
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA coordination polymer with a silylene-supported Pd6 core as an efficient heterogeneous hydrogenation catalyst†
Taiga
Mitomo
b,
Yoshimasa
Wada
 ab,
Tetsuro
Suda
b,
Atsushi
Tamura
a,
Shunsuke
Yagi
a,
Soichi
Kikkawa
c,
Seiji
Yamazoe
c and
Yusuke
Sunada
*ab
ab,
Tetsuro
Suda
b,
Atsushi
Tamura
a,
Shunsuke
Yagi
a,
Soichi
Kikkawa
c,
Seiji
Yamazoe
c and
Yusuke
Sunada
*ab
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan
bInstitute of Industrial Science, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan
cDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo, 192-0397, Japan
First published on 30th January 2025
Abstract
A hexanuclear palladium cluster supported by two silylene units was readily linked by molecules of a linear ditopic isocyanide to afford a coordination polymer that retained the core Pd6(SiPh2)2Cl2 framework. The obtained coordination polymer exhibited good performance as a heterogeneous catalyst in the hydrogenation of various alkenes in common organic solvents and in protic solvents such as H2O. Furthermore, the obtained coordination polymer showed sufficient stability during the hydrogenation in order for it to be recycled and reused.
Introduction
Heterogeneous catalysts such as metal nanoparticles immobilized on solid supports are fundamental materials in a wide range of fields in modern materials science.1 For example, Pd/C, which is composed of Pd nanoparticles embedded on charcoal, serves as a good catalyst in various organic transformations.2 Considering the superior catalytic performance of certain metal clusters such as Pdn clusters,3 solid-supported metal clusters are another class of heterogeneous catalysts that have attracted attention.4 More recently, coordination polymers with 2D or 3D network architectures consisting of molecular-based metal species as the core and bridging organic ligands as linkers have attracted increasing attention as heterogeneous catalysts. A typical example of such coordination polymers is metal–organic frameworks, which have been intensively investigated and applied as catalysts.5 In these materials, the judicious selection of appropriate core metal-containing molecules and bridging organic linkers is crucial for developing effective catalyst systems.Considering the superior catalytic performance of Pd(0) aggregates, the construction of coordination polymers by incorporating Pd species as the core can be envisaged as a straightforward approach to synthesizing effective heterogeneous catalysts. However, this strategy remains scarcely explored.6 In fact, McPherson, Pedersen, and coworkers have recently reported the only example of a coordination polymer consisting of Pd(0) cluster molecules, namely, a triangular Pd3 cluster-based organometallic 2D-coordination polymer, which was synthesized via the reaction of trinuclear [Pd(CNXyl)2]3 (Xyl = 2,6-Me2-C6H3) with a linear ditopic isocyanide. However, the obtained coordination polymer showed limited catalytic performance in the hydrogenation of styrene, with the conversion reaching only 5% when the reaction was performed at 40 °C for 4 h under 1 atm of H2 with 0.5 mol% catalyst loading.7 Thus, a more sophisticated strategy to construct Pd-based coordination polymers as catalysts needs to be established.
We have recently focused on the synthesis of Pd-based cluster molecules supported by organosilicon ligands and their application as catalysts.8,9 For instance, a planar tetranuclear Pd cluster that functioned as an effective homogeneous catalyst in the hydrogenation of various alkenes was synthesized via the reaction of [Pd(CNtBu)2]3 with a cyclic tetrasilane (Si4R8; R = iPr, cyclopentyl).9a Motivated by the high catalytic performance of silylene-bridged Pd clusters, we planned to develop a heterogeneous catalyst by linking organosilicon-supported Pd clusters with appropriate bridging linkers to construct structurally rigid 3D coordination polymers. Herein, we wish to report the facile synthesis of a coordination polymer by linking a silylene-supported Pd6 cluster having an edge-sharing tetrahedron framework with a linear ditopic isocyanide and its application as a heterogeneous catalyst in the hydrogenation of alkenes. A characteristic feature of this catalyst system is that it was effective in common organic solvents and protic solvents such as H2O, enabling the hydrogenation of alkenes bearing polar substituents such as –SO3− and –B(OH)2. Furthermore, the high stability of the framework structure of the heterogeneous catalyst even during the reaction in H2O allowed it to be recycled and reused.
