product. Nevertheless, we tested the chemoselectivity of 6w under conditions that would favor the isoindolin-1-imine formation.65,66,78
Hence, H-Trp-OH (2.0 eq.) was added to a solution of 6w in borate buffer pH 9 and EtOH (10%) and stirred for 1 hour at 19–21 °C (Table 2, entry 1). We then monitored the reaction progress by observing the signal at 230 nm, given that all of our chromophores absorb at this wavelength. (Fig. S46–48† with peak analysis by MS). Under this condition, we saw no formation of amine-only adducts 9a and 9b. We then treated 6w with larger excess H-Trp-OH (5.0 eq.) and stirred it for 5 hours (Table 2, entry 2) and only trace amounts of material (16%) corresponding to either imine 10a or isoindolinone 10b was formed. Given this low yield, full characterization was not pursued, yet subsequent experimentation hinted at the formation of 10a (vide infra).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Equiv. | Reaction time | Conversiona (%) | ||||
| Trp | Thiol | ||||||
| 8 | 9a | 9b | 10a/10b | ||||
| a Percent conversion is calculated as the area under the curve of the chromatogram observed at 230 nm relative to 1 equivalent of 6w. Other peaks observable at this wavelength is the DMSO co-solvent and this was not included in peak integration calculations. H-Trp (2.0 equivalents) also not included in percent conversion calculation because it remained in excess. All traces were obtained using the same HPLC method. Solvent A: 40 mM NH4OH/HCOOH pH 8, solvent B: MeCN. 5–100% over 12 minutes. | |||||||
| 1 | 2.0 | — | 1 hour | 0 | 0 | 0 | 0 |
| 2 | 5.0 | — | 5 hours | 0 | 0 | 0 | 16 |
| 3 | 2.0 | 1.3 | 5 min | 62 | 0 | 0 | 0 |
| 4 | 2.0 | 1.3 | 10 min | 81 | 0 | 0 | 0 |
| 5 | 2.0 | 1.3 | 1 hour | 90 | 0 | 0 | 0 |
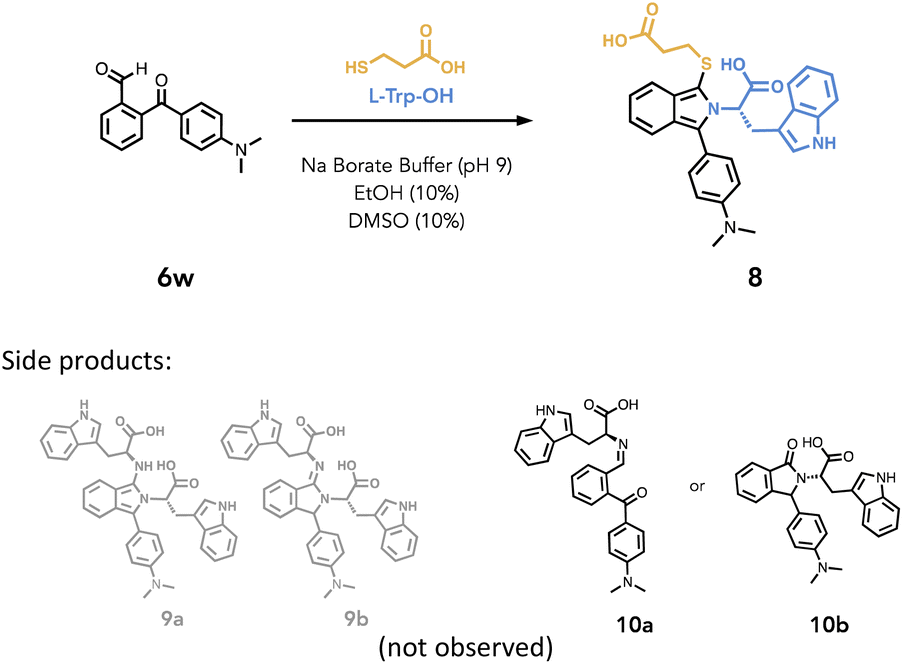
Next, the reaction of 6w with H-Trp-OH (2.0 eq.) and mercaptopropionic acid (1.3 eq.) resulted in good conversion to isoindole 8 (62%) within 5 minutes (Table 2, entry 3) and fluorescence was readily observed under hand-held UV lamp at 365 nm. Furthermore, having 6w reacting with H-Trp-OH for 1 hour prior to the addition of mercaptopropionic acid still resulted in the formation of 8 (81%) within 10 minutes (Table 2, entry 4). This observation strongly suggests that imine 10a rather than isoindolinone 10b is the only intermediate formed in this reaction as it is capable of reacting with the Cys-thiol to give the isoindole. Furthermore, this work suggests that under mildly basic conditions, the ketone does not readily undergo rearrangement to the lactam via 1,3-hydride shift, which often prevails in OPA condensation, leading to an isoindolinone.79,80 This contrasts with previous studies on the reaction of 2-benzoylbenzaldehyde with excess amine in acidic conditions, that favoured the isoindolinone.80 While these results demonstrate greater chemoselectivity for a three-component reaction with amine and thiol using 6w, we posit that similar selectivity likely extends to a number of other ArKBCHOs featured in this study however we did not confirm this to be the case for all ArKBCHOs reported.
Equipped with these results, we then set out to explore the scope and limitations of using 6w for stapling linear unprotected peptides with competing Lys–Cys pairs along the peptidic backbone. Positionally selective stapling within a peptide sequence often demands strategic sidechain protection or selective replacement of certain amino acids with those that can react via orthogonal stapling reactions,41 a classic example of which is the Ru-catalyzed ring-closing metathesis (RCM) reaction that results in olefinic staples. In designing FlICkbased helical staples, we looked to such antecedent examples to guide our designs.
To wit, Verdine and co-workers11,12,81 applied Grubbs' pioneering RCM to peptides by incorporating alkene-modified amino acids via SPPS and spacing them within one (i, i + 4) or two (i, i + 7) helical turns from each other.7 They extensively studied the optimal staple length that resulted in increased helicity and determined that a single helical turn could be enforced via a staple of at least 8–9 atoms.82,83 Other stapling methodologies have also capitalized on this atom count to favour highly stable α-helical turns.18,84 Coincidentally, a FlICk between Lys and Cys also satisfies this atom-count demand. Moreover, molecular modelling predicted that a FlICk could mimic the rigidity that is otherwise imparted by an olefinic staple (Fig. S85 and S86†). Hence, we explored the regioselective stapling of an unprotected peptide at positions i, i + 4 in the presence of a competing Lys or Cys residue situated i + n along the sequence.
