
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePronounced electronic modulation of geometrically-regulated metalloenediyne cyclization†
Sarah E.
Lindahl
a,
Erin M.
Metzger
a,
Chun-Hsing
Chen
 b,
Maren
Pink
b and
Jeffrey M.
Zaleski
*a
b,
Maren
Pink
b and
Jeffrey M.
Zaleski
*a
aDepartment of Chemistry, Indiana University, Bloomington, IN 47405, USA. E-mail: zaleski@iu.edu
bMolecular Structure Center, Indiana University, Bloomington, IN 47405, USA
First published on 12th November 2024
Abstract
Using a diverse array of thermally robust phosphine enediyne ligands (dxpeb, X = Ph, Ph-pOCH3, Ph-pCF3, Ph-m2CH3, Ph-m2CF3, iPr, Cy, and tBu) a novel suite of cisplatin-like Pt(II) metalloenediynes (3, Pt(dxpeb)Cl2) has been synthesized and represents unique electronic perturbations on thermal Bergman cyclization kinetics. Complexes 3e (Ph-m2CF3) and 3f (iPr) are the first of this structure type to be crystallographically characterized with inter alkyne termini distances (3e: 3.13 Å; 3f: 3.10 Å) at the lower end of the widely accepted critical distance range within which enediynes should demonstrate spontaneous ambient temperature cyclization. Despite different electronic profiles, these metalloenediynes adopt a rigid, uniform structure suggesting complexes of the form Pt(dxpeb)Cl2 have orthogonalized geometric and electronic contributions to thermal Bergman cyclization. Kinetic activation parameters determined using 31P NMR spectroscopy highlight the dramatic reactivity and thermal tunability of these complexes. At room temperature, the half-life (t1/2) of cyclization spans a range of ∼35 hours and for the aryl phosphine derivatives, cycloaromatization rates are 10–30 times faster for complexes with electron donating substituents (3b: Ph-pOCH3; 3d: Ph-m2CH3) compared to those with electron withdrawing substituents (3c: Ph-pCF3; 3e: Ph-m2CF3). Computational interrogation of the aryl phosphine metalloenediynes 3a–3e reveals that the origin of this precise electronic control derives from electronic withdrawing group-mediated alkyne carbon polarization that amplifies coulombic repulsion increasing the cyclization barrier height. Additionally, mixing between the in-plane π-orbitals and the phosphine aryl ring system is pronounced for complexes with electron donating substituents which stabilizes the developing C–C bond and lowers the activation barrier. This π-orbital mixing is negligible however, for complexes with electron withdrawing substituents due to an energetic mismatch of the orbital systems. Overall, this work demonstrates that for geometrically rigid frameworks, even remote enediyne functionalization can have pronounced effects on activation barrier.
Introduction
Over 30 years ago, the discovery of the unprecedented, enediyne-containing natural products1–6 introduced the world to the biologically active 1,5-diyne-3-ene functionality whose seemingly esoteric cyclization reactivity was described by Masamune7 and Bergman8 over a decade prior. Currently, 14 enediynes and 5 cycloaromatized compounds have been structurally characterized,9–17 verifying the unique diyne-ene motif that generates the reactive 1,4-phenyl diradical species responsible for biological potency. Despite being known for decades, enediynes are still on an upward trajectory and have not yet reached their peak impact. As evidence, two second-generation antibody-conjugated forms of calicheamicin (Mylotarg and Besponsa) have been approved by the FDA (Mylotarg reapproved) in 2017 and show low nanomolar-to-picomolar activity against acute myeloid and lymphoblastic leukemias.10,18,19 Other enediyne-containing conjugates have been approved outside the US or are currently in development and show cytotoxicity limits in comparable ranges.20–22 The success of these natural product conjugates is a triumph for a class of molecules that has struggled to maintain a presence in mainstream medicinal chemistry. Yet, there are still extensive unmet challenges: many natural product enediynes react too quickly at ambient temperature to be directly drugable,10 suites of synthetic analogues with delicately tuned reactivity do not yet exist, and the role of the enediyne unit in nature is not even well-defined. Many of these shortcomings survive because researchers have yet to fully understand and harness the geometric and electronic structural parameters responsible for the fundamental reactivity of the 1,5-diyne-3-ene unit to effectively put constructs beyond natural product enediyne-conjugates to appropriate use.One of the most fundamental paradigms describing the base reactivity of enediynes was established by Nicolaou et al. early on in the field's development and stated that a critical distance (d) between alkyne termini existed at which spontaneous enediyne cyclization would occur (d: 3.2–3.31 Å).23,24 Computational analysis by Schreiner later supplemented these findings and expanded the critical distance range, especially the lower limit, to 2.9–3.4 Å.25 Work by Snyder,26,27 and later in conjunction with Magnus,28 also added that differential strain between the ground and transition states contributes significantly to the kinetics of cycloaromatization for more complex ring-closed systems.
Electronic perturbations of the enediyne framework have also been shown to influence Bergman cyclization kinetics. Modulation of orbital contributions and energetic dependencies are inherently system-specific and can lead to complex trends in reactivity outcomes. For example, Semmelhack et al. demonstrated that incorporation of the –ene– unit into an aromatic ring stabilizes the base enediyne framework significantly and reduces cyclization reactivity.29,30 In a similar vein, Jones and others have shown that functionalization of the vinylic positions with σ-donating or σ/π-withdrawing groups leads to enhancement or retardation of cyclization, respectively,31,32 due to differential electron repulsion in the developing diradical transition state. This result is at first glance counterintuitive since electron withdrawing substituents, inductively or through space, should decrease the observed electrostatic interaction. It can only be rationalized by the structural disposition of the lone pairs and their field effect32 relative to the developing diradical and C2⋯C7 bond. An analogous polarization argument was employed by Schmittel et al. to explain differential barrier heights as a function of alkyne termini substituent. Here, however, distal p-phenyl substituted electron withdrawing groups play a more traditional inductive role in reducing the electron repulsion in the transition state thus lowering the activation barrier to Bergman cyclization.33 This result was later supported and enhanced by Schreiner et al. who showed that σ-withdrawing functionalities (or π-donors) directly at the alkyne termini engender a parallel stabilizing influence on the transition state structure by facilitating the formation of the benzenoid diradical intermediate leading to rate acceleration.34 It is clear from these demonstrations that electronic effects are complex and influenced by both pure electronic (i.e., inductive) as well as spatial (i.e., proximity) interactions between populated orbitals. These effects, while significant, are superimposed upon a given geometric structural reactivity base and further modulate the observed Bergman cyclization rate.
To further harness the geometric component of the activation, metal fragment complexation was first realized in the mid-1990s to provide a highly dynamic, conformational platform for activating bound enediyne substrates. Buchwald and coworkers developed the acyclic enediyne 1,2-bis((diphenylphosphino)ethynyl)benzene (dppeb) as a ligand for d8 Pd(II) and Pt(II) which generates a square planar geometry and is computationally determined to have a significantly reduced inter alkyne termini separation of 3.3 Å.35 This translates to spontaneous thermal reactivity in solution at ambient temperature for these complexes in situ, consistent with the original cyclization paradigm put forth by Nicolaou.
While an ensemble of geometrically-modulated, metal-mediated Bergman cyclization demonstrations exist, there are far fewer examples of electronic influences on metal-modulated enediyne frameworks.36,37 The thermal cyclization of a Mo(IV) dithiolate–enediyne complex is one of them. Here the high-valent metal fragment stabilizes the sulfur lone pairs which eliminates electron repulsion with the developing diradical species in the transition state. This stabilization lowers the activation barrier to cyclization as had been previously demonstrated for the purely organic examples by an analogous mechanism.37 Although this example relies directly on the electronics of the metal fragment to influence the cyclization rate, what we have learned from the organic enediyne reactivity demonstrations is that substitution of the ligand framework can also have significant electronic contributions that modulate the geometry-controlled reactivity set point.
More than a quarter century has passed since Nicolaou,23,24 Schreiner,25 Buchwald,35 and others demonstrated that there exists a crude geometric critical distance for seemingly disparate enediyne structures to react spontaneously, and this paradigm has held ever since. Similarly, it has been nearly as long since electronic influences on enediyne cyclization have been established on somewhat independent frameworks.26–34 Herein we show that for a Pt(dxpeb)Cl2 derivatized suite, these two control mechanisms can be selectively merged to enhance the rate of thermally stable constructs or stabilize otherwise spontaneously reactive molecules. A key outcome of this systematic approach is that molecular structures that fall at the lower end of the historical critical distance threshold now can be crystallized and their structures elucidated in detail, while those that should be reactive according to that same tenet are stabilized for extended periods due to the electronic counterbalance.
Results and discussion
Functionalized phosphine enediynes and oxide derivatives
We set to synthesizing a diverse array of phosphine enediyne ligands bearing both aryl (a: Ph, b: Ph-pOCH3, c: Ph-pCF3, d: Ph-m2CH3, and e: Ph-m2CF3) and alkyl (f: iPr, g: Cy, and h: tBu) substituents to better understand the ways in which electronic substitutions at the alkyne termini work to modulate the activation barrier to thermal Bergman cyclization. The ligands 1b–1h were synthesized from a modified procedure established for the phenyl-substituted phosphine enediyne 1a.35 Generally, to a solution of the TMS-protected precursor enediyne is added n-BuLi and the reaction mixture heated to achieve deprotection of the alkyne. Subsequently, addition of the appropriate ClPR2 reagent leads to isolation of the crude R-group-substituted phosphine enediyne (Scheme 1). Purification with silica gel flash chromatography affords these ligands as either pale yellow solids (1a–1b and 1d–1e) or as yellow oils (1c and 1f–1h). The phosphine oxide analogues (2a–2h) were intentionally synthesized by the addition of hydrogen peroxide to a solution of enediyne 1a–1h and their spectroscopic signatures identified, providing a valuable tool to monitor the oxidation of the enediyne ligands 1a–1h.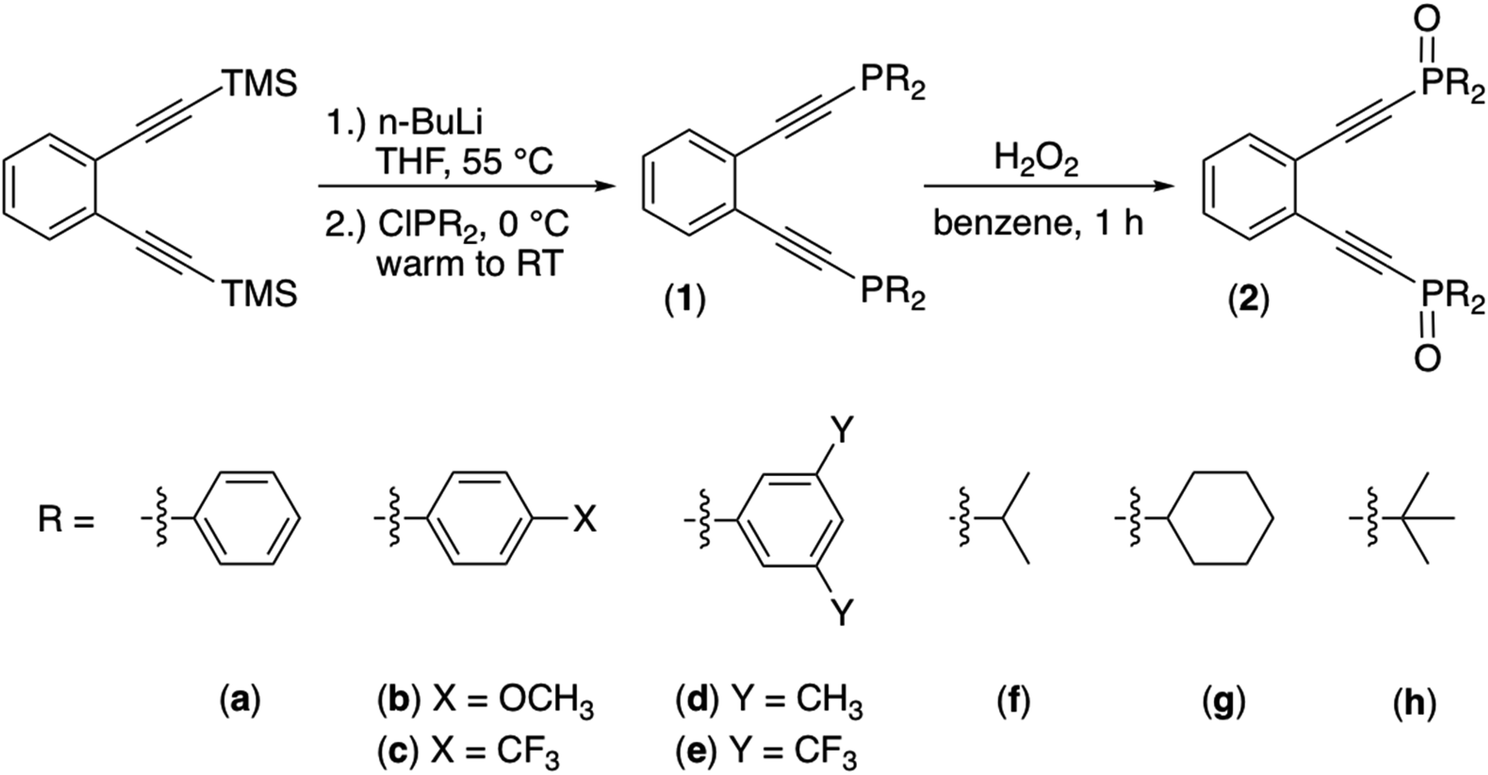 | ||
| Scheme 1 Synthesis of phosphine enediynes (1a–1h) and phosphine oxide derivatives (2a–2h). | ||
Evaluating the 31P NMR chemical shifts of the enediyne ligands allows us to visualize the extent to which functionalization of the alkyne termini perturbs the chemical environment of the phosphine. Phosphorus chemical shifts are influenced by the distribution of electron density in the σ-bonds between the phosphorus atom and its substituents, the degree of π-bonding, and variations in the C–P–C angles, which provide information about phosphine basicity.38 Clearly, derivatization with aromatic (1a–1e) and aliphatic (1f–1h) functional groups yields a wide range of 31P NMR resonances (Δδ ∼50 ppm). Despite this breadth, all enediynes bearing aryl substituents show 31P NMR shifts < 5 ppm of one another suggesting that the presence of electron donating and withdrawing groups at various positions on the terminal phenyl rings does not modulate the basicity of the phosphine to a considerable extent.
Reducing the flexibility of the phosphine by forming phosphine oxide analogues 2a–2h suppresses the variations in the C–P–C angles, minimizing steric influences on chemical shift, and allowing for isolation of electronic influences.38 As with enediynes 1a–1h, large chemical shift differences (>35 ppm) are observed between the aromatic (2a–2e) and aliphatic substituents (2f–2h), with the most electron donating R-groups appearing downfield. For the suite of aryl-substituted ligands, differences in 31P NMR chemical shift are small (<7 ppm); however, a clear trend is observed as a function of electron donating ability which is a mixture of both inductive (σ) and resonance (π) effects: 2e < 2c < 2a < 2b < 2d. Functionalization of the aromatic rings at the alkyne termini with electron donating groups minimally increases the basicity of the phosphine while the presence of electron withdrawing groups decreases basicity. While these effects are not as pronounced as those observed between aromatic and aliphatic R-groups, they do demonstrate that aryl ring substitution can be used to subtly tune the electronic nature of the phosphine.
The phosphine oxide ligands 2a–2h are stable when stored under ambient conditions while the enediyne ligands 1a–1h show no degradation over >9 months when stored as solids or dilute solutions in air at 0 °C. The robustness of these phosphine enediynes is reflected in their Bergman cyclization temperatures as measured by differential scanning calorimetry (DSC). For enediyne frameworks, endothermic events are consistent with melting while exothermic events are indicative of cycloaromatization.35,39–42 Though these studies were performed with neat material in the absence of a radical quenching agent, they provide a relative measure of the activation barrier to cycloaromatization, and when coupled with other structural information, can be used to predict the reactivity of enediyne-containing constructs towards Bergman cyclization in solution.
DSC traces have been obtained for aryl substituted enediynes 1a–1b and 1d–1e which melt between 78–144 °C and then cyclize in the liquid phase between 264–275 °C (Fig. 1S†). Though the range of melting is large (>60 °C), the cyclization window is quite narrow (11 °C). In this case, the presence of electron donating or withdrawing groups on the aromatic rings does not lead to a substantial change in the thermal activation barrier to Bergman cyclization. However, ligands whose phosphine termini have been functionalized at both meta positions (1d–1e) show higher melting temperatures than those bearing no functionalization (1a) or substitution only in the para position (1b), suggesting that differences in solid-state packing may inhibit melting of these phosphine enediynes. The cyclization temperatures of the phosphine oxide analogues are markedly higher than those of the non-oxidized ligands. In general, the melting temperature increases between 20–80 °C upon oxidation and no cyclization events are observed below 280 °C for ligands 2a–2b and 2d–2e.
Alkyne termini separations from X-ray crystallography can be used to predict relative activation barriers to diradical generation in simple enediyne systems. Ligands 1d–1e and 2d–2e were obtained as crystalline products from vapor diffusion of hydrocarbon solvents into saturated dichloromethane solutions at −20 °C and analyzed using X-ray crystallography (Fig. 1). Though 1a has been modeled computationally,351d–1e and 2d–2e represent the first phosphine enediyne ligands to be crystallographically characterized.