Results and discussion
Ligand exchange reaction of silylene-supported Pd6 cluster 1
Hexanuclear Pd cluster 1 was selected as the building block of the coordination polymer because the six Pd atoms in 1 are arranged into a 3D-shaped edge-sharing tetrahedron architecture.10 First, ligand-exchange reactions involving the eight CNtBu ligands in 1 were examined; however, no reaction occurred between 1 and 8 equiv. of nitrogen-containing ligands such as pyridine, 4,4′-bipyridine, and 1,4-diazabicyclo[2.2.2]octane at room temperature. In addition, complete decomposition of 1 was observed during the reaction of 1 with 8 equiv. of PMe2Ph or N-heterocyclic carbene iPrIMMe (iPrIMMe = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) at room temperature. In contrast, the ligand-exchange reaction between 1 and 8 equiv. of CNMes (Mes = 2,4,6-Me3-C6H2) readily proceeded in toluene at room temperature to afford cluster 2 in 47% isolated yield after 2 h of reaction (Scheme 1). | ||
| Scheme 1 Synthesis of 2. | ||
Although the data are not sufficient with an R1 value of 8.69%, presumably due to the small and low quality of the obtained crystals, the molecular structure of 2 was determined by an X-ray diffraction (XRD) analysis (Fig. 1). Consistent with the solid-state structure, the 1H spectrum of 2 at room temperature showed four singlets assignable to the methyl groups on the mesityl group of CNMes at 1.82, 1.93, 2.06, and 2.38 ppm with an integral ratio of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12:24:24 (Fig. S20†). The 13C NMR signals derived from the methyl groups of the mesityl groups appeared at 18.77, 19.24, 20.74 and 20.90 ppm (Fig. S21†). In the 29Si NMR spectrum, a singlet appeared at 104.70 ppm, which is slightly shifted to a lower field compared to that of 1 (98.8 ppm) (Fig. S22†). In the infrared (IR) spectrum, two strong absorption bands derived from the C
:12:24:24 (Fig. S20†). The 13C NMR signals derived from the methyl groups of the mesityl groups appeared at 18.77, 19.24, 20.74 and 20.90 ppm (Fig. S21†). In the 29Si NMR spectrum, a singlet appeared at 104.70 ppm, which is slightly shifted to a lower field compared to that of 1 (98.8 ppm) (Fig. S22†). In the infrared (IR) spectrum, two strong absorption bands derived from the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond were observed at 2054 and 2104 cm−1 (Fig. S23†). The elemental analysis of 2 was consistent with the theoretical values.
N bond were observed at 2054 and 2104 cm−1 (Fig. S23†). The elemental analysis of 2 was consistent with the theoretical values.
