
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrain-release enables access to carbonyl conjugated allylic diborons and alkenyl boronates having multiple contiguous stereocenters in a one-pot process†
Het
Vyas
 ,
Ashvin J.
Gangani
,
Aiswarya
Mini
,
Melissa
Pathil
,
Austin
Ruth
and
Abhishek
Sharma
*
,
Ashvin J.
Gangani
,
Aiswarya
Mini
,
Melissa
Pathil
,
Austin
Ruth
and
Abhishek
Sharma
*
Department of Chemistry and Chemical Biology, Stevens Institute of Technology, Hoboken, NJ 07307, USA. E-mail: abhishek.sharma@stevens.edu
First published on 2nd December 2024
Abstract
Allylic diboronates are highly valuable reagents in organic synthesis. Existing methods predominantly yield alkyl-substituted allylic diboronates, while the incorporation of electrophilic carbonyl groups conjugated to these allylic systems remains unknown. We present a strain-release promoted cycloaddition-based strategy that enabled access to unprecedented carbonyl conjugated secondary allylic diborons. This mild base-free method also facilitated a one-pot multicomponent cycloaddition–allylboration sequence for a highly diastereoselective installation of contiguous quaternary, tertiary and secondary carbon centers on a scaffold featuring valuable β-hydroxy ester, β-vinyl ester and vinyl boronate motifs. The synthetic utility of these densely functionalized products was demonstrated through their transformation into other rare and sterically congested alkenylborons such as borylated spiro-fused β-lactones and bicyclic γ-butyrolactones. Detailed 11B NMR, deuterium labeling and mass spectrometry studies provided insights on an unexpected base-free deboronative allylic shift reaction of conjugated allylic diboronates.
Introduction
Allylic boronates represent one of the most important classes of organoborons due to their unique ability to participate in stereoselective addition reactions with aldehydes and ketones.1 These allylboration reactions provide homoallylic alcohols which are ubiquitous motifs in natural products and bioactive molecules. It is, therefore, not surprising that development of novel methods to access allylic boronates and their synthetic applications has attracted significant interest.1 Several creative methods have been reported for preparation of substituted allylic boronates and expand the scope of allylboration reaction. The Szabo2 and Gong3 groups disclosed in situ formation of mono allylborons via palladium catalyzed borylation followed by nucleophilic addition to carbonyl compounds. Hall and coworkers reported remarkable rate acceleration of allylboration by using Lewis acid catalysts.4 They also developed monoallyl boronates containing a 2-alkoxy carbonyl motif for activation of the boron atom.4,5Continued progress in this area has led to the emergence of allylic geminal diborons (Scheme 1A) as allylating agents for simultaneous installation of alkenyl boronate units on homoallylic alcohols. Murakamai6,7 and Cho8,9 made early contributions to the synthesis of allylic diborons (Scheme 1B). Their strategy involved Pd,6 Ru7 or Ir8 catalyzed double bond transposition to give E-α-boryl crotyl boronates. A copper catalyzed cross-coupling of bis-boryl methyl zinc halide with vinyl iodonium salts provided γ-aryl/alkyl substituted allylic diboron.9 Meek and coworkers developed palladium catalyzed cross-coupling of lithiated bis-boryl methane and alkenyl halides to prepare γ,γ-dialkyl allylic diborons (Scheme 1B).10–13 These reagents enabled the construction of borylated homoallylic alcohols and amines having quaternary stereocenters. Chen and coworkers disclosed rhodium14 and nickel15 catalyzed alkene isomerization methods to afford α-methyl, γ-methyl allylic diboron or Z-α-boryl crotyl boronates (Scheme 1B). They also developed a nickel-catalyzed 1,4-diboration approach to Z-α,γ-diboryl allylic borons.16 Allylboration of aldehydes using these reagents provided facile stereoselective access to various syn and anti homoallylic alcohols. Recently, the Pd catalyzed Heck coupling route to allylic diborons was employed in one-pot allylboration to furnish homoallylic alcohols having adjacent secondary stereocenters (Scheme 1B).17 Addition of boron to hydrazones has been used to prepare allylic monoborons and a single example of allylic diboron was reported.18 An atom transfer radical annulation (ATRA) approach provided allylic diborons having a non-conjugated diester group (Scheme 1B).19 Allylboration of these intermediates led to homoallylic alcohols having one stereogenic center while the remote location of the ester motif from allylic alcohol precludes their intramolecular reaction.19
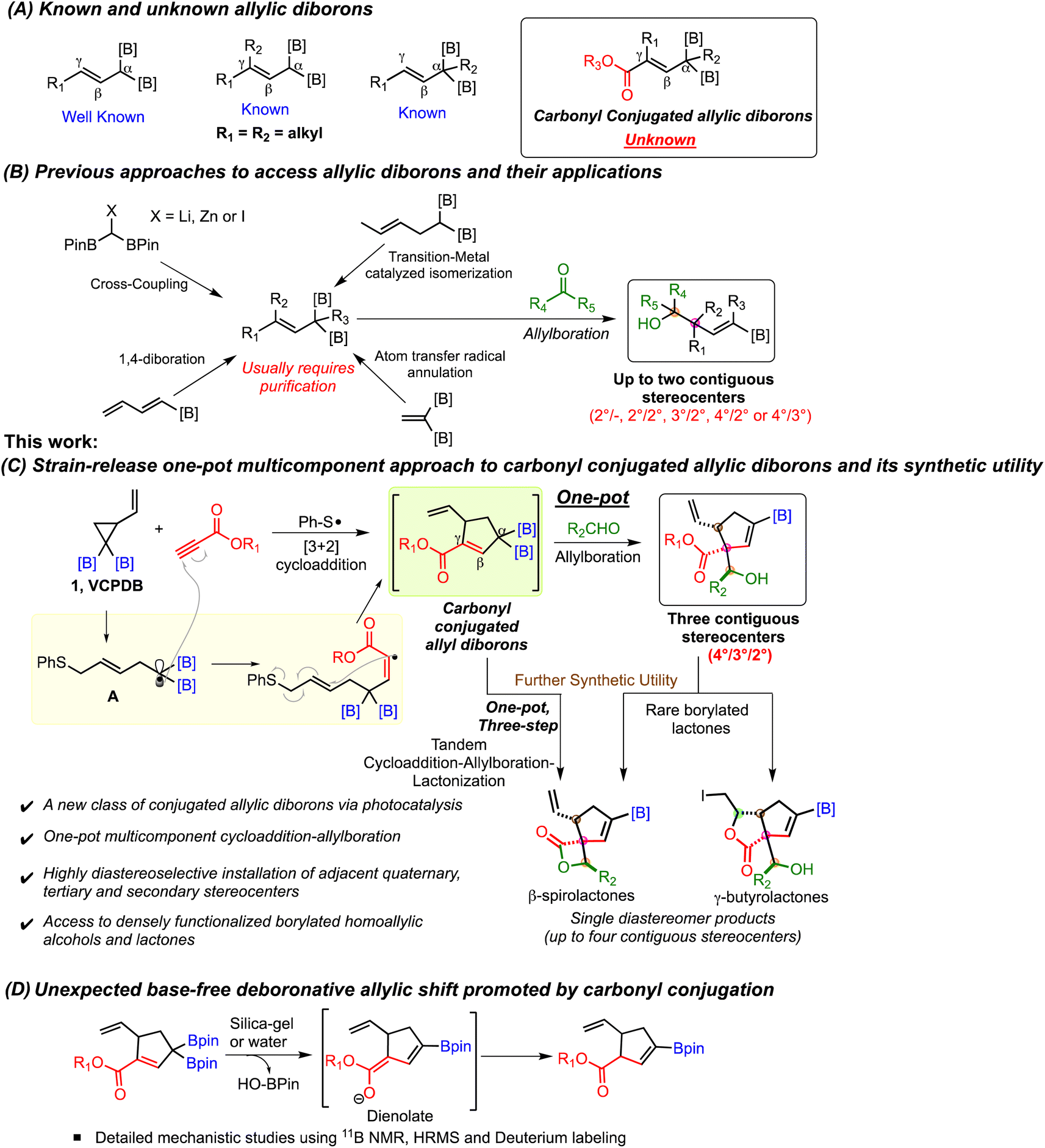 | ||
| Scheme 1 Strategies for synthesis and application of allylic diboronates. | ||
One of the key factors governing the versatility of the allylboration reaction is the availability of new types of substituted allylic diborons. In this context, the synthesis and allylboration of γ,γ-disubstituted10–13 and α-alkyl14 allylic diborons (Scheme 1A) to enable stereoselective formation of quaternary carbon centers has garnered much attention. Despite the significant progress made in the synthesis of allylic diborons, there are still challenges that hinder the broader application of these geminal dimetallic reagents. The majority of the existing approaches allow synthesis of alkyl substituted allylic diborons while incorporation of electrophilic groups such as a carbonyl motif conjugated to the allylic system has remained elusive (Scheme 1A). Such conjugated systems could reveal new reactivity patterns besides opening up access to previously inaccessible highly functionalized organoborons. Furthermore, the allylboration reaction of previously known allylic diborons installs only up to two contiguous stereocenters (Scheme 1B). Finally, a metal-free one-pot strategy involving formation of functionalized γ,γ-disubstituted allyl diborons followed by their nucleophilic addition to carbonyls is unknown.
