DOI:
10.1039/D4SC06354F
(Edge Article)
Chem. Sci., 2025,
16, 3114-3123
Self-sensitized Cu(II)-complex catalyzed solar driven CO2 reduction†
Received
19th September 2024
, Accepted 7th January 2025
First published on 9th January 2025
Abstract
Developing a self-sensitized catalyst from earth-abundant elements, capable of efficient light harvesting and electron transfer, is crucial for enhancing the efficacy of CO2 transformation, a critical step in environmental cleanup and advancing clean energy prospects. Traditional approaches relying on external photosensitizers, comprising 4d/5d metal complexes, involve intermolecular electron transfer, and attachment of photosensitizing arms to the catalyst necessitates intramolecular electron transfer, underscoring the need for a more integrated solution. We report a new Cu(II) complex, K[CuNDPA] (1[K(18-crown-6)]), bearing a dipyrrin amide-based trianionic tetradentate ligand, NDPA (H3L), which is capable of harnessing light energy, despite having a paramagnetic Cu(II) centre, without any external photosensitizer and photocatalytically reducing CO2 to CO in acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (19:1 v/v) with a TON as high as 1132, a TOF of 566 h−1 and a selectivity of 99%. This complex also shows hemilability in the presence of water, which not only plays a role in the proton relay mechanism but also helps stabilize a crucial Cu(I)-NDPA intermediate. The hemilability was justified by the formation of N3O (2) and N2O2 (3) coordinated congeners of the N4 bound complex 1. The overall mechanism was further investigated via spectroscopic techniques such as EPR, UV-vis, and spectroelectrochemistry, culminating in the justification of a single electron-reduced Cu(I)NDPA species as a proposed intermediate. In the next step, the binding of CO2 to the Cu(I) complex and subsequent electron transfer to form Cu(II)–COO·− was indirectly probed by a radical trapping experiment via the addition of p-methoxy-2,6-di-tert-butylphenol that led to the formation of a phenoxyl radical. This work provides new strategies for designing earth-abundant robust molecular catalysts that can function as photocatalysts without the aid of any external photosensitizers.
:water (19:1 v/v) with a TON as high as 1132, a TOF of 566 h−1 and a selectivity of 99%. This complex also shows hemilability in the presence of water, which not only plays a role in the proton relay mechanism but also helps stabilize a crucial Cu(I)-NDPA intermediate. The hemilability was justified by the formation of N3O (2) and N2O2 (3) coordinated congeners of the N4 bound complex 1. The overall mechanism was further investigated via spectroscopic techniques such as EPR, UV-vis, and spectroelectrochemistry, culminating in the justification of a single electron-reduced Cu(I)NDPA species as a proposed intermediate. In the next step, the binding of CO2 to the Cu(I) complex and subsequent electron transfer to form Cu(II)–COO·− was indirectly probed by a radical trapping experiment via the addition of p-methoxy-2,6-di-tert-butylphenol that led to the formation of a phenoxyl radical. This work provides new strategies for designing earth-abundant robust molecular catalysts that can function as photocatalysts without the aid of any external photosensitizers.
Introduction
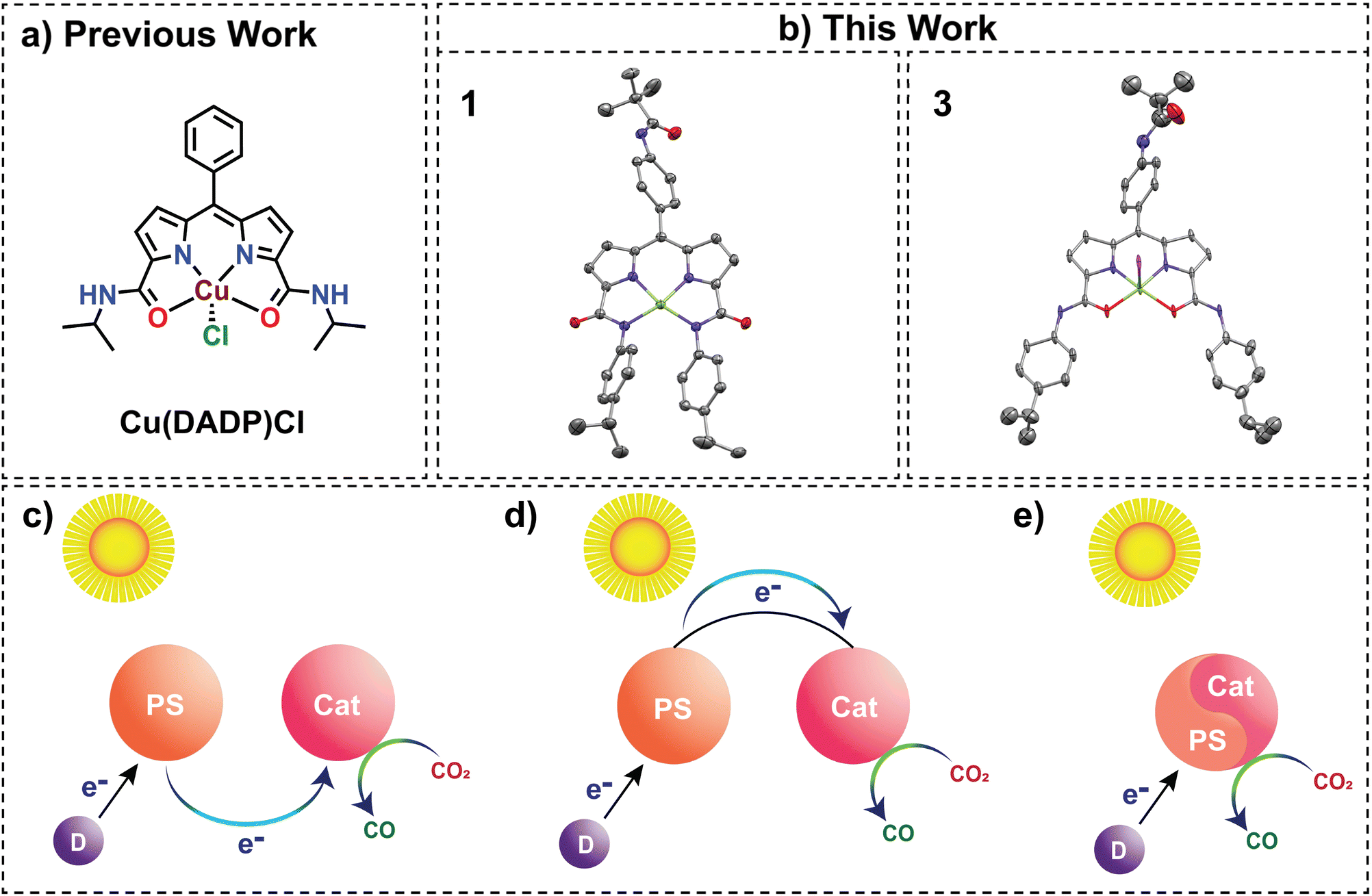
In recent years, selective and energy-effective conversion of CO2 to value-added chemicals has become a burdensome challenge.1 Sunlight-driven reactions have attracted much attention lately since solar energy is one of the few clean and enduring energy sources presently available. Biological processes such as photosynthesis showcase a compelling model for demonstrating remarkably efficient utilization of solar energy for H2O oxidation, which indirectly helps convert CO2 into chemical energy2,3 in the dark phase. Replicating this feat is chemically demanding due to the energetically uphill nature of CO2 reduction.4 Most metal complexes that have been reported so far to catalyse light-driven CO2 reduction necessitate the presence of external photosensitizers (Fig. 1c). External photosensitizers have an inevitable intermolecular electron transfer step between the photosensitizer and the catalyst, which may limit the efficiency of the process. Additionally, these external photosensitizers predominantly comprise noble metal complexes,5–7 which are scarcely available in the Earth's crust. In contrast, in photosystem II, the various subunits involved in either light harvesting, electron transfer, or the primary catalytic centre for the oxygen evolution reaction are in close proximity to maximize efficiency. This concept has inspired chemists to engineer self-photosensitizing systems, where photosensitizing arms are integrated into catalysts (Fig. 1d), eliminating the dependence on intermolecular electron transfer kinetics. For example, dyads containing a photosensitizing arm and a transition metal complex have been reported for solar-driven CO2 reduction.8–13 Here, there is a dependence on photoinduced electron transfer from the sacrificial electron donor to the photoexcited photosensitizer moiety and intramolecular electron transfer from the reduced photosensitizer to the catalyst moiety.14 However, the quest for photosensitizer-free solar-driven CO2 reduction using a self-sensitized catalyst,10 as illustrated in Fig. 1e, remains less explored with 3d transition metals despite being a valuable alternative for photocatalytic CO2 reduction. Most examples reported are based on 4d and 5d transition metal complexes.14,15
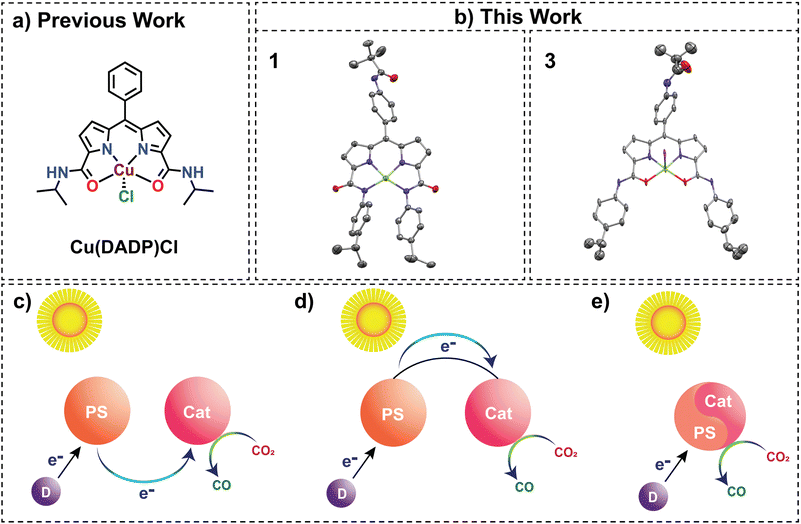 |
| | Fig. 1 (a) Previously reported literature on a similar diamidodipyrrin-based ligand. (b) This work on the Cu-based modified diamidodipyrrin complex shows the N4 (1[K(18-crown-6)]) and N2O2 (3) congeners in the perspective view of their crystal structures with 60% and 20% thermal ellipsoid probabilities, respectively. Hydrogen atoms, 18-crown-6, and the solvent molecules are omitted for clarity. Schematic illustration of photocatalytic systems for CO2 reduction. (c) PS and Cat as distinct functional units. (d) PS and Cat linked (e) self-sensitized system. (PS: photosensitizer, Cat: catalyst, and D: sacrificial donor). | |
