
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2-broken Ti–O bonds in the TiO6 octahedron of CaTiO3 for greatly enhanced room-temperature ferromagnetism†
Yuqi
Ouyang‡
a,
Bo
Gao‡
a,
Yaozheng
Tang
a,
Lianyu
Li
a and
Qun
Xu
 *ab
*ab
aCollege of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450052, P. R. China. E-mail: qunxu@zzu.edu.cn
bHenan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450052, P. R. China
First published on 11th December 2024
Abstract
Preparation of two-dimensional (2D) ferromagnetic nanomaterials and the study of their magnetic sources are crucial for the exploration of new materials with multiple applications. Herein, two-dimensional room-temperature ferromagnetic (FM) CaTiO3 nanosheets are successfully constructed with the assistance of supercritical carbon dioxide (SC CO2). In this process, the SC CO2-induced strain effect can lead to lattice expansion and introduction of O vacancies. More importantly, experimentally it can be found out that the breakage of the Ti–O2 bond by CO2 directly results in the equatorial plane of the TiO6 octahedron being exposed. This leads to more opportunities for oxygen vacancies and low-valent titanium to appear, where Ti3+ can optimize the spin structure, releasing the macroscopic magnetization. Greatly improved room-temperature ferromagnetic behavior, with an optimal magnetization of 0.1661 emu g−1 and a high Curie temperature (Tc) of 300 K can be achieved.
Introduction
With the advancement of nanotechnology, magnetic nanomaterials have shown great potential in magnetic recording and data storage due to their unique physical properties.1–3 Two-dimensional magnetic nanomaterials have attracted increasing research interest for fundamental physical studies as well as their huge potential applications in spintronic devices such as magnetic tunnel junctions, spin tunnel field-effect transistors, and spin valves.4–6 2D room-temperature ferromagnetic materials offer substantial potential for various advanced technological applications, particularly in fields like microelectronics, spintronics, non-volatile memory, and sensor technologies, and the discovery of ferromagnetism at room temperature in two-dimensional materials provides new possibilities for next-generation low-power, high-density electronic and information storage devices.7,8Among the plethora of magnetic materials discovered, perovskite oxides stand out for their intriguing array of physical properties, including ferroelectricity, ferromagnetism, superconductivity, and magnetoresistance, and this multifaceted behavior has sparked a surge of interest in both fundamental and applied investigations, leading to a significant global research endeavor over the past few decades.9–11 The robust correlation between the atomic structure and magnetic properties has been well documented in bulk perovskites.12,13 The general chemical formula for a chalcogenide oxide is ABO3, with A being the larger cation that forms the cubic sublattice and B being the smaller cation that is body-centered and cubic; in most cases, the B ion is coordinated with six O2− anions in corner-connected BO6 octahedra.14 Epitaxial strain can have a profound effect on the BO6 bonding environment, resulting in BO6 distortion and/or rotations.15,16 Given that magnetic ordering and transition temperatures in perovskites are sensitive to the B–O lengths and the B–O–B angles, modifying octahedral distortions and rotations provides a means of engineering magnetism in perovskite oxides.17 Furthermore, extrinsic effects such as oxygen vacancies, local non-stoichiometry, and cation intermixing are always present to some degree at real interfaces, and in some cases these effects dominate magnetic behavior.18–21 Nevertheless, the current development of spintronic devices is still hindered by limitations in both mechanisms and suitable materials. Finding stable two-dimensional ferromagnetic materials with room-temperature Curie temperatures and investigating their magnetic modulation mechanisms are key scientific issues for the development of spintronic devices, but challenges remain.
As a typical perovskite oxide, CaTiO3 is of great interest due to its high dielectric constant, low dielectric losses, and wide band gap.20 In the field of ferromagnetism, Hosseini prepared CaTiO3 nanoparticles by the sol–gel method, which exhibits room-temperature ferromagnetism after calcination at high temperature in air.22 Sun et al. found that room temperature ferromagnetism is present in the nanocrystalline CaTiO3 plates that are annealed at 1000 °C for 1 h and reduces after vacuum annealing, suggesting that the room temperature FM originates from the cation vacancies.23 Xu et al. reported that the spontaneous formation of a hole polaron in CaTiO3 could induce the coexistence of ferroelectricity and magnetism.24
In contrast to conventional techniques, SC CO2 displays superior capabilities in the synthesis and optimization of room-temperature ferromagnetic materials,25,26 owing to its excellent performance, such as high diffusion coefficients, outstanding wetting of surfaces, and low interfacial tension.27,28 In this work, we report SC CO2-assisted fabrication of 2D CaTiO3 nanosheets. In the presence of SC CO2, bulk CaTiO3 was exfoliated into nanosheet layers. The strain effect of SC CO2 at different pressures induces modulation and selective breakage of covalent bonds, which leads to the specific surface exposure of the prepared 2D CaTiO3. This results in an increase in the concentration of O vacancies and Ti3+, inducing 2D CaTiO3 to exhibit room-temperature ferromagnetism (Tc = 300 K). The saturation magnetization (Ms) value of 2D CaTiO3 is 0.1661 emu g−1, which is 32 times higher than that of the pristine CaTiO3 prepared by the sol–gel method.22
Results and discussion
Structural characterization of CaTiO3
Two-dimensional layered CaTiO3 nanosheets were prepared from pristine CaTiO3 powder with the aid of SC CO2 (Fig. 1a), and the preparation details are shown in the ESI.† The effect of SC CO2 in CaTiO3 is shown in Fig. 1b, where CO2 carries external physical pressure into the crystal structure, stretching the TiO6 octahedra, producing strain effects, destroying the original chemical coordination, exposing (101) the crystal plane and forming localized defects.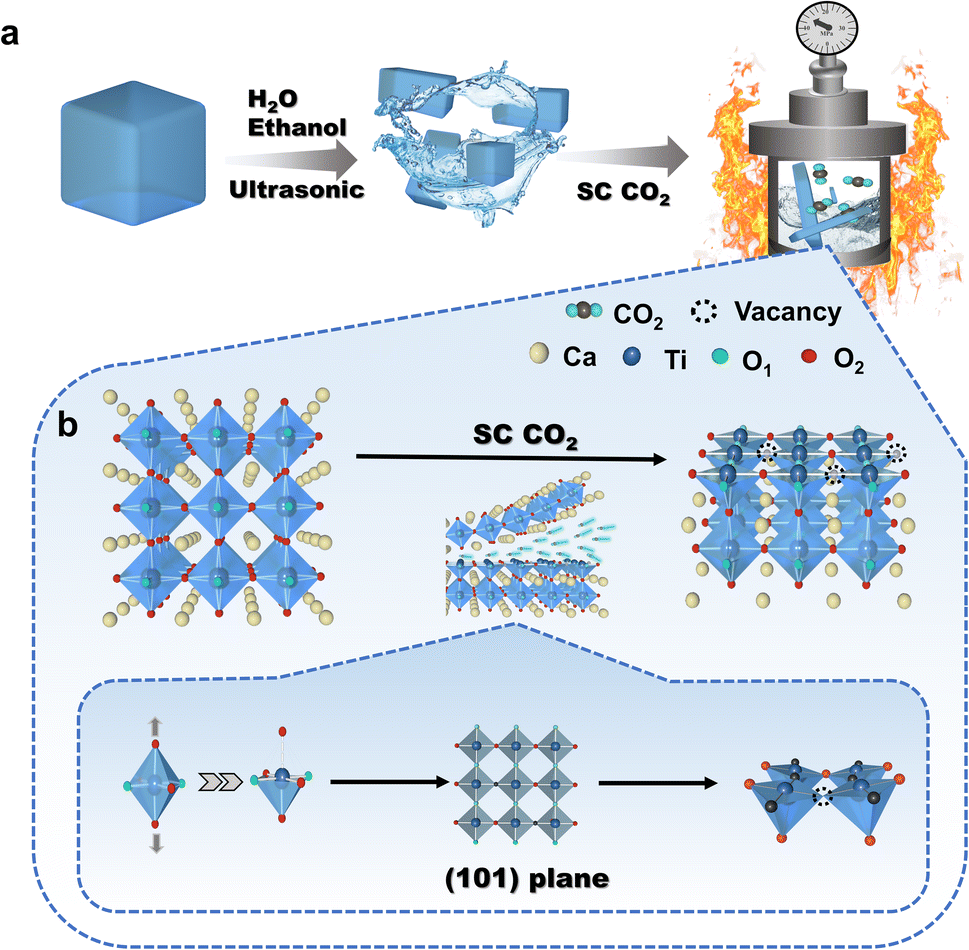 | ||
| Fig. 1 (a) The process flow of SC CO2 stripping on bulk phase CaTiO3. (b) Schematic diagram of the effect of SC CO2 on CaTiO3. | ||
The samples were observed by atomic force microscopy (AFM), and it was found that the SC CO2-treated 2D CaTiO3 nanosheets had a thickness of approximately ≈3.65 nm (Fig. 2a and b). The statistical analysis based on the AFM measurements demonstrated that the CaTiO3 nanosheets had various thicknesses, while 6–10 layers were found to be the major components (73.68%) (Fig. S2†).
