DOI:
10.1039/D4SC06494A
(Edge Article)
Chem. Sci., 2025,
16, 5565-5572
Multiple effects of aromatic substituents on excited-state properties and singlet fission process in azaquinodimethane systems†
Received
27th December 2024
, Accepted 14th February 2025
First published on 17th February 2025
Abstract
Singlet fission (SF) could offset the thermalization loss of high-energy photons via multiexciton generations, thus holding great potential in improving the power conversion efficiency of solar cells. However, the development of SF-based devices has basically remained stagnant so far owing to the limited scope of practical SF materials. Therefore, designing and developing practical SF material systems have been imperative, yet challenging so far. In this work, we comprehensively investigated the effects of aromatic substituents on excited-state properties and SF process of azaquinodimethane systems. Results indicated that the aromatic substituents have a significant influence on molecular diradical characters, thereby determining the excited-state energetics of the SF material system, including optical band gaps and triplet energy. Moreover, the aromatic substituents influenced charge transfer coupling interactions by adjusting molecular packing in the aggregate state to shunt the excited-state population to exert SF process or trap in excimer species. These results not only offer a deep insight into the multiple regulatory effects of the aromatic substituents on excited-state properties and SF process but also provide a practical SF material system, which could lay the foundation for the discovery of new SF-active chromophores and practical applications of new-generation light-harvesting materials.
Introduction
Singlet fission (SF) is an ultrafast multiexciton (ME) generation process in which organic semiconductors transform singlet excitons into double triplet excitons by absorbing a single photon.1–3 Such a process could effectively offset the thermalization loss of high-energy photons via ME generations, thus holding great potential for improving the power conversion efficiency of solar cells.4,5 The successful fabrication of an SF-based photovoltaic device with an external quantum efficiency of over 100% sufficiently manifested such potential in the next-generation light-harvesting applications.6 However, development of SF-based devices basically remains stagnant owing to the limited scope of practical SF materials.1–3,7 For light-harvesting applications, SF chromophores must possess a high extinction coefficient, suitable excited-state energetics, excellent SF properties and good stability.1–3,7,8 According to the requirements of SF process and the complementary semiconductor (i.e. Si ∼1.1 eV) sensitizers, the desirable optical bandgap (Eg) and triplet energy (ET) of a practical SF chromophore should be greater than 2.0 and 1.1 eV, respectively. For meeting these requirements, the most widely studied acene derivatives have shown significant advances in SF mechanism and exciton dynamics, yet these polycyclic compounds seem to be less explored owing to their drawbacks, such as low optical bandgap, triplet energy mismatch, slow SF process and poor stability against air and light, which limits their practical application in devices.1–3 Therefore, designing and developing practical SF material systems are imperative, yet challenging so far.8–16
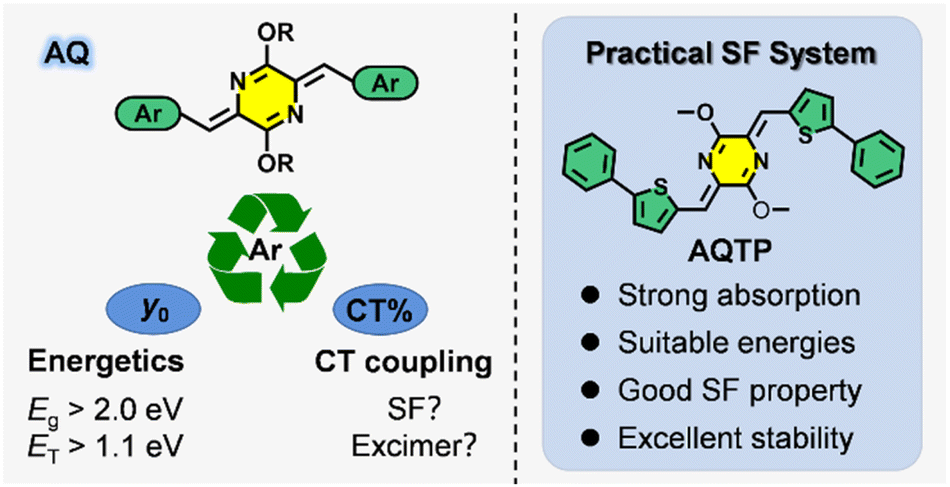
Azaquinodimethane (AQ)-core compounds feature distinctive optoelectronic properties, which have been successfully applied in high-mobility semiconductor devices, polyelectrolytes and photothermal therapy.17–20 In the previous work, we have proposed a donor-/acceptor-decorated quinoidal structure design strategy, followed by the fabrication of a new SF chromophore within an azaquinodimethane (AQ) skeleton, which exhibited strong visible-light absorption, efficient SF process and excellent stability.8 For practical applications, the AQ system still suffers from some drawbacks similar to the SF model molecule, pentacene, such as the relatively low optical bandgap and triplet energy.1–3,7,8 Moreover, the ambiguous SF mechanism of the AQ system also impedes efficient triplet exciton harvesting in practical device applications. Therefore, it is of great importance to carry out the fundamental structure–property relationship studies to design and develop practical SF material systems. In this work, we investigated the effects of aromatic substituents on excited-state properties and SF process in the AQ system in order to construct a practical SF material system (Scheme 1). Based on the comprehensive theoretical analysis and spectroscopic studies, we revealed that the aromatic substituents have a significant influence on molecular diradical characters and then determined the excited-state energetics of the SF material system, including optical bandgap and triplet energy. Moreover, the aromatic substituents could exert influence on charge transfer coupling interactions via adjusting molecular packing in the aggregate state to shunt the excited-state population to exert SF process or trap in excimer species. A practical SF material system based on a phenylthienyl-substituted AQ molecule, AQTP, was successfully fabricated with a high extinction coefficient, suitable energy levels, good SF property and excellent stability (Scheme 1). Deep insights into the multiple regulatory effects of the aromatic substituents on excited-state properties and SF process could promote the development of new SF-active chromophores and practical applications of new-generation light-harvesting materials.
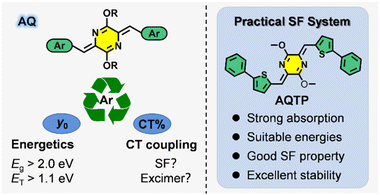 |
| | Scheme 1 Practical SF materials based on AQ molecular systems and the multiple regulatory effects of the aromatic substituents. | |
Results and discussion
Theoretical molecular preliminary screening
We first focused on the theoretical calculations of the AQ system to investigate the effects of aromatic substituents and perform preliminary molecular screening of practical SF candidates (Fig. 1). We selected a series of two-ring-based aromatic substituents including biphenyl, phenylthienyl, phenylfuranyl, bithienyl, bifuranyl and phenylfluorenyl groups and named the corresponding AQ derivatives as AQPP, AQTP, AQFP, AQFF, AQTT and AQWP, respectively. Then we investigated the diradical characters (y0) and excited-state energetics of these AQ derivatives. Based on the optimized ground-state (S0) molecular geometries, the geometries and adiabatic excitation energies of the triplet and singlet states (E(T1) and E(S1)) were evaluated using density functional theory (DFT) and time-dependent density functional theory (TD-DFT) methods, respectively (for computation details, see the Experimental section and Section 2 of the ESI†). The results indicated that there is a significant inverse relationship between the molecular diradical characters and excited-state energetics. As the molecular diradical character gradually increases from 0.194, 0.303, 0.314, 0.320, 0.349 to 0.422, the E(S1) and E(T1) decrease from 2.60, 2.28, 2.31, 2.28, 2.15 to 1.99 eV, and 1.35, 1.17, 1.14, 1.10, 1.07 to 0.72 eV, respectively. We could conclude that the aromatic substituents have a significant influence on molecular diradical characters and then determine the excited-state energetics including optical bandgap and singlet/triplet energy in the studied AQ system. Moreover, we found that all derivatives met the basic requirements for SF energetics, E(S1) ≈ 2E(T1). Among them, AQPP, AQTP, AQFP and AQFF molecules turn out to be the practical SF candidates, given that these AQ derivatives further fulfill the requirements of the optical bandgap and triplet energy. These results clearly suggest that via the systematic aromatic substituent modification engineering, we could tune the molecular diradical characters and then rationally adjust the excited-state energetics to achieve the design and screen the practical SF candidates. Then we selected AQPP and AQTP molecules as model systems to further verify our molecular design and study the SF mechanism of the current AQ system.
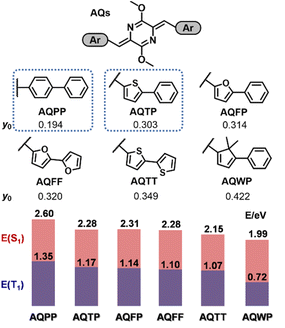 |
| | Fig. 1 Theoretical analysis of the AQ molecular systems. The diradical characters (y0) and excited-state energy distribution of the AQ derivatives with different aromatic substituents. | |
Molecular synthesis and steady-state characterizations
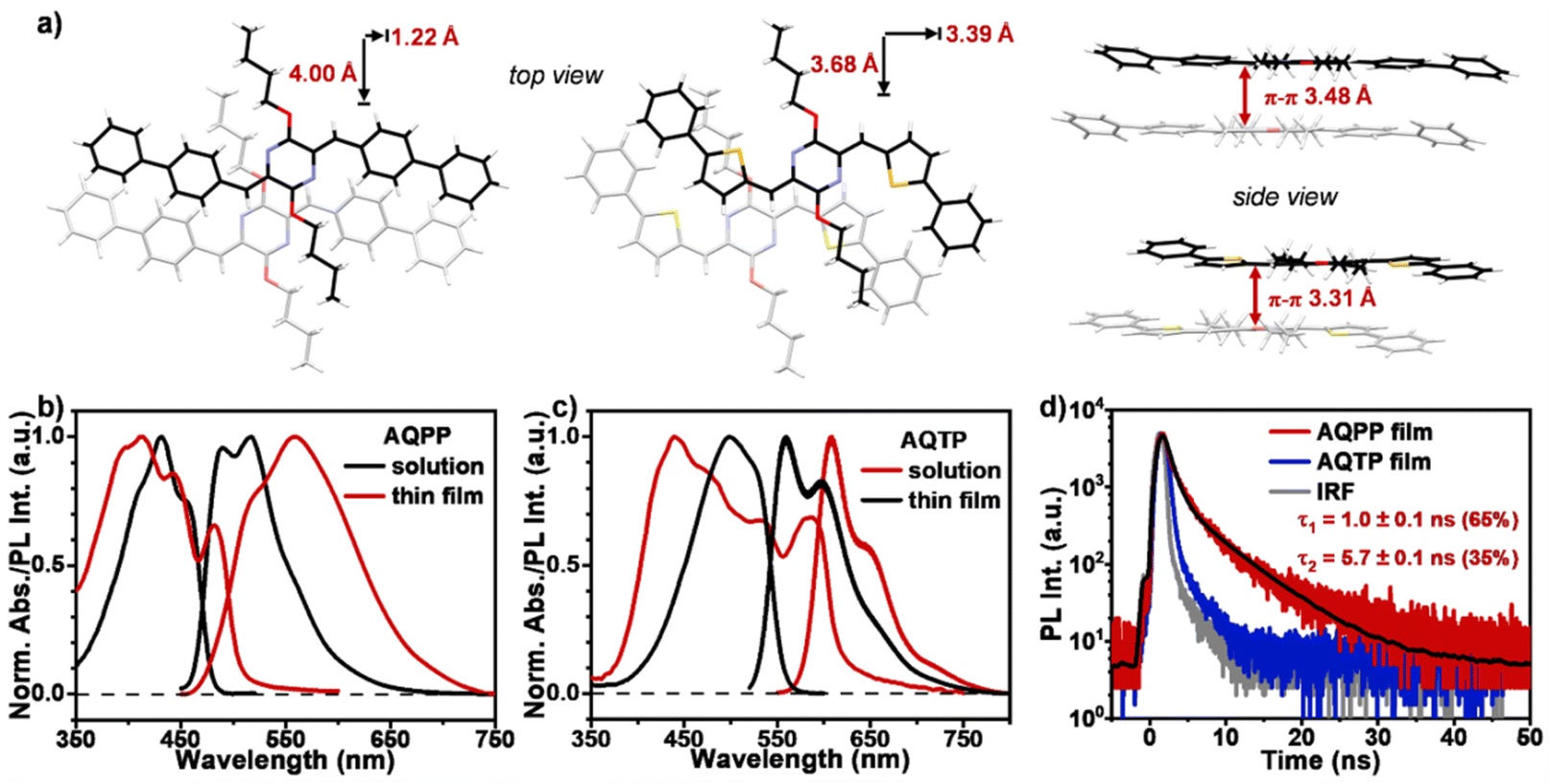
The molecular synthetic routes of two screened AQ derivatives, AQPP and AQTP, were provided in Section 3 of the ESI.† In order to reveal the photophysical properties, steady-state characterizations were performed for two AQ derivatives (Fig. 2). The single-crystal X-ray diffraction data are shown in Fig. 2a. The results indicated that both molecules hold an almost planar molecular structure, and exhibit slip-stacked molecular arrangements in the most closely associated dimer unit from the crystal structure. Two molecules display a similar longitudinal shift of ∼4 Å. The major differences lie in the transverse offsets and the π–π distances. AQPP molecules show a smaller transverse shift of 1.22 Å and a π–π distance of 3.48 Å, while AQTP molecules exhibit a relatively large transverse shift of 3.39 Å and a closer π–π distance of 3.31 Å. The UV/vis absorption and photoluminescence (PL) spectra were then investigated for AQPP and AQTP molecules in both dilute solutions and thin films (Fig. 2b and c). In dilute solutions, AQPP and AQTP molecules exhibit strong blue-green light absorption at 430 and 500 nm with molar extinction coefficients of 4.9 × 104 and 8.0 × 104 M−1 cm−1, respectively (Fig. S2†), which are consistent with the theoretical results. High molar extinction coefficients of SF materials are known to effectively control and minimize the active layer thickness conducive to practical device fabrication.1,2,8 Two molecules display strong fluorescence emission peaks at 490 and 560 nm in dilute solutions with lifetimes of 1.16 and 1.43 ns, and quantum yields of 0.53 and 0.55, respectively (Fig. S3 and S4†).
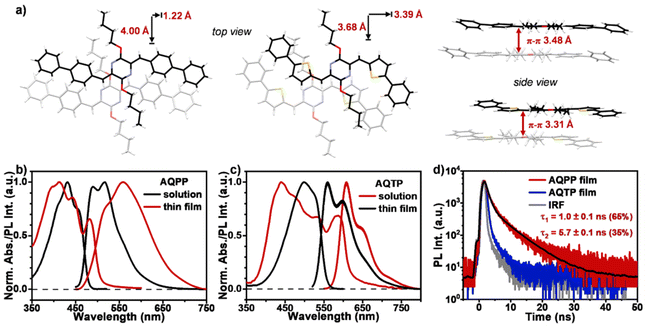 |
| | Fig. 2 Steady-state characterizations. (a) Molecular packing motifs in single crystals of AQPP and AQTP molecules. Steady-state absorption and PL spectra of (b) AQPP and (c) AQTP in dilute DCM solutions (black lines, 10−5 M) and thin films (red lines). (d) Time-resolved PL decay curves for AQPP (red line) and AQTP (blue line) films. | |
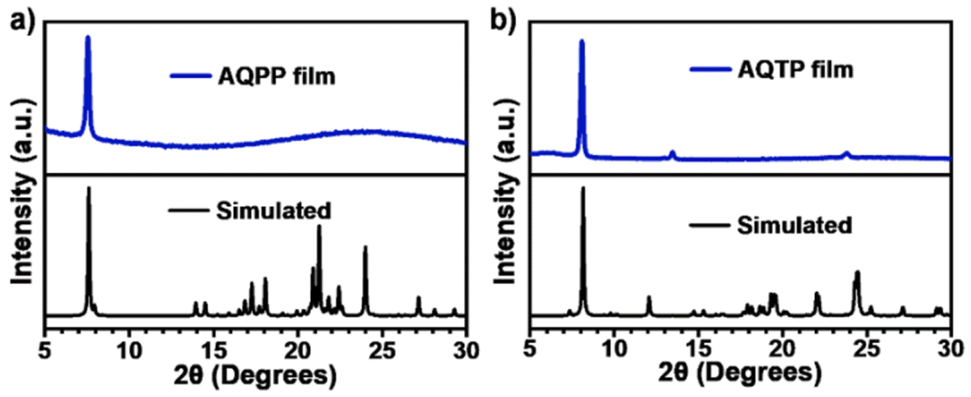
The solid thin films of these AQ derivatives were prepared by a vacuum deposition method. The results from X-ray diffraction (XRD) measurements show that these thin films are polycrystalline in nature and adopt a molecular packing mode similar to that in crystal structures (Fig. 3). In the thin films, the absorption spectra display obvious broadening and red shift compared to these in solutions due to the strong intermolecular interaction in the aggregate state (Fig. 2b and c). The absorption enhancement in the ultraviolet and deep-blue light regions is conducive to the practical application of the SF process for harvesting high-energy photons.1,2 The low-energy absorption was then attributed to the strong mixing between the molecular singlet localized excited (LE) and intermolecular CT states as well as the corresponding vibronic coupling effect. Compared to AQPP thin films, AQTP thin films show a more significantly red-shifted absorption band tail due to the different molecular packing motifs as discussed above (Fig. 2a). Such a solid aggregation difference also leads to totally different PL behaviors in the thin films of two molecules. AQPP molecules feature a broad and featureless excimer PL emission peak around 558 nm with biexponential fluorescence lifetimes of τ1 = 1.0 ns and τ2 = 5.7 ns, and a quantum yield of 0.12 (Fig. 2b and d). By contrast, AQTP molecules display a characteristic PL peak around 608 nm with an obvious vibrational fine-structure (Fig. 2c). However, the weak and short-lived fluorescence emission appears within the instrument response function (IRF) and has an extremely low quantum yield of 0.003, which excludes the accurate characterization of its lifetime (Fig. 2d). The results clearly indicate that an ultrafast photophysical process induces the rapid excited-state deactivation in the AQTP thin films.
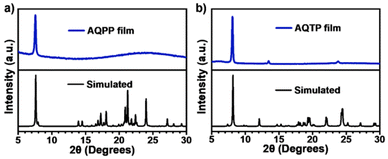 |
| | Fig. 3 XRD diffractograms (blue) and simulated powder patterns (black) of AQPP (a) and AQTP (b) thin films. | |
Excited-state dynamics in dilute solutions
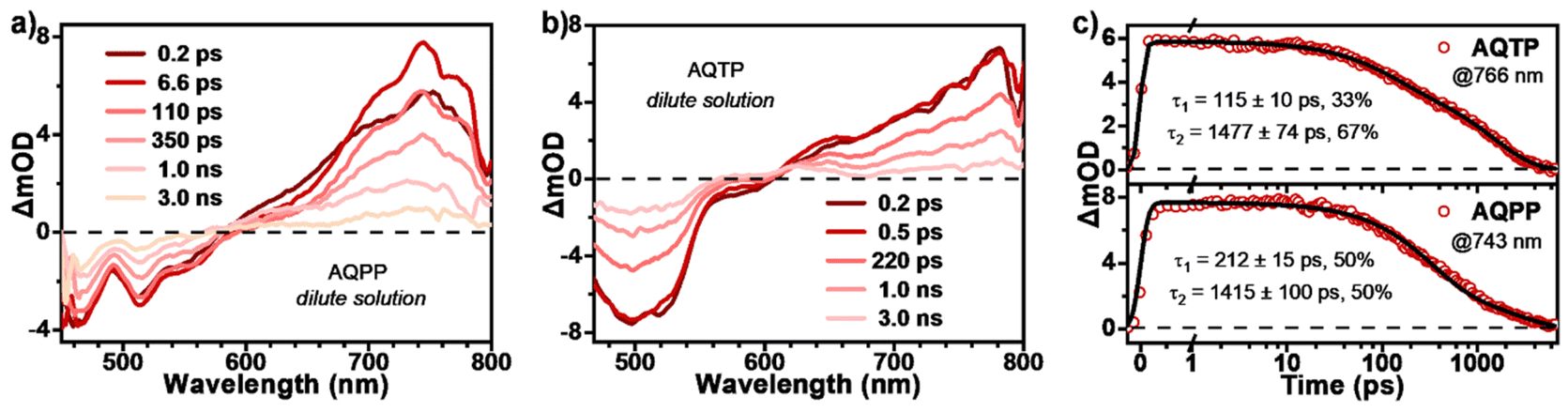
In order to reveal the excited-state photophysics of two AQ derivatives, transient absorption (TA) measurements were then performed in dilute solutions (Fig. 4). The TA data present a similar spectral evolution featuring singlet-dominated excited-state deactivation, namely radiative transition process, for both molecules in monomer state, which are consistent with the results from steady-state characterizations. Specifically, upon 450 nm photo-excitation, the TA spectra of AQPP molecules display negative ground state bleaching (GSB) around 470 nm, characteristic stimulated emission (SE) signals around 506 nm and a positive excited-state absorption (ESA) band around 600–800 nm (Fig. 4a). The TA spectra of AQTP present the positive ESA band around 600–800 nm and the negative GSB and SE signals around 500–600 nm (Fig. 4b). As the time delay increases, the TA signals of both molecules start to decay without any obvious spectral change, featuring the ESA band gradually attenuating, concurrent with the GSB and SE signals recovering. Then the TA data could be fit to biexponential decay with τ1 = 115 ps (33%) and τ2 = 1.5 ns (67%) for AQPP and τ1 = 212 ps (50%) and τ2 = 1.4 ns (50%) for AQTP molecules (Fig. 4c). These two lifetimes correspond to the structural relaxation and radiative decay processes in the singlet excited-state, respectively. Based on the steady-state and transient-state characterizations, singlet radiative transition dominates the excited-state deactivation process in the dilute solution of two AQ derivatives.
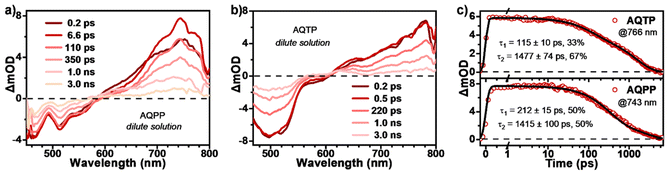 |
| | Fig. 4 Excited-state dynamics of AQs in dilute solutions. TA spectra of (a) AQPP and (b) AQTP in dilute DCM solutions (excited at 450 nm). (c) Selected kinetic curves and corresponding fitting decay constants for AQPP and AQTP solution measurements. | |
Excited-state dynamics in solid aggregates
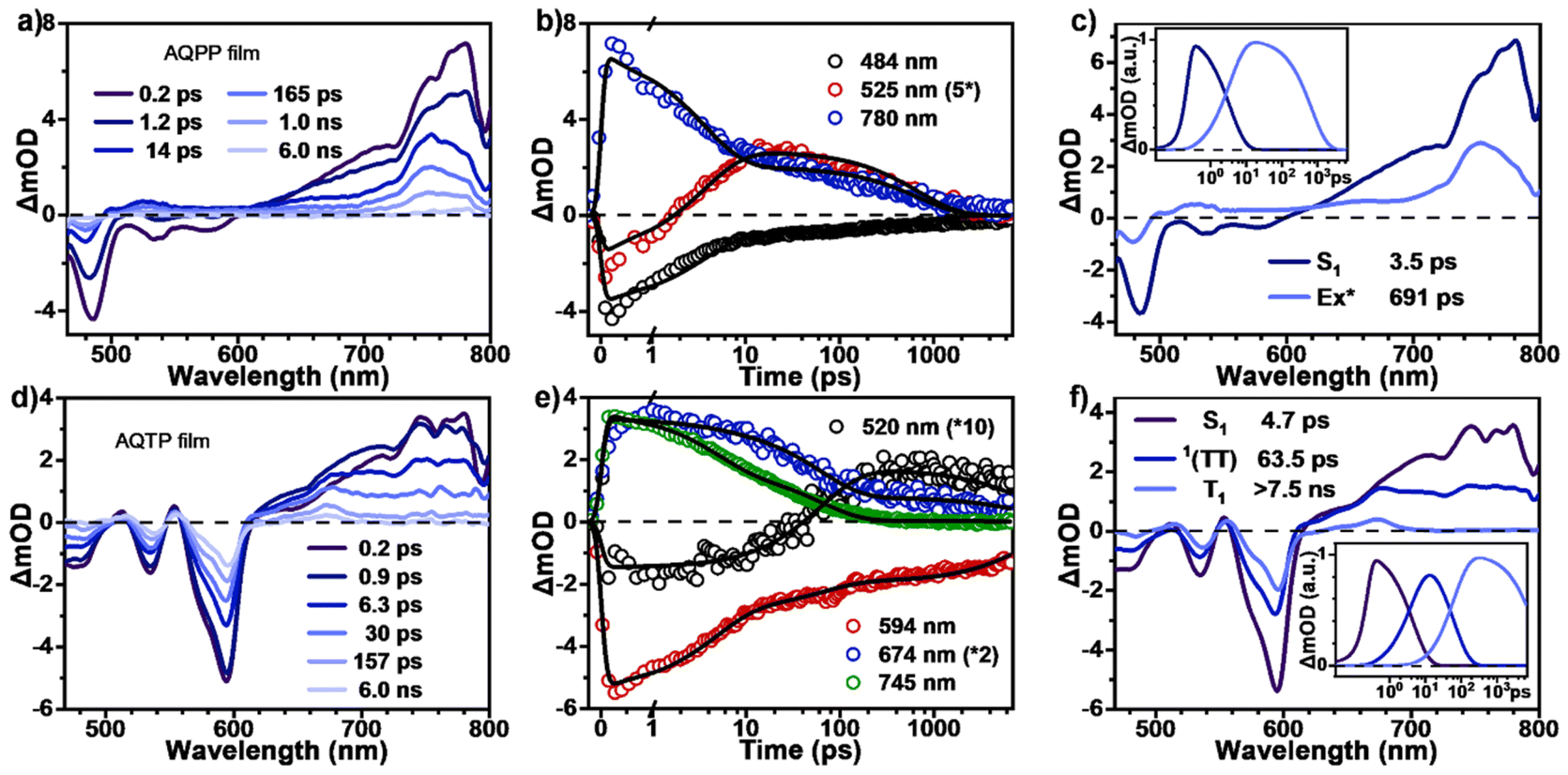
Subsequently, TA measurements were also carried out in the thin films of two AQ derivatives to study their excited-state dynamics in solid aggregates (Fig. 5). The TA data clearly indicate that the excited-state populations trap in excimer in the AQPP thin films, while they undergo the SF process to generate long-lived triplet excitons in the AQTP thin films. After photo-excitation, the TA spectra of AQPP thin films are similar to those of dilute solutions at early times, which are composed of the negative GSB around 490 nm, the SE signals around 500–600 nm and the positive ESA band around 600–800 nm (Fig. 5a). Within first few picoseconds, the ESA band decays and the GSB signals decrease substantially. Concurrent with these spectral changes, a new ESA band appears around 500–600 nm overlapping with the original GSB and SE signals, suggesting the formation of a new species. The selected kinetic decay curves clearly show this spectral evolution (Fig. 5b). At the later time delay, the new ESA band decays to baseline without further spectral changes. Then global analysis was performed and the TA data were well fit to a two-state kinetic model with time constants of 3.5 and 695 ps (Fig. 5c). As characterized by the steady-state PL spectra, AQPP molecules feature a broad and featureless excimer PL emission peak around 450–750 nm in thin films (Fig. 2b), which coincides with the new ESA band, leading to the substantial cancellation of the two transient signals. Therefore, the two transient species were then assigned to the optically populated S1 and the newly formed excimer (Ex*) states, respectively. That is, the excited-state populations rapidly trap in excimer in the AQPP thin films.
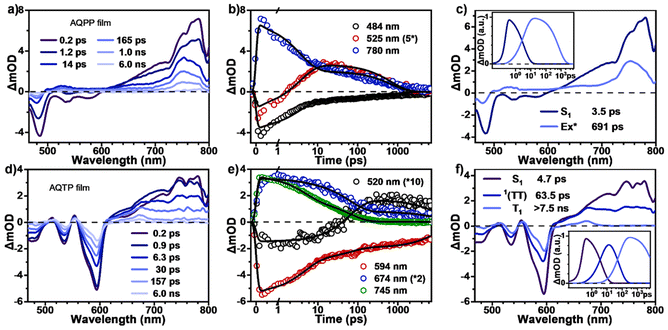 |
| | Fig. 5 Excited-state dynamics of AQs in solid aggregates. TA spectra and selected kinetic decay curves as well as global analysis results for (a–c) AQPP and (d–f) AQTP thin films (excited at 450 nm). | |
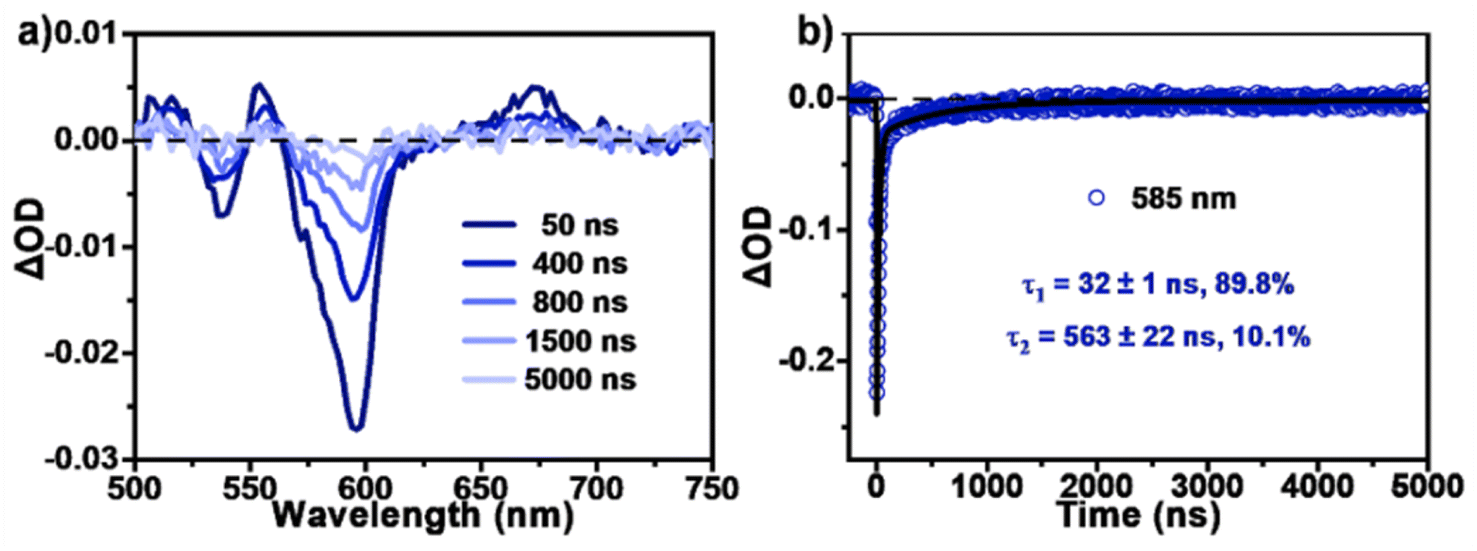
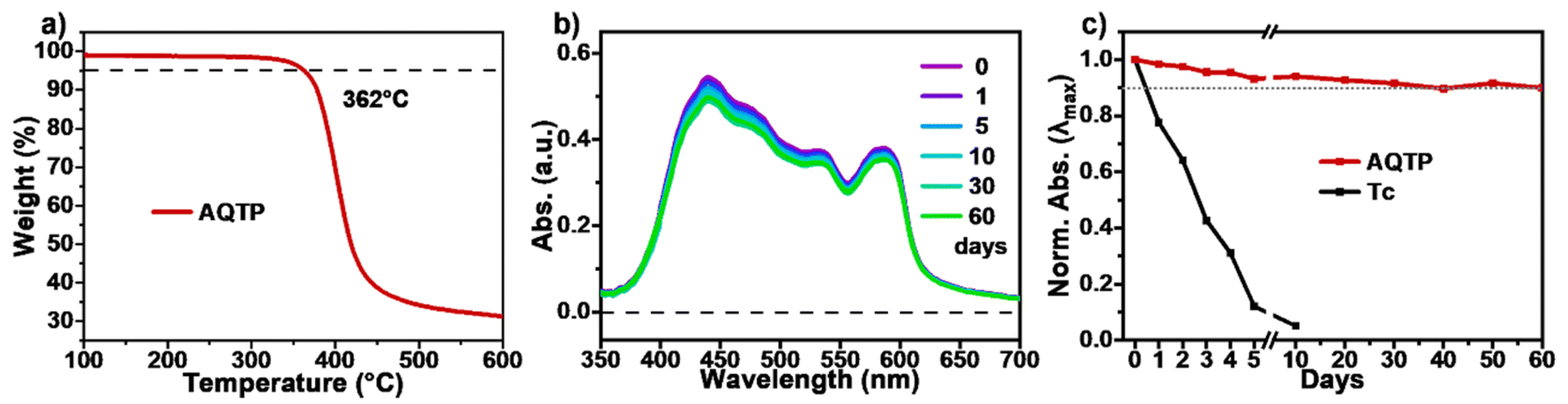
By contrast, the TA spectra of AQTP thin films present more significant spectral evolution changes (Fig. 5d–f). Upon 450 nm photo-excitation, the spectra present negative GSB signals around 470–600 nm and a positive broad ESA band around 500–800 nm (Fig. 5d). Subsequently, as the long-wavelength ESA band gradually becomes weak, the amplitude of the GSB signals shows a large cancellation concurrent with a new ESA band appearing around 470–750 nm within tens of picoseconds, indicating the emergence of a new species (Fig. 5d and e). After that, the TA signals decay without further spectral changes beyond the detection time window of our apparatus. In order to track the decay dynamics of the end transient species in the TA spectra, we performed nanosecond TA (ns-TA) measurements for the same thin-film sample (Fig. 6). The results indicate that the absorption line shape of the ns-TA spectra remains constant and overlaps well with the end transient signals in the TA spectra (Fig. 6a). Moreover, the species turns out to be a long-lived transient population that could persist over several microseconds. We assigned it to the long-lived triplet exciton in the thin films based on triplet sensitization experiment (for details, see Section 6.4 of the ESI†). The singlet depletion method was applied to determine the triplet yield for the AQTP thin films (for details, see Section 6.5 of the ESI†).8,21 A triplet yield of 166% ± 30% was then estimated. Considering the ultrafast formation rate and high triplet yield, we conclude that an efficient SF process dominates the excited-state deactivation, resulting in the long-lived triplet populations observed in the TA spectra of AQTP thin films. Then the global analysis on TA data was performed using a three-state sequential kinetic model providing the transient species that the time constants of 4.7 and 63.5 ps, and >7.5 ns, respectively (Fig. 5f). That is, the excited-state populations could undergo an ultrafast SF process to generate long-lived triplet excitons in the AQTP thin films, which is well consistent with the extremely short fluorescence lifetime in the transient PL characterization. Then the SF-generated triplet excitons decay with a biexponential kinetics with lifetimes τ1 = 32 ns (89.8%) and τ2 = 563 ns (10.1%) (Fig. 6b). We attribute these two different decay components to the lifetimes of the triplet states that undergo triplet–triplet annihilation in situ and the triplet states that diffuse into each other until annihilation, respectively.8,21 In addition, the stability tests show that the AQTP molecule has excellent thermal stability and photostability (Fig. 7). The thermogravimetric analysis (TGA) results indicate that the molecule has a high thermo-decomposing temperature of 362 °C (Fig. 7a). The long-period storage tests exhibit that the film of the conventional SF model molecule, tetracene (Tc), loses almost all of its initial absorbance after 5 days, while the AQTP film maintains over 90% of its original absorbance after 60 days and exhibits excellent photostability (Fig. 7b and c). Thus, the AQTP molecule turns out to be a practical SF material system, given its high extinction coefficient, suitable energy levels, excellent SF property and good stability.1,2,8
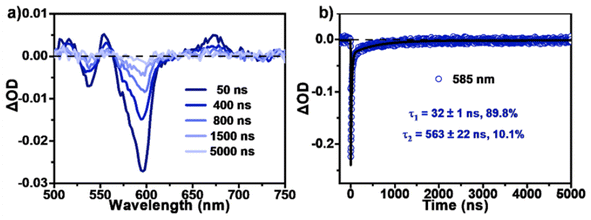 |
| | Fig. 6 Triplet-state decay dynamics. (a) ns-TA spectra and (b) selected kinetic decay curves of AQTP films (excited at 532 nm). | |
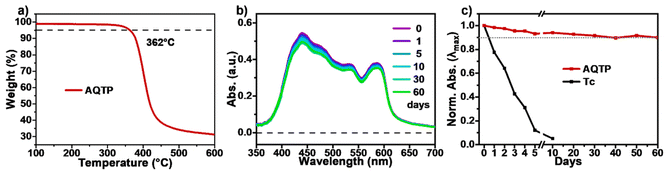 |
| | Fig. 7 Stability tests. (a) TGA curve of AQTP molecules. (b) Changes in the UV-vis absorption spectra of AQTP thin films over 60 days. (c) Normalized decay profiles of the absorption maximum of AQTP compared to Tc films. | |
Role of the CT state in the SF mechanism of the AQ system
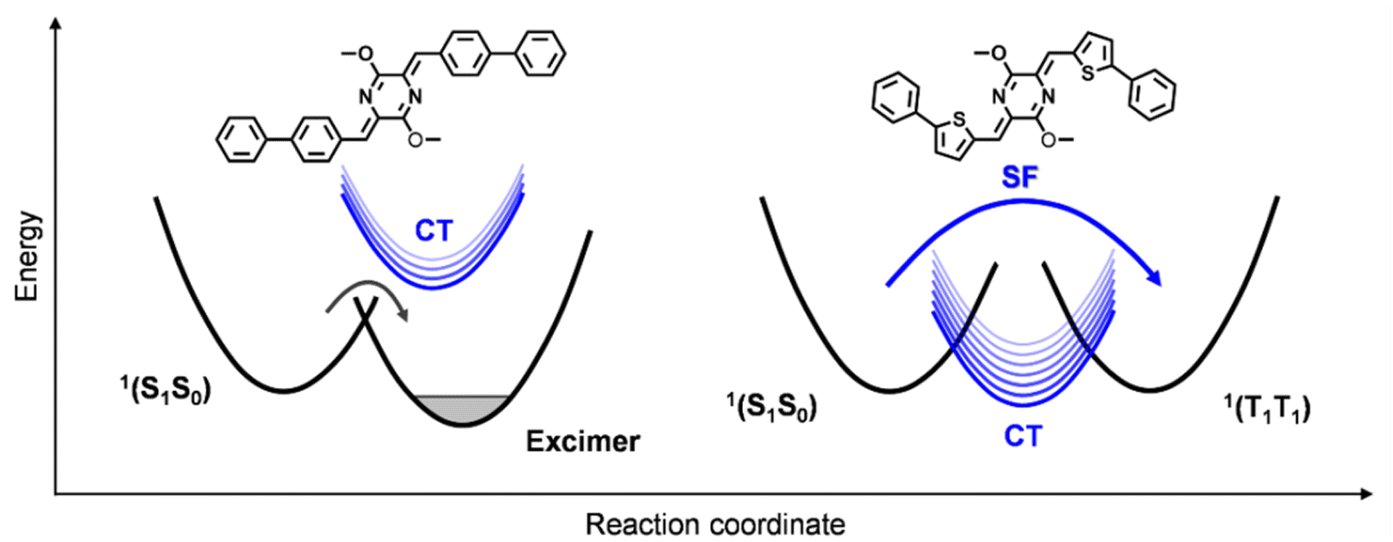
The results from the steady-state and TA characterizations clearly indicate that the excited-state populations undergo an ultrafast SF process to generate long-lived triplet excitons in the AQTP thin films, while they trap in excimer species in the AQPP thin films. The crystal structure data indicate that two molecules have similar slip-stacked molecular arrangements but different transverse offsets and π–π distances in the most closely associated dimer unit (Fig. 2a). The previous work supposed that the CT state might be involved in the SF process of the AQ system.8 In order to unravel the SF mechanism, we performed theoretical calculations to the effect of molecular arrangement on excited-state energy landscape and SF process.21–25 The results show that the CT state was estimated to be 2.85 eV and 0.37 eV above the singlet state (2.48 eV), and the CT coupling was calculated to be −1147 cm−1 in the dimer unit of the AQPP system. Thus, the high-lying CT state and weak CT coupling interaction make the excited-state populations trap in excimer species in the AQPP thin films (Scheme 2). By contrast, the AQTP molecules holds a CT state at 2.10 eV close to the singlet state of 2.07 eV, leading to a strong CT coupling of −5746 cm−1 in the dimer unit. The suitable energy level and strong coupling interaction enable the CT state to mediate the ultrafast SF process via superexchange mechanism in the AQTP thin films.26 We conclude that the aromatic substituents could exert influence on CT coupling interactions via adjusting molecular packing in the aggregate state to shunt the excited-state population to exert an SF process or trap in excimer species in the AQ system.
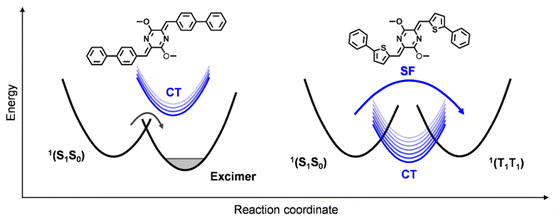 |
| | Scheme 2 Schematic of the excimer formation and SF processes in the AQ systems. | |
Conclusions
Based on the comprehensive theoretical analysis and spectroscopic studies, we investigated the multiple effects of aromatic substituents on excited-state properties and SF process in the AQ system. The results indicate that the aromatic substituents not only have a significant influence on molecular diradical characters and then determine the excited-state energetics of the SF material system but also exert influence on CT coupling interactions to shunt the excited-state population to exert SF process or trap in excimer species. Moreover, the results indicated that the CT state-mediated superexchange mechanism is responsible for the ultrafast SF process in the studied AQ system. More importantly, a practical SF material system based on a phenylthienyl-substituted AQ molecule, AQTP, has been successfully fabricated with a high extinction coefficient, suitable energy levels, excellent SF property and good stability. Deep insights into the multiple regulatory effects of the aromatic substituents on excited-state properties and SF process could promote the development of new SF-active chromophores and practical applications of new-generation light-harvesting materials.
Experimental section
Computational details
All the calculations were performed in the gas phase using the Gaussian 16 program package.27 The geometries of the ground state (S0) were optimized at the PBE0/6-311G(d) level.28 The geometries and the excitation energies of the singlet and triplet states were optimized and calculated using TD-DFT and DFT methods, respectively. The multiple diradical characters (y0) of these molecules were based on the spin-projected UHF theory.29 The symmetry breaking function of the molecular system was calculated at the theoretical UHF/6-31G* guess = mix level, and the double radical eigen overlap integral T based on the double orthogonalization orbital was calculated using the Multiwfn program,30 with the UHF wavefunction diradical characters expressed as y0 = 1 − (2 × T)/(1 + T2).
Spectroscopic measurements
UV-vis absorption and PL spectra were recorded using Shimadzu UV-3600 and Hitachi F-4500 spectrometers, respectively. Transient PL attenuation tests were performed using an Edinburgh FLS980 spectrometer equipped with an EPL-375 picosecond pulsed diode laser. PL quantum yields (PLQY) of the molecules in dilute solution and thin film were measured by the relative method and integrating sphere, respectively. The test was performed using an absolute PLQY measurement system, C9920-02G (Hamamatsu photonics). Femtosecond and nanosecond transient absorption spectroscopy measurements were all performed using previously described instruments and experimental conditions.31–34 The analysis of the kinetic traces derived from time-resolved spectra was performed using nonlinear least-square fitting to a general sum-of-exponentials function after the deconvolution of IRF. All the spectroscopic measurements were carried out at room temperature, unless specified otherwise.
Computational details of electronic coupling
The calculation was performed using a theoretical method previously demonstrated to be effective in modeling electronic coupling in SF systems of perylenediimide, diketopyrrolopyrrole and flavanthrene.21–25 The side chains are not expected to substantially change the excited-state energies as well as the electronic coupling values compared to the effect of slip-stacking patterns, and thus butyl substituents are simplified to methyl groups in molecular geometry for all calculations.21 The monomer molecular geometry was optimized and then placed in the corresponding dimer unit of crystal structures. The CT state energy, E(CT), was calculated using a Weller-like equation,35E(CT) = IP + EA + Esol + Eelec, where IP is the ionization potential, EA is the electron affinity, Esolv is the solvation energy for the dimer unit in the crystal structure, and Eelec is the electrostatic energy between the ion pair formed in the CT state. All calculations were performed using the Amsterdam Density Functional (ADF) package (B3LYP/TZ2P).36 The CT coupling values were calculated by the theoretical methods.23–25
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for AQPP and AQTO have been deposited at the CCDC under numbers 2381000 and 2381001 and can be obtained from https://www.ccdc.cam.ac.uk/data_request/cif.
Author contributions
L. W. conceptualized and conceived the project. Z. X. Z. and S. H. W. synthesized the compounds, conducted the photophysical characterizations and performed the data analysis, which were supervised by X. M. S., L. W. and H. B. F., Z. X. Z., L. W. wrote the manuscript and all the authors participated in the data analysis and discussions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22479107 and 22005210) and by the Fundamental Research Program of Shanxi Province (No. 202203021224004 and 20210302124469).
Notes and references
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- J. L. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. P. Liu, L. M. Campos and T. L. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef PubMed.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS PubMed.
- D. N. Congreve, J. Y. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485–3518 RSC.
- L. Wang, X. Shi, S. Feng, W. Liang, H. Fu and J. Yao, CCS Chem., 2022, 4, 2748–2756 CrossRef CAS.
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. W. Zhou, R. M. Young, Y. L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed.
- F. S. Conrad-Burton, T. Liu, F. Geyer, R. Costantini, A. P. Schlaus, M. S. Spencer, J. Wang, R. H. Sánchez, B. Zhang, Q. Xu, M. L. Steigerwald, S. Xiao, H. Li, C. P. Nuckolls and X. Zhu, J. Am. Chem. Soc., 2019, 141, 13143–13147 CrossRef CAS PubMed.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- L. Wang, W. Jiang, S. Guo, S. Wang, M. Zhang, Z. Liu, G. Wang, Y. Miao, L. Yan, J.-Y. Shao, Y.-W. Zhong, Z. Liu, D. Zhang, H. Fu and J. Yao, Chem. Sci., 2022, 13, 13907–13913 RSC.
- L. Mencaroni, B. Carlotti, F. Elisei, A. Marrocchi and A. Spalletti, Chem. Sci., 2022, 13, 2071–2078 RSC.
- Z. Liu, L. Wang, Q. Deng, J. Zhu, J. Sun, H. Wang, H. Fu and J. Yao, ACS Mater. Lett., 2023, 5, 3010–3016 CrossRef CAS.
- S. Wang, X.-Y. Liu, M. Zhang, L. Wang, G. Cui, H. Fu and J. Yao, CCS Chem., 2024, 6, 2142–2149 CrossRef CAS.
- K. Wang, X. You, X. Miao, Y. Yi, S. Peng, D. Wu, X. Chen, J. Xu, M. Y. Sfeir and J. Xia, J. Am. Chem. Soc., 2024, 146, 13326–13335 CrossRef CAS PubMed.
- X. Liu, B. He, C. L. Anderson, J. Kang, T. Chen, J. Chen, S. Feng, L. Zhang, M. A. Kolaczkowski, S. J. Teat, M. A. Brady, C. Zhu, L.-W. Wang, J. Chen and Y. Liu, J. Am. Chem. Soc., 2017, 139, 8355–8363 CrossRef CAS PubMed.
- C. L. Anderson, N. Dai, S. J. Teat, B. He, S. Wang and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 17978–17985 Search PubMed.
- H. Liang, C. Liu, Z. Zhang, X. Liu, Q. Zhou, G. Zheng, X. Gong, L. Xie, C. Yang, L. Zhang, B. He, J. Chen and Y. Liu, Adv. Funct. Mater., 2022, 32, 2201903 Search PubMed.
- X. Liu, C. L. Anderson and Y. Liu, Acc. Chem. Res., 2023, 56, 1669–1682 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 Search PubMed.
- N. Renaud, P. A. Sherratt and M. A. Ratner, J. Phys. Chem. Lett., 2013, 4, 1065–1069 Search PubMed.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed.
- X. Fei, S. Zhang, D. Zhai, Z. Wang, J.-L. Lin, Q. Xiao, C.-L. Sun, W. Deng, C. Zhang, W. Hu and H.-L. Zhang, Chem. Sci., 2022, 13, 9914–9920 RSC.
- C. E. Miller, M. R. Wasielewski and G. C. Schatz, J. Phys. Chem. C, 2017, 121, 10345–10350 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- K. Yamaguchi, Chem. Phys. Lett., 1975, 33, 330–335 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. Wang, S. Bai, Y. Wu, Y. Liu, J. Yao and H. Fu, Angew. Chem., Int. Ed., 2020, 59, 2003–2007 Search PubMed.
- L. Wang, X. Liu, X. Shi, C. L. Anderson, L. M. Klivansky, Y. Liu, Y. Wu, J. Chen, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 17892–17896 Search PubMed.
- L. Wang, T.-S. Zhang, L. Fu, S. Xie, Y. Wu, G. Cui, W.-H. Fang, J. Yao and H. Fu, J. Am. Chem. Soc., 2021, 143, 5691–5697 Search PubMed.
- L. Wang, L. Lin, T.-S. Zhang, S. Guo, Z. Liu, M. Zhang, S. Wang, G. Cui, W.-H. Fang, J. Zhu, H. Fu and J. Yao, CCS Chem., 2023, 5, 2264–2276 Search PubMed.
- H. Oevering, M. N. Paddon-Row, M. Heppener, A. M. Oliver, E. Cotsaris, J. W. Verhoeven and N. S. Hush, J. Am. Chem. Soc., 1987, 109, 3258–3269 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Long
Wang
c and
Long
Wang