
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilization of reactive rare earth alkyl complexes through mechanistic studies†
Elias
Tanuhadi
 ab,
Anna S.
Bair
ab,
Mary
Johnson
ab,
Philip
Fontaine
c,
Jerzy
Klosin
*c,
Sudipta
Pal
c and
Polly L.
Arnold
*ab
ab,
Anna S.
Bair
ab,
Mary
Johnson
ab,
Philip
Fontaine
c,
Jerzy
Klosin
*c,
Sudipta
Pal
c and
Polly L.
Arnold
*ab
aDept of Chemistry, University of California, Berkeley, Berkeley, CA 94720, USA. E-mail: pla@berkeley.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cCorporate R&D, The Dow Chemical Company, Midland, Michigan 48674, USA. E-mail: jklosin@dow.com
First published on 25th November 2024
Abstract
Rare earth tris(alkyl) complexes such as M(CH2SiMe3)3(sol)n are widely used as precursors for many compounds and as homogeneous catalysts for alkene polymerization and alkane functionalization. However, the thermal instability of those most conveniently made from the commercially available lithium salt of the neosilyl anion, LiCH2SiMe3, Li(r), restricts their utility. We present a new range of synthetically useful, more kinetically stable rare earth neosilyl solvates, derived from a full kinetic study of the various possible decomposition mechanisms of 7 known and 12 new solvated rare earth neosilyl complexes M(CH2SiMe3)3(sol)n M = Sc(III), Y(III), Lu(III), Sm(III), and sol = THF; TMEDA; DMPE; diglyme ((CH3)2(OCH2CH2)2O, G2), triglyme ((CH3)2(OCH2CH2)3O, G3). Surprisingly, simply using higher-denticity donors to sterically disfavor neosilyl γ-H elimination is not effective. While Sc(r)3((CH3)2(OCH2CH2)2O) has a half-life, t1/2, of 258.1 h, six times longer than for Sc(r)3(C4H8O)2 (t1/2 = 43 h), Lu(r)3((CH3)2(OCH2CH2)2O) and Y(r)3((CH3)2(OCH2CH2)2O) do not show the expected, analogous increased t1/2. This is because new decomposition pathways appear for poorly fitting donors. Finally, kinetic studies demonstrate the impact of small, and increasing amounts of LiCl on the kinetics of the reactivity of the smaller alkyls Y(r)3(THF)2 and Lu(r)3(THF)2; molecules used in hydrocarbon chemistry and catalysis for fifty years. A new route to pure Y(r)3(THF)2, which avoids the traditional use of Li(r), is presented.
Introduction
Since their discovery in 1973,1 neutral rare-earth metal complexes of the type RE(CH2SiMe3)3(THF)x, (RE = Sc, Y, Sm–Lu; x = 2, 3) featuring trimethylsilylmethyl ligands2,3 have been valuable reagents for (pre)catalyst design, in particular for olefin- and diene polymerization,4–7 and hydrocarbon C–H bond functionalization catalysis.8,9 More recently, RE(CH2SiMe3)3(donor)x (RE = Y, Dy, Er, Ho; donor = THF, quinuclidine, lutidine and OPCy3) have shown promise as single molecule magnets.10 Given that the Li and Mg salts of the trimethylsilylmethyl anion are commercially available, it is frustrating that all neosilyl complexes exhibit varying degrees of thermal instability, both in solution and in the solid state.2 The larger –CH(SiMe3)2 alkyl11 generates complexes with improved thermal stability, but its salts are not commercially available.The employment of donor-ligands to enhance kinetic stability, but yet can be easily substituted by protic reagents,12 should pave the way for novel RE-alkyl reactivity in these simple compounds.