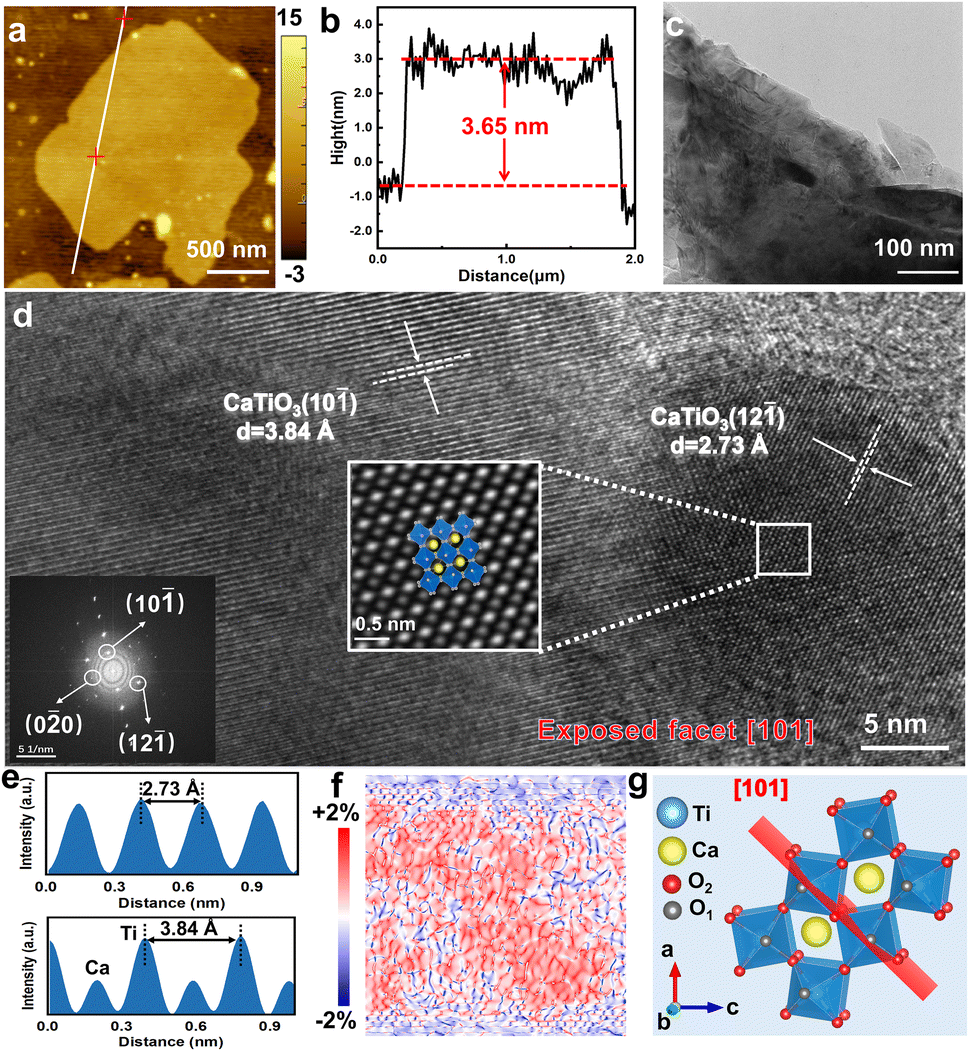 | ||
| Fig. 2 Characterization of CaTiO3 nanosheets treated with SC CO2 at 16 MPa. (a) AFM image and (b) height profile of the line in (a). (c) TEM image. (d) HRTEM image. (e) The line contour of the plane spacing in (d). (f) The strain map demonstrate on the HRTEM image by geometric phase analysis (GPA). Strain-values from −2% to +2% can be represented when color changes from blue to red. (g) Atomic structure diagram of the (101) plane. | ||
To obtain information on the microscopic morphology and structure, the samples were observed by transmission electron microscopy (TEM), and the images (Fig. 2c and S3†) also verified the low-layer CaTiO3 nanosheets, which turned slightly transparent to the electron beam. The atomic arrangement was characterized using high-resolution TEM (HRTEM) images and Fast Fourier transform (FFT) analysis. As shown in Fig. 2d, the lattice spacings of the SC CO2-treated CaTiO3 at 16 MPa are 0.384 and 0.273 nm, respectively, which are similar to the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (12) lattice spacings of standard CaTiO3 (0.382 and 0.272 nm), but with a slight increase, suggesting an increase in the lattice spacings of CaTiO3 after the SC CO2 treatment. Since SC CO2 has been reported to induce strain effects in oxide perovskites, we performed geometric phase analysis (GPA) on HRTEM images to assess the lattice strain of CaTiO3.29 The diffraction points (10−) and (0−
) and (12) lattice spacings of standard CaTiO3 (0.382 and 0.272 nm), but with a slight increase, suggesting an increase in the lattice spacings of CaTiO3 after the SC CO2 treatment. Since SC CO2 has been reported to induce strain effects in oxide perovskites, we performed geometric phase analysis (GPA) on HRTEM images to assess the lattice strain of CaTiO3.29 The diffraction points (10−) and (0−![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) in the inset of Fig. 2d were considered as the reflection sets. The strain effect of CaTiO3 nanosheets can be observed in Fig. 2f. Subsequently, the variation in strain was confirmed along the y-axis (εyy in Fig. S8, ESI†). The strain maps illustrate that lattice strain existed in CaTiO3 after the SC CO2 treatment. Moreover, the FFT-filter atomic resolution image is further utilized to visualize the domain transformation of the white-boxed region in Fig. 2d, and by fitting with the crystal models, the exposed surface of CaTiO3 at this pressure was determined to be the (101) crystal surface.
