
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReacon: a template- and cluster-based framework for reaction condition prediction†
Zihan
Wang‡
a,
Kangjie
Lin‡
 a,
Jianfeng
Pei
*b and
Luhua
Lai
*ab
a,
Jianfeng
Pei
*b and
Luhua
Lai
*ab
aBNLMS, Peking-Tsinghua Center for Life Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: lhlai@pku.edu.cn
bCenter for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, 100871, China. E-mail: jfpei@pku.edu.cn
First published on 6th December 2024
Abstract
Computer-assisted synthesis planning has emerged as a valuable tool for organic synthesis. Prediction of reaction conditions is crucial for applying the planned synthesis routes. However, achieving diverse suggestions while ensuring the reasonableness of predictions remains an underexplored challenge. In this study, we introduce an innovative method for forecasting reaction conditions using a combination of graph neural networks, reaction templates, and clustering algorithm. Our method, trained on the refined USPTO dataset, excels with a top-3 accuracy of 63.48% in recalling the recorded conditions. Moreover, when focusing solely on recalling reactions within the same cluster, the top-3 accuracy increases to 85.65%. Finally, by applying the method to recently published molecule synthesis routes and achieving an 85.00% top-3 accuracy at the cluster level, we demonstrate our approach's capability to deliver reliable and diverse condition predictions.
Introduction
The rapid development of machine learning has significantly advanced its application in aiding chemists with synthesis.1–4 In recent years, computer-assisted synthetic planning (CASP) has gained significant attention and demonstrated its value in drug synthesis5–9 and natural product synthesis.10,11 As indispensable components of chemical reactions, reaction conditions (catalysts, solvents, and reagents) also need to be accurately predicted.12 Reaction conditions are essential for forward prediction models,13,14 as the same reactants may yield completely different products under different conditions. Thoughtful consideration of reaction conditions not only enhances the selection of more feasible routes in synthetic planning algorithms15 but also aids chemists in grasping the model's underlying logic. This, in turn, facilitates the practical experimental implementation of the predicted routes.16,17Efforts have been made to predict conditions for specific reaction types. Struebing et al.18 employed quantum chemistry calculations in designing solvents for the Menschutkin reaction. Machine learning methods are also widely applied. Marcou et al.19 used multiple models to build an expert system to predict conditions for the Michael reaction. Maser et al.20 used a relational graph convolutional neural network to predict conditions for four high-value reaction types. Afonina et al.21 introduced an artificial neural network that ranks reaction conditions by their efficacy for hydrogenation reactions. Kwon et al.22 applied a graph-augmented variational autoencoder to predict feasible reaction conditions for cross-coupling reactions. Angello et al.23 developed a workflow utilizing machine learning and experimental robotics to accomplish the selection of general conditions for the heteroaryl Suzuki–Miyaura coupling reaction. Attempts such as utilizing high-throughput datasets with active learning methods have also been made.24–26
Apart from specific reaction types, there are also studies that focus on predicting conditions for general reactions. Gao et al.27 integrated molecular fingerprinting with fully connected neural networks to predict reaction conditions, ensuring cohesive connectivity between different condition components.
In addition to molecular fingerprinting and graph-based approaches, transformer-based models have also been widely applied to reaction condition prediction tasks. Schwaller et al.8 used a transformer model to make concurrent predictions of conditions and reactants for a given target molecule. Jaume-Santero et al.14 used a transformer model to predict reaction conditions for given reactants and products. Kreutter et al.28 also applied a transformer model for reagent prediction in their proposed triple transformer loop framework. Andronov et al.29 subdivided and sequenced reaction condition components and then trained a transformer model to predict conditions. Wang et al.30 developed a prediction model for reaction conditions based on a transformer architecture and incorporated a pretraining strategy that leverages reaction domain knowledge. Qian et al.31 utilized text retrieval methods to pinpoint relevant textual information for given reactions, thereby improving the accuracy of predicting conditions.
Nonetheless, many challenges remain to be solved in predicting general reaction conditions. A well-performing predictive model should provide reasonable recommendations for complete reaction conditions, as well as ensure compatibility among the different components (catalysts, solvents, and reagents). Since feasible reaction conditions for transforming reactants into products are usually not unique, an effective predictive model should present all possible reaction conditions, which has been overlooked in previous studies.
Given the intricate connections between different elements of reaction conditions, we propose a holistic approach that treats catalysts, solvents, and reagents as an integrated system when offering suggestions. Compared to reactions with different templates, reactions sharing the same reaction template often involve more similar reaction mechanisms. Herein, we introduce Reacon, a reaction template-driven framework for predicting reaction conditions using directed message passing neural networks (D-MPNN).32 This method leverages reaction conditions recorded under the same template to narrow down the model selection scope. Additionally, we present a label-based clustering algorithm that groups similar predicted conditions together to enhance the diversity of top-ranking predictions and simplify condition selection for experimental chemists. The workflow of our approach is illustrated in Fig. 1. We further validate our approach on several recently published synthesis routes and corresponding conditions.
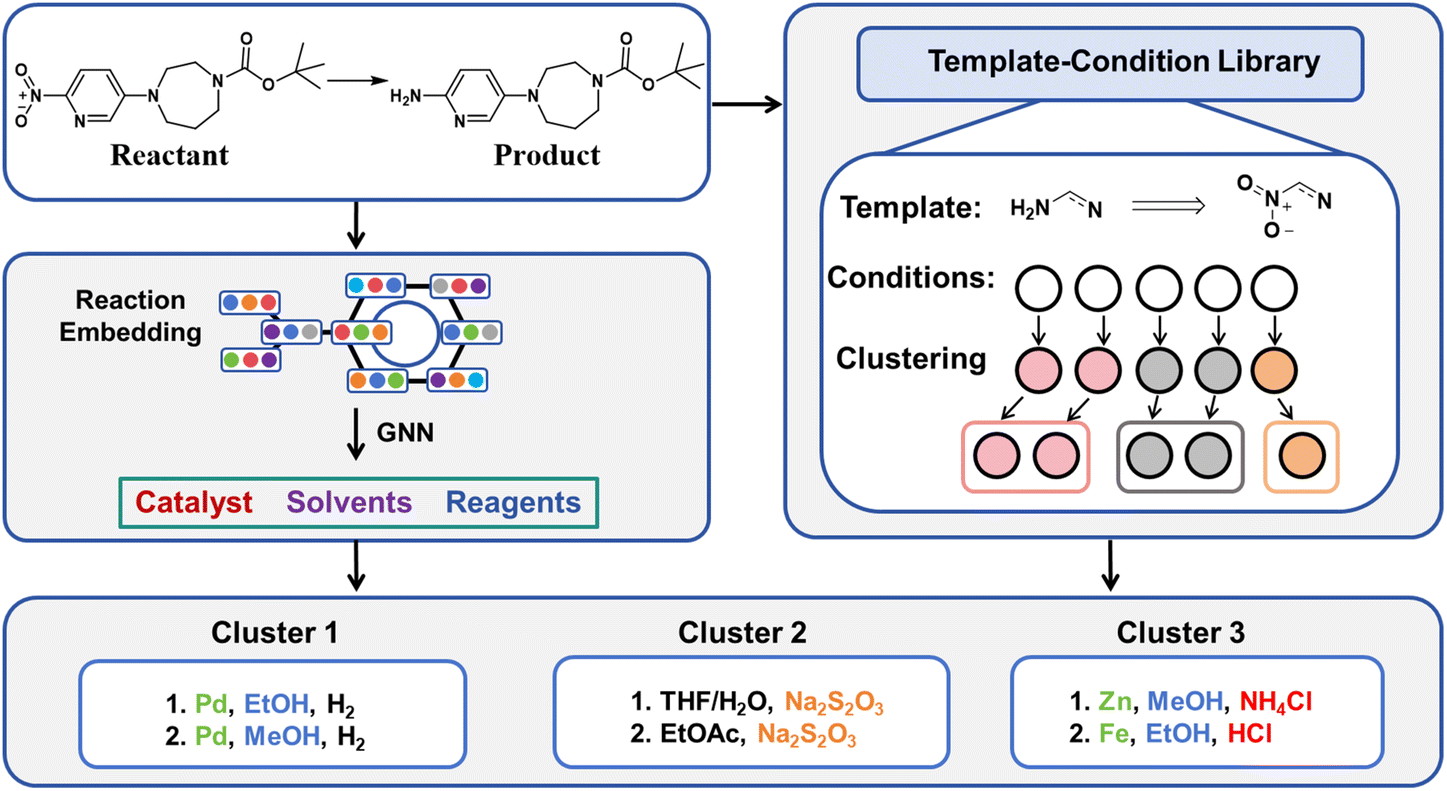 | ||
| Fig. 1 Schematic diagram of the complete condition prediction workflow. | ||
Methods
Data preparation and preprocessing
In the present study, we utilized the USPTO patent dataset,33 which is currently the largest and widely employed open-access dataset for organic reactions. We categorized the reaction conditions into three parts: catalyst, solvents, and reagents. The original dataset includes labeling of catalysts and solvent information, but it does not further distinguish between reactants and reagents. We defined molecules that contain atomic mapping as reactants and those without atomic mapping as condition components. However, considering the specificity of oxidizing agents, if only oxygen atoms are mapped in a molecule, we also classify it as a reagent. The comprehensive workflow for dataset processing is outlined as follows:(a) Reactions with SMILES34 that cannot be parsed by RDKit35 are removed.