Scheme 1 illustrates the three potential mechanisms for the elimination of SiMe4 from RE(CH2SiMe3)3(THF)2. The first mechanism, Scheme 1a, involves the attack by a coordinated neosilyl on an α-CH2 group. This results in the formation of a RE-alkylidene. However, this product has not been observed and is expected to be unstable. An early study of Er(CH2SiMe3)3(THF)3 suggested this decomposition mechanism, via silylalkylidene [Er(CH2SiMe3)(CHSiMe3)]n, but no evidence was provided to substantiate this, although this reactivity is well-documented for Ti.13,14 The second mechanism, denoted as β-H in Scheme 1b, involves the attack by a neosilyl on a β-CH2 group of a coordinated THF molecule, forming a metal vinyl alkoxide and ethylene, also anticipated to have low kinetic stability.15,16 The third mechanism, Scheme 1c, involves the attack of the CH2 carbon on a SiCH3, forming a metallacycle. Deuterolysis experiments with Lu(CH2SiMe3)3(THF)2 suggested a γ-H elimination pathway to a metalacyclic intermediate Me2Si(μ-CH2)2LuCH2SiMe3 although this was not observed directly.15
 | ||
| Scheme 1 The three possible mechanisms for elimination of SiMe4 from RE(r)3(THF)2 (r = CH2SiMe3): (a) α-H elimination; (b) β-H elimination, and (c) γ-H elimination. | ||
The addition of diglyme, (CH3)2(OCH2CH2)2O, G2, and glyme (CH3OCH2)2, G1, to Lu(CH2SiMe3)3(THF)2 generated the unanticipated mixed solvates Lu(CH2SiMe3)3(κ2-G2)(THF) and Lu(CH2SiMe3)3(κ2-G1)(THF) which the authors reported to be thermally robust but without providing data.15 Most recently, N,N tetramethyl ethylene diamine (TMEDA) or bis-dimethylphosphino ethane (DMPE) have been reported to enhance the kinetic inertness of [Y(CH2SiMe3)3], with no further data.17
Herein, we report a comprehensive study of the relative thermal stabilities of nineteen rare earth complexes under conditions widely applied for protonolysis reactions, identifying the decomposition mechanisms, characterizing the important intermediates, and highlighting the importance of compound purity. We have prepared a library of seven known and twelve new solvated rare earth neosilyl complexes M(r)3(donor)x for r = CH2SiMe3 (Fig. S5–S30, S55, and S56†) that cover a range of rare earth radii, between Sc (rcov, 6-coord = 0.745 Å) and Sm (rcov, 6-coord = 0.958 Å);18 tris(CH2SiMe3) adducts with measurable stability at room temperature have not been made yet for any RE larger than Sm.2,19 Previously reported complexes are: M(r)3(THF)2; M = Sm(III),19 Y(III),10 Lu(III),15 Sc(III);20 Y(r)3(donor);17 donor = DMPE/THF, TMEDA. The new complexes added here are: M(r)3(G2); M = Sm(III), Y(III), Lu(III), Sc(III); M(r)3(G3); M = Sm(III), Y(III), Lu(III); M(r)3(TMEDA); M = Sm(III), Lu(III), Sc(III); M(r)3(DMPE)(THF); M = Lu(III), Sc(III); G2 = diglyme, (CH3)2(OCH2CH2)2O, G3 = triglyme (CH3)2(OCH2CH2)3O (Table 1). For simplicity we have abbreviated them here to a label that describes the metal and donor solvent, i.e. M(r)3(donor) = M-donor. Single crystal X-ray data for all new compounds (except Sm-G2 and Sm-G3) are discussed in the ESI (Tables S1–S10 and Fig. S31–S40).†
| RE(r)3(donor)x | Label | t 1/2 [h] | Ref. |
|---|---|---|---|
| Sm(CH2SiMe3)3(triglyme) | Sm-triglyme | 0.7 | This work |
| Sm(CH2SiMe3)3(THF)3 | Sm-THF | 2.8 | 19 |
| Sm(CH2SiMe3)3(diglyme) | Sm-diglyme | 3.7 | This work |
| Y(CH2SiMe3)3(DMPE)(THF) | Y-DMPE | 5.8 | 17 |
| Y(CH2SiMe3)3(triglyme) | Y-triglyme | 7.8 | This work |
| Sc(CH2SiMe3)3(DMPE) | Sc-DMPE | 9 | This work |
| Sm(CH2SiMe3)3(TMEDA) | Sm-TMEDA | 11.7 | This work |
| Y(CH2SiMe3)3(diglyme) | Y-diglyme | 24.4 | This work |
| Y(CH2SiMe3)3(TMEDA) | Y-TMEDA | 36.5 | 17 |
| Sc(CH2SiMe3)3(THF)2 | Sc-THF | 43.9 | 20 |
| Sc(CH2SiMe3)3(TMEDA) | Sc-TMEDA | 73.4 | This work |
| Lu(CH2SiMe3)3(DMPE)(THF) | Lu-DMPE | 74.1 | This work |
| Y(CH2SiMe3)3(THF)2 | Y-THF | 119.6 | This work |
| Li[YCl(CH2SiMe3)3(THF)2] | LiClY-THF | 213 | 10 |
| Lu(CH2SiMe3)3(triglyme) | Lu-triglyme | 190.7 | This work |
| Sc(CH2SiMe3)3(diglyme) | Sc-diglyme | 258.1 | This work |
| Lu(CH2SiMe3)3(diglyme) | Lu-diglyme | 278.2 | This work |
| Lu(CH2SiMe3)3(THF)2 | Lu-THF | 482.6 | 15 |
| Lu(CH2SiMe3)3(TMEDA) | Lu-TMEDA | 487.8 | This work |
Results and discussion
Thermal stability and mechanistic studies
In a typical thermolysis experiment, 9.1 mM of a M complex is dissolved in C6D6 with hexamethylbenzene as an internal standard, in a J. Young NMR tube and the solution monitored by 1H NMR spectroscopy for at least two half-lives during incubation at 30 °C.Each compound's thermal stability is expressed as a half-life t1/2 [h] of the complex in solution. Since all three mechanisms result in the formation of SiMe4 (Scheme 1), the half-life (time for 50% of the compound to be converted into SiMe4) is determined from linearized plots of the relative integration of the SiMe4 resonance (Table 1) against hexamethylbenzene (Fig. S41–S45†). We first examined the half-life as a function of the metal center's ionic radius in M-THF and found a stability trend in the order Sm3+ < Sc3+ < Y3+ < Lu3+ (Table 1). The low thermal stability of Sm-THF is in line with its large size, and metals with ionic radii larger than Sm do not afford isolable neosilyl complexes.17,21
Notably, the stability trend Sc < Y < Lu is in line with the metals' Lewis-acidity rather than size.22
We examined the thermal decomposition mechanism of diamagnetic Lu-THF in greater depth. The compound decomposes with a half-life of t1/2 = 482.6 h. Kinetic studies using initial rates methods are first-order in [Lu-THF] suggesting an intramolecular decomposition process. We prepared the d2-r, d8-r, and d9-THF labelled isotopomers of Lu-THF (Fig. S48†).