0) in the inset of Fig. 2d were considered as the reflection sets. The strain effect of CaTiO3 nanosheets can be observed in Fig. 2f. Subsequently, the variation in strain was confirmed along the y-axis (εyy in Fig. S8, ESI†). The strain maps illustrate that lattice strain existed in CaTiO3 after the SC CO2 treatment. Moreover, the FFT-filter atomic resolution image is further utilized to visualize the domain transformation of the white-boxed region in Fig. 2d, and by fitting with the crystal models, the exposed surface of CaTiO3 at this pressure was determined to be the (101) crystal surface.
The crystal structures of the as-prepared nanosheets were examined using X-ray diffraction (XRD), as shown in Fig. 3. The characteristic peaks for all samples matched well with the orthorhombic structure of CaTiO3 (PDF card no. 22-0153, space group Pnma). Meanwhile, partially enlarged details of the (101), (121), and (042) planes (Fig. 3b) indicate that the diffraction peak of CaTiO3 after SC CO2 treatment shifted toward a lower angle, which is essential evidence for the increased lattice spacing originated from the strain effect due to the SC CO2 treatment, as observed by TEM characterization. The Williamson–Hall method was used to calculate the micro-strains in the samples, and the results are shown in Fig. S9,† which indicate that different strains can be obtained upon pressure variation.30 The degrees of sample strain at different pressures indicate that CO2 at a pressure of 16 MPa exerts the most significant influence on the strain effect of CaTiO3, which reaches 1.324%. Furthermore, the Bragg equation was employed to calculate the increased proportion of the lattice spacings and lattice constants (Tables 1 and S1†). Notably, the largest increase in lattice spacing corresponding to the (101) plane was ≈1.5313%, suggesting that the (101) crystal plane may be more susceptible to being stretched.
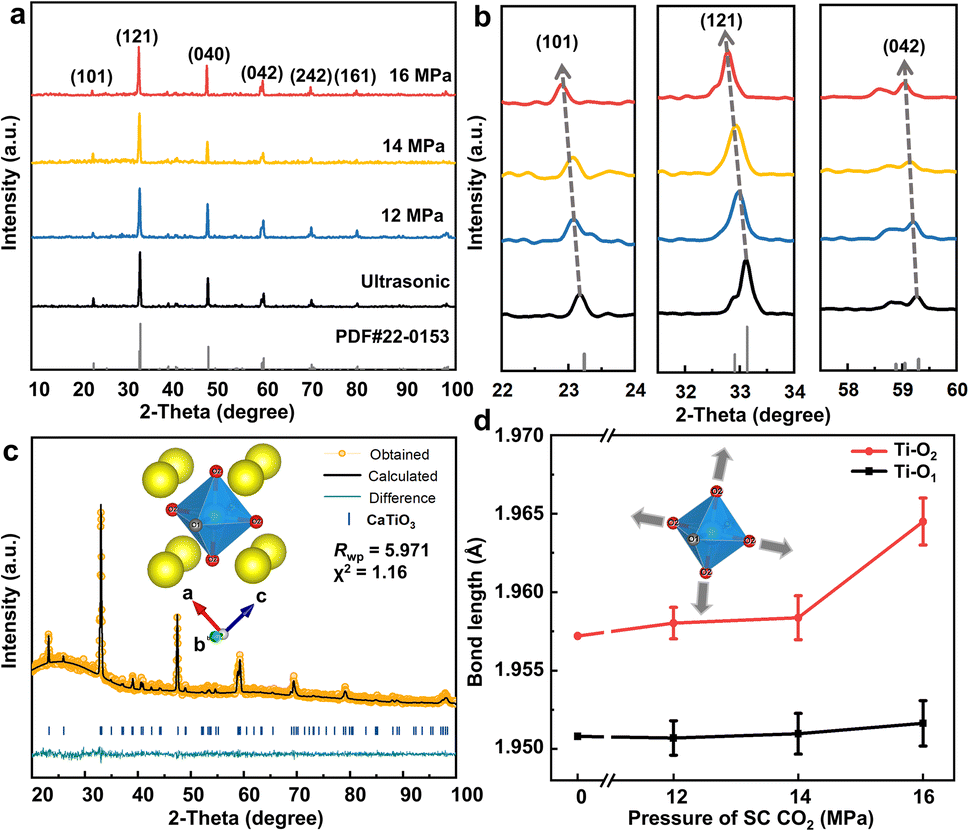 | ||
| Fig. 3 Characterization of CaTiO3 under different pressure treatments. (a) XRD. (b) Partially enlarged details of some planes. (c) The Rietveld-refined XRD patterns of CaTiO3 under 16 MPa. (d) Variation in the bond length value of TiO6 octahedra obtained by XRD refinement with increasing SC CO2 pressure. | ||
| Plane | Standard D (nm) | D (nm) | Δd (nm) | Strain% |
|---|---|---|---|---|
| (101) | 3.8240 | 3.8826 | 0.0586 | 1.5313 |
| (121) | 2.7010 | 2.7298 | 0.0288 | 1.0649 |
| (040) | 1.9110 | 1.9216 | 0.0106 | 0.5552 |
| (042) | 1.5570 | 1.5635 | 0.0065 | 0.4132 |
To obtain more structural information on CaTiO3 crystals under the strain effect, the GSAG II & EXPGUI software was used for Rietveld refinement analyses based on the XRD patterns of the sample under SC CO2 treatment. As shown in Fig. 3c, the reliability factor of the weighted patterns (Rwp) was less than 10%, indicating the high credibility of the fitting results. The results of the refinement demonstrate that as the CO2 pressure increases, the coordination environment of Ti ([TiO6] octahedra) was significantly distorted, including an increase in the average bond length, octahedral volume, and distortion index (Table S2†).
In the case of an octahedron centered on a Ti atom, the Ti atom is bonded to six neighboring O atoms. Two distinct types of Ti–O bonds are established, featuring varying bond lengths.31 The shorter bond is denoted as Ti–O1, with a length of 1.9508 Å, while the longer bond is referred to as Ti–O2, with a length of 1.9572 Å.32 In the refinement results, the Ti–O2 bond was observed to undergo a greater degree of stretching than the Ti–O1 bond when subjected to the same SC CO2 pressure (Fig. 3d). This phenomenon can be attributed to the inverse relationship between bond length and interatomic interactions, whereby longer bond lengths result in weaker interatomic forces. At a CO2 pressure of 16 MPa, there was a notable increase in the Ti–O2 bond length. Furthermore, at 16 MPa, the crystal surface exposed in CaTiO3 is the (101) crystal surface, and both Ti–O1 and Ti–O2 are present in the plane. However, only Ti–O2 is perpendicular to the direction of the (101) crystal surface, which is out of the plane. This difference between the in-plane and out-of-plane directions is also the reason for the different degrees of stretching of the two bonds.32 Given that the bond length is inversely proportional to the strength of interatomic interactions, this expansion in the Ti–O2 bond length resulted in a corresponding weakening of the interactions between Ti and O2 atoms. Consequently, the Ti–O2 bonds were more susceptible to breakage when external factors were introduced to create exposure of (101) crystal surfaces, which aligns with the observations reported in the HRTEM results.
