
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hollow-SiO2@CuxZnyMgzAl-LDHs as catalyst precursors for CO2 hydrogenation to methanol†
Tomasz
Kondratowicz
 a,
Marta
Gajewska
b,
Jiangtong
Li
c,
Molly Meng-Jung
Li
c,
Zoë R.
Turner
a,
Chunping
Chen
*a and
Dermot
O'Hare
*a
a,
Marta
Gajewska
b,
Jiangtong
Li
c,
Molly Meng-Jung
Li
c,
Zoë R.
Turner
a,
Chunping
Chen
*a and
Dermot
O'Hare
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: chunping.chen@chem.ox.ac.uk; dermot.ohare@chem.ox.ac.uk; Tel: +44 (0)1865 272686
bAcademic Centre for Materials and Nanotechnology, AGH University of Krakow, Mickiewicza 30, 30-059 Krakow, Poland
cDepartment of Applied Physics, The Hong Kong Polytechnic University, P. R. China
First published on 17th December 2024
Abstract
We report a new synthetic strategy for preparing well-organised, spherical and mesoporous, mixed-metal, hollow-core@layered double hydroxides. Hollow-SiO2@CuxZnyMgzAl-LDHs (x + y + z = 2.32 ± 0.06) were prepared by exploiting a unique “memory effect” feature of LDH materials. The reconstruction with simultaneous incorporation of Cu2+ and Zn2+ into the LDH shell was achieved by exposing hollow-SiO2@Mg2Al-LDO to an aqueous solution containing Cu2+ and Zn2+ cations. The effect of a single reconstruction step with various concentrations of Cu2+ and Zn2+ solutions (20–80 mM), as well as the implementation of five successive cycles of calcination–reconstruction on the chemical composition, morphology, texture and structure of the resulting materials are described. Hollow-SiO2@CuxZnyMgzAl-LDHs are precursors to active catalysts for CO2 hydrogenation to methanol. The most active catalyst exhibits a space-time yield for methanol of 1.68 gMeOH gCu−1 h−1 at 270 °C (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2:H2, 30 bar) which represents a 1.7-fold increase in space-time yield compared to commercial Cu/ZnO/Al2O3 catalyst under the same conditions.
:1 CO2:H2, 30 bar) which represents a 1.7-fold increase in space-time yield compared to commercial Cu/ZnO/Al2O3 catalyst under the same conditions.
Introduction
Over several decades, rapid development of civilisation and industrial expansion have sharply increased carbon dioxide (CO2) emissions, reaching approximately 35 billion tons in 2021.1 This has led to serious environmental issues including global warming, acid rain, ocean acidification and rising sea levels. The primary challenge now is to mitigate anthropogenic CO2 emissions by (i) improving energy efficiencies to reduce CO2 production, and (ii) implementing carbon capture and storage (CCS) and/or carbon capture and utilisation (CCU) technologies to manage the emissions effectively.2,3 CCS technology typically involves capturing CO2 from gas streams (e.g., flue gas) followed by storing it underground. However, this requires high investment and poses risks such as potential leakage of CO2.4 Unlike CCS, CCU upgrades CO2 waste into valuable products, such as polymers, chemicals and fuels.3–5 In particular, methanol has been proposed as a promising product from CO2 hydrogenation. Methanol has the potential to play a dual role in both the energy and chemicals sectors as highlighted by “methanol economy” concept.6 It can be used directly as fuel or blended with conventional fuels. It is also a key feedstock for the production of chemicals such as formaldehyde, dimethyl ether and acetic acid.7 As one of the ten most important industrial chemicals, global methanol production exceeded 110 million tons in 2018, with expectation to double by 2030.8 Since the 1960s, methanol has been produced on an industrial scale from CO-rich syn-gas (CO/CO2/H2) generated from fossil fuels, using a Cu/ZnO/Al2O3 catalyst at 3–5 MPa pressure and 200–300 °C.9 This well-established process can be readily used for the CO2 hydrogenation to produce green methanol, using captured CO2 and green hydrogen. Such approach has already been demonstrated at pilot-scale plants in Europe and Asia.2,10,11It is widely accepted that metallic copper (Cu0) produces the active sites for CO2 hydrogenation to methanol, with a well-established relationship between the addressable specific Cu0 surface area and catalyst performance.12,13 The Cu/ZnO/Al2O3 combination remains the most widely used catalyst system for methanol production via CO2 hydrogenation. ZnO acts as Cu promoter which can enhance and stabilise the Cu dispersion and adsorption of CO2, while Al2O3 can enhance thermal and chemical stability. In addition to these core components, various promoters such as Cr, Ga, Zr, Mg, Sr, Ba, and others have been extensively studied to improve catalytic performance by enhancing Cu dispersion, reducing Cu0 particle size, increasing Cu stability and reducibility.14–18 Among these, Mg has garnered particular attention due to its ability to modify the basicity for CO2 adsorption and stabilise the Cu/ZnO/Al2O3 interface. Recently, layered double hydroxides (LDHs) have been shown to offer great potential as precursors in making robust Cu/ZnxAlOy catalysts. Excellent metal dispersions are achieved by the incorporation of Cu, Zn, and Al into the LDH metal hydroxide layers.19–21 LDHs are a large family of anionic layer materials, the most frequently observed formulation can be written as [MII1−xMIIIx(OH)2]x+[(An−)x/n]·mH2O, consisting of positively-charged mixed metal hydroxide layers ([MII1−xMIIIx(OH)2]x+) intercalated with hydrated charge compensating anions ([(An−)x/n·mH2O]). LDHs offer advantages such as compositional flexibility and atomic-scale dispersion of metal ions within the layers.22 Upon calcination between 300–600 °C, LDHs transform into homogeneous mixed metal oxides, also known as layered double oxides (LDOs),23 which exhibit an unique “memory effect” that may allow reconstruction of the original layered structure in the presence of water, either in the gas or liquid phase.24,25 This behavior has allowed incorporation of new metal cations into the LDH matrix, simply via exposing the LDO to aqueous cations solutions.26–28 Despite the well-established, cost-effective methods for LDH synthesis, conventional techniques often result in aggregated powders with poor morphology and irregular particle size (so-called stone-like morphology), limiting their porosity and practical applications.29 One approach to mitigate these limitations is the decoration of LDH platelets on well-defined core inorganic or organic supports, e.g. SiO2,30 FexOy,31 TiO2,32 Cu2O,33 zeolites,34 MOF35 and carbon36 to create core–shell hybrids with shapes including spheres,30 cubes,33 wires37 and rods.38 Notably, core–shell structures typically contain vertically oriented LDH platelets forming a three-dimensional (3D) honeycomb-like LDH shell that exhibits enhanced textural properties and active site accessibility compared to unsupported LDH materials.39,40 Additionally, such hybrids can serve as templates for hollow structures,40–42 which have demonstrated enhanced properties over the parent core–shell materials such as higher porosity and concentration of basic sites.42
Previously, we have reported the synthesis of silica@CuxZnAl-LDH (x = 0.8–4.0) core–shell hybrids, using commercially available ES757 silica, mesoporous MCM-48, and SBA-16 as a core, with various CuxZnAl-LDHs used as a shell.20 Upon thermal activation, these core–shell materials exhibited significantly improved catalytic performance for CO2 hydrogenation to methanol compared to their equivalent unsupported CuxZnAl LDH precursors. Hollowed core@shell materials offer the advantage of greater catalytic activity per mass of catalyst as a result of reduction in inactive core, as well as potential for improved gas diffusion through the catalyst. Herein, we report a new advance by developing a novel strategy for the synthesis of hollow-SiO2@CuxZnyMgzAl-LDH catalyst precursors for CO2 hydrogenation to methanol.