First, α-H elimination can be ruled out based on the decomposition studies on Lu(d2-r)3(THF)2 and Lu(d9-r)3(THF)2 since NMR spectra show resonances corresponding to d2-and d10-labelled SiMe4 as decomposition products, respectively (Fig. 1, and S48, inset†). This strongly suggests γ-H elimination as the primary decomposition mechanism, Scheme 1c. We measured a primary KIE = 2.09 for Lu(d9-r)3(THF)2 (Fig. S53†) and no discernible KIEs for Lu(r)3(d8-THF)3 or Lu(d2-r)3(THF)2.
 | ||
| Fig. 1 Proposed γ-H elimination route converting Lu(d2-r)3(THF)2 to the metallacycle that is visible in NMR spectra of the d2-congener, with sections of the 1H NMR spectra below measured at t = 0 h (red), 78 h (orange), 103 h (purple), 127 h (green), 165 h (violet) in C6D6, 30 °C. The resonance circled in green and red show the increase of SiMe3(CD2H) and the –CH2 group of a d2-metallacyclic species, respectively. | ||
The thermolysis of Lu-THF, determined from the disappearance of the CH2 neosilyl resonance, proceeds with a rate (9 × 10−4 h−1) that is similar to that of SiMe4 formation (11 × 10−4 h−1), in agreement with decomposition via the liberation of 1 equiv. of SiMe4 per Lu-THF. The slight discrepancy between the rates of Lu-THF consumption and SiMe4 formation can be explained by an increasing resonance at δ = −0.894 in 1H NMR spectrum that overlaps with the CH2 – neosilyl resonance at δ = −0.891 (Fig. 1, and S49†). We assigned the growing resonance to the μ-CH2 of the formed metallacycle Me2Si(μ-CH2)2LuCH2SiMe3, (circled red in Fig. 1 and S49†), since the resonance is almost coincident with the LuCH2SiMe3 resonance, and can be observed in the 1H NMR spectrum of Lu(d2-r)3(THF)2.
Donor – dependence of RE-alkyl thermal stability
We initially synthesized Lu-G2, which is fac-pseudo octahedral at Lu in solution and the solid-state (Fig. S9, S10, and S35†) to target an increased thermal stability of Lu-THF by sterically disfavoring γ-H elimination. However, a kinetic study reveals that decomposition occurs via γ-H elimination from the glyme rather than from the neosilyl, which is reasonable considering the greater acidity of the glyme CH2 groups, decreasing the compound's half-life to t1/2 = 278.2 h (Table 1, and Scheme 2). The same destabilizing effect is observed for Y-G2. The size-fit correlation trend is supported by the observed increase in thermal stability for Sc-G2, which showed a negligible activation of the glyme CH2 groups (∼1.48% glyme decomposition by release of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHOMe23 into solution, Fig. S46, and S47†) granting the compound with an approximately six-fold increased half-life (t1/2 = 258.1 h) than Sc-THF (t1/2 = 43.9 h). Somewhat surprisingly to us, the destabilization is even more pronounced for Sm, Y, and Lu adducts of the larger glyme M-G3 (Table 1). To target stable complexes of larger Ln that are desirable synthesis precursors, we made M-donor for M = Sm, Y, Lu, Sc and donor = TMEDA, DMPE, recognizing both have less acidic CH2 groups than G2 and G3. This increases the half-life by a factor of up to four for Sm-TMEDA (t1/2 = 11.7 h). Notably, the kinetic study shows that Sm-TMEDA decomposes cleanly with release of one equiv. of SiMe4 (Fig. 2). Therefore we expect this will be an excellent precursor for protonolysis reactions. In contrast to M-TMEDA, all the M-DMPE congeners showed shorter half-lives compared to their THF counterparts (Table 1).
CHOMe23 into solution, Fig. S46, and S47†) granting the compound with an approximately six-fold increased half-life (t1/2 = 258.1 h) than Sc-THF (t1/2 = 43.9 h). Somewhat surprisingly to us, the destabilization is even more pronounced for Sm, Y, and Lu adducts of the larger glyme M-G3 (Table 1). To target stable complexes of larger Ln that are desirable synthesis precursors, we made M-donor for M = Sm, Y, Lu, Sc and donor = TMEDA, DMPE, recognizing both have less acidic CH2 groups than G2 and G3. This increases the half-life by a factor of up to four for Sm-TMEDA (t1/2 = 11.7 h). Notably, the kinetic study shows that Sm-TMEDA decomposes cleanly with release of one equiv. of SiMe4 (Fig. 2). Therefore we expect this will be an excellent precursor for protonolysis reactions. In contrast to M-TMEDA, all the M-DMPE congeners showed shorter half-lives compared to their THF counterparts (Table 1).
