
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reassessing the role and lifetime of Qx in the energy transfer dynamics of chlorophyll a†
Erika
Keil
 a,
Ajeet
Kumar
a,
Lena
Bäuml
b,
Sebastian
Reiter
b,
Erling
Thyrhaug
a,
Simone
Moser
c,
Christopher D. P.
Duffy
d,
Regina
de Vivie-Riedle
b and
Jürgen
Hauer
*a
a,
Ajeet
Kumar
a,
Lena
Bäuml
b,
Sebastian
Reiter
b,
Erling
Thyrhaug
a,
Simone
Moser
c,
Christopher D. P.
Duffy
d,
Regina
de Vivie-Riedle
b and
Jürgen
Hauer
*a
aTechnical University of Munich, TUM School of Natural Sciences, Department of Chemistry, Lichtenbergstrasse 4, 85748 Garching, Germany. E-mail: juergen.hauer@tum.de
bDepartment of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstr. 11, 81377 Munich, Germany
cInstitute of Pharmacy, Department of Pharmacognosy, University of Innsbruck, Austria
dDigital Environment Research Institute, Queen Mary University of London, London E1 4NS, UK
First published on 27th November 2024
Abstract
Chlorophylls are photoactive molecular building blocks essential to most photosynthetic systems. They have comparatively simple optical spectra defined by states with near-orthogonal transition dipole moments, referred to as Bx and By in the blue/green spectral region, and Qx and Qy in the red. Underlying these spectra is a surprisingly complex electronic structure, where strong electronic-vibrational interactions are crucial to the description of state characters. Following photoexcitation, energy-relaxation between these states is extremely fast and connected to only modest changes in spectral shapes. This has pushed conventional theoretical and experimental methods to their limits and left the energy transfer pathway under debate. In this work, we address the electronic structure and photodynamics of chlorophyll a using polarization-controlled static – and ultrafast – optical spectroscopies. We support the experimental data analysis with quantum dynamical simulations and effective heat dissipation models. We find clear evidence for B → Q transfer on a timescale of ∼100 fs and identify Qx signatures within fluorescence excitation and transient spectra. However, Qx is populated only fleetingly, with a lifetime well below our ∼30 fs experimental time resolution. Outside of these timescales, the kinetics are determined by vibrational relaxation and cooling. Despite its ultrashort lifetime, our theoretical analysis suggests that Qx plays a crucial role as a bridging state in B → Q energy transfer. In summary, our findings present a unified and consistent picture of chlorophyll relaxation dynamics based on ultrafast and polarization-resolved spectroscopic techniques supported by extensive theoretical models; they clarify the role of Qx in the energy deactivation network of chlorophyll a.
Introduction
Photosynthetic systems efficiently harness sunlight by absorbing it and transferring the excitation energy to a reaction center where charge separation occurs.1,2 Globally, the vast majority of light-harvesting and charge-separation functionality relies on chlorophylls (Chls) or bacteriochlorophylls (BChls). They play essential roles in energy transfer and charge separation and serve as the principal cofactors in the early steps of photosynthesis. For this reason, much effort has been devoted to understanding their electronic structure and ultrafast relaxation dynamics using various experimental and theoretical methods.3–8Despite their structural diversity, all Chls share two prominent spectral bands: the lowest energy Q-band (550–720 nm) and the Soret- or B-band (350–480 nm). Historically, these bands have been characterized using the Gouterman four-orbital model of the π–π* transitions in porphyrins.9,10 In this model, the Q-band consists of two distinct electronic states, which we name Qx,el and Qy,el. Their indices relate to the direction of the respective transition dipole moment within the plane of the macrocycle (Fig. 1a). Later, increasingly sophisticated quantum chemical calculations and experiments aimed to refine the assignment of the observed absorption bands to the two electronic states. However, the energetic position of the Qx,el state remained ambiguous. In 2013, Reimers et al.3 proposed a new band assignment based on vibronic coupling, mixing the states within the Q-band. We refer to the states after incorporating such coupling effects as Qx and Qy. This coupling strongly influences the spectral properties of Chl a. For example, the angle between the Qx and Qy transition dipole moments would naively be expected to be close to 90°, but the experimentally measured value for Chl a is ∼70–78°.11,12 In several subsequent two-dimensional electronic spectroscopy (2DES) studies, vibronic coupling was discussed as the basis of fast signal oscillations,13,14 the vibronic origin of which was proven by polarization-controlled studies.7 A similar conclusion was reached from theoretical studies, where it was shown that the Qx and Qy potential energy surfaces do not cross in an energetically accessible region, and non-adiabatic coupling facilitates ultrafast population transfer across the potential energy surfaces.5 Previous to 2DES studies, results from transient absorption (TA) spectroscopy were interpreted so that Qx → Qy transfer shows a strong and unusual solvent dependence, with Qx-lifetimes ranging from 100–250 fs in different solvents.8,15
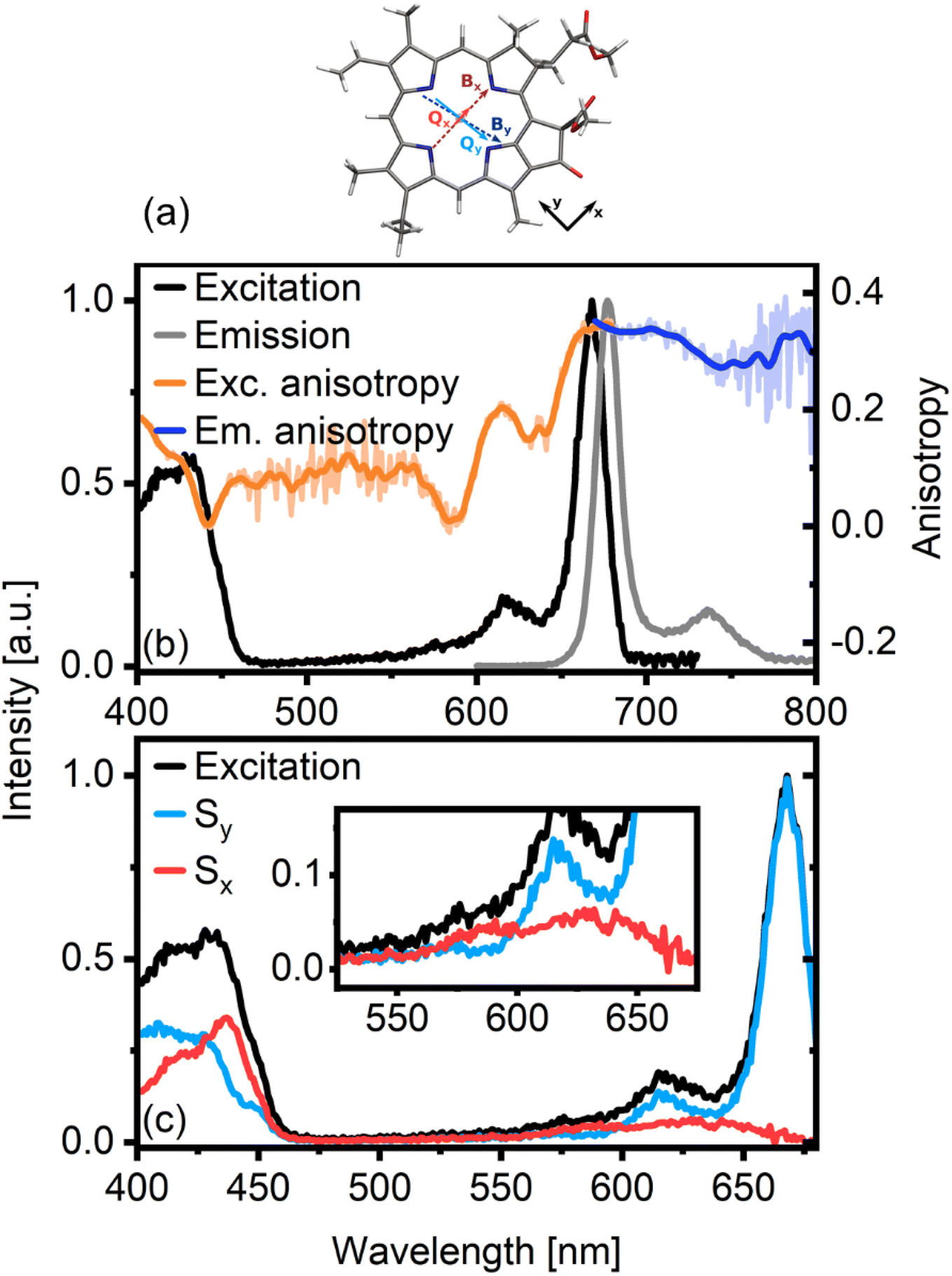 | ||
| Fig. 1 Molecular structure of Chl a including the relative orientations of the main transition dipole moments along the porphyrin rings (a). There are two sets of optical transitions (B and Q), each comprising two almost perpendicular TDMs (subscripts x and y). Panel (b) shows the excitation and emission anisotropy of Chl a in an isopropanol glass. The emission anisotropy (rem, blue line) was measured for λexc = 660 nm, while the excitation anisotropy (rexc, orange line) was measured for λem = 690 nm. The excitation anisotropy clearly shows the presence of differently oriented transitions. We therefore decompose it into x-polarized (Sx) and y-polarized (Sy) excitation spectra (c) for β = 17°. | ||
In this work, we reinterpret the internal conversion dynamics of Chl a, as determined by TA spectroscopy with ∼20 fs excitation and ultra-broadband probing. To offer a comprehensive picture of the relevant deactivation pathways in Chl a, we perform TA experiments exciting the B and the Q-band selectively and compare our results with theoretical models. We can unambiguously determine a B → Q transfer time of ∼100 fs from our experimental data and corroborate our findings using theoretical models. We also show that coupling effects within the Q bands lead to almost instantaneous intraband relaxation.