Results and discussion
Synthetic strategy to obtain hollow-SiO2@CuxZnyMgzAl-LDHs
The concept for developing mixed-metal hollow-SiO2@CuxZnyMgzAl-LDHs is schematically shown in Fig. 1. In the first step, solid SiO2@Mg2Al-LDH core–shell particles (S@MA-LDH) were synthesised by the pH controlled coprecipitation of Mg2Al–CO3 LDH in the presence of non-porous spherical SiO2 particles. The silica core from SiO2@Mg2Al-LDH was then selectively etched under static conditions by treatment with a NaOH solution to yield hollow-SiO2@Mg2Al-LDH spheres (H-S@MA-LDH), followed by calcination to produce hollow-SiO2@Mg2Al-LDO spheres (H-S@MA-LDO), according to our previous protocol.42 Afterwards, the hollow-SiO2@Mg2Al-LDO spheres were added to aqueous Cu and Zn nitrate solutions, facilitating ion exchange between the transition metals and Mg2+ cations during reconstruction of the Mg2Al-LDO into CuxZnyMgzAl-LDH through the memory effect, to obtain hollow-SiO2@CuxZnyMgzAl-LDH (H-S@CZMA-LDH_X, where X depends on applied reconstruction conditions). The impact of a single reconstruction step using various concentrations of Cu and Zn solutions, as well as the implementation of repeated calcination and reconstruction cycles on the chemical composition, morphology, structural and textural properties of the hollow sphere materials has been studied. | ||
| Fig. 1 Synthetic scheme used to obtain mixed-metal (Cu, Zn) hollow-SiO2@CuxZnyMgzAl-LDH spheres. | ||
Synthesis of hollow-SiO2@Mg2Al-LDH spheres
Fig. 2 shows the transmission electron microscope (TEM) images of pristine solid S@MA-LDH core–shell and the H-S@MA-LDH hollow spheres obtained after the leaching of the SiO2 core. It can be clearly observed that sample before etching (Fig. 2A) exhibits typical core–shell structure, with a dark solid core sphere surrounded by a bright shell composed of vertically grown hierarchical nanosheets of LDH. The average diameter of the core–shell particles is 562 ± 25 nm, of which the SiO2 core diameter is 340 ± 9 nm and the LDH shell thickness is 112 ± 22 nm, consistent with S@MA-LDH prepared previously.37 The EDX elemental maps in Fig. 2C clearly demonstrate that Si is mainly distributed in the core while Mg and Al are uniformly dispersed across the shell, confirming the formation of core–shell structure with SiO2 as core and Mg2Al-LDH as shell. After 4 h of immersion in an alkali solution, the SiO2 core was removed as evidenced in Fig. 2B, resulting in hollow shell structure with a clearly visible void inside the sphere while retaining the original particle size and spherical shape of the parent S@MA-LDH. The disappearance of contrast between the core and the shell confirms the high efficiency of the core removal. The elemental mapping analysis (Fig. 2D) further support this, as Si is absent in the core region but appears in the shell, likely due to partial deposition of Si species after leaching process. | ||
| Fig. 2 TEM images of S@MA-LDH solid core–shell (A), H-S@MA-LDH hollow spheres (B) and Mg, Al, Si elemental EDX mapping of S@MA-LDH (C) and H-S@MA-LDH (D). | ||
The elemental compositions were further quantitatively analysed using elemental microanalysis (CHN), energy dispersive X-ray spectroscopy (EDX), inductively coupled plasma optical emission spectroscopy (ICP-OES) and thermogravimetric analysis (TGA). As shown in Table S1,† both EDX and ICP data reveal that Mg/Al molar ratio remained similar before and after the leaching process. However, the molar ratio of Si/Al significantly drops from 3.77 to 0.68, indicating that most SiO2 has been removed. This is consistent with TGA analysis showing that the SiO2 content decreases from 48.00 wt% to 17.07 wt%. The presence of SiO2 in the hollow spheres, even at longer leaching times (up to 48 h) or at much harsh leaching conditions (2 M, 50 °C, 20 h),42 is likely related to the formation of Si–O–Al (Mg) bonds.43 Using the EDX, TGA and CHN data, we can estimate the chemical formula of S@MA-LDH and H-S@MA-LDH as [SiO2]0.81@[Mg2.18Al1.00(OH)6.36(CO3)0.13(OH)0.74(H2O)3.64]0.19 and [SiO2]0.44@[Mg2.16Al1.00(OH)6.32(CO3)0.25(OH)0.50(H2O)0.58]0.56, respectively.
The powder X-ray diffraction (XRD) patterns are shown in Fig. S1.† The virgin core–shell sample presents a combination of the characteristic features of both silica (amorphous structure, broad feature at 2θ = 22°) and crystalline LDH (Bragg peaks at 2θ = 11.5°, 23.2°, 34.8°, 39.3°, 46.7°, 60.8°, and 62.1°), which could be assigned to the (003), (006), (012), (015), (018), (110), and (113) reflections, typical of the double-layered structure in the trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.44 After 4 h of leaching, the broad feature at 2θ = 22° disappears, while the positions of the LDH Bragg reflections remain unchanged. However, the relative intensities slightly increase in H-S@MA-LDH, due to its higher proportion of crystalline LDH phase. The unit cell parameters a, c, LDH basal spacing d(003), and the crystallite domain size in the layer stacking direction, D(003) and in the ab-plane D(110) of H-S@MA-LDH are similar to that of S@MA-LDH (Table S2†), which confirms that applied core leaching process presents minimal impact on the structural properties of the LDH phase.
m space group.44 After 4 h of leaching, the broad feature at 2θ = 22° disappears, while the positions of the LDH Bragg reflections remain unchanged. However, the relative intensities slightly increase in H-S@MA-LDH, due to its higher proportion of crystalline LDH phase. The unit cell parameters a, c, LDH basal spacing d(003), and the crystallite domain size in the layer stacking direction, D(003) and in the ab-plane D(110) of H-S@MA-LDH are similar to that of S@MA-LDH (Table S2†), which confirms that applied core leaching process presents minimal impact on the structural properties of the LDH phase.