We hypothesized that positionally selective FlICking would occur owing to the induced proximity caused by the propensity of peptides to adopt helices. Once stapled, the peptide's helicity could be gauged by circular dichroism (CD). Conversely, an “undesired” FlICk between i and i + n will result in lower apparent helicity relative to the i, i + 4 FlICked regioisomer. For this, we focused our study on the use of 6w owing to the aforementioned chemoselectivity and photophysical attributes and applied it to a small library of peptides 11–18, with varied positioning of Lys and Cys (Fig. 4).
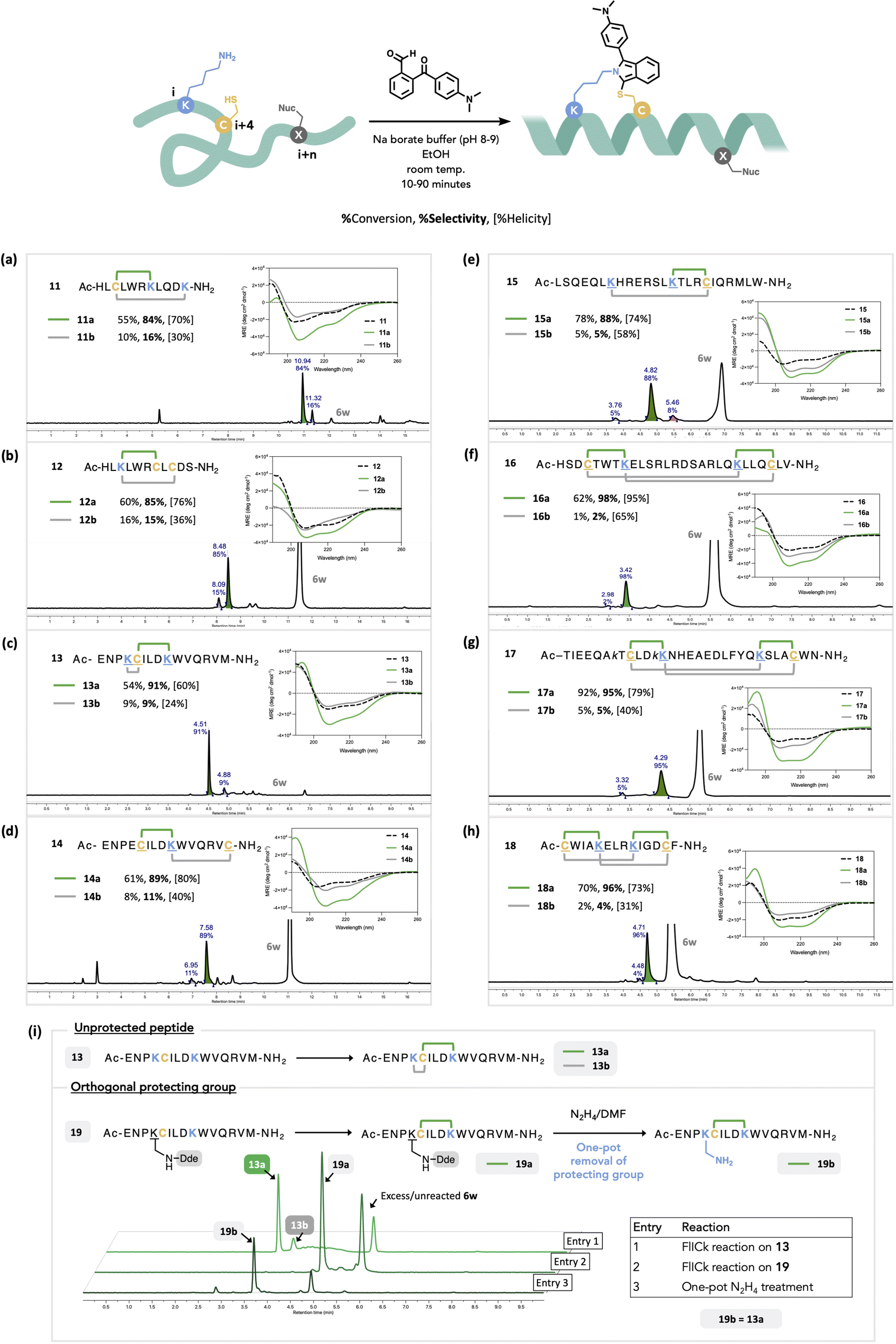 | ||
| Fig. 4 Scope and limitations of intramolecular FlICk reaction with 2-ketobenzaldehyde 6w to synthesize a regioselective staple on a linear peptide (a–h). The amount of 6w used ranged from 1.0–5.0 equiv., see ESI† for each entry. Each linear peptide afforded two stapled regioisomers a (i, i + 4; in green) and b (i, i + n; in grey), determined by relative % helicity based on given CD data. See ESI† for % helicity of the linear precursors. The ratio of respective isomers was determined by peak integration of crude LC trace observed at 360 nm, pertaining to the isoindole staple, while % conversion was determined by LC trace observed at 290 nm. For other wavelengths and details on each HPLC method, see ESI.† Excess and unreacted 6w is shown on the LC trace. (i) Orthogonal protecting group strategy in proving the positional selectivity of the FlICking reaction with 6w. | ||
Each sequence was derived from well-studied peptide inhibitors of PPIs, wherein all previously reported staples had been positioned in the hydrophobic region of the peptide.85 To expand this study, we started with short, 11-residue peptides 11 and 12, derived from Steroid Receptor Coactivator-2.86 Accordingly, these were designed with a competing amine or thiol nucleophile (Lys or Cys) placed distally to the anticipated i, i + 4 staple at positions i + 6 or i + 8. Peptide 11 contained two lysines, at i + 4 and i + 8, each of which could form imine anchor points prior to the addition of the thiol at the ith position (Fig. 4a). Intramolecular competition on 11 afforded good regioselectivity (84:16) with the major product 11a showing higher helicity as measured by CD (70%). This product was presumed to be stapled at i, i + 4, enforcing the characteristic α-helical turn. The minor product 11b showed relatively lower helical content (30%), which was presumed to be the i, i + 8 staple. We also tested the competition of two Cys-thiols; in peptide 12, the resulting staple could occur between i + 4 or i + 6 (Fig. 4b). This poses a greater inherent challenge as the competing reaction site is now situated two residues closer relative to that seen in peptide 11. Nevertheless, two regioisomers, 12a (85%) and 12b (15%) still formed with good positional selectivity, with the major isomer 12a showing a higher percent helicity, whilst minor isomer 12b showed helicity comparable to the linear precursor 12. The positional selectivity of this method is further corroborated by MS/MS, NMR analysis, and resynthesis of 12 employing orthogonal cysteine protection (disulfide), followed by FlICk stapling and then TCEP-mediated deprotection in one pot (Fig. S65D–L†). Next, we contemplated whether FlICk formation is sensitive to competing nucleophiles placed proximally with respect to the preferred i, i + 4. Noting a peptide's propensity to adopt a helical structure particularly for longer sequences, in shorter one (<15 residues), especially in a predominantly aqueous environment, helicity is usually transitory.87 Hence, we evaluated competition by a proximal nucleophile using longer peptides and thus synthesized 13, a 15-mer peptide that carries a cysteine at the ith-position along with two lysines, at positions i + 1 and i + 4, to compete for the same thiol (Fig. 4c). This peptide was designed to ask whether a FlICk at i, i + 1 will triumph over one at i, i + 4 owing to the residue proximity of Cys–Lys at i, i + 1. To our delight, we observed excellent positional selectivity (91:9) favouring 13a (i, i + 4) that possessed 60% helicity as measured by CD. By contrast, the minor isomer 13b showed CD characteristics almost identical to that of the linear starting material 13. This