 | ||
| Fig. 1 Molecular structure of 2 with thermal ellipsoids at 50% probability. All carbon atoms except for those in the coordinated –CN moieties are shown in wireframe style, and all hydrogen atoms are omitted for clarity. | ||
Construction of coordination polymer 3via the ligand-exchange reaction between 1 and a linear ditopic aryl isocyanide
Having demonstrated that 1 undergoes ligand-exchange reactions with aryl isocyanides, we selected a linear ditopic aryl isocyanide, i.e., 3,3′,5,5′-tetramethyl biphenyl-4,4′-bisocyanide (BXyDI), as the bridging linker to construct a coordination polymer based on the Pd6 cluster framework. Treatment of 1 with 4 equiv. of BXyDI in toluene at room temperature for 2 h furnished 3 as a red powder in 71% yield. Unfortunately, several attempts to obtain single crystals of 3 were unsuccessful, which prevented the determination of the molecular structure of 3 by a single crystal XRD analysis.Compound 3 is insoluble in organic solvents including THF, toluene, Et2O, pentane, CH3CN, EtOH, and MeOH as well as in H2O. Its elemental analysis was consistent with the theoretical value calculated for the coordination polymer consisting of a Pd6(SiPh2)2Cl2 core unit supported by eight surrounding BXyDI ligands (Scheme 2). In other words, two central Pd atoms are connected to two isocyanide ligands, and each of the four Pd atoms located on the edge bears one BXyDI ligand. In addition, an X-ray fluorescence (XRF) microanalysis of a powder sample of 3 indicated that the Pd:Si:Cl molar ratio was 29.90:3.44:2.36, which agrees well with the theoretical value (Pd:Si:Cl = 30.18:3.35:2.66). To gain further insight into the structure and electronic state of 3, an X-ray photoelectron-spectroscopy (XPS) analysis of 1–3 was performed (Fig. S12–S17†). First, the elemental composition of 3 was estimated to be Pd:Cl:N = 6:2.05:7.94, which is consistent with the formula shown in Scheme 2. The XPS spectrum of 1 and 2 showed Pd 3d5/2 signals at 335.8 eV for 1 and 336.1 eV for 2, respectively. Because these signals could not be deconvoluted, the oxidation state of all palladium atoms in 1 and 2 could be considered identical due to the electronic delocalization. Clusters 1 and 2 consist of six palladium atoms surrounded by two silylenes, eight isocyanides, and two chloride ligands. Among them, both silylene11 and isocyanide could be regarded as neutral ligands, and thus, the oxidation state of the Pd6 core could be regarded as +2. The Pd 3d5/2 signals in 1 (335.8 eV) and 2 (336.1 eV) are located in the region between the signal for Pd(0) metal (335.2 eV)12 and that of palladium(II) oxide (337.1 eV).13 These spectral features agree well with the slightly electron deficient nature of Pd atoms in 1 and 2 compared with the Pd(0) oxidation state.
 | ||
| Scheme 2 Synthesis of 3via the reaction of 1 with linear ditopic aryl isocyanide BXyDI. | ||
The XPS spectrum of 3 showed Pd 3d signals at 335.20 eV (3d5/2) and 340.52 eV (3d3/2), which are slightly shifted to lower energies compared with those of the parent cluster 1 (335.8 and 340.9 eV). This result suggests that the Pd center in 3 might be slightly electron rich compared with 1, but the electronic environment around Pd atoms is almost maintained upon the formation of the coordination polymer. It should be noted here that the Pd(0) center in a recently reported silylyne-bridged tetranuclear Pd cluster (335.8 and 340.8 eV).14 In the IR spectrum of 3, a relatively broad absorption band derived from the coordinated BXyDI appeared at around 2064 and 2095 cm−1.
Subsequently, 1 and 3 were subjected to a Pd K-edge X-ray absorption spectroscopy (XAS) analysis to obtain more insight into the structure of 3. It should be emphasized here that the XANES spectrum of a powder sample of 3 was almost identical to that of a powder sample of 1, indicating that the solid-state structure and electronic state of the Pd6 cluster core were maintained in 3. A curve-fitting analysis of the Pd K-edge Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectrum of 1 revealed that each of the Pd atoms is bound to adjacent Pd, Si, and Cl atoms with Pd–Pd, Pd–Si, Pd–Cl, and Pd–C (isocyanide) bond distances of ca. 2.78, 2.33, 2.50, and 1.96 Å, respectively (Table 1). The coordination numbers (CNs) of the Pd–Pd, Pd–Si, Pd–Cl, and Pd–C interactions were determined to be ca. 2.7, 0.9, 0.8, and 1.0, respectively. These bond distances and CNs are in accordance with those of the single-crystal XRD structure of 1.8 Based on the FT-EXAFS spectrum of 3, the Pd–Pd, Pd–Si, and Pd–C bond distances were estimated to be 2.74, 2.52, and 2.00 Å, and the CNs for Pd–Pd, Pd–Si, and Pd–C were ca. 3.1, 0.5, and 1.1 (Table 1). The estimated Pd–Pd and Pd–C bond distances are in good agreement with those estimated based on the FT-EXAFS spectrum of 1. In contrast, the Pd–Si bond in 3 is relatively longer than that in 1, and no apparent Pd–Cl bonding interaction was observed. Instead, the CN of Pd–Pd increased and that of Pd–Si decreased upon the formation of coordination polymer 3. Considering the results of the XRF, XPS, and elemental analyses, which indicate the presence of Cl atoms in 3 in a Pd:Cl ratio of 6:2 (vide supra), the XAFS results suggest that the disappearance of the Pd–Cl bond and the change in the CN value of the Pd–Pd and Pd–Si bonds in 3 might stem from the slightly shrunken hexapalladium-cluster-framework core with relatively weakened Pd–Cl and Pd–Si bonds. Considering these results, we concluded that 3 is composed of a Pd6(SiPh2)2Cl2 core architecture similar to cluster molecule 2 and that this core unit is linked by molecules of the linear ditopic isocyanide BXyDI.