These complexes play a dual role in harvesting light energy and subsequently catalyzing the reduction of CO2. Thus, a reaction without an electron transfer event between the photosensitizing and catalytic units is feasible. Some Ir-based complexes have also been reported previously, which can reduce CO2 to formic acid through the formation of an Ir–H intermediate.16 Masaoka and co-workers reported a self-sensitized Ru(II)P complex with a TOF of only 14.7 h−1.14 Similar studies were undertaken by Papish and co-workers, using Ru and Ni–CNC pincer-based complexes as self-sensitized systems for CO production.15,17 A previous report using Os(III)18 is shown to behave like a self-sensitized system using red light, while its Cr(III)19 congener with a slightly modified framework has been reported to achieve photocatalytic CO2 reduction using an external photosensitizer, thus highlighting the challenges in self-sensitization associated with moving from 3rd row to 1st row transition metals. Robert and co-workers explored the field of self-sensitized photocatalytic CO2 reduction using Fe-based complexes such as Fe-p-TMA and FeTPP having 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) and triethylamine as sacrificial electron donors respectively.20,21 Additionally, an Fe2Na3 purpurin complex was reported for self-sensitized CO2 reduction by Han and co-workers.22 Although the same group reported a Cu(II) purpurin-based complex for photocatalytic CO2 reduction, it showed a very low TON of 4.4 due to the limited stability of the complex under reaction conditions.23 Unlike Cu(II), Cu(I) has been well explored for self-sensitized CO2 reduction.24 Electrocatalytic and external photosensitizer-mediated photocatalytic CO2 reduction utilizing Cu(II), Cu(I) and Cu(0) systems have also been explored previously.25–29 Despite the advantages mentioned above, back electron transfer28 is a drawback that slows down the kinetics of these photosensitizer-free systems. This results in significantly lower TONs for these systems. A detailed comparative analysis of TONs of self-sensitized systems in the first-row transition metal-based complexes (Table S14†) bolsters us to claim that, to the best of our knowledge, a copper(II)-based, self-sensitized photocatalyst that combines both high-efficiency and high selectivity for CO production has not yet been reported. In this study, we describe a novel tri-anionic tetra-dentate redox-active ligand, NDPA (H3L), and its anionic copper complex (1) for solar-driven CO2 reduction to CO with excellent selectivity and high yields. The NDPA ligand was designed to incorporate a BODIPY-like (dipyrrin) backbone together with aromatic carboxamides. Our group has previously explored aromatic carboxamide-based ligands for redox catalysis.30,31 Such aromatic carboxamide-based Cu(II) systems were explored by Llobet and co-workers as well.32,33 Copper(II) complexes of ligands containing dipyrrin moieties are fluorescent.34 Additionally, amide-based complexes, particularly those derived from monoamides, have been utilized in the CO2 reduction reaction.35 This is due to their electron-donating capability facilitated by the anionic amidate moiety. Such anionic complexes containing N−-donors are electron-rich, thereby contributing significantly to their efficacy in CO2 reduction. Therefore, we hypothesized that designing a multi-dentate ligand containing both the dipyrrin and carboxamide units could form a Cu(II) complex capable of harvesting light and catalysing CO2 reduction. Herein, we report a trianionic dipyrrin–dicarboxamide ligand-based Cu(II) complex, which plays the dual role of a photosensitizer and a catalyst for selective CO2 reduction to CO. The complex has been extensively characterized experimentally (UV-vis, HRMS, EPR, and SC-XRD) and theoretically (optimization and TD-DFT) as we endeavour to unlock new possibilities in the realm of photosensitizer-free CO2 reduction.
Results and discussion
Synthesis and characterization of H3L and complex 1
A previous report of a dipyrrin carboxamide ligand led to the formation of a monoanionic ligand-based Cu(II) complex [Cu(DADP)Cl] with N2O2 coordination (Fig. 1a).36 We envisioned that the dipyrrin–carboxamide ligand could bind with N4 coordination to form a trianionic ligand-based Cu(II) complex if the aliphatic carboxamides are replaced with their aromatic analogues. The reduced electron density on the N donor in aromatic carboxamides is better suited to bind Cu(II) to form a trianionic complex. In fact, enhanced acidity of the aromatic amide group was observed in 1H NMR (Fig. S7†), where introducing an aromatic group in the coordinating amide side reduced the electron density on the nitrogen atom, enhancing the acidity of the amide hydrogen, and displayed a greater deshielding character compared to the DADP ligand. The NDPA (H3L) ligand synthesis has been achieved through a modified procedure of the previously reported methodology (Scheme S1†).36 In the synthesis of complex 1[K(18-crown-6)], deprotonation of H3L with 3 eq. of base (KH) in tetrahydrofuran induced a distinct change in solution colour from red to dark pink (Scheme S2†). This L3− was also characterized by 1H NMR (Fig. S10†). Subsequent addition of anhydrous CuCl2 produced a bluish-purple solution. Purple-coloured crystals were obtained after the addition of 1 eq. of 18-crown-6, followed by slow diffusion of ether into a saturated acetonitrile solution of the complex. The absorption spectra in acetonitrile exhibited distinctive peaks at 556 nm and 598 nm for 1[K(18-crown-6)], differing notably from the 482 nm peak observed for the ligand (Fig. 3a). Furthermore, High-Resolution Mass Spectrometry (HRMS) in negative ion mode revealed a prominent peak at m/z 701.2762, confirming the formation of complex 1 (Fig. S11†). The observed axial EPR spectrum in X-band for 1 (Fig. S26†) is similar to that for the previously reported [Cu(DADP)Cl] complex.36 The corresponding g-values for a frozen solution of 1 in acetonitrile:toluene (3:1; v/v) at 80 K were observed as g⊥ = 2.052 and gǁ = 2.185 (Fig. S26†).
Structural studies of complex 1
The structural elucidation of complex 1[K(18-crown-6)] reveals a four-coordinated Cu ion with exclusive nitrogen donors: two from the amides (N4 and N5) and two from the dipyrrin pyrrole moieties (N1 and N3) (Fig. 1b). The average Cu–Namide (2.007 Å) and Cu–Npyrrole (1.940 Å) bond distances (Table S2†) aligned closely with previously reported similar Cu(II) species,36 confirming the presence of Cu(II) in 1. Distinct deviation of the trans angles N1–Cu1–N4 = 164.92°(6) and N3–Cu1–N5 = 166.14°(6) from perfect square planarity was observed, yielding a structural index parameter (τ4) value of 0.2 (Table S3†).37 Hence, the Cu(II) ion in 1 adopts a slightly distorted square planar geometry. Notably, in contrast to the square pyramidal structure of the [Cu(DADP)Cl] complex, where a Cl− ion occupies the axial site, the observed geometry in 1 implies a stronger ligand field compared to [Cu(DADP)Cl]. Moreover, the metric parameters (Table S4†) associated with the ligand backbone in 1 reveal intriguing structural aspects. The respective C–C and C–N bond distances in both the pyrrole-amide halves around Cu(II) are almost similar. Remarkably, the average C–N amide bond length (1.358 Å) in 1 falls between typical C–N single and double bond values.38,39 Additionally, in contrast to the expectation, both the pyrrole moieties have one C–N single bond (1.382 Å) and one C–N double bond (1.328 Å) at identical positions.36,40 These findings collectively indicate extensive conjugation within the ligand framework of 1, suggestive of a high likelihood of charge delocalization.