result showed that the inherent potential for helicity nucleates stapling to favour a FlICk at i, i + 4 and the nascent helicity of longer peptides improves selectivity for i, i + 4 stapling. Based on previous reports on helical peptides stapled by RCM, we then synthesized peptides 1488 and 15,89 both of which were FlICked with excellent positional selectivity to give products of high helicity suggestive of the i, i + 4 staple (Fig. 4d and e). Finally, we demonstrated the feasibility of installing multiple FlICks using peptide precursors 16–18 with near exclusivity for i, i + 4 staples, which compare favorably with traditional staples.90–92 All major regioisomers for this set of peptides (16a–18a) were obtained in ca. 90% selectivity (Fig. 4f–h).
To further confirm the positional selectivity of this FlICk stapling method, we synthesized 19, an orthogonally protected derivative of 13 where one of the competing Lys residues was protected by a Dde group (Fig. 4i). We then treated this peptide under standard FlICk stapling reaction with 6w in Na-borate buffer, thus affording a singly stapled peptide 19a. In the same pot, we treated 19a with hydrazine, successfully cleaving the Dde protecting group while leaving 19b with a liberated Lys side chain. HRMS and co-injection of 19b and 13a on HPLC resulted in the same eluting peak, thus confirming the identity and positional selectivity of FlICk stapling on α-helix (Fig. S78A and B†).
In validating this stapling approach with respect to potential biological applications, we recapitulated the helicity of well-known olefin-stapled helical peptides by FlICking. For this purpose, we chose the all-hydrocarbon stapled BIMBH3 20a as a comparator for the FlICk-stapled 20b (Fig. 5). The design of 20b was based on 20a (BIM SAHBA1),42,81,93 an 11-carbon stapled peptide that mimics the α-helices of BCL-2 domains; Walensky and coworkers used RCM to staple (R)-7-octenylalanine and (S)-4-pentenylalanine to afford increased α-helicity and cell permeability, notwithstanding that such stapled peptides are usually obtained as an unresolved mixture of cis- and trans-olefinic stapled isomers.11,81,93 Extensive studies into its bioactivity have shown that 20a induces cell death in cultured cells and in vivo.94 Hence, we synthesized 20a as our control peptide via standard Fmoc-SPPS chemistry and performed RCM on-resin.81
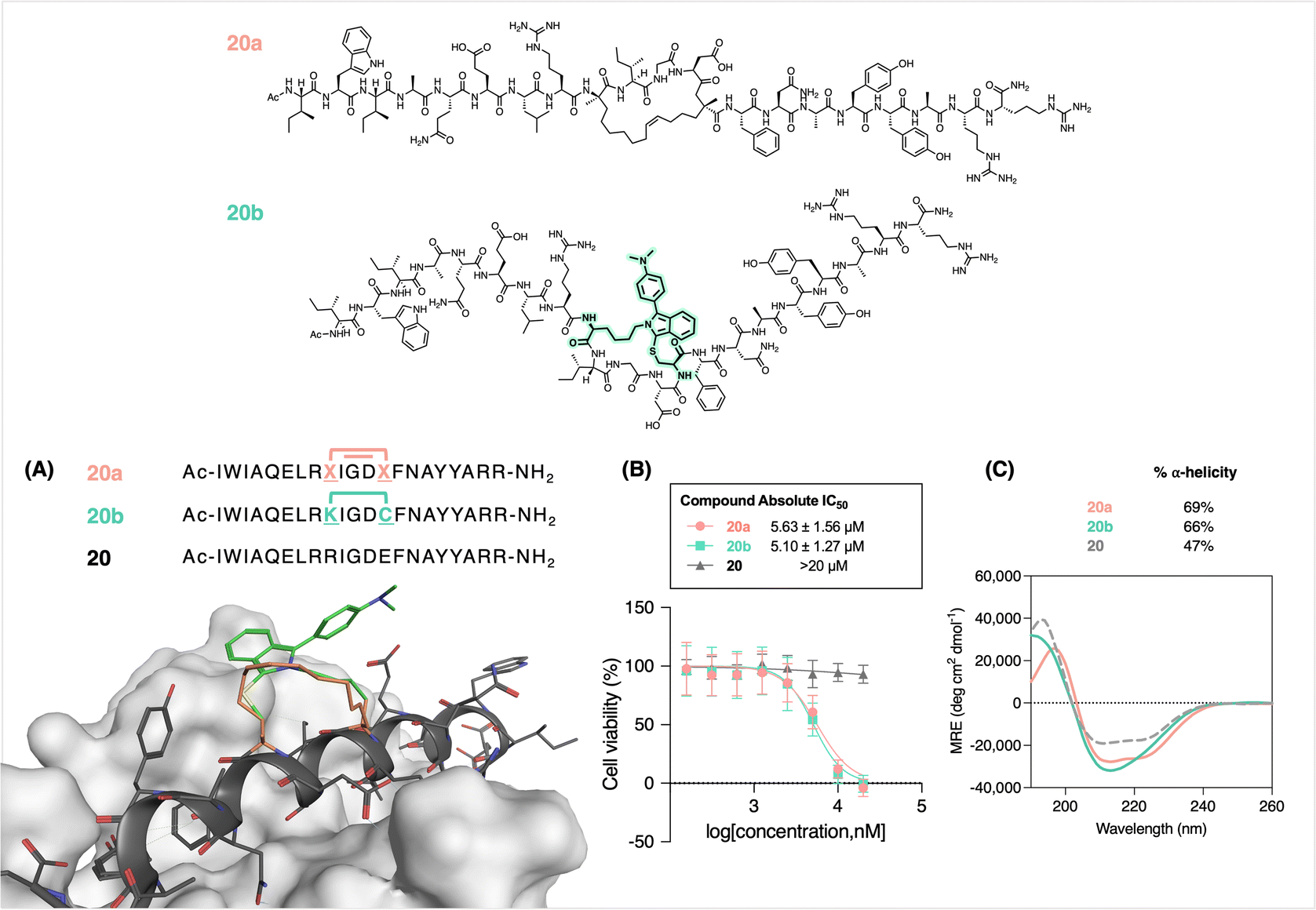 | ||
| Fig. 5 (A) Modelling 20a with respect to 20b binding to a target. (B) Comparative cell viability of Jurkat cells upon treatment of 20b and positive control 20a after a 24 hours dosing period. (C) Comparative CD. Samples were prepared as 50 μM solutions in 2:8 TFE/H2O. | ||
Accordingly, 20b was synthesized and FlICked with 6w in 94% apparent conversion (Fig. S79†). Modelling suggested very similar stapling scaffold and helicity (Fig. 5A). Subsequently, an MTS assay was conducted to evaluate the dose–response cytotoxicity of 20b on Jurkat cells (obtained from B. C. Cancer Agency). To our delight, 20b demonstrated cytotoxicity (IC50 = 5.1 ± 1.3 μM) that was equipotent to that of control 20a (IC50 = 5.6 ± 1.6 μM), while the absence of any staple in 20 showed no toxicity (IC50 > 20 μM). This similarity is further corroborated in CD analysis, where peptide 20b (66%) showed comparable helicity to that of 20a (69%). Notably, 20b showed a slight difference in the overall shape of the curve owing to a tighter helical turn where, in this case, the FlICk is three carbons shorter than the 11-atom all-hydrocarbon staple found in 20a. In addition, molecular modelling also shows adequate shape similarity with respect to staple flexibility and rigidity (Fig. 5A).