The composition and chemical state of the as-prepared CaTiO3 were characterized by X-ray photoelectron spectroscopy (XPS) as shown in Fig. 4 and S11†. Fig. 4a illustrates that the Ti 2p1/2 and Ti 2p3/2 XPS lines of CaTiO3 exhibit two peaks at 463.90 and 458.06 eV, respectively, under a range of experimental conditions, including SC CO2 treatment and ultrasonic treatment, and these peaks are attributed to the Ti4+ in CaTiO3.33 Following SC CO2 treatment, the Ti 2p XPS lines exhibited two additional characteristic peaks centered at 458.09 and 457.58 eV (red area), which were attributed to Ti3+.34,35 The XPS spectrum of O is shown in Fig. 3b. The O 1s spectrum of pristine CaTiO3 is deconvoluted into four peaks. The peak at 529.49 eV is ascribed to the crystal lattice oxygen (OL), the peak at 530.53 eV is attributed to the oxygen vacancy (OV) (red area), the peak at 531.27 eV is attributed to OH− and CO32− groups (OA2) and the peak at 532.2 eV is attributed to the surface adsorbed oxygen (OA1).35–37
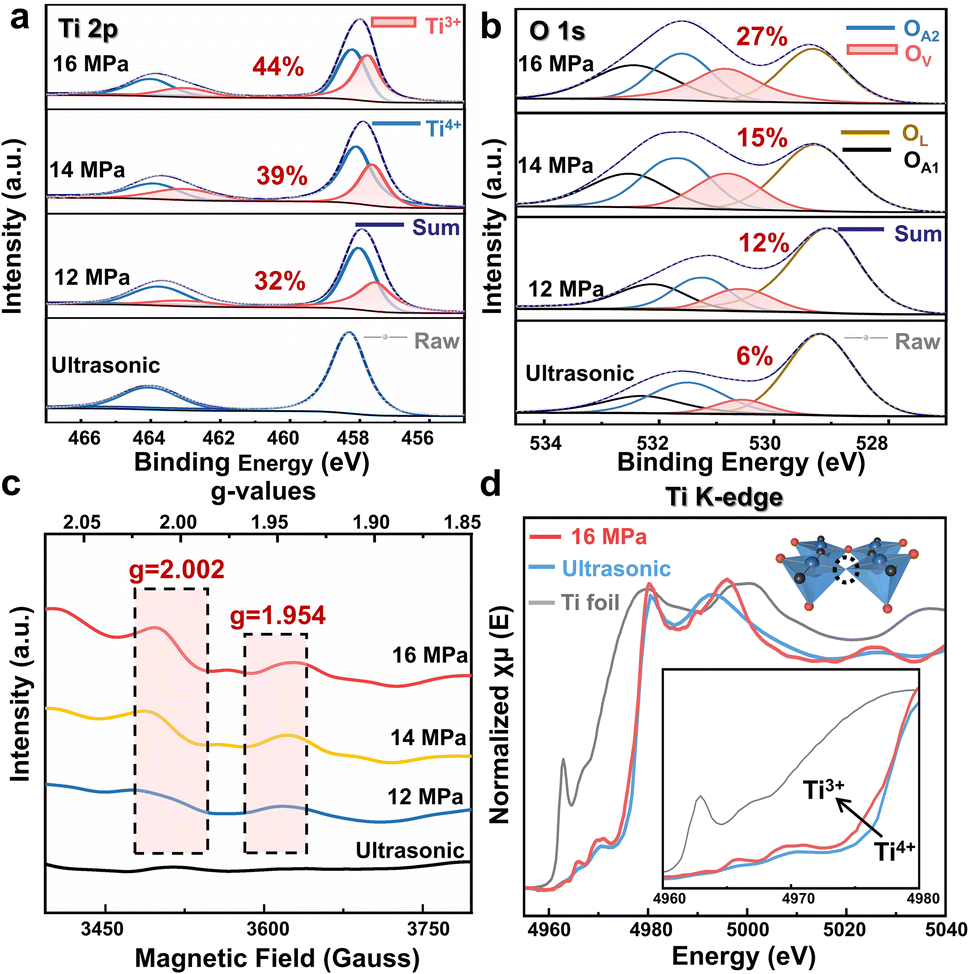 | ||
| Fig. 4 High-resolution XPS characterization under different CO2 pressures. (a) Ti 2p. (b) O 1s. The percentage in Fig. 4a and b corresponds to the content of Ti3+ and Ov, respectively. (c) Cryo-electron paramagnetic resonance (EPR) spectra. (d) Ti K-edge XANES spectra of Ti foil and CaTiO3 under different treatments. (Inset) A schematic diagram of the formation of the O-vacancy in non-stoichiometric CaTiO3 under the treatment with SC CO2 at 16 MPa. The blue balls represent Ti atoms, the red balls represent O1 atoms, the black balls represent O2 atoms, and the dashed circle represents the oxygen vacancy. | ||
According to Fig. S11,† the two peaks at 349.87 and 346.43 eV in the Ca 2p1/2 and Ca 2p3/2 XPS lines of CaTiO3 are attributed to the Ca2+ in CaTiO3.38 After SC CO2 treatment, two new characteristic peaks emerged at 355.49 and 346.96 eV (red area), which are assigned to the Ca at the surface, such as CaCO3 and Ca (OH)2.36
Low-temperature electron paramagnetic resonance (EPR) of the CaTiO3 samples is shown in Fig. 4c. After the SC CO2 treatment, a signal appears at approximately g = 2.002, which is assigned to the electrons trapped in the OV. Moreover, CaTiO3 treated with SC CO2 exhibits a signal at g = 1.954, which can be attributed to the d1 electron of Ti3+.39–41 The appearance of these two signal peaks further confirms the effect of the SC CO2 pressure on the surface OVs and Ti3+ concentration, which is in line with the XPS studies.
In order to elucidate the local order structure around the Ti atom, Ti K-edge X-ray absorption near-edge structure (XANES) spectra were measured. As illustrated in Fig. 4d, the red curve (16 MPa SC CO2 treated CaTiO3) exhibits a slightly higher absorption intensity near the Ti3+ peak in comparison to the blue curve (Ultrasonic). This suggests that high-pressure synthesis may result in a greater extent of Ti4+ being partially reduced to Ti3+ in CaTiO3, which is in accordance with the XPS analysis (Fig. 4a).42,43 Furthermore, the physical origin of the pre-edge feature at approximately 4965 eV is the transition of the metallic 1s electron to an unfilled d state.34 This peak area was attributed to the five-fold coordination (TiO5) concentration, and as shown in Fig. 4c, the area of this peak was observed to vary with the pressure of SC CO2, indicating that the CaTiO3 sample synthesized under high pressure may have undergone a crystal restructuring process, accompanied by an increase in the TiO5 content, which could affect the overlap of Ti and O orbitals.44,45 Moreover, the intensity of first features beyond the edge (between 4980 and 5010 eV) is observed to increase with the increase in pressure of CO2. The higher intensity of these peaks indicates that the local environment of the Ti atom is non-centrosymmetric, which distorts the octahedral configuration.46,47 It has been established that oxygen vacancies play a crucial role in the ferromagnetic ordering of otherwise non-magnetic materials, whereby they facilitate carrier-mediated ferromagnetic interactions between Ti ions.45 In the case of CaTiO3, the presence of oxygen vacancies has been observed to promote spin-polarized charge carriers, thereby enhancing the overall magnetic moment of the material.
It is evident that the concentration of Ti3+ and OV are elevated upon the introduction of SC CO2, which corroborates the presence of defects in the SC CO2 treated CaTiO3 samples. These defects resulted in the formation of non-stoichiometric CaTiO3. A substantial body of previous studies has demonstrated a robust correlation between non-stoichiometry and material magnetism. A comparison of the variation of Ti3+ and Ov with SC CO2 pressure revealed that the OV and Ti3+ contents reached their maximum values at 16 MPa (Fig. S12†), which may be related to the exposure surface. The breakage of the Ti–O2 bond, which occurs when the (101) crystalline surface of CaTiO3 is exposed, results in the equatorial plane of the TiO6 octahedron being exposed. This leads to an increase in the occupation of Ti and O atoms on the surface, thereby increasing the probability of the formation of Ti3+ and Ov.
Magnetic properties
The magnetic properties of the as-obtained 2D CaTiO3 were subsequently investigated by using a SQUID magnetometer. The ferromagnetic nature was confirmed by the appearance of magnetic hysteresis, saturation magnetization (Ms), and coercivity (Hc). Compared to the combination of ferromagnetism and diamagnetism of ultrasonicated CaTiO3 in Fig. 5a, CO2-treated CaTiO3 exhibited significantly enhanced ferromagnetism with saturation magnetizations of 0.1611, 0.0603, and 0.0229 emu g−1, respectively (Fig. 5b). This value is higher than that in previously reported CaTiO3 nanoparticles prepared by the sol–gel method (0.005 emu g−1) or the high-temperature solid-phase synthesis method (0.13 emu g−1).22,23 The enlarged part of the curve near H = 0 (Fig. 5c) shows an obvious residual magnetization (0.0117 emu g−1) and coercive field (71.44 Oe). Additionally, the room-temperature ferromagnetic signature of the CaTiO3 nanosheets with SC CO2 treatment was validated by analyzing the temperature dependence of magnetization measurements. Under a constant magnetic field (H = 500 Oe), field cooling (FC) and zero-field cooling (ZFC) processes were applied to measure the temperature dependence of magnetization (M–T) from 5 to 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K (Fig. 5d). The FC magnetization curve exhibited a downward trend during heating, whereas the ZFC magnetization curve increased with increasing temperature. Importantly, the FC and ZFC lines intersected at approximately 300 K, indicating that the transition temperature of the sample was approximately room temperature. Because no magnetic impurities were observed according to the XPS survey (Fig. S13†), it can be concluded that the observed room-temperature ferromagnetism originates from the CaTiO3 sample under SC CO2 treatment. Furthermore, following a one-year period of storage at room temperature and pressure, the M−H curve was reassessed, and the results are shown in Fig. S14;† these results demonstrate that Ms of the samples exhibited only minor reductions, confirming the robust stability of ferromagnetism in SC CO2-induced ferromagnetic CaTiO3.
K (Fig. 5d). The FC magnetization curve exhibited a downward trend during heating, whereas the ZFC magnetization curve increased with increasing temperature. Importantly, the FC and ZFC lines intersected at approximately 300 K, indicating that the transition temperature of the sample was approximately room temperature. Because no magnetic impurities were observed according to the XPS survey (Fig. S13†), it can be concluded that the observed room-temperature ferromagnetism originates from the CaTiO3 sample under SC CO2 treatment. Furthermore, following a one-year period of storage at room temperature and pressure, the M−H curve was reassessed, and the results are shown in Fig. S14;† these results demonstrate that Ms of the samples exhibited only minor reductions, confirming the robust stability of ferromagnetism in SC CO2-induced ferromagnetic CaTiO3.
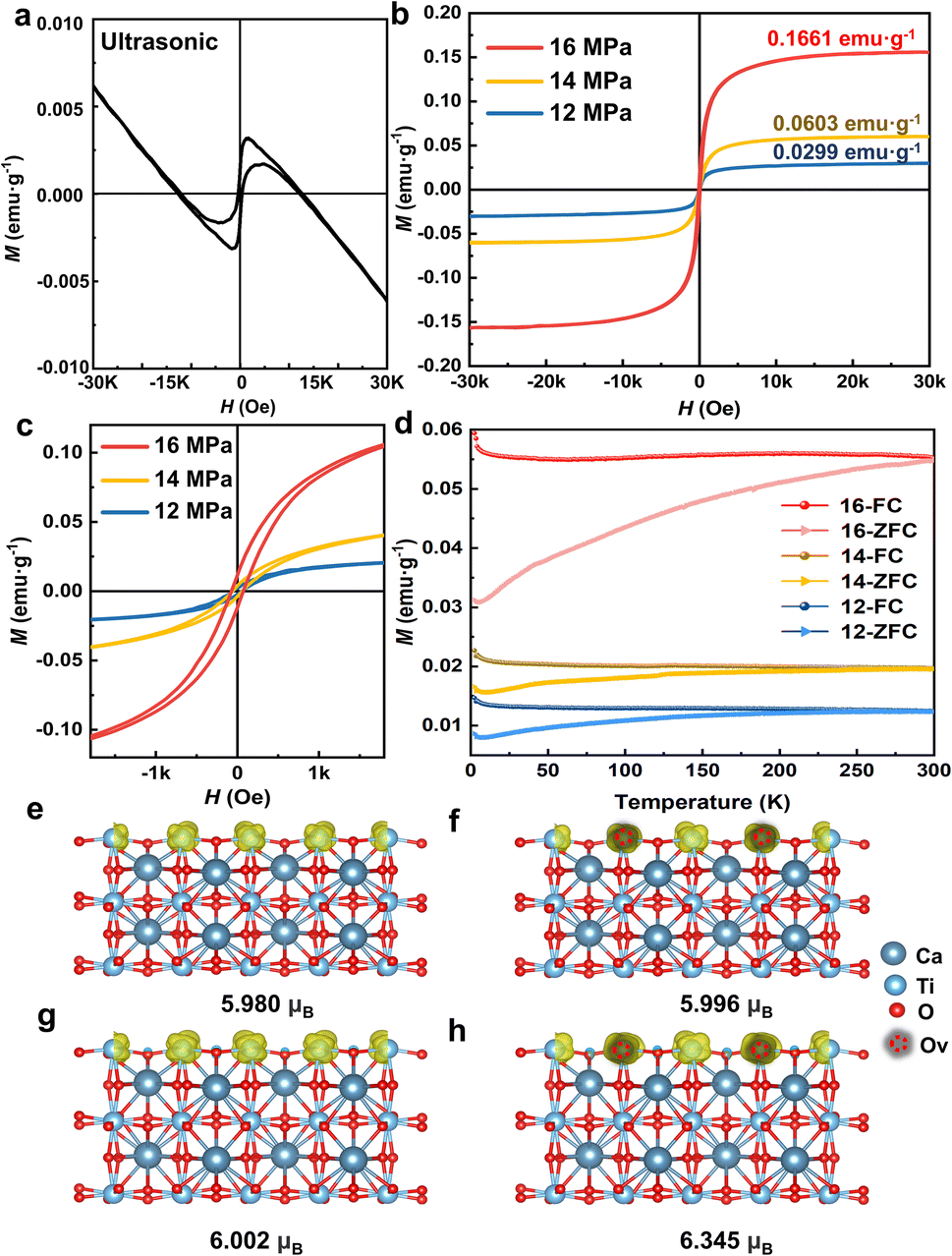 | ||
| Fig. 5 Ferromagnetic behavior. (a and b) M−H curves at 300 K. (c) Magnified curves near H = 0 Oe. (d) FC-ZFC magnetization curve of CaTiO3 treated with SC CO2 in an external magnetic field of 500 Oe. (e) Spin charge density distribution of the (101) plane without the strain effect or oxygen vacancies. (f) Spin charge density distribution with oxygen vacancies. (g) Spin charge density distribution with the strain effect. (h) Spin charge density distribution with the strain effect and oxygen vacancies. | ||
In order to investigate the origin of room-temperature ferromagnetism in SC CO2-treated CaTiO3, density-functional theory (DFT) calculation was conducted. According to experimental results, the breakage of Ti–O2 bonds in the TiO6 octahedra and the exposure of (101) facets occurred in SC CO2-treated CaTiO3 as a consequence of strain effects. These strain effects, induced by lattice distortions, can significantly modify magnetic interactions by altering the electronic structure, bond lengths and orbital overlap.48,49 Moreover, experimental results reveal that oxygen vacancies in CaTiO3 are generated in conjunction with the strain effect. Numerous studies on perovskite oxide have established that oxygen vacancies can disrupt the local electronic structure and generate unpaired electron spins, leading to localized magnetic moments.21,50 Consequently, the spin charge density distributions of the (101) plane in CaTiO3 were modelled using DFT, with a focus on separately investigating the effects of oxygen vacancies and tensile strain to ascertain which factor exerts a more dominant influence on the magnetic properties of CaTiO3. As illustrated in Fig. 5e, the magnetic moment of CaTiO3 without oxygen vacancies or tensile strain was 5.980 μB. Upon the introduction of oxygen vacancies and tensile strain, the magnetic moment increased to 5.996 μB and 6.002 μB, respectively (Fig. 5f and g). It is evident that tensile strain has a more pronounced impact on the magnetic properties of CaTiO3 than the presence of vacancies. Furthermore, according to the calculations, the magnetic moment of the (101) plane in CaTiO3 increases by 6% to 6.354 μB as a result of the combined effect of oxygen vacancies and tensile strain. However, due to the limitation of the number of atoms (eight CaTiO3 formula units), the promotion of CaTiO3 ferromagnetism by the surface strain effect and oxygen vacancies is constrained during the calculation. In contrast, experimental evidence demonstrates that there is a considerably larger amount of surface strain effect and defect content in the SC CO2-treated CaTiO3. Nevertheless, the calculation results still indicate that on the basis of surface formation, the presence of oxygen vacancies and strain has a further enhancing effect on the observed ferromagnetism. Therefore, based on both experimental and theoretical investigations, the origin of ferromagnetism in CaTiO3 can be attributed to the combined effect of strain and oxygen vacancies induced by surface exposure during SC CO2 treatment.
Experimental
Materials
Commercial CaTiO3 powder (99%) was purchased from Sigma Aldrich and can be used without further purification, and 99.99% pure CO2 was provided by Zhengzhou Shuang yang Gas Company as the receiver. All the experimental ethanol was purchased from China Medical Chemical Reagent Co., Ltd, and deionized water was prepared with double-distilled water.Exfoliation process
The SC CO2 apparatus used for this experiment was composed mainly of a stainless steel autoclave (50 mL) with a heating jacket and a temperature controller. In this study, 60 mg bulk CaTiO3 was dispersed in 30 mL of ethanol/water (Vethanol:Vwater = 1:1) solution and subjected to ultrasonic treatment for 4 hours. The resulting suspension was labeled as ultrasonicated CaTiO3. Meanwhile, the autoclave was heated up to 40 °C. Then the ultrasonicated CaTiO3 was immediately transferred into the autoclave, and CO2 was subsequently charged into the autoclave to the desired pressure (12/14/16 MPa) under stirring. During the exfoliation process, the temperature and pressure of the autoclave were kept constant. After reaction for 4 hours under SC CO2 conditions, the system was slowly depressurized and the sample was collected. The resulting dispersion was centrifuged at 4500 rpm for 10 minutes to remove the aggregates, and the precipitated supernatant was collected at 10000 rpm for 15 minutes. Finally, the precipitates were dried in an oven at 60 °C.
Characterization
Transmission electron microscope (TEM) images were recorded on a FEI Tecnai G2-F20 at an acceleration voltage of 200 kV. The thickness of nanosheets was measured by using an atomic force microscope (Bruker Dimension Icon). X-ray diffraction (XRD) patterns were collected on a Bruker D8 Focus diffractometer (Bruker AXS, Germany) using Cu K radiation. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha+ system. Raman measurements were performed using a LabRAM HR Evolution with a laser wavelength of 830 nm. The electron paramagnetic resonances were obtained by using an electron paramagnetic resonance spectrometer (EMX-9.5/12). The magnetic measurement was carried out with a physical property measurement system (quantum design, PPMS-9) and the magnetic hysteresis loop is obtained in the range of −30k Oe < H < 30k Oe at room temperature. K-edge analysis was performed with Si(111) crystal monochromators at the BL11B beamlines at the Shanghai Synchrotron Radiation Facility (SSRF) (Shanghai, China). Before the analysis at the beamline, the samples were pressed into thin sheets 1 cm in diameter and sealed using a Kapton tape film. The XAFS spectra were recorded at room temperature using a 4-channel Silicon Drift Detector (SDD) Bruker 5040. Negligible changes in the line-shape and peak position of K-edge XANES spectra were observed between two scans taken for a specific sample. The spectra were processed and analyzed by the software codes Athena.51Computational methods
The theoretical calculation based on density functional theory was completed by the VASP software package,52 and the projector augmented wave (PAW) method53 was used to describe the ion–electron interaction. Furthermore, the Perdew–Burke–Ernzerhof (PBE) functional54 was used to describe the exchange–correlation energy of the simulation system, and DFT-D355 was used to improve the calculation accuracy of dispersion force. The cutoff energy of plane wave basis was 500 eV, and the Monkhorst Pack scheme was used to generate k-points with a density of 0.4 per Angstrom for Brillouin zone sampling. The self-consistent field (SCF) calculation was kept within the energy convergence criterion of 1.0 × 10−5 eV. EDIFFG: this parameter is used to control the force convergence criteria for structural optimization. The optimization ends when the force on all atoms is less than 0.01 eV Å−1. In addition, a correction based on the Hubbard U model was used for the d orbitals of Ti atoms to obtain more accurate correlation energy, and the U value was 3.4 eV. For the slab model,56 a vacuum layer with a thickness of 15 Å was established to avoid layer-to-layer interaction.The data obtained by XRD were imported into Origin software, and then the diffraction peak 2θ values of (121) (040) (042) were obtained by Lorentz fit. The Brag eqn (1) was used to obtain the plane spacing, and the Williamson–Hall (W–H) eqn (2) was used to calculate the strain.