Acid–base induced hemilability of [CuNDPA]1− (1)
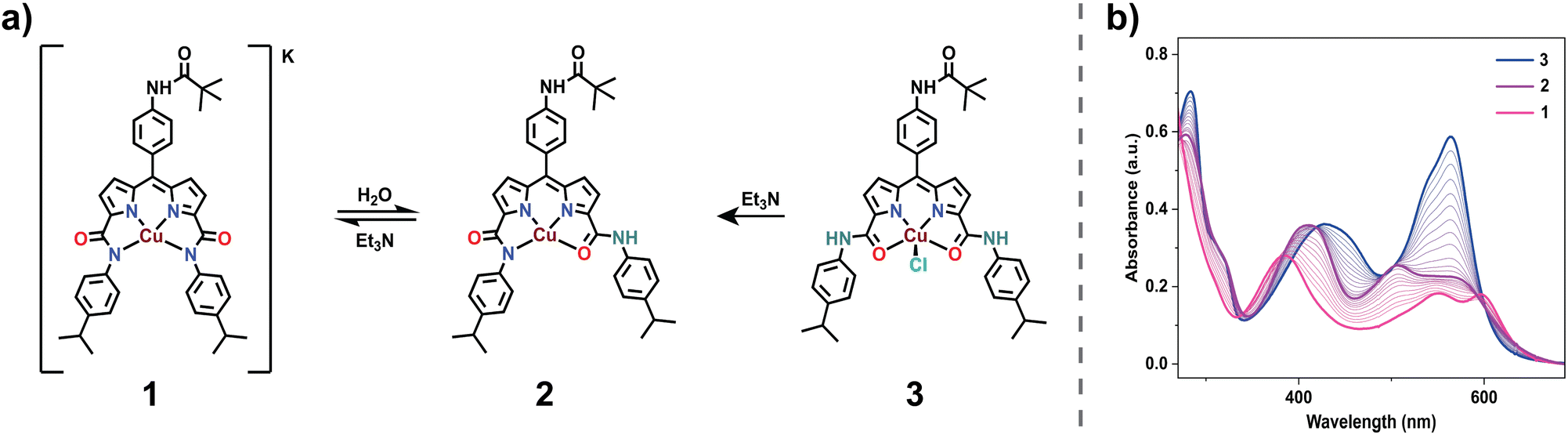
The introduction of water into an acetonitrile solution of complex 1 initiates a notable coordination switch, setting up a transition between the trianionic N4 to the monoanionic N2O2via the dianionic N3O coordination (Fig. S20†). At first, this transformation from N4 into N3O coordination is distinctly evidenced by changes observed in the UV-vis spectra, notably by the diminishment of the peak at 598 nm (Fig. S20†). Further High-Resolution Mass Spectrometry (HRMS) shows the disappearance of the peak at 701.2762 (negative ion-mode, m/z of 1) and the subsequent appearance of a new peak at m/z 703.2584 (1+H+1) in the positive-ion mode (Fig. S17†). Additionally, when D2O is introduced into the acetonitrile solution of complex 1, a distinct one-unit enhanced peak at m/z 704.2648 (1+D+1) (Fig. S18†) appears in the positive ion mode. These observations strongly suggest a protonation event at one of the coordinated deprotonated amides, followed by a subsequent coordination switch leading to the formation of complex 2. EPR studies of complex 2 were performed in a frozen acetonitrile:toluene (3:1; v/v) solution. An axial signal obtained at 80 K was (g⊥ = 2.052 and gǁ = 2.271) evidently different from that of 1 (Fig. S27†). We observed that over time (>3 days), the solution of complex 2 in acetonitrile kept in an open atmosphere converts into a distinctly different species. We hypothesized this to be the consequence of a second coordination switch event (N3O to N2O2), resulting in the formation of complex 3. To prove this hypothesis, we separately prepared the N2O2 coordinated CuNDPA species (complex 3) by refluxing a THF solution of H3L with anhydrous CuCl2 for 8 h. The absorption spectrum of 3 in acetonitrile shows a sharp peak at 563 nm (Fig. S14†), notably different from that of both 1 and 2. Slow evaporation of saturated methanol:acetonitrile solution of 3 afforded single crystals, which, upon XRD analysis, revealed an N2O2 coordination with a square pyramidal geometry (Fig. 1b). Positive ion mode HRMS displayed a peak at m/z 703.2533, in agreement with the expected mass of complex 3 (Fig. S13†). These observations conclusively prove the hemilability of complex 1 accompanied by a stepwise N to O coordination change. Of notable interest is the reversibility of this coordination switch. Initial titration of an acetonitrile solution of 3 with Et3N yielded a UV-vis absorbance spectrum conforming precisely to that of 2 (Fig. 2b). Further addition of Et3N led to the formation of 1 (Fig. S19†), providing additional evidence for the proposed coordination switch facilitated by the sequential deprotonation of the two amide groups (Fig. 2a). Such hemilability has been previously proposed in carboxamide-based complexes with terminal amides and is relatively rare for secondary amide-based ligands.41,42
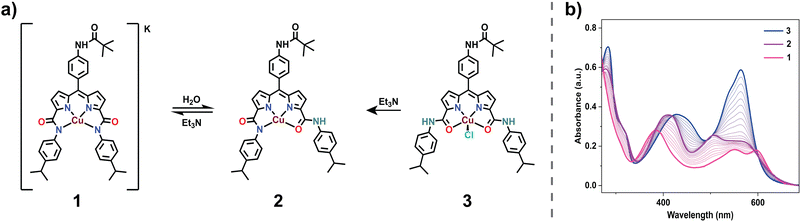 |
| | Fig. 2 (a) Structural representation of the coordination switch from N4(1) to N2O2(3) and vice versa in the presence of water and Et3N, respectively, in acetonitrile. (b) Absorption spectral changes accompanying the coordination switch from N2O2(3) to N4(1) via N3O(2) in the presence of Et3N. | |
Ground state absorption
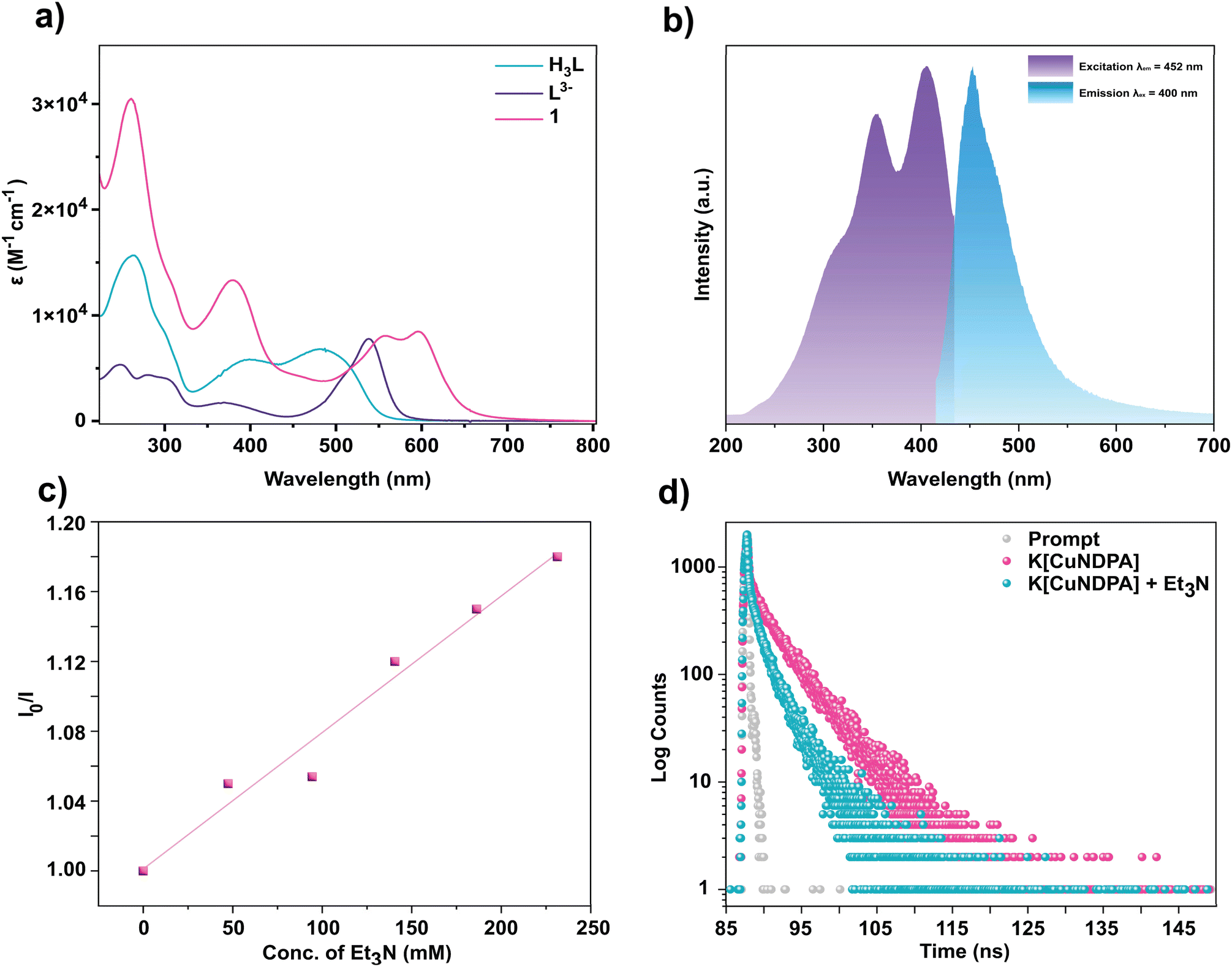
The ground state absorption characteristics of complex 1 were comprehensively examined using UV-vis spectroscopy (Fig. 3a). To identify the anionic ligand-based transitions, the absorption spectrum of the deprotonated ligand L3− was compared to that of 1. Notably, while H3L shows absorption bands at 390 nm and 482 nm, the deprotonated ligand L3− displayed peaks at 370 nm and 538 nm (Fig. 3a). 