In expanding the scope of peptides and cellular targets, we chose an Axin-mimicking peptide95 on which we modelled the FlICked peptide (Fig. 6D). Previously, confocal fluorescence microscopy showed an 8-carbon olefin-stapled Axin peptidomimetic that was highly penetrant towards DLD-1 cells;96 these peptides had been conjugated to FITC via a PEG1 linker, and their cell permeability was qualitatively evaluated in terms of fluorescence distributed throughout the cytosol and nucleus. Using 6w as the stapling agent, we generated a new fluorescent Axin-mimicking helical peptide 21b (also with an 8-atom FlICk) for direct testing on cells, thus obviating the need to subsequently append a known fluorophore (Fig. 6). DLD-1 colon cancer cells (obtained from ATTC) were incubated with 21b at 5 μM for 4 h, followed by standard fixation, and then by TO-PRO-3 iodide nuclear staining. To our delight, intracellular fluorescence was observed using standard excitation at 405 nm (Fig. 6C). Consistent with this, a positive control – namely an olefin-stapled fluoresceinated peptide 21a – showed comparable results (Fig. 6B), thus demonstrating the convenience of FlICking with 6w to deliver a useful fluorophore for confocal imaging studies. Moreover, peptide 21b showed good stability after storage as lyophilized powder at −20 °C over a period of at least 3 months (96% purity, Fig. S82†).
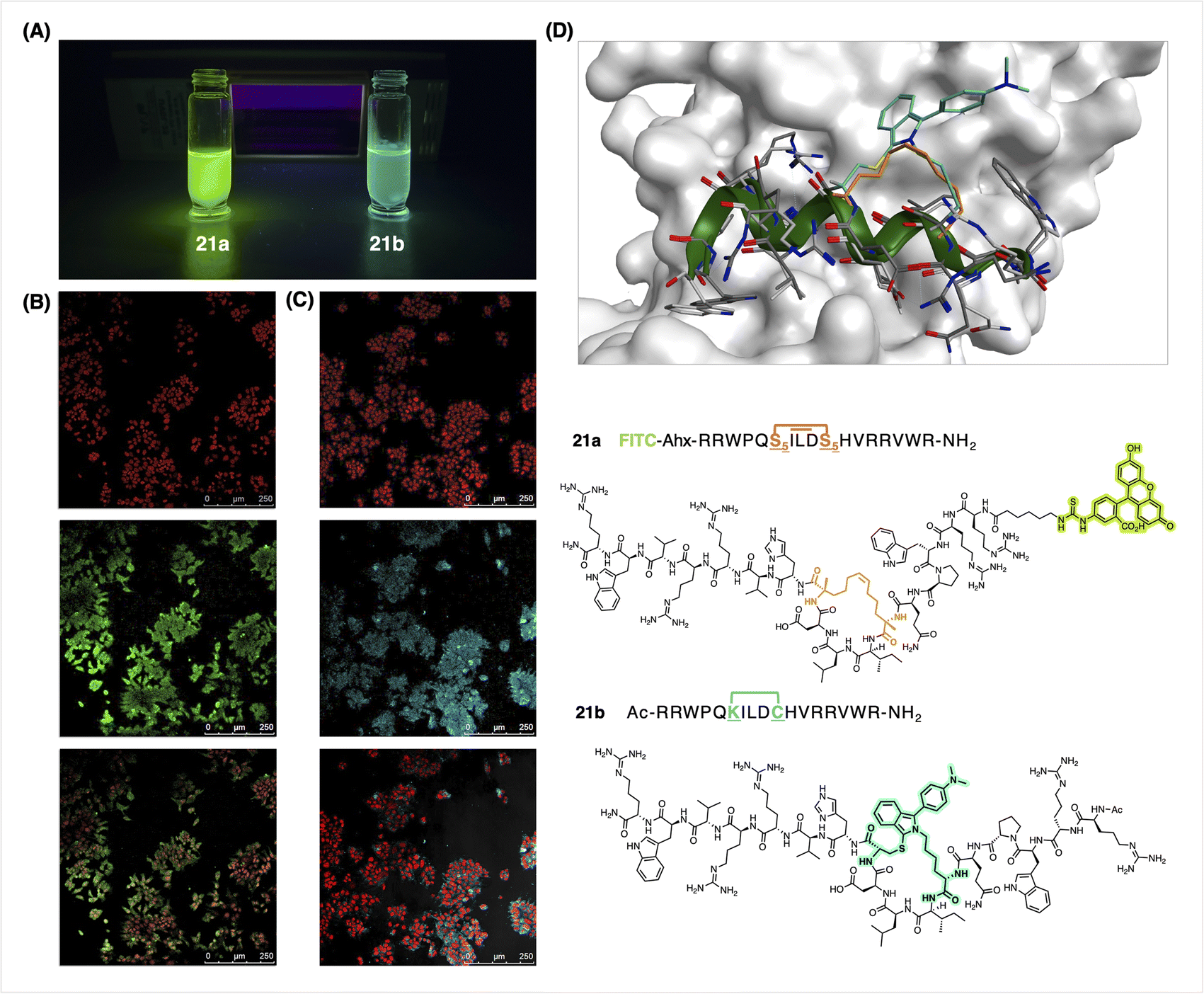 | ||
| Fig. 6 (A) Solution of 21a and 21b in PBS Buffer (pH 7.4), 10% EtOH. (B) Imaging of cellular uptake (DLD-1 cells) of 21a FITC-labeled peptide (7.5 μM, 1 h) and (C) 21b FlICked-peptide (5 μM, 1 h) by confocal fluorescence microscopy. Overlaid images, in red: nuclear TO-PRO-3 Iodide, in green: FITC-labeled 21a positive control, in blue: 21b FlICked peptide. TO-PRO-3 was excited at 663 nm, FITC at 488 nm, and FlICk-ed peptide at 405 nm. (D) Molecular modelling of 21a and 21b (FITC not shown in the model). Orange bonds represent the olefin staple found in 21a, and teal bonds represent the FlICk found in 21b. | ||
In summary, we have expanded access to new isoindole staples with an eye to regioselectivity in isoindole synthesis, expanded chromicity (fluorescent and visible) and moderate-to-high positional selectivity. The synthetic route featured herein provides access to an array of ArKBCHO's with a high degree of modularity and functional group tolerance with the added potential for tuning various chemical attributes and readily generating new ArKBCHOs. In turn, these extend the utility and ease of FlICking for simultaneous peptide stapling and fluorophore installation. More importantly, the 2-arylketobenzaldehyde motif featured in this study affords a regioselective and chemoselective isoindole stapling that circumvents the regioselectivity problems seen with OPA-derived isoindoles and further suppresses undesired N-terminus capping, i.e., isoindolinone. We submit that this synthetic access and the consequent chemoselective applications will be particularly important for generating single isomers of FlICked peptides for the generation of drug conjugates as detailed in the following discussion.
To qualitatively assess cell permeability, we chose a highly fluorescent stapling agent from our generated library and stapled a native, unprotected peptide with excellent conversion. Following purification, the FlICked peptide was directly tested for cellular uptake via confocal fluorescence microscopy. We envisioned that such workflow could streamline the process of staple scanning that is commonly employed in determining the optimal staple position along a peptide backbone to impart helicity and reasonably potent biological activity.
With regards to chemoselectivity, where Lys–Lys stapling and isoindolinone side product is suppressed, we imagine the potential for using ArKBCHOs to further diversify the chemical space pertinent to DNA-based libraries and peptide-displayed libraries, while negating the use of excess reagents.64,97 This synthetic route also allows for the incorporation of azides, nitriles, alkynes, and halogens into ArKBCHOs, thereby setting the stage for further functionalization e.g. click-type conjugations (Fig. S87–90†). The improved fluorescence quantum yields and red-shifted excitation maxima on a number of isoindoles demonstrated compatibility with common laser sources used in confocal microscopy (405 nm). We also anticipate these fluorogenic staples to find use in high-throughput fluorescence polarization assays where peptide–protein binding can be readily assessed. As we recapitulated the cytotoxicity of the all-hydrocarbon stapled BIM SAHBA1, we suggest that FlICking will find utility on a number of helical peptidomimetics of biological interest, while obviating the use of exotic building blocks or the need to graft an additional fluorophore to confirm uptake.98 In the same vein, the use of a non-fluorescent nitro-isoindole (7q) may find utility for detecting hypoxia in cells since reduction to the amino-FlICk will result in fluorescence.