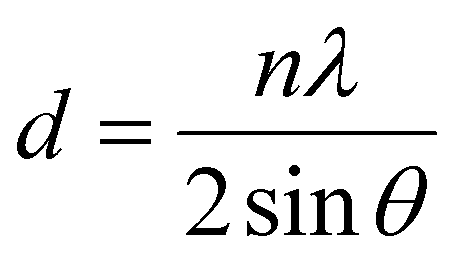 | (1) |
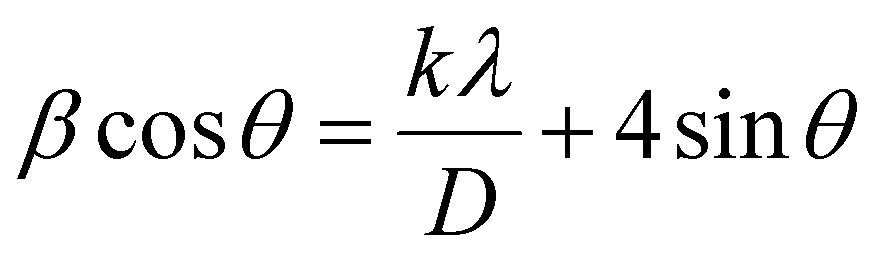 | (2) |
Geometric Phase Analysis (GPA) of HRTEM images used the GMS plug-in FRWR tools.
Conclusions
In summary, 2D CaTiO3 with room-temperature ferromagnetism has been prepared using SC CO2. Experimental studies have demonstrated the critical synergistic role of SC CO2 induced strain effects and O vacancies in ferromagnetic manipulation. Specifically, GPA illustrates the presence of strain effects, and XRD confirms the increase in crystal spacing, in particular the elongation of the Ti–O2 bonds in TiO6 octahedron, and the breakage of the Ti–O2 bonds at the surface of CaTiO3 forms the exposition of (101) plane observed in HRTEM, which confers more opportunities for oxygen vacancies and low-valent titanium to appear, where Ti3+ can optimize the spin structure, releasing the macroscopic magnetization, which leads to weak ferromagnetic ordering. To the best of our knowledge, this is the first report of increasing the intrinsic magnetic properties of CaTiO3 by an external stress field approach. This approach can achieve 2D ferromagnetism at room temperature Tc, in contrast to the conventional doping with magnetic impurities or building defects. Significantly, this method has been demonstrated to be effective in other perovskite,26 offering a promising avenue for investigating room-temperature ferromagnetism in perovskite materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. Ouyang conducted experiments, collected data and wrote the manuscript. B. Gao conducted computational investigations. Y. Tang and L. Li interpreted the data. Q. Xu designed the project and experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 51173170, 21703207, 21773216), the joint project from the Henan-Provincial and the China-National Natural Science Foundation (Project No. U2004208), and the Central Plains Science and Technology Innovation Leading Talent Project (Project No. 234200510008).Notes and references
- S. P. Gubin, Y. A. Koksharov, G. B. Khomutov and G. Y. Yurkov, Russ. Chem. Rev., 2005, 74, 489 CrossRef CAS.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao, T. Hu, Z. Sun and S. Wei, J. Am. Chem. Soc., 2015, 137, 2622 CrossRef CAS.
- Y. Sun, H. Lv, H. Yao, Y. Gao and C. Zhang, Carbon Energy, 2024, 6, e575 CrossRef.
- Q. H. Wang, A. Bedoya-Pinto, M. Blei, A. H. Dismukes, A. Hamo, S. Jenkins, M. Koperski, Y. Liu, Q.-C. Sun, E. J. Telford, H. H. Kim, M. Augustin, U. Vool, J.-X. Yin, L. H. Li, A. Falin, C. R. Dean, F. Casanova, R. F. L. Evans, M. Chshiev, A. Mishchenko, C. Petrovic, R. He, L. Zhao, A. W. Tsen, B. D. Gerardot, M. Brotons-Gisbert, Z. Guguchia, X. Roy, S. Tongay, Z. Wang, M. Z. Hasan, J. Wrachtrup, A. Yacoby, A. Fert, S. Parkin, K. S. Novoselov, P. Dai, L. Balicas and E. J. G. Santos, ACS Nano, 2022, 16, 6960 CrossRef CAS PubMed.
- X. Zhang, X. Wang, T. He, L. Wang, W.-W. Yu, Y. Liu, G. Liu and Z. Cheng, Sci. Bull., 2023, 68, 2639 CrossRef.
- Y. Jiao, Y. Li, Y. Zhou, P. Cen, Y. Ding, Y. Guo and X. Liu, Chin. Chem. Lett., 2024, 35, 109082 CrossRef CAS.
- Y. Guo, B. Wang, X. Zhang, S. Yuan, L. Ma and J. Wang, InfoMat, 2020, 2, 639 CrossRef CAS.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265 CrossRef CAS.
- N. C. Bristowe, J. Varignon, D. Fontaine, E. Bousquet and P. Ghosez, Nat. Commun., 2015, 6, 6677 CrossRef CAS.
- M. Namba, H. Takatsu, R. Mikita, Y. Sijia, K. Murayama, H.-B. Li, R. Terada, C. Tassel, H. Ubukata, M. Ochi, R. Saez-Puche, E. P. Latasa, N. Ishimatsu, D. Shiga, H. Kumigashira, K. Kinjo, S. Kitagawa, K. Ishida, T. Terashima, K. Fujita, T. Mashiko, K. Yanagisawa, K. Kimoto and H. Kageyama, J. Am. Chem. Soc., 2023, 145, 21807 CrossRef CAS.
- A. C. Garcia-Castro, Y. Ma, Z. Romestan, E. Bousquet, C. Cen and A. H. Romero, Adv. Funct. Mater., 2022, 32, 2107135 CrossRef CAS.
- P. G. Radaelli, G. Iannone, M. Marezio, H. Y. Hwang, S. W. Cheong, J. D. Jorgensen and D. N. Argyriou, Phys. Rev. B:Condens. Matter Mater. Phys., 1997, 56, 8265 CrossRef CAS.
- M. A. Subramanian, A. P. Ramirez and W. J. Marshall, Phys. Rev. Lett., 1999, 82, 1558 CrossRef CAS.
- H. Arandiyan, S. S. Mofarah, C. C. Sorrell, E. Doustkhah, B. Sajjadi, D. Hao, Y. Wang, H. Sun, B.-J. Ni, M. Rezaei, Z. Shao and T. Maschmeyer, Chem. Soc. Rev., 2021, 50, 10116 RSC.