1 exhibited absorption bands at 378 nm (13042 M−1 cm−1), 556 nm (8079 M−1 cm−1), and 598 nm (8437 M−1 cm−1) (Fig. S21–S23†). To further elucidate the origin of these transitions, we performed the TD-DFT analysis for complex 1. The structure was optimized using the B3LYP function (Fig. S48†). The LANL2DZ basis set was employed for Cu and 6-311+G* for all other atoms. The calculated transitions from TD-DFT analysis at 370 nm, 542 nm, and 596 nm closely correlated with the experimental peaks at 378, 556, and 598 nm, respectively. The transition at 370 nm was attributed primarily to the Ligand-to-Metal Charge Transfer (LMCT) transition with a minor contribution from Intra Ligand Charge Transfer (ILCT) (Fig. S50†), while the peak at 542 nm was assigned to a predominantly ILCT transition with minor contributions from Metal-to-Ligand Charge Transfer (MLCT) (Fig. S51†). The transition at 596 nm was primarily due to an MLCT transition (Fig. S52†).
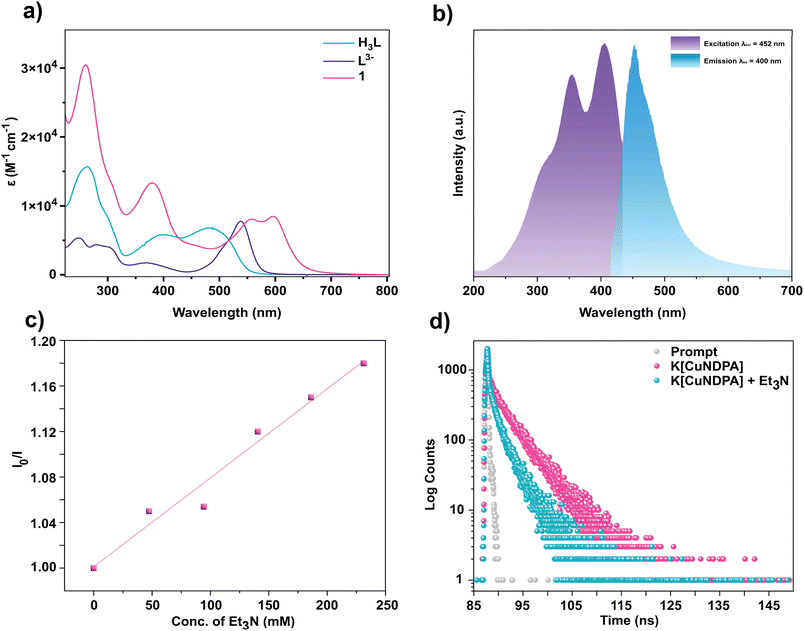 |
| | Fig. 3 (a) Absorption spectra of 1, H3L and L3− in acetonitrile. (b) Normalized ground state emission and excitation spectra of 1 in acetonitrile. (c) Stern–Volmer plot of the emission quenching of 1 with Et3N at 400 nm excitation. (d) Emission decay of 1 in acetonitrile at 298 K. | |
Electrochemical studies of complex 1
Cyclic voltammetry experiments were conducted in an acetonitrile solution of 1 (0.5 mM) employing tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte. A glassy carbon electrode was used as the working electrode, an aqueous Ag/AgCl electrode as the reference electrode, and a Pt-wire as the counter electrode (Fig. S24†). During oxidation, a reversible peak at E11/2ox = 0.178 V (vs. Fc+/Fc) in the anodic potential was observed. A second irreversible oxidation peak was observed at a higher potential, E21/2ox = 0.568 V (vs. Fc+/Fc). Upon reduction, one irreversible peak was noted at −1.120 V and another reversible peak at −1.378 V (vs. Fc+/Fc). Assigning the origin of cathodic peaks is thoroughly discussed in the spectroelectrochemistry section (below).
Spectroelectrochemical studies of 1
Electrochemical reduction by controlled potential electrolysis (CPE) at −1.25 V (vs. Fc+/Fc) generated an absorption peak at 517 nm (Fig. S31†). The decrease in the LMCT band at 378 nm indicates the plausible formation of Cu(I) from Cu(II). To validate the formation of a Cu(I)-based reduced species, Cu(I)-NDPA, a solution of deprotonated H3L in dry, degassed tetrahydrofuran (THF) was treated with anhydrous CuCl, which yielded a reddish-pink complex through subsequent workup procedures. Notably, the UV-vis spectral profile (Fig. S44†) in an anaerobic acetonitrile solution of Cu(I)-NDPA exhibited distinct differences from that of 1 and H3L, with a λmax at 517 nm, which matches with the electrochemically generated one electron-reduced species after the CPE at −1.25 V (vs. Fc+/Fc). Furthermore, the addition of 1 eq. of decamethylcobaltocene to 1 generates a UV-vis spectrum similar to the one obtained upon electrochemical reduction and is indicative of the formation of Cu(I)-NDPA (Fig. S44†). All the chemically and electrochemically generated batches of Cu(I)-NDPA are EPR silent, aligning with the anticipated behaviour of having Cu(I) in the one-electron reduced state. Cu(I)-NDPA is unstable under aerobic conditions, as evidenced by an immediate colour change from reddish-pink to reddish-purple upon exposure to air. This aerial oxidation-induced alteration is effectively monitored through UV-vis spectra, during which the transformation of Cu(I)-NDPA into the corresponding Cu(II) species of complex 2 is observed (Fig. S45†). The formation of the N3O-coordinated Cu(II) (2) upon oxidation suggests the possibility of an N3O-coordination in the Cu(I) species formed after 1 electron reduction of N4-coordinated 1. This coordination switch from N4 to N3O upon 1-electron reduction of 1 can be rationalized due to the preference for a tetrahedral geometry for Cu(I). The feasibility of the coordination change from N4 to N3O in this class of NDPA ligands described here has already been demonstrated in the case of Cu(II) complexes (1–3) earlier. We believe that switching from N4 to N3O in the Cu(I) state is more feasible since the N3O ligand will be less electron-rich due to a change in the coordination (neutral O instead of the deprotonated N of amide).