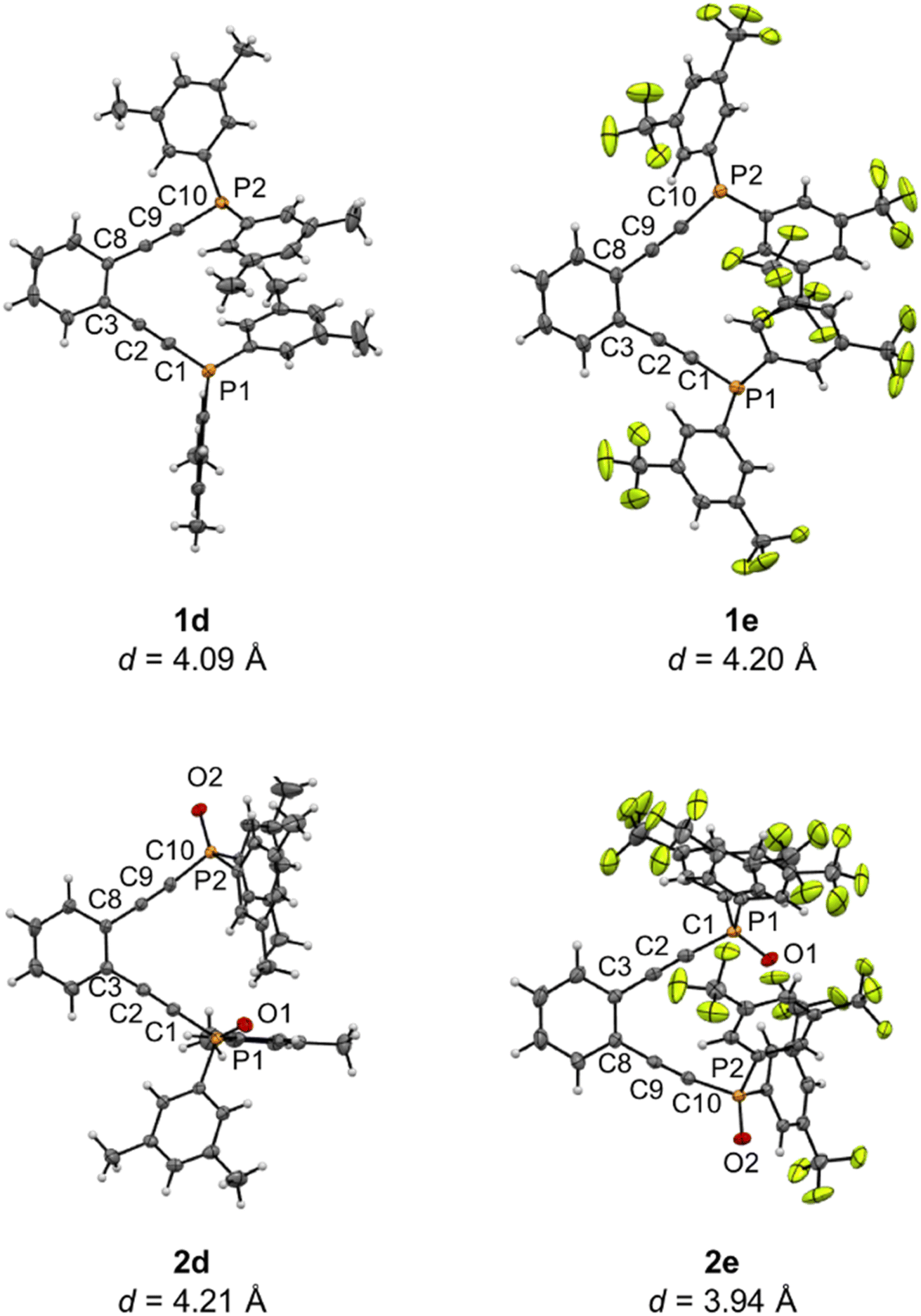 | ||
| Fig. 1 X-ray crystallographic structures of phosphine enediynes (1d and 1e) and phosphine oxide analogues (2d and 2e). Thermal ellipsoids are illustrated at 50% probability and interalkynyl distances (d) are given below structures. | ||
Enediynes 1d and 1e feature aryl rings at the phosphine termini which have been substituted in the meta positions with –CH3 and –CF3 groups, respectively. Both ligands show a high degree of π-stacking between the aromatic rings bound to the phosphorus atoms, with the closest points of contact between these stacked rings being ∼3.7 Å (1d) and 3.9 Å (1e). The presence of these non-covalent interactions supports the proposal that differences in solid-state interactions lead to variance in the DSC melting temperatures of these enediyne ligands. The two arms of the enediyne moiety are symmetric in 1e showing identical C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and CC–P angles while those of 1d show a high degree of asymmetry (Table 1S†). For both ligands, the alkyne termini separations are large (1d: 4.09 Å; 1e: 4.20 Å) and compare favorably with the computed distance of 1a which has been reported as 4.1 Å.35 The structures of the phosphine oxide analogues 2d and 2e have also been crystallographically determined and, though similar to their non-oxidized counterparts, show some unique differences from 1d and 1e. The inter alkyne termini distance of 2d is greater than that of 1d by ∼0.12 Å. Oxidation at the phosphine leads to changes in structure at the alkyne termini resulting in a widening of the enediyne framework. Interestingly, the interalkynyl distance of 2e is reduced by ∼0.26 Å relative to 1e. Here, one of the alkyne arms has rotated the bulky –CF3 substituted rings to the exterior of the enediyne framework lessening the steric clash between the large aromatic substituents. Overall, these structural data are consistent with the high cyclization temperatures observed in the DSC analyses and highlight the geometric similarities between these substituted phosphinoenediynes.
C and CC–P angles while those of 1d show a high degree of asymmetry (Table 1S†). For both ligands, the alkyne termini separations are large (1d: 4.09 Å; 1e: 4.20 Å) and compare favorably with the computed distance of 1a which has been reported as 4.1 Å.35 The structures of the phosphine oxide analogues 2d and 2e have also been crystallographically determined and, though similar to their non-oxidized counterparts, show some unique differences from 1d and 1e. The inter alkyne termini distance of 2d is greater than that of 1d by ∼0.12 Å. Oxidation at the phosphine leads to changes in structure at the alkyne termini resulting in a widening of the enediyne framework. Interestingly, the interalkynyl distance of 2e is reduced by ∼0.26 Å relative to 1e. Here, one of the alkyne arms has rotated the bulky –CF3 substituted rings to the exterior of the enediyne framework lessening the steric clash between the large aromatic substituents. Overall, these structural data are consistent with the high cyclization temperatures observed in the DSC analyses and highlight the geometric similarities between these substituted phosphinoenediynes.
Isolation of sensitive Pt(II) phosphine enediyne dichloride complexes
Using this suite of functionalized phosphine enediynes, we synthesized a series of square planar platinum(II) complexes, 3a–3h, bearing these ligands. Buchwald's original report for the in situ formation of 3a showed spontaneous solution cyclization at room temperature.35,43 Within this well-defined geometric framework, we recognized the opportunity to investigate how unexplored electronic modifications at the phosphine termini can lead to acceleration or retardation of ambient temperature Bergman cyclization kinetics.Compounds 3a–3g have been successfully synthesized and spectroscopically characterized, representing the first Pt(II) phosphine enediyne suite isolated to date. For all compounds except 3d, addition of 1.2 equivalents of phosphine enediyne to a solution of dichloro(1,5-cyclooctadiene)platinum(II), Pt(cod)Cl2, in dichloromethane affords the desired metalloenediynes as pale-yellow solids (Scheme 2). In the synthesis of 3d, a slight excess of metal precursor is required to isolate the intended Pt(II) phosphine enediyne product. Successful synthesis of 3a–3g required optimization of reaction times and temperatures in all cases. These synthetic changes suggest that R-group modifications of the phosphine ligands may have substantial consequences on metalloenediyne reactivity as some complexes such as 3b were unstable if isolated at room temperature while others such as 3f required prolonged reaction times at 25 °C. Though complex 3h bearing the tert-butyl-substituted phosphine enediyne was observed in situ by NMR spectroscopy, attempts to isolate this complex proved unsuccessful.
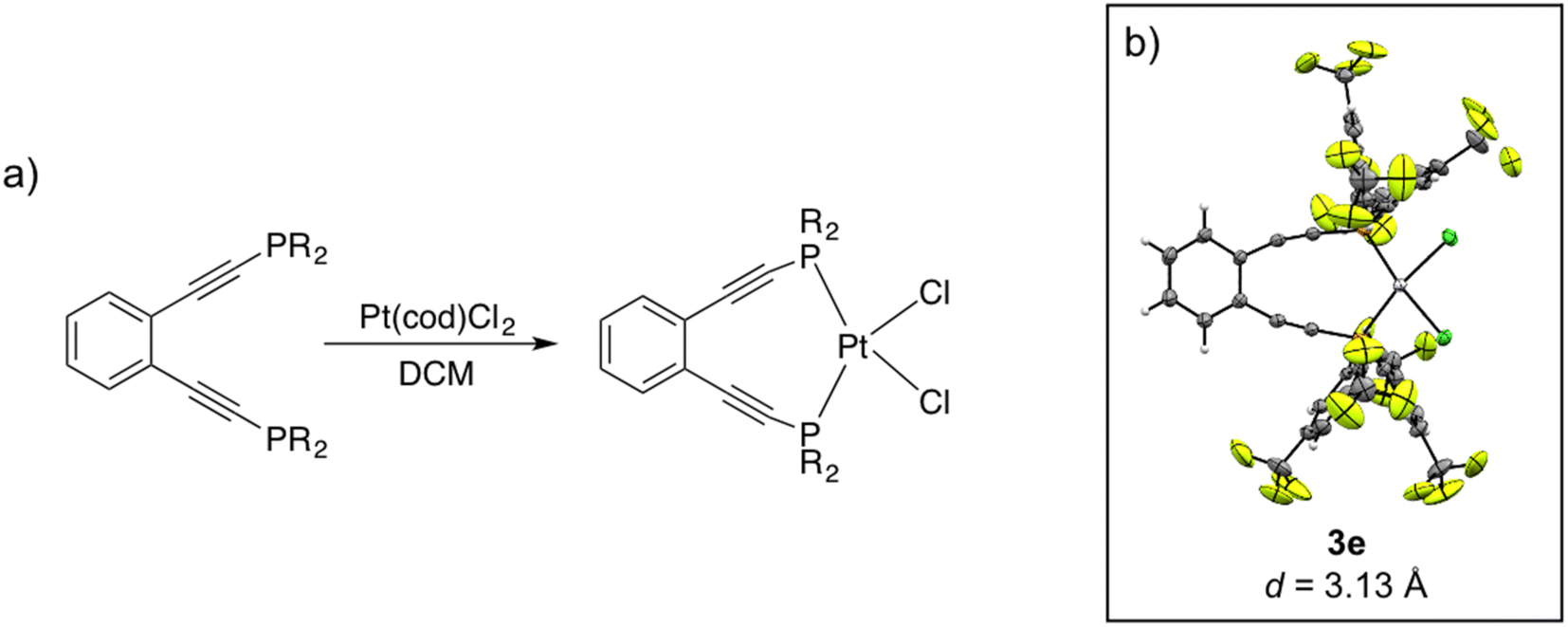 | ||
| Scheme 2 (a) General synthesis of Pt(II) phosphine enediyne dichloride complexes (3a–3g). (b) X-ray crystallographic structure of 3e with interalkynyl distance (d) shown below. Thermal ellipsoids are drawn at 50% probability. | ||
The crude solids of 3a–3g isolated directly from the reaction mixtures contained Pt(cod)Cl2 impurities by 1H NMR spectroscopy. Alteration of the synthetic conditions to eliminate this impurity was ineffective for all complexes, and in many instances resulted in the formation of intractable material if reaction times and/or temperatures were increased. Consequently, these metalloenediynes were purified to give pale-yellow solids either through preparatory silica chromatography (3a–3b, 3d, and 3f–3g) or repeated washings with diethyl ether (3c and 3e). Attempts to purify the fluorinated metalloenediynes 3c and 3e using chromatography led to the degradation of these complexes on the chromatographic substrate. This was unanticipated as the fluorinated ligands 1c and 1e are repeatedly purified by this method with no evidence of enediyne degradation. Once purified, compounds 3c and 3e–3g are stable for ∼10 days at −20 °C. In contrast, metalloenediynes 3a–3b and 3d degrade significantly upon standing at −20 °C for ∼48 h.
Interestingly, addition of a slight excess of 1d to a solution of Pt(cod)Cl2 in dichloromethane affords not the monomeric complex 3d (31P δ = −16.53 ppm), but rather the dimeric complex 5d (31P δ = −2.61 ppm) (Scheme 3). X-ray crystallography reveals 5d to have two equivalents of PtCl2 bridged by two molecules of the phosphine enediyne 1d generating a C2-symmetric dimer. The inter alkyne termini distance of 5d which has the methylated ligand 1d bridging two platinum centers is exceptionally large at 4.53 Å which suggests that this dinuclear complex is thermally robust (Table 2S†). There is significant distortion from linearity in the C–CC and CC–P bond angles which measure 168 and 163°, respectively, to accommodate bridging both platinum centers. The differential scanning calorimetry trace of 5d shows no exothermic event indicative of cyclization up to 280 °C which is consistent with the large alkyne termini separation. Incubation of 5d with excess 1,4-cyclohexadiene (1,4-CHD) at room temperature shows no reactivity over the course of 72 h when monitored by NMR spectroscopy, confirming that this dinuclear complex has a high activation barrier to diradical generation due to the increased alkyne termini separation. Buchwald reported a similar enediyne-bridged Pd(II) dimer of 1a on the basis of a saturation transfer experiment and suggested that the dimeric and monomeric Pd(II) complexes of 1a were in equilibrium with one another.35,43 Our observations with 5d, however, do not indicate interconversion between this dinuclear species and the mononuclear complex 3d, likely due to the slower ligand exchange kinetics observed for platinum complexes relative to their lighter congeners.
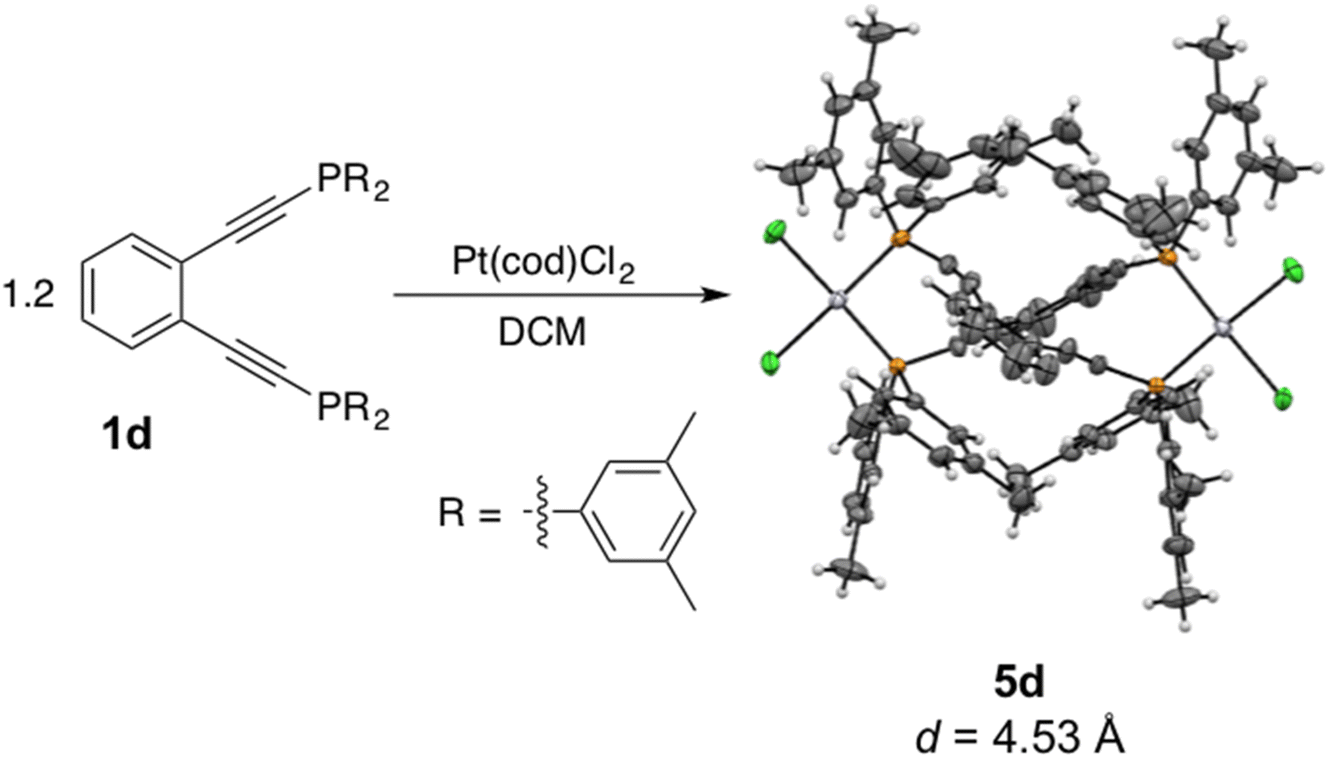 | ||
| Scheme 3 Synthesis and X-ray crystallographic structure of the Pt(II) phosphine enediyne-bridged dimer 5d. Thermal ellipsoids are illustrated at 50% probability and the interalkynyl distance (d) is provided below. | ||
The 31P NMR chemical shifts of 3a–3g show a similar trend to that observed for the free ligands 1a–1g. There is a large discrepancy (>30 ppm) in the chemical shifts of the aromatic- (3a–3e) and aliphatic-substituted (3f–3g) metalloenediynes; however, all aryl complexes give 31P NMR resonances within a 3 ppm range. From the chemical shift data alone, the remote substitutions at the meta or para positions of the phenyl rings at the phosphine termini do not appear to drastically change the chemical environment about the phosphine. However, the rapid degradation of 3a–3b and 3d relative to 3c and 3e at −20 °C suggests that the aryl substituted metalloenediynes functionalized with electron donating groups are more thermally sensitive than those bearing electron withdrawing groups.
Crystalline material was obtained from vapor diffusion of hexane or pentane into saturated dichloromethane solutions of 3e and 3f, respectively, at −20 °C and X-ray crystallographic analysis confirms these complexes as square planar Pt(II) phosphine enediyne dichloride complexes. Deviations from linearity of ∼15° and 7° are observed in the C–CC and CC–P angles for both 3e and 3f; however, these deviations are anticipated as bent acetylenic units are commonly observed for enediyne complexes and have been attributed to in-plane π–π repulsions between the alkyne moieties (Table 3S†).31 The inter alkyne termini distances are found to be 3.13 and 3.10 Å for 3e and 3f, respectively, which are considerably shorter than those computationally predicted for the Pd(II) and Pt(II) analogues of 1a (3.3 Å).35 The alkyne termini separation of ligand 1e is significantly reduced (>1.0 Å) upon platination, clearly demonstrating the drastic structural changes that can accompany metal ion complexation. The interalkynyl distances of 3e and 3f are well within the critical distance experimentally proposed by Nicolaou23 (3.2–3.31 Å) and computationally by Schreiner25 (2.9–3.4 Å) suggesting that these divalent complexes, and derivatives thereof, should cyclize spontaneously at ambient temperature. It is important to note that the solid-state structures of 3e and 3f are quite similar in all bond lengths and angles despite having notably different R-groups at the phosphine termini (3e: R = Ph-m2CF3; 3f: R = iPr). These data demonstrate that complexation of free ligands 1a–1g with Pt(II) has generated a suite of metalloenediyne complexes with a nearly uniform, rigidly defined structure. As a result, changes in geometric influences on Bergman cyclization are minimized allowing for clearer interrogation of electronic effects caused by variations in phosphine termini functionalization.
Differential scanning calorimetry has been used to probe the cyclization temperatures of 3a–3g. Although these metalloenediynes all demonstrate solid-state cyclization, complexes 3a–3d and 3f–3g show exothermic maxima between 106–177 °C, while 3e cyclizes at a considerably higher temperature, 236 °C (Table 1). The cyclization temperatures of aryl-substituted metalloenediynes increase as electron donating substituents are replaced with electron withdrawing groups suggesting that the former are more thermally reactive than the latter. The elevated cyclization temperature of 3e relative to the other derivatives likely results from solid-state packing differences (i.e., π-stacking between aryl rings, hydrogen bonding with fluorine atoms, etc.) not experienced by the other metalloenediynes.
| Bergman cyclization temperature/°C | ||
|---|---|---|
| Onset | Max | |
| 3a | 75 | 116 |
| 3b | 70 | 120 |
| 3c | 110 | 177 |
| 3d | 65 | 106 |
| 3e | 208 | 236 |
| 3f | 100 | 139 |
| 3g | 91 | 140 |
It is challenging to directly compare the cyclization temperatures of 3a–3b and 3d–3e to their corresponding free ligands, 1a–1b and 1d–1e due to differences in their phases during DSC analysis in which solid-state cyclization temperatures are often upwards of 70 °C higher than those of neat liquids. However, the data clearly show that metalation leads to a drastic reduction in the thermal activation barrier to diradical generation and suggests that these metalloenediynes should demonstrate facile cycloaromatization under ambient solution conditions. These results are unsurprising as the use of coordination chemistry to geometrically modulate Bergman cyclization has been well established.35,41,44–51 Moreover, the range of cyclization temperatures across this series of metalloenediynes indicates that electronic substitutions on the ligand framework, while seemingly inert in the free ligands, make dramatic impacts on the electronic component of the cyclization barrier in these well-defined, square geometric complexes.