The N2 adsorption isotherms of S@MA-LDH and H-S@MA-LDH are shown in Fig. S2,† and the determined textural parameters are listed in Table S3.† The shape of both isotherms is similar to IVa type, according to the IUPAC classification, with H3 hysteresis loops characteristic for mesoporous materials with slit-shape pores.45 Importantly, leaching of the predominant amount of the SiO2 template from S@MA-LDH leads to an increase in the porosity for the H-S@MA-LDH spheres, the mesopore (Vmeso) and total pore (Vtotal) volume increase from 0.38 to 0.66 cm3 g−1 and 0.50 to 0.89 cm3 g−1, respectively. Furthermore, the N2 BET surface area (SBET) doubles from 108 to 224 m2 g−1.
Synthesis of hollow-SiO2@CuxZnyMgzAl-LDHs through use of the “memory effect”
:M3+ brucite layer composition manipulated through use of the memory effect. The reconstruction and simultaneous incorporation of Cu2+ and Zn2+ into the brucite layers of the LDH are achieved by treating H-S@MA-LDO with aqueous solutions containing Cu2+ and Zn2+ cations at total concentrations of 20, 40, 60 and 80 mM (molar ratio of Cu to Zn = 1.30) at room temperature, which promotes a slow rehydration/reconstruction process. The reconstructed samples were designated as H-S@CZMA-LDH_x mM, where x is the concentration (mM) of the solution used. The elemental composition of the samples before and after a single modification were analysed using the ICP-OES. The results, expressed as Mg/Al, (Cu + Zn + Mg)/Al, and (Cu + Zn)/Al molar ratios, are presented in Fig. 3. A clear trend shows that as the concentration of metal (Cu2+ + Zn2+) ions increases, the (Cu + Zn)/Al molar ratio in the reconstructed samples gradually increases, reaching values of 0.86, 1.52, 1.82 and 1.84, for 20, 40, 60 and 80 mM, respectively. Conversely, the Mg/Al ratio decreases from 2.28 to 1.52, 0.85, 0.53 and 0.41, respectively. These changes strongly suggest a more extensive exchange of divalent metal ions with higher concentration solutions. Notably, the (Cu + Zn)/Al ratio appears to reach a plateau after modification with 60 mM, indicating that further increasing the concentration of metal solution will not result in additional ion exchange as a maximum has been reached. Interestingly, the (Cu + Zn + Mg)/Al ratio, representing the total M2+/Al3+ ratio, remains relatively constant (between 2.27 and 2.38), regardless of the metal concentration solutions. Moreover, these values are remarkably close to the initial Mg/Al ratio prior to the reconstruction process (Mg/Al = 2.28), suggesting that the exchange ratio between Mg2+ and (Cu2+ + Zn2+) is nearly equal to 1:1.
 | ||
| Fig. 3 Ratio of Mg/Al, (Cu + Zn + Mg)/Al and (Cu + Zn)/Al in H-S@CZMA-LDHs as a function of the initial aqueous [Cu2+] + [Zn2+]. | ||
Importantly, after LDH reconstruction and simultaneous incorporation of Cu2+ and Zn2+ into the MgzAl layers, the particles still maintain their spherical morphology with a visible hollow core and hierarchical organised platelets on the thin SiO2 shell (Fig. 4A–D). A slight morphological degradation occurred when thicker and aggregated particles formed at high metal concentration (80 mM) (Fig. 4D). EDX elemental mapping has been carried out for H-S@CZMA-LDH_20 mM (Fig. 4E); this sample represents an optimised balance between structural integrity with desired spherical morphology and elemental incorporation. A uniform elemental distribution (Mg, Al, Cu, Zn) is observed in the platelets across the spheres, confirming the incorporation of Cu2+ and Zn2+ ions into the LDH shell layer.
 | ||
| Fig. 4 TEM images of H-S@CZMA-LDHs obtained after immersing H-S@MA-LDO in solutions containing Cu2+ + Zn2+ at a concentration of 20 mM (A), 40 mM (B), 60 mM (C) and 80 mM (D) and Mg, Al, Cu, Zn elemental EDX mapping of H-S@CZMA-LDH_20 mM (E). | ||
The XRD patterns (Fig. S4†) show that after reconstruction of H-S@MA-LDO in 20 mM solution, characteristic LDH Bragg diffraction features appear. However, when the concentration of the reconstruction solution increased beyond 40 mM, a less crystalline LDH phase becomes apparent, accompanied by a series of intense reflections corresponding to layered metal hydroxy salts (LHS), specifically Cu2(OH)3NO3 (rouaite) with the monoclinic P21 space group.47,48 The formation of the LHS phase during reconstruction process is consistent with the findings of Wu et al.,46 who studied the reconstruction of the bulky Mg3Al-LDO (Mg/Al = 3) using Zn-nitrate solutions of different concentrations (12–108 mM). The presence of the LHS phase was also detected in samples when the concentration of reconstruction solution reaches 24 mM or higher. Moreover, the intensity of reflections attributed to the LHS phase progressively increases with increasing the concentration of used solutions. A detailed phase analysis using LeBail refinement of the XRD data was performed using TOPAS software (Fig. S5, S6 and Table S4†). Good agreement was obtained using a multi-phase model containing CuxZnyMgzAl-LDH, Cu2(OH)3NO3 and silica. In general, a gradual increase in the content of the LHS phase is observed with the increase in the concentration of CuZn-containing solutions. The content of both LHS and LDH phases in the discussed H-S@CZMAl-LDHs are 14.7, 23.9, 38.6 at% and 73.3, 62.5, 50.1 at%, respectively (Fig. S6†). Moreover, the silica content is around 11.3–13.6 at% which is lower than what we found from EDX result probably due to the existing of amorphous SiO2 phase.
The IR spectra of H-S@CZMA-LDHs as shown in Fig. S7† exhibit the characteristic bands of LDH: O–H stretching at 3440 cm−1 from the hydroxide group and bending vibration from water molecules (1637 cm−1), indicating successful rehydration of LDO through the memory effect. It is worth pointing out that the pure LDH phase is reconstructed with the incorporation of CO32− anions in the interlayer space at low CuZn concentration (20 mM), which is confirmed by the strong absorption band at 1360 cm−1 (associated with ν3 mode of CO32−).49 However, at higher concentrations of Cu and Zn metal ion solutions (40–80 mM), new bands are observed at 1336 and 1417 cm−1, which correspond to the ν3 mode of NO3−. Such signals can be assigned to Cu2(OH)3NO3 phase50,51 as well as the anion exchanged nitrate in LDH phase. At the same time, the CO32− signal at 1360 cm−1 disappears or overlaps with the neighbouring bands, which does not exclude the presence of CO32− ions in the samples.