Fluorescence studies of 1
Drawing inspiration from prior studies on a Cu(II) complex of a dipyrrin ligand,34 the photophysical properties of complex 1 were explored. Notably, the complex exhibited an emission band at 452 nm upon excitation at 400 nm (Fig. 3b). However, H3L also displayed emission closely resembling that of 1 under excitation at 400 nm (Fig. S28†). Furthermore, the possibility of emission occurring after photobleaching of complex 1 (leading to the formation of H3L) was ruled out since no change in the fluorescence intensity of the complex was observed even after 10 min of continuous irradiation with 390 nm light. The HPLC peak profile of the complex before and after irradiation remained unaltered (Fig. S40 and S41†). To elucidate the quenching mechanism of this CT state, triethylamine (Et3N) was introduced as a sacrificial donor. Increasing concentrations of Et3N led to a subsequent decrease in emission intensity. The linearity of the S–V plot indicates the occurrence of a dynamic-state quenching process (Fig. 3c). Additionally, Time-Correlated Single Photon Counting (TCSPC) measurements were performed to determine the lifetime of the corresponding excited state upon excitation at 400 nm (Fig. 3d). The average lifetime of 1 was found to be tavg = 4.22 ns from a bi-exponential fit (see the ESI†). Interestingly, the addition of Et3N led to a decrease in lifetime to tavg = 2.33 ns, correlating with the quenching of the emission from the corresponding excited state in the presence of a reductive quencher. The thermodynamic parameters justifying the feasibility of this photoinduced electron transfer have been calculated (see the ESI). The emission at ∼452 nm, despite having the lowest energy absorption of ∼596 nm, is rare yet not unusual for first-row transition metal complexes.43 Interestingly, a second emission at ∼633 nm for excitation at ∼575 nm is also observed. The emissions (at 452 & 633 nm) are most likely unrelated as they have distinctly different excitation profiles (Fig. S29† and 3b) and arise from different charge transfer process-related emissive states, namely LMCT and MLCT. Such emissions were reported to be Kasha-like,44 a conclusion we can also reach upon varying the excitation wavelength between 350–420 nm without observing any changes in the emission maxima (Fig. S30†). More detailed photophysical characterization to elucidate the dynamics of the excited state is under investigation.
Photocatalytic CO2 reduction
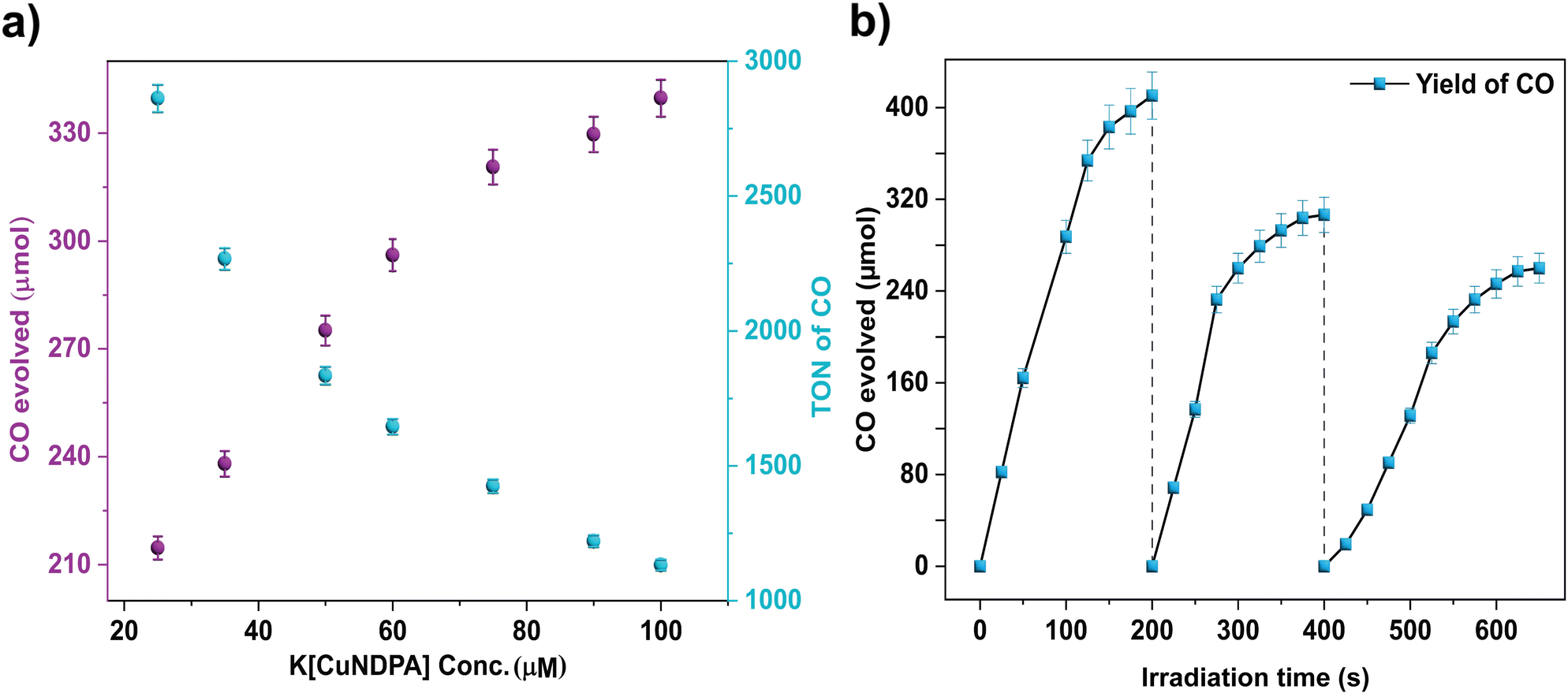
In order to study the photocatalytic CO2 reduction, a solution of the catalyst (1, 0.1 mM) in acetonitrile was exposed to 390 nm light in the presence of water (Table S9†) as a proton source and triethylamine (Table S10†) as a sacrificial reductant for 2 hours under a CO2 (1 atm) atmosphere at room temperature. An aliquot of gas from the headspace was analysed by gas chromatography (GC). The results of CO2 reduction to CO are tabulated in Table 1. Notably, augmenting the water concentration did not correspondingly enhance H2 production,45 indicating superior selectivity for CO2 reduction, achieving approximately 99% selectivity in acetonitrile. An increase in CO production was observed upon increasing the amount of water to 5%(v/v) (Fig. S36†). However, a further increase to 10% (v/v) led to a decrease in the yield of CO, possibly due to competition between CO2 and H2O to coordinate with the metal centre, resulting in decreased CO production (Table S9†) for coordination to the metal centre. Control reactions performed without catalyst 1 or under dark conditions exhibited no observable CO2 reduction, emphasizing the pivotal role of light in the reaction. A minute yield of CO (Table 1) was observed in a control reaction with no triethylamine but with 5% H2O. This is probably due to the potential of H2O to act as a sacrificial donor, as seen in earlier reports.46,47 Furthermore, no HCOOH formation was observed when solvent-phase products were checked (Fig. S47†). Interestingly, irradiation at 426 nm (visible range) yielded a significant CO yield (Table S11†). Despite the yield being lower than that observed under 390 nm light, complex 1 is capable of activating CO2 under visible light. This corresponds with what is expected from the fact that the excitation peak maximum (Fig. 3b) is closer to 390 nm than to 426 nm. To suit the needs of the day, we carried out our reactions in direct sunlight (11 am–3 pm, 11 Nov 2023, [22.9638°N, 88.5245°E], IISER Kolkata) and under a solar simulator (Xe lamp: 400 nm ≥ λ ≥ 750 nm) and got TONs of 920 and 929, respectively (Table S11†). Dynamic Light Scattering (DLS) studies confirmed the homogeneity of the reaction mixture post-reaction (Fig. S37†). Using ferric oxalate actinometry (λ = 450 nm), the quantum yield (ϕCO) for CO production under optimal conditions was determined to be 2.33%. The rate of CO production demonstrated a direct correlation with the increasing catalyst concentration, indicating the involvement of the Cu complex in the rate-limiting step for CO2 reduction, likely via a single Cu site (Fig. 4a). Additionally, the proton concentration played a secondary role, potentially establishing the effects of a proton relay due to a coordination switch by means of a proton channel formed by the de-coordinated N–H of the amide. This was supported by a classical secondary kinetic isotope effect observed and validated through comparative studies using D2O (kH/kD = 1.66) (Fig. S38†). Comparison of the UV-vis spectra pre- and post-reaction confirmed the preserved integrity of the catalyst during the reaction cycle (Fig. S39†) and the fact that under the reaction conditions, the active catalyst is predominantly complex 1. This is also complemented by similar data from the spectro-electrochemical and cyclic voltammetric studies for complex 1 under reaction conditions (95:5 CH3CN:H2O v/v, 70 mM TEA) and in pure CH3CN (Fig. S33–S35†). Higher concentrations (>95 mM) of triethylamine (Et3N) resulted in decreased CO production, presumably due to competitive binding between CO2 and Et3N to the Cu complex. The TONCO dropped to 33 without Et3N, with a turnover frequency (TOF) of 16 h−1. Furthermore, substantial CO production was evident in subsequent catalytic cycles, highlighting the catalytic stability and efficacy of complex 1 (Fig. 4b).