Electronic modulation of the activation barriers to thermal Bergman cyclization
While solid-state cyclization temperatures and crystallographically-determined inter alkyne termini distances provide an approximation of the thermal reactivity of complexes 3a–3g, kinetic studies in solution on the cyclization of these metalloenediynes can accurately measure activation barriers to Bergman cyclization and determine rates of cycloaromatization. Comparison of kinetic activation parameters across this series of metalloenediynes reveals the extent to which phosphine functionalization electronically influences thermal Bergman cyclization rate.To that end, 31P NMR spectroscopy was used to monitor the conversion of metalloenediynes 3a–3g to their corresponding cyclized products 4a–4g in the presence of 1,4-CHD in deuterated chloroform (Scheme 4). Though a reliance of cyclization rate on H-atom donor concentration has been observed for benzannelated enediynes, this dependence is less pronounced at high radical quench concentrations.30,42,52–62 Additionally, analysis of Bergman cyclization kinetics for the original in situ cycloaromatization of 1a with a soluble PdCl2 source found this reaction to be essentially zero order in 1,4-CHD.35,43 Therefore, only a 100-fold excess of 1,4-CHD was used in this study to establish pseudo-first order conditions. Metalloenediynes 3a–3g were purified directly before use to ensure the integrity of the samples. All studies were performed in triplicate, under ambient conditions, and in the presence of triphenylphosphine oxide (Ph3PO) which served as an inert internal standard. The disappearance of 3a–3g was followed to at least 90% completion by integration of the 31P NMR signal of the starting metalloenediyne vs. that of Ph3PO. Cyclization was monitored in 5 °C increments for each complex: 3b (5–20 °C); 3a, 3d (10–25 °C); 3c (15, 25–35 °C); 3e–3g (25–40 °C), and all samples were observed to have well-behaved pseudo-first order kinetics (R2 > 0.98) at all temperatures (Fig. 2S†). These data were then used to determine rate constants and derive the free energy of activation (ΔG‡) for this radical-mediated transformation.
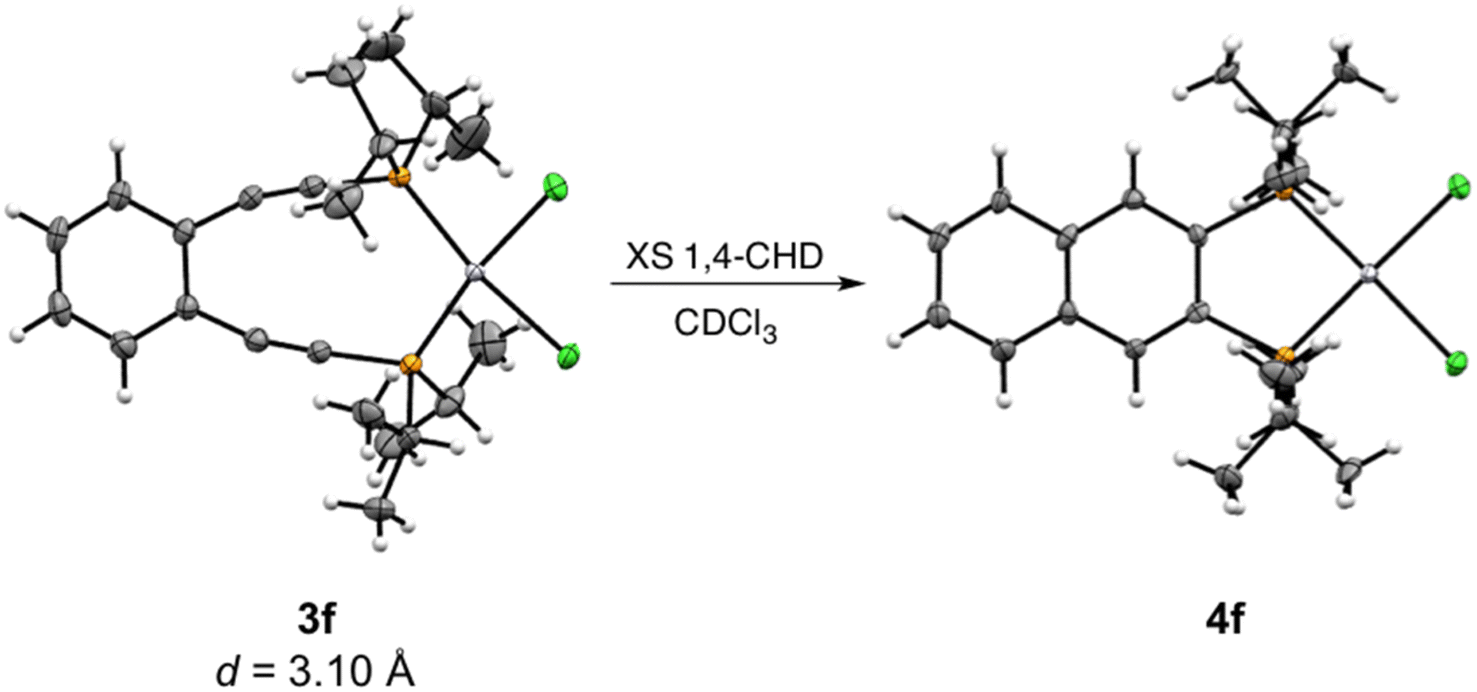 | ||
| Scheme 4 Ambient temperature Bergman cyclization of 3f to generate 4f. Thermal ellipsoids are illustrated at 50% probability and the interalkynyl distance (d) is given below. | ||
By 31P NMR spectroscopy, the cyclization of all complexes except 3g proceeds cleanly to the corresponding cyclized product in >90% yield (Fig. 2). In the case of 3g, though the cyclized product 4g is identifiable by 31P NMR, other phosphorus-containing species are also observed suggesting that self-quenching of the diradical intermediate with internal C–H bonds from the cyclohexyl substituents may occur leading to the formation of multiple products. Following the kinetics measurement, the reaction mixtures from all trials for a given compound were combined and purified using preparatory silica gel chromatography allowing for isolation and characterization of the cyclized compounds 4a–4f. Unfortunately, attempts to isolate 4gvia this same purification method proved unsuccessful due to the high quantities of side products. Complexes 4a and 4c–4f were isolated as crystalline materials and X-ray crystallography shows that these Bergman cyclized products adopt nearly idealized square planar geometries about the divalent platinum center (Fig. 3S†). The Pt–P and Pt–Cl bond lengths are conserved across the series and are consistent with other Pt(II) phosphine dihalide complexes (Table 4S†).41,63 Comparison of 4e and 4f to their metalloenediyne counterparts 3e and 3f shows slight bond angle changes, the most notable being the P1–Pt1–P2 angle which is reduced ∼17° upon closing of the inter alkyne termini distance.
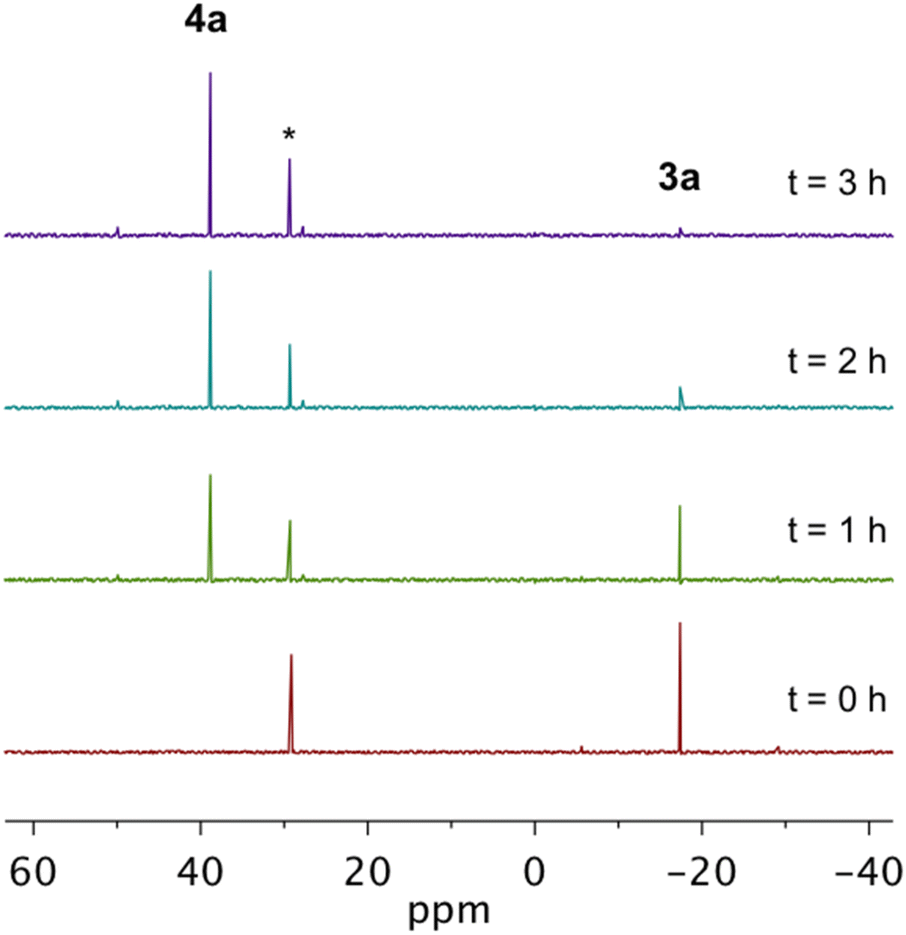 | ||
| Fig. 2 31P NMR spectroscopy demonstrating the cyclization of 3a (δ = −17.44 ppm) in the presence of 1,4-CHD to afford 4a (δ = 39.42 ppm) in near quantitative yield at 25 °C. These data are representative of the cyclizations of 3b–3f which proceed cleanly to the cyclized products 4b–4f. The designated (*) signal corresponds to triphenylphosphine oxide, the internal standard. | ||
Analysis of the room temperature kinetic studies reveals that the rate of Bergman cyclization of these Pt(II) metalloenediynes increases on the order of 3f < 3e < 3g < 3c < 3a ∼ 3d < 3b (Fig. 3a–b). In fact, cyclization of 3b proceeds so rapidly at room temperature that an accurate assessment of the rate could not be made. Consequently, cyclization studies had to be performed at or below 20 °C to obtain reliable kinetic data which could then be extrapolated to gain insights into the reactivity of this complex at 25 °C. Similarly, complexes 3a and 3d show rapid cycloaromatization at room temperature requiring all subsequent kinetic studies to be performed at low temperature.
 | ||
| Fig. 3 Summary of Bergman cyclization kinetics for complexes 3a–3g (a) at 25 °C and (b) at 15 °C. Sample Eyring plots of (c) 3f and (d) 3a used to calculate activation parameters. | ||
At 25 °C, the half-life (t1/2) of cyclization for 3a–3g spans a range of almost 35 hours, highlighting the dramatic thermal tunability that is afforded through functionalization of the phosphine enediyne with various electron donating and withdrawing substituents (Table 2). The temperature-dependent cyclization rates allow for the generation of Eyring plots and the elucidation of the free energy of activation, ΔG‡, at room temperature (Fig. 3c, d and 4S†). Cyclization parameters have been experimentally determined for both aryl (3a–3e) and aliphatic (3f–3g) substituents and in nearly all cases, the aliphatic substituents show slower cyclization kinetics and higher activation barriers to cycloaromatization than their aryl counterparts (Table 2). For acyclic enediynes, Schreiner has shown that direct functionalization of the alkyne termini with alkyl groups increases the endothermicity of Bergman cyclization due to stabilization of the enediyne core orbitals.25 We observe a similar increase in endothermicity upon alkyl substitution at the phosphine termini; however competing internal C–H bond quenching of the diradical intermediate in 3g suggests that the rate-determining pathway for the alkyl substituent constructs may differ from the mechanistic consistency of the aryl family.
| Experimental kinetic activation parameters at 25 °C kcal−1 mol−1 | ||||
|---|---|---|---|---|
| Compound | t 1/2/h | k/h−1 | ΔG‡ | |
| a Complex 3c contained ∼5% Pt(cod)Cl2 impurity. b Data in ( ) extrapolated from Eyring plot. | ||||

|
3f | 33.3 | 0.02 | 19.72 |

|
3e | 27.1 | 0.03 | 19.62 |

|
3g | 11.3 | 0.06 | 19.10 |

|
3c | 4.4 | 0.16 | 18.45 |

|
3a | 0.73 | 0.95 | 17.47 |

|
3d | 0.70 | 0.99 | 17.47 |

|
3b | (0.36) | (1.90) | 17.07 |
With regards to the aryl phosphines (3a–3e), the presence of electron donating substituents (3b and 3d) clearly accelerates Bergman cyclization while electron withdrawing groups (3c and 3e) retard cyclization. In some cases, the rate enhancement for structurally analogous metalloenediynes is minimal and in others, it is quite dramatic. For example, complex 3b (R = Ph-pOCH3) cyclizes approximately 10× faster than 3c (R = Ph-pCF3) whereas 3d (R = Ph-m2CH3) has a cyclization rate >30 times faster than the analogous 3e (R = Ph-m2CF3). As expected, these trends are mirrored in the ΔG‡ data, with the activation barrier varying by ∼2.6 kcal mol−1 across the series of aryl-substituted metalloenediynes (Table 2). Specifically, for the para- (3b and 3c) and meta-substituted (3d and 3e) derivatives, the free energy of activation decreases by ∼1.4 and ∼2.2 kcal mol−1, respectively, when electron withdrawing –CF3 groups are replaced with electron donating substituents. Overall, these data clearly highlight the role that electronic substitutions can play in controlling activation barriers to Bergman cyclization.
Probing the origin and mechanism of electronic control for aryl-substituted metalloenediynes
The rate dependence for Bergman cyclization on electron donating and withdrawing substituents for acyclic enediynes has been established by Schmittel33 and Schreiner.34 Here, as electron withdrawing substituents are added at the alkyne termini positions, the activation barrier for the reaction decreases due to reduced electron–electron repulsion of the in-plane pπ-orbitals in the transition state which facilitates C–C bond formation. This result is intuitive in that distributing electron density across multiple atoms via electron withdrawing functionalities decreases charge repulsion, whereas electron donors increase it.Broadly, our results are consistent with this literature precedence. Complexes with categorical electron withdrawing aryl substituents (3a–3e) generally have shorter half-lives and lower activation barriers to cycloaromatization than do those with electron donating alkyl substituents (3f and 3g) (Table 2). However, the rate enhancement within the series of the aryl-substituted metalloenediynes with electron donating groups (3b and 3d) relative to those with electron withdrawing groups (3c and 3e) is counter to this paradigm. The additional complexity of 3a–3e, i.e., the presence of an insulating heteroatom (P) and a PtCl2 fragment, diversify the electronic structures of these constructs globally, and within the geometrically equivalent metalloenediyne frameworks. This invariance is supported by the X-ray structures of Bergman cyclized Pt(II) products 4a and 4c–4e (vida supra) suggesting that there exists minimal geometric differences amongst these complexes. Thus, an important feature of this molecular suite is that we have in essence, independently accounted for the geometric and electronic contributions to thermal Bergman cyclization, which can be readily evaluated using computational methods.
The reaction profiles of simple organic enediynes25,31,34,64 and metalloenediynes37,65–67 to the diradical intermediate have been successfully modeled with density functional theory (DFT) and yield activation barriers consistent with experimental findings. The choice of theoretical approach plays a key role in modeling transition state structures and predicting activation energies. Many have noted that hybrid Hartree–Fock DFT methods such as B3LYP do not effectively model the diradical intermediate leading to spurious results, which at times can overestimate barrier heights by upwards of 8 kcal mol−1.25,31,68,69 It has been demonstrated however, that pure density functionals such as BLYP and BPW91 give computational activation parameters that are consistent with those obtained experimentally.34,37
Bergman cyclization of 3a–3e was modeled with DFT using Gaussian 16 (ref. 70) to confirm the geometric similarities across this suite of metalloenediynes, obtain activation parameters, and to visualize the molecular orbitals that play key roles in the formation of the reactive diradical intermediate. Both B3LYP71–73 and B3PW91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 72 yielded activation barriers that were consistently 5–10 kcal mol−1 higher in energy than those obtained experimentally, even when correcting for empirical dispersion via Grimme's GD3BJ74 damping function. The results from the pure BPW9171,75–79 functional however, were consistent with experimental findings within <2 kcal mol−1. Therefore, all structures were optimized using the (U)BPW91 functional with the 6-31G** basis set and the LANL2DZ80–82 pseudopotential was applied to transition metal atoms. The spin-unrestricted approach83 was used to compute diradical intermediates which were found to be open-shell singlet in character. Vibrational frequency calculations were performed at the (U)BPW91/6-31G**/LANL2DZ level of theory to confirm convergence to minima for ground state geometries and first-order saddle points for transition state structures, respectively, as well as provided zero-point energy (ZPE) corrections at room temperature. After performing all calculations in the gas phase, solvated single point energies were evaluated using the PCM model84–88 at the same level of theory to account for energy differences due to solvation in chloroform (ε = 4.71).
72 yielded activation barriers that were consistently 5–10 kcal mol−1 higher in energy than those obtained experimentally, even when correcting for empirical dispersion via Grimme's GD3BJ74 damping function. The results from the pure BPW9171,75–79 functional however, were consistent with experimental findings within <2 kcal mol−1. Therefore, all structures were optimized using the (U)BPW91 functional with the 6-31G** basis set and the LANL2DZ80–82 pseudopotential was applied to transition metal atoms. The spin-unrestricted approach83 was used to compute diradical intermediates which were found to be open-shell singlet in character. Vibrational frequency calculations were performed at the (U)BPW91/6-31G**/LANL2DZ level of theory to confirm convergence to minima for ground state geometries and first-order saddle points for transition state structures, respectively, as well as provided zero-point energy (ZPE) corrections at room temperature. After performing all calculations in the gas phase, solvated single point energies were evaluated using the PCM model84–88 at the same level of theory to account for energy differences due to solvation in chloroform (ε = 4.71).
Bergman cyclization reactions encounter multiple transition states: the first involves conversion of the enediyne to the diradical intermediate while subsequent barriers describe the H-atom abstraction steps to yield the benzannelated product. Relative to parent (Z)-hex-3-ene-1,5-diyl frameworks, incorporation of the enediyne backbone into an aromatic ring is known to increase the endothermicity of diradical formation while concomitantly lowering the barrier to retro-Bergman ring opening due to reduced aromatic stability of the naphthyl product.30,42,53,54,57–60 As such, the rate of cyclization of benzannelated enediyne complexes depends not only on formation of the diradical intermediate (k1) but also on the retrocyclization (k−1), while the H-atom abstraction (k2) pathways are considered comparable for structurally analogous compounds. However, when pseudo-first order conditions are achieved using sufficiently high concentrations of radical quenching agent, cyclization becomes the rate-limiting step.60,61
The interplay between k1 and k−1 on the observed rate can be gleaned from the computational barrier heights for the forward and retrocyclization reactions. Here, for the fastest cyclizing compound 3b, the forward barrier height is one of the lowest of the set 3a–3e by 1.8 kcal mol−1, while the barrier to retrocyclization is the highest by 1.2 kcal mol−1, therefore contributing to the largest observed rate. Conversely, 3e has the most significant barrier to Bergman cyclization and the smallest to retrocyclization, thereby retarding the observed reaction rate appreciably. Thus, the two barriers have complementary effects on the rate of the overall cyclization reaction. The first activation barrier leading to formation of the reactive diradical intermediate, however, is the more energetically dominant of the two contributors to the overall rate constant for these Bergman cyclization reactions.
These variations in state energies and rates must derive from geometric and/or electronic structural changes at the molecular level.56 Comparison of the computed structures for the metalloenediynes 3a–3e finds that all compounds have nearly identical enediyne core geometries, to within ∼0.05 Å and 3° in bond lengths and angles, respectively. Furthermore, the transition state geometries, 3a-TS–3e-TS, are also consistent in bond and angle metrics between all complexes investigated (Fig. 4). These computational findings agree with the experimental crystallographic data and support the proposal that the observed rate enhancements or retardations result from electronic rather than structural differences that manifest upon R-group modification at the phosphine termini. For complexes 3a–3e, geometric rearrangements between the reactants and products are minimal due to the structure enforced by the square planar metal center. Consequently, electronic reorganization may exhibit a greater level of control over the cyclization event than what has been observed previously in simple enediyne systems.