To test our hypothesis of near-instantaneous Qx → Qy transfer, we go beyond global analysis of TA data and search for Qx-signals using polarization-selective spectroscopy, followed by isolation of polarization-associated spectra.16 Given reported Qx lifetimes of up to several hundreds of femtoseconds,15 we should observe x-polarized stimulated emission (SE) from Qx, along with Qx ground-state bleach (GSB) and excited-state absorption (ESA). While we successfully isolate the GSB signature of Qx, we find no sign of x-polarized SE or ESA features to indicate a transiently populated Qx state. We attribute this to vibronic mixing and rule out the conventional interpretation of Qx as a state with up to 300 fs lifetime.15 Instead, we offer an interpretation of the kinetic components found in TA based on intra-molecular vibrational redistribution and subsequent vibrational cooling, as described by an effective model.
The paper is structured as follows: to identify GSB signatures of Qx, we first analyze polarization-associated spectra derived from cryogenic fluorescence excitation spectra (“Assignment of Qx and Qy transitions”). Direct excitation of Qx is a non-ideal starting ground for studying dynamics, as the absorption features of Qx and Qy overlap strongly. Instead, we excite the B-band and try to isolate transient Qx features. We compare these results with TA data after direct Qx/Qy excitation (“Ultrafast relaxation dynamics of Chl a”).
Similarly to the dissection of linear spectra, we attempt to isolate Qx- and Qy-related excited state absorption features in transient absorption anisotropy (TAA) (“Isolating Qx-features by polarization control”) and compare our measured results to theoretical predictions (“System and system-bath relaxation dynamics at different timescales”). We show that neither TA experiments with sub 20 fs pump pulses nor polarization-controlled experiments can establish a timescale for Qx → Qy transfer. Instead, our work offers a new perspective on the energy transfer dynamics in Chl a. We relate the experimentally observed ultrafast dynamics to solvent relaxation processes and obtain a robust and consistent picture supporting strong mixing between Q-states.
Results
Assignment of Qx and Qy transitions
Fig. 1b shows the normalized excitation (black line) and emission (grey line) spectra of Chl a in an isopropanol glass. The small Stokes shift of the fluorescence spectrum indicates a minor displacement of the excited state potential surface, as also discussed in earlier work.14,17 Accordingly, the emission and absorption spectra are similar in shape, although not exactly symmetric. The differences are mainly in the sidebands, where the breakdown in mirror-image symmetry has been connected to vibronic coupling interactions.17,18 The excitation spectrum in the Q band region has one dominant peak at ca. 670 nm and two shoulders at ca. 620 nm and 570 nm. Based on previous work, the main peak can be assigned to the Qy transition.19 However, assigning the shoulders – particularly determining the position of the Qx transition – is not straightforward.6,17 The locations of the Qx and Qy transitions in Chl a vary with the solvent and the coordination of the molecule. In isopropanol, where the system is penta-coordinated,20 the Qx band overlaps with Qy vibrational bands,17 making a definite assignment of the shoulder bands challenging. Several groups have investigated the transition energy of Qx and its vibronic sidebands, either by calculation4,5 or experiment.3,17 In this work, we employ polarized excitation and emission spectroscopy of Chl a in an isopropanol glass.Alongside the excitation and emission spectra, Fig. 1b also shows fluorescence anisotropy traces. The emission anisotropy (rem, blue line) was measured for λexc = 660 nm, while the fluorescence excitation anisotropy (rexc, orange line) was measured for λEm = 690 nm. In the Q band region, rexc of the main transition centered at 670 nm is almost flat. It reaches a value of 0.35, indicating near-parallel emission and absorption dipoles. As emission occurs from Qy, this band must be y-polarized. The same high value is obtained from rem upon excitation at 660 nm, proving the measurements to be consistent. While the main transition at 670 nm belongs to Qy, rexc shows clearly that the high-energy side of the Q band region contains contributions from electronic states with different polarizations, as anisotropy values here are as low as zero.
Several approaches have been developed to decompose optical spectra by leveraging the information on transition dipole moment (TDM) directions available from anisotropies.21–23 Most commonly, in the absence of a macroscopically oriented sample, these methods “project” the isotropic spectrum into contributions either parallel or orthogonal to a reference TDM. For excitation anisotropy measurements, this reference is the TDM of the emissive transition, while for emission anisotropy, the reference TDM is that of the pumped transition. In many cases, however, the TDMs of a given molecule are not exactly orthogonal, and the decomposition of the spectra in orthogonal components is not ideal. As earlier work has shown that the Chl a Qx and Qy TDMs are not orthogonal,11 this type of spectral decomposition will not result in optimal separation of the contributions. In a recent study, we developed a slightly modified decomposition approach, which is suitable for systems where TDMs are not orthogonal.16 In this approach, we can cleanly separate spectral contributions not only when the underlying TDMs are at an angle θ = 90°, but at any angle θ = 90° − β. The parameter angle β introduced here can be thought of as the rotation angle of an orthogonal molecular coordinate system defined by the reference TDM. For details, we refer to the corresponding publications.16,24
In Fig. 1c we show the decomposition of the Chl a excitation spectrum into components polarized parallel (here: Sy, blue in Fig. 1c) and orthogonal (here: Sx, red) to the emissive transition (Qy fluorescence) according to the procedure outlined above. We note that besides Qx features, Sx also contains signals with a TDM parallel to Bx, while Sy contains both Qy and By contributions. If we assume orthogonal Qx and Qy transitions (corresponding to β = 0), we clearly achieve sub-optimal separation of the spectrum with noticeable contamination of the Sx spectrum by features associated with the y-polarized transitions (see Fig. S1†). Increasing the value of β to 17°, however, maximizes the 670 nm peak in the Sy spectrum and simultaneously achieves optimal separation of the spectrum into “pure” but overlapping Qx and Qy contributions, each with their respective vibronic progressions. Note that both the Sx spectral maximum at approximately 640 nm and the relative angle between Qx and Qy of θ = 90 − β = 73° are in good agreement with previously reported results.4 In essence, polarization-associated analysis of Chl a excitation spectra is a straightforward and effective way of isolating overlapping Qx and Qy ground state transitions.