| Compound | Atom | Coordination number | Bond length (Å) | Debye–Waller factor | R factor (%)a |
|---|---|---|---|---|---|
| a R = (Σ(k3χdata(k) − k3χfit(k))2)1/2/(Σ(k3kdata(k))2)1/2. | |||||
| 1 | C | 1.0 ± 0.3 | 1.96 ± 0.07 | 0.005 ± 0.004 | 13.9 |
| Si | 0.9 ± 0.2 | 2.33 ± 0.05 | 0.004 ± 0.001 | ||
| Cl | 0.8 ± 0.2 | 2.50 ± 0.08 | 0.006 ± 0.005 | ||
| Pd | 2.7 ± 0.3 | 2.78 ± 0.04 | 0.010 ± 0.005 | ||
| 3 (as prepared) | C | 1.1 ± 0.3 | 2.00 ± 0.06 | 0.003 ± 0.002 | 12.9 |
| Si | 0.5 ± 0.4 | 2.52 ± 0.12 | 0.027 ± 0.026 | ||
| Pd | 3.1 ± 0.4 | 2.74 ± 0.05 | 0.014 ± 0.014 | ||
| 3 (recycled) | C | 0.8 ± 0.3 | 1.99 ± 0.07 | 0.003 ± 0.003 | 14.8 |
| Si | 0.4 ± 0.2 | 2.46 ± 0.10 | 0.009 ± 0.008 | ||
| Pd | 2.5 ± 0.3 | 2.73 ± 0.04 | 0.010 ± 0.006 | ||
Unfortunately, several attempts to obtain single crystals of 3 suitable for single crystal X-ray diffraction analysis were unsuccessful. Thus, a powder X-ray diffraction analysis of 1 and 3 was performed. The powder X-ray diffraction pattern of 1 might be comparable to the simulated pattern, which was estimated from a molecular structure determined by a single crystal X-ray diffraction analysis, but it is difficult to draw conclusions due to the low signal to noise ratio of the experimental diffractogram (Fig. S18†). In contrast, powder X-ray diffraction analysis of 3 showed only background noise and a halo pattern, suggesting that 3 was obtained as an amorphous material.