After the reconstruction and incorporation with Cu2+ and Zn2+ cations, the adsorption–desorption isotherms (Fig. S8†) of the H-S@CZMA-LDHs remained type IVa with an H3 hysteresis loop according to IUPAC classification. However, the total pore volume and BET surface area (Table S3†) vary. The N2SBET values for samples immersed in 20 mM and 40 mM solutions are comparable to those of the parent H-S@MA-LDH (225 and 237 vs. 224 m2 g−1) while Vtotal slightly decreases (0.81 and 0.78 vs. 0.89 cm3 g−1). However, the samples modified in more concentrated solutions (60 and 80 mM) exhibit a significant reduction in SBET and Vtotal (156 and 113 m2 g−1, 0.55 and 0.46 cm3 g−1, respectively). A similar trend is observed for the mesopore volume which gradually decreases from 0.67 to 0.37 cm3 g−1 with increasing metal concentration. This porosity reduction is likely due to the increasing presence of the highly crystalline Cu2(OH)3NO3 phase (Fig. S4–S6†). In addition, gradually changing morphology of the samples (Fig. 4) may contribute to partial pore blockage.
The elemental composition of the samples after each calcination–reconstruction cycle was analysed by ICP-OES as shown in Fig. S9.† The (Cu + Zn)/Al molar ratio in the reconstructed samples steadily increases with each cycle, reaching 0.18, 0.36, 0.53, 0.74 and 0.96, while the Mg/Al ratio decreases from 2.28 to 2.11, 1.98, 1.78, 1.57 and 1.25. These trends suggest a stepwise exchange of divalent metal ions during the calcination–reconstruction cycle. In addition, the ratio of (Cu + Zn + Mg)/Al remained relatively constant (2.21–2.34), regardless of the number of cycles. Given the initial Mg/Al of 2.28, the exchange ratio between Mg2+ and Cu2+ + Zn2+ appears to be 1 to 1, which is the same as that found in the single immersion approach. The Cu/Zn ratio across all sample was 1.42–1.44, closely matching the theoretical value of 1.30. Based on these results, the metal composition of the LDH in H-S@CZMA-LDHs after successive cycles is determined as follows: Cu0.10Zn0.07Mg2.11Al, Cu0.21Zn0.15Mg1.98Al, Cu0.31Zn0.22Mg1.78Al, Cu0.44Zn0.31Mg1.57Al1.00, Cu0.56Zn0.39Mg1.25Al, respectively.
The XRD patterns of the samples after each cycle are shown Fig. 5. In all cases, only distinct characteristic Bragg reflections for a crystalline LDH structure are observed. Notably, a systematic shift in the positions of (110) and (113) Bragg reflections toward lower 2θ values is observed as highlighted in the inset of Fig. 5. This shift is attributed to the differing ionic radii of Mg2+ compared to the Cu2+ and Zn2+ ions that replace it (octahedral ionic radii: Mg2+ 0.72 Å, Cu2+ 0.73 Å and Zn2+ 0.74 Å).52 The substitution affects the lattice parameters (Table 1). Particularly, the a-lattice parameter (the metal–metal distance in brucite-like layers) increases gradually from 0.3049 nm in the parent H-S@MA-LDH to 0.3073 nm after five cycles. This increase reflects the progressive replacement of Mg2+ ions with Cu2+ and Zn2+ ions, consistent with the ICP-OES analysis in Fig. S9.† The c-lattice parameter remains within 2.302–2.318 nm, corresponding to an LDH basal spacing of 7.67–7.73 Å with CO32− ions in the interlayer space.53 The presence of CO32− anions in the interlayer space were further confirmed by IR spectroscopy (Fig. S10†). All reconstructed samples, regardless of the number of the calcination–reconstruction cycles, possess a similar spectral shape with a clearly distinct ν(C–O) stretching mode at 1360 cm−1.
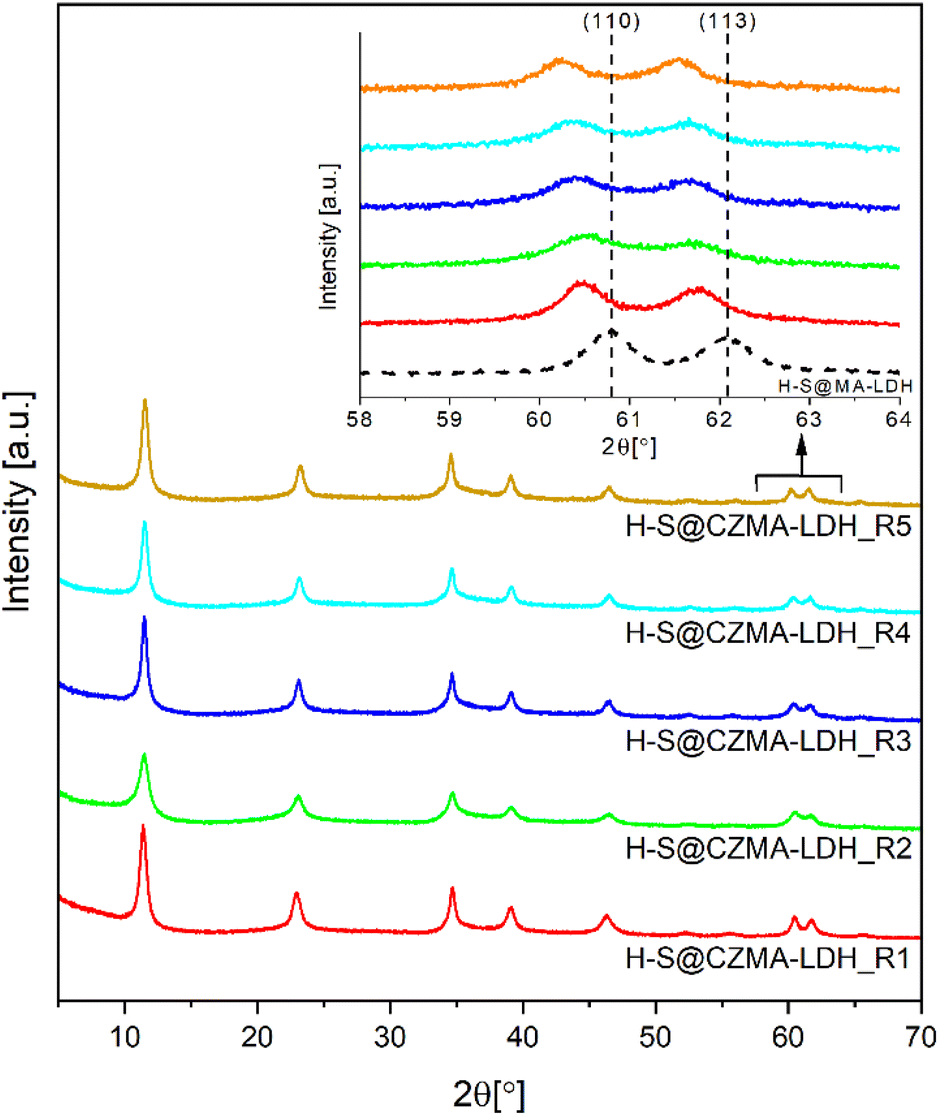 | ||
| Fig. 5 XRD patterns for H-S@CZMA-LDHs after five successive iterations of calcination–reconstruction with a 15 mM CuZn-containing solution. | ||
| Sample | LDH lattice parametersa [nm] | LDH d(003) [nm] | LDH crystallite sizeb [nm] | S BET [m2 g−1] | V micro [cm3 g−1] | V meso [cm3 g−1] | V total [cm3 g−1] | ||
|---|---|---|---|---|---|---|---|---|---|
| a | c | D (003) | D (110) | ||||||
|
a
a = 2d110, c = 3d003.