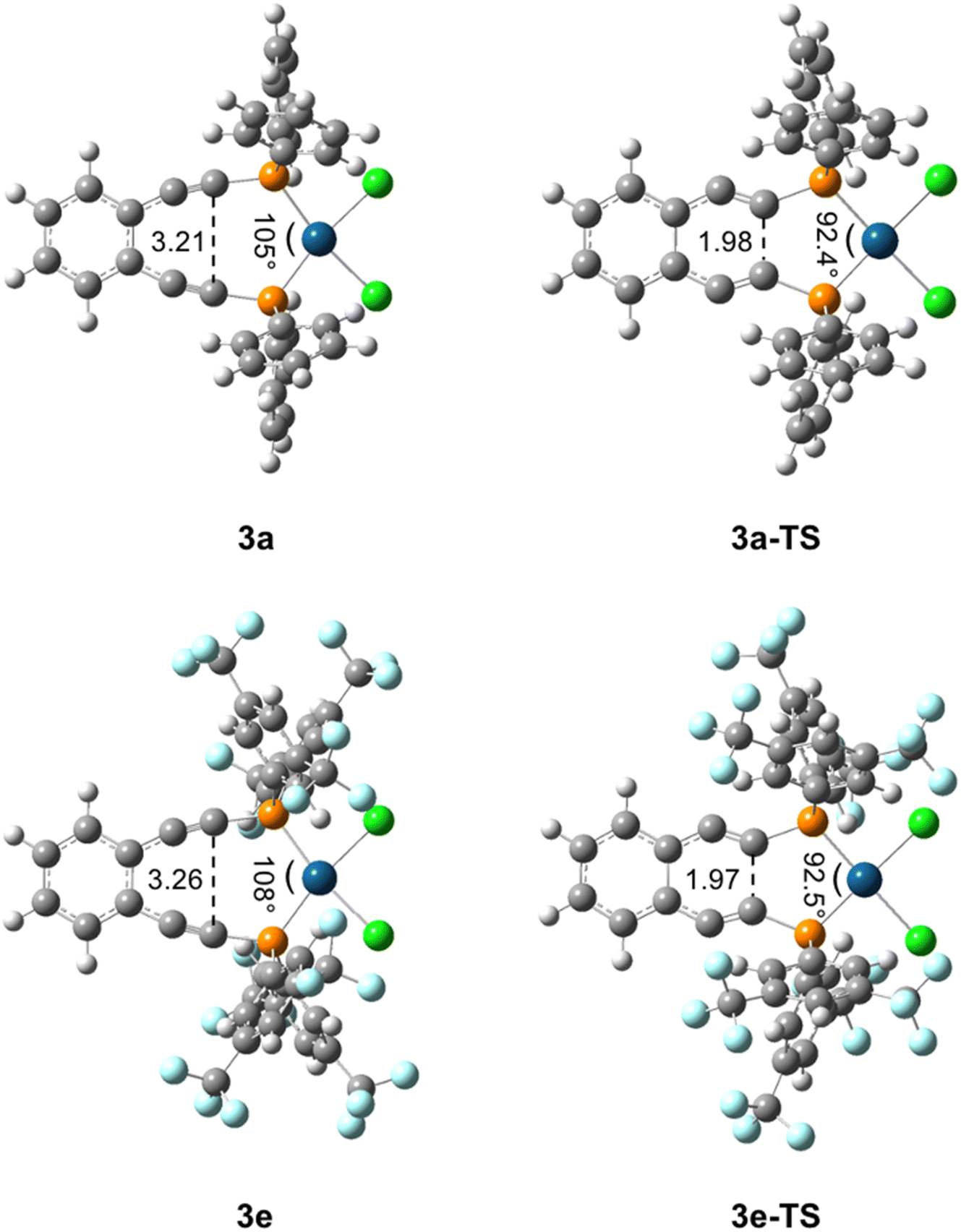 | ||
| Fig. 4 Key structural parameters of (U)BPW91/6-31G**/LANL2DZ optimized metalloenediynes 3a and 3e and their transition states 3a-TS and 3e-TS. These bond length (Å) and angle (°) changes are representative of those observed for all metalloenediynes 3a–3e and transition states 3a-TS–3e-TS. | ||
For enediyne frameworks, electron repulsion between filled in-plane π-orbitals accounts for considerable destabilization upon bending of the alkynes to facilitate sigma C–C bond formation.56 Removal of electron density from this in-plane π-system promotes formation of the cycloaromatized products by lowering the energy of the transition state. The relationship between 13C NMR chemical shifts and atomic charges has been studied both experimentally and computationally. Although effects may vary depending on hybridization, the general consensus is that for a series of closely related compounds, charge-shift trends can provide a good approximation of electronic polarization.89–92 Specifically, 13C NMR spectroscopy has been shown to be a sensitive reporter of electron density in acetylenic moieties, with a correlation demonstrated between the shielding of an alkyne carbon and the π-electron density on that atom.93–98 The distribution of electron density in the alkyne carbons of a ground state structure has important consequences on the electronic rearrangement that occurs upon formation of the reactive diradical intermediate. Much like crystallographic analysis, 13C NMR investigations of the reactive metalloenediyne complexes 3a–3e were prohibited due to the long acquisition times needed for data collection and the highly reactive nature of the complexes. However, with computational support that structural differences amongst 3a–3e are minute, the geometrically-locked phosphine oxide free enediyne ligands, 2a–2e, can be used as model systems to experimentally interrogate alkyne charge distribution in the ground state.
It is clear from the 13C NMR shifts of 2a–2e that the alkyne carbon adjacent to the phosphine atom, CA, supports considerably more electron density than the alkyne carbon closer to the benzannelated backbone, CB (Fig. 5). The degree of shielding (CA) and deshielding (CB) of these signals reveals that all phosphine oxide ligands, regardless of aryl ring R-group substitution, are polar in nature, with the dipole moment situated such that CA is more electron-rich than CB. However, there appears to be a correlation between the identity of the R-group and the magnitude of alkyne polarization, with ligands bearing electron withdrawing substituents being more highly polarized than those supporting electron donating substituents (Δδ(CC): 2e = 19.6 ppm; 2b = 12.6 ppm). The polar nature of the alkyne carbons of the free ligand phosphine oxides demonstrates that subtle changes in the enediyne framework itself can be used to tune the electronic properties of ligand. Moreover, it suggests that there likely exist differential charges in the acetylenic moieties of the reactive metalloenediyne complexes 3a–3e that, over the course of the cyclization reaction, may play a role in governing barrier heights to diradical formation. This 13C NMR charge-shift analysis may not be indiscriminately applied to all acetylenic systems; however, correlations can be drawn in this case due to the highly conserved geometry across the series of phosphine oxide enediyne ligands and corresponding metalloenediynes.
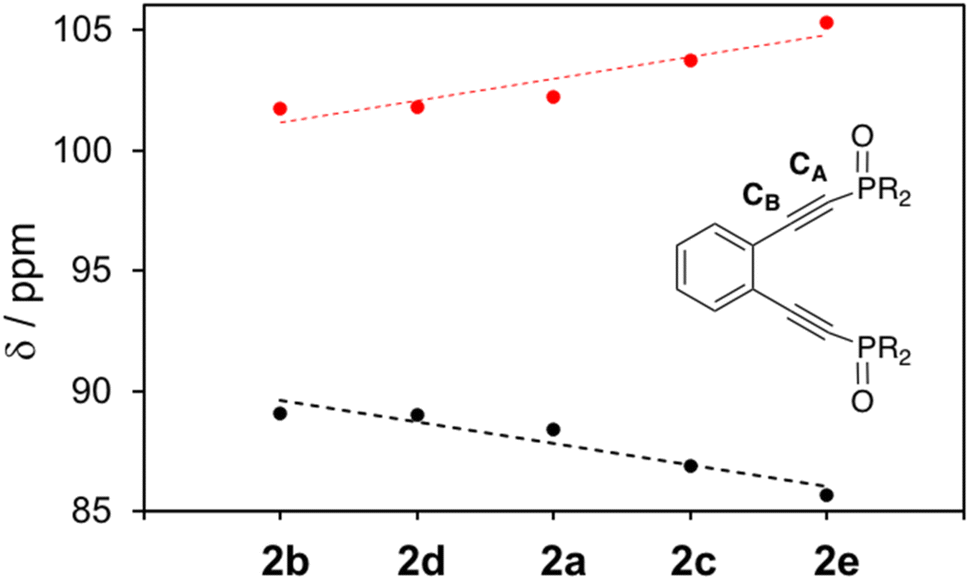 | ||
| Fig. 5 13C NMR resonances of phosphine oxide analogues 2a–2e revealing the polar nature of the alkyne carbons, CA (black) and CB (red), the magnitude of which is enhanced in the presence of electron withdrawing substituents. | ||
Although 13C NMR spectroscopy provides information about the relative distribution of electron density in the alkyne carbons of the organic-only enediyne oxide frameworks, in-depth charge analysis of the computed ground state (GS) and transition state (TS) structures of the reactive metalloenediynes 3a–3e can more definitively establish whether there exists a correlation between the polarization of the enediyne framework and activation barriers to Bergman cyclization. Nuclear rearrangement (i.e., bending of the alkynes) necessitates concomitant electronic reorganization to both facilitate σ-bond formation and to promote development of the diradical intermediate. Despite the importance of CA in C–C bond formation, consideration of only these differential charges may be an oversimplification of the electronic parameters that govern barriers to thermal Bergman cyclization. Indeed CB, though not directly involved in bond formation, is the site of diradical generation, and as such, likely experiences electronic perturbations which may also modulate activation barriers to cycloaromatization.
Bearing this in mind, natural bond order (NBO) charges from the computed GS structures of 3a–3e and those of corresponding transition states, 3a-TS–3e-TS, can be used to calculate the dipole moment of each alkyne (CA–CB) according to eqn (1) (Fig. 6a). In this case, the magnitude of the dipole, 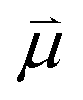 , is given by the product of the differential charges of the alkyne carbons (qA – qB) and the separation between those charges, rAB. It is important to consider not only the polarization of a single acetylenic unit but also the dipole interaction energy which is operative as geometric rearrangement brings the two arms of the enediyne framework in close proximity to initiate C–C bond formation. Eqn (2) shows that the dipole interaction energy is dependent on the dipole moments,
, is given by the product of the differential charges of the alkyne carbons (qA – qB) and the separation between those charges, rAB. It is important to consider not only the polarization of a single acetylenic unit but also the dipole interaction energy which is operative as geometric rearrangement brings the two arms of the enediyne framework in close proximity to initiate C–C bond formation. Eqn (2) shows that the dipole interaction energy is dependent on the dipole moments,  and
and 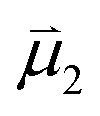 , of the alkyne fragments, the separation between the center of the two dipoles, r12, the permittivity of free space, εo, and the projection of the dipoles on one another, θ1, θ2, and θ12 (Fig. 6b). It is clear from the structures of 3a–3e and 3a-TS–3e-TS and the orientation of the CA–CB dipoles that there are both attractive and repulsive interactions between the two opposing alkynes; undoubtedly, the magnitude of these coulombic forces influences the electronic contributions to the activation energy for the reaction.
, of the alkyne fragments, the separation between the center of the two dipoles, r12, the permittivity of free space, εo, and the projection of the dipoles on one another, θ1, θ2, and θ12 (Fig. 6b). It is clear from the structures of 3a–3e and 3a-TS–3e-TS and the orientation of the CA–CB dipoles that there are both attractive and repulsive interactions between the two opposing alkynes; undoubtedly, the magnitude of these coulombic forces influences the electronic contributions to the activation energy for the reaction.
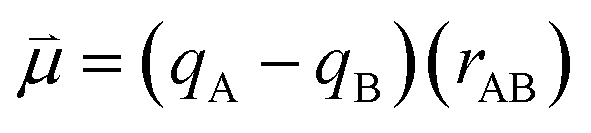 | (1) |
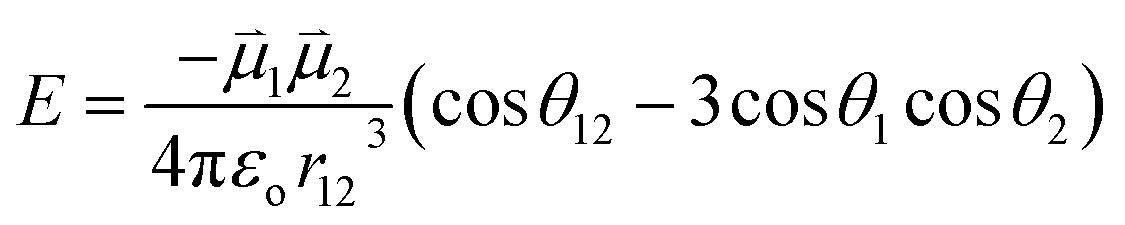 | (2) |
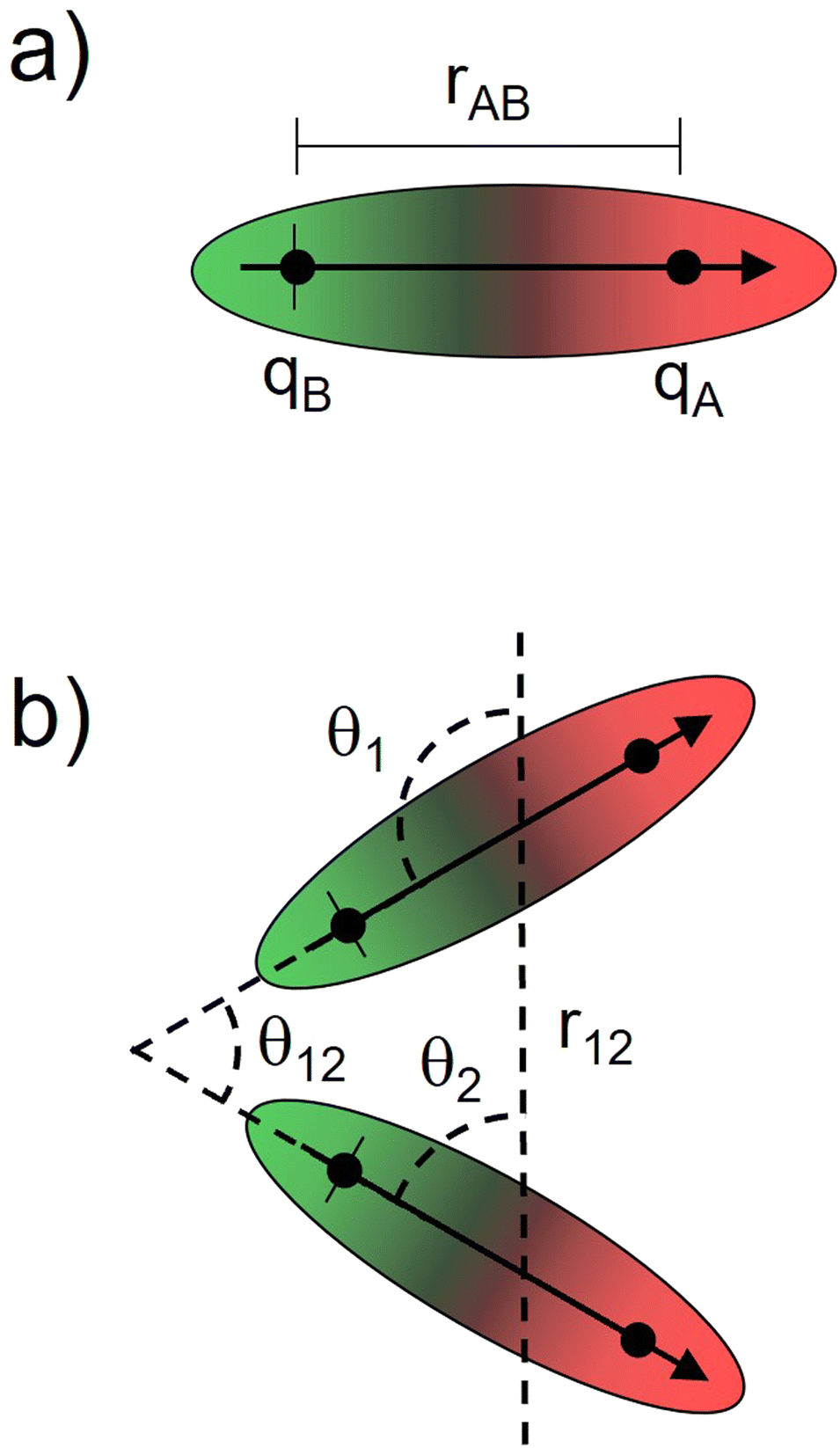 | ||
| Fig. 6 Global distribution of charge throughout the enediyne framework can be approximated by considering (a) the net polarization of alkyne carbons CA and CB and (b) the magnitude of coulombic interactions between opposing acetylenic dipoles. | ||
For all complexes in the ground state, CA and CB are negatively and positively polarized, respectively, resulting in a net dipole moment in the direction of CA (Fig. 7a). The magnitude of this CA–CB dipole increases in the presence of electron withdrawing substituents such that complexes 3c and 3e show the greatest charge separation between the alkyne carbons, consistent with the data from the 13C NMR analysis of the phosphine oxide enediyne ligands 2a–2e. Comparison of the ground state dipole interaction energies to the experimentally determined barrier height reveals an empirical correlation between the free energy of activation and the R-group of the phosphine-bound aryl rings (Fig. 7b). The complex with the highest activation barrier (3e: R = Ph-m2CF3) has the greatest coulombic repulsion between the alkyne fragments while the complex with the most facile cycloaromatization (3b: R = Ph-pOCH3) has the smallest dipole interaction energy.
 | ||
| Fig. 7 Plots of ΔG‡ as a function of alkyne dipole interaction energy, evaluated using (a) NBO charge analysis (representative example of 3a shown) for (b) enediyne ground states (3a–3e) and (c) calculated transition states (3a-TS–3e-TS) demonstrating the relationship between alkyne polarity (CA–CB) and experimental barrier height to thermal Bergman cyclization. | ||
NBO charge analysis of the computed transition state structures (3a-TS–3e-TS) shows that as geometric rearrangement begins, electron density flows from CB towards the site of σ-bond formation resulting in an increased negative charge on CA (Fig. 7a). The dipole moment (CA–CB) in the transition state lies in the same direction as that of the ground state and an overall increase in the polarization of the alkyne carbons is observed. Plotting the experimental activation barriers as a function of TS dipole interaction energy reveals the same correlation between enediyne R-group and free energy of activation as was observed with the GS structures (Fig. 7c). For complexes 3a-TS–3e-TS, those with electron withdrawing substituents are overall more polar in the transition state than those with electron donating groups. This NBO charge analysis provides a global picture of electron distribution within the enediyne framework and indicates that the presence of electron withdrawing substituents on the terminal phosphine aryl rings increases the coulombic repulsions in the transition state between the two acetylenic moieties in these metalloenediynes leading to an increase in barrier height for Bergman cyclization.
In addition to the dipole–dipole interaction energy contributions to the barrier, interrogation of the frontier molecular orbitals (FMO) of the enediyne core may give a more detailed perspective on how the distribution of electron density across the acetylenic carbons as a function of R-group influences the energetics for the cycloaromatization reaction. For the representative cyclization reaction involving (Z)-enediynes, Alabugin has computationally shown destabilization of the out-of-phase combination of the in-plane π-orbitals to be the largest energetic penalty paid to reach the transition state and achieve p-benzyne formation. This destabilizing effect, however, is offset by stabilization of the in-phase, in-plane π-orbitals which contribute to the formation of the new carbon–carbon sigma bond.56,69 A minor, secondary source of stabilization comes from the out-of-plane alkyne π-system which yields a conjugated aromatic product upon H-atom abstraction.56 Since the activation barrier to Bergman cyclization is simply the difference between the energies of the reactants and the transition state,69 the degree of stabilization or destabilization of the in-plane symmetric and antisymmetric π-orbitals, respectively, should predominantly govern the barrier height and thus thermal cycloaromatization of enediyne-containing constructs.
Examination of the molecular orbital manifolds of 3a–3e and their associated transition states reveals several orbitals with in-plane π-orbital density indicating that it is an orbital ensemble rather than a singular stabilization/destabilization effect that contributes to the overall acceleration or retardation of Bergman cyclization kinetics. Regardless, some of these in-plane π-orbitals show markedly greater energy differences between the ground and transition states suggesting that they contribute significantly to the overall barrier height. The antisymmetric in-plane π-orbitals, which become the site of diradical formation, are destabilized in the transition state for all complexes 3a–3e. For a set of complimentary orbitals in the ground state, electron density is delocalized across the in-plane π-system as well as the platinum center and the two chloride ligands. In the transition state however, the electron density of these orbitals is now almost exclusively localized in the in-plane π*-orbitals. The magnitude of this destabilization varies as a function of R-group, with complexes bearing electron withdrawing groups, 3e, being destabilized to a greater extent than those with electron donating substituents, 3b (Fig. 8). When viewed collectively with the NBO charge analysis, this electronic reorganization in the transition state results in differentially increased electron–electron repulsions between the approaching acetylenic carbons.
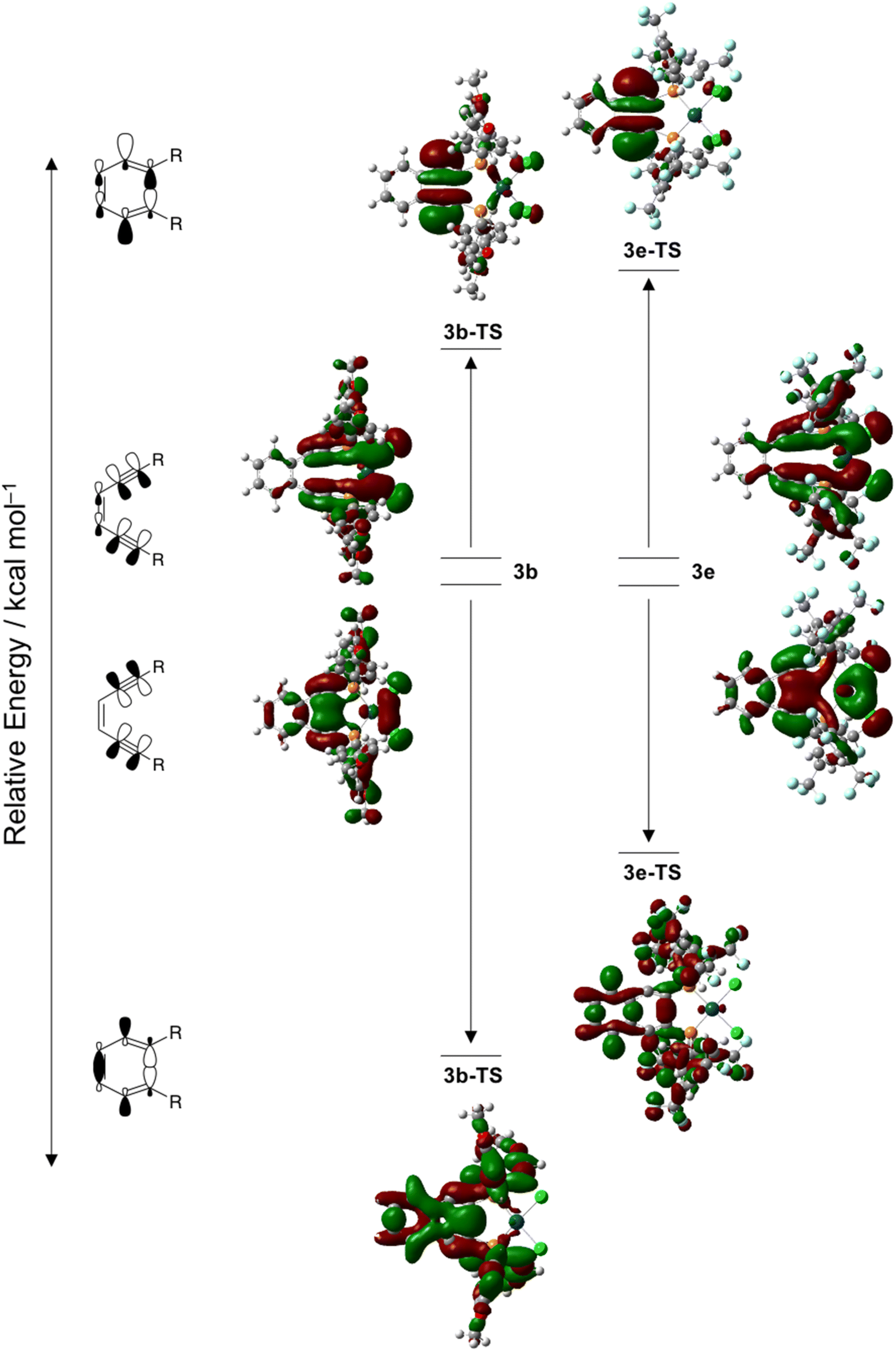 | ||
| Fig. 8 Walsh diagram showing the in-plane alkyne π/π*-orbitals most stabilized and destabilized by the cyclization reaction of complexes 3b and 3e and their associated transition states 3b-TS and 3e-TS. The degree of stabilization or destabilization of these orbitals in the transition state varies significantly due to the presence of electron donating (3b: R = Ph-pOCH3) or electron withdrawing (3e: R = Ph-m2CF3) groups at the alkyne termini. | ||
While this destabilization of the antisymmetric in-plane π-orbital is significant for all complexes 3a–3e, it is offset by the stabilization of the symmetric in-plane π-orbitals. Within this orbital manifold, complexes with electron withdrawing substituents generally have a distribution of orbitals with similar character compared to those with electron donating substituents, where electron density is localized to a single orbital. This aggregation of orbitals makes pinpointing the stabilization effect to a single orbital challenging. Nevertheless, comparison of a set of complimentary in-phase π-orbitals for complexes 3b and 3e finds the ground state orbitals to be similar in character to those of the antisymmetric set, with electron density distributed predominantly within the square planar core of the metalloenediyne. In the transition state, electron density about the PtCl2 fragment has decreased and there remains overlap between the in-phase alkyne π-orbitals, although the magnitude of this interaction appears R-group dependent (Fig. 8). In general, complexes with electron donating groups on the aryl phosphine rings have substantially greater stabilization of these in-phase, in-plane π-orbitals compared to those complexes bearing electron withdrawing substituents. This is consistent with the NBO charge analysis which finds complexes with less polar acetylenic bonds have lower barriers to cycloaromatization.
An additional noteworthy point: the symmetric in-plane π-FMOs for 3a-TS, 3b-TS, and 3d-TS have considerably more electron density within the aryl ring system which are predominately pσ-character, than do the corresponding orbitals of 3c-TS and 3e-TS, mainly pπ- and –CF3 lone pair character. The orbital parentage for complexes with electron donating substituents suggests that there exists π-orbital mixing between the acetylenic orbitals and the aryl ring system which substantially lowers the energy of the transition state orbitals involved in the formation of the developing C–C bond. Since the aryl ring system is chiefly low-lying pσ-character, this π-orbital mixing is reflected in a net stabilization of the transition state which affords a lower activation barrier to Bergman cyclization. The presence of electron withdrawing substituents on the terminal aryl rings decreases the energy of these pσ-orbitals such that they are no longer capable of interacting with those of the enediyne core. Consequently, only minimal stabilization of the developing σ-bond occurs which results in an overall increase in the activation barrier to cycloaromatization.
Finally, the degree with which the alkyne pσ- and pπ-density mixes with that of the PtCl2 fragment cannot be ignored. DSC data, experimental cyclization kinetics, and crystallographic analysis (vida supra) confirm that the significant rate enhancement or retardation of Bergman cyclization with the aryl-substituted enediynes is electronic in nature and not operable until platination occurs. In general, metalloenediynes with electron donating groups at the para or meta positions of the aryl rings have a higher percentage of frontier molecular orbitals that show mixing of the PtCl2 fragment with the alkyne orbitals relative to the complexes with electron withdrawing groups in these remote positions. The increased mixing of the alkyne and PtCl2 orbitals stabilizes the transition state which facilitates rapid Bergman cyclization at ambient temperature. Overall, for this suite of reactive metalloenediyne complexes, it appears that the R-group strongly influences the orbital mixing between the enediyne backbone, aryl ring system, and PtCl2 fragment which either promotes (EDGs) or impedes (EWGs) C–C bond formation which is directly reflected in the overall barrier height due to stabilization or destabilization of the transition state, respectively.
Conclusions
The original paradigms illustrating the critical distance for spontaneous Bergman cyclization by Nicolaou (d ∼3.2–3.3 Å), and significant geometric rate acceleration using square planar Pt(II) by Buchwald, have stood for more than a quarter century. Our development of a suite of functionalized phosphine enediynes bearing either aliphatic (iPr, Cy, tBu) or aromatic (Ph, Ph-pOCH3, Ph-pCF3, Ph-m2CH3, Ph-m2CF3) substituents with donor/acceptor functionality has moved the frontiers of both of these benchmarks simultaneously. With these ligands, we have synthesized a series of cisplatin-like complexes featuring an array of phosphine enediynes that represent the first divalent platinum phosphine enediyne complexes isolated as discrete structures to date. X-ray crystallography indicates that these Pt(II) dichloride complexes have greatly reduced inter alkyne termini distances relative to the free ligands, suggesting that these metalloenediynes should demonstrate very rapid Bergman cyclization under modest conditions. While these structures are primed to cyclize at 25 °C, their half-lives vary considerably (t1/2 ∼0.6–35 h) depending upon the donating or withdrawing nature of the substituents, even though experimentally, they are structurally analogous.The geometries and activation parameters of the aryl-substituted complexes are computationally consistent with the experimental findings suggesting that the electronic functionalization is the driving force controlling the cyclization rates and thus the stabilization of this otherwise geometrically reactive framework. The origin of this modulation derives from two coincident phenomena: (1) NBO charge analysis reveals that differential charges between the alkyne carbons contribute to an overall polarization of the in-plane π-system which either promotes (EDGs) or inhibits (EWGs) cycloaromatization and (2) complexes with electron donating substituents experience substantial orbital mixing between the acetylenic π-orbitals, the phosphine-bound aryl ring system, and the PtCl2 fragment resulting in a significant stabilization of the developing σ-bond and transition state. This, concomitant with reduced polarization of the alkyne carbons, lowers the activation barrier dramatically relative to those complexes bearing EWGs where orbital energies are disparate, and mixing is unfavorable. Together these new parameters push the boundaries of enediyne reactivity space well beyond what was originally deemed possible, and thus encourage creative structural manipulations to further advance the reaches of diradical utility.
Materials and methods
Materials
Reactions to synthesize enediyne ligands and platinum(II) phosphine enediyne dichloride complexes were carried out using standard dry box and Schlenk techniques under inert nitrogen atmosphere. All chemicals and solvents were purchased from commercial sources and used as received. Solvents were dried and distilled according to standard protocols unless otherwise noted. Deuterated solvents were dried over 4 Å molecular sieves and used without further purification. Flash and preparative chromatography were performed on silica gel (Sorbtech, 60 Å porosity, 230 × 400 mesh) and (Analtech, 1000 μm), respectively.Physical measurements
NMR spectra were collected on a Varian Inova 400 or 500 MHz NMR spectrometer using the residual proton resonance of the solvent as an internal reference. All 31P and 19F NMR spectra were externally referenced using 85% H3PO4 and 100% trifluoroacetic acid standards, respectively. Mass spectrometry data were acquired on an Agilent 1200 HPLC-6130 MSD mass spectrometer. Differential scanning calorimetry (DSC) measurements were made on a TGA Q10 DSC system with a TA Instruments thermal analyzer at a heating rate of 10 °C min−1. Infrared spectra were measured on a Nicolet 6700 FT-IR spectrometer equipped with OMNIC software. Elemental analyses were obtained from Robertson Microlit Laboratories, Inc. X-ray crystallographic data were obtained by the Indiana University Molecular Structure Center on a Bruker APEX II Kappa Duo diffractometer using Mo Kα radiation (graphite monochromator). Summaries of X-ray crystallographic analyses can be found in the ESI.†Computational methods
All calculations were carried out using Density Functional Theory (DFT) as implemented in Gaussian 16.70 Geometry optimizations were performed with the (U)BPW91 functional and 6-31G** basis set for all structures.71,75–79 The LANL2DZ effective core potential was used to model transition metal atoms.80–82 Calculation of the open-shell, diradical intermediates used the broken-symmetry, broken-spin approach which allows for the α and β electrons to be localized on separate carbon centers.99 Vibrational frequency calculations were performed at the (U)BPW91/6-31G**/LANL2DZ level of theory to confirm proper convergence to minima and maxima for equilibrium and transition state geometries, respectively, and to derive zero-point energy (ZPE) and entropy corrections at room temperature (298.15 K). Single point energy calculations in chloroform (dielectric constant ε = 4.71) were performed using the PCM model84–88 at the same level of theory to account for energetic contributions from solvation. The energy components were calculated following the standard protocol.67 Isosurface contour plots of all molecular orbitals were created with Gaussview 6.1.1 with isovalues at 0.02. Coordinates for all optimized geometries can be found in the ESI.†1,2-Bis((diphenylphosphino)ethynyl)benzene, dppeb (1a). This enediyne has been synthesized using a modified literature procedure35,43 to yield the ligand as a solid rather than an oil as previously reported. To a solution of EDTMS (2.17 g, 8.00 mmol) in 150 mL THF at 0 °C was added 1.6 M n-BuLi in hexanes (10.50 mL, 16.81 mmol) and the reaction mixture allowed to warm slowly to 55 °C. The reaction mixture was stirred at elevated temperature during which time the clear, dark brown solution lightened considerably and became milky. After 3 h, the solution was cooled to 0 °C and chlorodiphenylphosphine (3.16 mL, 17.61 mmol) was added via syringe. The mixture was slowly warmed to RT and allowed to stir for 3 h resulting in a clear, dark brown solution. The product was isolated as a pale yellow solid after purification by silica gel flash chromatography with a 15
:1 hexanes:ethyl acetate eluent. Yield: 52%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.69–7.64 (m, 8H), 7.61–7.59 (m, 2H), 7.39–7.36 (m, 2H), 7.31–7.29 (m, 12H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 136.6 (d, J = 6.7 Hz), 133.1 (d, J = 1.8 Hz), 133.0 (d, J = 21.1 Hz), 129.4 (d, J = 38.0 Hz), 129.3, 129.3, 125.7, 106.6 (d, J = 3.7 Hz), 91.0 (d, J = 9.2 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −33.11 (s). HRMS-ESI: m/z 495.1426 (M + H)+. IR (KBr) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1): 630, 808, 873 (P–CC), 2161 (CC). DSC: 78 °C (melt), 266 °C (maximum). Elemental analysis, calcd for C34H24P2: C, 82.58; H, 4.89. Found: C, 82.18; H, 4.74.
(cm−1): 630, 808, 873 (P–CC), 2161 (CC). DSC: 78 °C (melt), 266 °C (maximum). Elemental analysis, calcd for C34H24P2: C, 82.58; H, 4.89. Found: C, 82.18; H, 4.74.
1,2-Bis((bis(4-methoxyphenyl)phosphino)ethynyl)benzene, dppeb-pOCH3 (1b). To a solution of EDTMS (1.30 g, 4.80 mmol) in 125 mL THF at 0 °C was added 1.6 M n-BuLi in hexanes (6.29 mL, 10.07 mmol), resulting in an immediate color change from pale yellow to dark brown. The mixture was slowly warmed to 55 °C leading to a gradual color change to light, milky brown. After stirring for 2 h at elevated temperature the mixture was again cooled to 0 °C and a solution of chlorobis(4-methoxyphenyl)phosphine (2.69 g, 10.55 mmol) in 25 mL THF was added. The solution gradually became dark brown and cleared upon phosphine addition. The mixture was warmed to RT and stirred for 3 h. The product was isolated as an off-white solid after purification using silica gel flash chromatography with a 100% benzene eluent. Yield: 63%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.60–7.55 (m, 10H), 7.36–7.34 (m, 2H), 6.85–6.81 (m, 8H), 3.76 (s, 12H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 160.1, 133.7, 133.5, 132.0 (d, J = 3.1 Hz), 126.8 (d, J = 6.3 Hz), 124.8, 113.9 (d, J = 13.9 Hz), 105.0 (d, J = 5.2 Hz), 91.1 (d, J = 15.2 Hz), 54.7. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −36.72. HRMS-ESI MS: m/z 615.1850 (M + H)+. IR (KBr)
(cm−1): 624, 809, 871 (P–CC), 2161 (CC). DSC: 96 °C (melt), 264 °C (maximum). Elemental analysis, calcd for C38H32O4P2: C, 74.26; H, 5.25. Found: C, 73.97; H, 5.34.
1,2-Bis((bis(4-(trifluoromethyl)phenyl)phosphino)ethynyl)benzene, dppeb-pCF3 (1c). To a solution of EDTMS (1.88 g, 6.93 mmol) in 125 mL THF at 0 °C was added 1.6 M n-BuLi in hexanes (9.09 mL, 14.56 mmol), resulting in an immediate color change from pale yellow to dark brown. The mixture was slowly warmed to 55 °C leading to a gradual color change to light, milky brown. After stirring for 2 h at elevated temperature the mixture was again cooled to 0 °C and a solution of chlorobis(4-(trifluoromethyl)phenyl)phosphine (5.00 g, 14.02 mmol) in 10 mL THF was added. The solution became clear, dark brown immediately upon phosphine addition. The mixture was warmed to RT and stirred for 3 h. The product was isolated as a yellow oil after purification using silica gel flash chromatography with a 10
:1 hexanes:ethyl acetate eluent followed by a second purification using a 2:1 hexanes:dichloromethane eluent. Yield: 20%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.78–7.74 (m, 8H), 7.66–7.63 (m, 2H), 7.54–7.51 (m, 8H), 7.46–7.43 (m, 2H). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −32.75. 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −63.33. HRMS-ESI MS: m/z 767.0926 (M + H)+. IR (KBr) (cm−1): 628, 813, 873 (P–CC), 2163 (CC).
1,2-Bis((bis(3,5-dimethylphenyl)phosphino)ethynyl)benzene, dppeb-m2CH3 (1d). To a solution of EDTMS (2.09 g, 7.74 mmol) in 150 mL THF at to 0 °C was carefully added 1.6 M n-BuLi in hexanes (10.16 mL, 16.26 mmol) resulting in an immediate color change from pale yellow to dark brown. The reaction mixture was stirred for 2 h after slowly warming to 55 °C during which time the solution turned light brown and became milky. After 2 h, the reaction was cooled again to 0 °C and a solution of chlorobis(3,5-dimethylphenyl)phosphine (5.00 g, 18.07 mmol) in 20 mL THF was added. After slowly warming to RT, the mixture was stirred for 3 h during which time the color darkened leaving a clear brown solution. The product was obtained as a white solid after purification by silica gel flash chromatography using a 20
:1 hexanes:ethyl acetate eluent. Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of heptane into a saturated solution of dichloromethane at −20 °C. Yield: 63%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.60–7.57 (m, 2H), 7.37–7.35 (m, 2H), 7.23 (d, J = 11.2 Hz, 8H), 6.93 (br s, 4H), 2.24 (s, 24 H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 138.7 (d, J = 8.2 Hz), 136.2 (d, J = 6.5 Hz), 132.8, 131.3, 130.8, 130.6, 129.1, 125.9, 106.4 (d, J = 3.8 Hz), 91.7 (d, J = 10.0 Hz), 21.6. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −33.59. HRMS-ESI MS: m/z 607.2682 (M + H)+. IR (KBr) (cm−1): 630, 808, 873 (P–CC), 2161 (CC). DSC: 121 °C (melt), 271 °C (maximum). Elemental analysis, calcd for C42H40P2: C, 83.14; H, 6.64. Found: C, 82.97; H, 6.55.
1,2-Bis((bis(3,5-bis(trifluoromethyl)phenyl)phosphino)ethynyl)benzene, dppeb-m2CF3 (1e). To a solution of EDTMS (1.88 g, 6.93 mmol) in 100 mL THF at 0 °C was added 1.6 M n-BuLi in hexanes (9.09 mL, 14.55 mmol) resulting in an immediate color change from yellow to dark brown. The solution was allowed to warm to 55 °C and was stirred at elevated temperature for 2 h during time which time the reaction mixture turned light brown and became milky. The solution was then cooled to 0 °C and bis(3,5-bis(trifluoromethyl)phenyl)chlorophosphine (7.51 g, 15.25 mmol) in 20 mL THF was added. The reaction mixture turned dark blue and was allowed to stir at RT for 3 h. The crude reaction mixture was filtered through a silica gel plug and washed with copious amounts of benzene and dichloromethane to remove impurities yielding a yellow/brown solution. The product was then isolated as an off-white solid after purification by silica gel flash chromatography with a 5
:1 hexanes:ethyl acetate eluent. Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of hexane into a saturated dichloromethane solution at −20 °C. Yield: 52%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.07 (d, J = 7.6 Hz, 8H), 7.89 (s, 4H), 7.68–7.66 (m, 2H), 7.52–7.50 (m, 2H). 13C{1H} NMR (125 MHz, 298 K, CD2Cl2) δC (ppm): 138.8 (d, J = 13.7 Hz), 133.0 (d, J = 38.8 Hz), 133.0 (d, J = 22.1 Hz), 132.9 (q of d, J = 33.5 Hz, J = 7.3 Hz), 130.6, 124.9, 124.5 (m), 123.6 (q, J = 273.1 Hz), 109.5 (d, J = 6.2 Hz), 86.9 (d, J = 20.2 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −31.96. 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −63.28. HRMS-ESI MS: m/z 1073.0009 (M + Cl)–. IR (KBr) (cm−1): 634, 806, 877 (P–CC), 2167 (CC). DSC: 144 °C (melt), 275 °C (maximum). Elemental analysis, calcd for C42H16F24P2: C, 48.58; H, 1.55. Found: C, 48.40; H, 1.56.
1,2-Bis((diisopropylphosphino)ethynyl)benzene, dipeb (1f). To a solution of EDTMS (2.14 g, 7.92 mmol) in 125 mL of THF at 0 °C was added 1.6 M n-BuLi in hexanes (10.39 mL, 16.63 mmol) resulting in an immediate color change from pale yellow to dark brown. The mixture was slowly warmed to 55 °C and stirred for 2 h during which time the reaction color lightened to milky brown. After stirring at elevated temperature, the solution was cooled to 0 °C and a solution of chlorodiisopropylphosphine (2.77 mL, 17.42 mmol) in 10 mL THF was added. Upon slow warming to RT, the reaction mixture became a clear, dark brown solution which was stirred for 3 h. The product was isolated as a yellow oil after purification with silica gel flash chromatography using a 5
:1 hexanes:benzene eluent. Yield: 81%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.47–7.44 (m, 2H), 7.27–7.25 (m, 2H), 2.02–1.95 (m, 4H), 1.26 (d, J = 11.2 Hz, 6H), 1.24 (d, J = 11.2 Hz, 6H), 1.16 (d, J = 16.8 Hz, 6H), 1.15 (d, J = 17.2 Hz, 6H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 133.1, 128.5, 126.0, 104.1 (d, J = 4.7 Hz), 92.3 (d, J = 27.3 Hz), 24.5 (d, J = 8.8 Hz), 20.2 (d, J = 19.7 Hz), 19.9 (d,J = 6.8 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −11.79. HRMS-ESI MS: m/z 359.2042 (M + H)+. IR (KBr) (cm−1): 636, 802, 871 (P–CC), 2158 (CC).
1,2-Bis((dicyclohexylphosphino)ethynyl)benzene, dcpeb (1g). To a solution of EDTMS (2.33 g, 8.61 mmol) in 150 mL THF at 0 °C was added 1.6 M n-BuLi in hexanes (11.30 mL, 18.08 mmol) and the reaction mixture allowed to warm slowly to 55 °C. The reaction was stirred at elevated temperature for 2 h during which time the clear, dark brown solution lightened considerably and became milky. After 2 h, the solution was cooled to 0 °C and a solution of chlorodicyclohexylphosphine (4.18 mL, 18.94 mmol) in 10 mL THF was added. The mixture was slowly warmed to RT during which time the color darkened resulting in a clear brown solution after stirring for 3 h. The product was isolated as a yellow oil after purification by silica gel flash chromatography with a 10
:1 hexanes:benzene eluent. Yield: 74%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.46–7.43 (m, 2H), 7.27–7.24 (m, 2H), 2.02–2.00 (m, 4H), 1.82–1.80 (m, 18H), 1.71–1.68 (m, 4H), 1.42–1.24 (m, 18H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 133.1, 128.4, 126.2, 104.4 (d, J = 3.64 Hz), 92.6 (d, J = 26.4 Hz), 33.7 (d, J = 8.7 Hz), 30.8 (d, J = 18.0 Hz), 30.2 (d, J = 4.9 Hz), 27.7 (d, J = 18.7 Hz), 27.7 (d, J = 2.3 Hz), 27.0. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −20.80. HRMS-ESI MS: m/z 519.3329 (M + H)+. IR (KBr) (cm−1): 626, 800, 885 (P–CC), 2159 (CC).
1,2-Bis((di-tert-butylphosphino)ethynyl)benzene, dtpeb (1h). To a solution of EDTMS (2.02 g, 7.48 mmol) in 150 mL THF at 0 °C was added 1.6 M n-BuLi in hexanes (9.81 mL, 15.71 mmol). The solution immediately changed color from pale yellow to dark brown. The reaction mixture was heated to 50 °C and allowed to stir for 2 h during which time the solution color lightened, and the consistency became milky. The mixture was then cooled to 0 °C and a solution of di-tert-butylchlorophosphine (3.12 mL, 16.45 mmol) in 20 mL THF was added, resulting in an instantaneous color change to dark brown and a clearing of the cloudy solution. The mixture was slowly warmed to RT and stirred for 3 h. The product was isolated as a yellow oil after purification by silica gel flash chromatography using a 5
:1 hexanes:benzene eluent. Yield: 64%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.49–7.47 (m, 2H), 7.28–7.26 (m, 2H), 1.29 (d, J = 12.4 Hz, 36H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 133.3, 128.4, 126.0, 104.7 (d, J = 3.6 Hz), 93.8 (d, J = 25.3 Hz), 33.5 (d, J = 17.1 Hz), 30.2 (d, J = 14.6 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 12.33. HRMS-ESI MS: m/z 415.2700 (M + H)+. IR (KBr) (cm−1): 640, 802, 867 (P–CC), 2159 (CC).
General procedure for the synthesis of phosphine oxide enediynes. Syntheses for the phosphine oxide enediyne ligands were adapted from a published literature procedure100 and performed under ambient conditions. To a solution of enediyne (1b–1h) (1 eq.) in 8 mL benzene was added excess hydrogen peroxide (30% by wt). The mixture was stirred vigorously at RT for 1 h after which time Na2SO4 was added. The reaction mixture was filtered and then evaporated to dryness to yield the final product (2b–2h). The phosphine oxide ligand 1,2-phenylenebis(ethyne-2,1-diyl)bis(diphenylphosphine oxide) (2a) has been reported previously.41
1,2-Phenylenebis(ethyne-2,1-diyl)bis(bis(4-methoxyphenyl)phosphine) oxide (2b). The product was isolated as a yellow oil. Yield: 81%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.77–7.72 (m, 8H), 7.69–7.67 (m, 2H), 7.50–7.47 (m, 2H), 6.94–6.91 (m, 8H), 3.81 (s, 12H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 163.3, 134.1, 133.3 (d, J = 12.7 Hz), 130.9, 125.7, 124.4, 114.8 (d, J = 14.6 Hz), 101.7 (d, J = 28.5 Hz), 89.1 (d, J = 160.4 Hz), 55.9. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 7.56. HRMS-ESI MS: m/z 647.1756 (M + H)+. IR (KBr)
(cm−1): 626, 819, 875 (P–CC), 1203 (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2177 (CC). Elemental analysis, calcd for C38H32P2O6·H2O: C, 68.67; H, 5.15. Found: C, 68.73; H, 5.19.
O), 2177 (CC). Elemental analysis, calcd for C38H32P2O6·H2O: C, 68.67; H, 5.15. Found: C, 68.73; H, 5.19.
1,2-Phenylenebis(ethyne-2,1-diyl)bis(bis(4-(trifluoromethyl)phenyl)-phosphine) oxide (2c). The product was isolated as a yellow oil. Yield: 67%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.06–8.01 (m, 8H), 7.76–7.71 (m, 10H), 7.59–7.57 (m, 2H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 137.0 (d, J = 121.2 Hz), 134.6 (dd, J = 33.1 Hz, J = 3.3 Hz), 134.3 (d, J = 1.9 Hz), 132.0 (d, J = 11.7 Hz), 131.8, 126.5 (q, J = 3.6 Hz), 126.4 (q, J = 4.0 Hz), 125.4, 123.4, 122.7, 103.7 (d, J = 30.0 Hz), 86.9 (d, J = 169.4). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 4.16. 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −64.56. HRMS-ESI MS: m/z 799.0818 (M + H)+. IR (KBr)
(cm−1): 630, 823, 879 (P–CC), 1215 (PO), 2179 (CC).
1,2-Phenylenebis(ethyne-2,1-diyl)bis(bis(3,5-dimethylphenyl)phosphine) oxide (2d). The product was isolated as a shiny white solid. Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of pentane into a saturated dichloromethane solution at −20 °C. Yield: 93%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.71–7.69 (m, 2H), 7.51–7.49 (m, 2H), 7.45 (d, J = 16.0 Hz, 8H), 7.09 (br s, 4H), 2.26 (s, 24H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 139.1 (d, J = 14.2 Hz), 134.4 (d, J = 3.1 Hz), 134.2, 134.1, 132.9, 130.9, 128.8 (d, J = 11.1 Hz), 123.9, 101.8 (d, J = 27.8 Hz), 89.0 (d, J = 158.9 Hz), 21.5. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 8.42. HRMS-ESI MS: m/z 639.2575 (M + H)+. IR (KBr)
(cm−1): 640, 809, 875 (P–CC), 1199 (PO), 2177 (CC). DSC: 153 °C (melt), no maximum up to 280 °C. Elemental analysis, calcd for C42H40P2O2·C6H6·H2O: C, 78.46; H, 6.59. Found: C, 78.13; H, 6.28.
1,2-Phenylenebis(ethyne-2,1-diyl)bis(bis(3,5-bis(trifluoromethyl)phenyl)-phosphine) oxide (2e). Due to the low solubility of 1e in benzene, a 1
:1 solvent mixture of benzene:dichloromethane was utilized. The product was isolated as an off-white solid. Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of pentane into a saturated dichloromethane solution at −20 °C. Yield: 88%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.37 (d, J = 15.6 Hz, 8H), 8.10 (s, 4H), 7.82–7.80 (m, 2H), 7.66–7.64 (m, 2H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 135.4 (d, J = 123.4 Hz), 134.0, 133.0 (q of d, J = 34.2 Hz, J = 13.9 Hz), 132.3, 131.8 (d, J = 12.9 Hz), 127.4, 124.7, 122.0, 105.3 (d, J = 31.9 Hz), 85.7 (d, J = 176.4 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 1.83. 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −64.26. HRMS-ESI MS: m/z 1071.0302 (M + H)+. IR (KBr) (cm−1): 646, 809, 885 (P–CC), 1216 (PO), 2181 (CC). DSC: 164 °C (melt), no maximum up to 280 °C. Elemental analysis, calcd for C42H16F24P2O2: C, 47.12; H, 1.51. Found: C, 46.87; H, 1.49.
1,2-Phenylenebis(ethyne-2,1-diyl)bis(diisopropylphosphine oxide) (2f). The product was isolated as a yellow oil. Yield: 66%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.62–7.60 (m, 2H), 7.46–7.44 (m, 2H), 2.19–2.10 (m, 4H), 1.30 (d, J = 7.2 Hz, 6H), 1.28 (d, J = 6.8 Hz, 6H), 1.25 (d, J = 7.2 Hz, 6H), 1.24 (d, J = 6.8 Hz, 6H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 134.0, 130.6, 124.0, 100.2 (d, J = 20.3 Hz), 86.4 (d, J = 129.4 Hz), 28.1, 27.3, 16.6 (d, J = 2.7 Hz), 15.3 (d, J = 3.2 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 42.65. HRMS-ESI MS: m/z 391.1954 (M + H)+. IR (KBr)
(cm−1): 667, 813, 873 (P–CC), 1180 (PO), 2177 (CC).
1,2-Phenylenebis(ethyne-2,1-diyl)bis(dicyclohexylphosphine oxide) (2g). The product was isolated as a yellow oil. Yield: 94%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.63–7.60 (m, 2H), 7.46–7.43 (m, 2H), 2.04–1.83 (m, 20H), 1.74–1.71 (m, 4H), 1.56–1.46 (m, 8H), 1.36–1.23 (m, 12H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 133.9 (d, J = 1.8 Hz), 130.5, 124.1 (m), 100.0 (d, J = 20.0 Hz), 87.3 (d, J = 128.4 Hz), 37.3 (d, J = 78.5 Hz), 26.8 (d, J = 7.2 Hz), 26.7 (d, J = 6.7 Hz), 26.5 (d, J = 3.2 Hz), 26.4 (d, J = 5.0 Hz), 25.3 (d, J = 3.3 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 35.80. HRMS-ESI MS: m/z 551.3213 (M + H)+. IR (KBr)
(cm−1): 632, 808, 873 (P–CC), 1211 (PO), 2177 (CC).
1,2-Phenylenebis(ethyne-2,1-diyl)bis(di-tert-butylphosphine oxide) (2h). The product was isolated as a yellow oil. Yield: 75%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.63–7.61 (m, 2H), 7.47–7.45 (m, 2H), 1.37 (d, J = 15.2 Hz, 36H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 134.2, 130.5, 124.0, 100.4 (d, J = 17.2 Hz), 87.3 (d, J = 121.1 Hz), 37.1, 36.3, 26.8. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 49.41. HRMS-ESI MS: m/z 447.2585 (M + H)+. IR (KBr)
(cm−1): 644, 815, 869 (P–CC), 1220 (PO), 2175 (CC). Elemental analysis, calcd for C26H40P2O2·H2O2: C, 64.98; H, 8.81. Found: C, 64.77; H, 8.63.
Dichloro-(1,2-bis((diphenylphosphino)ethynyl)benzene)platinum(II), Pt(dppeb)Cl2 (3a). This complex has been generated in situ previously.35,43 Using the procedure detailed here, the complex has now been isolated and characterized. To a solution of Pt(cod)Cl2 (64.1 mg, 0.171 mmol) in 6 mL DCM was added a solution of 1a (102 mg, 0.206 mmol) in 6 mL DCM. The mixture was allowed to stir at RT for 30 min, then the solvent was removed in vacuo. The residue was suspended in minimal DCM and filtered to remove insoluble material. To the filtrate was added excess hexanes. The resulting off-white precipitate was collected, washed with hexanes, and dried. Crude yield: 53%. The crude precipitate was purified by preparatory chromatography on silica gel with a 50
:1 DCM:acetone eluent (Rf = 0.57). Purified yield: 31%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.93–7.87 (m, 8H), 7.53–7.42 (m, 16H). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −17.44 (s, JP–Pt = 3823 Hz). HRMS-ESI MS: m/z 725.0702 (M–Cl)+. IR (KBr) (cm−1): 637, 820, 882 (P–CC), 2171 (CC). DSC: 116 °C (maximum).
Dichloro-(1,2-bis((bis(4-methoxyphenyl)phosphino)ethynyl)benzene) platinum(II), Pt(dppeb-pOCH3)Cl2 (3b). To a solution of Pt(cod)Cl2 (57.2 mg, 0.153 mmol) in 10 mL DCM at 0 °C was added a solution of 1b (113 mg, 0.184 mmol) in 5 mL DCM, also at 0 °C. The mixture was allowed to stir on ice for 30 min after which time the solvent was removed by rotary evaporation at 0 °C. The resulting residue was dissolved in minimal DCM and excess hexanes added. The off-white precipitate was isolated by filtration, washed with hexanes, and dried. Crude yield: 43%. The crude precipitate was purified by preparatory chromatography on silica gel with a 20
:1 DCM:acetone eluent (Rf = 0.65). Purified yield: 20% 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.83–7.78 (m, 8H), 7.52–7.46 (m, 4H), 6.96–6.93 (m, 8H), 3.85 (s, 12H). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −19.13 (s, JP–Pt = 3846 Hz). HRMS-ESI MS: m/z 844.1100 (M–Cl)+. IR (KBr) (cm−1): 620, 823, 881 (P–CC), 2167 (CC). DSC: 120 °C (maximum).
Dichloro-(1,2-bis((bis(4-(trifluoromethyl)phenyl)phosphino)ethynyl)benzene) platinum(II), Pt(dppeb-pCF3)Cl2 (3c). To a solution of Pt(cod)Cl2 (25.7 mg, 0.069 mmol) in 15 mL DCM was added a solution of 1c (63.2 mg, 0.083 mmol) in 10 mL DCM. The mixture was stirred at RT for 30 min and then the solvent was removed in vacuo. The residue was dissolved in minimal DCM, followed by the addition of excess hexanes. The off-white precipitate was filtered, washed with hexanes, and dried. Crude yield: 45%. The crude solid was stirred in cold Et2O for 2 min and then filtered resulting in an off-white solid. Purified yield: 17% 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.05–8.00 (m, 8H), 7.73–7.71 (m, 8H), 7.58 (s, 4H). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −17.65 (s, JP–Pt = 3797 Hz). 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −63.59. HRMS-ESI MS: m/z 996.0226 (M–Cl)+. IR (KBr)
(cm−1): 630, 829, 883 (P–CC), 2175 (CC). DSC: 177 °C (maximum).
Dichloro-(1,2-bis((bis(3,5-dimethylphenyl)phosphino)ethynyl)benzene) platinum(II), Pt(dppeb-m2CH3)Cl2 (3d). To a solution of Pt(cod)Cl2 (82.1 mg, 0.219 mmol) in 10 mL DCM was added a solution of 1d (111 mg, 0.183 mmol) in 5 mL DCM. The mixture was stirred at RT for 30 min and then solvent was removed under reduced pressure. The remaining residue was dissolved in minimal DCM and excess hexanes added to induce precipitation. An off-white solid was filtered, washed with hexanes, and dried. Crude yield: 92%. The product was purified by preparatory chromatography on silica gel with a 2
:1 hexanes:acetone eluent (Rf = 0.63). Purified yield: 56%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.54–7.52 (m, 2H), 7.49–7.43 (m, 10H), 7.11 (br s, 4H), 2.31 (s, 24H). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −16.53 (s, JP–Pt = 3818 Hz). HRMS-ESI MS: m/z 837.1972 (M–Cl)+. IR (KBr) (cm−1): 635, 814, 883 (P–CC), 2163 (CC). DSC: 106 °C (maximum).
Dichloro-(1,2-bis((bis(3,5-bis(trifluoromethyl)phenyl)phosphino)ethynyl)-benzene)platinum(II), Pt(dppeb-m2CF3)Cl2 (3e). To a solution of Pt(cod)Cl2 (61.2 mg, 0.164 mmol) in 10 mL DCM was added a solution of 1e (204 mg, 0.196 mmol) in 10 mL DCM. The mixture was stirred at RT for 30 min followed by solvent removal by rotary evaporation. The residue was dissolved in minimal DCM and filtered. To the filtrate was added excess hexanes to induce precipitation. The off-white precipitate was filtered, washed with hexanes and cold Et2O, and dried. Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of hexane into a saturated dichloromethane solution at −20 °C. Yield: 43%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.38 (d, J = 11.6 Hz, 8 H), 8.14 (s, 4H), 7.63 (s, 4H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 134.3, 132.9 (q of d, J = 34.4 Hz, J = 12.8 Hz), 132.6, 131.7 (d, J = 80.9 Hz), 131.6, 127.0, 124.1, 123.2 (q, J = 273.3 Hz), 115.3 (d, J = 16.2 Hz), 84.1 (d, J = 112.9 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −18.17 (s, JP–Pt = 3812 Hz). 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −64.23. HRMS-ESI MS: m/z 1268.9723 (M–Cl)+. IR (KBr)
(cm−1): 655, 817, 885 (P–CC), 2175 (CC). DSC: 236 °C (maximum). Elemental analysis, calcd for C42H16 F24P2PtCl2: C, 38.67; H, 1.24. Found: C, 38.99; H, 1.07.
Dichloro-(1,2-bis((diisopropylphosphino)ethynyl)benzene)platinum(II), Pt(dipeb)Cl2 (3f). To a solution of Pt(cod)Cl2 (280 mg, 0.748 mmol) in 12 mL DCM was added a solution of 1f (320 mg, 0.893 mmol) in 5 mL DCM. The mixture was allowed to stir at RT for 75 min after which time the solvent was removed by rotary evaporation. The resulting residue was dissolved in minimal DCM and excess hexanes added. The off-white precipitate was isolated by filtration, washed with hexanes, and dried. Crude yield: 80%. The crude precipitate was purified by preparatory chromatography on silica gel with a 10
:1 benzene:acetone eluent (Rf = 0.58). Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of pentane into a saturated dichloromethane solution at −20 °C. Purified yield: 49%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.59–7.57 (m, 2H), 7.53–7.51 (m, 2H), 3.01–2.97 (m, 4H), 1.45 (d, J = 7.0 Hz, 6H), 1.41 (d, J = 7.0 Hz, 6H), 1.37 (d, J = 7.0 Hz, 6H), 1.34 (d, J = 7.0 Hz, 6H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 130.1, 129.7, 125.1, 110.1, 85.3 (d, J = 5.7 Hz), 84.7 (d, J = 5.5 Hz), 27.9 (d, J = 42.4 Hz), 20.3, 19.0. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 13.46 (s, JP–Pt = 3815 Hz). HRMS-ESI MS: m/z 589.1321 (M–Cl)+. IR (KBr) (cm−1): 634, 827, 881 (P–CC), 2163 (CC). DSC: 139 °C (maximum). Elemental analysis, calcd for C22H32P2PtCl2: C, 42.32; H, 5.17. Found: C, 42.04; H, 4.89.
Dichloro-(1,2-bis((dicyclohexylphosphino)ethynyl)benzene)platinum(II), Pt(dcpeb)Cl2 (3g). To a solution of Pt(cod)Cl2 (115 mg, 0.307 mmol) in 10 mL DCM was added a solution of 1g (191 mg, 0.369 mmol) in 5 mL DCM. The mixture was allowed to stir at RT for 2 h after which time the solvent was removed by rotary evaporation. The resulting residue was dissolved in minimal DCM and excess hexanes added. The off-white precipitate was then filtered, washed with hexanes, and dried. Crude yield: 72% The crude precipitate was purified by preparatory chromatography on silica gel with a 10
:1 benzene:acetone eluent (Rf = 0.75). Purified yield: 46%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.59–7.57 (m, 2H), 7.52–7.50 (m, 2H), 2.77–2.69 (m, 4H), 2.39 (br s, 4H), 1.99–1.96 (m, 4H), 1.81 (br s, 8H), 1.70–1.65 (m, 8H), 1.57–1.51 (m, 6H), 1.40–1.21 (m, 10H). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 4.35 (s, JP–Pt = 3822 Hz). HRMS-ESI MS: m/z 749.2589 (M–Cl)+. IR (KBr) (cm−1): 642, 827, 889 (P–CC), 2163 (CC). DSC: 140 °C (maximum).
General procedure for the Bergman cyclization of Pt(II) phosphine enediyne complexes and the determination of cycloaromatization rate. Pt(II) phosphine enediyne dichloride complexes were purified as described above directly before use. Metalloenediynes (3a–3g) were dissolved in CDCl3, dried over 4 Å molecular sieves, to give solutions of 3–30 mM, depending on solubility. A slight molar excess of triphenylphosphine oxide, Ph3PO, was then added as an internal 31P NMR standard followed by 100 equivalents of 1,4-cyclohexadiene. The solution was sealed in an NMR tube under ambient conditions and maintained in a temperature-controlled bath or in the NMR spectrometer. 31P NMR spectra were acquired at t = 0, 2, 4, 8, 12, 24, 36 h… or t = 0, 1, 2, 3, 4 h… time intervals for complexes with cyclization t1/2 > 12 h and t1/2 < 12 h, respectively. The disappearance of the starting material was followed to at least 90% completion by integration of the Pt(II) phosphine enediyne dichloride 31P NMR signal (3a–3g) vs. that of Ph3PO. Cyclization was monitored in 5 °C increments for each complex: 3b (5–20 °C); 3a, 3d (10–25 °C); 3c (15, 25–35 °C); 3e–3g (25–40 °C), and data were collected in triplicate. Complex 3c contained ∼5% Pt(cod)Cl2 impurity. Rate constants were obtained from highly linear first order kinetic plots (R2 > 0.98) and the free energy of activation (ΔG‡) was calculated from standard Eyring plots. The cyclization of all complexes except of 3g proceeds cleanly to the corresponding cyclized product in >90% yield. Due to the low concentrations used during the kinetic experiments (3–30 mM), the reaction mixtures from all trials for a given compound were combined prior to purification with preparatory silica gel chromatography. Following purification, all cyclized compounds were fully characterized.
Dichloro-(2,3-bis(diphenylphosphino)naphthalene)platinum(II), Pt(dppnap)Cl2 (4a). This complex has been reported previously using a different synthetic protocol.43 After completion of the kinetic experiments described above (3.9–7.4 mM solutions), the product was isolated as a white solid using silica gel preparatory chromatography with a 50
:1 DCM:acetone eluent (Rf = 0.45). Crystals suitable for characterization by X-ray crystallography were grown from slow evaporation of a saturated solution in CD2Cl2 at room temperature. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.12–8.09 (m, 2H), 7.89–7.87 (m, 2H), 7.81–7.75 (m, 8H), 7.67–7.64 (m, 2H), 7.55–7.51 (m, 4H), 7.47–7.43 (m, 8H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 134.6, 134.5, 134.4, 132.4, 130.2, 129.4, 129.3, 129.2, 129.1. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 39.42 (s, JP–Pt = 3597 Hz). HRMS-ESI MS: m/z 726.0851 (M–Cl)+. Elemental analysis, calcd for C34H26P2PtCl2: C, 53.56; H, 3.44. Found: C, 53.75; H, 3.23.
Dichloro-(2,3-bis(bis(4-methoxyphenyl)phosphino)naphthalene)platinum(II), Pt(dppnap-pOCH3)Cl2 (4b). After completion of the kinetic experiments described above (3.4–8.9 mM solutions), the product was isolated as a white solid using silica gel preparatory chromatography with a 20
:1 DCM:acetone eluent (Rf = 0.56). 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.08–8.05 (m, 2H), 7.90–7.87 (m, 2H), 7.71–7.66 (m, 10H), 6.98–6.95 (m, 8H), 3.84 (s, 12H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 163.0, 136.2, 136.1, 136.0, 129.9, 129.2, 115.0, 114.9, 114.8, 56.0. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 37.19 (s, JP–Pt = 3638 Hz). HRMS-ESI MS: m/z 847.1293 (M–Cl)+.
Dichloro-(2,3-bis(bis(4-(trifluoromethyl)phenyl)phosphino)naphthalene) platinum(II), Pt(dppnap-pCF3)Cl2 (4c). After completion of the kinetic experiments described above (3.6–4.7 mM solutions), the product was isolated as an off-white solid using silica gel preparatory chromatography with a 2
:1 hexanes:ethyl acetate eluent (Rf = 0.57). Crystals suitable for X-ray crystallographic analysis were grown from vapor diffusion of heptane into a saturated solution of dichloromethane. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.17–8.14 (m, 2H), 7.97–7.90 (m, 10H), 7.77–7.74 (m, 10H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 136.4 (d, J = 18.9 Hz), 135.3 (d, J = 6.8 Hz), 134.9 (d, J = 11.8 Hz), 134.5 (d, J = 33.0 Hz), 133.7 (d, J = 29.9 Hz), 132.5 (d, J = 67.0 Hz), 131.2, 129.4, 126.6–126.4 (m), 124.0 (q, J = 273.8 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 38.93 (s, JP–Pt = 3553 Hz). 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −64.51. HRMS-ESI MS: m/z 998.0370 (M–Cl)+.
Dichloro-(2,3-bis(bis(3,5-dimethylphenyl)phosphino)naphthalene) platinum(II), Pt(dppnap-m2CH3)Cl2 (4d). After completion of the kinetic experiments described above (9.7–16.3 mM solutions), the product was isolated as an off-white solid using silica gel preparatory chromatography with a 2
:1 hexanes:acetone eluent (Rf = 0.56). Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of cyclohexane into a saturated dichloromethane solution at −20 °C. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.04–8.01 (m, 2H), 7.89–7.87 (m, 2H), 7.65–7.63 (m, 2H), 7.35 (d, J = 12.0 Hz, 8H), 7.15 (br s, 4H), 2.28 (s, 24H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 139.1 (d, J = 12.7 Hz), 139.1, 134.1, 132.0 (d, J = 11.8 Hz), 132.0, 129.8, 129.2, 128.9, 128.2, 21.7. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 39.58 (s, JP–Pt = 3614 Hz). HRMS-ESI MS: m/z 838.2087 (M–Cl)+.
Dichloro-(2,3-bis(bis(3,5-(trifluoromethyl)phenyl)phosphino)naphthalene) platinum(II), Pt(dppnap-m2CF3)Cl2 (4e). After completion of the kinetic experiments described above (2.8–3.3 mM solutions), the product was isolated as a white solid using silica gel preparatory chromatography with a 2
:1 hexanes:acetone eluent (Rf = 0.49). Crystals suitable for characterization by X-ray crystallography were grown directly from the cyclization reaction mixture. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.25–8.17 (m, 14H), 8.04–8.01 (m, 2H), 7.87–7.85 (m, 2H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 137.2 (d, J = 18.4 Hz), 135.5 (d, J = 6.9 Hz), 134.1, 134.0, 133.5 (q of d, J = 21.9 Hz, J = 12.8 Hz), 132.4, 130.6 (d, J = 68.2 Hz), 129.7, 127.6, 123.0 (q, J = 273.5 Hz). 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 39.87 (s, JP–Pt = 3519 Hz). 19F NMR (400 MHz, 298 K, CD2Cl2) δF (ppm): −64.35. HRMS-ESI MS: m/z 1327.9443 (M + Na)+. Elemental analysis, calcd for C42H18F24P2PtCl2: C, 38.61; H, 1.39. Found: C, 38.33; H, 1.13.
Dichloro-(2,3-bis(diisopropylphosphino)naphthalene)platinum(II), Pt(dipnap)Cl2 (4f). After completion of the kinetic experiments described above (21.1–30.0 mM solutions), the product was isolated as a white solid using silica gel preparatory chromatography with a 25
:5:1 benzene:acetone:hexanes eluent (Rf = 0.48). Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of heptane into a saturated dichloromethane solution at −20 °C. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 8.28 (d, J = 9.2 Hz, 2H), 8.05–8.03 (m, 2H), 7.76–7.74 (m, 2H), 3.14–3.04 (m, 4H), 1.43 (d, J = 6.8 Hz, 6H), 1.39 (d, J = 6.8 Hz, 6H), 1.25 (d, J = 7.2 Hz, 6H), 1.21 (d, J = 7.2 Hz, 6H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 134.5 (dd, J = 53.5 Hz, J = 24.9 Hz), 134.1 (m), 132.7 (m), 129.3, 128.6, 27.5 (d, J = 36.3 Hz), 19.1, 18.5. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): 60.73 (s, JP–Pt = 3576 Hz). HRMS-ESI MS: m/z 590.1482 (M–Cl)+.
Dichloro-(1,2-bis((bis(3,5-dimethylphenyl)phosphino)ethynyl)benzene) platinum(II) dimer, Pt2(μ2-dppeb-m2CH3)2Cl4 (5d). To a solution of Pt(cod)Cl2 (51.6 mg, 0.138 mmol) in 7 mL DCM was added a solution of 1d (101 mg, 0.166 mmol) in 5 mL DCM. The mixture was allowed to stir at RT for 30 min after which time the solvent was removed in vacuo. The resulting residue was dissolved in minimal DCM and excess hexanes added. The off-white precipitate was filtered, washed with hexanes, and dried. Crystals suitable for characterization by X-ray crystallography were grown from vapor diffusion of hexane into a saturated dichloromethane solution at −20 °C. Yield: 61%. 1H NMR (400 MHz, 298 K, CD2Cl2) δH (ppm): 7.69 (d, J = 13.6 Hz, 8H), 7.28–7.26 (m, 4H), 7.06 (br s, 4H), 6.99–6.95 (m, 12H), 6.26 (s, 4H), 2.31 (s, 24H), 1.84 (s, 24H). 13C{1H} NMR (101 MHz, 298 K, CD2Cl2) δC (ppm): 139.1–139.0 (m), 138.1–138.0 (m), 133.8, 133.2, 133.1 (t, J = 6.6 Hz), 133.0, 131.8 (d, J = 77.1 Hz), 130.4 (d, J = 5.4 Hz), 129.4, 123.4 (d, J = 75.1 Hz), 121.3, 110.4 (d, J = 19.8 Hz), 88.0 (dd, J = 112.8 Hz, 6.8 Hz), 21.7, 21.1. 31P{1H} NMR (161 MHz, 298 K, CD2Cl2) δP (ppm): −2.61 (s, JP–Pt = 3787 Hz). HRMS-ESI MS: m/z 1709.3569 (M–Cl)+. DSC: no maximum observed up to 280 °C. Elemental analysis, calcd for C82H80P4Pt2Cl4: C, 57.80; H, 4.62. Found: C, 57.98; H, 4.28.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. E. Lindahl and E. M. Metzger carried out the experimental work. Crystallographic data were solved by M. Pink and C.-H. Chen. S. E. Lindahl wrote the manuscript with input from all authors. J. M. Zaleski supervised the work.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The generous support of the National Institutes of Health (R01-CA196293) and National Science Foundation (CHE-1265703, CHE-2247314) is gratefully acknowledged. Additionally, this research was supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute. The authors would also like to thank Dr Aurora Clark, Dr Krystyna Kirschner, and Stephen Ratvasky for thoughtful discussions.References
- K. Edo, M. Mizugaki, Y. Koide, H. Seto, K. Furihata, N. Otake and N. Ishida, The Structure of Neocarzinostatin Chromophore Possessing a Novel Bicyclo-[7,3,0]Dodecadiyne System, Tetrahedron Lett., 1985, 26, 331–334 CrossRef CAS.
- J. Golik, J. Clardy, G. Dubay, G. Groenewold, H. Kawaguchi, M. Konishi, B. Krishnan, H. Ohkuma, K. Saitoh and T. W. Doyle, Esperamicins, A Novel Class of Potent Antitumor Antibiotics. 2. Structure of Esperamicin X, J. Am. Chem. Soc., 1987, 109, 3461–3462 CrossRef CAS.
- J. Golik, G. Dubay, G. Groenewold, H. Kawaguchi, M. Konishi, B. Krishnan, H. Ohkuma, K. Saitoh and T. W. Doyle, Esperamicins, A Novel Class of Potent Antitumor Antibiotics. 3. Structures of Esperamicins A1, A2, and A1b, J. Am. Chem. Soc., 1987, 109, 3462–3464 CrossRef CAS.
- M. Konishi, H. Ohkuma, K. Matsumoto, T. Tsuno, H. Kamei, T. Miyaki, T. Oki, H. Kawaguchi and A. Dynemicin, A Novel Antibiotic with the Anthraquinone and 1,5-diyn-3-ene Subunit, J. Antibiot., 1989, 42, 1449–1452 CrossRef CAS.
- M. D. Lee, T. S. Dunne, C. C. Chang, G. A. Ellestad, M. M. Siegel, G. O. Morton, W. J. McGahren and D. B. Borders, Calichemicins, A Novel Family of Antitumor Antibiotics. 2. Chemistry and Structure of Calichemicin g1, J. Am. Chem. Soc., 1987, 109, 3466–3468 CrossRef CAS.
- M. D. Lee, T. S. Dunne, M. M. Siegel, C. C. Chang, G. O. Morton and D. B. Borders, Calichemicins, A Novel Family of Antitumor Antibiotics. 1. Chemistry and Partial Structure of Calichemicin g1, J. Am. Chem. Soc., 1987, 109, 3464–3466 CrossRef CAS.
- N. Darby, C. U. Kim, J. A. Salaün, K. W. Shelton, S. Takada and S. Masamune, Concerning the 1,5-Didehydro[10]annulene System, Chem. Commun., 1971, 1516–1517 RSC.
- R. G. Bergman, Reactive 1,4-Dehydroaromatics, Acc. Chem. Res., 1973, 6, 25–31 CrossRef CAS.
- S. G. Van Lanen and B. Shen, Biosynthesis of Enediyne Antitumor Antibiotics, Curr. Top. Med. Chem., 2008, 8, 448–459 CrossRef CAS.
- B. Shen, Hindra, X. Yan, T. Huang, H. Ge, D. Yang, Q. Teng, J. D. Rudolf and J. R. Lohman, Enediynes: Exploration of Microbial Genomics to Discover New Anticancer Drug Leads, Bioorg. Med. Chem. Lett., 2015, 25, 9–15 CrossRef CAS PubMed.
- Z. J. Low, G.-L. Ma, H. T. Tran, Y. Zou, J. Xiong, L. Pang, S. Nuryyeva, H. Ye, J.-F. Hu, K. N. Houk and Z.-X. Liang, Sungeidines from a Non-canonical Enediyne Biosynthetic Pathway, J. Am. Chem. Soc., 2020, 142, 1673–1679 CrossRef CAS PubMed.
- M. Igarashi, R. Sawa, M. Umekita, M. Hatano, R. Arisaka, C. Hayashi, Y. Ishizaki, M. Suzuki and C. Kato, Sealutomicins, New Enediyne Antibiotics from the Deep-Sea Actinomycete Nonomuraea sp. MM565M-173N2, J. Antibiot., 2021, 74, 291–299 CrossRef CAS PubMed.
- L. Liu, J. Pan, Z. Wang, X. Yan, D. Yang, X. Zhu, B. Shen, Y. Duan and Y. Huang, Ribosome Engineering and Fermentation Optimization Leads to Overproduction of Tiancimycin A, a New Enediyne Natural Product from Streptomyces sp. CB03234, J. Ind. Microbiol. Biotechnol., 2018, 45, 141–151 CrossRef CAS.
- J. H. Im, D. Shin, Y. H. Ban, W. S. Byun, E. S. Bae, D. Lee, Y. E. Du, J. Cui, Y. Kwon, S.-J. Nam, S. Cha, S. K. Lee, Y. J. Yoon and D.-C. Oh, Targeted Discovery of an Enediyne-Derived Cycloaromatized Compound, Jejucarboside A, from a Marine Actinomycete, Org. Lett., 2022, 24, 7188–7193 CrossRef CAS.
- S. Y. Ma, Y. S. Xiao, B. Zhang, F. L. Shao, Z. K. Guo, J. J. Zhang, R. H. Jiao, Y. Sun, Q. Xu, R. X. Tan and H. M. Ge, Amycolamycins A and B, Two Enediyne-Derived Compounds from a Locust-Associated Actinomycete, Org. Lett., 2017, 19, 6208–6211 CrossRef CAS.
- D.-C. Oh, P. G. Williams, C. A. Kauffman, P. R. Jensen and W. Fenical, Cyanosporasides A and B, Chloro- and Cyano-cyclopenta[a]indene Glycosides from the Marine Actinomycete “Salinispora pacifica”, Org. Lett., 2006, 8, 1021–1024 CrossRef CAS PubMed.
- X. Yan, J.-J. Chen, A. Adhikari, D. Yang, I. Crnovcic, N. Wang, C.-Y. Chang, C. Rader and B. Shen, Genome Mining of Micromonospora yangpuensis DSM 45577 as a Producer of an Anthraquinone-Fused Enediyne, Org. Lett., 2017, 19, 6192–6195 CrossRef CAS PubMed.
- Y. Fu and M. Ho, DNA Damaging Agent-Based Antibody-Drug Conjugates for Cancer Therapy, Antibody Ther., 2018, 1, 43–53 CrossRef CAS.
- N. K. Damle and P. Frost, Antibody-Targeted Chemotherapy with Immunoconjugates of Calicheamicin, Curr. Opin. Pharmacol., 2003, 3, 386–390 CrossRef CAS PubMed.
- H. Maeda, SMANCS and Polymer-Conjugated Macromolecular Drugs: Advantages in Cancer Chemotherapy, Adv. Drug Delivery Rev., 2001, 46, 169–185 CrossRef CAS PubMed.
- H. Maeda, K. Greish and J. Fang, The EPR Effect and Polymeric Drugs: A Paradigm Shift for Cancer Chemotherapy in the 21st Century, Adv. Polym. Sci., 2006, 193, 103–121 CrossRef CAS.
- N. Joubert, A. Beck, C. Dumontet and C. Denevault-Sabourin, Antibody-Drug Conjugates: The Last Decade, Pharmaceuticals, 2020, 13, 245–275 CrossRef CAS.
- K. C. Nicolaou, G. Zuccarello, Y. Ogawa, E. J. Schweiger and T. Kumazawa, Cyclic Conjugated Enediynes Related to Calicheamicins and Esperamicins: Calculations, Synthesis, and Properties, J. Am. Chem. Soc., 1988, 110, 4866–4868 CrossRef CAS.
- K. C. Nicolaou, G. Zuccarello, C. Riemer, V. A. Estevez and W.-M. Dai, Design, Synthesis, and Study of Simple Monocyclic Conjugated Enediynes. The 10-Membered Ring Enediyne Moiety of the Enediyne Anticancer Antibiotics, J. Am. Chem. Soc., 1992, 114, 7360–7371 CrossRef CAS.
- P. R. Schreiner, Monocyclic Enediynes: Relationships between Ring Sizes, Alkyne Carbon Distances, Cyclization Barriers, and Hydrogen Abstraction Reactions. Singlet-Triplet Separations of Methyl-Substituted p-Benzynes, J. Am. Chem. Soc., 1998, 120, 4184–4190 CrossRef CAS.
- J. P. Snyder, The Cyclization of Calicheamicin-Esperamicin Analogues: A Predictive Biradicaloid Transition State, J. Am. Chem. Soc., 1989, 111, 7630–7632 CrossRef CAS.
- J. P. Snyder, Monocyclic Enediyne Collapse to 1,4-Diyl Biradicals: A Pathway under Strain Control, J. Am. Chem. Soc., 1990, 112, 5367–5369 CrossRef CAS.
- P. Magnus, S. Fortt, T. Pitterna and J. P. Snyder, Synthetic and Mechanistic Studies on Esperamicin A1 and Calichemicin g1. Molecular Strain Rather than p-Bond Proximity Determines the Cycloaromatization Rates of Bicyclo[7.3.1] Enediynes, J. Am. Chem. Soc., 1990, 112, 4986–4987 CrossRef CAS.
- M. F. Semmelhack, T. Neu and F. Foubelo, Arene 1,4-Diradical Formation from o-Dialkynylarenes, Tetrahedron Lett., 1992, 33, 3277–3280 CrossRef CAS.
- M. F. Semmelhack, T. Neu and F. Foubelo, Arene 1,4-Diradical Formation from o-Dialkynylarenes, J. Org. Chem., 1994, 59, 5038–5047 CrossRef CAS.
- G. B. Jones and P. M. Warner, Electronic Control of the Bergman Cyclization: The Remarkable Role of Vinyl Substitution, J. Am. Chem. Soc., 2001, 123, 2134–2145 CrossRef CAS.
- G. W. Plourde II, P. M. Warner, D. A. Parrish and G. B. Jones, Halo-Enediynes: Probing the Electronic and Stereoelectronic Contributions to the Bergman Cycloaromatization, J. Org. Chem., 2002, 67, 5369–5374 CrossRef PubMed.
- M. Schmittel and S. Kiau, Polar Effects in the Transition State of the Bergman Cyclization, Chem. Lett., 1995, 953–954 CrossRef CAS.
- M. Prall, A. Wittkopp, A. A. Fokin and P. R. Schreiner, Substituent Effects on the Bergman Cyclization of (Z)-1,5-Hexadiyne-3-enes: A Systematic Computational Study, J. Comput. Chem., 2001, 22, 1605–1614 CrossRef CAS.
- B. P. Warner, S. P. Millar, R. D. Broene and S. L. Buchwald, Controlled Acceleration and Inhibition of Bergman Cyclization by Metal Chlorides, Science, 1995, 269, 814–816 CrossRef CAS.
- S. Bhattacharyya, D. F. Dye, M. Pink and J. M. Zaleski, A Geometric Switching Approach toward Thermal Activation of Metalloenediynes, Chem. Commun., 2005, 5295–5297 RSC.
- S. Bhattacharyya, M. Pink, M.-H. Baik and J. M. Zaleski, A Unique Approach to Metal-Induced Bergman Cyclization: Long-Range Enediyne Activation by Ligand-to-Metal Charge Transfer, Angew. Chem., Int. Ed., 2005, 44, 592–595 CrossRef CAS PubMed.
- T. T. Derencsenyi, Phosphorus-31 Nuclear Magnetic Resonance as a Method of Predicting Ligand Basicity and Rates of Homogeneous Catalysis, Inorg. Chem., 1981, 20, 665–670 CrossRef CAS.
- N. L. Coalter, T. E. Concolino, W. E. Streib, C. G. Hughes, A. L. Rheingold and J. M. Zaleski, Structure and Thermal Reactivity of a Novel Pd(0) Metalloenediyne, J. Am. Chem. Soc., 2000, 122, 3112–3117 CrossRef CAS.
- R. H. Grubbs and D. Kratz, Highly Unsaturated Oligomeric Hydrocarbons: a-(Phenylethynyl)-w-phenylpoly[1,2-phenylene(2,1-ethynediyl)], Chem. Ber., 1993, 126, 149–157 CrossRef CAS.
- S. E. Lindahl, H. Park, M. Pink and J. M. Zaleski, Utilizing Redox-Mediated Bergman Cyclization toward the Development of Dual-Action Metalloenediyne Therapeutics, J. Am. Chem. Soc., 2013, 135, 3826–3833 CrossRef CAS.
- J. D. Spence, A. C. Rios, M. A. Frost, C. M. McCutcheon, C. D. Cox, S. Chavez, R. Fernandez and B. F. Gherman, Syntheses, Thermal Reactivities, and Computational Studies of Aryl-Fused Quinoxalenediynes: Effect of Extended Benzannelation on Bergman Cyclization Energetics, J. Org. Chem., 2012, 77, 10329–10339 CrossRef CAS.
- B. P. Warner, Doctor of Philosophy in Organic Chemistry, Massachusetts Institute of Technology, 1995 Search PubMed.
- P. J. Benites, D. S. Rawat and J. M. Zaleski, Metalloenediynes: Ligand Field Control of Thermal Bergman Cyclization Reactions, J. Am. Chem. Soc., 2000, 122, 7208–7217 CrossRef CAS.
- B. König and W. Pitsch, Synthesis, Structure, and Reactivity of Enediyne Macrocycles, J. Org. Chem., 1996, 61, 4258–4261 CrossRef PubMed.
- B. König, H. Hollnagel, B. Ahrens and P. G. Jones, Activation of Macrocyclic Biaryl-Enediynes by Metal Ion Coordination, Angew. Chem., Int. Ed., 1995, 34, 2538–2540 CrossRef.
- M. R. Porter, S. E. Lindahl, A. Lietzke, E. M. Metzger, Q. Wang, E. Henck, C.-H. Chen, H. Niu and J. M. Zaleski, Metal-Mediated Diradical Tuning for DNA Replication Arrest via Template Strand Scission, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7405–E7414 CAS.
- D. S. Rawat and J. M. Zaleski, Mg2+ Induced Thermal Enediyne Cyclization at Ambient Temperature, J. Am. Chem. Soc., 2001, 123, 9675–9676 CrossRef CAS PubMed.
- D. S. Rawat, P. J. Benites, C. D. Incarvito, A. L. Rheingold and J. M. Zaleski, The Contribution of Ligand Flexibility to Metal Center Geometry Modulated Thermal Cyclization of Conjugated Pyridine and Quinoline Metalloenediynes of Copper(I) and Copper(II), Inorg. Chem., 2001, 40, 1846–1857 CrossRef CAS.
- A. Basak and J. C. Shain, Synthesis and Thermal Reactivity of a Novel Macrocyclic Enediyne and its Copper(II) Complex, Tetrahedron Lett., 1998, 39, 3029–3030 CrossRef CAS.
- A. Basak, J. C. Shain, U. K. Khamrai, K. R. Rudra and A. Basak, Nitrogen Substituted Cyclic Enediynes: Synthesis, Thermal Reactivity, and Complexation with Metal Ions, J. Chem. Soc., Perkin Trans. 1, 2000, 1955–1964 RSC.
- D. L. Boger and J. Zhou, CDPI3-Enediyne and CDPI3-EDTA Conjugates: A New Class of DNA Cleaving Agents, J. Org. Chem., 1993, 58, 3018–3024 CrossRef CAS.
- T. Kaneko, M. Takahashi and M. Hirama, Benzannelation Alters the Rate Limiting Step in Enediyne Cycloaromatization, Tetrahedron Lett., 1999, 40, 2015–2018 CrossRef CAS.
- S. Koseki, Benzannelation Effect on Enediyne Cycloaromatization: An ab Initio Molecular Orbital Study, J. Phys. Chem. A, 1999, 103, 7672–7675 CrossRef CAS.
- T. P. Lockhart, P. B. Comita and R. G. Bergman, Kinetic Evidence for the Formation of Discrete 1,4-Dehydrobenzene Intermediates. Trapping by Inter- and Intramolecular Hydrogen Atom Transfer and Observation of High-Temperature CIDNP, J. Am. Chem. Soc., 1981, 103, 4082–4090 CrossRef CAS.
- R. K. Mohamed, P. W. Peterson and I. V. Alabugin, Concerted Reactions That Produce Diradicals and Zwitterions: Electronic, Steric, Conformational, and Kinetic Control of Cycloaromatization Processes, Chem. Rev., 2013, 113, 7089–7129 CrossRef CAS PubMed.
- M. Prall, A. Wittkopp and P. R. Schreiner, Can Fulvenes Form From Enediynes? A Systematic High-Level Computational Study on Parent and Benzannelated Enediyne and Enyne-Allene Cyclizations, J. Phys. Chem. A, 2001, 105, 9265–9274 CrossRef CAS.
- M. F. Semmelhack and R. Sarpong, Kinetic Analysis of a Reactive Model Enediyne, J. Phys. Org. Chem., 2004, 17, 807–813 CrossRef CAS.
- E. C. Sherer, K. N. Kirschner, F. C. Pickard IV, C. Rein, S. Feldgus and G. C. Shields, Efficient and Accurate Characterization of the Bergman Cyclization for Several Enediynes Including an Expanded Substructure of Esperamicin A1, J. Phys. Chem. B, 2008, 112, 16917–16934 CrossRef CAS.
- T. A. Zeidan, M. Manoharan and I. V. Alabugin, Ortho Effect in the Bergman Cyclization: Interception of p-Benzyne Intermediate by Intramolecular Hydrogen Abstraction, J. Org. Chem., 2006, 71, 954–961 CrossRef CAS.
- T. A. Zeidan, S. V. Kovalenko, M. Manoharan and I. V. Alabugin, Ortho Effect in the Bergman Cyclization: Comparison of Experimental Approaches and Dissection of Cycloaromatization Kinetics, J. Org. Chem., 2006, 71, 962–975 CrossRef CAS.
- S. A. Valenzuela, A. J. Cortés, Z. J. E. Tippins, M. H. Daly, T. E. Keel, B. F. Gherman and J. D. Spence, Effect of Extended Benzannelation Orientation on Bergman and Related Cyclizations of Isomeric Quinoxalenediynes, J. Org. Chem., 2017, 82, 13297–13312 CrossRef CAS.
- T. G. Appleton, H. C. Clark and L. E. Manzer, Trans-Influence. Its Measurement and Significance, Coord. Chem. Rev., 1973, 10, 335–422 CrossRef CAS.
- E. Kraka and D. Cremer, CCSD(T) Investigation of the Bergman Cyclization of Enediyne. Relative Stability of o-, m-, and p-Didehydrobezene, J. Am. Chem. Soc., 1994, 116, 4929–4936 CrossRef CAS.
- S. Bhattacharyya, A. E. Clark, M. Pink and J. M. Zaleski, Isolation of Electronic from Geometric Contributions to Bergman Cyclization of Metalloenediynes, Chem. Commun., 2003, 1156–1157 RSC.
- A. E. Clark, S. Bhattacharyya and J. M. Zaleski, Density Functional Analysis of Ancillary Ligand Electronic Contributions to Metal-Mediated Enediyne Cyclization, Inorg. Chem., 2009, 48, 3926–3933 CrossRef CAS.
- L. J. K. Boerner, S. Mazumder, M. Pink, M.-H. Baik and J. M. Zaleski, Conformational and Electronic Consequences in Crafting Extended, p-Conjugated, Light-Harvesting Macrocycles, Chem. –Eur. J., 2011, 17, 14539–14551 CrossRef CAS.
- E. Kraka and D. Cremer, Enediynes, Enyne-Allenes, Their Reactions, and Beyond, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 285–324 CAS.
- I. V. Alabugin and M. Manoharan, Reactant Destabilization in the Bergman Cyclization and Rational Design of Light- and pH-Activated Enediynes, J. Phys. Chem. A, 2003, 107, 3363–3371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Mrenich, J. Bloino, B. J. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Rangasinghe, V. G. Zakrewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc. , Wallingford, CT., 2019 Search PubMed.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Function of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Burke, J. P. Perdew and Y. Wang, Electronic Density Functional Theory: Recent Progress and New Directions, Plenum, 1998 Search PubMed.
- J. P. Perdew, Electronic Structure of Solids '91, Akademie Verlag, Berlin, 1991 Search PubMed.
- J. P. Perdew, K. Burke and Y. Wang, Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-Electron System, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Erratum: Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4978 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- C. J. Cramer, Bergman, Aza-Bergman, and Protonated Aza-Bergman Cyclizations and Intermediate 2,5-Arynes: Chemistry and Challenges to Computation, J. Am. Chem. Soc., 1998, 120, 6261–6269 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab Initio Study of Solvated Molecules: A New Implementation of th Polarizable Continuum Model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of Ab Initio Molecular Potentials for the Prevision of Solvent Effects, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- S. Miertus and J. Tomasi, Approximate Evaluations of the Electrostatic Free Energy and Internal Energy Changes in Solution Processes, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- J. L. Pascual-Ahuir, E. Silla and I. Tuñón, GEPOL: An Improved Description of Molecular Surfaces. III. A New Algorithm for the Computation of a Solvent-Excluding Surface, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS.
- M.-T. Béraldin, E. Vauthier and S. Fliszár, Charge Distributions and Chemical Effects. XXVI. Relationships Between Nuclear Magnetic Resonance Shifts and Atomic Charges for 17O Nuclei in Ethers and Carbonyl Compounds, Can. J. Chem., 1982, 60, 106–110 CrossRef.
- S. Fliszár, G. Cardinal and M.-T. Béraldin, Charge Distributions and Chemical Effects. 30. Relationship Between Nuclear Magnetic Resonance Shifts and Atomic Charges, J. Am. Chem. Soc., 1982, 104, 5287–5292 CrossRef.
- H. Henry and S. Fliszár, Charge Distributions and Chemical Effects. 13. Ab Initio Study of the Charge Density-13C Nuclear Magnetic Resonance Shift Correlation for Ethylenic Carbon Atoms, J. Am. Chem. Soc., 1978, 100, 3312–3315 CrossRef CAS.
- I. V. Alabugin, in Stereoelectronic Effects: A Bridge Between Structure and Reactivity, 2016, pp. 322–375, DOI:10.1002/9781118906378.ch12.
- U. Rosenthal, G. Oehme, V. V. Burlakov, P. V. Petrovskii, V. B. Shur and M. E. Vol’pin, Carbon-13 and Proton NMR Studies of Selected Transition Metal Alkyne Complexes, J. Organomet. Chem., 1990, 391, 119–122 CrossRef CAS.
- M. Karplus and J. A. Pople, Theory of Carbon NMR Chemical Shifts in Conjugated Molecules, J. Chem. Phys., 1963, 38, 2803–2807 CrossRef CAS.
- K. M. Kirschner, S. C. Ratvasky, M. Pink and J. M. Zaleski, Anion Control of Lanthanoenediyne Cyclization, Inorg. Chem., 2019, 58, 9225–9235 CrossRef CAS.
- M. Rubin, A. Trofimov and V. Gevorgyan, Can Polarization of Triple Bond in Tolanes Be Deduced from 13C NMR Shifts? Re-evaluation of Factors Affecting Regiochemistry of the Palladium-Catalyzed Hydrostannation of Alkynes, J. Am. Chem. Soc., 2005, 127, 10243–10249 CrossRef CAS.
- E. Kleinpeter and A. Schulenburg, Quantification of the Push-Pull Effect in Tolanes and a Revaluation of the Factors Affecting the 13C Chemical Shifts of the Carbon Atoms of the C-C Triple Bond, J. Org. Chem., 2006, 71, 3869–3875 CrossRef CAS PubMed.
- N. Nardangeli, N. Topolovčan, R. Simionescu and T. Hudlicky, Polarization Effect on Regioselectivity of Pd-Catalyzed Cyclization of 2-Alkynylbenzaldehydes, Eur. J. Org Chem., 2020, 227–233 CrossRef CAS.
- J. Gräfenstein, E. Kraka, M. Filatov and D. Cremer, Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals?, Int. J. Mol. Sci., 2002, 3, 360–394 CrossRef.
- W. E. Slinkard and D. W. Meek, Co-ordination Properties of Methylenebis(diphenylphosphine chalcogenides) with Cobalt(II), Nickel(II), and Palladium(II), J. Chem. Soc., Dalton Trans., 1973, 1024–1027 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The data supporting this article including crystallographic data, DSC traces, kinetic measurements, and Cartesian coordinates and vibrational frequencies of computed structures. CCDC 2083580–2083585 and 2083637–2083642. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05396f |
| This journal is © The Royal Society of Chemistry 2025 |