(b) Reaction templates (radii = 1) for each reaction are extracted using RDChiral36 and reactions for which templates could not be extracted or occurred fewer than 5 times are removed.
(c) Reactions containing catalysts, solvents, or reagents that appeared fewer than 5 times are eliminated.
(d) For reactions with multiple solvents and reagents, we sort them according to their frequency of occurrence under the corresponding template. Reaction conditions that occurred only once or had more than one catalyst, two solvents, and three reagents are removed.
(e) The dataset is randomly divided into training, validation, and testing sets with a 0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1:0.1 ratio.
:0.1:0.1 ratio.
The final dataset consists of 690872 data points; encompassing 439 catalysts, 542 solvents, and 2746 reagents.
Following the initial data screening, we proceeded to construct our template-condition library using the training dataset. As shown in Fig. 2A, for each reaction data, we extracted three different types of templates: r1, r0, and r0*. Here, r1 and r0 represent templates extracted using RDChiral36 with different radii, while r0* represents the simplest form obtained by retaining only atoms and bonds from the r0 template. We obtained a total of 26228 r1 templates, 9755 r0 templates and 7106 r0* templates. Templates with less information cover a larger chemical space but exhibit lower specificity. Thus, candidate reaction conditions provided by identical r1 templates should be the most accurate, followed by r0 templates and r0* templates.
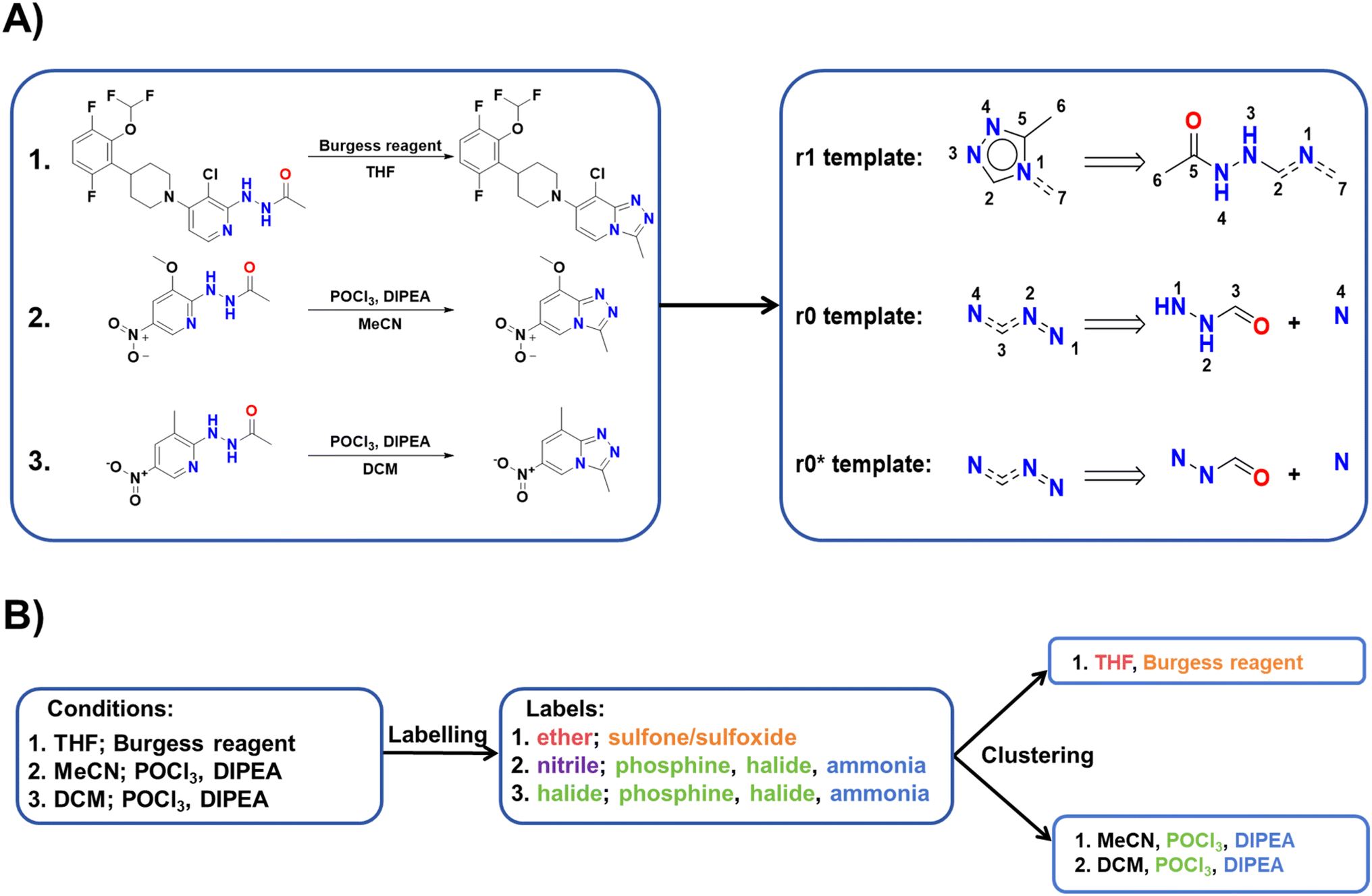 | ||
| Fig. 2 (A) Examples of r1, r0, and r0* templates. (B) Example of the condition clustering algorithm. | ||
Condition clustering algorithm
Drawing inspiration from the way chemists classify reaction conditions, we conducted feature extraction for each component using 31 labels, as illustrated in Table 1. These labels encompass: (a) presence of specific functional groups, such as alkenes, alcohols, and carboxylic acids. (b) Presence of specific elements, primarily referring to metals such as transition metals, main group metals, and reducing metals (alkali metals and alkaline earth metals). (c) Featuring specific functionalities, such as oxidants, reductants, and acids. This determination is based on the presence of corresponding constituents, such as high-valence metals or hydride ions. (d) Additionally, an “ionic” label was assigned to compounds exhibiting charge separation, while those not falling into any category were labeled as “other.” Except for the ‘other’ label, all labels are non-exclusive, allowing each component to have multiple labels. The detailed criteria for assessment are presented in Table S1.†| Feature types | Number | Labels |
|---|---|---|
| Functional group | 21 | Alkene, alkyne, alcohol, ether, aldehyde, ketone, carboxylic acid, ester, amide, nitro, amine, halide, acid chloride, anhydride, nitrile, aromatic, sulfone/sulfoxide, phosphine, metal alkyl, silane, sulfide |
| Element | 3 | Transition metal, reducing metal, main group metal |
| Function | 5 | Oxidizer, reductant, acid, Lewis acid, base |
| Else | 2 | Ionic, other |
Given the frequently encountered ambiguous distinction between solvents and reagents in the dataset, two conditions must meet the following criteria to be classified into the same cluster: (1) share a common catalyst label (or both have no catalyst label). (2) If the total number of solvent and reagent labels exceeds 2, there should be a minimum of two overlaps; otherwise, the labels must be entirely identical. The detailed clustering process is given in Fig. S1.† An example of condition labelling and clustering is shown in Fig. 2B.