We recently developed vinyl cyclopropyl diborons (VCPDBs) as a source of homoallylic α,α-diboryl radicals via diboron-directed regioselective ring opening.20 These radicals were found to undergo diastereoselective [3 + 2] cycloaddition reaction with a variety of alkenes, including acrylates. We wondered if the diboryl stabilized radicals (A, Scheme 1C) would participate in cycloaddition with propiolates under photocatalytic conditions. If successful, this approach would offer a direct pathway to carbonyl-conjugated allylic diborons (Scheme 1C) which would be difficult to access using conventional methods. These conjugated allyldiborons would also incorporate the desirable γ,γ-disubstituted and α-alkyl motifs within the same structure. Additionally, we reasoned that the mild base-free reaction conditions enabled by thiyl radical catalysis should allow the allyldiborons to react with aldehydes in a one-pot multicomponent process. The resulting products would be densely functionalized borylated homoallylic alcohols, featuring adjacent quaternary, tertiary, and secondary carbon centers, with their relative stereochemistry defined during the allylboration step (Scheme 1C). These unique allylboration products bearing strategically located β-hydroxy ester and β-vinyl ester motifs were expected to have significant synthetic potential for construction of structurally complex organoborons via intramolecular reactions (Scheme 1C). Herein, we report the development of carbonyl conjugated allylic diborons as novel building blocks for direct synthesis of rare and highly functionalized organoborons and their unexpected base-free deboronative allylic shift reaction (Scheme 1D).
Results
We began our studies with a reaction of VCPDB (1a) and methyl propiolate (2a) using diphenyl disulfide as a catalyst under a 365 nm LED. Surprisingly, purification of this reaction mixture via silica-gel chromatography provided a substituted cyclopentene (4a, Scheme 2) having a vinyl boronate group instead of the expected allyl diboron (3a). 1H NMR analysis of the crude reaction mixture confirmed the presence of the desired carbonyl conjugated allylic diboron product (3a) in excellent yield (80% based on NMR, see the ESI† for details). Replacement of silica-gel with neutral alumina or use of oven-dried silica-gel for chromatographic purification also provided the deboronated product (4a). Further detailed investigations (see the ESI,†Table 1) showed that crude 3a is stable at room temperature under a nitrogen atmosphere up to 72 hours but it converts to vinylboronate (4a) in the presence of silica-gel, aq. acid/aq. base or even water. Such a facile deboronation under mild or neutral conditions is unusual in light of a previous report21 that showed the requirement of fluoride ions or acetic acid for deborylation of allylic boronates. Interestingly, silica-gel chromatography of the reaction mixture obtained by treatment of 1a with phenyl acetylene (5a) afforded the γ–γ-substituted allylic diboronate (6a, Scheme 2) instead of the vinyl boronate.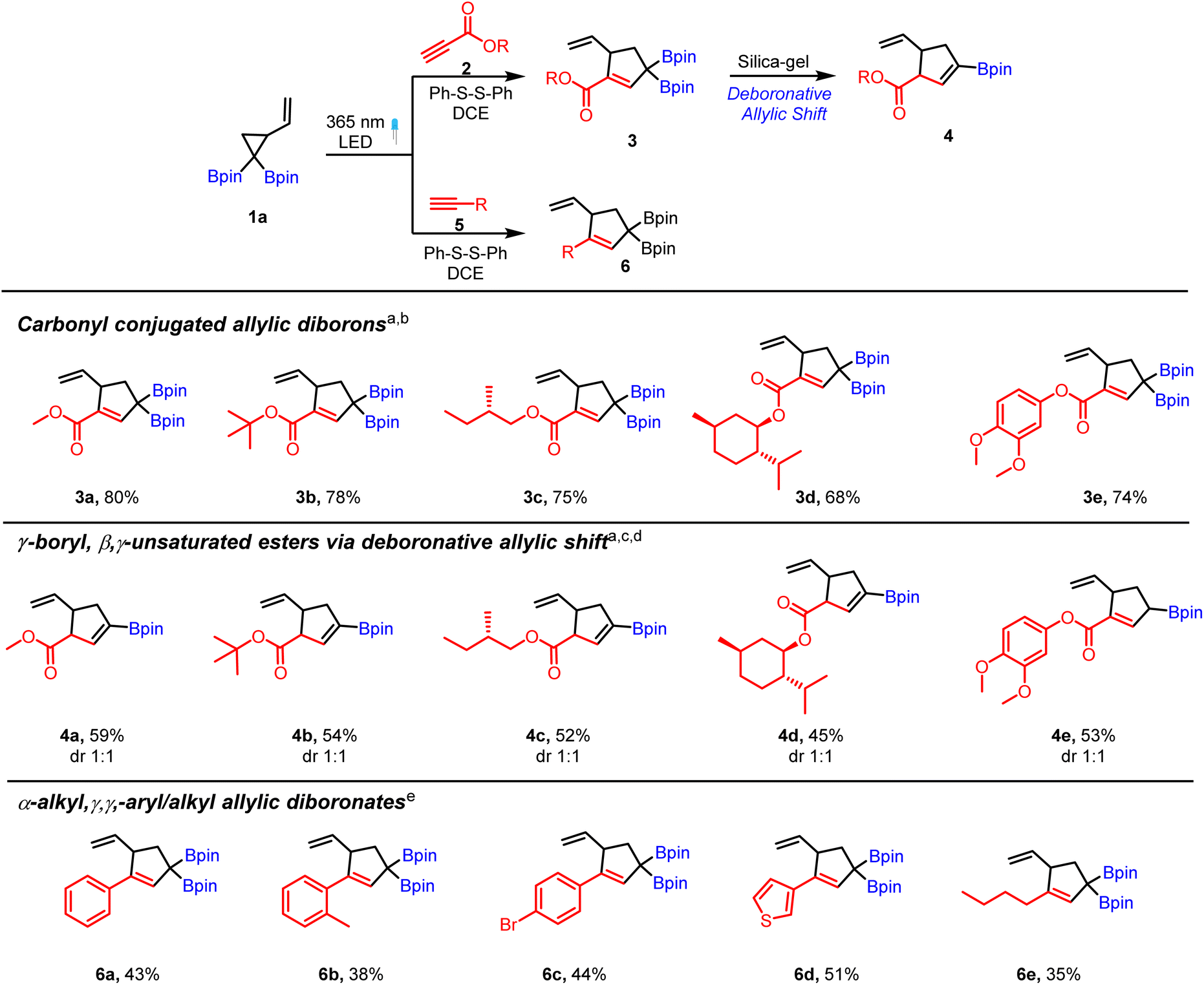 | ||
| Scheme 2 Substrate scope of propiolates and alkynes for photoreaction with VCPDB. a VCPDB (1a, 0.31 mmol), propiolate (2, 0.43 mmol, 1.3 equiv.), diphenyl disulfide (0.062 mmol, 20 mol%) and DCE (1.2 mL, 0.25 M). b Yield based on 1H NMR. c Reaction mixture was passed through silica gel. d both the diastereomers were isolated for 4a–4e. e VCPDB (1a, 0.31 mmol), alkyne (5, 0.93 mmol, 3 equiv.), diphenyl disulfide (0.37 mmol, 1.2 equiv.) and DCE (1.2 mL, 0.25 M). | ||
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield (%) |
| a Standard reaction conditions: VCPDB (1a, 0.31 mmol), methyl propiolate (2a, 0.43 mmol, 1.3 equiv.), diphenyl disulfide (0.062 mmol, 20 mol%) and solvent (1.2 mL, 0.25 M), under a photoreactor for 5 h followed by removal of solvent, addition of benzaldehyde (0.62 mmol, 2 equiv.) and toluene (1.2 mL, 0.25 M) to the same pot and heating at 80 °C for 48 h. b Reaction time: 60 h. c Temp. 90 °C. d Temp. 23 °C. e Temp. 50 °C. f Reaction time: 24 h. | ||
| 1 | DCE as solvent in step-2 | 61b |
| 2 | None | 62 |
| 3 | 10 mol% diphenyl disulfide | 40 |
| 4 | Toluene as solvent in step-1 | 51c |
| 5 | 23 °C temperature in step-2 | Trace |
| 6 | With 4 Å MS | 61 |
| 7 | 10 mol% Sc(OTf)3 in step-2 | 23d |
| 8 | 20 mol% BF3OEt2 in step-2 | 20 |
| 9 | 5 mol% R-TRIP in step-2 | 28e |
| 10 | 110 °C using Dean Stark apparatus | 35f |
| 11 | Toluene as solvent in step-1 and benzaldehyde added in the first step (three-component reaction) | 36c |
The above results suggest that the carbonyl group is the primary driver of the unusual deboronation observed in 3a. Given the immense synthetic utility of allylic diborons and vinyl boronates, we investigated the scope of the thiyl radical catalyzed [3 + 2] cycloaddition reaction of VCPDB (1a) and various alkynes. A brief survey of reaction conditions showed 20 mol% phenyl disulfide catalyst in DCE as solvent to be optimal for complete consumption of the starting material. A variety of alkyl and aryl propiolates were compatible reaction partners with VCPDB (1a) to provide the carbonyl conjugated allylic diboronates in good yield based on 1H NMR analysis (3a–3e, Scheme 2). NMR analysis of the reaction mixture showed a very clean reaction with mainly the product and catalyst present at the end of the photoreaction (see the ESI†). Further scope of the propiolates including derivatives of bioactive compounds was evaluated and the results are discussed during their application for allylboration reactions (see Scheme 4). Alkynes bearing aromatic, heteroaromatic and alkyl groups were also tolerated and the resulting allylic diborons were found to be stable to silica gel chromatography (Scheme 2, 6a to 6e). Overall, the silica-gel induced deboronative allylic shift reaction of propiolate-derived allylic diborons provided a mild strategy to access cyclic vinyl boronates containing an ester and terminal alkene functional groups. It is worth mentioning that synthesis of these cyclic γ-boryl, β–γ-unsaturated carbonyl compounds (4a to 4e) would be difficult using the hydroboration approach due to regioselectivity and chemoselectivity issues. Furthermore, the dr of these products can be increased in favor of the thermodynamically more stable trans-isomer by treatment of alkenyl boronates (e.g.4a) or allylic diboron (e.g.3a) with a base (see the ESI† for details).
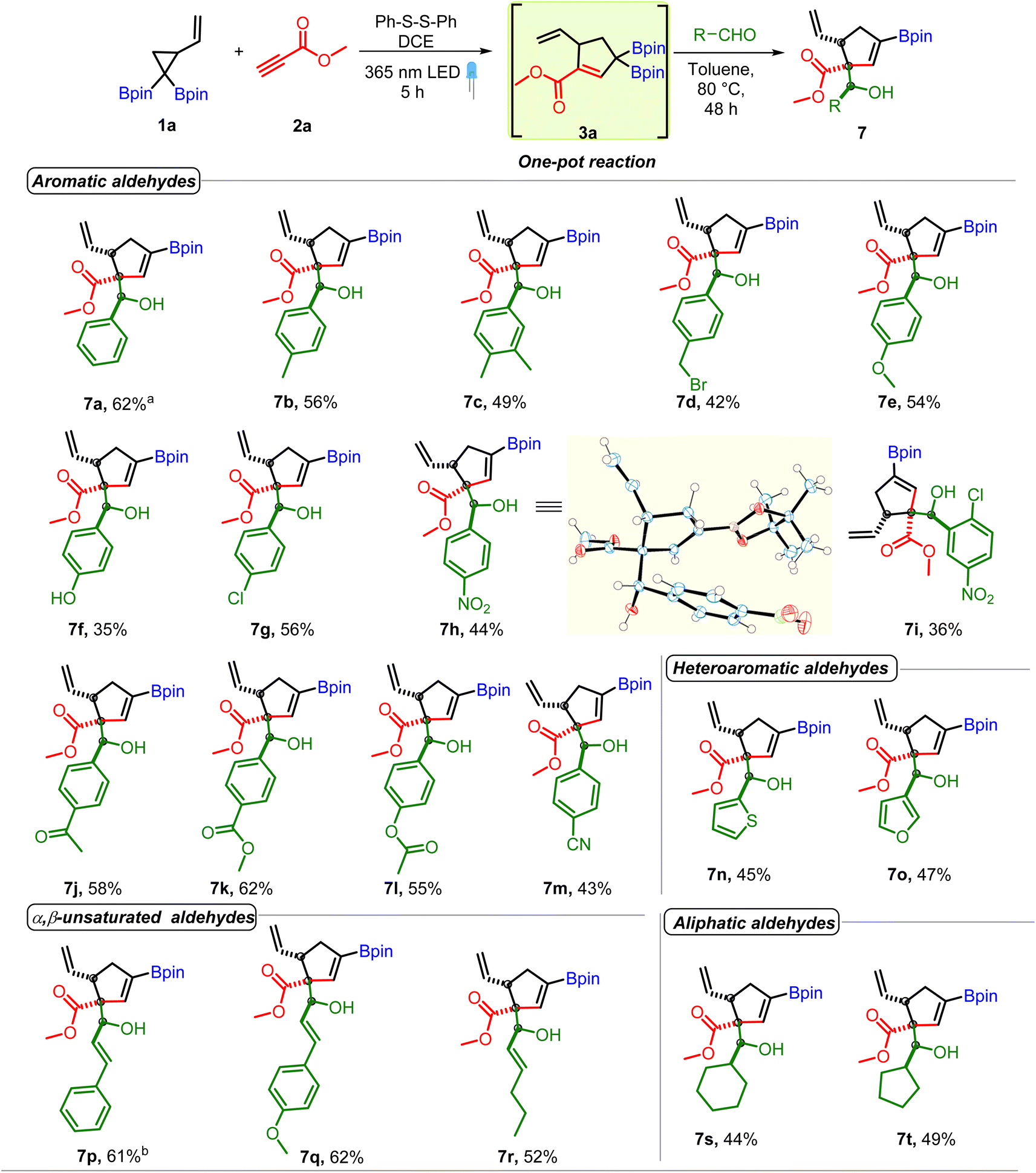 | ||
| Scheme 3 Substrate scope of aldehydes. Reaction conditions: VCPDB (1a, 0.31 mmol), methyl propiolate (2a, 0.43 mmol, 1.3 equiv.), diphenyl disulfide (0.062 mmol, 20 mol%) and DCE (1.2 mL, 0.25 M), under a 365 nm LED for 5 h followed by removal of DCE and addition of aldehyde (0.62 mmol, 2 equiv.), toluene (1.2 mL, 0.25 M) to the same pot and heating at 80 °C for 48 h. a Average yield of four reactions. b Average yield of two reactions. | ||
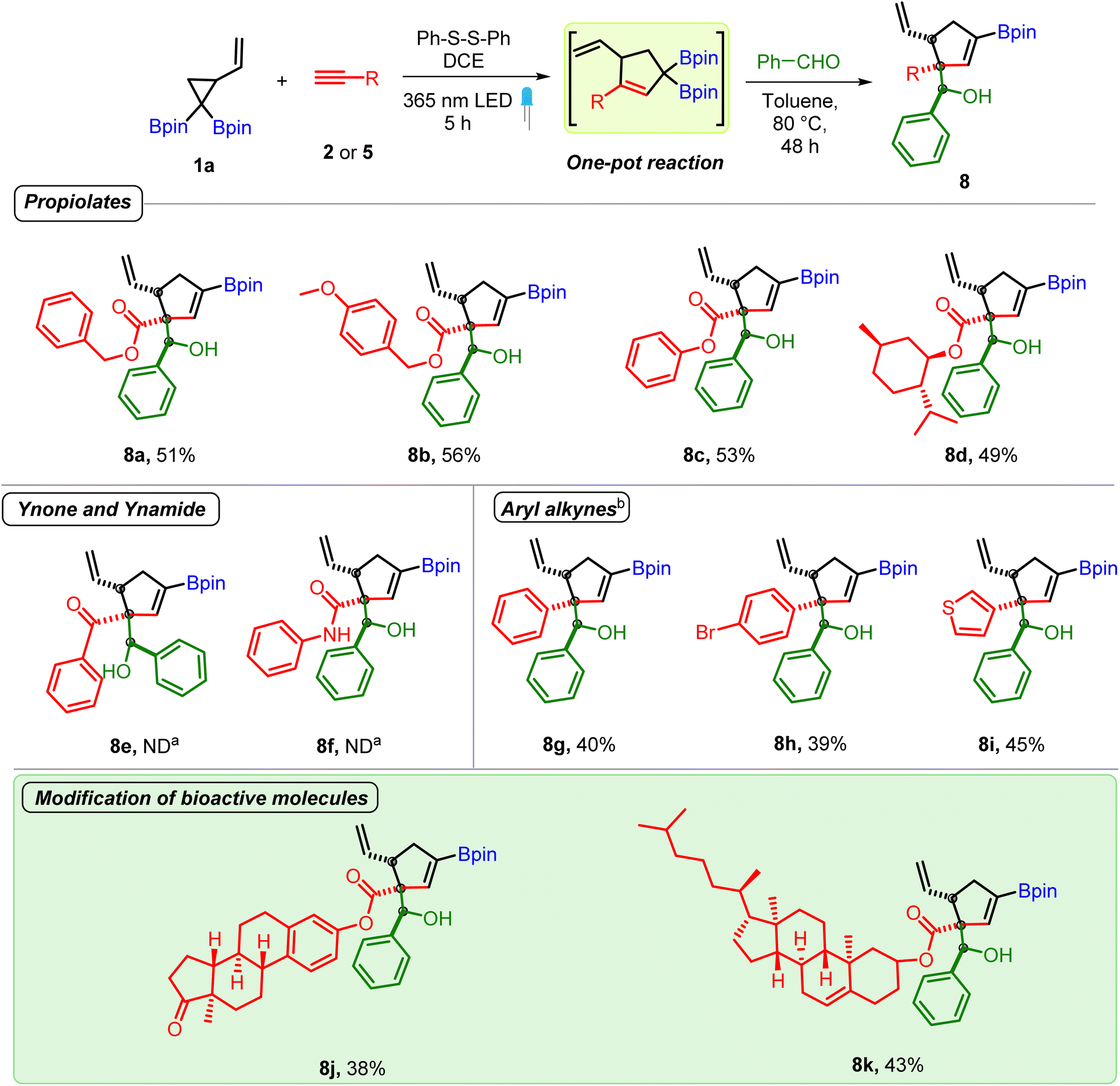 | ||
| Scheme 4 Substrate scope of alkynes. Reaction conditions: VCPDB (1a, 0.31 mmol), propiolates (2, 0.43 mmol, 1.3 equiv.), diphenyl disulfide (0.062 mmol, 20 mol%) and DCE (1.2 mL, 0.25 M), under a 365 nm LED for 5 h followed by DCE removal, addition of aldehyde (0.62 mmol, 2 equiv.) and toluene (1.2 mL, 0.25 M) to the same pot and heating at 80 °C for 48 h. a ND = not detected. b 1.2 equiv. of diphenyl disulfide and 3 equiv. of alkyne (5) were used. | ||
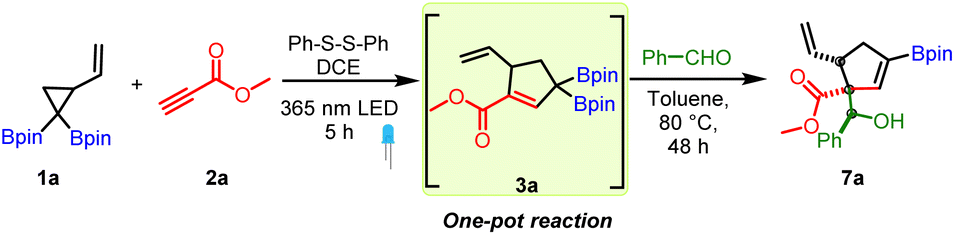
We next turned our attention to establish the synthetic utility of the photocatalytic route to conjugated allylic diborons. We wondered if the mild reaction conditions involving thiyl radical catalysis could allow for a sequential one-pot allyl diboron formation and addition reaction with aldehydes. To test this hypothesis, 1a was treated with 2a in the presence of diphenyl disulfide as a catalyst under a 365 nm LED to form the conjugated allylic diboron (3a). Thereafter, benzaldehyde was added to the same pot and the reaction mixture heated to 80 °C. This one-pot reaction successfully provided the desired allylboration product in very good diastereoselectivity (Table 1, entry 1). Motivated by this result, a detailed optimization of the one-pot reaction was conducted. Reducing the disulfide catalyst loading to 10 mol% or changing the solvent in the photocatalytic step to toluene lowered the yield (entries 3 and 4). Conducting the allylboration reaction at room temperature resulted in unreacted allylic diboron (entry 5). However, replacing DCE with toluene as solvent before the addition of benzaldehyde reduced the reaction time while maintaining good yield (entry 2). The addition of molecular sieves or Lewis acids to promote the allylboration reaction was not beneficial (entries 6–8). Use of a chiral Brønsted acid provided the desired product (entry 9) but without inducing enantioselectivity likely because these chiral catalysts typically need low reaction temperature. Increasing the reaction temperature for the allylboration step to 110 °C and use of Dean–Stark apparatus lowered the yield (entry 10). A three-component reaction done by treating a mixture of 1a, 2a and benzaldehyde and disulfide catalyst in a photoreactor followed by heating to 90 °C afforded 7a in 36% yield (entry 11). This lower yield is likely due to reduced efficiency of the photoreaction using toluene as solvent. In comparison to the allylboration reaction of previously known allylic diborons, the longer reaction time needed for reaction of 3a with aldehyde is likely due to significant steric hindrance resulting from the presence of the γ,γ-disubstituted α,α-diboryl non-terminal allylic motif in 3a.
The above optimized conditions were used to examine the substrate scope of the one-pot multicomponent cycloaddition–allylboration. A very broad range of aromatic and aliphatic aldehydes were found to be compatible for one-pot reaction with carbonyl conjugated allyl diboron (3a, Scheme 3) with excellent diastereoselectivity. Benzaldehydes having electron donating groups such as alkyl, alkoxy (7b to 7f) or electron withdrawing motifs like halogens or nitro at o, p or m-positions (7g to 7i) were well tolerated. Furthermore, synthetically useful electrophilic handles such as a ketone (7j), ester (7k and 7l) and nitrile (7m) could be used on benzaldehydes without compromising the yield. Heteroaromatic aldehydes (7n and 7o) were compatible. The multicomponent reaction was also amenable to α,β-unsaturated and aliphatic aldehydes (7p–7t). Other electrophiles such as ketones, imines or indoles didn't participate in the allylboration reaction with 3a (see the ESI†). Of note, the reaction showed excellent diastereoselectivity and a single diastereomer was obtained in most cases. The relative stereochemistry was found to be anti on the basis of X-ray crystallography (entry 7h, Scheme 3).
Next, we investigated the scope of alkynes. Several aryl and alkyl propiolates underwent smooth reaction thereby demonstrating the ability of highly congested carbonyl conjugated allyl diborons to participate in allylboration reaction (entries 8a–8d, Scheme 4). In contrast, reaction of allyl diborons obtained from a ynone or ynamide didn't give the desired allylboration product (8e and 8f) even as the crude reaction mixture after the photoreaction showed the formation of corresponding carbonyl conjugated allylic diboronates. On the other hand, both aromatic and heteroaromatic acetylenes proved to be competent reaction partners (8g–8i). To showcase the utility of the developed method for late-stage modification of complex bioactive molecules, alkynes having cholesterol and estrone cores were employed. These reactions successfully provided the corresponding allylboration products (8j and 8k) bearing homoallylic alcohol/ester and steroidal motifs as a single diastereomer. Collectively, the above results demonstrate the benefit of the mild VCPDB-based approach to open up access to unprecedented and structurally complex conjugated allylic diborons and their one-pot conversion into densely functionalized products having contiguous quaternary, tertiary and secondary carbon centers.
To determine the scope of the VCPDB component, vinyl cyclopropanes having (+) pinanediol boronate, MIDA boronate and neopentyl boronate motifs were tested (Scheme 5). The presence of the geminal bis-pinanediol boronate group or unsymmetrical diboron motif (pinanediol boronate/Bpin and neopentyl boronate/Bpin) on VCP (1c and 1e) allowed the formation of corresponding carbonyl conjugated allylic diboron via cycloaddition with 2a which reacted smoothly with benzaldehyde to give the desired 7a and 9a albeit without chirality transfer from (+) pinanediol (Scheme 5). In contrast, VCP having geminal BMIDA/BPin groups didn't participate in the multicomponent reaction (9b). In addition to the allylic diborons, the formation of carbonyl conjugated monoboronates was also feasible as the VCP containing a single Bpin group (1f) led to the desired allylboration product (9c). The above results suggest that while one Bpin group (sp2 – hybridized B) on VCP is sufficient to induce the regioselective ring opening of VCP, the presence of a bulky BMIDA group (sp3 – hybridized B, 9b) inhibits the VCP ring opening and/or the addition of a homoallylic radical (A, Scheme 1C) to an alkyne.
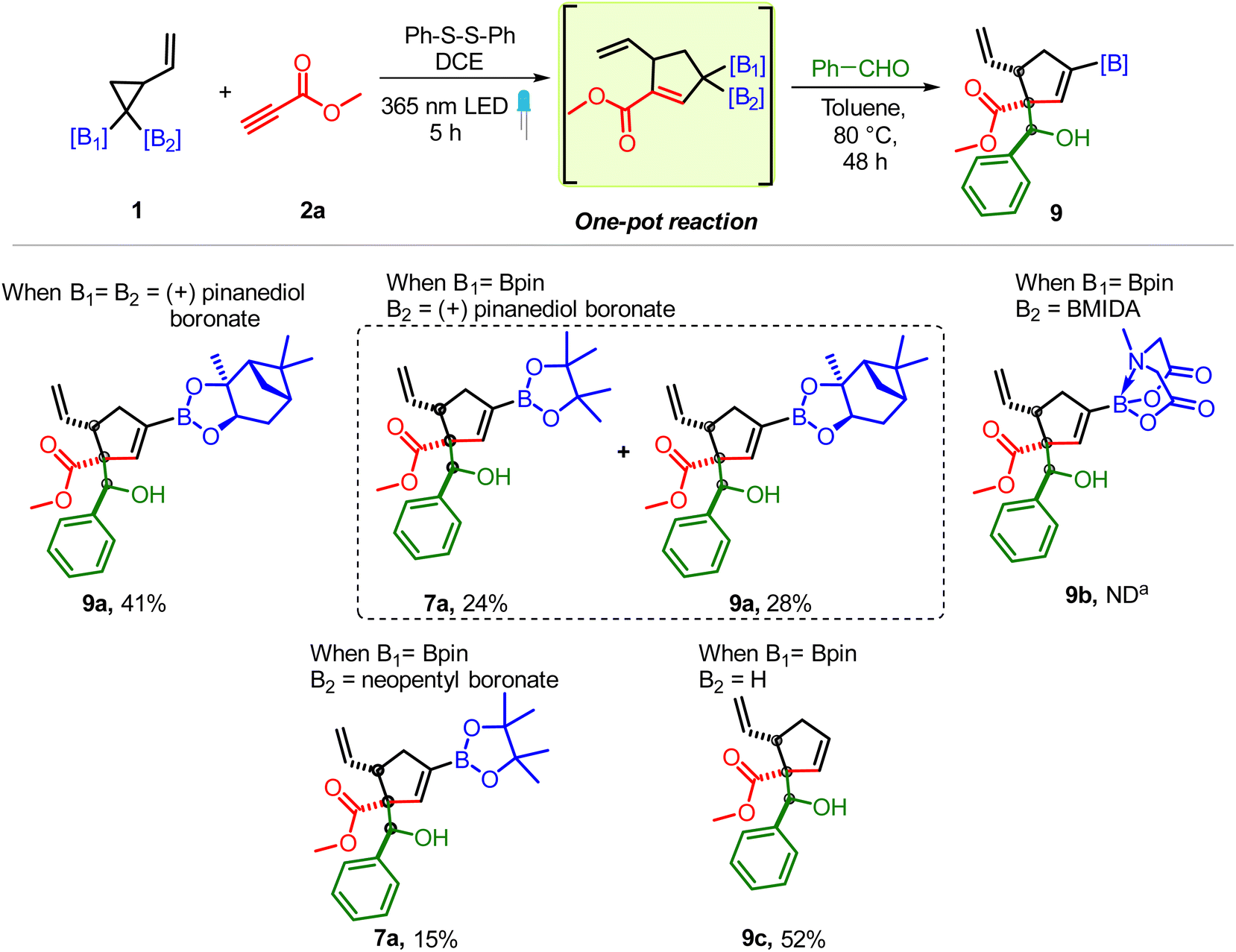 | ||
| Scheme 5 Substrate scope of vinyl cyclopropanes. Reaction conditions: VCPDB (1b–1e, 0.31 mmol), methyl propiolates (2a, 0.43 mmol, 1.3 equiv.), diphenyl disulfide (0.062 mmol, 20 mol%) and DCE (1.2 mL, 0.25 M), under a 365 nm LED for 5 h followed by removal of DCE, addition of aldehyde (0.62 mmol, 2 equiv.) and toluene (1.2 mL, 0.25 M) to the same pot and heating at 80 °C for 48 h. a ND = not detected. | ||
The robustness of the method was confirmed by a scale-up experiment (1 mmol scale) on two different substrates (7a and 8b, Scheme 6). The anti diastereoselectivity observed in allylboration of 3 can be rationalized by the rigid six-membered transition state model1,22 (Scheme 7) where the aldehyde proton occupies the pseudo axial position. The presence of an allylic diboron motif within the cyclic structure imposes further conformational constraints leading to very high anti selectivity.
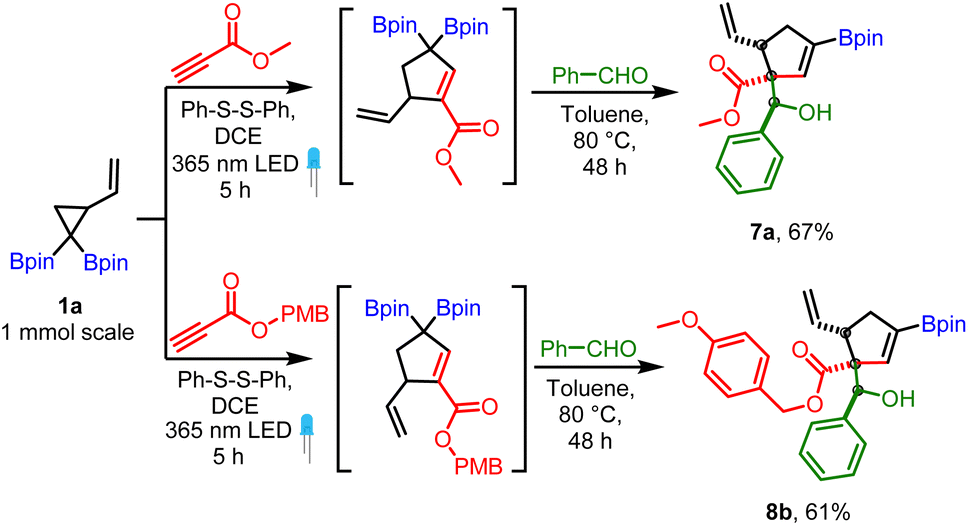 | ||
| Scheme 6 Scale-up reactions (1 mmol scale); reaction conditions: VCPDB (1a, 1 mmol), propiolates (1.3 mmol, 1.3 equiv.), diphenyl disulfide (0.2 mmol, 20 mol%) and DCE (4 mL, 0.25 M), under a 365 nm LED for 5 h followed by removal of DCE and addition of aldehyde (2 mmol, 2 equiv.) and toluene (4 mL, 0.25 M) to the same pot and heating at 80 °C for 48 h. | ||
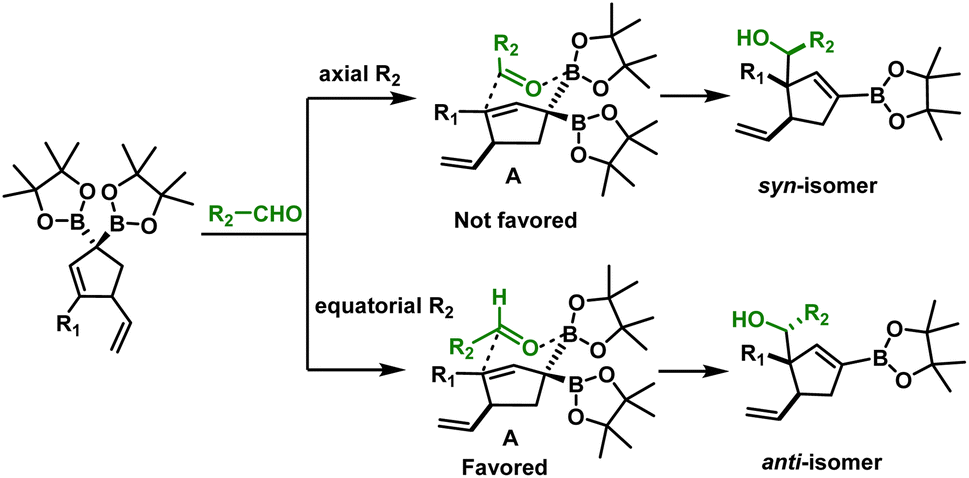 | ||
| Scheme 7 Proposed transition state model for allylboration. | ||
Our next goal was to establish the synthetic utility of allylboration products (7–8). In particular, we wondered if the beta-hydroxy ester motif in 7 or 8 would allow the construction of borylated spiro-fused β-lactone (Scheme 8). β-Lactones are structural frameworks of bioactive natural products and also serve as valuable synthetic intermediates due to inherent strain in the lactone ring.23–26 In particular, the spiro β-lactones display potent proteasome inhibitory and GABA antagonistic activity;27,28 therefore, methods for stereoselective construction of these lactones are in high demand. We reasoned that the allyboration products obtained from p-methoxy benzyl (PMB) propiolates could be converted into corresponding β-hydroxy carboxylic acid under mild conditions without disturbing the boronate or vinyl group. This acid could then be activated with BOP-Cl29 for lactone formation. As a test of this hypothesis, 8b was treated with BCl3 for deprotection of PMB ester. To our surprise, instead of the expected acid, the above reaction provided the β-spiro lactone (10a) along with some other side products which were difficult to remove by column chromatography. Motivated by the above result, several other acidic conditions for one-pot ester deprotection, spirolactonization, were evaluated.
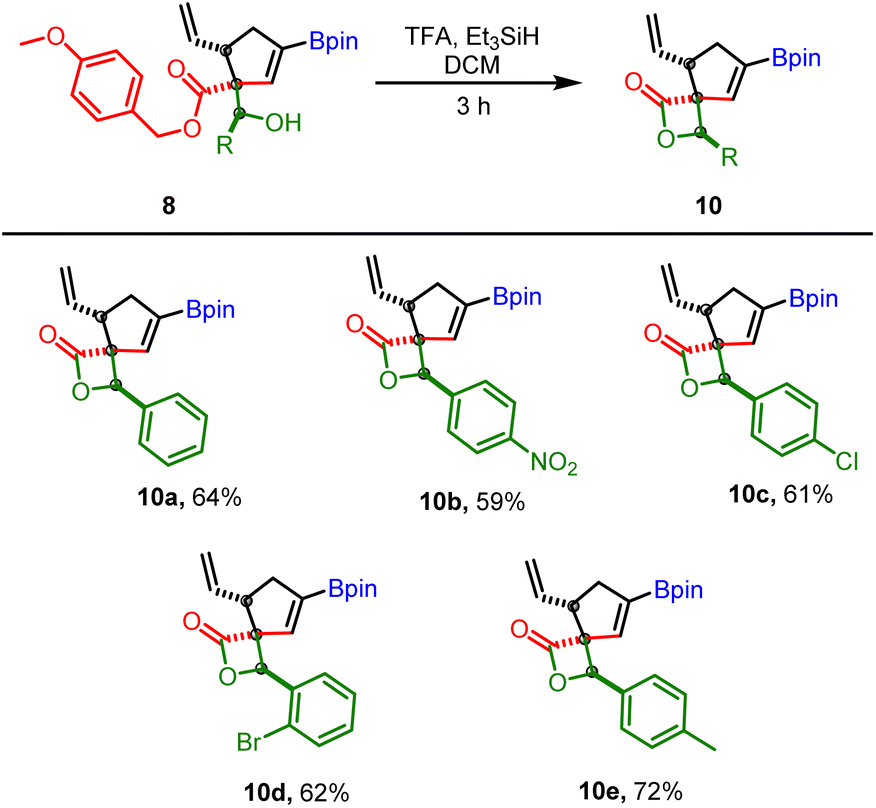 | ||
| Scheme 8 Application of allylboration products for spirolactonization; reaction conditions: 8 (0.25 mmol), triethyl silane (2.5 mmol, 10 equiv.) TFA (1.5 mmol, 5 equiv.) in DCM (1 mL, 0.25 M). | ||
A combination of trifluoracetic acid (TFA) and Et3SiH was found to provide the desired β-spiro lactone in good yield within 3 h (Scheme 8). The substrate scope of this reaction was broad as several other allylboration products containing electron withdrawing groups (nitro, halogen) or electron donating groups (methyl) furnished the bicyclic β-spiro lactone as single diastereomers (Scheme 8). Significantly, the direct conversion of VCPDB (1a) into borylated spiro-fused β-lactone was also feasible via a one-pot three step reaction (4 C–C bonds formed) with an overall yield (43%, Scheme 9) better than that obtained by two-pot reaction (10a, Scheme 8, overall yield = 36%). These results highlight the robustness and versatility of the developed VCPDB-based approach for a concise and stereoselective construction of densely functionalized biologically important molecules containing multiple contiguous stereogenic centers and the synthetically useful vinyl boronate group.
 | ||
| Scheme 9 One-pot tandem cycloaddition–allylboration–lactonization. | ||
To further demonstrate the utility of allylboration products obtained from conjugated allyl diborons, the cis-orientation of β-vinyl and ester groups in 7 was harnessed for iodo-lactonization reaction. This approach enabled facile access to bicyclic γ-butyrolactones having a boronate group which can be used for further structural diversification (Scheme 10). It is pertinent to mention that while the γ-butyrolactone30,31 scaffold is ubiquitous in several bioactive molecules and FDA approved drugs, the exocyclic hydroxyl group present in lactones 11 is also a recurring motif in natural products that regulate antibiotic biosynthesis.32
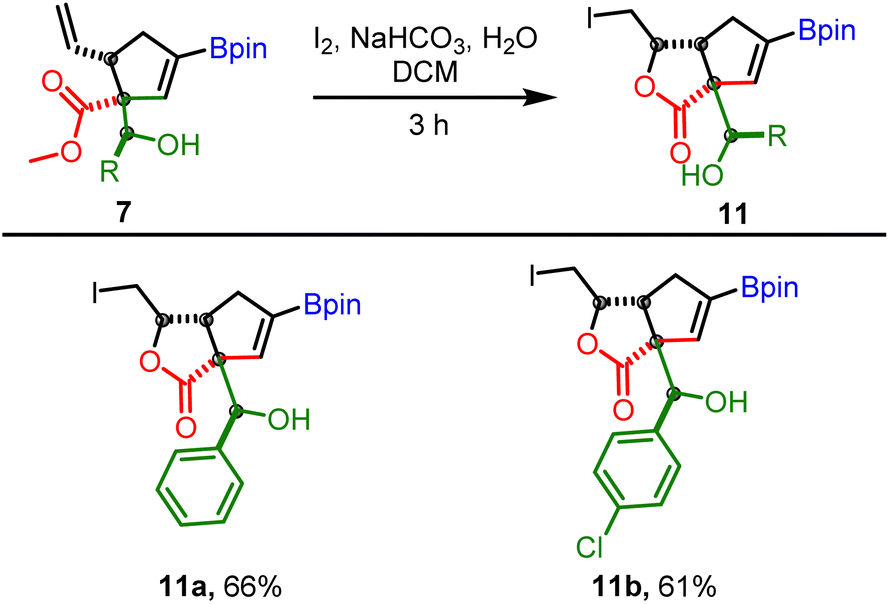 | ||
| Scheme 10 Application of allylboration products for iodolactonization. Reaction conditions: 7 (0.25 mmol), iodine (0.3 mmol, 1.2 equiv.), NaHCO3 (0.3 mmol, 1.2 equiv.), H2O (200 μL) in DCM (1 mL, 0.25 M). | ||
Having established the synthetic utility of conjugated allyldiborons, mechanistic studies were conducted to better understand the light-promoted thiyl radical catalyzed VCP ring opening and the unusual base-free deboronative allylic shift of 3a. Based on our previous studies,20 a plausible mechanism involves the initial generation of thiyl radicals from homolysis of disulfide. This thiyl radical (I) adds to the VCPDB (1) to give the cyclopropyl carbinyl radical (II, Fig. 1A) which can revert back to 1via β-fragmentation or undergo ring opening to form the homoallylic radical (III). The presence of a geminal diboron group in II favors the ring opening pathway due to the stabilization of the ensuing carbon-centered radical (III) via conjugation with the empty p-orbitals of two boron atoms. The homoallylic radical (III) undergoes addition to an alkyne which is followed by ring closure and concomitant regeneration of the thiyl radical. Light-on/off experiments indicated the need for continuous irradiation of the reaction mixture (Fig. 1B). This result suggests that the thiyl radical released from the catalytic cycle participates in two competing pathways: recombination to give the disulfide or addition to VCPDB (1).
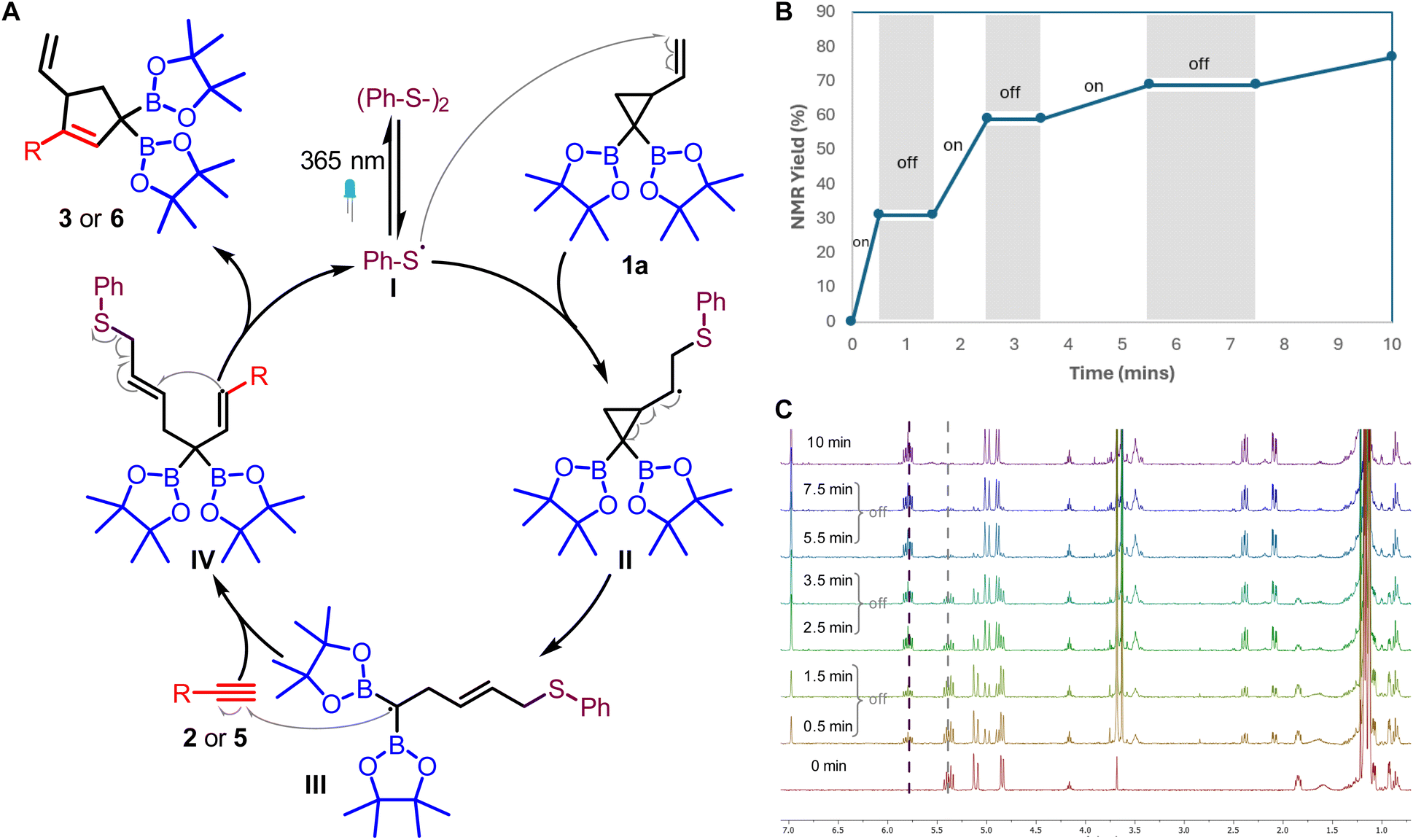 | ||
| Fig. 1 (A) A plausible mechanism for [3 + 2] radical cycloaddition; (B) light “on/off” experiment; (C) stacked 1H NMR from “on/off” experiments at different time intervals. | ||
The High Resolution Mass Spectrum (HRMS) of crude 3a using the ASAP probe showed the presence of 3a, vinyl boronate 4a and the Bpin-OH species (Fig. 2A) which indicates deboronation during the mass spec analysis. The deuterium labelling experiment performed by treating 3a in D2O showed 75% deuterium incorporation at the alpha-position of the ester (Fig. 2B).
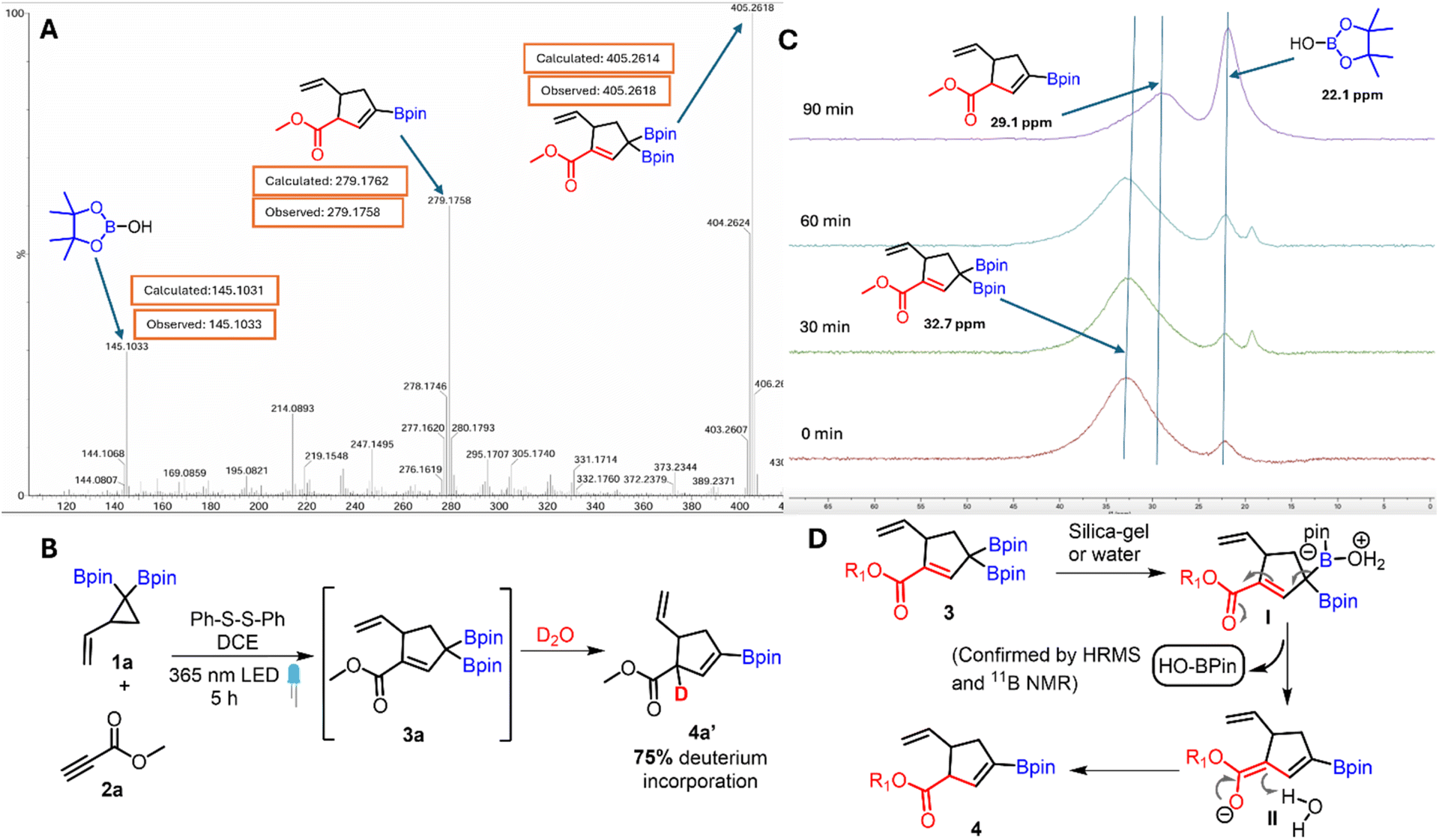 | ||
| Fig. 2 Mechanistic studies of the base-free deboronative allylic shift. (A) HRMS analysis of carbonyl conjugated allylic diboron. (B) Deuterium labeling experiment. (C) 11B NMR analysis of silica-gel induced deboronation. (D) Proposed mechanism. | ||
The silica-gel mediated deboronation was monitored by 11B NMR spectroscopy (Fig. 2C) in CDCl3 as solvent. During the first few minutes of this experiment, the allyl diboron was the major species (δ = 32.7 ppm); however, the intensity of this signal progressively decreased with a concomitant increase in the intensity of the resonance at δ = 29.1 and 22.1 ppm which was assigned to vinyl boronate and Bpin-OH33 byproducts respectively. Similar 11B NMR spectral data was obtained when silica gel was replaced with water (see the ESI†). In view of the above data, a plausible mechanism (Fig. 2D) involves coordination of water molecules with the BPin to generate the boronate complex (I). The presence of an α,β-unsaturated ester motif in this intermediate induces water mediated deboronation resulting in the formation of dienolate (II) which undergoes protonation to afford the vinyl boronate (4).
Conclusion
In conclusion, this work discloses the first examples of carbonyl conjugated allylic diborons and metal-free one-pot formation-nucleophilic addition of allylic diborons. A photocatalytic [3 + 2] cycloaddition of vinyl cyclopropyl diborons (VCPDB) with propiolates provided highly substituted allyldiborons conjugated to an ester group. This mild base- and transition metal-free approach enabled a tandem cycloaddition–allylboration reaction involving an in situ formation of allyl diborons followed by their nucleophilic addition to aldehydes. The above one-pot multicomponent reaction showed broad substrate scope and excellent diastereoselectivity to afford highly functionalized borylated homoallylic alcohols featuring the β-hydroxy ester and β-vinyl ester motifs across contiguous quaternary, tertiary and secondary stereogenic centers. These unique allylboration products opened up access to other rare and congested alkenyl borons such as borylated spiro-fused β-lactones and bicyclic γ-butyrolactones which are components of bioactive molecules. Of note, the direct conversion of VCPDB into spiro β-lactones via a one-pot three step process was also feasible. The carbonyl conjugated allylic diborons also displayed an unexpected deboronative allylic shift reaction in the presence of silica gel or water. Detailed mechanistic studies involving 11B NMR, deuterium labelling and mass spec analysis suggested the involvement of a dienolate intermediate via water-mediated deboronation.Data availability
The data that support the findings of this study are available within the article and its ESI.† Crystallographic data for compounds 4a and 7h are available free of charge from the Cambridge Crystallographic Data Centre under references CCDC 2377383 and 2377384. All other data are available in the main text or the ESI.†Author contributions
A. S. and H. V. designed the research. A. S., H. V., A. G. and A. M. analyzed the data. H. V., A. G., A. M., M. P., A. R. conducted the experiments. A. S. wrote the manuscript with input from all the coauthors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Support of this research from NIH-NIGMS (R35GM143091 and R15GM135891) is gratefully acknowledged. We thank Dr Athula Attygalle for HRMS analysis and Dr Michelle C. Neary for X-ray studies. The X-ray machine was funded by the Air Force Office of Scientific Research under award number FA9550-20-1-0158.References
- C. Diner and K. J. Szabo, J. Am. Chem. Soc., 2017, 139, 2–14 CrossRef.
- H. P. Deng, L. Eriksson and K. J. Szabo, Chem. Commun., 2014, 50, 9207–9210 RSC.
- Z. L. Tao, X. H. Li, Z. Y. Han and L. Z. Gong, J. Am. Chem. Soc., 2015, 137, 4054–4057 CrossRef.
- J. W. Kennedy and D. G. Hall, J. Am. Chem. Soc., 2002, 124, 11586–11587 CrossRef.
- J. W. Kennedy and D. G. Hall, J. Org. Chem., 2004, 69, 4412–4428 CrossRef PubMed.
- T. Miura, J. Nakahashi and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 6989–6993 CrossRef PubMed.
- T. Miura, J. Nakahashi, W. Zhou, Y. Shiratori, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2017, 139, 10903–10908 CrossRef.
- J. Park, S. Choi, Y. Lee and S. H. Cho, Org. Lett., 2017, 19, 4054–4057 CrossRef.
- M. Shin, M. Kim, C. Hwang, H. Lee, H. Kwon, J. Park, E. Lee and S. H. Cho, Org. Lett., 2020, 22, 2476–2480 CrossRef.
- J. M. Zanghi and S. J. Meek, Angew. Chem., Int. Ed., 2020, 59, 8451–8455 CrossRef.
- M. Z. Liang and S. J. Meek, J. Am. Chem. Soc., 2020, 142, 9925–9931 CrossRef PubMed.
- J. C. Green, J. M. Zanghi and S. J. Meek, J. Am. Chem. Soc., 2020, 142, 1704–1709 CrossRef PubMed.
- E. Wheatley, J. M. Zanghi and S. J. Meek, Org. Lett., 2020, 22, 9269–9275 CrossRef PubMed.
- S. Gao, M. Duan, Q. Shao, K. N. Houk and M. Chen, J. Am. Chem. Soc., 2020, 142, 18355–18368 CrossRef PubMed.
- S. Gao, J. Chen and M. Chen, Chem. Sci., 2019, 10, 3637–3642 RSC.
- M. Wang, S. Gao and M. Chen, Org. Lett., 2019, 21, 2151–2155 CrossRef PubMed.
- Y. Wei, X.-Y. Xie, J. Liu, X. Liu, B. Zhang, X.-Y. Chen, S.-J. Li, Y. Lan and K. Hong, Angew. Chem., Int. Ed., 2024, 63, e202401050 CrossRef PubMed.
- H. Li, X. Shangguan, Z. Zhang, S. Huang, Y. Zhang and J. Wang, Org. Lett., 2014, 16, 448–451 CrossRef PubMed.
- D. Circule, F. Dénès and P. Renaud, Adv. Synth. Catal., 2024, 366, 2945–2955 CrossRef.
- H. Vyas, A. J. Gangani, A. Mini, S. Lin, J.-M. Chu, C. O. Agee, J. Gabriel, R. T. Williamson, Y. Zhang and A. Sharma, Chem.–Eur. J., 2024, 30, e202303175 CrossRef PubMed.
- M. J. Hesse, C. P. Butts, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2012, 51, 12444–12448 CrossRef PubMed.
- R. W. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 555–566 CrossRef.
- J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 6358–6360 CrossRef CAS PubMed.
- C. Schneider, Angew. Chem., Int. Ed., 2002, 41, 744–746 CrossRef CAS.
- Y. Ge, F. Ye, J. Liu, J. Yang, A. Spannenberg, H. Jiao, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 21585–21590 CrossRef CAS PubMed.
- S. L. Robinson, J. K. Christenson and L. P. Wackett, Nat. Prod. Rep., 2019, 36, 458–475 RSC.
- M. Groll, E. P. Balskus and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 14981–14983 CrossRef CAS PubMed.
- E. P. Balskus and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 6810–6812 CrossRef CAS.
- L. R. Reddy, P. Saravanan and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 6230–6231 CrossRef CAS PubMed.
- B. Mao, M. Fananas-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502–10566 CrossRef CAS PubMed.
- N. Winssinger and W. Liu, Synthesis, 2021, 53, 3977–3990 CrossRef.
- L. E. Wilbanks, H. E. Hennigan, C. D. Martinez-Brokaw, H. Lakkis, S. Thormann, A. S. Eggly, G. Buechel and E. I. Parkinson, ACS Chem. Biol., 2023, 18, 1624–1631 CrossRef CAS PubMed.
- X. Li and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 10304–10308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for synthesis of compounds, spectral and X-ray data. CCDC 2377383 and 2377384. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06514j |
| This journal is © The Royal Society of Chemistry 2025 |