b Crystallite size of LDH in stacking or plane direction (D(003) and D(110), respectively), calculated according to Scherrer equation: D(hkl) = 0.9 × λ/(β × cosθ), λ = 0.154 nm, θ is the position (deg.), and β is the FWHM (rad.) of the Bragg diffraction peaks.
|
|||||||||
| H-S@MA-LDH | 0.3049 | 2.308 | 0.769 | 7.9 | 17.9 | 224 | 0.008 | 0.66 | 0.89 |
| H-S@CZMA-LDH_R1 | 0.3062 | 2.318 | 0.773 | 13.9 | 15.0 | 163 | 0.009 | 0.68 | 0.67 |
| H-S@CZMA-LDH_R2 | 0.3061 | 2.318 | 0.773 | 9.8 | 9.2 | 157 | 0.012 | 0.70 | 0.66 |
| H-S@CZMA-LDH_R3 | 0.3065 | 2.312 | 0.771 | 15.8 | 9.9 | 158 | 0.012 | 0.74 | 0.73 |
| H-S@CZMA-LDH_R4 | 0.3067 | 2.306 | 0.769 | 14.8 | 10.9 | 158 | 0.010 | 0.75 | 0.74 |
| H-S@CZMA-LDH_R5 | 0.3073 | 2.302 | 0.767 | 14.2 | 13.5 | 179 | 0.002 | 0.66 | 0.74 |
The H-S@CZMA-LDHs exhibited similar porosity with nearly identical adsorption–desorption type IVa isotherms and H3 hysteresis loops (Fig. S11†). Their specific BET surface area and pore volumes are summarised in Table 1. The SBET and Vtotal are in the range of 158–179 m2 g−1 and 0.66–0.74 cm3 g−1, respectively. The mesopore volumes are between 0.68–0.75 cm3 g−1, with the highest volume obtained after the fourth reconstruction cycle. Although, no clear correlation was found between porosity and the number of modifications, the calcination–reconstruction process had a minimal impact on the overall porosity of the materials.
Spherical hollow-SiO2@CuxZnyMgzAl as catalyst precursors
To deconvolute the relationship between catalyst structure/composition (including copper phase and size) and catalytic CO2-to-methanol activity, three samples (H-S@CZMA-LDH_20 mM, H-S@CZMA-LDH_80 mM and H-S@CZMA-LDH_R5) were selected as promising catalyst precursors. As shown by elemental analysis (Table S5†), H-S@CZMA-LDH_80 mM possesses two times the Cu + Zn content of H-S@CZMAl-LDH_20 mM but contains two phases of Cu2(OH)3NO3 and CuZnMgAl-LDH. In turn, H-S@CZMA-LDH_R5, developed after five calcination–reconstruction cycles, retains a pure LDH structure with a similar metal content to H-S@CZMAl-LDH_20 mM. All samples were calcined in air at 330 °C and reduced in H2 at 290 °C.The relationship between CO2 conversion, selectivity to methanol and CO, and reaction temperature is detailed in Fig. S12A–C.† All catalysts exhibit an increase in CO2 conversion and CO selectivity and a decrease in methanol selectivity with increasing reaction temperature. This is consistent with thermodynamic control for CO2 hydrogenation.5 CO is the main by-product generated via a reverse water gas shift process (RWGS, CO2 + H2 ⇌ CO + H2O) which is an endothermic reaction (ΔH0 = 41.2 kJ mol−1). When the temperature increases, the RWGS becomes more predominant, leading to an increased CO selectivity and a decreased methanol selectivity. The catalysts with comparable Cu + Zn loading (H-S@CZMA_20 mM and H-S@CZMA_R5) show comparable CO2 conversion, reaching 16.5 and 17.5% at 290 °C, respectively. In contrast, the Cu-rich catalyst (H-S@CZMA_80 mM) shows poor activity for CO2 hydrogenation with a CO2 conversion rate below 2.4%. As the temperature increases, MeOH selectivity drops from 70.6 to 21.3% and from 65.4 to 19.7% for H-S@CZMA_20 mM and H-S@CZMA_R5, respectively. However, H-S@CZMA_80 mM exhibits a more moderate decrease in methanol selectivity (from 85.2 to 58.2%) over the same temperature range.
Given the wide range of reaction conditions used by different research groups, a direct comparison of the catalytic performance of our catalysts with those reported in the literature is challenging. However, we have summarised the performance of various Cu-containing catalysts with respect to the reaction conditions in Table S6.† We have been able to compare the catalytic efficiency of our catalysts with the commercial Cu/ZnO/Al2O3 catalyst, under the identical reaction conditions (Fig. S12D†). Although on a gram catalyst basis, the commercial Cu/ZnO/Al2O3 catalyst showed better CO2 hydrogenation efficiency to methanol with CO2 conversion of 9.4–22.2% and selectivity of 52.9–17.1% to MeOH in the temperature range of 230–290 °C, the situation changes dramatically when we normalise on a per Cu wt% basis. After normalisation, the space-time yield of methanol (STYMeOH) per gram Cu (gCu) is shown in Fig. 6. At low temperature (230 °C), the commercial Cu/ZnO/Al2O3 catalyst is more effective than our catalysts. However, at higher temperatures, H-S@CZMA_20 mM and H-S@CuZnMgAl_R5 showed significantly higher STYMeOH. At 270 °C, the STYMeOH for H-S@CZMA_20 mM and H-S@CZMA_R5 are 1.4 and 1.7 times higher than the commercial Cu/ZnO/Al2O3 catalyst (1.41 and 1.68 vs. 1.00 gMeOH gCu−1 h−1, respectively). This advantage became even more pronounced at the higher temperature, where determined values of STYMeOH was 1.56 and 1.68 versus 0.67 gMeOH gCu−1 h−1, respectively.