Additionally, FlICking to enforce additional peptide secondary structures, e.g. β-turns, remains a subject of ongoing interest. Not addressed here is the ability to fine-tune the staple length with easily accessible lysine analogs e.g. ornithine, diaminobutyric acid as well as other thiols e.g. homocysteine, penicillamine and mercapto-phenylalanine. Applications to phage display,99 and for the production of ADCs are also readily contemplated. It is further anticipated that new fluorescent proteins can be engineered provided that proteins can be designed or evolved to bind to and react specifically with a given ArKBCHO. The results herein portend the synthesis of an array of ArKBCHOs to probe active-site labeling with greater specificity for applications in proteomic probing or in screens for new anticancer agents and antibiotics.100–103
Curiously, pestalone,104 a natural product produced by a marine fungus in response to bacterial challenge, is a highly substituted ArKBCHO that shows moderate in vitro cytotoxicity against tumor cells and potent antibiotic activity against methicillin-resistant S. aureus and vancomycin-resistant E. faecium.105 Although the basis for pestalone's activity is unknown, its reaction with an isolated amine under mildly acidic conditions results in the isoindolinone, suggesting that pestalone might act by modifying N-terminal peptides and/or critical lysine side chains.80,106 In light of our results, its cytocidal properties may instead arise from an irreversible reaction with a Lys/Cys couple, a motif that is known to be present in a large number of protein families, including hydrolases, amidases, dehydrogenases, and ubiquitous redox-switches.107–112
Conclusion
We report a class of molecular linchpins called ArKBCHOs, which has found utility as a chemoselective, regioselective, and positionally selective fluorogenic stapling agent on unprotected peptides. Salient points of this work are as follows: (1) ArKBCHOs serve as a scaffold for a diverse set of isoindole staples, of which several show improved excitation–emission properties relative to isoindoles formed from modified ortho-phthalaldehydes; (2) use of ArKBCHOs eliminates the question of regioisomers as a single regioisomeric isoindole staple is formed without competitive reaction with lysine-amines; (3) positional-selective stapling favouring a helical turn in unprotected linear peptides is readily achievable in the presence of competing amine and thiol nucleophiles; (4) this methodology is applied to peptides of biological interest, where stapling confers cytocidal properties consistent with similar peptidomimetics stapled with traditional methods; (5) FlICk stapled peptides show good cellular uptake and we have shown that it can be directly imaged by virtue of the FlICk itself; (6) facile synthetic access to a large array of ArKBCHOs is envisioned to support the production of new fluorogenic/chromogenic reagents for peptide stapling and more broadly, towards the discovery of amine/thiol-reactive biological probes.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. M. P. and N. D. designed the project and developed the hypothesis, N. D., performed primary experimental work, and analyzed all of the data. N. D. authored the first draft of the manuscript and revisions were done in collaboration with D. M. P. All biological work was performed by J. F. Synthesis of compounds reported in this manuscript was done by N. D. with the assistance of P. R.Conflicts of interest
UBC has filed a provision patent on which N. D. and D. M. P. are named as inventors, which claims compositions matter detailed herein.Acknowledgements
The authors are grateful for the financial support from NSERC RGPIN-2020-05461. We thank Ms Seja Elgadi for assistance with quantum yield measurements, Kristina W. Rothchild for their assistance with MS/MS acquisition, and Dr Maria Ezhova for NMR acquisition. We also extend our gratitude to Ms Katherine Li, Ms Areesha Rizwan, Ms Frannie Dai, and Mr Matthieu Lebeau for assistance in the scale-up of starting materials.Notes and references
- P. A. Cistrone, A. P. Silvestri, J. C. J. Hintzen and P. E. Dawson, ChemBioChem, 2018, 19, 1031–1035 CrossRef PubMed.