Table 1 Control and other photocatalytic CO2 reduction experiments
| Entry |
1 (μM) |
[TEA] (mM) |
[H2O] (% v/v) |
Irradiation time (h) |
CO (μmol) |
H2 (μmol) |
TONCO |
TOFCO (h−1) |
TONH2 |
Selectivity for CO (%) : H2 (%) |
| 1 |
100 |
0 |
5 |
2 |
10.1 |
1.9 |
34 |
17 |
6 |
81:19 |
| 2 |
100 |
70 |
0 |
2 |
76.8 |
2.5 |
256 |
128 |
8 |
97:3 |
| 3 |
100 |
70 |
5 |
2 |
339.7 |
2.2 |
1132 |
566 |
7 |
99:1 |
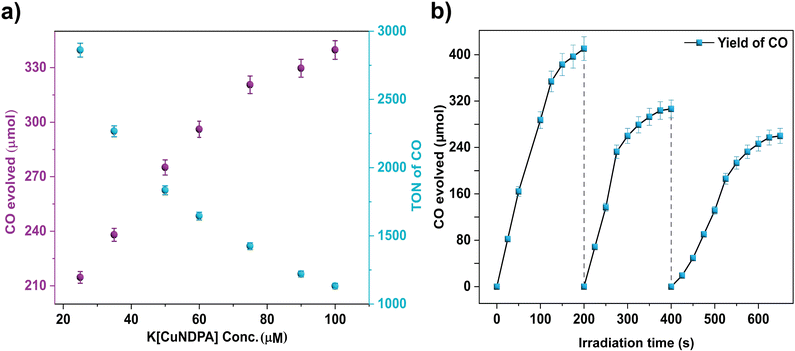 |
| | Fig. 4 (a) Photocatalytic CO2 reduction [TON (cyan) and amounts (purple)] of CO irradiated for 10 h in CO2-saturated acetonitrile solution containing varying amounts of 1, 70 mM Et3N and 1% H2O. (b) Recycling tests of 1 for 10 h in 3 cycles as indicated above. For each cycle, 70 mM of TEA and 1% H2O were freshly added to the existing reaction mixture with the light source of 390 nm lamp. | |
Mechanistic studies of the catalytic cycle
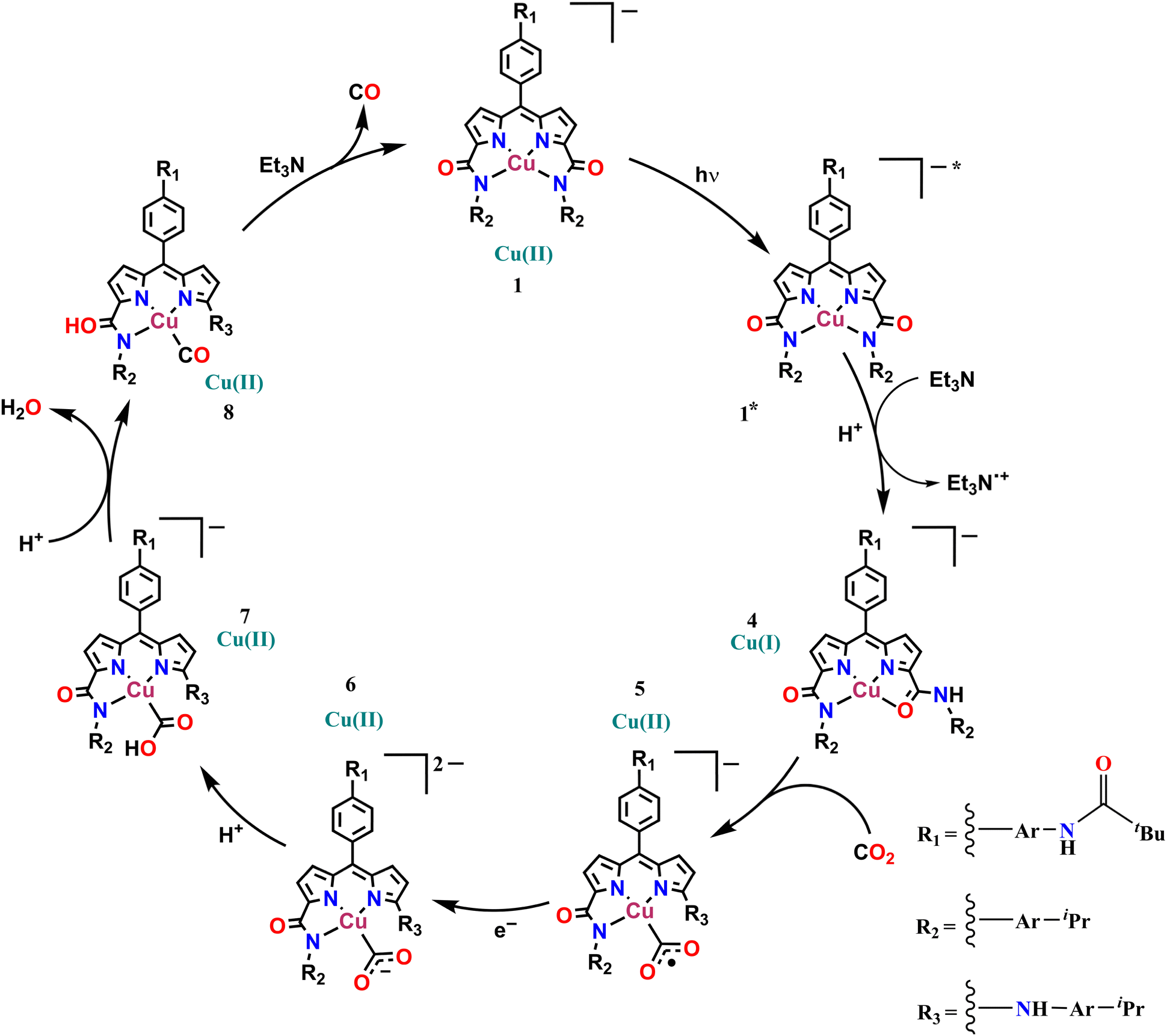
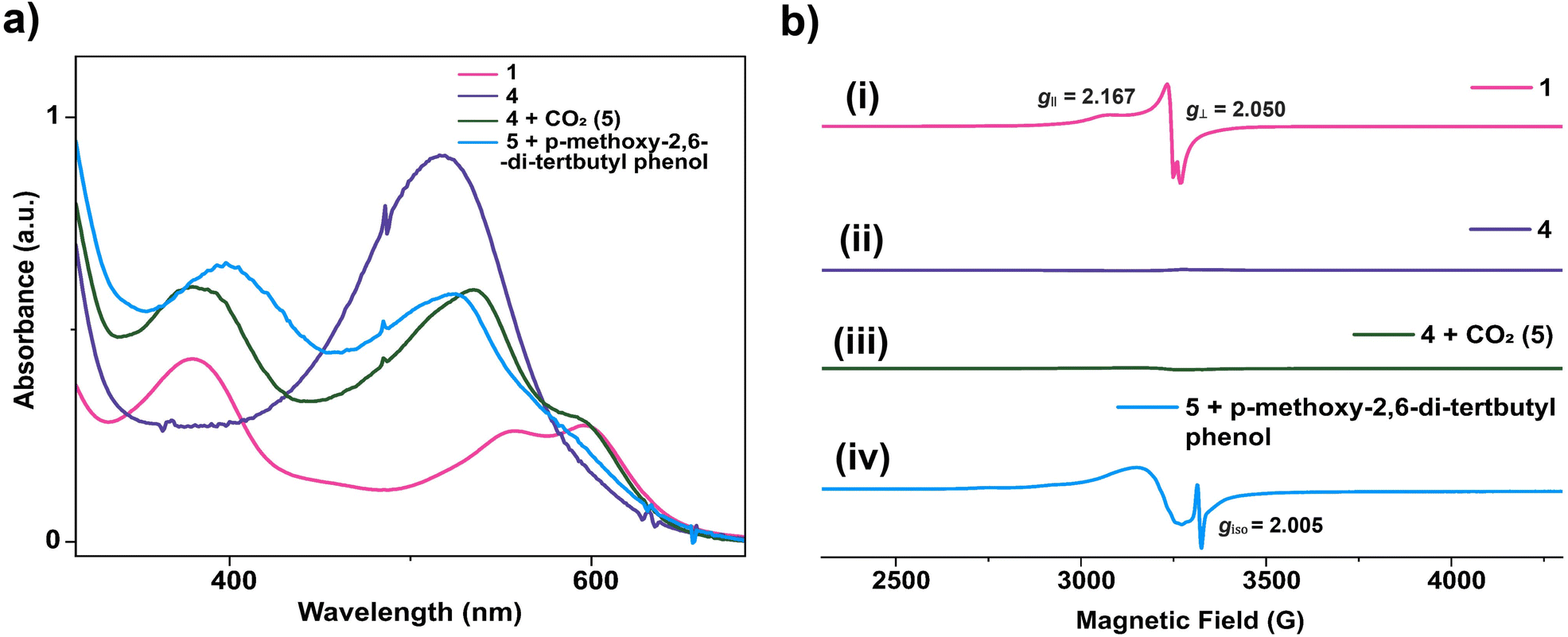
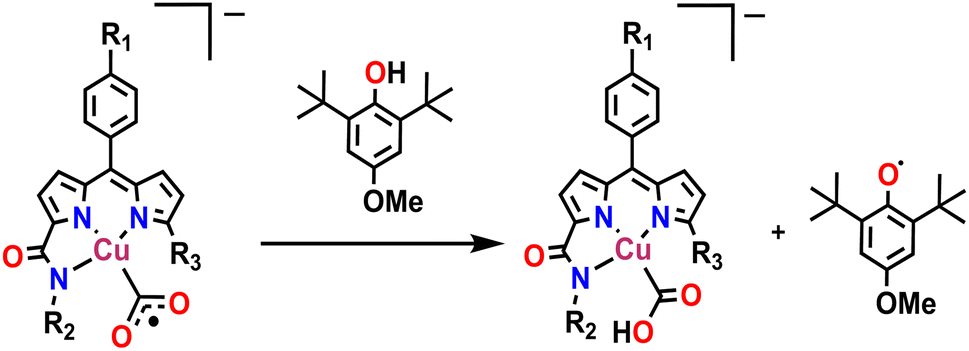
Possibly, upon shining light, a ligand-centred electron gets excited and transferred to the metal centre, which reduces the Cu(II) to Cu(I). Ishitani et al. proposed a similar electron transfer between a photosensitizer and photocatalyst in a bimetallic Ru–Re supramolecular complex for CO2 reduction.10 The possibility of a coordination switch in the Cu(I) species from N4 to N3O is considered due to the electronic and geometric preferences of N3O for Cu(I), as discussed earlier. Thus, the single electron reduced species 4 is formed (Scheme 1). In the next step, CO2 binds to the Cu(I) centre by substituting the weakly bound amide-O. This is supported by cyclic voltammetry studies, where, under CO2-saturated conditions, the emergence of a catalytic wave is observed, suggesting the involvement of the reduced species in CO2 reduction (Fig. S42†). A similar cyclic voltammogram of 1 under the catalytic conditions indicates the presence of 1 in the reaction mixture (Fig. S33†). Similarly, in the reaction cycle, the Cu(I) complex transfers a single electron to CO2 to form a Cu(II)–COO·− species (5). To support this single electron transfer step, we purged a solution of 4 (generated as described earlier) with CO2. The reappearance of the 378 nm LMCT transition confirms metal centre oxidation from Cu(I) to Cu(II) and the likely formation of Cu(II)–COO·− (5) due to single electron transfer (SET) from Cu(I) to CO2 (Fig. 5a). The EPR spectrum was also silent after CO2 addition to Cu(I), signifying the SET and formation of 5 (a Cu(II)-coordinated radical species). However, upon adding 