Ultrafast relaxation dynamics of Chl a
Although Chl a has been studied extensively, open questions remain concerning the exact mechanism of energy transfer dynamics within this molecule. The most common scheme employed to explain Chl a energy relaxation is a sequential one, i.e., Bx → Qx → Qy. B → Q transfer seems to occur within 100–150 fs, as inferred by time-resolved fluorescence depletion spectroscopy15 or UV pump, NIR probe TA.25 A time-constant in the 100–250 fs range with notable strong and unusual solvent dependence has been attributed to the subsequent Qx → Qy transfer.15To better elucidate the mechanism of energy transfer dynamics in Chl a, we perform TA experiments under different excitation conditions (B- and Q-band excitation) and in various solvents (acetone, ethanol (EtOH), and benzonitrile (BN)). In particular, we expect to observe direct evidence of B → Q transfer and to understand the origin of the reported solvent dependence of the Qx → Qy energy transfer step. In the following, we show a representative set of data for Chl a in acetone. The corresponding plots for the other two solvents are shown in ESI Fig S2 and S3.† Absorption and pump pulse spectra are shown in Fig. 2a (Fig. S2†). Chirp-corrected TA data in magic angle (MA) configuration after B- and Q-band are shown in Fig. 2b and c, and the global analysis results are shown in Fig. 2d and e (Fig. S3†). The lifetimes corresponding to the individual spectral components are reported in each panel. Only the evolution-associated spectra (EAS) are shown for simplicity, while the decay-associated spectra for all solvents are shown in ESI Fig. S4.†
 | ||
| Fig. 2 Absorption spectrum of Chl a in acetone plotted against the pump spectra for B-band (blue line) and Q-band (red line) excitation (a). Chirp-corrected TA data for both excitation conditions are shown in (b) and (c). Visible changes in the dynamics are present after B excitation, with a clear increase of the Q-band GSB signal over time, but this is not true after Q excitation. The EAS and lifetimes extracted from global analysis of Chl a in acetone are shown in (d) after excitation in the B-band and in (e) after excitation in the Q band. The corresponding plots for Chl a in EtOH and benzonitrile are shown in Fig. S2.† | ||
In the case of B-band excitation (Fig. 2b and d), the best fit is achieved with four kinetic components. The shortest component has a lifetime of ∼110 fs (black line), with negligible solvent dependence. It is characterized by a decrease and blue shift of the signal in the B-band region (430 nm) and a concomitant increase of the main Q-band signal (660 nm). This behavior is consistent with B → Q transfer, and the B → Q transfer time agrees with previously reported values.8 The second component (dark blue line) shows minimal spectral evolution; the only noticeable feature is a slight broadening of the Qy band. From the global fits, it appears to be strongly solvent-dependent, varying from ca. 400 fs (BN, cf. Fig. S3†) to 2 ps (acetone). Notably, the amplitude of this component is near-negligible, and it is not associated with any spectral evolution typical of state-to-state energy transfer. As changes to small-amplitude components do not substantially affect the goodness-of-fit, the lifetime associated with this component is inevitably highly uncertain. As such, although this component appears to exhibit some solvent dependence, we do not make strong quantitative claims about this behavior. We emphasize, however, that this component does not contain significant changes to stimulated emission or excited state absorption, implying that it is not related to population transfer. The third kinetic component (red line), with a lifetime of about 7–11 ps, correlates with a narrowing of the Qy band and an increase in its amplitude. Finally, the last kinetic component (light blue line, fixed to 5 ns) is readily assigned to the excited state lifetime of Chl a as known from fluorescence lifetime measurements.26
In the case of Q-band excitation (Fig. 2c and e), only three components are necessary to obtain a satisfactory data fit. We again observe an ultrafast component of 100–300 fs (black line) that was previously assigned to Qx → Qy transfer.8,15 If this component was associated with population transfer, we would expect a signal decrease in the 520–630 nm Qx-region concomitant with an increase in the Qy ground-state bleach (GSB)/stimulated emission (SE) amplitude around 660 nm. As in the B-band excitation case, however, we do not observe any such features, leaving this component inconsistent with a population transfer process. An intermediate 1.5–15 ps component (red line) with extremely small amplitude similarly does not relate to significant spectral changes, suggesting a small structural reorganization process. The longest component is again the excited state lifetime of Chl a, fixed at 5 ns (light blue line).
Isolating Qx features by polarization control
General problems with the analysis of the Qx features of Chl a are the weakness of the transition and its strong spectral overlap with the Qy-related signals (cf.Fig. 1). It could then be conceivable that the lack of Qx → Qy population transfer features in MA-TA stems from their smaller amplitudes being covered by the larger Qy signals.To isolate Qx-signatures and to determine their transient behavior, we have measured transient absorption anisotropy (TAA) data for Chl a in acetone after B-band excitation. With these data, we can decompose the TA signals into polarized components in an analogous procedure to that described for excitation anisotropy.16,24 In this case, a value of β = 32° is necessary to fully suppress the Qy peak. In the employed representation, Sx contains signals with a TDM parallel to that of Bx, including all Qx features, while Sy contains the Qy and By contributions. Representative spectra are shown for a time delay Δt = 120 fs in Fig. 3a. At this delay time, effects due to pump-and-probe pulse overlap are negligible, and there is already population in the Q-band resulting from B → Q transfer (cf.Fig. 2b and d).