Catalytic hydrogenation of alkenes mediated by coordination polymer 3
As 3 was obtained as an insoluble powder, we turned our attention to its potential use as a heterogeneous catalyst for the hydrogenation of alkenes. For that purpose, the catalytic performance of 3 was initially examined in the hydrogenation of styrene, which revealed that the reaction completed within 6 h under 1 atm of H2 at room temperature in toluene in the presence of 2 mol% (for Pd) of 3 to afford ethylbenzene as the single product (Table 2, entry 2). Similarly, the hydrogenation of 9-vinylanthracene also resulted in complete conversion of the substrate (entry 7). In contrast, the conversion of styrene reached only 20% when the reaction was conducted with 2 mol% (for Pd) of 1 (entry 1). During the reaction mediated by 1, a black insoluble material was formed after 18 h, suggesting that decomposition of 1 occurred. Conversely, 3 was sufficiently stable under the applied hydrogenation conditions, and no dissolved BXyDI linker was observed in the 1H NMR spectra of the reaction mixture.|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Time (h) | Solvent | Alkene | Yield (%)b |
| a All reactions were carried out using 1 mmol of alkene in the presence of a catalytic amount of catalyst 1, 3 or Pd/C in the solvent indicated in Table 1 (2 mL). The catalyst loading of 2 mol% indicates the total catalyst loading of Pd (0.02 mmol of Pd) in all experiments. b The product yield was determined by 1H NMR spectroscopy in the presence of 1,4-dioxane as the internal standard, which was used to determine the conversion of the starting material. c The formation of a black insoluble material was observed. d Values in parentheses refer to the isolated yield. | |||||
| 1 | 1 | 18 | Toluene |
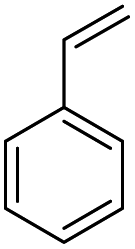
|
20c |
| 2 | 3 | 6 | Toluene | >99 | |
| 3 | Pd/C | 6 | Toluene | >99 | |
| 4 | 1 | 6 | H2O | 25c | |
| 5 | 3 | 6 | H2O | >99 | |
| 6 | Pd/C | 6 | H2O | 21 | |
| 7 | 3 | 18 | Toluene |

|
>99 |
| 8 | 1 | 18 | H2O |

|
10c |
| 9 | 3 | 18 | H2O | >99 (92)d | |
| 10 | 3 | 18 | CH3OH |

|
>99 (80) d |
| 11 | 3 | 18 | CH3OH |

|
>99 (72) d |

The hydrogenation of alkenes catalyzed by the conventional Pd/C catalyst is generally conducted in organic solvents or alcohols. However, the development of organic reactions performed in H2O has recently received much attention because H2O is a safe, nonflammable, inexhaustible, and naturally abundant solvent.15 Interestingly, 3 could be used in protic solvents such as H2O and MeOH. For instance, styrene underwent complete hydrogenation in H2O under atmospheric pressure of H2 at room temperature catalyzed by 3 (entry 4), whereas the immediate decomposition of 1 occurred under the same reaction conditions, affording the product in only 25% yield (entry 5). It is noteworthy that hydrogenation of styrene catalyzed by conventional Pd/C in H2O gave the product only in 21% yield (entry 6). This indicates that coordination polymer catalyst 3 showed superior catalytic performance to the conventional Pd catalyst in the reaction performed in H2O.
Next, we focused on the use of p-styrenesulfonic acid sodium salt as the substrate given that it shows limited solubility in common organic solvents but good solubility in H2O. It should be noted here that the hydrogenation of p-styrenesulfonic acid sodium salt in H2O has been reported to be difficult, even when using a water-soluble cationic Ru catalyst.16 The effective hydrogenation of this substrate has only been achieved using a Rh complex sorbed on the aluminophosphate molecular sieve VPI-5 (ref. 17) or a water-soluble polymer-bound metal catalyst.18 Nevertheless, treatment of p-styrenesulfonic acid sodium salt with 2 mol% of 3 in H2O at room temperature for 18 h furnished the hydrogenated product quantitatively in 92% isolated yield (entry 9). In stark contrast, the hydrogenated product was obtained in a low 10% yield under identical reaction conditions mediated by 1 (entry 8), most likely due to the high moisture sensitivity of 1. In addition, we confirmed that the catalysis mediated by 3 was applicable to the hydrogenation of alkenes bearing a –B(OH)2 group. Thus, the hydrogenation of 4-vinyl phenyl boronic acid and trans-2-phenyl boronic acid was realized in MeOH under 1 atm of H2 at room temperature (entries 10 and 11), and the corresponding products were isolated in 80% and 72% yields, respectively. These results indicate that 3 can be used in reactions conducted in both organic and protic solvents.
Interestingly, we discovered that catalyst 3 can be recycled multiple times in the hydrogenation of p-styrenesulfonic acid sodium salt (for details, see the ESI†). The first run of the catalytic hydrogenation was performed under 1 atm of H2 in H2O at room temperature for 18 h in the presence of 2 mol% (for Pd) of 3. Subsequently, the reaction mixture was centrifuged and catalyst 3 was recovered and subjected to the next run of the same hydrogenation process. After four cycles, the recovered catalyst retained high catalytic activity, and the desired product was obtained in quantitative yield in all runs. Thus, 3 could be reused at least four times by centrifugation without significant loss of the catalytic performance.