For each new reaction, we first determine whether it belongs to an existing reaction condition cluster. If it does, we add it to the corresponding group and update the labels. Otherwise, we establish a new cluster with the label of the new reaction. To ensure no intersection between different categories, when a reaction could be classified into multiple categories, we assign it to the cluster with the most label intersections. If the number of label intersections is equal, the category with a larger intersection of catalyst labels will have higher priority. Specific examples of clustering effects can be found in Table S2.†
GNN models
For the prediction of separate conditions, we utilized the D-MPNN (Directed Message Passing Neural Network)37 and GAT (Graph Attention Network)38 models. The detailed network descriptions are given in ESI Section 2†, and information on hyperparameter selection can be found in Tables S3 and S4.†As shown in Fig. 3A, the input of the model consists of two parts: the molecular graph of the reactant and the differences between the reactant and product. For each molecular graph, vertex information includes atom type, bond count, and charge, among other features. Bond information includes bond order, isomerism, and whether it forms a ring.32
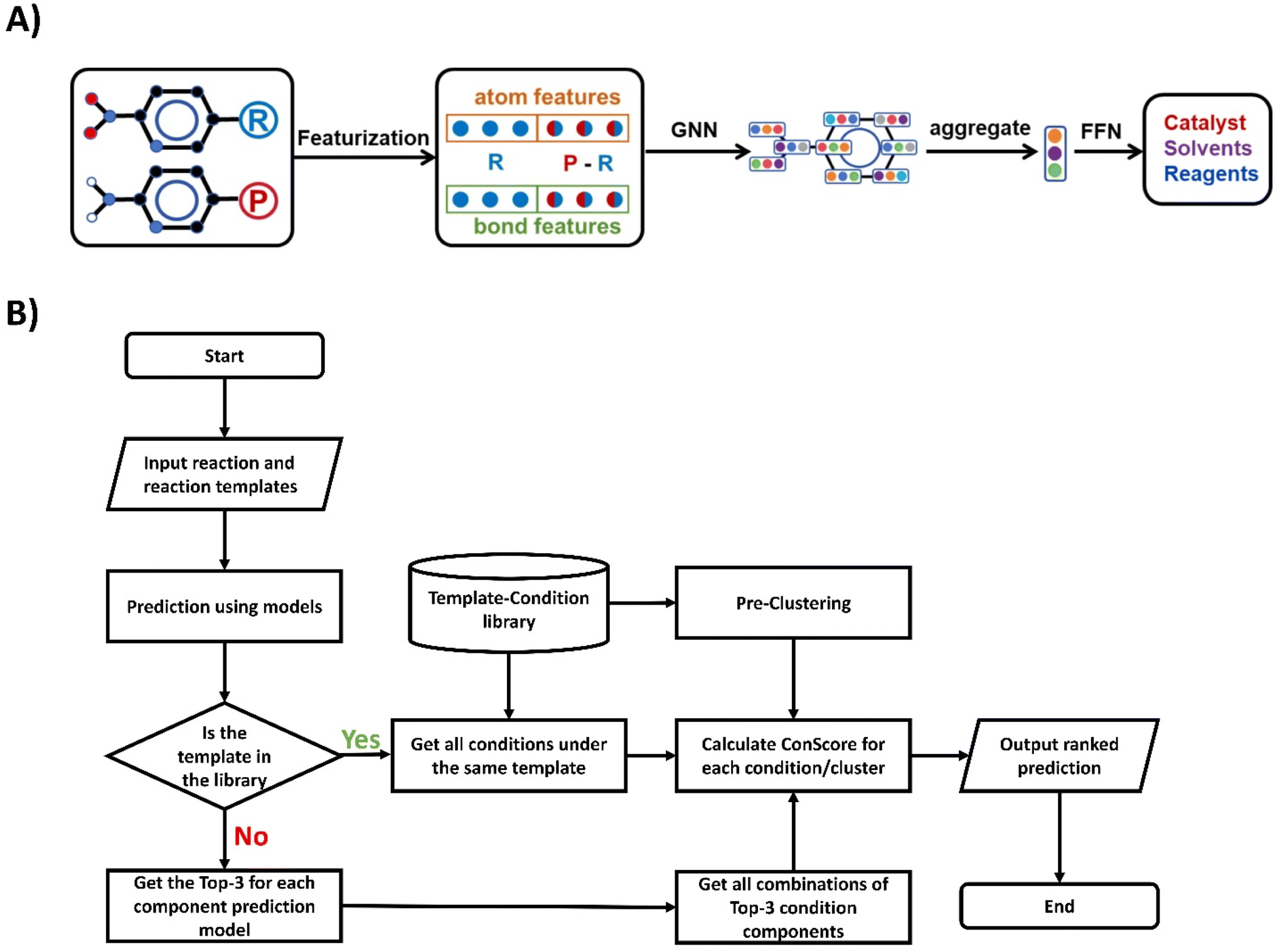 | ||
| Fig. 3 (A) Overview of the model architecture used for reaction condition prediction. (B) Flowchart of the condition prediction algorithm. | ||
Baseline models
To facilitate model performance comparison, we designed the following three baselines:(1) Popularity baseline: to assess the model's ability to differentiate between various conditions under the same template,39 we devised this popularity baseline method to identify the most frequently occurring reaction condition for each template.
(2) Similarity baseline: similar to the Retrosim approach,40 this model calculates the overall molecular similarity between the input reaction and reactions in the corresponding template-condition library, and then outputs the condition with the highest similarity score. The overall similarity is determined by the product similarity multiplied by the similarity of the reactants.
(3) Reaction fingerprint MLP: this encompasses six feed-forward neural network models, each with two hidden layers (256, 64). These models are utilized to forecast the catalyst, solvent 1, solvent 2, reagent 1, reagent 2, and reagent 3 for individual reactions. The input of this model comprises two components: the reagent fingerprint (1024 dimensions) and the reaction fingerprint (1024 dimensions), which are obtained by subtracting the reactant fingerprint from the product fingerprint.
(4) RCR: RCR (Reaction Condition Recommender) is a reaction condition prediction framework proposed by Gao et al.27 It used several neural network models for predicting each reaction condition component. For the complete reaction condition prediction, a stepwise prediction strategy was adopted, i.e., the catalyst information was predicted first, and the corresponding information was introduced into the solvent prediction, and the subsequent components were predicted in the same way. We reproduce the RCR model as we understand it using the same training hyperparameters as in the original literature.
Condition score metric
For a complete set of reaction conditions (including catalyst, solvent 1, solvent 2, reagent 1, reagent 2, and reagent 3), the condition score (ConScore) for these conditions can be obtained by multiplying the selection probabilities of each component.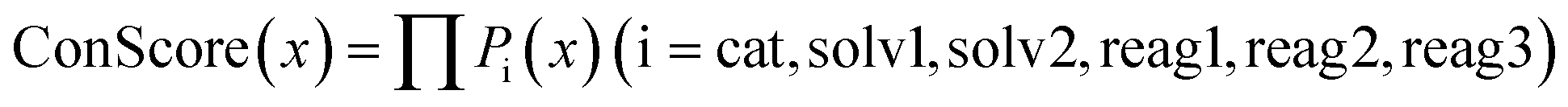 | (1) |
Complete condition prediction algorithm
The workflow of the condition prediction algorithm is shown in Fig. 3B. For each input reaction, we will first use the reaction condition prediction model to obtain the probability of each condition component. Then we will search the template-condition library for conditions recorded under the same template of the input reaction as candidates. During the process of generating candidate conditions, we first search for identical r1 templates in the template-condition library. If none are found, we then search for r0 templates and r0* templates. After obtaining the condition candidates, we use the previously obtained probabilities to calculate the ConScore for each candidate. These candidates are then ordered by their ConScore, and this ranked list constitutes our predictive output. The pseudo-code for the template-based reaction prediction is illustrated in Fig. S2.† For situations where the template of the predicted reaction does not appear in the template-condition library, we obtain the top-3 predictions for each reaction component model and generate all possible combinations of these as candidates.Results and discussion
Predicting individual components of reaction conditions
We first trained independent models for the prediction of catalyst, solvent 1, solvent 2, reagent 1, reagent 2, and reagent 3. Table 2 presents the performance of the GAT and D-MPNN models alongside other baselines. All models demonstrated high prediction accuracies for the catalyst, approximately 90%. This is because a large fraction of the reactions do not require catalysts, which simplifies model training, thereby minimizing performance disparities among models. The same reason applies to the prediction of solvent 2 and reagent 3. In contrast, predicting solvent 1, reagent 1, and reagent 2 proved to be relatively more challenging. Here, the GAT model attained prediction accuracies of 61.53%, 66.74%, and 78.24% respectively, while the D-MPNN model attained 61.93%, 68.23%, and 80.44% respectively, significantly outperforming the other baselines. The largest gaps, nearly 30% in prediction accuracies for solvent 1 and reagent 1, were found between the GNN models and other models.| Model | Catalyst | Solvent 1 | Solvent 2 | Reagent 1 | Reagent 2 | Reagent 3 |
|---|---|---|---|---|---|---|
| Top-1 (%) | Top-1 (%) | Top-1 (%) | Top-1 (%) | Top-1 (%) | Top-1 (%) | |
| Popularity baseline | 91.03 | 37.01 | 76.14 | 39.50 | 75.10 | 93.45 |
| MLP | 87.07 | 28.56 | 80.72 | 35.48 | 75.01 | 95.00 |
| RCR | 90.01 | 55.58 | 85.08 | 62.37 | 76.34 | 95.21 |
| Similarity baseline | 90.56 | 30.11 | 75.69 | 34.19 | 76.40 | 93.25 |
| GAT | 91.73 | 61.53 | 85.16 | 66.74 | 78.25 | 95.31 |
| D-MPNN | 93.12 | 61.93 | 86.61 | 68.23 | 80.44 | 96.05 |
| D-MPNN (multi-task) | 92.45 | 59.76 | 86.12 | 66.72 | 79.63 | 95.95 |
We also trained a multi-task D-MPNN model to predict all the conditions simultaneously. Compared to the separately trained models, the multi-task model can make predictions faster and it also performs well on all tasks. More model performance results can be found in Table S5.†
Predicting complete reaction conditions
After obtaining the predictive model for each component of the reaction conditions, we used it to calculate the ConScore for all complete reaction conditions in the template-condition library that correspond to the same template as the input reaction. The performance of complete condition prediction on the test set is listed in Table 3, where accuracy is calculated based on whether the predicted complete condition with the top-N ConScore matches the ground-truth conditions. GNN models surpassed other baseline models by 5–10% under this framework, suggesting that they can effectively learn the relationship between reactants and reaction conditions.| Metric | Model | Top-1 (%) | Top-3 (%) | Top-10 (%) |
|---|---|---|---|---|
| Exact accuracy | Popularity baseline | 36.02 | 56.19 | 73.01 |
| MLP | 34.43 | 54.19 | 68.21 | |
| RCR | 28.56 | 37.18 | 42.03 | |
| Similarity baseline | 36.01 | 54.98 | 71.23 | |
| GAT | 41.67 | 61.46 | 76.51 | |
| D-MPNN | 44.52 | 63.49 | 78.55 | |
| D-MPNN (multi-task) | 42.41 | 61.42 | 76.78 |
In comparison, the similarity baseline did not achieve the expected high accuracy, performing slightly below the popularity baseline. This discrepancy may be attributed to the USPTO dataset's abundance of similar reaction instances with varying conditions.