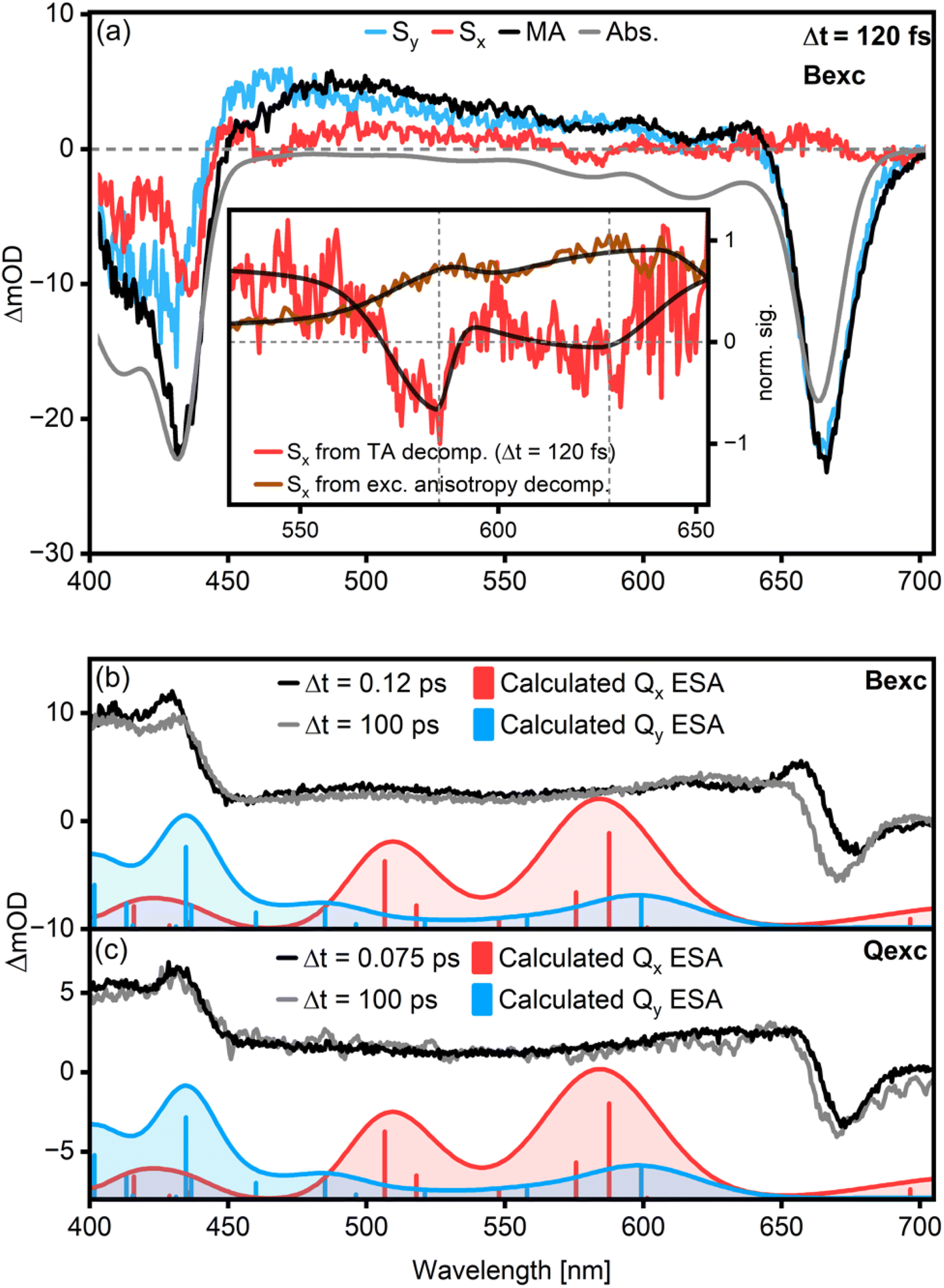 | ||
| Fig. 3 Results of the Chl a TA data decomposition into polarized components at Δt = 120 fs (a) for β = 32°. Here, Sx is chosen to indicate the direction parallel to the pumped TDM and contains all contributions with this polarization. Accordingly, the Qy transitions are suppressed and appear only in Sy. The Qx GSB transitions extracted from polarized TA and steady-state anisotropy match well (a, inset). They show an energy spacing of about 1100 cm−1 as calculated by fitting a bi-gaussian function to the peaks. Excited-state associated spectra of Chl a after excitation in the B-band (b) and the Q-band (c) were calculated for different time delays (grey and black lines) and are compared to theoretical Qx and Qy ESA band positions (blue and red histograms and curves, oscillator strength unscaled). The theoretical ESA band positions were calculated considering one acetone molecule coordinating to the central Mg of a Chl a molecule. Calculated ESA transitions from Qy match the experimental spectrum well, while Qx-ESA features are absent. | ||
In search for Qx-features, we identify two weak negative peaks at 580 nm (ca. 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 cm−1) and 625 nm (ca. 16000 cm−1), matching the Qx features observed in the excitation anisotropy measurements (Fig. 3a, red line and inset). They show an energy spacing of about 1100 cm−1 as calculated by fitting a bi-gaussian function to the peaks (Fig. 3a, inset, black lines), in excellent agreement with magnetic circular dichroism spectra for pentacoordinated Chl a.3
200 cm−1) and 625 nm (ca. 16000 cm−1), matching the Qx features observed in the excitation anisotropy measurements (Fig. 3a, red line and inset). They show an energy spacing of about 1100 cm−1 as calculated by fitting a bi-gaussian function to the peaks (Fig. 3a, inset, black lines), in excellent agreement with magnetic circular dichroism spectra for pentacoordinated Chl a.3
The lineshape of the Sx spectrum remains identical for time delays between 100 fs and several ps (Fig. S5†). This means that while we observe the Qx GSB, no Qx SE (or excited-state absorption (ESA)) features are discernible. We support this statement further by performing a global analysis of the polarization-decomposed TA maps shown in the ESI (Fig. S6†). Similar time constants are needed to fit Sx and Sy, and there is no evidence for a transient population of an excited Qx state.
We further examine the possibility of Qx ESA features by comparing the experimental excited state spectrum to calculations for both B-excitation (Fig. 3b) and Q-excitation (Fig. 3c). The experimental excited state spectrum is determined from the measured TA spectrum by subtracting the GSB.27,28 The latter is approximated by a scaled absorption spectrum. The resulting spectra only contain ESA and SE contributions. We then compare the lineshapes of these spectra at early and late times with calculations for the Qx- and Qy-associated ESA, considering one acetone molecule coordinating to the central Mg of a Chl a molecule. Notably, the excited-state spectrum at early times after B-band excitation has a positive feature at 655 nm which is absent at later times and in the case of Q-band excitation. We attribute this to ESA from the B-band, which is cancelled by the Q-band GSB, explaining why we do not observe this feature in the magic angle TA data in Fig. 2d. While the calculated Qy ESA-peaks correspond to measured features at both early and late times, the features related to Qx ESA are missing. In particular, two intense Qx-features are expected in the 16000–21000 cm−1 region at early times. However, the measured spectra in this region at early and late times are effectively identical. While ESA from Qy is clearly visible, the lack of excited-state signatures associated with Qx means that Qx is either not significantly involved in the energy relaxation network or its lifetime is shorter than the experimental time resolution.