1 eq. of p-methoxy 2,6-di-tert-butylphenol to this solution, a distinctive Cu(II) axial EPR signal (g⊥ = 2.062 and gǁ = 2.312), coupled with a sharp phenoxyl radical peak (giso = 2.005), had emerged (Fig. 5b). In UV-vis spectra, the sharp peak at ∼405 nm48 for the phenoxyl radical could not be separated as the broad 378 LMCT signal obscured it, but a 25 nm red shift was noticed (Fig. 5a). Additionally, a peak at 1632 cm−1 was observed via infrared spectroscopic analysis of the above solution (Fig. S43†), which corresponds to a previously reported adduct between a Cu centre and CO2.49 This confirms that species 5 was competent to abstract the H-atom from p-methoxy 2,6-di-tert-butylphenol and indirectly supports our proposition for the formation of Cu(II)–COO·− (5) and subsequent formation of 7. Similarly, in the catalytic cycle (Scheme 1), the Cu(II)–COO·− species then further undergoes a PCET process to form Cu(II)–COOH (7). Here, the second electron transfer could be possible from different sources, as discussed by both Masaoka & co-workers and the Saito group.14 Upon taking these deductions into account, we would like to propose the one electron photo-reduced species of our complex (4) as the likely source. In the final step, CO is released, and Et3N assists in the regeneration of complex 1 (Scheme 2).
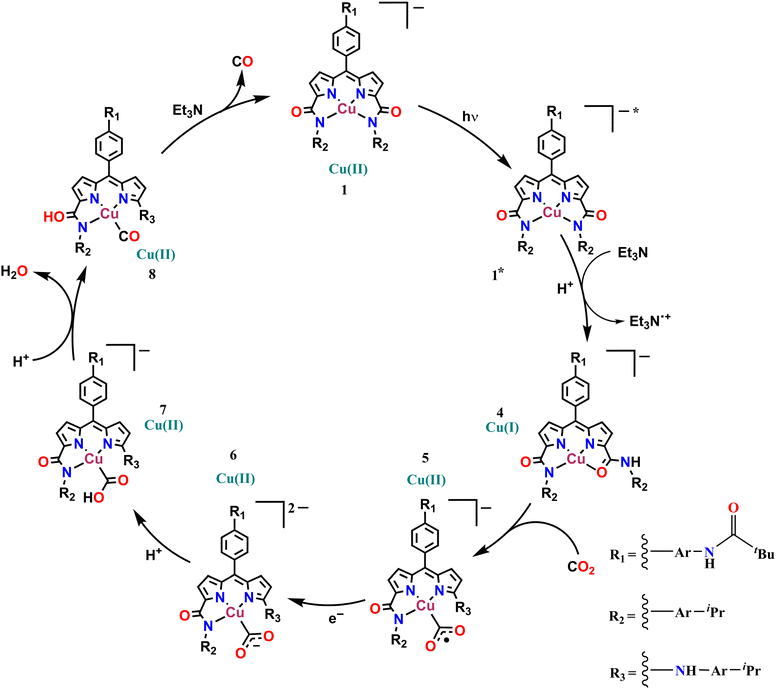 |
| | Scheme 1 The proposed catalytic mechanism for photocatalytic CO2 reduction by 1. | |
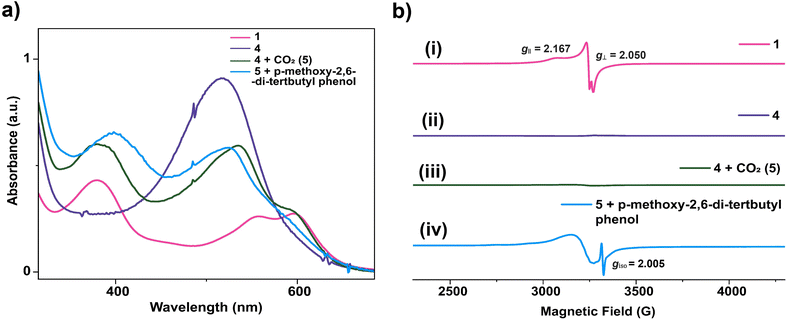 |
| | Fig. 5 (a) UV-vis experiment of complex 1 after reduction and reaction with CO2. (b) X-band (100 K) EPR experiment for the proposed intermediate involved in the photocatalytic CO2 reduction by 1 in acetonitrile. (i) EPR spectra of 1 (pink), (ii) EPR spectra of [CuNDPA]2− (4) (violet), (iii) EPR spectra after CO2 purging into the solution of 4 (5) (green), and (iv) EPR spectra after addition of p-methoxy-2,6-di-tert-butylphenol into the solution of 5 (blue). | |
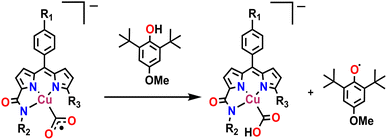 |
| | Scheme 2 Trapping the radical intermediate by HAT. | |
Before concluding, one important point is worth noting. When the reaction was carried out in CH3CN:H2O (95:5, v/v), conditions under which complex 2 is generated from complex 1, CO2 reduction to CO with a very low TONCO of 34 was observed (compared to TONCO = 1132 when TEA was present). Hence, there is a possibility that complex 2 might also catalyze the reduction of CO2 to CO, albeit with much less efficiency. The detailed photophysical study of complex 2 and its potential to reduce CO2 to CO is being explored in our laboratory.
Conclusion
This study showcases a new class of dipyrrin–dicarboxamide ligands (NDPA) formed by modification of the previously reported DADP ligand. This modification helps the NDPA ligand to form a unique N4-coordinated Cu(II) complex. The rational incorporation of the dipyrrin moieties into the NDPA ligand framework enables complex 1 to show light-harvesting capabilities with a lifetime of 4.36 ns, as evidenced by photophysical studies, highlighting the pivotal role of the metal centre. The NDPA ligand further supports two additional coordination modes with Cu(II), namely N3O and N2O2. All three coordinations (N4, N3O, and N2O2) have been proven to show both pH- and redox-dependent hemilability studied using XRD, UV-vis, EPR, and HRMS. The observed hemilability helps in the one-electron reduction of 1 as the reduced Cu(I) switches to an N3O coordination. It subsequently helps in substrate binding via a labile neutral O coordination in the reduced state. The reactivity towards CO2 under photo-irradiation results in the exclusive formation of CO with a TON of 1132. The mechanistic cycle was probed via UV-vis, EPR, spectroelectrochemistry, and IR spectroscopy, culminating in the proposal of a Cu(II)–COO·− as a reactive intermediate. Water serves as a proton source in the reaction and shows a secondary kinetic isotope effect, implicating a proton shuttle mechanism facilitated by the amide groups due to the coordination switch, potentially contributing to an increased reaction rate. The impressive TON achieved with this complex surpasses the efficiencies of previously reported Fe-p-TMA and Cu purpurin complexes.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†
Author contributions
S. D. and S. S. G. conceptualized the work. All the synthesis and characterization were done by A. R., N. C., and S. D. Theoretical calculation was done by S. D. S. D., N. C., and A. S. performed the photophysical experiments. N. C. and S. D. performed all the CO2 reduction experiments. S. D., N. C., and N. M. performed the intermediate trapping experiment. The manuscript was written by S. D., N. C., and S. S. G. The overall work was supervised by S. S. G. with valuable suggestions and ideas. All authors have approved the final version of the article before submission.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. D. and A. S. thank CSIR, New Delhi, for giving fellowship assistance. N. C. acknowledges INSPIRE for the scholarship. We are very thankful to Dr Noufal Kandoth, Prof. Jyotishman Dasgupta, Prof. Prasun Kumar Mandal, and Prof. Amitava Das for their photophysical inputs. We would also like to acknowledge Dr Dibyendu Das for allowing us to avail of his HPLC instrumental facility.
References
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- B. Mondal, J. Song, F. Neese and S. Ye, Curr. Opin. Chem. Biol., 2015, 25, 103–109 CrossRef CAS PubMed.
- G. Fuchs, Annu. Rev. Microbiol., 2011, 65, 631–658 CrossRef CAS PubMed.
- Q.-J. Wu, D.-H. Si, S. Ye, Y.-L. Dong, R. Cao and Y.-B. Huang, J. Am. Chem. Soc., 2023, 145, 19856–19865 CrossRef CAS PubMed.
- A. Rosas-Hernández, H. Junge and M. Beller, ChemCatChem, 2015, 7, 3316–3321 CrossRef.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
- K. Kosugi, C. Akatsuka, H. Iwami, M. Kondo and S. Masaoka, J. Am. Chem. Soc., 2023, 145, 10451–10457 CrossRef CAS PubMed.
- T. Ishizuka, A. Hosokawa, T. Kawanishi, H. Kotani, Y. Zhi and T. Kojima, J. Am. Chem. Soc., 2023, 145, 23196–23204 CrossRef CAS PubMed.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- S. K. Lee, M. Kondo, M. Okamura, T. Enomoto, G. Nakamura and S. Masaoka, J. Am. Chem. Soc., 2018, 140, 16899–16903 CrossRef CAS PubMed.
- S. Das, R. R. Rodrigues, R. W. Lamb, F. Qu, E. Reinheimer, C. M. Boudreaux, C. E. Webster, J. H. Delcamp and E. T. Papish, Inorg. Chem., 2019, 58, 8012–8020 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- S. Y. Manafe, N. Le, E. C. Lambert, C. Curiac, D. Nugegoda, S. Das, L. A. Hunt, F. Qu, L. M. Whitt, I. Fedin, N. I. Hammer, C. E. Webster, J. H. Delcamp and E. T. Papish, ACS Catal., 2024, 14, 6589–6602 CrossRef CAS.
- K. Kamada, J. Jung, C. Yamada, T. Wakabayashi, K. Sekizawa, S. Sato, T. Morikawa, S. Fukuzumi and S. Saito, Angew. Chem., Int. Ed., 2024, 63, e202403886 CrossRef CAS PubMed.
- T. Wakabayashi, Y. Kametani, E. Tanahashi, Y. Shiota, K. Yoshizawa, J. Jung and S. Saito, J. Am. Chem. Soc., 2024, 146, 25963–25975 Search PubMed.
- H. Rao, J. Bonin and M. Robert, Chem. Commun., 2017, 53, 2830–2833 RSC.
- J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 6, 3200–3207 CrossRef CAS.
- H. Yuan, J. Du, M. Ming, Y. Chen, L. Jiang and Z. Han, J. Am. Chem. Soc., 2022, 144, 4305–4309 CrossRef CAS PubMed.
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Nat. Commun., 2021, 12, 1835 CrossRef CAS PubMed.
- C. Bruschi, X. Gui, P. Rauthe, O. Fuhr, A. Unterreiner, W. Klopper and C. Bizzarri, Chem.–Eur. J., 2024, 30, e202400765 CrossRef CAS PubMed.
- J.-W. Wang, Z. Li, Z.-M. Luo, Y. Huang, F. Ma, S. Kupfer and G. Ouyang, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2221219120 CrossRef CAS PubMed.
- X. Feng, Y. Pi, Y. Song, C. Brzezinski, Z. Xu, Z. Li and W. Lin, J. Am. Chem. Soc., 2020, 142, 690–695 CrossRef CAS PubMed.
- N. Liu, S. Bartling, A. Springer, C. Kubis, O. S. Bokareva, E. Salaya, J. Sun, Z. Zhang, S. Wohlrab, A. M. Abdel-Mageed, H. Liang and R. Francke, Adv. Mater., 2024, 36, 2309526 CrossRef CAS PubMed.
- P. J. DeLaive, T. K. Foreman, C. Giannotti and D. G. Whitten, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- W. Liu, H. Huang, T. Ouyang, L. Jiang, D. Zhong, W. Zhang and T. Lu, Chem.–Eur. J., 2018, 24, 4503–4508 CrossRef CAS PubMed.
- C. Panda, A. Sarkar and S. Sen Gupta, Coord. Chem. Rev., 2020, 417, 213314 CrossRef CAS.
- C. Panda, M. Ghosh, T. Panda, R. Banerjee and S. S. Gupta, Chem. Commun., 2011, 47, 8016–8018 RSC.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- P. Garrido-Barros, D. Moonshiram, M. Gil-Sepulcre, P. Pelosin, C. Gimbert-Suriñach, J. Benet-Buchholz and A. Llobet, J. Am. Chem. Soc., 2020, 142, 17434–17446 CrossRef CAS PubMed.
- A. B. Scharf, S.-L. Zheng and T. A. Betley, Dalton Trans., 2021, 50, 6418–6422 RSC.
- R. Kanega, M. Z. Ertem, N. Onishi, D. J. Szalda, E. Fujita and Y. Himeda, Organometallics, 2020, 39, 1519–1531 CrossRef CAS.
- V. S. Thoi, J. R. Stork, E. T. Niles, E. C. Depperman, D. L. Tierney and S. M. Cohen, Inorg. Chem., 2008, 47, 10533–10541 CrossRef CAS PubMed.
- I. Verma, N. Mukhopadhyay, A. Sengupta and R. Mukherjee, J. Organomet. Chem., 2021, 956, 122121 CrossRef CAS.
- M. Ray, R. Mukherjee, J. F. Richardson, M. S. Mashuta and R. M. Buchanan, J.
Chem. Soc., Dalton Trans., 1994, 965–969 RSC.
- R. Mukherjee, Inorg. Chem., 2020, 59, 12961–12977 CrossRef CAS PubMed.
- S. S. Saund, S. L. Goldschmid, K. Ng, V. Stewart, M. A. Siegler and V. Sara Thoi, Chem. Commun., 2019, 55, 1825–1828 RSC.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- L. Rostami and H. Golchoubian, Inorg. Chim. Acta, 2017, 462, 215–222 CrossRef CAS.
- M. Dorn, J. Kalmbach, P. Boden, A. Päpcke, S. Gómez, C. Förster, F. Kuczelinis, L. M. Carrella, L. A. Büldt, N. H. Bings, E. Rentschler, S. Lochbrunner, L. González, M. Gerhards, M. Seitz and K. Heinze, J. Am. Chem. Soc., 2020, 142, 7947–7955 CrossRef CAS PubMed.
- J. C. del Valle and J. Catalán, Phys. Chem. Chem. Phys., 2019, 21, 10061–10069 RSC.
- Z. Guo, F. Yu, Y. Yang, C.-F. Leung, S.-M. Ng, C.-C. Ko, C. Cometto, T.-C. Lau and M. Robert, ChemSusChem, 2017, 10, 4009–4013 CrossRef CAS PubMed.
- M. Mifsud, S. Gargiulo, S. Iborra, I. W. C. E. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 3145 CrossRef PubMed.
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- A. Sarkar, S. Das, P. Mondal, B. Maiti and S. Sen Gupta, Inorg. Chem., 2023, 62, 20439–20449 Search PubMed.
- J. Bharti, L. Chen, Z. Guo, L. Cheng, J. Wellauer, O. S. Wenger, N. von Wolff, K.-C. Lau, T.-C. Lau, G. Chen and M. Robert, J. Am. Chem. Soc., 2023, 145, 25195–25202 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Aritra
Roy
,
Aritra
Roy