In addition to the random split, we also conducted a more challenging time split. We used data from 1976 to 2014 as the training set, data from 2015 as the evaluation set, and data from 2016 as the test set. Our model continued to outperform the other baselines on this dataset. The performance of the model is shown in Tables S6–S8.†
We also tested the performance of our model on the reaction condition dataset provided by Wang et al. and compared it to the performance of the parrot and RCR models reported in the article.30 Our model maintains the leading accuracy on this dataset, with performance details shown in Tables S9 and S10.†
Clustering reaction conditions
Despite the satisfactory accuracy demonstrated by our model, we also found some problems. As illustrated in Fig. 4, we have observed a high degree of similarity among the top-ranking conditions suggested by the model, frequently limited to substitutions in solvents or reagents. To give more diversified conditions, we further clustered similar predicted conditions together that better align with chemists' preferences. The clustering algorithm is described in the Methods section.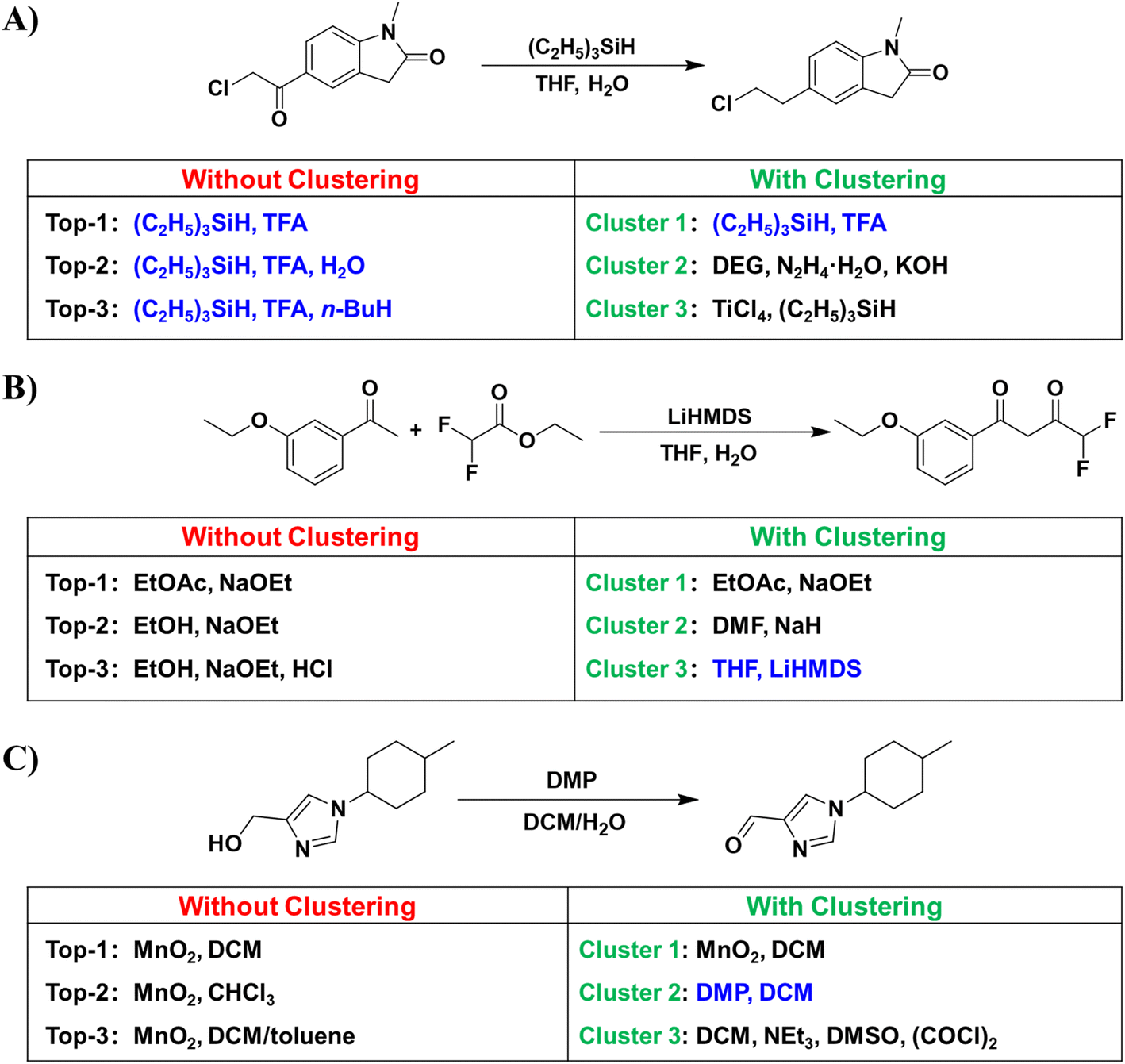 | ||
| Fig. 4 Comparison of the performance of the D-MPNN condition predictor with and without using the condition clustering algorithm. (A) Ketone reduction. (B) Ester condensation reaction. (C) Oxidation of alcohol. The same cluster of reaction conditions as the ground truth is highlighted in blue. | ||
Fig. 4A illustrates a reaction wherein a ketone is reduced to a methyl group. While the D-MPNN model successfully replicated the ground truth in the top-2 predictions, the predominant similarity in the top-ranking predictions gives limited information for users in selecting reaction conditions. After clustering, a variety of alternative conditions were ranked within the top-3 clusters, such as the Wolff-Kishner–Huang Minglong reduction41,42 or substituting acids with Lewis acids. As shown in Fig. 4B, the ground truth involved using a bulky base, which is not particularly common in ester condensation reactions. Consequently, the D-MPNN model without clustering failed to predict the corresponding conditions within its top-3 predictions. After clustering, similar conditions were integrated, leading to the identification of the corresponding condition within the top-3 clusters. Fig. 4C illustrates an oxidation reaction from alcohol to aldehyde. The top-3 predictions used MnO2 as the oxidant, differing only in the solvent. After clustering, in addition to correctly identifying the ground-truth Dess–Martin oxidation, the D-MPNN model also provided the Swern oxidation within the top-3 clusters.
