
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand protonation leads to highly fluorescent boronium cations†
Alexander E. R.
Watson
a,
Paul D.
Boyle
a,
Paul J.
Ragogna
 *ab and
Joe B.
Gilroy
*a
*ab and
Joe B.
Gilroy
*a
aDepartment of Chemistry, The University of Western Ontario, London, Ontario N6A 5B7, Canada. E-mail: pragogna@uwo.ca; joe.gilroy@uwo.ca
bSurface Science Western, London, Ontario N6G 0J3, Canada
First published on 26th December 2024
Abstract
Fluorophores that respond to external stimuli, such as changes in pH, have utility in bio-imaging and sensing applications. Almost all pH-responsive fluorophores rely on complex syntheses and the use of pH-responsive functional groups that are peripheral to the fluorophore framework. In this work, pH-responsive boron-containing heterocycles based on tridentate acyl pyridylhydrazone ligands were prepared. These non-emissive heterocycles were synthesized in three steps from inexpensive, commercially available reagents without the use of chromatography or air-sensitive reagents. Treatment with acid resulted in protonation of the boron-bound methylamine donor and efficient blue photoluminescence. Experimental and computational analysis revealed that protonation changed the geometric structure of the heterocycles and prevented photoluminescence quenching associated with photoinduced electron transfer. This work demonstrates a new approach for the design of fluorophores with potential applications in biological imaging.
Introduction
The ability of a molecule to respond to external stimuli, such as light, mechanical force, temperature, and pH, is desirable for the design of functional materials.1 This molecular change can be coupled to a macro-scale outcome, such as alterations in a material's morphology, colour, or photoluminescence, affording stimuli-responsiveness. Molecules that respond to changes in pH are well-known and relevant in the context of colorimetric pH-sensing and fluorescent probes for biological imaging.2–12 Boron-based fluorophores that are amenable to derivatization, such as boron dipyrromethenes (BODIPYs),13 are typically constructed with discrete fluorophores and peripheral pH-responsive functional groups to act as fluorescent pH sensors (Fig. 1).14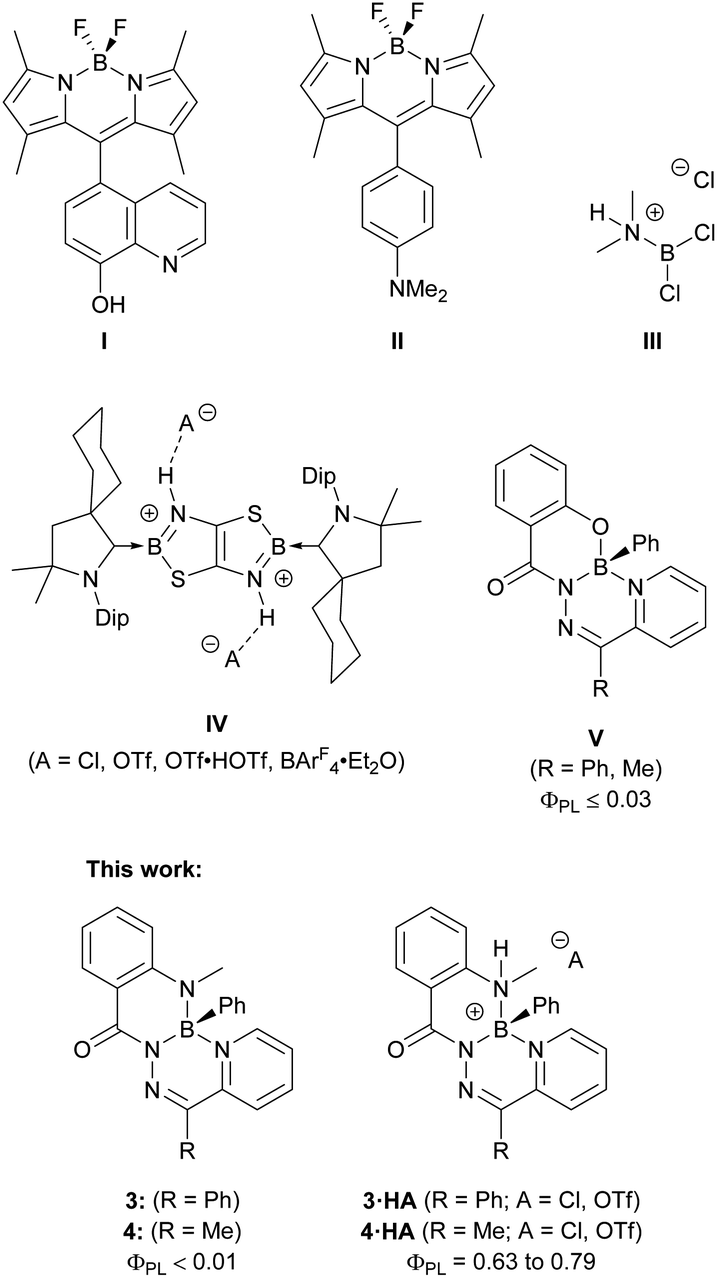 | ||
| Fig. 1 Chemical structures of pH-responsive fluorophores, boronium cations formed by treatment with a Brønsted acid, and boron complexes of tridentate hydrazone ligands. | ||
Protonation or deprotonation of the appended 6-hydroxyquinoline substituent in compound I quenched the fluorescence of the BODIPY chromophore.15 This quenching involved photo-induced electron transfer (PET), where protonation or deprotonation changed the energetics associated with the peripheral 6-hydroxyquinoline substituent to allow for electron transfer that quenched the excited state of the BODIPY chromophore. Compound II used a BODIPY chromophore to achieve the opposite response: the neutral compound was non-emissive due to PET quenching from the lone pair of the dimethylamino-substituent.16 This electron transfer was prevented upon protonation of the dimethylamino-group, and bright fluorescence was observed. Fluorescent pH-responsive probes based on boron heterocycles, such as these substituted BODIPYS, are numerous and are specifically constructed such that a change in protonation of a peripheral substituent alters the electronic properties of the fluorophore.17–24
Protonation at atoms bound to a boron centre, as opposed to an auxiliary substituent, modifies the coordination environment and potentially the charge of the boron centre, which can directly alter the electronic and structural properties of the fluorophore. The protonation of aminoboranes is one of the oldest methods for the generation of boron cations.25,26 Wiberg and Schuster treated dichlorodimethylaminoborane with HCl to protonate the dimethylamine ligand and generate borenium cation III.27 This approach to boron cation formation remains viable today, as demonstrated by the CBS catalyst, which can be protonated by triflic acid to generate a borenium cation as the active species.28,29 Analogous transformations to a fluorophore to alter its photoluminescence have been uncommon, though significant changes to the electronic structure of the fluorophore can be imposed.30,31 Braunschweig and colleagues synthesized boron-doped thiazolothiazoles (IV) and reported unique responses to changes in pH.31 While the neutral starting material was non-emissive, treatment with acid protonated both nitrogen atoms bound to the boron centres and enabled photoluminescence that could be altered based on the nature of the hydrogen-bonding interaction of the dicationic heterocycle with different anions.
Previous work on boron complexes of tridentate N-acyl pyridylhydrazones (V) with an N,N′,O coordination mode resulted in poorly emissive compounds with photoluminescence quantum yields no higher than 0.03.32 In this work, non-emissive boron heterocycles based on π-conjugated N-acyl pyridylhydrazones with N,N′,N′′ coordination modes were synthesized. When treated with a Brønsted acid, strongly fluorescent boronium cations were formed. These cations were isolated and their photophysical properties were thoroughly investigated. The origins of the substantial change in photoluminescence resulting from such a simple chemical change were explored by experimental and computational means. The described compounds highlight the dramatic change in electronic structure and photophysical properties that can result from a simple protonation reaction at a site that is integrated into, instead of peripheral to, the fluorophore core.
Results and discussion
2-(Methylamino)benzohydrazide was prepared according to a published procedure33 and treated with the requisite 2-pyridylketone to afford N-acylhydrazones 1 and 2 (Fig. S1–S8†). Compounds 1 and 2 were combined with phenylboronic acid to yield boron heterocycles 3 and 4 in an 85% and 81% yield, respectively (Fig. S9–S14†). The synthesis of 3 and 4 was accompanied by a disappearance of two broad NH resonances that were observed in the 1H NMR spectra of the respective hydrazone ligands (1: 14.91 ppm and 8.81 ppm; 2: 15.62 and 9.01 ppm). A sharp resonance was observed in the 11B NMR spectra of 3 (3.5 ppm) and 4 (3.4 ppm). Both observations were consistent with the incorporation of a tetracoordinate boron centre into the ligand binding pocket.Heterocycles 3 and 4 were observed to be non-emissive in solution and in the solid-state, similar to the structurally similar compounds V.32 However, when CH2Cl2 solutions of 3 and 4 were irradiated with a handheld UV light (254 nm), bright blue photoluminescence, which increased in intensity over time, was observed by eye. Closer study of this phenomenon revealed that photoluminescence was turned-on through irradiation in halogenated solvents CH2Cl2 and CHCl3, but irradiation in non-halogenated solvents, such as CH3CN, did not result in photoluminescence (Fig. S30–S32†). Irradiation of neat CH2Cl2 or CHCl3, followed by addition of compound 3 produced identical photoluminescence to previous irradiation experiments, and ruled out the possibility of a photochemical reaction of 3 leading to photoluminescence (Fig. S33 and S34†). Halogenated solvents like CH2Cl2 and especially CHCl3 are known to produce HCl when irradiated with UV light.34–36 It was observed that a Brønsted acid was needed to turn-on photoluminescence when the addition of anhydrous HCl to dilute solutions of 3 and 4 resulted in photoluminescence at the same wavelength as the irradiated solutions (Fig. S35†). The addition of NEt3 quenched this photoluminescence. Other acids, including H2SO4, HNO3, HBr, and trifluoroacetic acid, also resulted in photoluminescence at the same wavelength as the addition of HCl (Fig. S36†).
The observation of bright photoluminescence prompted us to investigate the structure of the highly emissive species produced in the presence of Brønsted acids. The addition of anhydrous HCl in dioxane to a solution of 3 or 4 under an inert atmosphere afforded compounds 3·HCl and 4·HCl in near quantitative yields (Scheme 1 and Fig. S15–S20†). A resonance in the 1H NMR spectrum of heterocycle 3, associated with the methylamine functional group, changed from a singlet (2.96 ppm) to a doublet (3.06 ppm, 3JHH = 4.6 Hz) upon formation of compound 3·HCl, and an analogous observation was made upon formation of compound 4·HCl. The signal in the 11B NMR spectrum of both 3·HCl and 4·HCl shifted downfield (11B: 3·HCl: 3.2 ppm; 4·HCl: 2.9 ppm) relative to the neutral heterocycles 3 and 4. Both observations were consistent with protonation at the methylamine donor to form a tetracoordinate boronium cation.
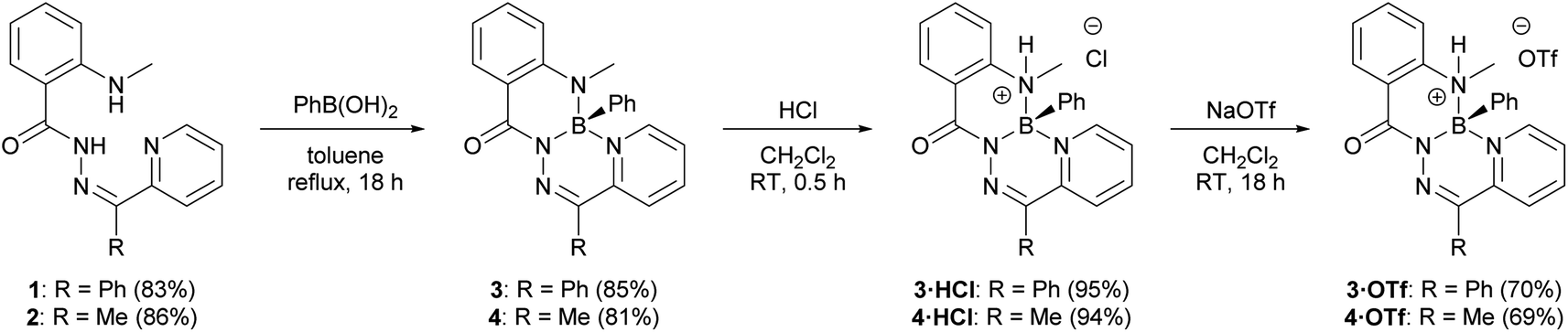 | ||
| Scheme 1 Preparation of boron heterocycles 3 and 4 by treatment with phenylboronic acid. Compounds 3·HCl and 4·HCl were afforded by treatment with dry HCl, followed by metathesis with NaOTf to afford compounds 3·HOTf and 4·HOTf. | ||
Compounds 3·HCl and 4·HCl were amenable to anion metathesis with NaOTf to afford compounds 3·HOTf and 4·HOTf (Scheme 1 and Fig. S21–S28†). Attempts at similar metatheses with other weakly-coordinating anion sources (i.e., NaPF6, NaBF4, K[B(C6F5)4]) resulted in the formation of several inseparable species, as observed by multinuclear NMR spectroscopy.
Compounds 3·HCl and 4·HCl were observed to exist in an equilibrium with their neutral starting materials 3 or 4. When a solution of compound 3·HCl was placed under vacuum at room temperature and monitored by 1H NMR spectroscopy, starting material 3 reformed over the course of several hours (Fig. 2). Similar observations were made for 3·HOTf and 4·HOTf, although the triflate salts reverted much more slowly than their chloride counterparts. The isolation of these compounds had to be performed through trituration with pentane and filtration to avoid placing the compounds under high vacuum.
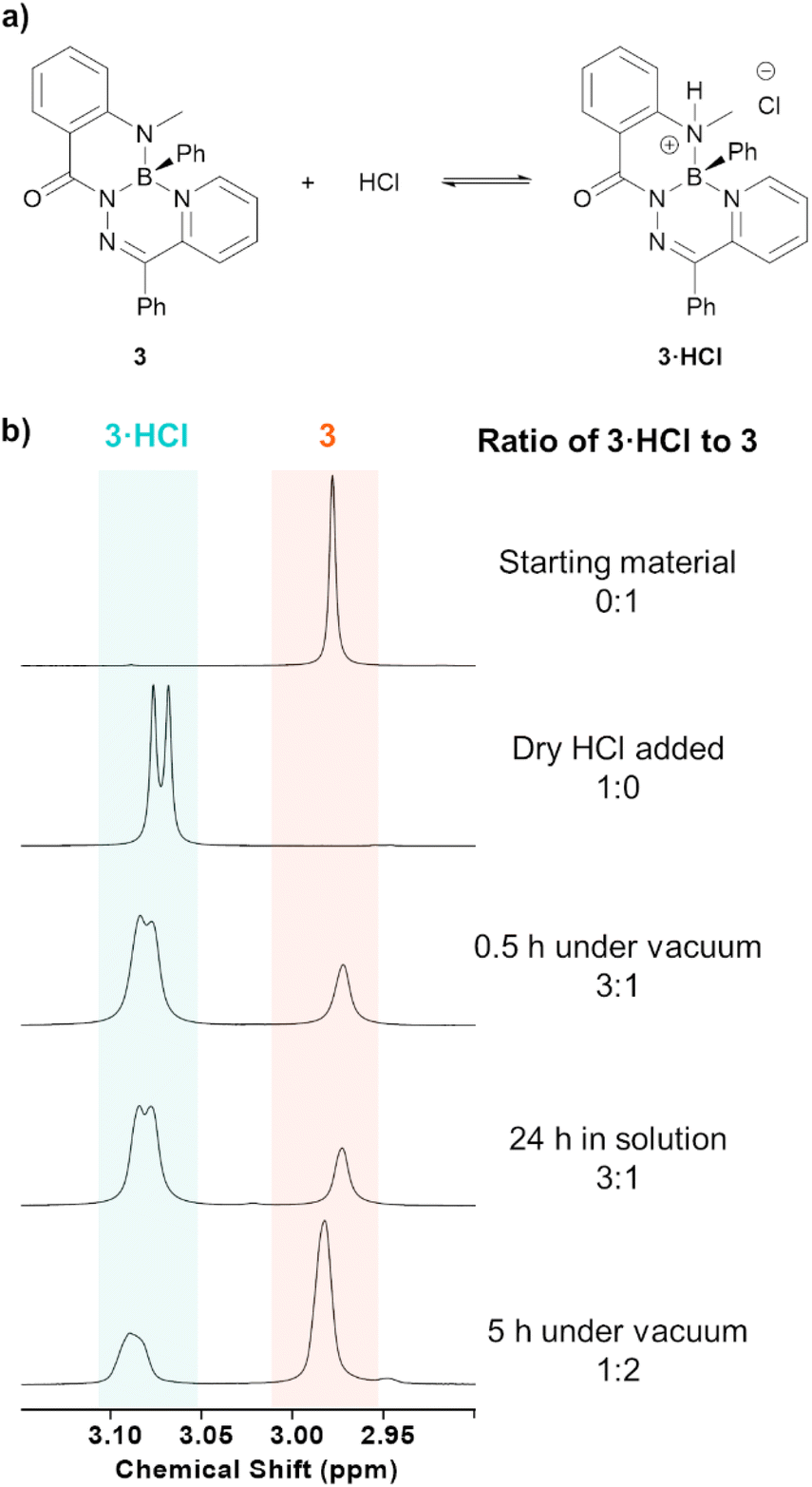 | ||
| Fig. 2 (a) Equilibrium of neutral heterocycle 3 with compound 3·HCl upon treatment with HCl. (b) 1H NMR spectra (600 MHz, CDCl3) of the N–CH3 protons of the above equilibrium before and after treatment with HCl, as well as the effects of having the reaction mixture under high vacuum. | ||
The structures of compounds 3 and 3·HCl were confirmed by single crystal X-ray diffraction (Fig. 3 and Table S1†). Both structures featured a tetrahedral boron centre chelated in an N,N′,N′′ fashion to the N-acylhydrazone ligand. The planarity of these compounds was quantified by comparing the dihedral angle between the planes defined by selected pyridyl (coloured in blue) and phenylene (coloured in red) ring systems. Compound 3 was saddle-shaped, and the dihedral angle of the phenylene and pyridyl rings was calculated to be 77.45(5)°. The salt 3·HCl had a substantially reduced phenylene-pyridyl dihedral angle of 8.26(6)°, exemplifying its more planar structure. Compound 3·HCl was observed to have a 0.173(1) Å displacement of its boron atom from the 6-membered hydrazone ring (C1–N2–N1–B1–N3–C2), compared to a value of 0.363(1) Å for the neutral compound 3. This represented a significant alteration in the geometry of the π-system of the compounds following protonation of the methylamine nitrogen. Compared to neutral compound 3, the solid-state structure of compound 3·HCl saw a lengthening of the methylamine bond to boron (B1–N4 3: 1.497(2) Å, 3·HCl: 1.593(2) Å) and a contraction of the boron–pyridyl nitrogen bond (B1–N3 3: 1.639(2) Å, 3·HCl: 1.587(2) Å). The boron centre's bonds to the hydrazone and phenyl substituent were not appreciably changed upon protonation (B1–N1 3: 1.532(1) Å, 3·HCl: 1.533(2) Å; B1–C20 3: 1.611(2) Å, 3·HCl: 1.609(2) Å).
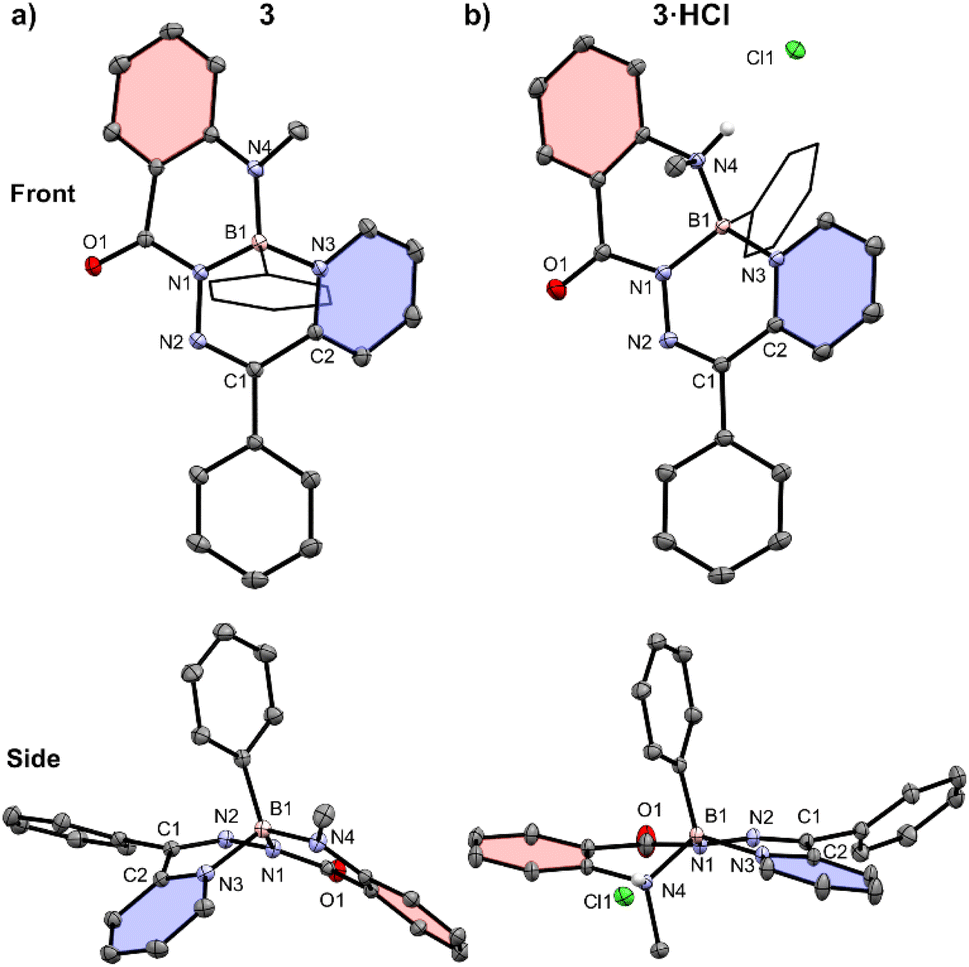 | ||
| Fig. 3 Front and side views of the solid-state structures of (a) compound 3 and (b) compound 3·HCl. Solvent molecules and hydrogen atoms, except those bound to heteroatoms, are omitted for clarity. Anisotropic displacement ellipsoids are displayed at a 50% probability level. Select phenyl substituents are shown in wireframe. A pyridyl ring (C2–N3–C3–C4–C5–C6) of each structure is coloured blue, and a phenylene ring (C14–C15–C16–C17–C18–C19) is coloured in red. | ||
The synthesized neutral and cationic heterocycles were subjected to cyclic voltammetry (Fig. S40†). Neutral heterocycles 3 and 4 both featured a reversible reduction at similar voltages (Ered3: −1.26 V; 4: −1.23 V, relative to the Fc/Fc+ redox couple) and several irreversible oxidations. Compounds 3·HCl, 3·HOTf, 4·HCl, and 4·HOTf exhibited several new irreversible reductions and the loss of reversibility for the reduction at −1.26 or −1.23 V. Similar irreversible oxidations events to the parent neutral compounds were also observed.
The electronic structures of the reported boron heterocycles were investigated by UV-vis spectroscopy (Fig. 4 and Table 1). Compounds 3 and 4 possessed similar absorption spectra in CH2Cl2, with low-energy absorption maxima (3: 467 nm; 4: 451 nm) that were more weakly absorbing than high-energy maxima at ∼380 nm (3: 385 nm; 4: 368 nm). Changing the carbon-bound substituent on the heterocycle from a phenyl ring (3) to a methyl group (4) resulted in a slight hypsochromic shift. Neither compound was observed to exhibit appreciable fluorescence in solution or in the solid state. This was unexpected, as boron difluoride hydrazones (BODIHYs) and boron complexes of structurally similar N,N′,O chelated acyl pyridylhydrazones are fluorescent, especially in the solid state.32,37–44
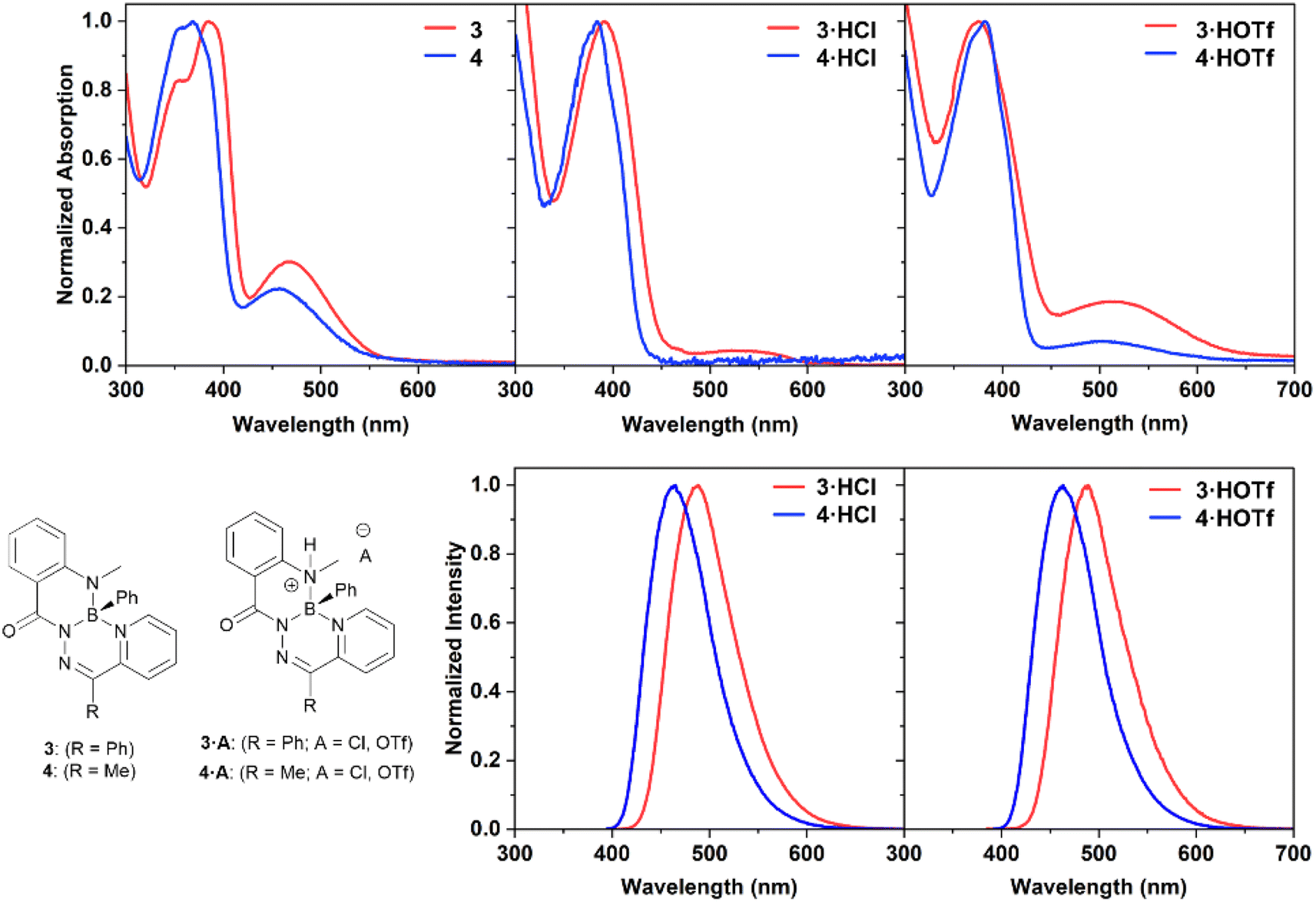 | ||
| Fig. 4 Normalized UV-vis absorption and photoluminescence spectra of compounds 3, 3·HCl, 3·HOTf, 4, 4·HCl, and 4·HOTf as 10 μM solutions in dry, degassed CH2Cl2. | ||
| Compound | λ max (nm) | ε (M−1 cm−1) | λ PL (nm) | Φ PL |
|---|---|---|---|---|
| a Compounds 3·HCl and 4·HCl were analyzed in acidic CH2Cl2 solutions with one molar equivalent of HCl added. | ||||
| 3 | 467 | 6500 | — | — |
| 385 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
— | — | |
| 3·HCl | 392 | 8400 | 488 | 0.71 |
| 3·HOTf | 510 | 1100 | — | — |
| 375 | 5900 | 489 | 0.63 | |
| 4 | 451 | 2200 | — | — |
| 368 | 7800 | — | — | |
| 4·HCl | 384 | 4700 | 465 | 0.79 |
| 4·HOTf | 503 | 608 | — | — |
| 382 | 8500 | 463 | 0.72 | |
Dilute solutions of salts 3·HCl and 4·HCl in CH2Cl2 exhibited absorption features that were indicative of a small quantity of neutral species being present, namely the presence of low energy absorbances around 480–550 nm (Fig. S37†). This was due to an equilibrium between the cationic species and neutral heterocycles 3 and 4 at the dilute concentrations necessary for UV-vis spectroscopy. The effect of excess acid on the properties of compound 3 was studied by NMR and UV-vis spectroscopy (Fig. S29, S38 and S39†). It was observed that excitation at the low-energy maxima did not lead to photoluminescence. There was no evidence of residual neutral species at NMR spectroscopy concentrations (∼30 mM), and no evidence for the formation of a doubly protonated species in any of the solutions.
The UV-vis absorbance spectra of salts 3·HCl and 4·HCl broadly featured the same changes when compared to the spectra of their parent heterocycles: a reduction or disappearance of the lower energy maxima and retention of strong absorbances at ∼380 nm (λmax3·HCl: 386 nm; 4·HCl: 369 nm). Compounds 3·HCl and 4·HCl also exhibited blue photoluminescence (λPL3·HCl: 485 nm; 4·HCl: 463 nm). The photoluminescence quantum yields of these compounds were high (ΦPL3·HCl: 0.71; 4·HCl: 0.79). Exchanging the anion for a triflate group in compounds 3·HOTf and 4·HOTf maintained the common ∼380 nm absorption maxima (λmax3·HOTf: 375 nm; 4·HOTf: 382 nm) as well as a weak low energy absorbance (λmax3·HOTf: 510 nm; 4·HOTf: 503 nm). Efficient blue photoluminescence at a similar wavelength as their chloride counterparts was observed (λPL3·HOTf: 489 nm; 4·HOTf: 463 nm), and high quantum yields were maintained (ΦPL3·HCl: 0.63; 4·HCl: 0.72). The independence of the photoluminescence wavelength from the identity of the anion contrasts the behaviour observed for compound IV, where the identity of the anion dictated the photoluminescence wavelength.31 Additionally, unlike compound IV, the fluorescence turn-on of 3 and 4 upon protonation was operated through geometric changes to the heterocycle and alterations to the frontier molecular orbitals energies (vide infra).
Density functional theory (DFT) calculations were performed to help rationalize the origin of the drastic change in photoluminescence upon protonation (Fig. 5 and Table S2†). Natural bond orbital (NBO) analysis showed a natural charge of approximately +1 on the boron atoms of [3+H]+ and [4+H]+, thus supporting their description as boronium cations (Table S3†). Neutral compounds 3 and 4 were modelled alongside their cationic derivatives [3+H]+ and [4+H]+. The frontier molecular orbitals of these compounds were modelled at the ωB97XD/def2-SVP (ω = 0.14) level of theory.
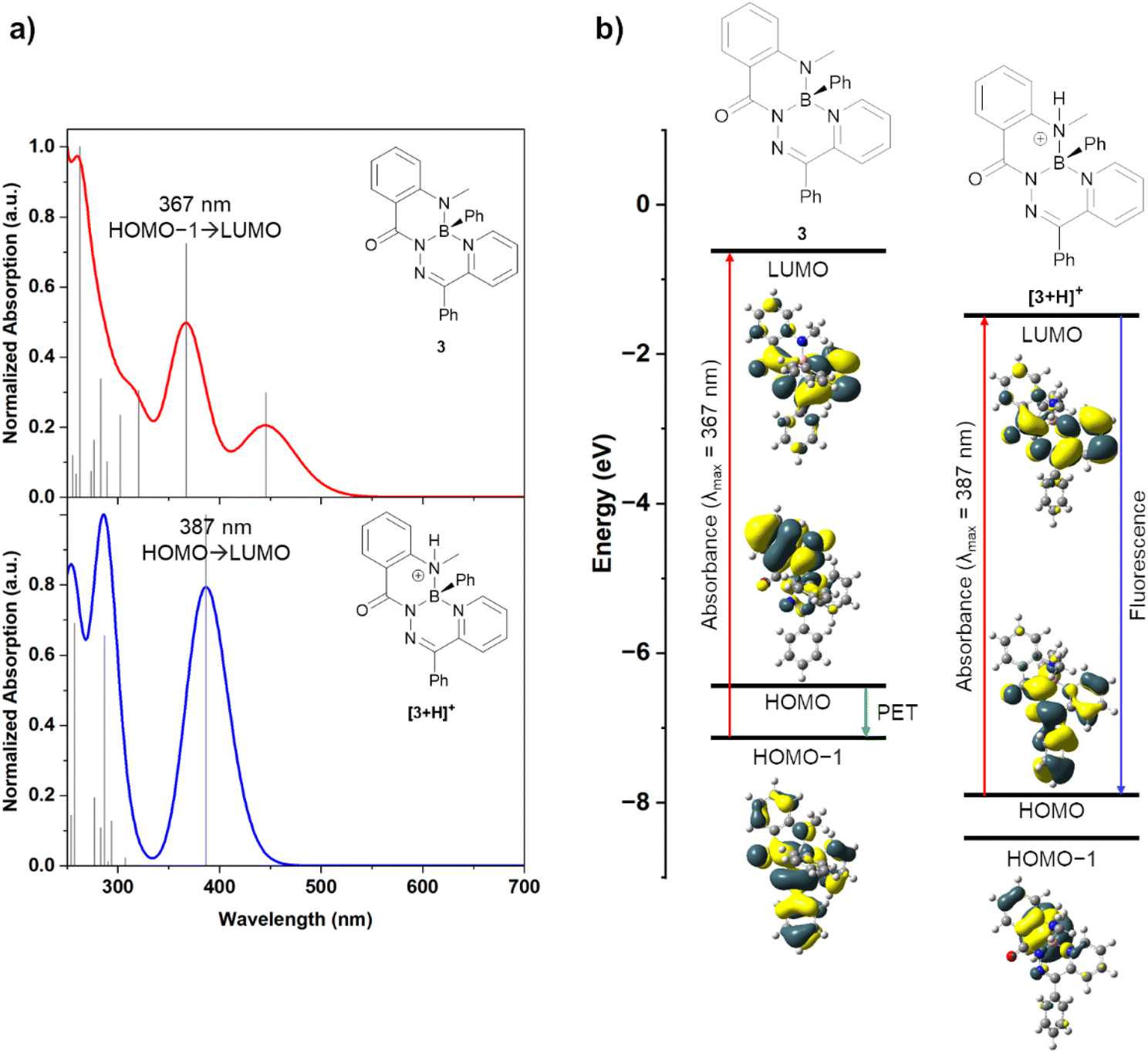 | ||
| Fig. 5 (a) Calculated UV-vis absorption spectra of compounds 3 and [3+H]+ by TD-DFT. (b) Modelled frontier molecular orbitals of compounds 3 and [3+H]+, molecular orbital energies, and associated photophysical processes. | ||
Experimental UV-vis spectra of both compound 3 and its salts 3·HCl and 3·HOTf were observed to have an absorbance peak at ∼375 nm. Time-dependent DFT (TD-DFT) calculations of compound 3 revealed an absorbance at 367 nm (experimental: 354, 385 nm) that was primarily associated with a HOMO−1 → LUMO transition (84%) (Fig. 5a). The HOMO−1 and LUMO molecular orbitals had electron density delocalized over the entire molecule or the annulated hydrazone and pyridine ring systems, respectively. When a photon is absorbed to populate the LUMO, photoinduced electron transfer (PET) from the HOMO, which had significant electron density on the methylamine moiety, can occur to quench the excited state and prevent photoluminescence (Fig. 5b). This type of photoluminescence quenching is well-described for other pH-responsive fluorophores.1,15 Cation [3+H]+ was modelled with a protonated methylamine donor (such as in compounds 3·HCl and 3·HOTf). In TD-DFT experiments of [3+H]+, an absorbance at 387 nm was calculated (experimental: 385 nm). This transition was associated with a HOMO → LUMO transition (94%), and both molecular orbitals had significant electron density on the central hydrazone and pyridine ring systems. The molecular orbital with significant electron density on the protonated methylamine moiety, now the HOMO−1, was lowered in energy upon protonation, relative to compound 3. Efficient photoluminescence can be realized without the possibility of PET quenching. This demonstrated that the protonation of the methylamine donor of compound 3 enabled photoluminescence. A similar ordering of orbital energies and transitions was calculated for compound 4 and its associated cation [4+H]+ (Fig. S41†).
Conclusions
A pair of boron heterocycles 3 and 4, based on a tridentate pyridylhydrazone ligand, were prepared through a facile procedure. While these neutral compounds were non-emissive, protonation at the methyl amine donor induced efficient blue photoluminescence. This contrasts the behaviour of other pyridylhydrazone-based boron heterocycles described in the literature which possess appreciable photoluminescence only in the solid state.37,39 Examining the solid-state structures of the neutral and cationic heterocycles showed significant planarization upon protonation. Computational analysis revealed a pathway for PET quenching in non-emissive compounds 3 and 4, while their protonated counterparts possessed distinct frontier orbital energies that prevented PET photoluminescence quenching. The use of boron-bound Brønsted basic amine donor atoms to turn-on photoluminescence is a unique strategy among emissive boron heterocycles and represents an accessible synthetic modification toward the preparation of other efficient, pH-responsive fluorophores for sensing and bio-imaging applications. Tuning the photoluminescence wavelength to lower energies and achieving water solubility through structural variation would position these fluorophores well for use in imaging biological systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. E. R. W. prepared and characterized the described compounds, performed the DFT calculations, and wrote the manuscript. P. D. B. performed X-ray crystallography studies. P. J. R. and J. B. G. secured funding for this research, supervised the project, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank The University of Western Ontario, the Natural Science and Engineering Research Council (NSERC) of Canada (PGS-D scholarship to A. E. R. W.; Grant DG, RGPIN-2021-04377 to P. J. R.; Grant DG, RGPIN-2023-03318 to J. B. G.), the Ontario Ministry of Research and Innovation (Grant ER14-10-147 to J. B. G.), the Canada Foundation for Innovation (Grant JELF 33977 to J. B. G.), and the Digital Research Alliance of Canada for supporting this work.Notes and references
- S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC.
- M. Kneen, J. Farinas, Y. Li and A. S. Verkman, Biophys. J., 1998, 74, 1591–1599 CrossRef PubMed.
- S. Charier, O. Ruel, J.-B. Baudin, D. Alcor, J.-F. Allemand, A. Meglio and L. Jullien, Angew. Chem., Int. Ed., 2004, 43, 4785–4788 CrossRef PubMed.
- K. M.-C. Wong, W.-S. Tang, X.-X. Lu, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2005, 44, 1492–1498 CrossRef.
- A. S. Vasylevska, A. A. Karasyov, S. M. Borisov and C. Krause, Anal. Bioanal. Chem., 2007, 387, 2131–2141 CrossRef PubMed.
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef PubMed.
- M. Tian, X. Peng, J. Fan, J. Wang and S. Sun, Dyes Pigm., 2012, 95, 112–115 CrossRef.
- T. Berbasova, M. Nosrati, C. Vasileiou, W. Wang, K. S. S. Lee, I. Yapici, J. H. Geiger and B. Borhan, J. Am. Chem. Soc., 2013, 135, 16111–16119 CrossRef.
- J. Qi, D. Liu, X. Liu, S. Guan, F. Shi, H. Chang, H. He and G. Yang, Anal. Chem., 2015, 87, 5897–5904 CrossRef PubMed.
- J. Shangguan, D. He, X. He, K. Wang, F. Xu, J. Liu, J. Tang, X. Yang and J. Huang, Anal. Chem., 2016, 88, 7837–7843 CrossRef PubMed.
- S. Takahashi, Y. Kagami, K. Hanaoka, T. Terai, T. Komatsu, T. Ueno, M. Uchiyama, I. Koyama-Honda, N. Mizushima, T. Taguchi, H. Arai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2018, 140, 5925–5933 CrossRef PubMed.
- M. Shamsipur, A. Barati and Z. Nematifar, J. Photochem. Photobiol., C, 2019, 39, 76–141 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211–10220 CrossRef CAS.
- Y. Chen, H. Wang, L. Wan, Y. Bian and J. Jiang, J. Org. Chem., 2011, 76, 3774–3781 CrossRef CAS PubMed.
- M. Kollmannsberger, T. Gareis, S. Heinl, J. Daub and J. Breu, Angew Chem. Int. Ed. Engl., 1997, 36, 1333–1335 CrossRef CAS.
- M. Baruah, W. Qin, N. Basarić, W. M. De Borggraeve and N. Boens, J. Org. Chem., 2005, 70, 4152–4157 CrossRef PubMed.
- M. Tian, X. Peng, F. Feng, S. Meng, J. Fan and S. Sun, Dyes Pigm., 2009, 81, 58–62 CrossRef.
- Q. Liu, X. Wang, H. Yan, Y. Wu, Z. Li, S. Gong, P. Liu and Z. Liu, J. Mater. Chem. C, 2015, 3, 2953–2959 RSC.
- X. Zhu, H. Huang, R. Liu, X. Jin, Y. Li, D. Wang, Q. Wang and H. Zhu, J. Mater. Chem. C, 2015, 3, 3774–3782 RSC.
- B. Lee, B. G. Park, W. Cho, H. Y. Lee, A. Olasz, C.-H. Chen, S. B. Park and D. Lee, Chem.–Eur. J., 2016, 22, 17321–17328 CrossRef PubMed.
- K. K. Neena and P. Thilagar, ChemPlusChem, 2016, 81, 955–963 CrossRef PubMed.
- S. M. Barbon, J. V. Buddingh, R. R. Maar and J. B. Gilroy, Inorg. Chem., 2017, 56, 12003–12011 CrossRef PubMed.
- A. Sutter, M. Elhabiri and G. Ulrich, Chem.–Eur. J., 2018, 24, 11119–11130 CrossRef.
- T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef PubMed.
- W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef.
- E. Wiberg and K. Schuster, Z. Anorg. Allg. Chem., 1933, 213, 77–88 CrossRef.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS.
- E. J. Corey, T. Shibata and T. W. Lee, J. Am. Chem. Soc., 2002, 124, 3808–3809 CrossRef CAS PubMed.
- I. B. Lozada, R. J. Ortiz, J. D. Braun, J. A. G. Williams and D. E. Herbert, J. Org. Chem., 2022, 87, 184–196 CrossRef CAS.
- S. Hagspiel, F. Fantuzzi, M. Arrowsmith, A. Gärtner, M. Fest, J. Weiser, B. Engels, H. Helten and H. Braunschweig, Chem.–Eur. J., 2022, 28, e202201398 CrossRef CAS PubMed.
- A. E. R. Watson, P. D. Boyle, P. J. Ragogna and J. B. Gilroy, Eur. J. Org Chem., 2024, 27, e202400132 CrossRef CAS.
- M. Kuriakose, M. R. P. Kurup and E. Suresh, Spectrochim. Acta, Part A, 2007, 66, 353–358 CrossRef PubMed.
- I. Unger and G. P. Semeluk, Can. J. Chem., 1966, 44, 1427–1436 CrossRef CAS.
- Y. A. Mikheev, V. P. Pustoshnyi and D. Y. Toptygin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1976, 25, 1962–1964 CrossRef.
- I. I. Levina, O. N. Klimovich, D. S. Vinogradov, T. A. Podrugina, D. S. Bormotov, A. S. Kononikhin, O. V. Dement’eva, I. N. Senchikhin, E. N. Nikolaev, V. A. Kuzmin and T. D. Nekipelova, J. Phys. Org. Chem., 2018, 31, e3844 CrossRef.
- Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610–613 RSC.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83–87 CrossRef CAS.
- D. Cappello, D. A. B. Therien, V. N. Staroverov, F. Lagugné-Labarthet and J. B. Gilroy, Chem.–Eur. J., 2019, 25, 5994–6006 CrossRef CAS PubMed.
- D. Cappello, R. R. Maar, V. N. Staroverov and J. B. Gilroy, Chem.–Eur. J., 2020, 26, 5522–5529 CrossRef.
- D. Cappello, F. L. Buguis, P. D. Boyle and J. B. Gilroy, ChemPhotoChem, 2022, 6, e202200131 CrossRef.
- D. Cappello, F. L. Buguis and J. B. Gilroy, ACS Omega, 2022, 7, 32727–32739 CrossRef PubMed.
- A. N. Bismillah and I. Aprahamian, J. Phys. Org. Chem., 2023, 36, e4485 CrossRef.
- Q. Qi, S. Huang, X. Liu and I. Aprahamian, J. Am. Chem. Soc., 2024, 146, 6471–6475 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, X-ray diffraction methods, NMR spectra, computational methods, UV-vis methods and spectra. CCDC 2376336 and 2376337. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06392a |
| This journal is © The Royal Society of Chemistry 2025 |