- A. Marthinsen, C. Faber, U. Aschauer, N. A. Spaldin and S. M. Selbach, MRS Commun., 2016, 6, 182 CrossRef CAS.
- Q. Wang, Y. Gu, C. Chen, L. Han, M. U. Fayaz, F. Pan and C. Song, ACS Appl. Bio Mater., 2024, 16, 3726 CrossRef CAS PubMed.
- J. M. Rondinelli, S. J. May and J. W. Freeland, MRS Bull., 2012, 37, 261 CrossRef CAS.
- S. Jaiswar and K. D. Mandal, J. Phys. Chem. C, 2017, 121, 19586 CrossRef CAS.
- A. Bhattacharya and S. J. May, Annu. Rev. Mater. Res., 2014, 44, 65–90 CrossRef CAS.
- A. I. Waidha, H. K. Siddiqui, Y. Ikeda, M. Lepple, S. Vasala, M. Donzelli, A. D. Fortes, P. Slater, B. Grabowski, U. I. Kramm and O. Clemens, Chem. Eur. J., 2021, 27, 9763 CrossRef.
- P. Tan, C. Zhu, J. Yang, S. Zhao, T. Xia, M.-H. Zhao, T. Han, Z. Deng and M.-R. Li, Chin. Chem. Lett., 2024, 35, 108485 CrossRef CAS.
- S. A. Hosseini, J. Mater. Sci.:Mater. Electron., 2017, 28, 3703 CrossRef.
- L. Sun, Y. Zhang, L. Ju, C. Shi, H. Qin and J. Hu, IEEE Trans. Magn., 2014, 50, 1 Search PubMed.
- T. Xu, M. Mori, H. Hirakata, T. Kitamura and T. Shimada, Phys. Chem. Chem. Phys., 2024, 26, 842 RSC.
- B. Gao, S. Xu and Q. Xu, Angew. Chem., Int. Ed., 2022, 61, e202117084 CrossRef CAS PubMed.
- L. Li, B. Gao, S. Xu and Q. Xu, Small, 2023, 19, 2300765 CrossRef CAS.
- K. P. Johnston, K. L. Harrison, M. J. Clarke, S. M. Howdle, M. P. Heitz, F. V. Bright, C. Carlier and T. W. Randolph, Science, 1996, 271, 624 CrossRef CAS.
- P. Zhou, Q. Xu, H. Li, Y. Wang, B. Yan, Y. Zhou, J. Chen, J. Zhang and K. Wang, Angew. Chem., Int. Ed., 2015, 54, 15226 CrossRef CAS PubMed.
- M. J. Hÿtch, E. Snoeck and R. Kilaas, Ultramicroscopy, 1998, 74, 131 CrossRef.
- B. L. Choudhary, U. Kumar, A. M. Quraishi, P. M. Z. Hasan, R. Darwesh, S. Kumar, S. Dalela, S. Kumar, S. N. Dolia and P. A. Alvi, J. Mater. Sci.:Mater. Electron., 2022, 33, 6829 CrossRef CAS.
- K. S. Knight, J. Alloys Compd., 2011, 509, 6337 CrossRef CAS.
- M. D. Biegalski, L. Qiao, Y. Gu, A. Mehta, Q. He, Y. Takamura, A. Borisevich and L.-Q. Chen, Appl. Phys. Lett., 2015, 106, 162904 CrossRef.
- Y. Yan, H. Yang, X. Zhao, R. Li and X. Wang, Mater. Res. Bull., 2018, 105, 286 CrossRef CAS.
- G. K. Ribeiro, F. S. Vicente, M. I. B. Bernardi and A. Mesquita, J. Alloys Compd., 2016, 688, 497 CrossRef CAS.
- X. Zhang, L. Luo, R. Yun, M. Pu, B. Zhang and X. Xiang, ACS Sustainable Chem. Eng., 2019, 7, 13856 CrossRef CAS.
- S. Kačiulis, G. Mattogno, L. Pandolfi, M. Cavalli, G. Gnappi and A. Montenero, Appl. Surf. Sci., 1999, 151, 1 CrossRef.
- T. Watanabe and T. Ohba, Acs Sustain Chem Eng, 2021, 9, 3860 CrossRef CAS.
- J. Zhao, X. Cao, Y. Bai, J. Chen and C. Zhang, Opt. Mater., 2023, 135, 113239 CrossRef CAS.
- P. Boutinaud, E. Pinel, M. Dubois, A. P. Vink and R. Mahiou, J. Lumin., 2005, 111, 69 CrossRef CAS.
- M. Shivaram, R. H. Krishna, H. Nagabhushana, S. C. Sharma, B. M. Nagabhushana, B. S. Ravikumar, N. Dhananjaya, C. Shivakumara, J. L. Rao and R. P. S. Chakradhar, Mater. Res. Bull., 2013, 48, 1490 CrossRef CAS.
- M. Shi, B. Rhimi, K. Zhang, J. Xu, D. W. Bahnemann and C. Wang, Chemosphere, 2021, 275, 130083 CrossRef CAS.
- B. D. Begg, E. R. Vance, B. A. Hunter and J. V. Hanna, J. Mater. Res., 1998, 13, 3181 CrossRef CAS.
- L. R. Blackburn, L. T. Townsend, M. C. Dixon Wilkins, T. Ina, M. Kuman, S.-K. Sun, A. R. Mason, L. J. Gardner, M. C. Stennett, C. L. Corkhill and N. C. Hyatt, Sci. Rep., 2023, 13, 9329 CrossRef CAS.
- R. V. Vedrinskii, V. L. Kraizman, A. A. Novakovich, V. D. Ph and S. V. Urazhdin, J. Phys.: Condens. Matter, 1998, 10, 9561 CrossRef CAS.
- S. A. Chambers, T. C. Droubay, C. M. Wang, K. M. Rosso, S. M. Heald, D. A. Schwartz, K. R. Kittilstved and D. R. Gamelin, Mater. Today, 2006, 9, 28 CrossRef CAS.
- T. M. Mazzo, L. M. d. R. Oliveira, L. R. Macario, W. Avansi, R. S. Andre, I. L. V. Rosa, J. A. Varela and E. Longo, Mater. Chem. Phys., 2014, 145, 141 CrossRef CAS.
- S. de Lazaro, J. Milanez, A. T. de Figueiredo, V. M. Longo, V. R. Mastelaro, F. S. De Vicente, A. C. Hernandes, J. A. Varela and E. Longo, Appl. Phys. Lett., 2007, 90, 111904 CrossRef.
- J. Wei, H. Zhong, J. Liu, X. Wang, F. Meng, H. Xu, Y. Liu, X. Luo, Q. Zhang, Y. Guang, J. Feng, J. Zhang, L. Yang, C. Ge, L. Gu, K. Jin, G. Yu and X. Han, Adv. Funct. Mater., 2021, 31, 2100380 CrossRef CAS.
- G. Zhou, X. Wang, H. Ji, J. Zhang, P. Kang, Z. Li and X. Xu, J. Magn. Magn. Mater., 2020, 515, 167303 CrossRef CAS.
- X. Han, J. Lee and H.-I. Yoo, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 79, 100403 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05607h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |