
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-high molecular weight polymer synthesis via aqueous dispersion polymerization†
Cabell B.
Eades
 ,
Kaden C.
Stevens
,
Danyella E.
Cabrera
,
Micayla K.
Vereb
,
Megan E.
Lott
,
Jared I.
Bowman
and
Brent S.
Sumerlin
*
,
Kaden C.
Stevens
,
Danyella E.
Cabrera
,
Micayla K.
Vereb
,
Megan E.
Lott
,
Jared I.
Bowman
and
Brent S.
Sumerlin
*
George & Josephine Butler Polymer Research Laboratory, Center for Macromolecular Science & Engineering, Department of Chemistry, University of Florida, PO Box 117200, Gainesville, Florida 32611, USA. E-mail: sumerlin@chem.ufl.edu
First published on 27th February 2025
Abstract
The synthesis of ultra-high molecular weight (UHMW, Mn ≥ 106 g mol−1) polymers is generally complicated by the high viscosity of the resulting polymer solution. We report the synthesis of UHMW double-hydrophilic block copolymers (DHBCs) by leveraging polymerization-induced self-assembly (PISA) to obtain concentrated but free-flowing dispersions of UHMW water-soluble particles. By polymerizing N-acryloylmorpholine (NAM) from a poly(N,N-dimethylacrylamide) (PDMA) macroiniferter in the presence of a kosmotropic salt ((NH4)2SO4), the salt sensitivity of the resultant poly(NAM) (PNAM) block induced self-assembly to result in free-flowing dispersions of polymeric particles (η < 6 Pa·s), despite the UHMW and high concentration of the newly formed block copolymer. To retrieve the UHMW polymer products, simple dilution with water lowered the (NH4)2SO4 concentration sufficiently to resolubilize the PNAM chains, affording a highly viscous solution of fully dissolved DHBCs. The simplicity of this synthetic route has important implications for the facile production of UHMW materials on an industrial scale.
Introduction
Ultra-high molecular weight (UHMW) polymers (Mn ≥ 106 g mol−1) synthesized via reversible-deactivation radical polymerization (RDRP) techniques have recently become potentially important components in the development of next-generation materials.1–4 While polymers in this molecular weight regime are accessible through a select group of conventional polymerization approaches, these methods generally do not allow access to materials with targetable molecular weights, functionalized chain ends, narrow molecular weight distributions, or advanced polymer architectures.5,6 While RDRP techniques have been used to synthesize polymer materials bearing all these characteristics, achieving UHMW has traditionally proven challenging, requiring specialized reaction conditions, such as high pressures7–9 or catalyst loadings.10–13 Recent reports, however, have used more mild, accessible conditions to obtain UHMW materials.4,14–19Notably, our group has demonstrated that photoiniferter polymerization can be leveraged to access UHMW polymers with excellent control over chain length and dispersity.20–26 Hartlieb and coworkers have shown that the chain-end fidelity maintained during photoiniferter polymerizations can be exploited to permit the synthesis of block copolymers with 20 blocks and molecular weights exceeding 1800 kg mol−1.27 Despite the molecular weight range afforded by this photochemical approach, the synthesis of UHMW polymers ultimately yields viscous solutions that complicate purification and processing of the polymer product. In an attempt to remedy this issue, our research group has reported the synthesis of UHMW polymers in inverse miniemulsion conditions.28–30 This heterogeneous approach maintains low solution viscosity by confining the polymerization to water droplets dispersed in a nonpolar solvent. Despite the success of this technique for low-viscosity UHMW polymer synthesis, the large amount of surfactant necessary to stabilize the reaction droplets may preclude its application on an industrial scale.
Polymerization-induced self-assembly (PISA) is another well-reported, heterogeneous methodology that leverages in situ phase separation for polymer synthesis.31–37 PISA involves a solvophilic macroinitiator being chain-extended with a soluble monomer that polymerizes to form a solvophobic polymer. Self-assembly of the nascent block copolymers occurs when the extending block reaches a critical degree of polymerization (DP) such that it becomes sufficiently solvophobic to self-assemble.34,38In situ formation of polymeric nanoparticles, as opposed to molecularly dissolved block copolymers, keeps the reaction medium from becoming viscous during the polymerization.39,40 However, despite the utility of PISA as a technique for nanoparticle preparation, the controlled synthesis of UHMW polymers has remained relatively unexplored using this route.35 Recently, Armes and coworkers have reported on low-viscosity syntheses of high-molecular-weight hydrophilic block copolymers via PISA.41–44 Polymeric nanoparticles were obtained by chain-extending hydrophilic macro-chain transfer agents with monomers that yielded thermoresponsive core-forming blocks. Carrying out PISA in the presence of aqueous salt solutions caused the otherwise hydrophilic core-forming blocks to become hydrophobic as they grew, ultimately leading to self-assembly. This approach was able to access high molecular weight (Mn > 500 kg mol−1, 1.9 < Đ < 2.4) double-hydrophilic block copolymers (DHBCs) while maintaining a relatively low solution viscosity. However, the high amounts of exogenous initiator used resulted in many chains becoming permanently terminated, limiting the achievable molecular weights and resulting in polymers with relatively high dispersity.
We sought to combine these findings with our approach of leveraging PISA to simplify complex polymer synthesis45 to prepare hydrophilic narrow-dispersity UHMW polymers via photoiniferter PISA. By chain-extending poly(N,N-dimethylacrylamide) (PDMA) macroiniferters with N-acryloylmorpholine (NAM) in aqueous (NH4)2SO4, the nascent, salt-sensitive PNAM segments self-assembled into polymeric particles with eventual hydrodynamic diameters (Dh) of 500 nm–2.5 μm (Fig. 1). By using photoiniferter polymerization, we were able to reach molecular weights in excess of 106 g mol−1 without sacrificing dispersities. Dilution of the resultant particle solutions with water lowered the salt concentration, solubilizing the PNAM blocks and yielding a viscous solution of molecularly dissolved UHMW block copolymers. These results suggest PISA can be exploited as a tool to afford UHMW copolymers at high concentrations while avoiding the complication of high viscosity.
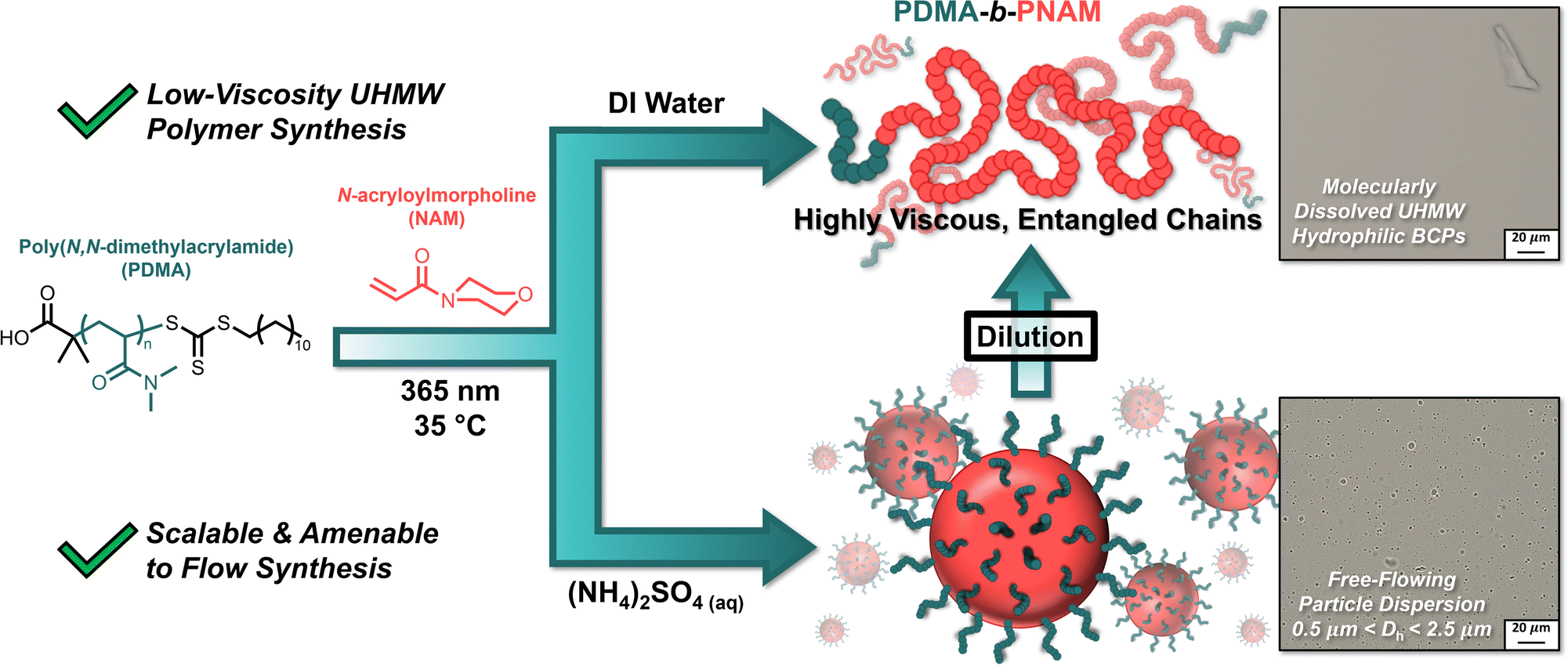 | ||
| Fig. 1 Synthesis of hydrophilic ultra-high molecular weight (UHMW) polymers can be simplified by leveraging the benefits of dispersion polymerizations. The polymerization-induced self-assembly (PISA) of salt-sensitive poly(N-acryloylmorpholine) (PNAM) chains in aqueous (NH4)2SO4 yields a free-flowing solution of UHMW polymer particles. Upon dilution with water, the (NH4)2SO4 concentration is lowered sufficiently to solubilize the PNAM chains, resulting in a highly viscous solution of molecularly dissolved UHMW block copolymers. | ||
Results and discussion
We first set out to synthesize three PDMA macroiniferters of varying molecular weights to assess the necessary coronal chain size for steric stabilization of UHMW polymer core chains. UV light-mediated photoiniferter polymerization (365 nm, 3.5 mW cm−2) afforded PDMA macroiniferters of 30.5 kg mol−1 (MI30k), 81.4 kg mol−1 (MI80k), and 124.1 kg mol−1 (MI120k).46 Size-exclusion chromatography (SEC) analysis indicated well-controlled polymerizations with close agreement between theoretical and experimental molecular weights and low dispersities (Table S1†). With macroiniferters in hand, we then began conducting PISA experiments to find the upper limit of the number-average degree of polymerization (DP) for the core block that each macroiniferter could stabilize before macroscopic precipitation of the polymer chains was observed. Informed by recent findings by Armes and coworkers, we chose to chain extend our macroiniferters with N-acryloylmorpholine (NAM) in 0.5 M (NH4)2SO4 (Fig. 2A).41,42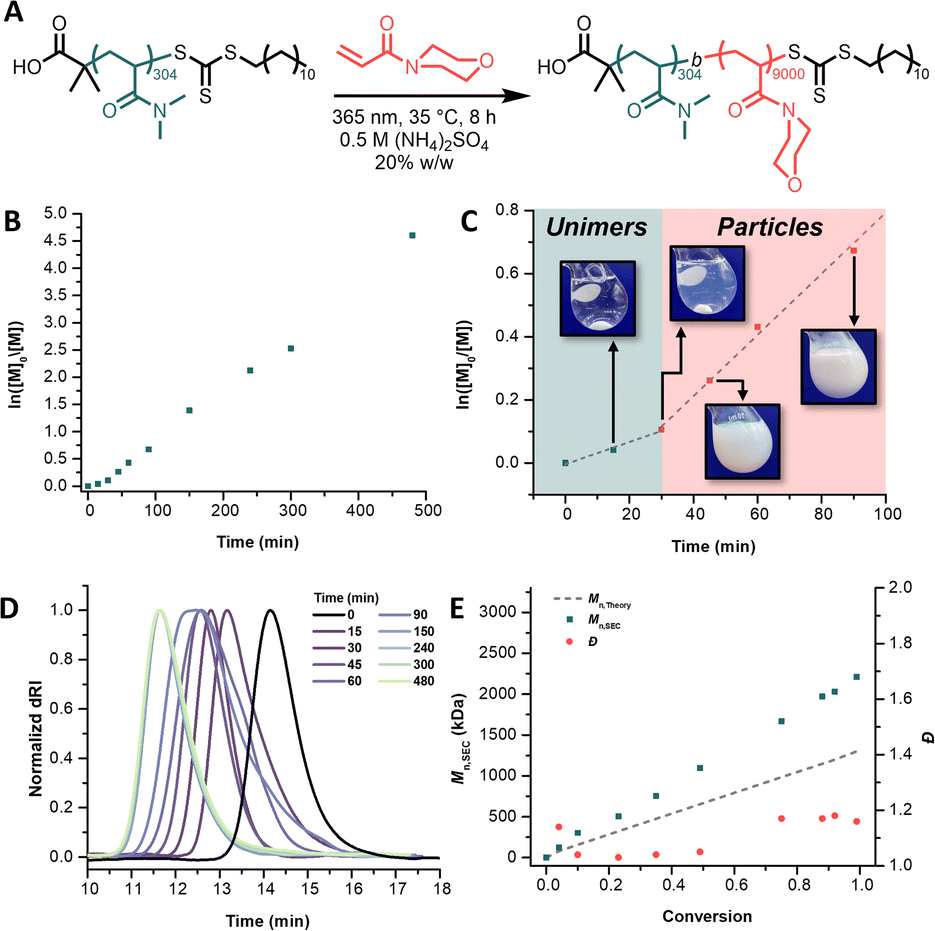 | ||
| Fig. 2 (A) Reaction scheme depicting chain extension of poly(N,N-dimethylacrylamide) (PDMA) with N-acryloylmorpholine (NAM) in 0.5 M (NH4)2SO4via UV light-mediated photoiniferter polymerization; (B) pseudo-first-order kinetic plot of PDMA-b-PNAM particle synthesis (target core degree of polymerization = 9000) displaying a constant radical concentration during the polymerization; (C) increase in apparent polymerization rate before (green region, kp,app = 0.00351 min−1) and after (red region, kp,app = 0.00969 min−1) assembly, along with images of the nascent polymer particles corresponding to 15, 30, 45, and 90 min (left to right); (D) size-exclusion chromatograms showing molecular weight evolution during PISA as indicated by peaks shifting to lower elution times; (E) close agreement between theoretical (gray dashed line) and experimental (green squares) molecular weights and low dispersities (red squares) indicating good control for the duration of the polymerization. | ||
To overcome the high extents of irreversible chain termination that had been previously observed, we chose to perform our PISA reactions via photoiniferter polymerization. This approach resulted in drastically increased chain-end fidelity, which consequently afforded access to UHMW PDMA-b-PNAM with excellent control over molecular weight distributions (Đ < 1.3) (Fig. 2). Linear pseudo-first-order kinetics were observed for the chain extension of MI30k with a target core DP of 9000 (Fig. 2B). Closer inspection revealed a marked increase in the apparent rate constant of propagation (kp,app) after 30 min, coinciding with when self-assembly was visually observed (Fig. 2C). A transition from a completely transparent, homogeneous solution to a slightly blue, turbid, heterogeneous one was a result of the PDMA-b-PNAM particles nucleating and scattering light.47–49 SEC analysis (Fig. 2D) showed shifts to lower elution times as the PDMA macroiniferter was chain extended with NAM, implying efficient chain extensions with high maintenance of chain-end fidelity. SEC kinetic analysis also indicated close agreement between theoretical and experimental molecular weights while maintaining low dispersities (Fig. 2E).
We then expanded our macroiniferter and target core DP scope. These experiments are summarized in Table 1. The concentration of (NH4)2SO4 and solids content were held at 0.5 M and 20% w/w, respectively, for all reactions. Chain extending MI30k with 9000 equiv. of NAM in 0.25 M (NH4)2SO4 resulted in a physically entangled, turbid gel. In 1.0 M (NH4)2SO2, the same polymerization conditions resulted in sedimentation of polymer product in under 1 h. Upon chain extending MI30k with a target core DP of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, sedimentation was again observed. MI80k and MI120k, however, were large enough to stabilize PNAM DPs up to 16000 and 18000, respectively, resulting in the desired free-flowing dispersions of polymer particles. These results highlight the importance of selecting an MI of sufficient length to stabilize growth of UHMW salt-responsive segments.
000, sedimentation was again observed. MI80k and MI120k, however, were large enough to stabilize PNAM DPs up to 16000 and 18000, respectively, resulting in the desired free-flowing dispersions of polymer particles. These results highlight the importance of selecting an MI of sufficient length to stabilize growth of UHMW salt-responsive segments.
| Macroiniferter | Target core DP | M n,Theory (kg mol−1)b | M n,SEC (kg mol−1)c | Đ |
|---|---|---|---|---|
| a All chain extensions conducted in 0.5 M (NH4)2SO4 unless otherwise noted. b Calculated from the conversion of monomer determined by 1H NMR spectroscopy. c Determined by size-exclusion chromatography (SEC) equipped with a multiangle light-scattering detector. | ||||
| MI30k (no (NH4)2SO4) | 9000 | 1300 | 1780 | 1.23 |
| MI30k | 9000 | 1300 | 2550 | 1.19 |
| 12000 |
1720 | 2550 | 1.17 | |
| MI80k | 9000 | 1350 | 1760 | 1.19 |
| 12000 |
1780 | 2420 | 1.13 | |
| 14000 |
2060 | 1970 | 1.21 | |
| 16000 |
2340 | 2410 | 1.21 | |
| MI120k | 9000 | 1390 | 1680 | 1.27 |
| 12000 |
1820 | 1880 | 1.21 | |
| 14000 |
2100 | 1970 | 1.23 | |
| 16000 |
2380 | 2560 | 1.15 | |
| 18000 |
2670 | 2080 | 1.11 | |
Resultant polymer particles were then characterized by dynamic light scattering (DLS) (Table 2). Interestingly, we observed that chain extending MI30k with 9000 equiv. of NAM resulted in polymer particles with Z-average hydrodynamic diameters (Dz) much larger than the rest of the formulations, despite it involving the lowest molecular weight macroiniferter and lowest core chain DP. The large size of these particles (Dz > 2 μm, Fig. S16†) permitted the use of visible light microscopy (VLM) for particle imaging (Fig. 3A). By VLM, we observed distinct spherical morphologies with sizes similar to those measured by DLS (Table 2). Upon dilution with water, the particles completely dissociated into molecularly dissolved chains invisible by VLM (Fig. 3B). To further investigate the structure of the PDMA304-b-PNAM9000 particles, we relied on transmission electron microscopy (TEM) (Fig. 3C). The resulting TEM images corroborated our observations by VLM, showing large particles with spherical morphologies.
| Macroiniferter | Target core DP | D z (nm) | PDI |
|---|---|---|---|
| a 0.1% w/w of particles in 0.5 M (NH4)2SO4. | |||
| MI30k | 9000 | 2165 | 0.002 |
| 12000 |
Sed. | Sed. | |
| MI80k | 9000 | 569 | 0.009 |
| 12000 |
835 | 0.204 | |
| 14000 |
873 | 0.053 | |
| 16000 |
1036 | 0.185 | |
| MI120k | 9000 | 527 | 0.034 |
| 12000 |
752 | 0.186 | |
| 14000 |
683 | 0.022 | |
| 16000 |
727 | 0.017 | |
| 18000 |
1278 | 0.076 | |
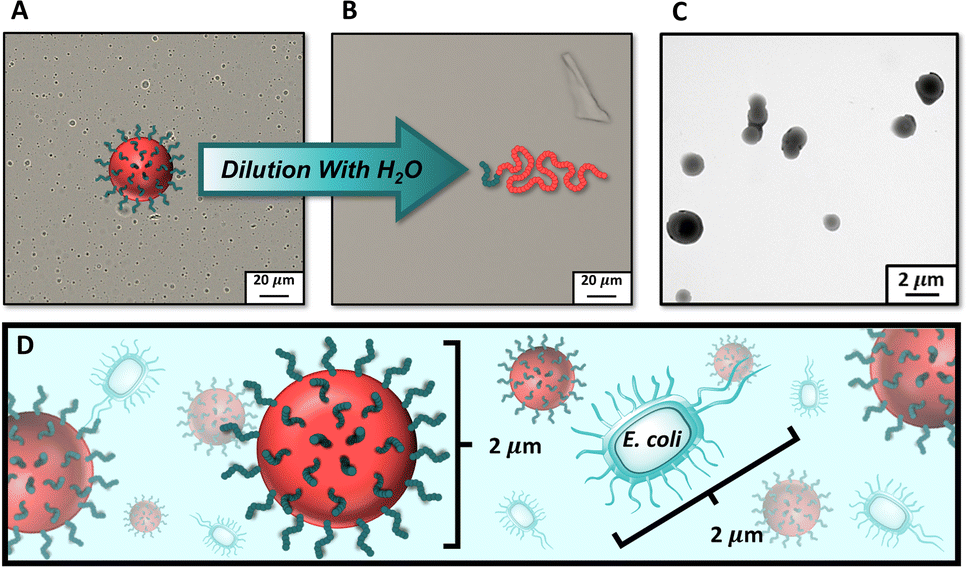 | ||
| Fig. 3 (A) Visible light microscopy (VLM) image of PDMA-b-PNAM particles made by chain extending MI30k with 9000 equiv. of NAM (PDMA304-b-PNAM9000); (B) VLM image of PDMA304-b-PNAM9000 particles after a 1:1 dilution with water, showing the complete dissociation of the particles into molecularly dissolved chains invisible by VLM; (C) transmission electron micrograph of PDMA304-b-PNAM9000 particles; (D) cartoon comparing the size of the PDMA304-b-PNAM9000 particles to that of Escherichia coli cells. | ||
We then employed oscillatory rheology to study the viscosity of the particle dispersions. We observed a decrease in solution viscosity with increasing shear rate, indicative of shear-thinning behaviors (Fig. 4). Furthermore, we compared the viscosity of the native PDMA304-b-PNAM9000 particle solution (0.5 M (NH4)2SO4) to that of a 1:1 dilution of the same solution with DI water (0.25 M (NH4)2SO4). Upon dilution, the completely opaque, white solution of particles becomes perfectly clear and homogeneous with an accompanying drastic increase in observed viscosity. This observation is consistent with dilution of the salt below the level needed to induce insolubility of the PNAM block. Once the core block becomes solvophilic, the block copolymer aggregates are disassembled to yield molecularly dissolved unimers. This occurrence leads to a counterintuitive increase in viscosity upon dilution.41,42
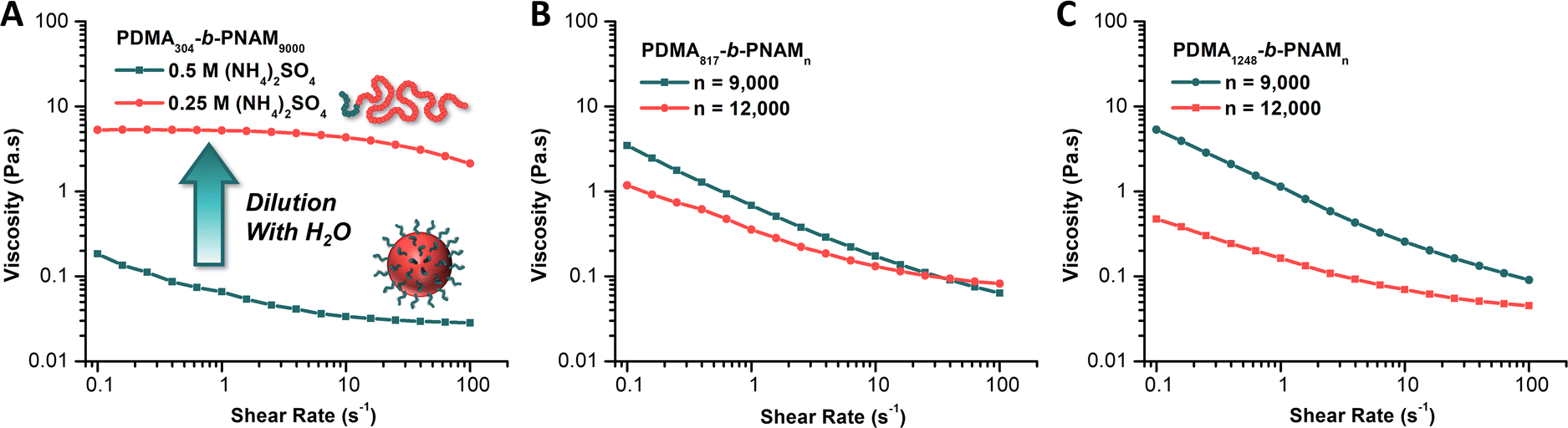 | ||
| Fig. 4 Oscillatory rheology data of: (A) PDMA304-b-PNAM9000 as particles in the native 0.5 M (NH4)2SO4 solution (green) and as molecularly dissolved chains after a 1:1 dilution of the native solution with pure water (red), (B) MI80k after chain extension with 9000 (green) and 12000 (red) equivalents of NAM, and (C) MI120k after chain extension with 9000 (green) and 12000 (red) equivalents of NAM, both in a 0.5 M (NH4)2SO4 solution. | ||
Rheological analysis gave viscosities of 0.185 Pa·s and 5.30 Pa·s for the native and diluted solution, respectively, at a shear rate of 0.1 s−1. At a shear rate of 100 s−1, these became 0.0283 Pa·s and 2.13 Pa·s for the native and diluted solutions, respectively, a difference of approximately two orders of magnitude (Fig. 4A). Further analysis of particle solutions made from chain extending MI80k (Fig. 4B) and MI120k (Fig. 4C) with 9000 and 12000 equiv. of NAM revealed similarly low viscosities for the respective native particle solutions. We observed that the solution viscosities were higher for particles of smaller Dz, which we reasoned was due to the higher surface area of the smaller particles resulting in more significant interparticle interactions.50 We also observed that for a given macroiniferter the viscosity decreased significantly when the core DP increased from 9000 to 12000 (MI80k, Fig. 4B; MI120k, Fig. 4C). While, outwardly, this may seem counterintuitive, we used the same rationale as before, such that larger particles (those with core DP = 12000) result in lower solution viscosities than the dispersions of smaller particles (those with core DP = 9000). Nonetheless, the low viscosities of all particle solutions measured relative to the diluted, molecularly dissolved polymer solution indicated the amenability of these reactions to continuous flow synthesis, a primary focus of ongoing research in our group.
Conclusions
Here, we report on the successful heterogeneous synthesis of UHMW hydrophilic polymers by photoiniferter PISA. By exploiting PISA, we developed a novel method towards UHMW polymer synthesis, wherein the viscosities of these concentrated, but free-flowing, PDMA-b-PNAM particle dispersions were kept relatively low. These results demonstrate a simple, tuneable method by which one can synthesize hydrophilic UHMW polymers in dispersed media by leveraging both the inherent low viscosities of polymer particle dispersions and the salt-dependent hydrophilicity of water-soluble polymers. These findings have the potential to make industrial-scale syntheses of hydrophilic UHMW polymers more facile and feasible, with the hope of bringing these unique materials to commercial prominence.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, C. B. E., K. C. S., B. S. S.; methodology, C. B. E., K. C. S., D. E. C., M. K. V., B. S. S.; investigation, C. B. E., K. C. S., D. E. C., M. K. V., M. E. L.; data curation, C. B. E., K. C. S., D. E. C., M. K. V.; writing, C. B. E., J. I. B., B. S. S.; supervision, B. S. S.; project administration, B. S. S.; funding acquisition, B. S. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Professor Steven P. Armes for helpful discussion and Justin Ellenburg for obtaining visible light microscopy images for this publication. M. E. L. was supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate (NDSEG) Fellowship Program. This material is based on work supported by the National Science Foundation (DMR-2404144) and the DoD through the ARO (W911NF2410050, W911NF2310260).References
- M. Streicher, V. Boyko and A. Blanazs, ACS Appl. Mater. Interfaces, 2023, 15, 59054 CrossRef PubMed.
- Y. Kamiyama, R. Tamate, T. Hiroi, S. Samitsu, K. Fujii and T. Ueki, Sci. Adv., 2022, 8, eadd0226 CrossRef CAS PubMed.
- J. K. D. Mapas, T. Thomay, A. N. Cartwright, J. Ilavsky and J. Rzayev, Macromolecules, 2016, 49, 3733–3738 CrossRef CAS.
- Y. Kamiyama, R. Tamate, K. Fujii and T. Ueki, Soft Matter, 2022, 18, 8582–8590 RSC.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS PubMed.
- J. Rzayev and J. Penelle, Angew. Chem., Int. Ed., 2004, 43, 1691–1694 CrossRef CAS PubMed.
- R. W. Simms and M. F. Cunningham, Macromolecules, 2007, 40, 860–866 CrossRef CAS.
- L. Mueller, W. Jakubowski, K. Matyjaszewski, J. Pietrasik, P. Kwiatkowski, W. Chaladaj and J. Jurczak, Eur. Polym. J., 2011, 47, 730–734 CrossRef CAS.
- P. Zhang, Q. Xu, W. Mao, J. Lv, H. Tang and H. Tang, Polymers, 2023, 15, 2501 CrossRef CAS PubMed.
- M. Yuan, L. Xu, X. Cui, J. Lv, P. Zhang and H. Tang, Polymers, 2020, 12, 2747 CrossRef CAS PubMed.
- S. K. De and M. Bhattacharjee, J. Polym. Sci., Part A:Polym. Chem., 2013, 51, 1540–1549 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- A. Reyhani, S. Allison-Logan, H. Ranji-Burachaloo, T. G. McKenzie, G. Bryant and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1922–1930 CrossRef CAS.
- R. Li, S. Zhang, Q. Li, G. G. Qiao and Z. An, Angew. Chem., Int. Ed., 2022, 61, e202213396 CrossRef CAS PubMed.
- Y. T. Chou, W. R. Lee and S. S. Yu, Macromolecules, 2024, 57, 9241–9249 CrossRef CAS.
- Z. An, ACS Macro Lett., 2020, 9, 350–357 CrossRef CAS PubMed.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097 RSC.
- R. Li and Z. An, Angew. Chem., Int. Ed., 2020, 59, 22258–22264 CrossRef CAS PubMed.
- E. K. Stacy, M. L. McCormick, K. C. Stevens, P. E. Jankowski, J. Aguinaga, D. L. Patton, B. S. Sumerlin and T. D. Clemons, ACS Macro Lett., 2024, 13, 1662–1669 CrossRef CAS PubMed.
- R. W. Hughes, M. E. Lott, R. A. S. Olson and B. S. Sumerlin, Prog. Polym. Sci., 2024, 156, 101871 CrossRef CAS.
- J. B. Young, S. L. Goodrich, J. A. Lovely, M. E. Ross, J. I. Bowman, R. W. Hughes and B. S. Sumerlin, Angew. Chem., Int. Ed., 2024, 63, e202408592 CrossRef CAS PubMed.
- L. Trachsel, K. A. Stewart, D. Konar, J. D. Hillman, J. A. Moerschel and B. S. Sumerlin, J. Am. Chem. Soc., 2024, 146, 16257–16267 CrossRef CAS PubMed.
- L. Trachsel, D. Konar, J. D. Hillman, C. L. G. Davidson and B. S. Sumerlin, J. Am. Chem. Soc., 2024, 146, 1627–1634 CrossRef CAS PubMed.
- R. W. Hughes, M. E. Lott, I. S. Zastrow, J. B. Young, T. Maity and B. S. Sumerlin, J. Am. Chem. Soc., 2024, 146, 6217–6224 CrossRef CAS PubMed.
- L. E. Diodati, A. J. Wong, M. E. Lott, A. G. Carter and B. S. Sumerlin, ACS Appl. Polym. Mater., 2023, 5, 9714–9720 CrossRef CAS.
- A. C. Lehnen, J. A. M. Kurki and M. Hartlieb, Polym. Chem., 2022, 13, 1537 RSC.
- R. A. Olson, M. E. Lott, J. B. Garrison, C. L. G. Davidson, L. Trachsel, D. I. Pedro, W. G. Sawyer and B. S. Sumerlin, Macromolecules, 2022, 55, 8451–8460 CrossRef CAS.
- C. G. L. Davidson, M. E. Lott, L. Trachsel, A. J. Wong, R. A. Olson, D. I. Pedro, W. G. Sawyer and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 1224–1230 CrossRef PubMed.
- M. E. Lott, L. Trachsel, E. Schué, C. L. G. Davidson, R. A. Olson S, D. I. Pedro, F. Chang, Y. Hong, W. G. Sawyer and B. S. Sumerlin, Macromolecules, 2024, 57, 4007–4015 CrossRef CAS.
- D. J. Rucco, B. E. Barnes, J. B. Garrison, B. S. Sumerlin and D. A. Savin, Biomacromolecules, 2020, 21, 5077–5085 CrossRef CAS PubMed.
- A. B. Korpusik, Y. Tan, J. B. Garrison, W. Tan and B. S. Sumerlin, Macromolecules, 2021, 54, 354–7363 CrossRef.
- J. Y. Rho, G. M. Scheutz, S. Häkkinen, J. B. Garrison, Q. Song, J. Yang, R. Richardson, S. Perrier and B. S. Sumerlin, Polym. Chem., 2021, 12, 3947–3952 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2024, 8, 1029–1054 CrossRef PubMed.
- H. Wang, Y. Huang, J. Y. Rho, A. B. Korpusik, M. Hoteit, J. B. Garrison and B. S. Sumerlin, Polym. Chem., 2024, 8, 5255–5446 Search PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- A. J. Wong, C. B. Eades, J. I. Bowman, C. L. G. Davidson IV and B. S. Sumerlin, Polym. Chem., 2025, 16, 620–625 RSC.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- R. J. McBride, J. F. Miller, A. Blanazs, H. J. Hähnle and S. P. Armes, Macromolecules, 2022, 55, 7380–7391 CrossRef CAS PubMed.
- R. J. McBride, E. Geneste, A. Xie, A. J. Ryan, J. F. Miller, A. Blanazs, C. Rösch and S. P. Armes, Macromolecules, 2024, 57, 2432–2445 CrossRef CAS PubMed.
- V. J. Cunningham, M. J. Derry, L. A. Fielding, O. M. Musa and S. P. Armes, Macromolecules, 2016, 49, 4520–4533 CrossRef CAS PubMed.
- C. György, J. S. Wagstaff, S. J. Hunter, E. U. Etim and S. P. Armes, Macromolecules, 2024, 57, 6816–6827 CrossRef PubMed.
- G. M. Scheutz, J. I. Bowman, S. Mondal, J. Y. Rho, J. B. Garrison, J. Korpanty, N. C. Gianneschi and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 454–461 CrossRef CAS PubMed.
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 14–19 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1800460 CrossRef PubMed.
- J. I. Bowman, C. B. Eades, M. A. Vratsanos, N. C. Gianneschi and B. S. Sumerlin, Angew. Chem., Int. Ed., 2023, 62, e202309951 CrossRef CAS PubMed.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, England, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00589b |
| This journal is © The Royal Society of Chemistry 2025 |