- S. Punna, J. Kuzelka, Q. Wang and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2215–2220 CrossRef.
- S. Cantel, A. L. C. Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D'Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef PubMed.
- J. Q. Zhang, M. Mulumba, H. Ong and W. D. Lubell, Angew. Chem., Int. Ed., 2017, 56, 6284–6288 CrossRef.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef PubMed.
- Y. Angell and K. Burgess, J. Org. Chem., 2005, 70, 9595–9598 CrossRef PubMed.
- J. M. Beierle, W. S. Horne, J. H. van Maarseveen, B. Waser, J. C. Reubi and M. R. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4725–4729 CrossRef PubMed.
- S. Ingale and P. E. Dawson, Org. Lett., 2011, 13, 2822–2825 CrossRef.
- M. Sousbie, M. Vivancos, R. L. Brouillette, E. Besserer-Offroy, J. M. Longpre, R. Leduc, P. Sarret and E. Marsault, J. Med. Chem., 2018, 61, 7103–7115 CrossRef.
- H. E. Blackwell, J. D. Sadowsky, R. J. Howard, J. N. Sampson, J. A. Chao, W. E. Steinmetz, D. J. O'Leary and R. H. Grubbs, J. Org. Chem., 2001, 66, 5291–5302 CrossRef PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef.
- L. D. Walensky, K. Pitter, J. Morash, K. J. Oh, S. Barbuto, J. Fisher, E. Smith, G. L. Verdine and S. J. Korsmeyer, Mol. Cell, 2006, 24, 199–210 CrossRef CAS.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef.
- G. J. Hilinski, Y. W. Kim, J. Hong, P. S. Kutchukian, C. M. Crenshaw, S. S. Berkovitch, A. Chang, S. Ham and G. L. Verdine, J. Am. Chem. Soc., 2014, 136, 12314–12322 CrossRef.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef.
- H. G. Lee, G. Lautrette, B. L. Pentelute and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 3177–3181 CrossRef PubMed.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 20, 2552–2554 RSC.
- A. M. Spokoyny, Y. K. Zou, J. J. Ling, H. T. Yu, Y. S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef PubMed.
- A. A. Aimetti, R. K. Shoemaker, C. C. Lin and K. S. Anseth, Chem. Commun., 2010, 46, 4061–4063 RSC.
- B. C. Zhao, Q. Z. Zhang and Z. G. Li, J. Pept. Sci., 2016, 22, 540–544 CrossRef PubMed.
- Y. Tian, J. X. Li, H. Zhao, X. Z. Zeng, D. Y. Wang, Q. S. Liu, X. G. Niu, X. H. Huang, N. H. Xu and Z. G. Li, Chem. Sci., 2016, 7, 3325–3330 Search PubMed.
- Y. X. Wang and D. H. C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 120–129 CrossRef PubMed.
- L. Mendive-Tapia, S. Preciado, J. Garcia, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 9 Search PubMed.
- S. P. Brown and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef PubMed.
- Y. B. Feng and K. Burgess, Chem.–Eur. J., 1999, 5, 3261–3272 CrossRef.
- M. R. Wang, D. Pan, Q. Zhang, Y. J. Lei, C. Wang, H. Y. Jia, L. Y. Mou, X. K. Miao, X. Y. Ren and Z. Q. Xu, J. Am. Chem. Soc., 2024, 146, 6675–6685 CrossRef.
- Q. Yu, L. Y. Bai and X. F. Jiang, Angew. Chem., Int. Ed., 2023, 62, 9 Search PubMed.
- L. H. Shen, O. Monasson, E. Peroni, F. Le Bideau and S. Messaoudi, Angew. Chem., Int. Ed., 2023, 62, 8 Search PubMed.
- F. J. Chen, N. Pinnette, F. Yang and J. M. Gao, Angew. Chem., Int. Ed., 2023, 62, 8 Search PubMed.
- E. H. Abdelkader, H. Qianzhu, J. George, R. L. Frkic, C. J. Jackson, C. Nitsche, G. Otting and T. Huber, Angew. Chem., Int. Ed., 2022, 61, 5 CrossRef.
- P. Guo, X. Chu, C. J. Wu, T. J. Qiao, W. L. Guan, C. Z. Zhou, T. Wang, C. L. Tian, G. He and G. Chen, Angew. Chem., Int. Ed., 2024, 63, 8 Search PubMed.
- Y. Zhang, R. J. Yin, H. Jiang, C. M. Wang, X. Wang, D. P. Wang, K. Zhang, R. L. Yu, X. C. Li and T. Jiang, Org. Lett., 2023, 25, 2248–2252 CrossRef PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef PubMed.
- J. Ceballos, E. Grinhagena, G. Sangouard, C. Heinis and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 9022–9031 CrossRef.
- J. R. Frost, C. C. G. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 CrossRef PubMed.
- M. G. Ricardo, D. Llanes, L. A. Wessjohann and D. G. Rivera, Angew. Chem., Int. Ed., 2019, 58, 2700–2704 CrossRef.