System and system-bath relaxation dynamics at different timescales
We compare our experimental results with quantum dynamics calculations (details in Fig. S7, S8 and Table T1†). Our XMS-CASPT2 results predict that the excited-state potentials of Chl a are nested on top of each other with minimal curve displacement. This is illustrated in Fig. 4a with a cut through the potential energy surface (PES) along the mode with the highest projection onto the non-adiabatic coupling (NAC) vector, with a vibrational frequency of 1489 cm−1 (mode 171 in the frequency analysis). Cuts along modes with weaker coupling and different frequencies show the same trend (Fig. S8†). We did not observe any crossings in the central region of the PES where most of the population resides. The temporal evolution of the population of the adiabatic states is depicted in Fig. 4b. We note that while the dynamics simulations include the non-adiabatic coupling terms, the depicted populations correspond to those of the electronic states within the Born–Oppenheimer approximation and not the vibronic states observed in experiment. After excitation to the B-band with an explicitly simulated pump pulse, a population transfer from B to Q occurs on a moderately longer timescale than the intra-band transfer. This is explained by the significant energy difference between the B and Q bands (Fig. 4a). The exact timescale of the B → Q transfer depends on the amount of initially excited vibrational levels contained in the wavepacket. On PES constructed with normal modes of lower overlap with the NAC vector and in different spectral ranges, the dynamics are generally slower than normal modes of high overlap (Fig. S8†). Nonetheless, all simulations have in common that the intra-band transfer in both the B and the Q band is almost instantaneous. In contrast, the B to Q transfer takes about 50–150 fs. This is in excellent agreement with our experimental data presented in the discussion of Fig. 2. Further, we find that the B → Q transfer heavily depends on coupling the B band with Qx. If this coupling is turned off, the B → Q transfer slows down significantly. On the contrary, if B/Qy coupling is turned off, the population dynamics do not change much (Fig. S8†). | ||
| Fig. 4 Cuts through the potential energy surfaces of Chl a along the mode with the strongest projection on the non-adiabatic coupling vector (a). The curve displacement between the excited states is negligible, resulting in nested potentials with no evident curve crossings. Bx and By, as well as Qx and Qy, show a small energy gap and therefore large coupling, which explains the almost instantaneous population transfer between them. The comparatively large energy gap between Bx and Qx, on the other hand, leads to predicted transfer times of ∼100 fs (b). At longer timescales, energy relaxation occurs via intra-molecular vibrational redistribution (IVR), followed by slower equilibration with the surrounding solvent (vibrational cooling VC). We capture the effects of biphasic energy relaxation in a simplified model for B-band (c) and Q-band (d) excitation scenarios and for different solvents. Since less energy is deposited into the Qy vibrational manifold, the peak local temperature is less than for B-band excitation. | ||
While we can readily explain ultrafast time constants with our quantum dynamics simulations, the dynamics at longer times are not practically accessible with this type of calculation. At these longer timescales, we expect energy redistribution and dissipation to the solvent to be the dominating contributions to the dynamics.28 As such, the bath must be considered when trying to reproduce the longer kinetic components observed in TA.
We construct a simplified numerical model to test our hypothesis of cooling dynamics on multiple timescales. Excitation of Chl a with a visible laser pulse always means that a large amount of energy is deposited into the vibrational modes of the molecule. Energy deposited – directly or indirectly – into a high-lying state of Qy will relax in a non-linear sequence of intra-molecular vibrational redistribution (IVR) events, solute-to-solvent energy transfer, and solvent equilibration. Using an effective molecular temperature Tm (details and modeling parameters in the ESI text, Table T2, Fig. S9 and S10†) that increases with the energy deposited into the system, we predict a molecule-to-solvation shell transfer time of ∼1 ps. This is consistent with observations from our previous work on carotenoids.29–31 The dynamics of Tm are determined by two timescales: τIVR ∼ 1 ps, which accounts for the rise in Tm due to thermalization of the Qy state, and τVC ∼ 7–9 ps, which is the relaxation of the solute due to vibrational cooling (VC) into the solvation shell. The exact values of the time constants vary with the solvent (Fig. 4c and d). The results indicate that the relaxation kinetics are almost independent of whether one excites Qy directly or indirectly via the B-band. However, the maximum Tm reached should be affected by excitation wavelength, as this defines the amount of energy that thermalizes (Fig. 4c and d). As τIVR is associated with heating and τVC with cooling, we expect to observe thermal broadening effects of the Qy-band over ∼1 ps as the maximum Tm is achieved, followed by spectral narrowing over ∼10 ps as the population of low-frequency vibrational modes drops while the solute and first solvation shell re-equilibrate with the bulk solvent. Careful inspection of the decay-associated and evolution-associated spectra (DAS and EAS, respectively) in the Q-band region (Fig. S11†) confirms these predictions for excitation in the B-band, although the observed lineshape changes are minimal. After Q-band excitation, IVR and cooling are much less pronounced. In particular, we do not observe a narrowing in ∼10 ps as expected. Instead, we observe a minimal but continuous redshift typical of Stokes shift dynamics (Fig. S11†).28
Discussion
The results presented in this work clarify some essential aspects of the mechanism of energy relaxation in Chl a. When excited in the B-band, population transfer to the Q band occurs within 120 fs, as demonstrated by TA experiments (Fig. 2d) and quantum dynamical calculations (Fig. 4b). The Q band contains Qx and Qy, two distinct but strongly coupled states with overlapping but discernible absorption spectra (Fig. 1). However, polarized TA measurements show no evidence of x-polarized excited state features (Fig. 3). As such, we can conclude that either Qx is not involved in B → Q energy relaxation in Chl a, or that the Qx → Qy transfer is faster than our time resolution (ca. 30 fs). Our quantum chemical calculations strongly support the latter interpretation, as the B → Q transfer was shown to slow significantly if the B/Qx coupling is turned off (Fig. S8†).An implication of these observations is that none of the time constants found in MA-TA (cf.Fig. 2) can be assigned to Qx → Qy transfer. The small amplitudes and only subtle influence on the spectra of the kinetic components other than the one related to B → Q transfer suggest instead an association with IVR and subsequent heat dissipation to the solvent.
In Fig. 5, we propose a comprehensive scheme for the internal conversion dynamics of Chl a. After the initial B → Q transfer, the state we detect is y-polarized. Therefore, if Qx is participating in the energy transfer as suggested by the quantum dynamics calculations, its relaxation towards a vibrationally hot, y-polarized state must be faster than our time resolution. The relaxation from there to the vibrationally relaxed Qy occurs through IVR, leading to heat exchange with the first solvation shell. Further relaxation to bulk solvent (VC) then occurs on slower timescales. Overall, IVR and VC are sufficient to explain the timescales in the evolution of the Chl a TA spectra beyond the first few hundred fs after B-band excitation: ∼1 ps for IVR to a hot pseudo-thermal state, ∼10 ps for relaxation of the hot thermal state (Fig. 4c and cf.Fig. 2d). The effect of IVR and VC on the lineshape is much less pronounced after Q-band excitation. In this case, the changes in the lineshape can be explained by Stokes shift dynamics.28
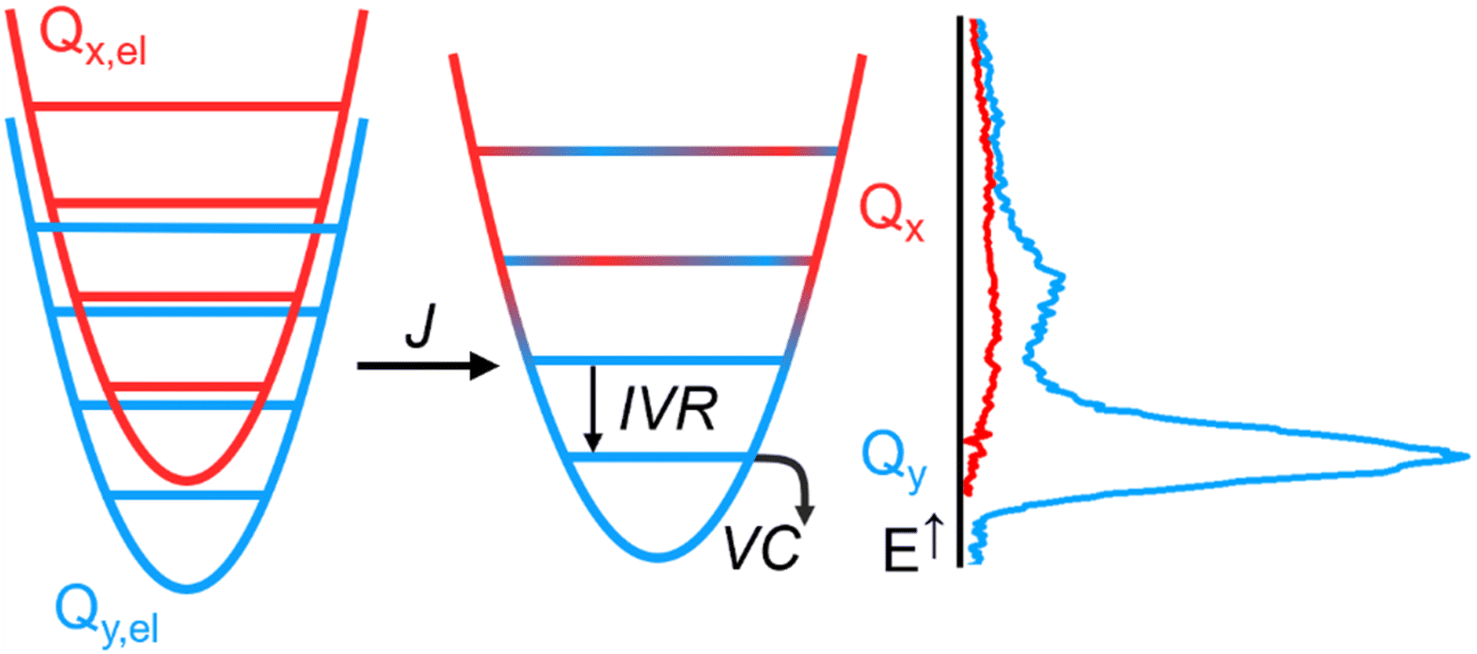 | ||
| Fig. 5 Vibronic mixing (J) in the Q band leads to strongly coupled states Qx (red) and Qy (blue). Ultrafast transfer via Qx populates a vibrationally excited Qy, which in turn relaxes via intra-molecular vibrational redistribution (IVR), followed by slower equilibration with the surrounding solvent (vibrational cooling VC). The right panel shows a sketch of the spectral profiles of Qx and Qy, as discussed in Fig. 1. | ||
In summary, our findings shed new light on the internal conversion dynamics in Chl a. Although the heating–cooling cycle on the Qy surface of Chl a is highly complicated and requires further investigation from both experimental and theoretical sides, we reach a good agreement between model and experiment. We present experimental and theoretical evidence for B → Q transfer on a ∼120 fs timescale. The relaxation dynamics in the Q band are dominated by the strong coupling of the Qx and Qy states and occur on a <30 fs timescale. This leads to the absence of x-polarized excited-state signatures in TAA experiments. We attribute this ultrafast Qx lifetime to the small and internal-coordinate-independent energy gap between Qx and Qy. Regardless of its short lifetime, Qx seems important for efficient B → Q relaxation, as the B → Q transfer rate slows down if B/Qx coupling is deactivated. Based on these results, we reinterpret the solvent-dependent, longer time constants found in TA measurements as a combination of IVR, VC, and possibly Stokes shift dynamics on Qy. Overall, our unique combination of extensive theoretical modeling, numerical methods, and highly sensitive TA experiments allows us to paint a clear and consistent picture of the energy transfer in Chl a under the excitation conditions relevant to photosynthetic systems and to shed light on the previously controversial role of the Qx state in the Chl a dynamics.
Materials and methods
Materials
HPLC and spectroscopy grade solvents acetonitrile (ACN), ethyl acetate (EtOAc), acetone, ethanol (EtOH), BN and methanol (MeOH) were obtained from VWR (Ismaning, Germany) and ultra-pure water (18 MΩ cm−1) from a Millipore S.A.S. Milli-Q Academic system (18.2 MΩ cm−1, Molsheim, France).Chromatography
Spectroscopy
HR-MS were measured at the MS facility of the Department of Chemistry, University of Munich. Data were processed using Xcalibur.UV-vis spectra were measured on a PerkinElmer Lambda 365 spectrometer. Emission spectra were measured on a Spectrofluorometer FS5 (Edinburgh Instruments). All spectra are background-corrected.
Extraction procedure for Chl a
Frozen spinach was purchased at a local supermarket (Edeka junger Spinat). 8 portions of frozen spinach (approximately 15 g each) were thawed by soaking the spinach in water; water was removed from the soaked spinach by squeezing the spinach with a table cloth. The spinach was then mixed with a blender in 100 mL of cold acetone/MeOH (9/1 v/v). The resulting slurry was centrifuged at 4 °C for 5 min at 1000 rpm. The supernatant was filtered; the filtrate was collected on ice and directly applied to semi-preparative HPLC. The Chl a fraction was collected on ice, and the solvents were evaporated in vacuo, yielding 9.7 mg of Chl a.HR ESI-MS (positive ion mode): m/z (found) = 893.5426; m/z (calculated) = 893.5426, Δ = 0.56 ppm.
TA spectroscopy
Chl a samples were prepared by dissolving the pigment in acetone, EtOH or BN to an OD of 0.2–0.3 and then degassing the solution with nitrogen for about one minute.TA experiments were performed for Chl a in acetone, ethanol (EtOH), and benzonitrile (BN). Chl a was excited in the B and Q bands in separate experiments for all solvents. The pulse duration of the two pump pulses was comparable, with the B-pump at 20 fs and the Q-pump at 15 fs as measured by SD-FROG and SHG-FROG, respectively. The measured and retrieved FROG traces, as well as the retrieved spectral and temporal phase profiles, are shown in the ESI (Fig. S12†). The TA setups used for B- and Q-band excitation have been described in detail before.33,34 Briefly, a 5 kHz, 2.5 mJ, 25 fs amplified laser system (Coherent Legend Elite Duo HE+ Ti:Sapphire MOPA) is used to seed a 1 m long, commercial hollow-core fiber (HCF) with a core diameter of 250 μm filled with 1 bar of Argon gas. The resulting supercontinuum is either frequency-doubled in an achromatic second-harmonic scheme to yield the B-band pump pulse33 or spectrally filtered to obtain the Q-band pump pulse.34 In the first case, the pump is compressed by a three-prism configuration, while in the second case, the compression is achieved by chirped mirrors. In both experiments, the probe pulse is given by a supercontinuum obtained by seeding a CaF2 crystal with the NIR part of the HCF output. For B-band excitation, a double-chopping scheme was employed to reduce scatter, and the pump energy was 60 nJ per pulse with a focal spot diameter of 200 μm; for Q-band excitation, the pump pulse was not chopped and its energy was 16 nJ per pulse with a focal spot of 140 μm. The data was recorded at MA (54.7°) configuration between pump and probe for B- and Q-pumping. Additionally, datasets with parallel and perpendicular polarization of pump and probe were recorded for B-band excitation of Chl a in acetone in order to calculate the TAA.
Global analysis was performed with the open-source software Glotaran.35,36
Quantum dynamics simulations
The quantum dynamics simulations were performed on 2D PES at the XMS-CASPT2 (ref. 37–39) level of theory, using the basis set ANO-RCC-VDZP40–42 and a CAS(6/6) active space in the gas phase. As coordinates, we chose the normal modes exhibiting the highest overlap with the non-adiabatic coupling vector at the Qy minimum geometry,5 optimized at the CAM-B3LYP/6-311G* level.43 These modes are 171 and 198, lying at 1489 cm−1 and 1639 cm−1, respectively. Other normal modes (239 and 103) with smaller overlap with the NAC vector and in a different spectral range were also tested for PES construction. Geometries are available in the ESI.† The resulting quantum dynamics is similar for all modes, but the timescales slow down with decreasing overlap with the NAC vector (Fig. S9†). At each point of the PES, a RASSCF calculation was performed with state averaging over four states, followed by one with state averaging over eight states (Fig. S8†). The ensuing XMS-CASPT2 calculation was performed for the states of interest, namely the S0, Qy, Qx, Bx, and By. An IPEA44 and an imaginary shift45 of 0.1 were applied, as in our previous study.5 With this approach, an energy difference between Qx and Qy at the FC point of 0.225 eV is obtained, which corresponds well with our reference value of 0.23 eV.5 The non-adiabatic coupling elements were computed at the XMS-CASPT2 level of theory. All PES, NAC, and TDM calculations were performed with the OpenMolcas 23.06 program package.46–48 The QDng software package49 was used for the subsequent quantum dynamic simulations. The propagation was initially performed by assuming a delta excitation into By, therefore populating this state's manifold of vibrational levels. Later, an explicitly simulated pump pulse with a maximum field strength of 0.0026 GV cm−1, an FWHM of 20 fs, and a central frequency of ω0 = 3.60 eV was also applied to excite the population from the ground state into the B Band. The laser parameters were chosen to excite a similar population (about 5%) into the B-band as in our experimental studies.Calculated ESA spectra
Geometries of the Qy and Qx states of Chl a with one axially coordinated acetone molecule were optimized using Gaussian 16C.02 (ref. 50) with the CAM-B3LYP density functional51 and the 6-311G* basis set.43,52,53 Bulk solvent effects were described with the polarizable continuum model (PCM).54,55 Five roots were included in the optimization. Optimized geometries were verified as energy minima by the absence of imaginary vibrational frequencies.Excited state absorption spectra for the optimized geometries were simulated at the double-hybrid TDDFT level using the ORCA 5.0.3 software package.56–58 The SCS-ωPBEPP86 density functional59 was used in the Tamm–Dancoff approximation60 along with the def2-TZVPD61,62 basis. The calculation was accelerated by employing the RIJCOSX approximation63,64 with the def2/J65 and def2-TZVPD/C66 auxiliary basis sets. A tighter-than-usual integration grid (ORCA keyword DefGrid3) and convergence criteria (ORCA keywords VeryTightSCF TightPNO) were employed. The bulk solvent was described implicitly via the linear-response conductor-like polarizable continuum model (LR-CPCM),67,68 assuming equilibrium solvation in the excited state.
Data availability
Data for this article, including absorption spectra, pump pulse spectra, transient absorption and transient absorption anisotropy maps are available at https://zenodo.org/records/14033283.Author contributions
EK and AK performed the experiments; SM provided the sample; SR, LB, CD, and RdVR performed calculations; EK, AK, ET, SR, LB, RdVR, CDPD, and JH analyzed and interpreted the data; EK and JH prepared the plots; EK, AK, ET and JH wrote the main text. All authors reviewed the manuscript before submission.Conflicts of interest
The authors declare no competing interests.Acknowledgements
J. H., A. K., S. R., L. B. and R. d. V.-R. gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the cluster of excellence e-conversion under Germany's Excellence Strategy – EXC 2089/1 – 390776260 and via project 514636421. S. M. would like to thank Rita Socher for technical support.References
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley/Blackwel, Chichester, West Sussex, 2nd edn, 2014 Search PubMed.
- R. Croce and H. van Amerongen, Natural strategies for photosynthetic light harvesting, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS PubMed.
- J. R. Reimers, Z.-L. Cai, R. Kobayashi, M. Rätsep and E. Krausz, Assignment of the Q-Bands of the Chlorophylls: Coherence Loss via Qx-Qy Mixing, Sci. Rep., 2013, 3, 2761 CrossRef PubMed.
- C. Zahn, T. Stensitzki and K. Heyne, Femtosecond anisotropy excitation spectroscopy to disentangle the Qx and Qy absorption in chlorophyll a, Chem. Sci., 2022, 13, 12426–12432 RSC.
- S. Reiter, L. Bäuml, J. Hauer and R. de Vivie-Riedle, Q-Band relaxation in chlorophyll: new insights from multireference quantum dynamics, Phys. Chem. Chem. Phys., 2022, 24, 27212–27223 RSC.
- Y. Song, A. Schubert, E. Maret, R. Burdick, B. Dunietz, E. Geva and J. Ogilvie, Vibronic structure of photosynthetic pigments probed by polarized two-dimensional electronic spectroscopy and ab initio calculations, Chem. Sci., 2019, 10, 8143–8153 RSC.
- E. Bukarté, A. Haufe, D. Paleček, C. Büchel and D. Zigmantas, Revealing vibronic coupling in chlorophyll c1 by polarization-controlled 2D electronic spectroscopy, Chem. Phys., 2020, 530, 110643 CrossRef.
- W. P. Bricker, P. Shenai, A. Ghosh, Z. Liu, M. Enriquez, P. Lambrev, H.-S. Tan, C. Lo, S. Tretiak, S. Fernandez-Alberti and Y. Zhao, Non-radiative relaxation of photoexcited chlorophylls: Theoretical and experimental study, Sci. Rep., 2015, 5, 13625 CrossRef PubMed.
- M. Gouterman, Spectra of porphyrins, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- M. Gouterman, G. H. Wagnière and L. C. Snyder, Spectra of Porphyrins Part II. Four Orbital Model, J. Mol. Spectrosc., 1963, 11, 108–127 CrossRef CAS.
- M. Linke, A. Lauer, T. Von Haimberger, A. Zacarias and K. Heyne, Three-dimensional orientation of the Qy electronic transition dipole moment within the chlorophyll a molecule determined by femtosecond polarization resolved VIS pump-IR probe spectroscopy, J. Am. Chem. Soc., 2008, 130, 14904–14905 CrossRef PubMed.
- R. Simonetto, M. Crimi, D. Sandonà, R. Croce, G. Cinque, J. Breton and R. Bassi, Orientation of chlorophyll transition moments in the higher-plant light- harvesting complex CP29, Biochemistry, 1999, 38, 12974–12983 CrossRef CAS PubMed.
- E. Meneghin, C. Leonardo, A. Volpato, L. Bolzonello and E. Collini, Mechanistic insight into internal conversion process within Q-bands of chlorophyll a, Sci. Rep., 2017, 7, 1–7 CrossRef CAS PubMed.
- V. Tiwari, W. K. Peters and D. M. Jonas, Electronic resonance with anticorrelated pigment vibrations drives photosynthetic energy transfer outside the adiabatic framework, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1203–1208 CrossRef CAS PubMed.
- Y. Shi, J. Y. Liu and K. L. Han, Investigation of the internal conversion time of the chlorophyll a from S3, S2 to S1, Chem. Phys. Lett., 2005, 410, 260–263 CrossRef CAS.
- Y. Xu, L. Mewes, E. Thyrhaug, V. Sláma, F. Šanda, H. Langhals and J. Hauer, Isolating Pure Donor and Acceptor Signals by Polarization-Controlled Transient Absorption Spectroscopy, J. Phys. Chem. Lett., 2023, 14, 5390–5396 CrossRef CAS PubMed.
- M. Rätsep, J. Linnanto and A. Freiberg, Mirror symmetry and vibrational structure in optical spectra of chlorophyll a, J. Chem. Phys., 2009, 130, 194501 CrossRef PubMed.
- J. R. Reimers, M. Rätsep, J. M. Linnanto and A. Freiberg, Chlorophyll Spectroscopy: Conceptual Basis, Modern High-Resolution Approaches, and Current Challenges, Proc. Est. Acad. Sci., 2022, 71, 127 CrossRef.
- C. Houssier and K. Sauer, Circular dichroism and magnetic circular dichroism of the chlorophyll and protochlorophyll pigments, J. Am. Chem. Soc., 1970, 92, 779–791 CrossRef CAS.
- S. Krawczyk, The effects of hydrogen bonding and coordination interaction in visible absorption and vibrational spectra of chlorophyll a, Biochim. Biophys. Acta, Bioenerg., 1989, 976(2–3), 140–149 CrossRef CAS.
- E. W. Thulstrup, J. Michl and J. H. Eggers, Polarization spectra in stretched polymer sheets, II.1 Separation of π–π* absorption of symmetrical molecules into components, J. Phys. Chem., 1970, 74, 3868–3878 CrossRef CAS.
- A. C. Albrecht, Polarizations and assignments of transitions: The method of photoselection, J. Mol. Spectrosc., 1961, 6, 84–108 CrossRef CAS.
- E. Thyrhaug, T. J. Sørensen, I. Gryczynski, Z. Gryczynski and B. W. Laursen, Polarization and symmetry of electronic transitions in long fluorescence lifetime triangulenium dyes, J. Phys. Chem. A, 2013, 117, 2160–2168 CrossRef CAS PubMed.
- Y. Xu, M. Peschel, M. Jänchen, R. Foja, G. Storch, E. Thyrhaug, R. de Vivie-Riedle and J. Hauer, Determining Excited-State Absorption Properties of a Quinoid Flavin by Polarization-Resolved Transient Spectroscopy, J. Phys. Chem. A, 2024, 128, 3830–3839 CrossRef CAS PubMed.
- P. M. Shenai, S. Fernandez-Alberti, W. P. Bricker, S. Tretiak and Y. Zhao, Internal Conversion and Vibrational Energy Redistribution in Chlorophyll a, J. Phys. Chem. B, 2016, 120, 49–58 CrossRef CAS PubMed.
- J. S. Connolly, F. A. Janzen and E. B. Samuel, Fluorescence lifetimes of chlorophyll a : solvent, concentration and oxygen dependence, Photochem. Photobiol., 1982, 36(5), 559–563 CrossRef CAS.
- D. A. Cherepanov, F. Gostev, I. Shelaev, A. Aibush, A. Semenov, M. Mamedov, V. Shuvalov and V. Nadtochenko, Visible and Near Infrared Absorption Spectrum of the Excited Singlet State of Chlorophyll a, High Energy Chem., 2020, 54, 145–147 CrossRef CAS.
- P. Martinsson, J. Oksanen, M. Hilgendorff, P. Hynninen, V. Sundström and E. Akesson, Dynamics of ground and excited state chlorophyll a molecules in pyridine solution probed by femtosecond transient absorption spectroscopy, Chem. Phys. Lett., 1999, 309(5–6), 386–394 CrossRef CAS.
- V. Šebelík, C. D. P. Duffy, E. Keil, T. Polívka and J. Hauer, Understanding Carotenoid Dynamics via the Vibronic Energy Relaxation Approach, J. Phys. Chem. B, 2022, 126(22), 3985–3994 CrossRef.
- V. Balevičius, A. Pour, J. Savolainen, C. Lincoln, V. Lukeš, E. Riedle, L. Valkunas, D. Abramavicius and J. Hauer, Vibronic energy relaxation approach highlighting deactivation pathways in carotenoids, Phys. Chem. Chem. Phys., 2015, 17, 19491–19499 RSC.
- V. Balevičius, C. Lincoln, D. Viola, G. Cerullo, J. Hauer and D. Abramavicius, Effects of tunable excitation in carotenoids explained by the vibrational energy relaxation approach, Photosynth. Res., 2018, 135(1–3), 55–64 CrossRef.
- S. M. Petrovic, J. B. Zvezdanović and D. Z. Marković, The identification of chlorophyll and its derivatives in the pigment mixtures: HPLC-chromatography, visible and mass spectroscopy studies, Adv. Technol., 2012, 1, 16–24 Search PubMed.
- E. Keil, P. Malevich and J. Hauer, Achromatic frequency doubling of supercontinuum pulses for transient absorption spectroscopy, Opt. Express, 2021, 29, 39042 CrossRef CAS PubMed.
- A. Kumar, P. Malevich, L. Mewes, S. Wu, J. Barham and J. Hauer, Transient absorption spectroscopy based on uncompressed hollow core fiber white light proves pre-association between a radical ion photocatalyst and substrate, J. Chem. Phys., 2023, 158, 144201 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, Glotaran: A Java-Based Graphical User Interface for the R Package TIMP, J. Stat. Software, 2012, 49(3), 1–22 Search PubMed.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Global and target analysis of time-resolved spectra, Biochim. Biophys. Acta, Bioenerg., 2004, 1657(2–3), 82–104 CrossRef CAS PubMed.
- T. Shiozaki, W. Győrffy, P. Celani and H.-J. Werner, Communication: Extended multi-state complete active space second-order perturbation theory: Energy and nuclear gradients, J. Chem. Phys., 2011, 135, 081106 CrossRef PubMed.
- A. A. Granovsky, Extended multi-configuration quasi-degenerate perturbation theory: The new approach to multi-state multi-reference perturbation theory, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, The multi-state CASPT2 method, Chem. Phys. Lett., 1998, 288(2–4), 299–306 CrossRef CAS.
- B. O. Roos, V. Veryazov and P.-O. Widmark, Relativistic atomic natural orbital type basis sets for the alkaline and alkaline-earth atoms applied to the ground-state potentials for the corresponding dimers, Theor. Chem. Acc., 2004, 111, 345–351 Search PubMed.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set, J. Phys. Chem. A, 2004, 108(15), 2851–2858 CrossRef CAS.
- P.-O. Widmark, P.-Å. Malmqvist and B. O. Roos, Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions, Theor. Chim. Acta, 1990, 77, 291–306 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2), Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- N. Forsberg and P.-Å. Malmqvist, Multiconfiguration perturbation theory with imaginary level shift, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- G. Li Manni, et al., The OpenMolcas Web: A Community-Driven Approach to Advancing Computational Chemistry, J. Chem. Theory Comput., 2023, 19, 6933–6991 CrossRef CAS PubMed.
- F. Aquilante, et al., Modern quantum chemistry with [Open]Molcas, J. Chem. Phys., 2020, 152, 214117 CrossRef CAS PubMed.
- I. Fernandez-Galván, et al., OpenMolcas: From Source Code to Insight, J. Chem. Theory Comput., 2019, 15(11), 5925–5964 CrossRef PubMed.
- M. Kowalewski and R. de Vivie-Riedle, QDng: A Grid Based Molecular Quantum Dynamics Package (1.0.0), Deposited 8 April 2024, Zenodo, DOI:10.5281/zenodo.10944497.
- M. J. Frisch, G. W. Trucks, H. B. Schlegelet al., Gaussian 16 Rev. C.02, Wallingford, CT, 2016 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- M. Casanova-Páez, M. B. Dardis and L. Goerigk, ωB2PLYP and ωB2GPPLYP: The First Two Double-Hybrid Density Functionals with Long-Range Correction Optimized for Excitation Energies, J. Chem. Theory Comput., 2021, 17, 5165–5186 CrossRef PubMed.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm-Dancoff approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Rappoport and F. Furche, Property-optimized Gaussian basis sets for molecular response calculations, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- R. Izsák and F. Neese, An overlap fitted chain of spheres exchange method, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- R. Cammi, B. Mennucci and J. Tomasi, Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethylaminobenzonitrile, J. Phys. Chem. A, 2000, 104, 5631–5637 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06441k |
| This journal is © The Royal Society of Chemistry 2025 |