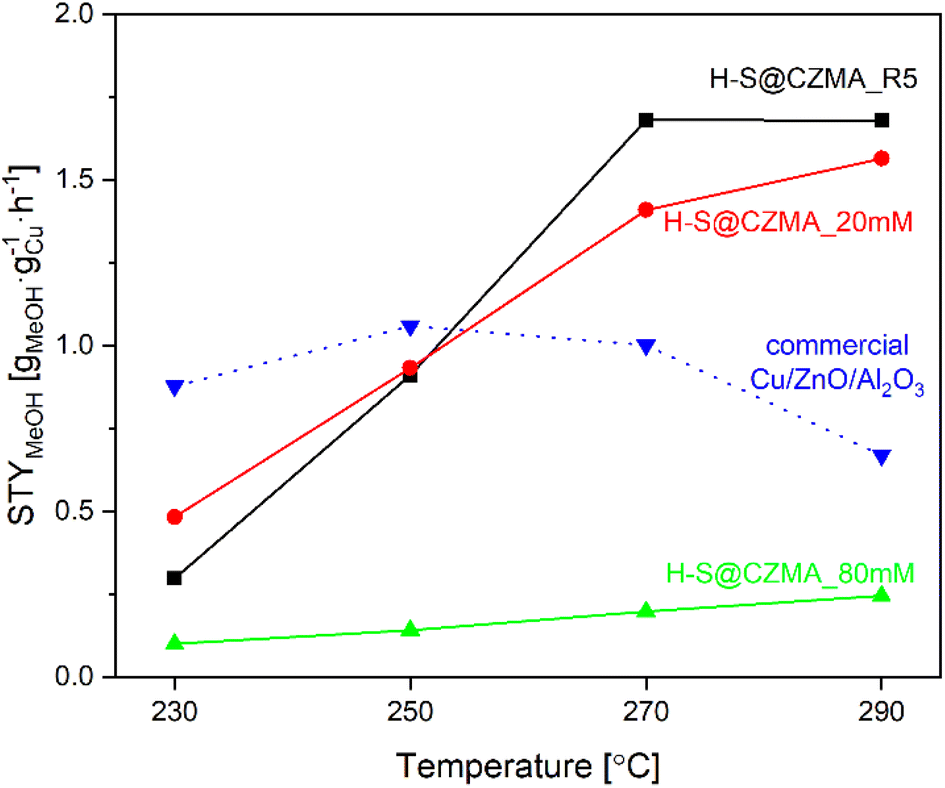 | ||
| Fig. 6 The space-time yield for methanol production (STYMeOH) of H-S@CZMA catalysts and commercial Cu/ZnO/Al2O3 as a function of temperature (CO2:H2 = 3, 30 bar). | ||
In order to understand the correlation between catalyst structure and catalytic CO2-to-methanol activity, comprehensive characterisation studies were conducted on the synthesised catalysts. XRD measurements were performed for thermally activated samples. After calcination at 330 °C (Fig. S13A†), the H-S@CZMA-LDH_20 mM and H-S@CZMA-LDH_R5 converted into equivalent amorphous LDO solids, with the crystalline LDH features disappearing due to dehydration, dehydroxylation and partially decarbonylation. Two broad scattering features are observed at ca. 2θ = 35.3 and 61.0° which are attributed to the formation of poorly-crystalline ZnO phase.54 In contrast, the H-S@CZMA-LDO_80 mM showed highly the presence of a crystalline CuO phase, most likely due to its high Cu content (approx. 39 at%) of Cu2(OH)3NO3 in H-S@CZMAl-LDH_80 mM, which facilitated CuO formation.47 After reduction in H2 at 290 °C (Fig. S13B†), an additional signal at 2θ = 43.3° appears in both H-S@CZMA_20 mM and H-S@CZMA_R5. This weak scattering feature is assigned to the (111) Bragg reflection of metallic nanocopper (Cu0). H-S@CZMA_80 mM presented two additional sharp Bragg reflections at 2θ = 43.4° and 50.6°, which could be assigned to the (111) and (200) Bragg reflections of Cu0.55 These differences in crystallinity of the metallic Cu phases in the catalysts were quantified using the Scherer equation. For H-S@CZMA_20 mM and H-S@CZMA_R5, the mean Cu crystallite domain lengths are 3.2 and 2.7 nm, respectively. While the highly crystalline H-S@CZMA_80 mM possesses Cu crystallites with much larger domain lengths of 33.9 nm.
TEM electron microscopy was used to evaluate the size distribution of metallic copper particles in our samples, as shown in Fig. S14.† The H-S@CZMA_20 mM sample presents a narrow Cu particle size distribution (2–6 nm) with an average size of 3.8 ± 0.7 nm. Similarly, the H-S@CZMA_R5 catalyst showed Cu nanoparticles predominantly within the same range, with a slightly broader average size of 3.6 ± 1.2 nm. In contrast, H-S@CZMA_80 mM catalyst displays significantly larger Cu particles, with 66% of the particles in the 10–40 nm range and an average size of 30.1 ± 18.5 nm.
These insights into the Cu particle size, supported by the results of XRD and TEM measurements, allow us to conclude that this parameter is strongly dependent on the conditions applied during the reconstruction process. Both of the more active methanol catalysts contains similarly small sized metallic copper nanoparticles incorporated into the catalyst precursor matrix (pure LDH) using less concentrated solutions, regardless of whether a single immersion or multiple calcination–reconstruction cycles were used. Conversely, the use of highly concentrated copper solution in single immersion process resulted in the formation of large particles/aggregates of metallic copper, leading to materials with the low catalytic efficiency.
Conclusions
We have developed a systematic synthesis approach to hollow-SiO2@CuxZnyMgzAl-LDHs where LDH nanosheets are anchored perpendicularly to the surface of thin SiO2 shells. Our approach leverages the memory effect of LDHs to regenerate hollow-SiO2@CuxZnyMgzAl-LDHs within a pre-formed hollow-SiO2@Mg2Al-LDH template. We have demonstrated that the concentration of Cu2+ and Zn2+ solution significantly influences the composition, crystallinity, and porosity of the reconstructed LDH. A pure CuxZnyMgzAl-LDH phase with desired morphology and porosity can be obtained in a single immersion approach using 20 mM. Higher concentration solutions greater than 40 mM result in the poorly crystalline LDH phase and a new phase with high crystallinity (Cu2(OH)3NO3), leading to lower porosity and poorer hollow morphology. Another approach using multiple calcination–reconstruction cycles allows gradual incorporation of Cu2+ and Zn2+ ions into the pure and crystalline LDH matrix, while maintaining good morphology and high porosity. Furthermore, our investigation underscored the significant influence of the reconstruction conditions on the formation of catalytically active phase. It was clearly demonstrated that a catalyst with a high content of the Cu0 metallic phase, but in the form of large particles, shows a significantly lower efficiency in methanol production compared to those with a lower Cu content, but smaller and well-dispersed Cu0 particle sizes. We believed that this research provides valuable insight into the design and synthesis of a new generation of hollow spherical material with promising properties and potential, particularly in fields related to climate protection and CO2 management.Data availability
General experimental details, synthetic protocols and additional characterising data supporting this article have been included as part of the ESI.†Author contributions
T. Kondratowicz performed the synthetic and experimental work as well as conceptualised the research; M. Gajewska assisted with the electron microscopy studies; M. Li and J. Li conducted the catalytic CO2 to MeOH experiments; C. Chen and Z. R. Turner supervised the work; D. O'Hare conceptualised and supervised the research and acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. Kondratowicz, Z. R. Turner and C. Chen would like to thank SCG Chemicals Public Co., Ltd (Thailand) for funding.Notes and references
- Z. Liu, Z. Deng, S. J. Davis, C. Giron and P. Ciais, Monitoring global carbon emissions in 2021, Nat. Rev. Earth Environ., 2022, 3, 217–219 CrossRef.