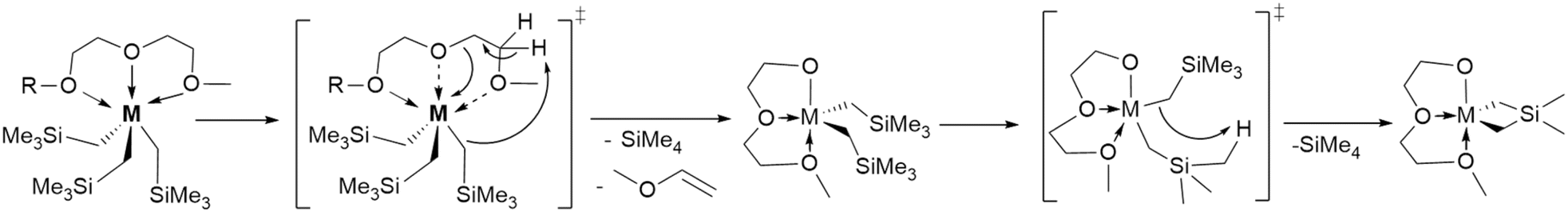 | ||
| Scheme 2 Suggested route to the hypothesized ethoxyether-stabilised Lu silametallacycle from Lu-diglyme. | ||
 | ||
| Fig. 2 1H NMR spectrum (600 MHz) of Sm-TMEDA in d6-benzene (t = 28 h) showing SiMe4 and TMEDA release. | ||
1H NMR studies on the d9-labelled isotopomers of Lu(r)3(sol) (sol = diglyme, TMEDA, DMPE) (Fig. S50–S53†) indicate thermal decomposition via γ-CH activation as shown by the emergence of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet resonance that can be assigned to SiMe3(CH2D).
:1:1 triplet resonance that can be assigned to SiMe3(CH2D).
Impact of LiCl on the thermal stability and reactivity of the RE alkyls
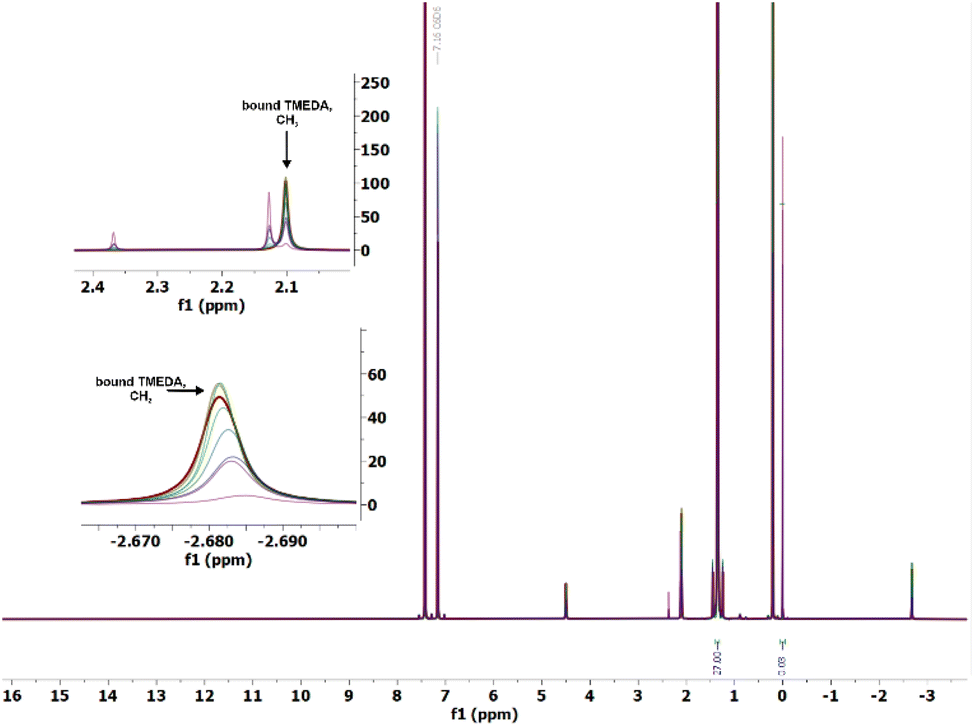
While M(r)3(THF)x (M = Sc, Lu; x = 2 and Sm; x = 3) were shown to decompose according to a first-order rate law in complex, our initial experiments indicated a second-order rate-law for decomposition of the YIII congener. This was supported by crossover studies on equimolar reaction mixtures of the d2-r and d9-r labelled complexes (Scheme S1, and Fig. S54†). All the reported routes to Y-THF originate from Li(r)6,10 (Table S11†). We note that crystal structures of LiCl – free, neutral homoleptic M(r)3(THF)x (M = Y, Lu, Yb, Er, Ho) have been reported.10,14,24 While the previous reports did not discuss whether 7Li NMR spectra were measured. However, we observed no resonance under normal conditions in 7Li NMR spectroscopic studies of solutions.The addition of 12-crown-4 ether (12-c-4) to a C6D6 solution of Y-THF results in a species we assign as Li(12-c-4)YCl(r)3(THF)·(12-c-4)1.75, which shows a 7Li NMR resonance at δ = −2.09 (Fig. S57†)23 and an 1H NMR spectrum that is different to the known Y(r)3(12-c-4) (Fig. S58, and S59†).25 In contrast, 12-c-4 addition to Lu(r)3(THF)2 solutions shows no change in the (silent) 7Li NMR spectrum.
ICP – OES analyses combined with titration for chloride content (Mohr's method) indicate that Y-THF prepared from the reagent Li(r) is contaminated with 7% LiCl. This content explains why samples isolated in previous reports were able to ‘pass’ elemental analyses for carbon and hydrogen content (Table S11†). The use of elemental analysis to confirm bulk purity of a sample of a new complex has been the subject of recent scrutiny.26,27
We examined the effect of increasing the amount of incorporated LiCl from zero to 1 equiv. on the properties of Lu-THF in benzene solution. We observe a factor of 2.5 increase in the half-life (t1/2) of the complex from t1/2 = 482 h for Lu-THF to t1/2 = 1122 h (Fig. S63†). It is notable that the chemical shifts of the THF CH2 groups are significantly shifted to higher frequencies in both the 1H – & 13C NMR spectra compared to those of the alkyl groups. We suggest that in benzene solution at higher concentrations the THF is preferentially binding to Li and the Cl to the RE center, (Fig. S61, and S62†). Although the LiCl impurity improves the complexes' half-lives,28,29 we expect it to hamper reactivity.
The inclusion of LiCl in s- and p-block alkyl complexes has been shown to significantly influence their stability, speciation, and reaction rates.28 Knochel and co-workers have demonstrated that organomagnesium and organozinc compounds, in the presence of alkali metals, exhibit increased basicity, allowing them to deprotonate otherwise inert aromatic and heteroaromatic C–H bonds.30 The combination of Grignard reagents with LiCl, known as turbo Grignard reagents, marks a significant advancement in main-group organometallic chemistry, leading to more accessible Grignard-like compounds that enable more powerful and controlled reactions. Recent ab initio molecular dynamics simulations suggest that LiCl aggregates form through μ2-Cl bridging ligands, creating Li4Cl4 cubane-like structures in THF solutions.29
The observation of second-order kinetics described above for the decomposition of our Li(r)-prepared Y-THF suggests the facile formation of a chloride bridged intermediate shown in Scheme 3, as a potential deviation from the mechanism found for the pure alkyls.
 | ||
| Scheme 3 Suggested route for the decomposition of LiCl-incorporated Y-THF that accounts for the observed kinetics. | ||
Finally, a new route to pure Y-THF from Na(r)31 is described in this work. As anticipated, ICP analyses and Mohr's method rule out LiCl or NaCl contamination of the complex, which decomposes following first order kinetics, with a t1/2 = 119.6 h, compared with t1/2 = 213 h for Y-THF prepared from Li(r) (Fig. S60, S64–S66,† and Table 1).
Conclusions
A new range of synthetically useful, more kinetically inert rare earth neosilyl solvates has been developed, based on the study of known and new solvates M(CH2SiMe3)3(sol)n, where M = Sc(III), Y(III), Lu(III), Sm(III), and sol = THF, TMEDA, DMPE, diglyme (G2), and triglyme (G3). The donor adduct with the longest half-life has been identified for each metal and is: Sc-G2, 258 h; Y-THF, 120 h, Lu-TMEDA, 488 h; Sm-TMEDA, 12 h at room temperature. Samarium is the largest rare earth cation that we have been able to stabilise as a tris(r) adduct by adding simple donors.Notably, simply increasing the denticity of donors to sterically hinder neosilyl γ-H elimination proves ineffective, as poorly fitting donors lead to new decomposition pathways. A precise size match with polydentate ligands can dramatically improve half-lives, as observed with Sc-G2. However, when the fit is less optimal, alternative decomposition mechanisms, such as γ-H activation at a glyme CH2 group, become more prominent. Thus, Sc-donor shows a significant increase in half-life when switching donor from THF (44 h) to diglyme (258 h), but Lu-donor shows a significant decrease in half-life when switching donor from THF (483 h) to diglyme (278 h). The stability trend of Sc < Y < Lu within the M-THF solvate series aligns more with Lewis acidity than size.
The incorporation of LiCl into the system results in an increase in half-life, particularly for Lu-THF by a factor of 2.5, although the effect on reactivity, particularly protonolysis, has not yet been evaluated.
Furthermore, while neutral complexes such as Lu(r)3(sol) (sol = THF, TMEDA, DMPE, G2) exhibit first-order kinetics involving γ-C–H activation, as confirmed by labelling experiments, ate complexes follow second-order kinetics, likely due to μ-Cl incorporation. Finally, a new route to pure Y-THF from Na(r) is described. While Y-THF exhibits a lower thermal stability compared to LiClY-THF, we expect it to open new perspectives for clean and fast protonolysis relevant to rare earth synthetic chemistry and catalysis.
Experimental
Materials and methods
All moisture and air sensitive materials were manipulated using standard high-vacuum Schlenk-line techniques and MBraun gloveboxes and stored under an atmosphere of dried and deoxygenated argon. All glassware items, cannulae and Fisherbrand 1.2 μm retention glass microfiber filters were dried in a 160 °C oven overnight before use. M(CH2SiMe3)3(THF)2; M = Y(III),10 Lu(III),15 Sc(III)20, Sm(CH2SiMe3)3(THF)3,19 and Y(CH2SiMe3)3(donor);17 donor = (DMPE)(THF), TMEDA and NaCH2SiMe3 (ref. 31) were prepared according to published literature procedures and characterized with 1H and 7Li NMR spectroscopy.n-Hexane, tetrahydrofuran (THF), diethyl ether (Et2O) and toluene for use with moisture and air sensitive compounds were dried using an MBRAUN SPS 800 Manual solvent purification system and stored over activated 3 Å molecular sieves. Benzene-d6 was purchased from Cambridge Isotope Laboratories and refluxed over potassium metal for 24 hours, freeze–pump–thaw degassed and purified by trap-to-trap distillation prior to use. THF-d8 was purchased from Cambridge Isotope Laboratories and dried over sodium/benzophenone before being freeze–pump–thaw degassed and purified by trap-to-trap distillation prior to use. Diglyme ((CH3)2(OCH2CH2)2O, G2) was dried over sodium metal before freeze–pump–thaw degassed and purified by trap-to-trap distillation prior to use. Triglyme (CH3)2(OCH2CH2)3O, G3) was dried over sodium metal before being purified by dynamic vacuum distillation prior to use. Tetramethylethylenediamine (TMEDA) was dried over sodium before being freeze–pump–thaw degassed and purified by trap-to-trap distillation prior to use. 1,2-Bis(dimethylphosphino)ethane (DMPE) was purified by trap-to-trap distillation and stored over 3 Å molecular sieves.
All solvents were purchased from Sigma-Aldrich or Fisher Scientific and stored over 3 Å molecular sieves for at least 12 hours before being used.
Selected synthetic procedures (all other data in the ESI†)
:1 mixture of Et2O and pentane. The resulting white suspension was cooled to −78 °C and NaCH2SiMe3 (ref. 31) (44 mg, 0.4 mmol, 3 eq.) was added portion wise as a solid. After stirring the resulting white suspension for 1 h at −78 °C, it was warmed to room temperature for 15 min. Filtration and removal of the solvent at −40 °C yielded the title compound as a white crystalline solid. Yield: 28 mg, 44% based on Y.
1H NMR (600 MHz, C6D6) δ 3.88 (s, 8H), 1.34–1.32 (m, 8H), 0.30 (s, 27H), −0.68 (d, J = 2.7 Hz, 6H). 13C NMR (151 MHz, C6D6) δ 70.60, 33.88, 33.65, 25.13, 4.63.
Anal. calcd for YC20H49Si3O2: C, 48.55; H, 9.98. Found: C, 48.43; H, 9.81.
1H NMR (600 MHz, C6D6) δ 3.07 (s, 6H), 2.97 (s, 4H), 2.68 (s, 4H), 0.44 (s, 27H), −0.42 (d, J = 2.9 Hz, 6H). 13C NMR (151 MHz, C6D6) δ 69.18, 67.97, 60.68, 35.42 (d, J = 36.5 Hz), 4.78.
Anal. calcd for YC18H47Si3O3: C, 44.60, H, 9.77. Found: C, 42.37; H, 9.16.
1H NMR (600 MHz, C6D6) δ 3.23 (t, J = 5.2 Hz, 3H), 3.02 (s, 6H), 2.76 (t, J = 5.3 Hz, 5H), 0.41 (s, 36H), 0.09 (s, 6H). 13C NMR (151 MHz, C6D6) δ 70.05, 69.71, 60.73, 4.33.
Anal. calcd for ScC18H47Si3O3: C, 49.05, H, 10.75. Found: C, 48.97; H, 10.66.
:1 mixture of pentane and Et2O (2.8 mL). The white suspension was cooled down to −78 °C and a solution of LiCH2SiMe3 (50.2 mg, 0.533 mmol, 3 eq.) in pentane (1.5 mL) was added dropwise. The resulting reaction mixture was stirred for 2 h at room temperature. The yellow filtrate was evacuated cold (−40 °C), the residues extracted with pentane (3 × 2 mL) and diglyme ((CH3OCH2CH2)2O, G2) (22.7 mg, 0.16 mmol) added to the filtrate to yield Sm(CH2SiMe3)3(C6H14O3) (Sm-G2) as a yellow crystalline solid after storage overnight at −40 °C. Yield: 41%, based on Sm.
1H NMR (600 MHz, C6D6) δ 5.46 (s, 6H), 5.29 (s, 6H), 0.44 (s, 27H), −0.14 (s, 4H), −1.53 (s, 4H). 13C NMR (151 MHz, C6D6) δ 131.79, 31.97, 23.05, 14.34, 2.84.
Anal. calcd for SmC18H47Si3O3: C, 39.58, H, 8.67. Found: C, 39.29; H, 8.30.