In addition to improving model performance, we found that clustering can help identify potential erroneous reactions in the dataset. This provides a convenient means to screen for erroneous reaction conditions, thus enabling optimization of the quality of the reaction condition dataset.29 The detailed examples can be found in Table S2.†
To evaluate the overall performance at the condition cluster level, we introduced a metric called cluster accuracy. This metric assesses whether the reaction conditions provided by the model belong to the top-N condition clusters as the ground truth. The ranking of different clusters is based on the highest ConScore within the same cluster. This metric can address many of the issues present in exact accuracy metrics. For instance, in practical experiments, several reagents may have similar effects and can be used interchangeably. The specific selection of reagents frequently depends on the preferences of chemists, laboratory inventory, and various other influencing factors. Therefore, expecting the model to provide complete reaction conditions that precisely match the ground truth in the test set is overly strict.
The performance of different models after clustering on the test set is shown in Table 4. Notably, after adding the clustering algorithm, the top-1 cluster accuracy of the D-MPNN model achieved an impressive 65.68%, surpassing baseline models by more than 10%. Furthermore, the top-10 cluster accuracy soared to an exceptional 96.11%. These results underscored the method's remarkable ability to effectively predict diverse reaction conditions. We also tested the effect of different factors on the final cluster size with cluster accuracy. The results showed that different clustering criteria had a large impact on the results, compared to the use of different template libraries. Additional discussion on the effect of cluster size on model performance is given in ESI Section 5 and Tables S11–S13.†
| Metric | Model | Top-1 (%) | Top-3 (%) | Top-10 (%) |
|---|---|---|---|---|
| Cluster accuracy | Popularity baseline | 54.04 | 79.55 | 91.89 |
| Reaction fingerprint MLP | 51.38 | 74.01 | 86.39 | |
| Similarity baseline | 53.63 | 77.34 | 91.70 | |
| GAT | 63.14 | 83.59 | 95.10 | |
| D-MPNN | 65.68 | 85.65 | 96.11 | |
| D-MPNN (multi-task) | 63.88 | 84.17 | 95.91 |
We ultimately selected the D-MPNN model, which demonstrated the best performance on the test set. By integrating it with the clustering algorithm, we constructed our reaction condition predictor Reacon.
Analysis of problematic predicted cases. In addition to highlighting the strengths of Reacon, we also analyzed instances where it diverged from the ground truth in assigning the correct condition cluster. Fig. 5 presents representative cases where the model failed to predict the ground truth cluster within the top-3 cluster predictions (more examples can be found in Fig. S3†). In many cases, discrepancies between the model predictions and the dataset records were not a result of unreasonable outputs, but rather were attributed to dataset issues or the presence of various potential reaction conditions.
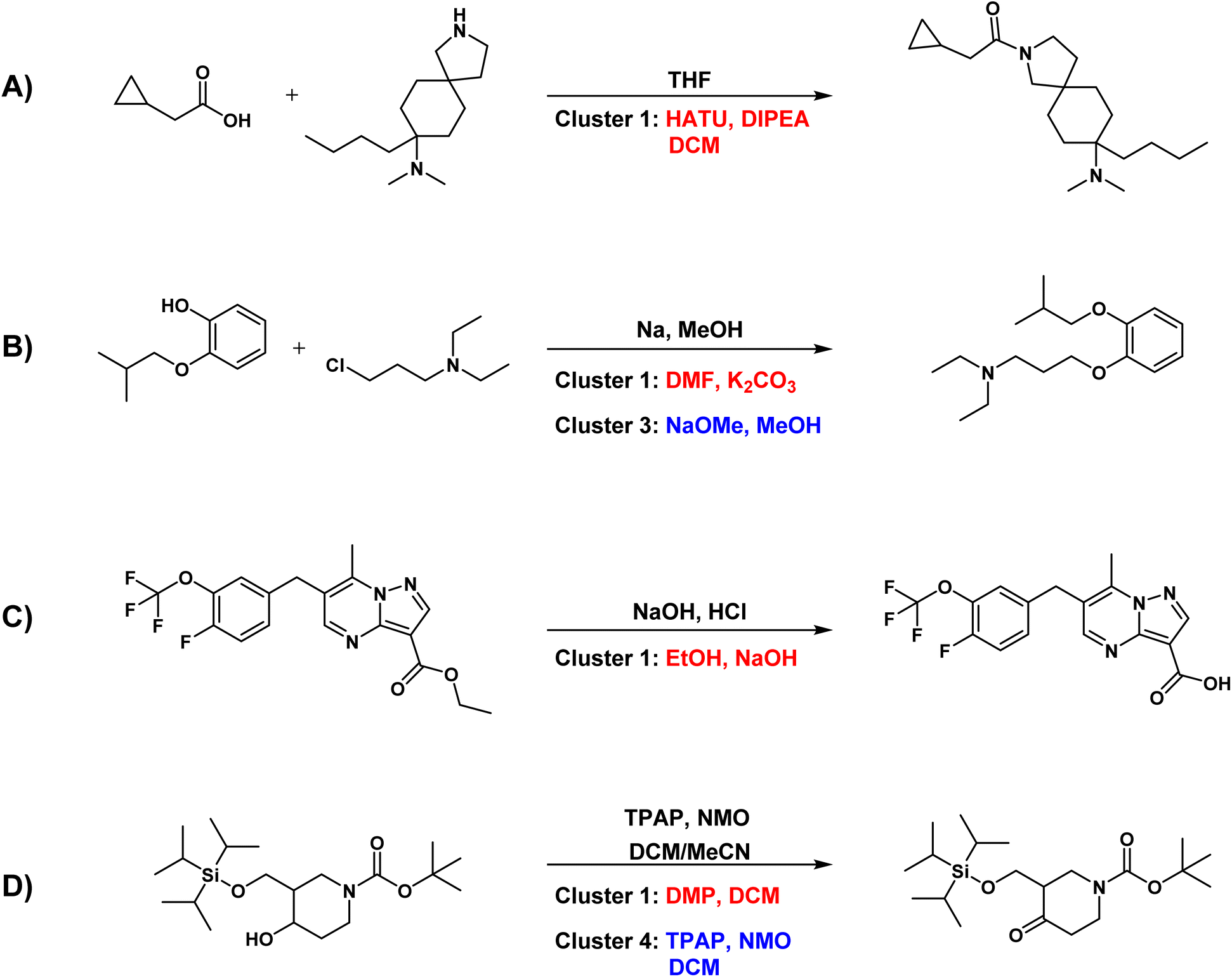 | ||
| Fig. 5 Reaction cases where the model's predictions do not agree with the ground truth. Conditions from the dataset or literature are marked in black, while predictions matching the ground truth type are marked in blue, and inconsistent predictions are marked in red. (A) Amide formation reaction. (B) Nucleophilic substitution reaction. (C) Hydrolysis of ester. (D) Oxidation of alcohol to ketone. | ||
Fig. 5A illustrates a very common problem in our dataset, where there are missing data for reaction conditions. Taking the amide formation reaction as an example, only THF was recorded as the condition. Yet, conducting this type of reaction without a carboxylate activating reagent often presents significant difficulties. In contrast, Reacon provided more comprehensive and accurate conditions. Erroneous recording of reaction condition data is also frequent in USPTO datasets. As shown in Fig. 5B, the direct use of sodium metal in this reaction is dangerous, and it is not a reasonable condition for this type of reaction. We checked the original patent record and found that sodium methoxide was actually used, but it was incorrectly recorded in the dataset as Na and methanol, which are the raw materials used to prepare sodium methoxide. Reacon not only predicted the ground-truth condition but also forecasted a commonly used non-nucleophilic base. Fig. 5C illustrates another common problem in the USPTO dataset, namely its inability to distinguish the order of multi-step operations. For example, for this ester hydrolysis reaction, the dataset recorded sodium hydroxide and hydrochloric acid as reaction conditions, with hydrochloric acid being used for post-processing rather than being added simultaneously. The dataset may inadvertently mix up the records, potentially causing challenges in the model's learning process. In this case, Reacon could still predict accurate conditions.
In the above cases, the differences between the model predictions and the ground truth records primarily stem from issues within the dataset. Specifically, in cases of missing or inaccurately recorded conditions, Reacon can effectively complete or correct the conditions, offering more logical conditions. We therefore believe that our model may have some potential for dataset optimization tasks as well.29
Apart from dataset issues, another challenge in predicting reaction conditions is the existence of multiple feasible outcomes. Fig. 5D depicts a reaction where an alcohol is oxidized to a ketone using TPAP (tetrapropylammonium perruthenate), an oxidizing agent classified within the fourth ranking cluster in Reacon's prediction. However, the Dess–Martin oxidation43 and Swern oxidation,44 recommended by Reacon in the top-2 clusters, are also proven oxidation methods with high yields. Even though the model fails to predict conditions consistent with those reported in the literature sometimes, it still offers feasible conditions.