- I. Dimitriou, P. García-Gutiérrez, R. H. Elder, R. M. Cuéllar-Franca, A. Azapagic and R. W. K. Allen, Carbon dioxide utilisation for production of transport fuels: process and economic analysis, Energy Environ. Sci., 2015, 8, 1775–1789 RSC.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, The reverse water gas shift reaction: a process systems engineering perspective, React. Chem. Eng., 2021, 6, 954–976 RSC.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, An overview of CO2 capture technologies, Energy Environ. Sci., 2010, 3, 1645–1669 RSC.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- C. Filosa, X. Gong, A. Bavykina, A. D. Chowdhury, J. M. R. Gallo and J. Gascon, Enabling the Methanol Economy: Opportunities and Challenges for Heterogeneous Catalysis in the Production of Liquid Fuels via Methanol, Acc. Chem. Res., 2023, 56, 3492–3503 CrossRef CAS PubMed.
- A. García-Trenco, A. Regoutz, E. R. White, D. J. Payne, M. S. P. Shaffer and C. K. Williams, PdIn intermetallic nanoparticles for the Hydrogenation of CO2 to Methanol, Appl. Catal., B, 2018, 220, 9–18 CrossRef.
- O. Bazaluk, V. Havrysh, V. Nitsenko, T. Baležentis, D. Streimikiene and E. A. Tarkhanova, Assessment of Green Methanol Production Potential and Related Economic and Environmental Benefits: The Case of China, Energies, 2020, 13, 3113 CrossRef CAS.
- K. Wei, H. Guan, Q. Luo, J. He and S. Sun, Recent advances in CO2 capture and reduction, Nanoscale, 2022, 14, 11869–11891 RSC.
- P. S. Murthy, W. Liang, Y. Jiang and J. Huang, Cu-Based Nanocatalysts for CO2 Hydrogenation to Methanol, Energy Fuels, 2021, 35, 8558–8584 CrossRef CAS.
- S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- C. Baltes, S. Vukojević and F. Schüth, Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis, J. Catal., 2008, 258, 334–344 CrossRef CAS.
- H. Lei, Z. Hou and J. Xie, Hydrogenation of CO2 to CH3OH over CuO/ZnO/Al2O3 catalysts prepared via a solvent-free routine, Fuel, 2016, 164, 191–198 CrossRef CAS.
- V. D. B. C. Dasireddy, S. Š. Neja and L. Blaž, Correlation between synthesis pH, structure and Cu/MgO/Al2O3 heterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol, J. CO2 Util., 2018, 28, 189–199 CrossRef CAS.
- V. D. B. C. Dasireddy, N. S. Štefančič, M. Huš and B. Likozar, Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction, Fuel, 2018, 233, 103–112 CrossRef CAS.
- H. Ren, C.-H. Xu, H.-Y. Zhao, Y.-X. Wang, J. Liu and J.-Y. Liu, Methanol synthesis from CO2 hydrogenation over Cu/γ-Al2O3 catalysts modified by ZnO, ZrO2 and MgO, J. Ind. Eng. Chem., 2015, 28, 261–267 CrossRef CAS.
- C. S. Santana, L. F. Rasteiro, F. C. F. Marcos, E. M. Assaf, J. F. Gomes and J. M. Assaf, Influence of Al, Cr, Ga, or Zr as promoters on the performance of Cu/ZnO catalyst for CO2 hydrogenation to methanol, Mol. Catal., 2022, 528, 112512 CrossRef CAS.
- K. Lee, H. Yan, Q. Sun, Z. Zhang and N. Yan, Mechanism-Guided Catalyst Design for CO2 Hydrogenation to Formate and Methanol, Acc. Mater. Res., 2023, 4, 746–757 CrossRef CAS.
- M. M. J. Li, C. Chen, T. Ayvalı, H. Suo, J. Zheng, I. F. Teixeira, L. Ye, H. Zou, D. O'Hare and S. C. E. Tsang, CO2 Hydrogenation to Methanol over Catalysts Derived from Single Cationic Layer CuZnGa LDH Precursors, ACS Catal., 2018, 8, 4390–4401 CrossRef CAS.
- M. Lyu, J. Zheng, C. Coulthard, J. Ren, Y. Zhao, S. C. E. Tsang, C. Chen and D. O'Hare, Core–shell silica@CuxZnAl LDH catalysts for efficient CO2 hydrogenation to methanol, Chem. Sci., 2023, 14, 9814–9819 RSC.
- A. M. H. Lim and H. C. Zeng, Controlling Nanosheet Spacing of ZnAl-Layered Double Hydroxide Assemblages for High-Efficiency Hydrogenation of CO2 to Methanol, Ind. Eng. Chem. Res., 2023, 62, 1877–1890 CrossRef CAS.
- S. Li, D. Wang, X. Wu and Y. Chen, Recent advance on VOCs oxidation over layered double hydroxides derived mixed metal oxides, Chin. J. Catal., 2020, 41, 550–560 CrossRef CAS.
- M. Mokhtar, A. Inayat, J. Ofili and W. Schwieger, Thermal decomposition, gas phase hydration and liquid phase reconstruction in the system Mg/Al hydrotalcite/mixed oxide: A comparative study, Appl. Clay Sci., 2010, 50, 176–181 CrossRef CAS.
- S. Abelló, F. Medina, D. Tichit, J. Pérez-Ramírez, J. C. Groen, J. E. Sueiras, P. Salagre and Y. Cesteros, Aldol Condensations Over Reconstructed Mg–Al Hydrotalcites: Structure–Activity Relationships Related to the Rehydration Method, Chem.–Eur. J., 2005, 11, 728–739 CrossRef.
- L. Dubnová, R. Daňhel, V. Meinhardová, V. Korolova, L. Smoláková, T. Kondratowicz, O. Kikhtyanin and L. Čapek, Reconstruction of the ZnAl Mixed Oxides Into the Layered Double Hydroxide Catalysts Active in the Aldol Condensation of Furfural: The Role of ZnO Particles, Front. Chem., 2022, 9, 803764 CrossRef.
- P. Liu, M. Derchi and E. J. M. Hensen, Promotional effect of transition metal doping on the basicity and activity of calcined hydrotalcite catalysts for glycerol carbonate synthesis, Appl. Catal., B, 2014, 144, 135–143 CrossRef CAS.
- S. K. Jana, Y. Kubota and T. Tatsumi, High activity of Mn-MgAl hydrotalcite in heterogeneously catalyzed liquid-phase selective oxidation of alkylaromatics to benzylic ketones with 1 atm of molecular oxygen, J. Catal., 2007, 247, 214–222 CrossRef CAS.
- P. Liu, Y. Guan, R. A. v. Santen, C. Li and E. J. M. Hensen, Aerobic oxidation of alcohols over hydrotalcite-supported gold nanoparticles: the promotional effect of transition metal cations, Chem. Commun., 2011, 47, 11540–11542 RSC.
- H. Suo, C. Chen, J.-C. Buffet and D. O'Hare, Dendritic silica@aqueous miscible organic-layered double hydroxide hybrids, Dalton Trans., 2018, 47, 16413–16417 RSC.