1H NMR (600 MHz, C6D6) δ 3.04 (s, 6H), 2.96 (t, J = 5.3 Hz, 4H), 2.61 (t, J = 5.3 Hz, 4H), 0.44 (s, 27H), −0.66 (s, 6H). 13C NMR (151 MHz, C6D6) δ 69.41, 68.43, 60.70, 41.60, 4.91.
Anal. calcd for LuC18H47Si3O3: C, 37.88, H, 8.30. Found: C, 37.54; H, 8.13.
1H NMR (400 MHz, C6D6) δ 3.47 (s, 6H), 3.09 (t, J = 4.8 Hz, 4H), 3.03–2.93 (m, 8H), 0.39 (s, 27H), −1.00 to −1.15 (m, 6H). 13C NMR (151 MHz, C6D6) δ 72.32, 71.98, 69.51, 68.37, 5.27, 4.92.
Anal. calcd for YC20H51Si3O4: C, 45.43, H, 9.72. Found: C, 45.38; H, 9.76.
:1 mixture of pentane and Et2O (8.4 mL). The white suspension was cooled down to −78 °C and a solution of LiCH2SiMe3 (146.5 mg, 1.556 mmol) in pentane (1.6 mL) was added dropwise. The resulting yellow reaction mixture was stirred for 2 h at room temperature. After stirring, it was cooled down to −78 °C at which temperature it was filtered. The yellow filtrate was evacuated cold (−40 °C), the residues were extracted with pentane (3 × 5 mL), and triglyme ((C8H18O4), G3) (92.4 mg, 0.52 mmol) was added to the combined fractions yielding Sm(CH2SiMe3)3((CH3)2(OCH2CH2)3O) (Sm-G3). Yield: 48%, based on Sm.
1H NMR (600 MHz, C6D6) δ 10.25 (s, 1H), 6.00 (s, 1H), 1.39 (s, 6H), −1.80 (s, 1H), −3.79 (s, 1H). 13C NMR (151 MHz, C6D6) δ 152.17, 86.66, 72.19, 70.77, 69.74, 67.62, 58.73, 3.61.
Anal. calcd for SmC20H51Si3O4: C, 40.7, H, 8.71. Found: C, 40.32; H, 8.43.
1H NMR (600 MHz, C6D6) δ 3.23 (t, J = 4.9 Hz, 4H), 3.13 (d, J = 8.5 Hz, 10H), 2.97 (t, J = 5.2 Hz, 4H), 0.42 (s, 27H), −0.82 (s, 6H). 13C NMR (151 MHz, C6D6) δ 71.53, 69.69, 69.26, 60.51, 38.44, 34.45, 22.73, 14.27, 5.06.
Anal. calcd for LuC20H51Si3O4: C, 39.07, H, 8.36. Found: C, 38.69; H, 8.11.
1H NMR (600 MHz, C6D6) δ 1.91 (s, 12H), 1.57 (s, 4H), 0.40 (s, 27H), 0.10 (s, 6H). 13C NMR (151 MHz, C6D6) δ 56.85, 46.69, 16.93, 4.41.
Anal. calcd for ScC18H49Si3N2: C,51.13, H, 11.68; N, 6.63. Found: C, 50.97; H, 11.59; N, 6.57.
1H NMR (600 MHz, C6D6) δ 4.50 (s, 6H), 2.10 (s, 12H), 0.21 (s, 27H), −2.68 (s, 4H). 13C NMR (151 MHz, C6D6) δ 58.45, 46.03.
1H NMR (600 MHz, C6D6) δ 1.82 (s, 12H), 1.48 (s, 4H), 0.41 (s, 27H), −0.61 (s, 6H). 13C NMR (151 MHz, C6D6) δ 56.60, 46.23, 45.51, 4.87.
Anal. calcd for LuC18H49Si3N2: C, 39.11, H, 8.93; N, 5.07. Found: C, 39.11; H, 8.97; N, 4.99.
1H NMR (600 MHz, C6D6) δ 0.92 (s, 4H), 0.76 (t, J = 1.6 Hz, 12H), 0.40 (s, 27H), 0.26 (s, 6H). 13C NMR (151 MHz, C6D6) δ 28.15, 14.04, 14.01, 13.97, 13.94. 31P NMR (243 MHz, C6D6) δ −36.30.
Anal. calcd for ScC18H49Si3P2: C,47.33; H, 10.81. Found: C, 46.95; H, 10.56.
:1 mixture of hexane and Et2O (2 mL). The resulting solution was stored at −40 °C, yielding diffraction quality crystals of Lu(CH2SiMe3)3(C6H16P2)(C4H8O) (Lu-DMPE) after three days. Yield: 41 mg, 60% based on Lu(CH2SiMe3)3(THF)2.
1H NMR (600 MHz, C6D6) δ 3.91 (s, 4H), 1.36–1.28 (m, 4H), 1.12 (t, J = 7.0 Hz, 4H), 0.78 (s, 12H), 0.36 (s, 27H), −0.63 (s, 6H). 13C NMR (151 MHz, C6D6) δ 25.13, 12.30, 4.77. 31P NMR (243 MHz, C6D6) δ −38.88.