To gain further insight into the stability of the structure of 3, the red powder of 3 recovered after one cycle of the hydrogenation of p-styrenesulfonic acid sodium salt under the conditions shown in entry 9, Table 2 was subjected to an XAS analysis. A comparison of the XANES spectra (Fig. S10†) revealed that the spectrum of the recovered catalyst was roughly identical to that of fresh 3. Based on the FT-EXAFS spectrum of the recovered catalyst, the Pd–Pd, Pd–Si, and Pd–C (isocyanide) bond distances were estimated to be ca. 2.73, 2.46, and 1.99 Å, respectively, and the CNs for Pd–Pd, Pd–Si, and Pd–C were 2.5, 0.4, and 0.8 (Table 1). It is important to note here that the signal of the Pd–Pd bond was not enhanced compared with that of fresh 3, indicating that no aggregation of Pd atoms to form Pd nanoparticles occurred. These spectral results suggest that the structure of 3 consisting of a Pd6(SiPh2)2Cl2 core unit linked by linear ditopic BXyDI ligands remains stable during the catalysis.
Conclusions
We have discovered that a hexanuclear Pd cluster supported by two silylene SiPh2 units can be readily linked by molecules of a linear ditopic isocyanide to afford coordination polymer 3, which can serve as a heterogeneous catalyst for the hydrogenation of alkenes with excellent catalytic activity. Interestingly, the catalysis could be performed in common organic solvents and in H2O, which allows the hydrogenation of water-soluble alkenes. Catalyst 3 exhibited sufficient stability under the applied reaction conditions and can be recycled and reused at least four times. X-ray fluorescence (XRF), X-ray absorption fine structure (XAFS), and elemental analyses revealed that 3 consists of a Pd6(SiPh2)2Cl2 core linked by linear ditopic isocyanide ligands and that the Pd cluster framework was retained during the catalytic hydrogenation in H2O. The results of this study suggest that linking silicon-bridged metal clusters provides an effective strategy to construct coordination polymers that can serve as heterogeneous catalysts with high catalytic performance and durability.Data availability
All experimental data are provided in the ESI.†Author contributions
T. M. carried out the synthesis, characterization, and catalysis. Y. W. and T. S. helped with the XPS measurements and their analysis. Y. S. conceptualized and supervised the project. A. T. and S. Y. performed the powder XRD measurements. S. K. and S. Y. performed the XAS measurements. The manuscript was written through contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by JST (Japan) in the form of the PRESTO grant JPMJPR20A9 and JSPS KAKENHI Grant Numbers JP24K01495. The synchrotron radiation experiments were performed at the BL01B1 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2022B1259).References
- (a) S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Coord. Chem. Rev., 2016, 312, 99–148 CrossRef CAS; (b) R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC; (c) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS; (d) M. J. Ndolomingo, N. Bingwa and R. Meijboom, J. Mater. Sci., 2020, 55, 6195–6241 CrossRef CAS; (e) A. Fukuoka and P. L. Dhepe, Chem. Rec., 2009, 9, 224–235 CrossRef CAS PubMed.
- (a) A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed; (b) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC; (c) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (d) Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC; (e) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (f) J. Guo, Y. Qin, Y. Zhu, X. Zhang, C. Long, M. Zhao and Z. Tang, Chem. Soc. Rev., 2021, 50, 5366–5396 RSC; (g) N. F. Suremann, B. D. McCarthy, W. Gschwind, A. Kumar, B. A. Johnson, L. Hammarström and S. Ott, Chem. Rev., 2023, 123, 6545–6611 CrossRef CAS PubMed; (h) Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed; (i) D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS; (j) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (k) L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS; (l) J. Fonseca, T. Gong, L. Jiao and H.-L. Jiang, J. Mater. Chem. A, 2021, 9, 10562–10611 RSC; (m) Z. Lin, J. J. Richardson, J. Zhou and F. Caruso, Nat. Rev., 2023, 7, 273–286 CAS; (n) J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS; (o) P. Sutar and T. K. Maji, Chem. Commun., 2016, 52, 8055–8074 RSC.