- M. Todorovic, K. D. Schwab, J. Zeisler, C. C. Zhang, F. Benard and D. M. Perrin, Angew. Chem., Int. Ed., 2019, 58, 14120–14124 CrossRef PubMed.
- Y. Zhang, Q. Zhang, C. T. T. Wong and X. C. Li, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef.
- A. L. Edwards, F. Wachter, M. Lammert, A. J. Huhn, J. Luccarelli, G. H. Bird and L. D. Walensky, ACS Chem. Biol., 2015, 10, 2149–2157 CrossRef PubMed.
- A. Afonso, L. Feliu and M. Planas, Tetrahedron, 2011, 67, 2238–2245 CrossRef.
- H. Brown, M. Chung, A. Ueffing, N. Batistatou, T. Tsang, S. Doskocil, W. Q. Mao, D. Willbold, R. C. Bast, Z. Lu, O. H. Weiergraeber and J. A. Kritzer, J. Am. Chem. Soc., 2022, 144, 14687–14697 CrossRef PubMed.
- T. X. Mi, S. Siriwibool and K. Burgess, Angew. Chem., Int. Ed., 2023, 62, 11 Search PubMed.
- O. Al Musaimi, L. Lombardi, D. R. Williams and F. Albericio, Pharmaceuticals, 2022, 15, 33 CrossRef PubMed.
- H. X. Luong, H. T. P. Bui and T. T. Tung, J. Med. Chem., 2022, 65, 3026–3045 CrossRef PubMed.
- R. Jimmidi, Eur. J. Org Chem., 2023, 26, 17 CrossRef.
- A. A. Komar, Molecules, 2023, 28, 13 CrossRef PubMed.
- Y. W. Zhang, J. B. Guo, J. J. Cheng, Z. H. Zhang, F. H. Kang, X. X. Wu and Q. Chu, J. Med. Chem., 2023, 66, 95–106 CrossRef PubMed.
- S. Ullrich and C. Nitsche, Pept. Sci., 2024, 116, 15 Search PubMed.
- F. J. Chen, W. Z. Lin and F. E. Chen, Nat. Rev. Chem, 2024, 8, 304–318 CrossRef PubMed.
- A. R. Paquette and C. N. Boddy, ChemBioChem, 2023, 24, 13 CrossRef.
- L. Costa, E. Sousa and C. Fernandes, Pharmaceuticals, 2023, 16, 35 CrossRef.
- J. Whisenant and K. Burgess, Chem. Soc. Rev., 2022, 51, 5795–5804 RSC.
- K. Tsuchiya, T. Kurohara, K. Fukuhara, T. Misawa and Y. Demizu, Processes, 2022, 10, 24 CrossRef.
- Y. T. Wu, J. Williams, E. D. D. Calder and L. J. Walport, RSC Chem. Biol., 2021, 2, 151–165 RSC.
- X. Li, S. Chen, W. D. Zhang and H. G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef PubMed.
- I. V. Smolyar, A. K. Yudin and V. G. Nenajdenko, Chem. Rev., 2019, 119, 10032–10240 CrossRef PubMed.
- Y. H. Lau, P. De Andrade, Y. T. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- X. Y. Liu, W. Cai, N. Ronceray, A. Radenovic, B. Fierz and J. Waser, J. Am. Chem. Soc., 2023, 145, 26525–26531 CrossRef PubMed.
- T. Thompson, T. Pewklang, P. Piyanuch, N. Wanichacheva, A. Kamkaew and K. Burgess, Org. Biomol. Chem., 2024, 22, 506–512 RSC.
- J. Liu, X. Liu, F. F. Zhang, J. J. Qu, H. Y. Sun and Q. Zhu, Chem.–Eur. J., 2020, 26, 16122–16128 CrossRef PubMed.
- Q. G. Nie, X. F. Fang, C. Y. Liu, G. Zhang, X. H. Fan, Y. F. Li and Y. Z. Li, J. Org. Chem., 2022, 87, 2551–2558 CrossRef PubMed.
- B. Li, L. Wang, X. X. Chen, X. Chu, H. Tang, J. Zhang, G. He, L. Li and G. Chen, Nat. Commun., 2022, 13, 9 CrossRef PubMed.
- X. Chu, B. Li, H. Y. Liu, X. W. Sun, X. C. Yang, G. He, C. Z. Zhou, W. M. Xuan, S. L. Liu and G. Chen, Angew. Chem., Int. Ed., 2023, 62, 9 Search PubMed.
- L. A. Sternson, J. F. Stobaugh and A. J. Repta, Anal. Biochem., 1985, 144, 233–246 CrossRef PubMed.
- F. Xiao, M. Z. Sun, L. Y. Zhang and X. G. Lei, J. Org. Chem., 2024, 89(20), 14619–14624 CrossRef PubMed.
- S. C. Beale, J. C. Savage, D. Wiesler, S. M. Wietstock and M. Novotny, Anal. Chem., 1988, 60, 1765–1769 CrossRef CAS PubMed.
- S. C. Beale, Y. Z. Hsieh, J. C. Savage, D. Wiesler and M. Novotny, Talanta, 1989, 36, 321–325 CrossRef CAS PubMed.
- J. P. Liu, Y. Z. Hsieh, D. Wiesler and M. Novotny, Anal. Chem., 1991, 63, 408–412 CrossRef CAS.
- A. Kotali, M. Papapetrou, V. Dimos and P. A. Harris, Org. Prep. Proced. Int., 1998, 30, 177–181 CrossRef CAS.
- A. Kotali and P. G. Tsoungas, Tetrahedron Lett., 1987, 28, 4321–4322 CrossRef CAS.
- A. Kotali, Curr. Org. Chem., 2002, 6, 965–985 CrossRef CAS.
- R. Hadsarung, S. Thongnest, S. Oekchuae, D. Chaiyaveij, J. Boonsombat and S. Ruchirawat, Tetrahedron, 2022, 121, 12 CrossRef.
- H. W. Ai, N. C. Shaner, Z. H. Cheng, R. Y. Tsien and R. E. Campbell, Biochemistry, 2007, 46, 5904–5910 CrossRef CAS.
- O. M. Subach, I. S. Gundorov, M. Yoshimura, F. V. Subach, J. H. Zhang, D. Gruenwald, E. A. Souslova, D. M. Chudakov and V. V. Verkhusha, Chem. Biol., 2008, 15, 1116–1124 CrossRef CAS.
- Y. D. Bao, M. Y. Xing, N. Matthew, X. H. Chen, X. Wang and X. J. Lu, Org. Lett., 2023, 26, 2763–2767 CrossRef.