Evaluation on actual synthetic routes
To further evaluate the practical performance of our model, we selected 12 drug molecule synthesis routes (100 reactions) from recently published articles in the Journal of Medicinal Chemistry45–56 and predicted their conditions using Reacon. This task is relatively challenging due to several factors. Firstly, some of the reagents are chosen by chance, depending on the preferences of the chemists or the laboratory inventory. Secondly, the conditions reported in the literature should have been purposefully optimized by the chemists, which are difficult to learn from general datasets. Overall, our predictor reached a top-3 exact accuracy of 39.00% and a top-3 cluster accuracy of 85.00%. The detailed results are shown in Table S14.† The predictions of two representative routes are shown in Fig. 6,46,47 and other predicted results can be found in Fig. S4–15.†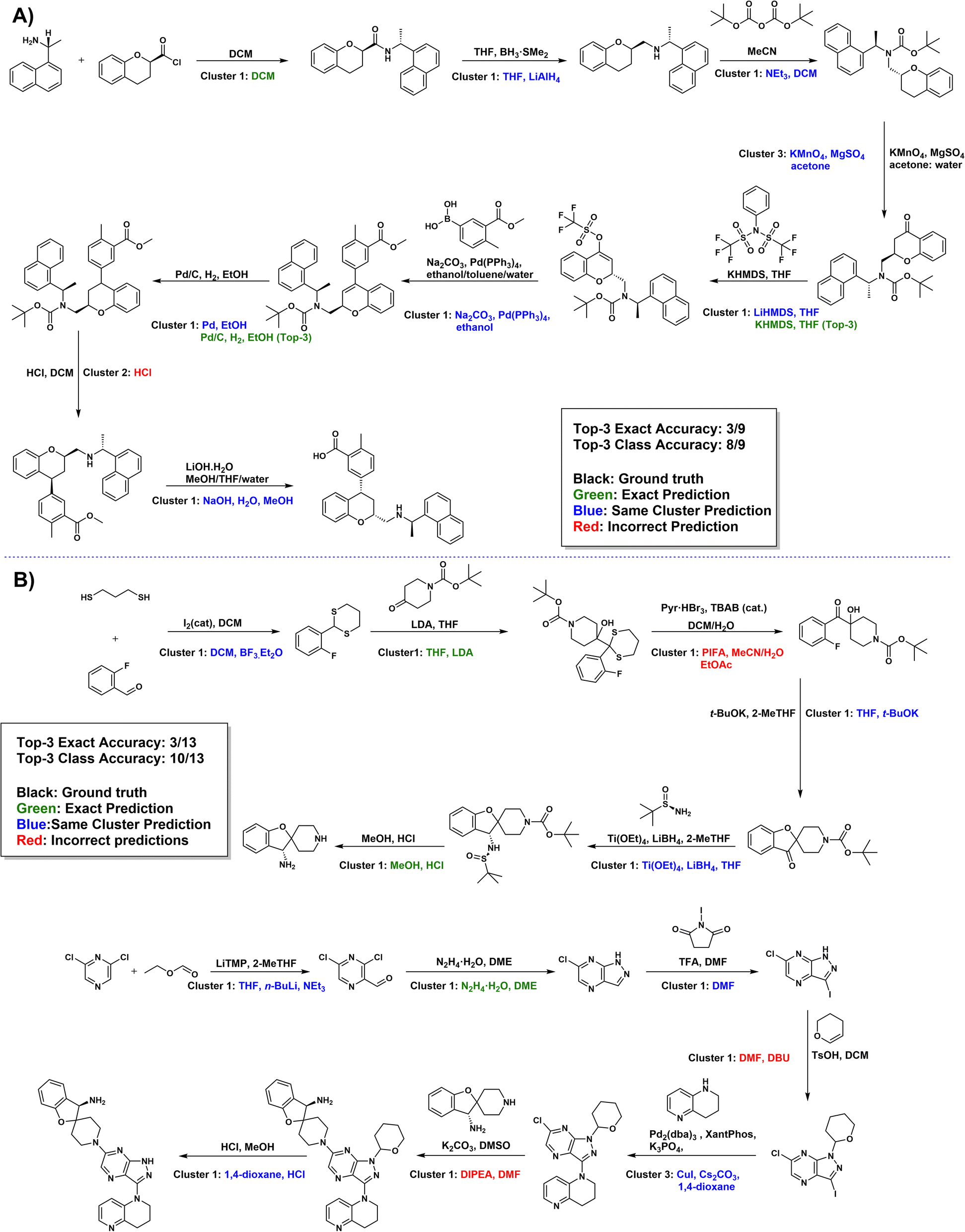 | ||
| Fig. 6 (A) Synthesis route of LNP1892 with actual and predicted reaction conditions. (B) Synthesis route of GDC-1971 with actual and predicted conditions. Ground truth conditions are marked in black. Consistent predictions are marked in green, while those matching the condition type but not the ground truth conditions are marked in blue, and inconsistent predictions are marked in red. The default display shows the top-1 results for each cluster unless the rank is specified within parentheses. | ||
In the route depicted in Fig. 6A, many of these predictions can be achieved by substituting similar reagents with conditions from the literature, often requiring just a single step. For example, the actual conditions in step 2 can be achieved by substituting the model's predictions of LiAlH4 with BH3·SMe2, and the conditions in step 5 can be obtained by replacing LiHMDS with KHMDS. Additionally, in step 6 of the route, Reacon accurately predicted the identical catalysts, reagents, and one of the three solvents reported in the literature.
In the synthesis route depicted in Fig. 6B, Reacon's predictions closely match the reported conditions in the literature. For example, the reported conditions in steps 4 and 5 can both be achieved by replacing the model-suggested THF with 2-MeTHF. Even predictions deviating from the ground truth remain rational. For instance, in step 3's dethioacetalization, where the literature favored pyridinium perbromide, Reacon proposed an alternative scenario using PIFA57 as an oxidizing agent. However, it was inevitable that Reacon occasionally provided unreasonable results, such as in step 10, where it erroneously interpreted a reaction involving 3,4-dihydropyran as the addition of an amino group to a double bond. This misinterpretation stems from the absence of comparable templates in our condition library.
Given that the primary objective of general reaction condition prediction models is to provide synthetic chemists with a reasonable starting point for condition optimization, this outcome is sufficiently informative and valuable as a reference.
Conclusions
We have developed a novel framework, Reacon, which encompasses a GNN model and predicts rational reaction conditions by integrating templates and clustering algorithms. Reacon achieves a top-1 accuracy of 44.52% in recalling ground truth conditions and 65.68% in predicting the corresponding cluster in the test dataset. Despite dataset inaccuracies leading to some errors, Reacon excels in providing more rational reaction conditions. Furthermore, the model exhibited satisfactory performance in predicting reaction conditions for actual synthesis routes. It successfully identified the reported condition clusters in the literature within the top-3 clusters with an accuracy of 85.00%, showcasing its capacity to aid chemists in screening reaction conditions. Overall, our work offers a reliable condition prediction tool that contributes to chemists in selecting conditions for new reactions and computer-assisted synthetic planning.Despite the promising results, the current approach can be further improved in the following directions. Firstly, the excellent performance of our framework relies heavily on the template-condition library, which enhances the performance of our models while limiting their scalability. This leads to difficulties for our model to make effective predictions for reaction conditions that do not appear in the training set. Therefore, further exploration should focus on expanding the predictor's capacity to predict new conditions. Secondly, temperature and reaction time are also important components of reaction conditions, which can be included in future work. Lastly, as inaccurate reaction conditions can negatively impact both model training and the clustering of reaction conditions, a high-quality dataset needs to be collected, especially from high-throughput experiments to further improve the model.
Code availability
Full code and trained models are available at: https://github.com/wzhstat/Reaction-Condition-Selector.Data availability
The dataset used for training is available at https://www.dropbox.com/scl/fo/v1rhyes2wvead9dz3x4fb/hrlkey=nqtst7azldcry3ixnoigmcv3v%26dl=0.Author contributions
Z. W. and K. L. designed the research, conducted the experiments, analysed the data and wrote the manuscript. J. P. and L. L. supervised the project and revised the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Key R&D Program of China (grant 2023YFF1205103) and the National Natural Science Foundation of China (22033001 and T2321001) and the Chinese Academy of Medical Sciences (2021-I2M-5-014).References
- C. W. Coley, W. H. Green and K. F. Jensen, Machine Learning in Computer-Aided Synthesis Planning, Acc. Chem. Res., 2018, 51(5), 1281–1289 CrossRef CAS.
- T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis, J. Med. Chem., 2020, 63(16), 8667–8682 CrossRef CAS PubMed.