Anal. calcd for LuC22H57Si3P2O: C,40.1; H, 8.72. Found: C, 39.89; H, 8.62.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data are deposited at CCDC, codes 2351142–2351150, 2369795 and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
ET, AB, MJ: experiments, analysis, writing & editing. PLA, JK, SP, PF: project conceptualization, funding acquisition, supervision, analysis, writing & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dow Chemical for support of this work. Some of the alkyl kinetic studies were also supported by the Catalysis Program of the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division at the Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231. We thank Dr Cooper Citek and the Catalysis Laboratory in the DOE Catalysis Program for resource and instrumentation used in this work. We acknowledge the National Institutes of Health (NIH) for funding the UC Berkeley College of Chemistry NMR facility under grant no. S10OD024998 and the NIH Shared Instrumentation Grant S10-RR027172 for funding the UC Berkeley College of Chemistry Small Molecule X-ray Crystallography Facility (CheXray).References
- M. F. Lappert and R. Pearce, J. Chem. Soc. Chem. Commun., 1973, 126, 10.1039/C39730000126
.
- M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194–6259 CrossRef CAS
- M. Zimmermann, J. Takats, G. Kiel, K. W. Tornroos and R. Anwander, Chem. Commun., 2008, 612–614, 10.1039/b713378b
- S. Arndt, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 5075–5079 CrossRef CAS PubMed
- C. Queffelec, F. Boeda, A. Pouilhès, A. Meddour, C. Kouklovsky, J. Hannedouche, J. Collin and E. Schulz, ChemCatChem, 2011, 3, 122–126 CrossRef CAS
- S. C. Kosloski-Oh, K. D. Knight and M. E. Fieser, Inorg. Chem., 2023, 63, 9464–9477 CrossRef
- J. Zhai, F. You, S. Xu, A. Zhu, X. Kang, Y.-M. So and X. Shi, Inorg. Chem., 2022, 61, 1287–1296 CrossRef CAS PubMed
- P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kuhn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS
- H. Liu, S. Saha and M. S. Eisen, Coord. Chem. Rev., 2023, 493, 215284 CrossRef CAS
- S.-M. Chen, Y.-Q. Zhang, J. Xiong, B.-W. Wang and S. Gao, Inorg. Chem., 2020, 59, 5835–5844 CrossRef CAS
-
M. F. Lappert, in Inorganic Compounds with Unusual Properties, American Chemical Society, 1976, ch. 21, vol. 150, pp. 256–265 Search PubMed
- D. J. H. Emslie, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2002, 21, 4226–4240 CrossRef CAS
- H. Schumann and J. Müller, J. Organomet. Chem., 1979, 169, C1–C4 CrossRef CAS
- J. J. Carbó, D. García-López, M. Gómez-Pantoja, J. I. González-Pérez, A. Martín, M. Mena and C. Santamaría, Organometallics, 2017, 36, 3076–3083 CrossRef
- K. A. Rufanov, D. M. M. Freckmann, H.-J. Kroth, S. Schutte and H. Schumann, Z. Naturforsch. B Chem. Sci., 2005, 60, 533–537 CrossRef CAS
- A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706–708 CrossRef CAS PubMed
- A. Mortis, C. Maichle-Mössmer and R. Anwander, Dalton Trans., 2022, 51, 1070–1085 RSC
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- H. Schumann, D. M. Freckmann and S. Dechert, Z. Anorg. Allg. Chem., 2002, 628, 2422–2426 CrossRef CAS
- A. G. B. Getsoian, B. Hu, J. T. Miller and A. S. Hock, Organometallics, 2017, 36, 3677–3685 CrossRef CAS
- A. Mortis, F. Kracht, T. Berger, J. Lebon, C. Maichle-Mössmer and R. Anwander, Dalton Trans., 2023, 52, 44–51 RSC
- R. R. Golwankar, T. D. Curry II, C. J. Paranjothi and J. D. Blakemore, Inorg. Chem., 2023, 62, 9765–9780 CrossRef CAS
- A. O. Tolpygin, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Organometallics, 2012, 31, 5405–5413 CrossRef CAS
- W. J. Evans, J. C. Brady and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7711–7712 CrossRef CAS
- S. Arndt, P. M. Zeimentz, T. P. Spaniol, J. Okuda, M. Honda and K. Tatsumi, Dalton Trans., 2003, 3622–3627 RSC
- F. P. Gabbaï, P. J. Chirik, D. E. Fogg, K. Meyer, D. J. Mindiola, L. L. Schafer and S.-L. You, Organometallics, 2016, 35, 3255–3256 CrossRef
- R. E. H. Kuveke, L. Barwise, Y. van Ingen, K. Vashisth, N. Roberts, S. S. Chitnis, J. L. Dutton, C. D. Martin and R. L. Melen, ACS Cent. Sci., 2022, 8, 855–863 CrossRef CAS PubMed
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS
- M. de Giovanetti, S. H. Hopen Eliasson, A. C. Castro, O. Eisenstein and M. Cascella, J. Am. Chem. Soc., 2023, 145, 16305–16309 CrossRef CAS
- B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and mechanistic details. CCDC 2351142–2351150, and 2369795. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05983b |
| This journal is © The Royal Society of Chemistry 2025 |