- S. Zhao, W.-C. Tsen, F. Hu, F. Zhong, H. Liu, S. Wen, G. Zheng, C. Qin and C. Gong, Layered double hydroxide-coated silica nanospheres with 3D architecture-modified composite anion exchange membranes for fuel cell applications, J. Mater. Sci., 2020, 55, 2967–2983 CrossRef CAS.
- M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, Preparation of Fe3O4@SiO2@Layered Double Hydroxide Core–Shell Microspheres for Magnetic Separation of Proteins, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS.
- Y. Dou, S. Zhang, T. Pan, S. Xu, A. Zhou, M. Pu, H. Yan, J. Han, M. Wei, D. G. Evans and X. Duan, TiO2@Layered Double Hydroxide Core–Shell Nanospheres with Largely Enhanced Photocatalytic Activity Toward O2 Generation, Adv. Funct. Mater., 2015, 25, 2243–2249 CrossRef CAS.
- C. Wang, B. Ma, S. Xu, D. Li, S. He, Y. Zhao, J. Han, M. Wei, D. G. Evans and X. Duan, Visible-light-driven overall water splitting with a largely-enhanced efficiency over a Cu2O@ZnCr-layered double hydroxide photocatalyst, Nano Energy, 2017, 32, 463–469 CrossRef CAS.
- R. Li, T. Xue, R. Bingre, Y. Gao, B. Louis and Q. Wang, Microporous Zeolite@Vertically Aligned Mg–Al Layered Double Hydroxide Core@Shell Structures with Improved Hydrophobicity and Toluene Adsorption Capacity under Wet Conditions, ACS Appl. Mater. Interfaces, 2018, 10, 34834–34839 CrossRef CAS PubMed.
- M. Lyu, C. Chen, J.-C. Buffet and D. O'Hare, A facile synthesis of layered double hydroxide based core@shell hybrid materials, New J. Chem., 2020, 44, 10095–10101 RSC.
- Y. Ni, L. Yao, Y. Wang, B. Liu, M. Cao and C. Hu, Construction of hierarchically porous graphitized carbon-supported NiFe layered double hydroxides with a core–shell structure as an enhanced electrocatalyst for the oxygen evolution reaction, Nanoscale, 2017, 9, 11596–11604 RSC.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Cu nanowires shelled with NiFe layered double hydroxide nanosheets as bifunctional electrocatalysts for overall water splitting, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- Q. Guo, Q. Zhang, H. Wang, Z. Liu and Z. Zhao, Core-shell structured ZnO@Cu-Zn–Al layered double hydroxides with enhanced photocatalytic efficiency for CO2 reduction, Catal. Commun., 2016, 77, 118–122 CrossRef CAS.
- C. Chen, R. Felton, J.-C. Buffet and D. O'Hare, Core–shell SiO 2 @LDHs with tuneable size, composition and morphology, Chem. Commun., 2015, 51, 3462–3465 RSC.
- T. Kondratowicz, O. Horký, S. Slang, L. Dubnová, M. Gajewska, L. Chmielarz and L. Čapek, Hollow @CuMgAl double layered hydrotalcites and mixed oxides with tunable textural and structural properties, and thus enhanced NH3-NOx-SCR activity, Nanoscale Adv., 2023, 5, 3063–3074 RSC.
- M. Li, P. Yuan, S. Guo, F. Liu and J. P. Cheng, Design and synthesis of Ni-Co and Ni-Mn layered double hydroxides hollow microspheres for supercapacitor, Int. J. Hydrogen Energy, 2017, 42, 28797–28806 CrossRef CAS.
- T. Kondratowicz, S. Slang, L. Dubnová, O. Kikhtyanin, P. Bělina and L. Čapek, Controlled silica core removal from SiO2@MgAl core-shell system as a tool to prepare well-oriented and highly active catalysts, Appl. Clay Sci., 2022, 216, 106365 CrossRef CAS.
- P. Kenyon, S. Roberts, Z. R. Turner, N. H. Rees and D. O'Hare, Influence of the Alumina Interface layer in Silica@Layered Double Hydroxide Core–Shell Particles, J. Phys. Chem. C, 2024, 128, 12249–12258 CrossRef CAS.
- G. Mitran, T. Cacciaguerra, S. Loridant, D. Tichit and I.-C. Marcu, Oxidative dehydrogenation of propane over cobalt-containing mixed oxides obtained from LDH precursors, Appl. Catal., A, 2012, 417–418, 153–162 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. Wu, J. Zhang, Y. Peng, J. Zhou, X. Ruan, J. Liu, Q. Liu, Y. Xi, R. Frost and G. Qian, An investigation into mechanism of cation adsorption by reconstruction of calcined layered double hydroxide, Microporous Mesoporous Mater., 2017, 242, 182–189 CrossRef CAS.
- T. N. Ramesh and T. L. Madhu, Thermal Decomposition Studies of Layered Metal Hydroxynitrates (Metal: Cu, Zn, Cu/Co, and Zn/Co), Int. J. Inorg. Chem., 2015, 2015, 536470 Search PubMed.
- M. R. Jalal, H. Hojjati, J. R. Jalal, S. Ebrahimi and M. R. Z. Bighashi, Synthesis of Copper Hydroxide Nitrate (Cu2(OH)3NO3) micro-sheets by plasma eletrolysis of Cu(NO3)2 aqueous solution in atmospheric air, JITL, 2018, 2, 109–112 Search PubMed.
- M. Smyrnioti, C. Tampaxis, T. Steriotis and T. Ioannides, Study of CO2 adsorption on a commercial CuO/ZnO/Al2O3 catalyst, Catal. Today, 2020, 357, 495–502 CrossRef CAS.
- C. Henrist, K. Traina, C. Hubert, G. Toussaint, A. Rulmont and R. Cloots, Study of the morphology of copper hydroxynitrate nanoplatelets obtained by controlled double jet precipitation and urea hydrolysis, J. Cryst. Growth, 2003, 254, 176–187 CrossRef CAS.
- E. A. Secco and G. G. Worth, Infrared spectra of unannealed and of annealed Cu4(OH)6(NO3)2, Can. J. Chem., 1987, 65, 2504–2508 CrossRef CAS.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- S. Miyata, Anion-Exchange Properties of Hydrotalcite-Like Compounds, Clays Clay Miner., 1983, 31, 305–311 CrossRef CAS.
- X. Fang, Y. Men, F. Wu, Q. Zhao, R. Singh, P. Xiao, T. Du and P. A. Webley, Improved methanol yield and selectivity from CO2 hydrogenation using a novel Cu-ZnO-ZrO2 catalyst supported on Mg-Al layered double hydroxide (LDH), J. CO2 Util., 2019, 29, 57–64 CrossRef CAS.
- M. Salavati-Niasari, F. Davar and N. Mir, Synthesis and characterization of metallic copper nanoparticles via thermal decomposition, Polyhedron, 2008, 27, 3514–3518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07292h |
| This journal is © The Royal Society of Chemistry 2025 |