- S. Szymkuc, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Computer-Assisted Synthetic Planning: The End of the Beginning, Angew Chem. Int. Ed. Engl., 2016, 55(20), 5904–5937 CrossRef CAS PubMed.
- J. Dong, M. Zhao, Y. Liu, Y. Su and X. Zeng, Deep learning in retrosynthesis planning: datasets, models and tools, Briefings Bioinf., 2022, 23(1), bbab391 CrossRef.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 2018, 4(3), 522–532 CAS.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning chemical syntheses with deep neural networks and symbolic AI, Nature, 2018, 555(7698), 604–610 CrossRef CAS.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, A robotic platform for flow synthesis of organic compounds informed by AI planning, Science, 2019, 365(6453), eaax1566 CrossRef CAS PubMed.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy, Chem. Sci., 2020, 11(12), 3316–3325 RSC.
- K. Lin, Y. Xu, J. Pei and L. Lai, Automatic retrosynthetic route planning using template-free models, Chem. Sci., 2020, 11(12), 3355–3364 RSC.
- B. Mikulak-Klucznik, P. Golebiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuc, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Computational planning of the synthesis of complex natural products, Nature, 2020, 588(7836), 83–88 CrossRef CAS PubMed.
- Y. Lin, R. Zhang, D. Wang and T. Cernak, Computer-aided key step generation in alkaloid total synthesis, Science, 2023, 379(6631), 453–457 CrossRef CAS PubMed.
- Z. Tu, T. Stuyver and C. W. Coley, Predictive chemistry: machine learning for reaction deployment, reaction development, and reaction discovery, Chem. Sci., 2023, 14(2), 226–244 RSC.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, Prediction of Organic Reaction Outcomes Using Machine Learning, ACS Cent. Sci., 2017, 3(5), 434–443 CrossRef CAS.
- F. Jaume-Santero, A. Bornet, A. Valery, N. Naderi, D. Vicente Alvarez, D. Proios, A. Yazdani, C. Bournez, T. Fessard and D. Teodoro, Transformer Performance for Chemical Reactions: Analysis of Different Predictive and Evaluation Scenarios, J. Chem. Inf. Model., 2023, 63(7), 1914–1924 CrossRef CAS.
- B. Zhang, X. Zhang, W. Du, Z. Song, G. Zhang, G. Zhang, Y. Wang, X. Chen, J. Jiang and Y. Luo, Chemistry-informed molecular graph as reaction descriptor for machine-learned retrosynthesis planning, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(41), e2212711119 CrossRef CAS.
- T. Gaich and P. S. Baran, Aiming for the ideal synthesis, J. Org. Chem., 2010, 75(14), 4657–4673 CrossRef CAS.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, The economies of synthesis, Chem. Soc. Rev., 2009, 38(11), 3010–3021 RSC.
- H. Struebing, Z. Ganase, P. G. Karamertzanis, E. Siougkrou, P. Haycock, P. M. Piccione, A. Armstrong, A. Galindo and C. S. Adjiman, Computer-aided molecular design of solvents for accelerated reaction kinetics, Nat. Chem., 2013, 5(11), 952–957 CrossRef CAS PubMed.
- G. Marcou, J. Aires de Sousa, D. A. R. S. Latino, A. de Luca, D. Horvath, V. Rietsch and A. Varnek, Expert System for Predicting Reaction Conditions: The Michael Reaction Case, J. Chem. Inf. Model., 2015, 55(2), 239–250 CrossRef CAS.
- M. R. Maser, A. Y. Cui, S. Ryou, T. J. DeLano, Y. Yue and S. E. Reisman, Multilabel Classification Models for the Prediction of Cross-Coupling Reaction Conditions, J. Chem. Inf. Model., 2021, 61(1), 156–166 CrossRef CAS PubMed.
- V. A. Afonina, D. A. Mazitov, A. Nurmukhametova, M. D. Shevelev, D. A. Khasanova, R. I. Nugmanov, V. A. Burilov, T. I. Madzhidov and A. Varnek, Prediction of Optimal Conditions of Hydrogenation Reaction Using the Likelihood Ranking Approach, Int. J. Mol. Sci., 2022, 23(1), 248 CrossRef CAS.
- Y. Kwon, S. Kim, Y.-S. Choi and S. Kang, Generative Modeling to Predict Multiple Suitable Conditions for Chemical Reactions, J. Chem. Inf. Model., 2022, 62(23), 5952–5960 CrossRef CAS PubMed.
- N. H. Angello, V. Rathore, W. Beker, A. Wołos, E. R. Jira, R. Roszak, T. C. Wu, C. M. Schroeder, A. Aspuru-Guzik, B. A. Grzybowski and M. D. Burke, Closed-loop optimization of general reaction conditions for heteroaryl Suzuki-Miyaura coupling, Science, 2022, 378(6618), 399–405 CrossRef CAS PubMed.
- Y. Kwon, D. Lee, J. W. Kim, Y. S. Choi and S. Kim, Exploring Optimal Reaction Conditions Guided by Graph Neural Networks and Bayesian Optimization, ACS Omega, 2022, 7(49), 44939–44950 CrossRef CAS PubMed.
- J. A. G. Torres, S. H. Lau, P. Anchuri, J. M. Stevens, J. E. Tabora, J. Li, A. Borovika, R. P. Adams and A. G. Doyle, A Multi-Objective Active Learning Platform and Web App for Reaction Optimization, J. Am. Chem. Soc., 2022, 144(43), 19999–20007 CrossRef CAS.
- K. Atz, D. F. Nippa, A. T. Müller, V. Jost, A. Anelli, M. Reutlinger, C. Kramer, R. E. Martin, U. Grether, G. Schneider and G. Wuitschik, Geometric deep learning-guided Suzuki reaction conditions assessment for applications in medicinal chemistry, RSC Med. Chem., 2024, 15(7), 2310–2321 RSC.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, Using Machine Learning To Predict Suitable Conditions for Organic Reactions, ACS Cent. Sci., 2018, 4(11), 1465–1476 CrossRef CAS PubMed.
- D. Kreutter and J.-L. Reymond, Multistep retrosynthesis combining a disconnection aware triple transformer loop with a route penalty score guided tree search, Chem. Sci., 2023, 14(36), 9959–9969 RSC.
- M. Andronov, V. Voinarovska, N. Andronova, M. Wand, D. A. Clevert and J. Schmidhuber, Reagent prediction with a molecular transformer improves reaction data quality, Chem. Sci., 2023, 14(12), 3235–3246 RSC.
- X. Wang, C.-Y. Hsieh, X. Yin, J. Wang, Y. Li, Y. Deng, D. Jiang, Z. Wu, H. Du, H. Chen, Y. Li, H. Liu, Y. Wang, P. Luo, T. Hou and X. Yao, Generic Interpretable Reaction Condition Predictions with Open Reaction Condition Datasets and Unsupervised Learning of Reaction Center, Research, 2023, 6, 0231 CrossRef CAS.
- Y. Qian, Z. Li, Z. Tu, C. Coley and R. Barzilay, Predictive Chemistry Augmented with Text Retrieval, Proceedings of the 2023 Conference on Empirical Methods in Natural Language Processing, Singapore, 2023, pp , pp 12731–12745 Search PubMed.
- E. Heid and W. H. Green, Machine Learning of Reaction Properties via Learned Representations of the Condensed Graph of Reaction, J. Chem. Inf. Model., 2022, 62(9), 2101–2110 CrossRef CAS PubMed.
- D. Lowe, Chemical reactions from US patents (1976-Sep2016), figshare, DOI:10.6084/m9.figshare.5104873.v1.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- RDKit: Open-Source Cheminformatics, https://www.rdkit.org Search PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, RDChiral: An RDKit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application, J. Chem. Inf. Model., 2019, 59(6), 2529–2537 CrossRef CAS PubMed.
- E. Heid, K. P. Greenman, Y. Chung, S. C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, Chemprop: A Machine Learning Package for Chemical Property Prediction, J. Chem. Inf. Model., 2024, 64(1), 9–17 CrossRef CAS PubMed.
- P. Velickovic, G. Cucurull, A. Casanova, A. Romero, P. Lio’ and Y. Bengio, Graph Attention Networks, arXiv, 2017, preprint, arXiv:1710.10903, DOI:10.48550/arXiv.1710.10903.
- W. Beker, R. Roszak, A. Wolos, N. H. Angello, V. Rathore, M. D. Burke and B. A. Grzybowski, Machine Learning May Sometimes Simply Capture Literature Popularity Trends: A Case Study of Heterocyclic Suzuki-Miyaura Coupling, J. Am. Chem. Soc., 2022, 144(11), 4819–4827 CrossRef CAS PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, Computer-Assisted Retrosynthesis Based on Molecular Similarity, ACS Cent. Sci., 2017, 3(12), 1237–1245 CrossRef CAS.