- R. Grigg, H. Q. N. Gunaratne and V. Sridharan, Chem. Commun., 1985, 1183–1184 RSC.
- D. Augner, D. C. Gerbino, N. Slavov, J. M. Neudorfl and H. G. Schmalz, Org. Lett., 2011, 13, 5374–5377 CrossRef CAS PubMed.
- G. H. Bird, W. C. Crannell and L. D. Walensky, Curr. Protoc. Chem. Biol., 2011, 3, 99–117 CrossRef PubMed.
- R. N. Chapman, G. Dimartino and P. S. Arora, J. Am. Chem. Soc., 2004, 126, 12252–12253 CrossRef CAS.
- Y. W. Kim and G. L. Verdine, Bioorg. Med. Chem. Lett., 2009, 19, 2533–2536 CrossRef CAS.
- M. Scrima, A. Le Chevalier-Isaad, P. Rovero, A. M. Papini, M. Chorev and A. M. D'Ursi, Eur. J. Org Chem., 2010, 2010, 446–457 CrossRef.
- M. L. Stewart, E. Fire, A. E. Keating and L. D. Walensky, Nat. Chem. Biol., 2010, 6, 595–601 CrossRef CAS.
- T. E. Speltz, S. W. Fanning, C. G. Mayne, C. Fowler, E. Tajkhorshid, G. L. Greene and T. W. Moore, Angew. Chem., Int. Ed., 2016, 55, 4252–4255 CrossRef CAS PubMed.
- N. E. Shepherd, H. N. Hoang, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 2974–2983 CrossRef CAS PubMed.
- T. N. Grossmann, J. T. H. Yeh, B. R. Bowman, Q. Chu, R. E. Moellering and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17942–17947 CrossRef CAS.
- K. Takada, D. Zhu, G. H. Bird, K. Sukhdeo, J. J. Zhao, M. Mani, M. Lemieux, D. E. Carrasco, J. Ryan, D. Horst, M. Fulciniti, N. C. Munshi, W. Q. Xu, A. L. Kung, R. A. Shivdasani, L. D. Walensky and D. R. Carrasco, Sci. Transl. Med., 2012, 4, 13 Search PubMed.
- J. Y. S. Chu, L. T. O. Lee, C. H. Lai, H. Vaudry, Y. S. Chan, W. H. Yung and B. K. C. Chow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15961–15966 CrossRef.
- A. D. de Araujo, J. Lim, K. C. Wu, Y. B. Xiang, A. C. Good, R. Skerlj and D. P. Fairlie, J. Med. Chem., 2018, 61, 2962–2972 CrossRef.
- S. Pomplun, M. Jbara, A. J. Quartararo, G. Zhang, J. S. Brown, Y.-C. Lee, X. Ye, S. Hanna and B. L. Pentelute, ACS Cent. Sci., 2021, 7, 156–163 CrossRef.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef PubMed.
- J. L. LaBelle, S. G. Katz, G. H. Bird, E. Gavathiotis, M. L. Stewart, C. Lawrence, J. K. Fisher, M. Godes, K. Pitter, A. L. Kung and L. D. Walensky, J. Clin. Invest., 2012, 122, 2018–2031 CrossRef PubMed.
- L. Dietrich, B. Rathmer, K. Ewan, T. Bange, S. Heinrichs, T. C. Dale, D. Schade and T. N. Grossmann, Cell Chem. Biol., 2017, 24, 958–968 CrossRef PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef PubMed.
- T. Y. Wei, D. F. Li, Y. Zhang, Y. B. Tang, H. Y. Zhou, H. Liu and X. C. Li, Small Methods, 2022, 6, 7 Search PubMed.
- S. O. Squire, S. Sebghati and M. C. Hammond, ACS Chem. Biol., 2023, 19, 3–8 CrossRef PubMed.
- H. Xiang, L. W. Bai, X. D. Zhang, T. Dan, P. Cheng, X. Q. Yang, H. L. Ai, K. Li and X. X. Lei, Chem. Sci., 2024, 15, 11847–11855 RSC.
- S. Sheikh and S. S. Katiyar, J. Enzyme Inhib., 1995, 9, 235–242 CrossRef PubMed.
- G. Matteucci, V. Lanzara, C. Ferrari, S. Hanau and C. M. Bergamini, Biol. Chem., 1998, 379, 921–924 CAS.
- R. N. Puri, D. Bhatnagar and R. Roskoski, Biochim. Biophys. Acta, 1988, 957, 34–46 CrossRef CAS PubMed.
- C. Y. Chen, F. A. Emig, V. L. Schramm and D. E. Ash, J. Biol. Chem., 1991, 266, 16645–16652 CrossRef CAS.
- M. Cueto, P. R. Jensen, C. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Nat. Prod., 2001, 64, 1444–1446 CrossRef CAS.
- N. Slavov, J. Cvengros, J. M. Neudörfl and H. G. Schmalz, Angew. Chem., Int. Ed., 2010, 49, 7588–7591 CrossRef CAS PubMed.
- D. Augner, O. Krut, N. Slavov, D. C. Gerbino, H. G. Sahl, J. Benting, C. F. Nising, S. Hillebrand, M. Krönke and H. G. Schmalz, J. Nat. Prod., 2013, 76, 1519–1522 CrossRef CAS.
- R. Stappen, T. Dierks, A. Broer and R. Kramer, Eur. J. Biochem., 1992, 210, 269–277 CrossRef CAS PubMed.
- T. Nakayama, H. Tanabe, Y. Deyashiki, M. Shinoda, A. Hara and H. Sawada, Biochim. Biophys. Acta, 1992, 1120, 144–150 CrossRef CAS PubMed.
- S. Jagtap and M. Rao, Biochem. Biophys. Res. Commun., 2006, 347, 428–432 CrossRef CAS PubMed.
- B. W. Weber, S. W. Kimani, A. Varsani, D. A. Cowan, R. Hunter, G. A. Venter, J. C. Gumbart and B. T. Sewell, J. Biol. Chem., 2013, 288, 28514–28523 CrossRef CAS PubMed.
- K. Van Laer, M. Oliveira, K. Wahni and J. Messens, Protein Sci., 2014, 23, 238–242 CrossRef CAS PubMed.
- F. R. von Pappenheim, M. Wensien, J. Ye, J. Uranga, I. Irisarri, J. de Vries, L. M. Funk, R. A. Mata and K. Tittmann, Nat. Chem. Biol., 2022, 18, 368–375 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04839c |
| This journal is © The Royal Society of Chemistry 2025 |