- (a) N. Jeddi, N. W. J. Scott and I. J. S. Fairlamb, ACS Catal., 2022, 12, 11615–11638 CrossRef CAS; (b) N. Jeddi, N. W. J. Scott, T. Tanner, S. K. Beaumont and I. J. S. Fairlamb, Chem. Sci., 2024, 15, 2763–2777 RSC.
- (a) B. C. Gates, Chem. Rev., 1995, 95, 511–522 CrossRef CAS; (b) B. C. Gates, Metal cluster catalysts dispersed on solid supports, in Catalysis by Di- and Polynuclear Metal Cluster Complexes, ed. R. D. Adams and F. A. Cotton, Wiley-VCH, New York, 1998, pp. 509–538 Search PubMed; (c) S. Hermans, Clusters and Immobilization, in Synthesis of Solid Catalysts, ed. K. P. de Jong, Wiley-VCH, Weinheim, 2009, pp. 153–169 Search PubMed; (d) H. Rong, S. Ji, J. Zhang, D. Wang and Y. Li, Nat. Commun., 2020, 11, 5884 CrossRef CAS; (e) B. C. Gates, J. Mol. Catal. A: Chem., 2000, 163, 55–65 CrossRef CAS; (f) J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS; (g) Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef; (h) C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS; (i) A. Kulkarni, R. J. Lobo-Lapidus and B. C. Gates, Chem. Commun., 2010, 46, 5997–6015 RSC.
- (a) J. G. de Vries and C. J. Elsevier, in Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) S. Nishimura, in Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley-VCH, New York, 2001 Search PubMed; (c) H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3–18 CrossRef CAS; (d) X. Liu and D. Astruc, Adv. Synth. Catal., 2018, 360, 3426–3459 CrossRef CAS; (e) F.-X. Felpin, Synlett, 2014, 25, 1055–1067 CrossRef.
- (a) T. He, X.-J. Kong, J. Zhou, C. Zhao, K. Wang, X.-Q. Wu, X.-L. Lv, G.-R. Si, J.-R. Li and Z.-R. Nie, J. Am. Chem. Soc., 2021, 143, 9901–9911 CrossRef CAS PubMed; (b) M. T. Payne, C. N. Neumann, E. Stavitski and M. Dincǎ, Inorg. Chem., 2021, 60, 11764–11774 CrossRef CAS PubMed; (c) W. Zhang, Z. Chen, M. Al-Naji, P. Guo, S. Cwik, O. Halbherr, Y. Wang, M. Muhler, N. Wilde, R. Gläser and R. A. Fischer, Dalton Trans., 2016, 45, 14883–14887 RSC; (d) C.-H. Wang, W.-Y. Gao, Q. Ma and D. C. Powers, Chem. Sci., 2019, 10, 1823–1830 RSC; (e) Y. Dong, J.-J. Jv, X.-W. Wu, J.-L. Kan, T. Lin and Y.-B. Dong, Chem. Commun., 2019, 55, 14414–14417 RSC; (f) S. Parshamoni, R. Nasani, A. Paul and S. Konar, Inorg. Chem. Front., 2021, 8, 693–699 RSC; (g) J. A. Navarro, E. Barea, J. M. Salas, N. Masciocchi, S. Galli, A. Sironi, C. O. Ania and J. B. Parra, Inorg. Chem., 2006, 45, 2397–2399 CrossRef CAS PubMed; (h) F. X. Llabrés i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef.
- X. Liu, J. N. McPherson, C. E. Andersen, M. S. Jørgensen, R. W. Larsen, N. J. Yutronkie, F. Wilhelm, A. Rogalev, M. Giménez-Marqués, G. M. Espallargas, C. R. Göb and K. S. Pedersen, Nat. Commun., 2024, 15, 1177 CrossRef CAS.
- Y. Sunada, K. Yamaguchi and K. Suzuki, Coord. Chem. Rev., 2022, 469, 214673 CrossRef CAS.
- (a) C. Yanagisawa, S. Yamazoe and Y. Sunada, ChemCatChem, 2021, 13, 169–173 CrossRef CAS; (b) K. Shimamoto and Y. Sunada, Chem. Sci., 2022, 13, 4115–4121 RSC; (c) R. Usui and Y. Sunada, Inorg. Chem., 2021, 60, 15101–15105 CrossRef CAS PubMed; (d) N. Kojima, M. Kato and Y. Sunada, Chem. Sci., 2022, 13, 7610–7615 RSC; (e) Y. Umehara, R. Usui, Y. Wada and Y. Sunada, Commun. Chem., 2023, 6, 93 CrossRef CAS PubMed; (f) K. Shimamoto and Y. Sunada, Chem. Commun., 2021, 57, 7649–7652 RSC; (g) R. Usui and Y. Sunada, Chem. Commun., 2020, 56, 8464–8467 RSC; (h) Y. Sunada, N. Taniyama, K. Shimamoto, S. Kyushin and H. Nagashima, Inorganics, 2017, 5, 84 CrossRef; (i) Y. Sunada, R. Haige, K. Otsuka, S. Kyushin and H. Nagashima, Nat. Commun., 2013, 4, 3014 CrossRef PubMed.
- K. Shimamoto and Y. Sunada, Chem.–Eur. J., 2019, 25, 3761–3765 CrossRef CAS PubMed.
- (a) R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS; (b) Y. Tsuchido, R. Abe, M. Kamono, K. Tanaka, M. Tanabe and K. Osakada, Bull. Chem. Soc. Jpn., 2018, 91, 858–864 CrossRef CAS; (c) K. Osakada, M. Tanabe and T. Tanase, Angew. Chem., Int. Ed., 2000, 39, 4053–4055 CrossRef CAS; (d) C. Watanabe, T. Iwamoto, C. Kabuto and M. Kira, Angew. Chem., Int. Ed., 2008, 47, 5386–5389 CrossRef CAS PubMed.
- (a) M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J.-Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed; (b) C. Li, Q. Yuan, B. Ni, T. He, S. Zhang, Y. Long, L. Gu and X. Wang, Nat. Commun., 2018, 9, 3702 CrossRef PubMed; (c) W. Jiao, C. Chen, W. You, J. Zhang, J. Liu and R. Che, Small, 2019, 15, 1805032 CrossRef PubMed; (d) W. Si, Z. Yang, X. Hu, Q. Lv, X. Li, F. Zhao, J. Hea and C. Huang, J. Mater. Chem. A, 2021, 9, 14507 RSC.
- A. Tressaud, S. Khairoun, H. Touhara and N. Watanabe, Z. Anorg. Allg. Chem., 1986, 540, 291 CrossRef.
- J. Sekiguchi, Y. Kazama, A. Ishii and N. Nakata, Chem. Commun., 2023, 59, 9844–9847 RSC.
- (a) R. Breslow, Acc. Chem. Res., 1991, 24, 159–164 CrossRef CAS; (b) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC; (c) T. Kitanosono and S. Kobayashi, Chem.–Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed; (d) C.-J. Li, Chem. Rev., 1993, 93, 2023–2035 CrossRef CAS; (e) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed; (f) N. A. Harry, S. Radhika, M. Neetha and G. Anilkumar, ChemistrySelect, 2019, 4, 12337–12355 CrossRef CAS; (g) U. M. Lindström, Chem. Rev., 2002, 102, 2751–2772 CrossRef PubMed.
- P. Csabai and F. Joó, Organometallics, 2004, 23, 5640–5643 CrossRef CAS.
- J. R. Anderson, E. M. Campi, W. R. Jackson and Z. P. Yang, J. Mol. Catal. A:Chem., 1997, 116, 109–115 CrossRef CAS.
- D. E. Bergbreiter and Y.-S. Liu, Tetrahedron Lett., 1997, 38, 3703–3706 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic details, and crystal data for 2. CCDC 2375542. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05663a |
| This journal is © The Royal Society of Chemistry 2025 |