- L. Wolff, Chemischen Institut der Universität Jena: Methode zum Ersatz des Sauerstoffatoms der Ketone und Aldehyde durch Wasserstoff, Justus Liebigs Ann. Chem., 1912, 394(1), 86–108 CrossRef.
- M. Huang, Reduction of Steroid Ketones and other Carbonyl Compounds by Modified Wolff-Kishner Method, J. Am. Chem. Soc., 1949, 71(10), 3301–3303 CrossRef.
- D. B. Dess and J. C. Martin, Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- K. Omura and D. Swern, Oxidation of alcohols by “activated” dimethyl sulfoxide. a preparative, steric and mechanistic study, Tetrahedron Lett., 1974, 15(16), 1465–1560 Search PubMed.
- D. B. Freeman, T. D. Hopkins, P. J. Mikochik, J. P. Vacca, H. Gao, A. Naylor-Olsen, S. Rudra, H. Li, M. S. Pop, R. A. Villagomez, C. Lee, H. Li, M. Zhou, D. C. Saffran, N. Rioux, T. R. Hood, M. A. L. Day, M. R. McKeown, C. Y. Lin, N. Bischofberger and B. W. Trotter, Discovery of KB-0742, a Potent, Selective, Orally Bioavailable Small Molecule Inhibitor of CDK9 for MYC-Dependent Cancers, J. Med. Chem., 2023, 66(23), 15629–15647 CrossRef CAS.
- A. M. Taylor, B. R. Williams, F. Giordanetto, E. H. Kelley, A. Lescarbeau, K. Shortsleeves, Y. Tang, W. P. Walters, A. Arrazate, C. Bowman, E. Brophy, E. W. Chan, G. Deshmukh, J. B. Greisman, T. L. Hunsaker, D. R. Kipp, P. Saenz Lopez-Larrocha, D. Maddalo, I. J. Martin, P. Maragakis, M. Merchant, M. Murcko, H. Nisonoff, V. Nguyen, V. Nguyen, O. Orozco, C. Owen, L. Pierce, M. Schmidt, D. E. Shaw, S. Smith, E. Therrien, J. C. Tran, J. Watters, N. J. Waters, J. Wilbur and L. Willmore, Identification of GDC-1971 (RLY-1971), a SHP2 Inhibitor Designed for the Treatment of Solid Tumors, J. Med. Chem., 2023, 66(19), 13384–13399 CrossRef CAS.
- M. R. Shukla, G. Sadasivam, A. Sarde, M. Sayyed, V. Pachpute, R. Phadtare, N. Walke, V. D. Chaudhari, R. Loriya, T. Khan, G. Gote, C. Pawar, M. Tryambake, N. Mahajan, A. Gandhe, S. Sabde, S. Pawar, V. Patil, D. Modi, M. Mehta, P. Nigade, V. Modak, R. Ghodke, L. Narasimham, M. Bhonde, J. Gundu, R. Goel, C. Shah, S. Kulkarni, S. Sharma, D. Bakhle, R. K. Kamboj and V. P. Palle, Discovery of LNP1892: A Precision Calcimimetic for the Treatment of Secondary Hyperparathyroidism, J. Med. Chem., 2023, 66(14), 9418–9444 CrossRef CAS.
- C. Mo, X. Xu, P. Zhang, Y. Peng, X. Zhao, S. Chen, F. Guo, Y. Xiong, X. J. Chu and X. Xu, Discovery of HPG1860, a Structurally Novel Nonbile Acid FXR Agonist Currently in Clinical Development for the Treatment of Nonalcoholic Steatohepatitis, J. Med. Chem., 2023, 66(14), 9363–9375 CrossRef CAS.
- Y. Wu, J. Xi, Y. Li, Z. Li, Y. Zhang, J. Wang and G. H. Fan, Discovery of a Potent and Selective CCR8 Small Molecular Antagonist IPG7236 for the Treatment of Cancer, J. Med. Chem., 2023, 66(7), 4548–4564 CrossRef CAS PubMed.
- M. R. Garnsey, A. C. Smith, J. Polivkova, A. L. Arons, G. Bai, C. Blakemore, M. Boehm, L. M. Buzon, S. N. Campion, M. Cerny, S. C. Chang, K. Coffman, K. A. Farley, K. R. Fonseca, K. K. Ford, J. Garren, J. X. Kong, M. R. M. Koos, D. W. Kung, Y. Lian, M. M. Li, Q. Li, L. A. Martinez-Alsina, R. O'Connor, K. Ogilvie, K. Omoto, B. Raymer, M. R. Reese, T. Ryder, L. Samp, K. A. Stevens, D. W. Widlicka, Q. Yang, K. Zhu, J. P. Fortin and M. F. Sammons, Discovery of the Potent and Selective MC4R Antagonist PF-07258669 for the Potential Treatment of Appetite Loss, J. Med. Chem., 2023, 66(5), 3195–3211 CrossRef CAS PubMed.
- M. E. Layton, J. C. Kern, T. J. Hartingh, W. D. Shipe, I. Raheem, M. Kandebo, R. P. Hayes, S. Huszar, D. Eddins, B. Ma, J. Fuerst, G. K. Wollenberg, J. Li, J. Fritzen, G. B. McGaughey, J. M. Uslaner, S. M. Smith, P. J. Coleman and C. D. Cox, Discovery of MK-8189, a Highly Potent and Selective PDE10A Inhibitor for the Treatment of Schizophrenia, J. Med. Chem., 2023, 66(2), 1157–1171 CrossRef CAS PubMed.
- B. Chen, J. Wu, Z. Yan, H. Wu, H. Gao, Y. Liu, J. Zhao, J. Wang, J. Yang, Y. Zhang, J. Pan, Y. Ling, H. Wen and Z. Huang, 1,3-Substituted beta-Carboline Derivatives as Potent Chemotherapy for the Treatment of Cystic Echinococcosis, J. Med. Chem., 2023, 66(24), 16680–16693 CrossRef CAS PubMed.
- L. Zhang, Y. Li, C. Tian, R. Yang, Y. Wang, H. Xu, Q. Zhu, S. Chen, L. Li and S. Yang, From Hit to Lead: Structure-Based Optimization of Novel Selective Inhibitors of Receptor-Interacting Protein Kinase 1 (RIPK1) for the Treatment of Inflammatory Diseases, J. Med. Chem., 2024, 67(1), 754–773 CrossRef CAS PubMed.
- J. Szychowski, R. Papp, E. Dietrich, B. Liu, F. Vallee, M. E. Leclaire, J. Fourtounis, G. Martino, A. L. Perryman, V. Pau, S. Y. Yin, P. Mader, A. Roulston, J. F. Truchon, C. G. Marshall, M. Diallo, N. M. Duffy, R. Stocco, C. Godbout, A. Bonneau-Fortin, R. Kryczka, V. Bhaskaran, D. Mao, S. Orlicky, P. Beaulieu, P. Turcotte, I. Kurinov, F. Sicheri, Y. Mamane, M. Gallant and W. C. Black, Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306, J. Med. Chem., 2022, 65(15), 10251–10284 CrossRef CAS PubMed.
- M. D. Hill, M. J. Blanco, F. G. Salituro, Z. Bai, J. T. Beckley, M. A. Ackley, J. Dai, J. J. Doherty, B. L. Harrison, E. C. Hoffmann, T. M. Kazdoba, D. Lanzetta, M. Lewis, M. C. Quirk and A. J. Robichaud, SAGE-718: A First-in-Class N-Methyl-d-Aspartate Receptor Positive Allosteric Modulator for the Potential Treatment of Cognitive Impairment, J. Med. Chem., 2022, 65(13), 9063–9075 CrossRef CAS.
- T. Inghardt, T. Antonsson, C. Ericsson, D. Hovdal, P. Johannesson, C. Johansson, U. Jurva, J. Kajanus, B. Kull, E. Michaelsson, A. Pettersen, T. Sjogren, H. Sorensen, K. Westerlund and E. L. Lindstedt, Discovery of AZD4831, a Mechanism-Based Irreversible Inhibitor of Myeloperoxidase, As a Potential Treatment for Heart Failure with Preserved Ejection Fraction, J. Med. Chem., 2022, 65(17), 11485–11496 CrossRef CAS.
- G. Stork and K. Zhao, A simple method of dethioacetalization, Tetrahedron Lett., 1989, 30(3), 287–290 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05946h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |