
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of (−)-flueggenine A and (−)-15′-epi-flueggenine D†
Seung Mo
Seo
 a,
Dongwook
Kim
ab,
Taewan
Kim
a and
Sunkyu
Han
*ab
a,
Dongwook
Kim
ab,
Taewan
Kim
a and
Sunkyu
Han
*ab
aDepartment of Chemistry, Korea Advanced Institute of Science & Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: sunkyu.han@kaist.ac.kr
bCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
First published on 12th December 2024
Abstract
Securinega alkaloids, known for their unique structures and neuroplasticity-inducing potential, are promising candidates for treating neurodegenerative diseases such as depression and substance use disorders (SUD). Herein, we delineate the total synthesis of two dimeric Rauhut–Currier (RC) reaction-based securinega alkaloids, (−)-flueggenine A and (−)-15′-epi-flueggenine D. The key step involved a novel reductive Heck dimerization strategy, utilizing a silyl-tethered enone coupling partner to ensure the desired reactivity and stereoselectivity. This dimerization method, combined with established chemistry explored en route to (−)-flueggenines C and D, offers a comprehensive synthetic approach for accessing all known RC-based oligomeric securinega alkaloids.
Introduction
Securinega alkaloids have fascinated the synthetic community for over 60 years due to their unique molecular architectures1 and potent biological activities.2 The tetracyclic monomeric (nor)securinane framework characterized by the butenolide moiety and the tertiary amine group has spurred the development of novel synthetic strategies and tactics.3,4 Recently, it has been reported that securinega alkaloids can act as neuroplastogens,5–7 rendering them promising candidates for the development of therapeutics against neurodegenerative diseases such as depression and SUD.8,9Organisms have evolved to synthesize diverse natural products from a common precursor, optimizing the production of secondary metabolites and thereby gaining selective advantages. To expand the structural repertoires of secondary metabolites, organisms often biosynthesize oligomeric natural products by conjugating a well-defined monomeric unit.10 The plant Flueggea virosa has also adopted this strategy by biosynthesizing various RC reaction-based oligomeric securinega alkaloids such as fluevirosines A (1, Fig. 1),11 D (2),12 and G (3),13 and fluevirosinines F (4) and H (5).14
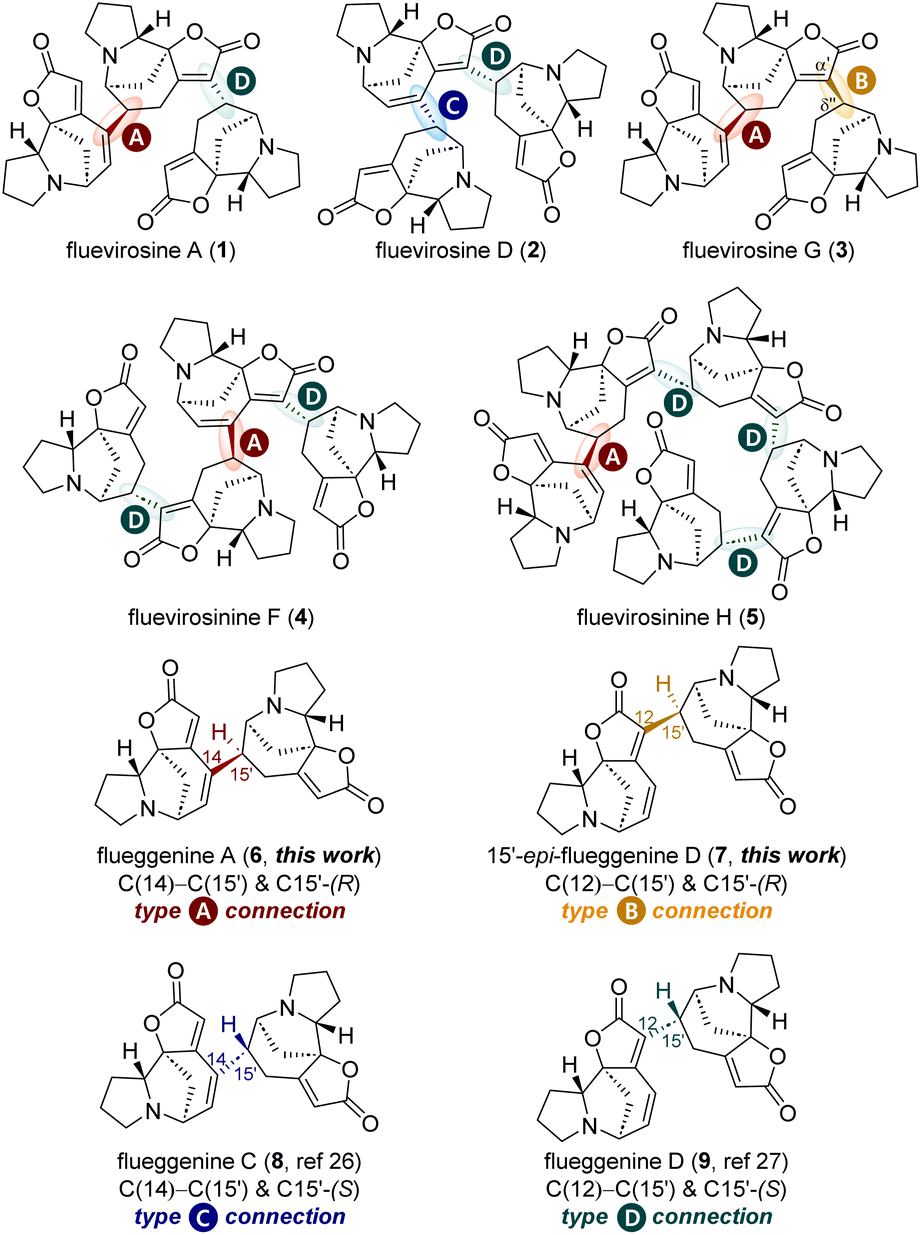 | ||
| Fig. 1 Representative Rauhut–Currier reaction-based high-order securinega alkaloids. | ||
Notably, the monomeric norsecurinine units that consist of these oligomeric securinega alkaloids are conjugated by four different types of connectivity that are exemplified in dimeric securinega alkaloid flueggenines A (6),15 C (8), and D (9),12 and 15′-epi-flueggenine D (7). For example, the C(14)–C(15′) bond with an (R)-configuration at C15′ present in flueggenine A (6, type A connection) and the C(12)–C(15′) bond with an (S)-configuration at C15′ present in flueggenine D (9, type D connection) are two connection types that conjugate three norsecurinine units in fluevirosine A (1). Similarly, norsecurinines that consist of trimeric fluevirosine D (2) are conjugated by types C and D connections found in dimeric flueggenines C (8) and D (9), respectively (Fig. 1). It is notable that fluevirosine G (3) is networked by type A connection and the connection that conjugates the C(α′) and C(δ′′) positions with an (R)-configuration at the C(δ′′) site. The latter connection (type B) is present in 15′-epi-flueggenine D (7), a presumed natural product yet to be discovered.16,17 An analysis of all RC-based high-order securinega alkaloids has shown that these four types of connections encompass all the conjugations among monomers found in this family of natural products.
Despite significant advancement in the total synthesis of monomeric securinega alkaloids,3,4 the synthesis of dimeric securinega alkaloids has been more challenging with fewer successful cases.18–23 Our group has been interested in the total synthesis of RC-based high-order securinega alkaloids.24,25 In 2017, we reported the total synthesis of (−)-flueggenine C (8) via an accelerated RC reaction strategy.26 In 2020, our group described the total synthesis of flueggenine D (9) enabled by a dimerization strategy involving a Stille cross-coupling reaction and a stereoselective conjugate reduction.27 However, these dimerization strategies could not be applied to the synthesis of flueggenine A (6) or the 15′-epimer of flueggenine D, the missing pieces needed to complete the conjugation network of RC-based oligomeric securinega alkaloids.28 Herein, we describe a new dimerization strategy that enables types A and B connections for the total synthesis of (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7).
Results and discussion
Retrosynthetic analysis of (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7) is presented in Scheme 1A. We envisioned to assemble the B-rings present in flueggenine A (6) and 15′-epi-flueggenine D (7) via parallel intramolecular N-alkylation reactions. For the construction of the butenolide D-rings, intramolecular Horner–Wadsworth–Emmons (HWE) reactions would be employed. We planned to apply a reductive Heck reaction for the key dimerization. While flueggenine A precursor 10 would be derived from a reductive Heck reaction between alkenyl iodide 11 and enone 12, epi-flueggenine D precursor 13 was designed to be accessed from a reductive Heck reaction between iodide 14 and the common enone 12. Iodides 11 and 14 were planned to be accessed from common enone 12via an α-iodination reaction and an iodo-Meyer–Schuster reaction, respectively.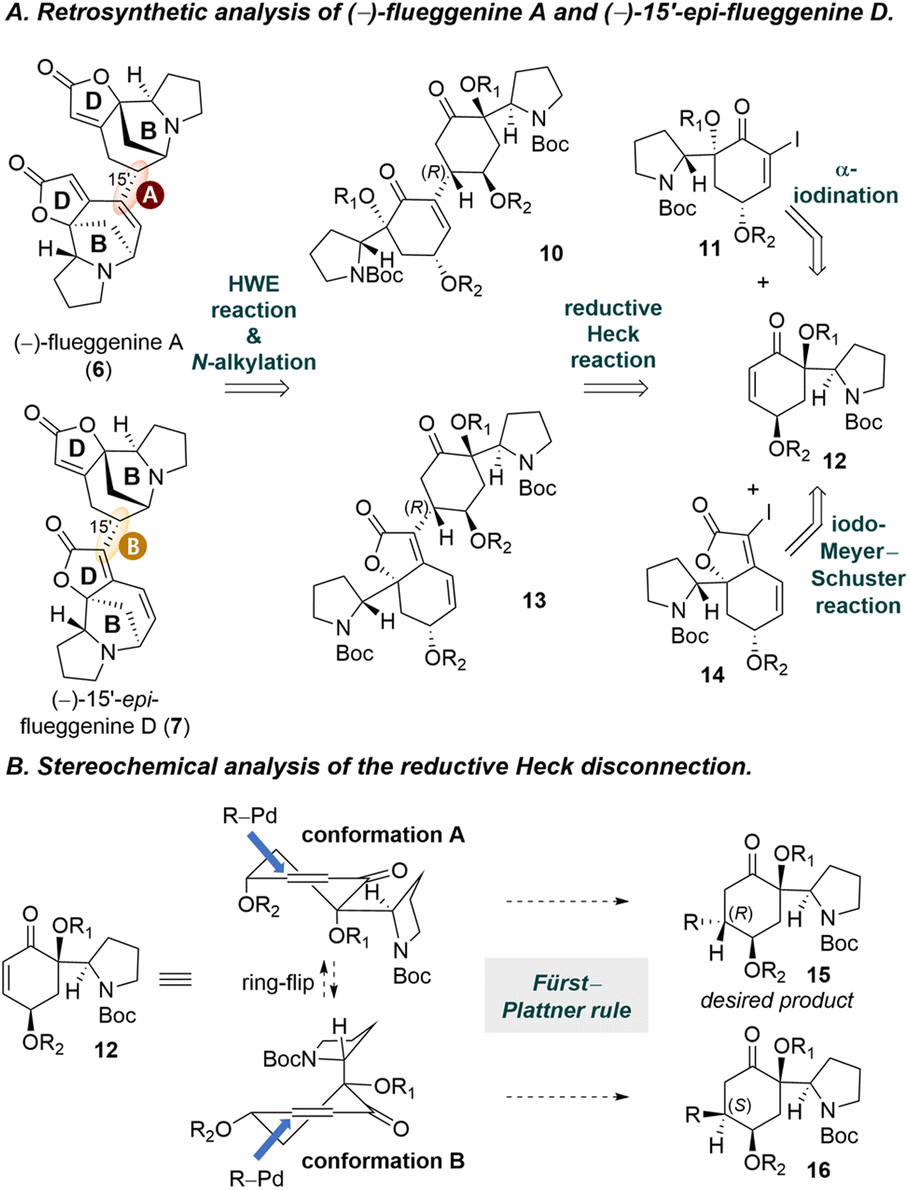 | ||
| Scheme 1 Synthetic design of (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7). | ||
We predicted that the stereochemical outcome of the reductive Heck reaction would be influenced by the conformation of enone 12. Enone 12 would adopt either conformation A or B (Scheme 1B). According to the Fürst–Plattner rule,29 when compound 12 assumes conformation A, the alkenylpalladium species approaches from the top face to avoid an unfavorable twist boat-like transition state, resulting in product 15 with the desired (R)-configuration at the connection junction. On the other hand, in conformation B, the alkenylpalladium intermediate would approach from the bottom face to yield compound 16 with an (S)-configuration at the connecting carbon. For the latter case, we anticipated that the protected allylic hydroxyl moiety may hinder the approach of the organopalladium species. Taking into consideration the sterically bulky nature of the Boc-protected pyrrolidine moiety, we initially anticipated that conformation A of enone 12 would be favored over conformation B.
To test the plausibility of the reductive Heck dimerization strategy, we initially attempted the reaction with alkenyl iodide 17 and cyclohexenone (18). Pleasantly, when iodide 17 and cyclohexenone (18) were heated in the presence of 5 mol% of palladium(II) trifluoroacetate (Pd(TFA)2) and diisopropylethylamine (DIPEA) in N-methyl-2-pyrrolidone (NMP),30 reductive Heck reaction product 19 was obtained in 76% yield as a 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers (Scheme 2). Encouraged by these results, we next attempted the reductive Heck reaction between iodide 17 and enone 20. However, even after extensive experimentation, the desired conjugated product 21 was not formed (Scheme 2). Deiodination of compound 17 was the major undesired pathway, yielding enone 20. This suggests that while the oxidative addition of the palladium catalyst to alkenyl iodide 17 was occurring, the migratory insertion of the resulting organopalladium intermediate into enone 20 was hindered.
:1 mixture of diastereomers (Scheme 2). Encouraged by these results, we next attempted the reductive Heck reaction between iodide 17 and enone 20. However, even after extensive experimentation, the desired conjugated product 21 was not formed (Scheme 2). Deiodination of compound 17 was the major undesired pathway, yielding enone 20. This suggests that while the oxidative addition of the palladium catalyst to alkenyl iodide 17 was occurring, the migratory insertion of the resulting organopalladium intermediate into enone 20 was hindered.
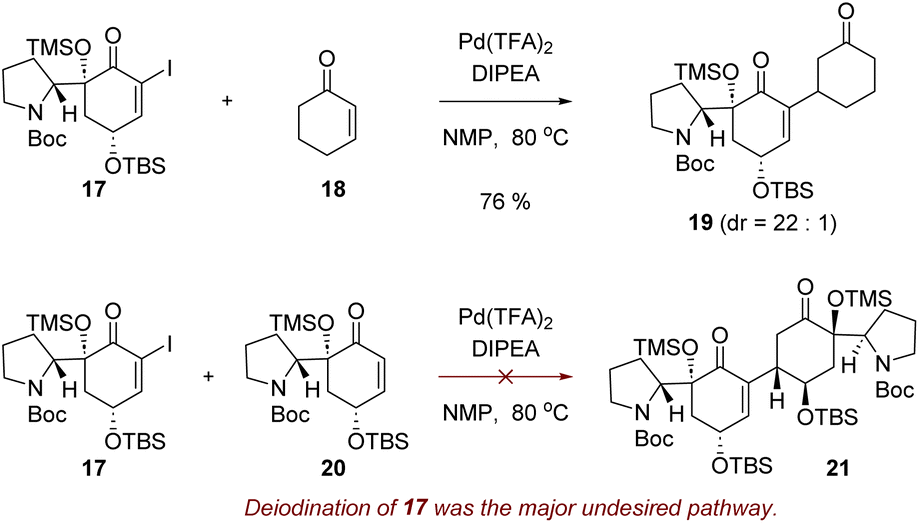 | ||
| Scheme 2 Initial attempts for the reductive Heck reaction. | ||
Further insights into the lack of the desired reactivity were gained through single-crystal X-ray diffraction (SCXD) analysis of enone 20 (CCDC number of compound 20: 2385997). Contrary to our initial predictions, the SCXD data revealed that compound 20 adopts a conformation where the Boc-protected pyrrolidine moiety is positioned pseudo-axially, while the two silyl ether groups are arranged pseudo-equatorially (Scheme 3). Solution phase DFT-calculations of enone 20 also corroborated this conformational preference (see the ESI for details†). In this conformation, the approach of the alkenylpalladium species from the top face would be inhibited by steric hindrance caused by the bulky Boc-protected pyrrolidine moiety and the unfavored twist boat-like transition state. The absence of the reductive Heck product with an (S)-configuration at the connection junction suggests that the approach of the organopalladium intermediate from the bottom face is also hindered, presumably due to the presence of the silylether group.
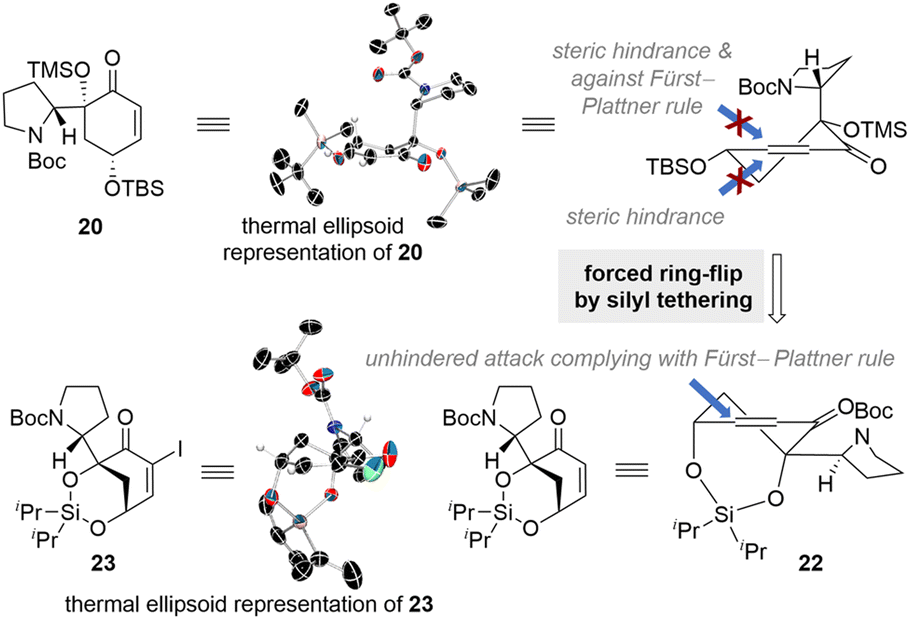 | ||
| Scheme 3 Conformational analysis of 20 and design of the silyl-tethered enone 22 for the reductive Heck reaction. | ||
These observations made it clear that we needed to induce a ring flip in the conformation of the reductive Heck acceptor enone to achieve the desired reactivity with the correct stereochemistry. Historically, there have been elegant examples where an energetically less favorable conformation was induced through transitory covalent bond formation to achieve the desired reactivity and selectivity.31–35 Inspired by these precedents, we envisioned tethering the two hydroxyl groups in compound 20 to enforce their axial positioning. To accomplish this, we designed silyl-tethered enone compound 22 to promote a ring flip. We anticipated that this silyl-tethered compound would adopt a conformation in which the Boc-protected pyrrolidine group is positioned pseudo-equatorially (Scheme 3). Indeed, SCXD analysis of the α-iodinated derivative of 22 confirmed that the silyl-tethered enone adopts the desired ring-flipped conformation (CCDC number of compound 23: 2385995). With this newly designed silyl-tethered reductive Heck acceptor enone 22, we predicted an unhindered approach of the alkenylpalladium species from the top face of it, complying with a Fürst–Plattner rule, to yield the desired types A and B connections present in flueggenine A (6) and 15′-epi-flueggenine D (7).
With the new design of tethered reductive Heck acceptor enone 22, we embarked on the total synthesis of both (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7). The synthesis of reductive Heck reaction coupling partners commenced with our previously accessed γ-hydroxyenone compound 24.26 For the synthesis of α-iodoenone 17, γ-hydroxyenone 24 was subjected to a two-step protocol involving TBS protection of the hydroxyl group (91% yield) and α-iodination of the enone moiety with iodine in pyridine and chloroform co-solvent (94% yield). The synthesis of silyl tethered enone 22 was achieved by first removing the TMS group of 24 in the presence of triethylamine trihydrofluoride to yield diol 25 in >99% yield (Scheme 4). Diol 25 was subsequently allowed to react with dichlorodiisopropylsilane in the presence of DIPEA to produce the key silyl tethered enone 22 in 94% yield. The synthesis of iodobutenolide compound 28 involved selective TBS protection of the secondary allylic hydroxyl group in diol 25. The resulting ketone compound 26 was reacted with lithium phenoxyacetylide generated in situ from phenoxy dichloroethene 27 to yield the propargylic alcohol intermediate. This 1,2-addition product was subsequently allowed to react with N-iodosuccinimide (NIS) to yield iodobutenolide 28via a presumed iodo-Meyer–Schuster rearrangement in 47% yield over two steps.27,36,37 The structure of iodide 28 was corroborated by SCXD analysis (CCDC number of compound 28: 2385999).
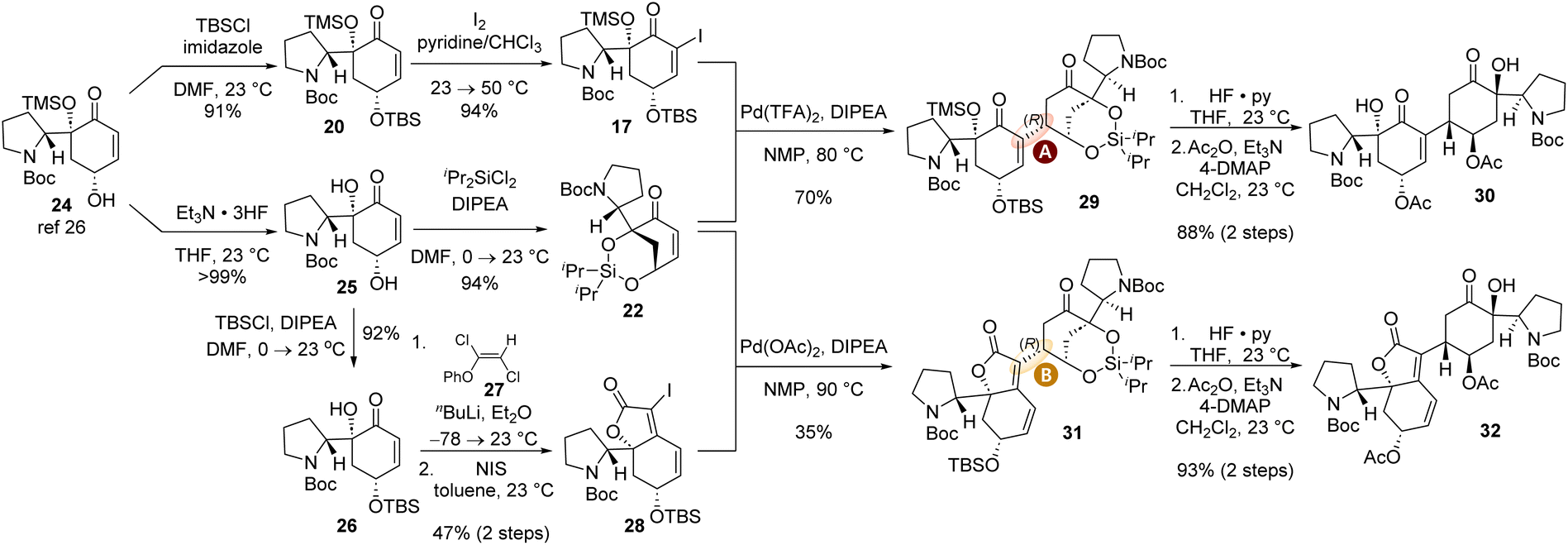 | ||
| Scheme 4 Synthesis of the coupling partners for the reductive Heck reaction and its successful execution toward the synthesis of (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7). | ||
With iodoenone 17 and silyl tethered enone 22 in hand, we executed the key reductive Heck reaction in the presence of Pd(TFA)2 and DIPEA. To our utmost delight, the desired reductive Heck product 29 with the (R)-configuration at the connection junction (type A connection) was obtained in 70% yield consistent with our stereochemical model (Schemes 3 and 4). HF·pyridine-mediated global desilylation of compound 29 and subsequent acetylation of the resulting product afforded diacetate 30 in 88% yield over two steps. To our pleasure, iodobutenolide 28 and silyl tethered enone 22 successfully furnished conjugated product 31 with type B connectivity in the presence of Pd(OAc)2 and DIPEA in 35% yield. When silyl ether 31 was subjected to the aforementioned desilylation and acetylation protocol, acetylated compound 32 was obtained in 93% yield over two steps. The structure of compound 32 was unambiguously conformed by its SCXD analysis (Scheme 5, CCDC number of compound 32: 2386004).
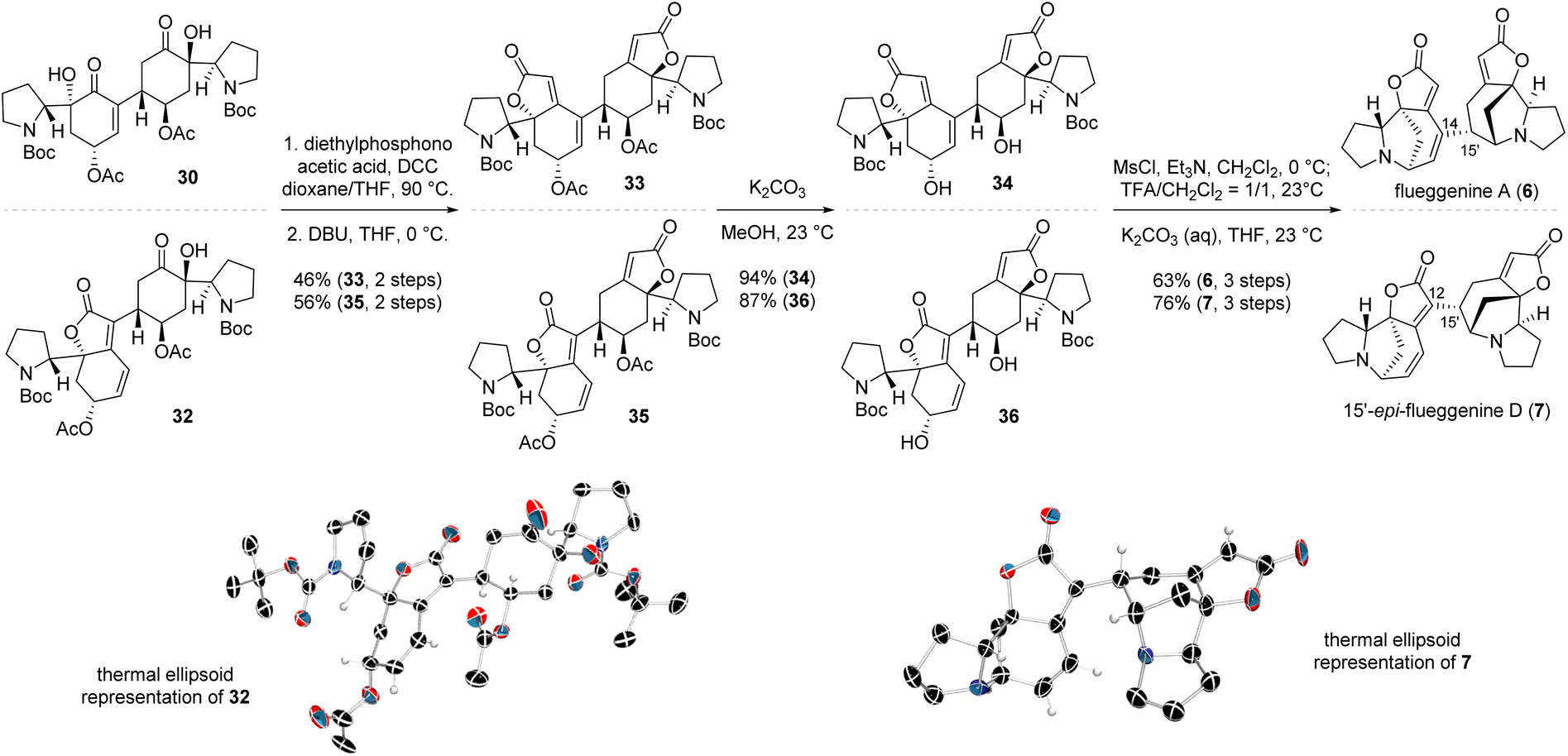 | ||
| Scheme 5 Completion of the total synthesis of (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7). | ||
The end-game of the synthetic campaign toward (−)-flueggenine A (6) involved parallel installation of the butenolide moiety and intramolecular N-alkylation to construct the octacyclic framework. The butenolide moiety was introduced by treating diol 30 with diethylphosphonoacetic acid in the presence of N,N′-dicyclohexylcarbodiimide (DCC), yielding the HWE precursor. Subsequent reaction of the resulting ester with 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) produced the butenolide compound 33 in 46% yield over two steps (Scheme 5). For the N-alkylation, acetate 33 was subjected to methanolysis, yielding diol 34 in 94% yield. The diol moiety was then activated by mesylation. Trifluoroacetic acid (TFA)-mediated Boc-deprotection of the resulting carbamate intermediate, followed by base treatment of the secondary amine, afforded the first synthetic sample of (−)-flueggenine A (6) in 63% yield over 3 steps. The spectral data of the synthetic compound matched those of the natural product.15 To our delight, applying the same end-game protocol to alcohol 32 resulted in the successful synthesis of 15′-epi-flueggenine D (7) in analogous efficiency compared to the route to (−)-flueggenine A (6). The structure of 15′-epi-flueggenine D (7) was unequivocally corroborated by its SCXD analysis (CCDC number of compound 7: 2387436).
Conclusions
In conclusion, we have completed the total synthesis of RC-based dimeric securinega alkaloids (−)-flueggenine A (6) and (−)-15′-epi-flueggenine D (7). The key dimerization step was achieved via a reductive Heck reaction. The design and employment of silyl-tethered reductive Heck acceptor enone was crucial to achieve the desired reactivity as well as stereoselectivity. This new dimerization strategy, enabling types A and B linkages between norsecurinine monomeric units, combined with existing conjugation chemistry for types C and D connections,26,27 provides a foundational synthetic solution to access all known RC-based oligomeric securinega alkaloids. Those studies are currently underway in our laboratory and will be the subject of forthcoming reports.Data availability
The experimental procedures and additional data can be found in the ESI.† Crystallographic data for the structure reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number 2387436 (7), 2385997 (20), 2385995 (23), 2385999 (28), and 2386004 (32). Copies of the data can be obtained free of charge from the CCDC viahttps://www.ccdc.cam.ac.uk/structures/.Author contributions
S. M. S. and S. H. conceived the study. S. H. supervised the project. S. M. S. played a key role in experimentation. D. K. performed the single crystal X-ray diffraction analysis. T. K. conducted computational studies. S. H. and S. M. S. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our sincere gratitude to Dr Jae-Sun Shin for his valuable discussions and expert guidance on the NMR analyses. Our thanks also extend to Dr Young Ho Jang for his insightful advice during the studies. We are grateful to Kyungbae Kim, an undergraduate student, for his experimental contribution to the project. This work was supported by the National Research Foundation of Korea (NRF-2021R1A2C2011203). Furthermore, we acknowledge support by the National Research Foundation of Korea (NRF-2018R1A5A1025208). This research was also supported by the KAIST UP project, KAIST Grand Challenge 30 project, and KAIST Cross-Generation Collaborative Lab Project.References
- E. Chirkin, W. Atkatlian and F.-H. Porée, in The Alkaloids: Chemistry and Biology, ed. H.-J. Knölker, Academic Press, London, 2015, ch. 1, pp. 1–120 Search PubMed.
- W. Hou, H. Huang, X.-Q. Wu and J.-X. Lan, Bioactivities and mechanism of action of securinega alkaloids derivatives reported prior to 2022, Biomed. Pharmacother., 2023, 158, 114190 CrossRef CAS PubMed.
- S. M. Weinreb, Total synthesis of the Securinega alkaloids, Nat. Prod. Rep., 2009, 26, 758–775 RSC.
- R. Wehlauch and K. Gademann, Securinega Alkaloids: Complex Structures, Potent Bioactivities, and Efficient Total Syntheses, Asian J. Org. Chem., 2017, 6, 1146–1159 CrossRef CAS.
- H. Xiao, Q. Zhang, P. Zhong, G. Tang, L. Tao, Z. Huang, D. Guo, Y. Liao, Y. Peng, Z.-L. Wu, Y. Wang, W.-C. Ye and L. Shi, Securinine Promotes Neuronal Development and Exhibits Antidepressant-like Effects via mTOR Activation, ACS Chem. Neurosci., 2021, 12, 3650–3661 CrossRef CAS PubMed.
- Z.-L. Wu, X.-J. Huang, M.-T. Xu, X. Ma, L. Li, L. Shi, W.-J. Wang, R.-W. Jiang, W.-C. Ye and Y. Wang, Flueggeacosines A–C, Dimeric Securinine-Type Alkaloid Analogues with Neuronal Differentiation Activity from Flueggea suffruticosa, Org. Lett., 2018, 20, 7703–7707 CrossRef CAS PubMed.
- Q.-F. He, Z.-L. Wu, L. Li, W.-Y. Sun, G.-Y. Wang, R.-W. Jiang, L.-J. Hu, L. Shi, R.-R. He, Y. Wang and W.-C. Ye, Discovery of Neuritogenic Securinega Alkaloids from Flueggea suffruticosa by a Building Blocks-Based Molecular Network Strategy, Angew. Chem., Int. Ed., 2021, 60, 19609–19613 CrossRef CAS PubMed.
- D. E. Olson, Psychoplastogens: A Promising Class of Plasticity-Promoting Neurotherapeutics, J. Exp. Neurosci., 2018, 12, 1179069518800508 CrossRef PubMed.
- H. N. Saeger and D. E. Olson, Psychedelic-inspired approaches for treating neurodegenerative disorders, J. Neurochem., 2022, 162, 109–127 CrossRef CAS PubMed.
- S. A. Snyder, A. M. ElSohly and F. Kontes, Synthetic approaches to oligomeric natural products, Nat. Prod. Rep., 2011, 28, 897–924 RSC.
- H. Zhang, C.-R. Zhang, K.-K. Zhu, A.-H. Gao, C. Luo, J. Li and J.-M. Yue, Fluevirosines A–C: A Biogenesis Inspired Example in the Discovery of New Bioactive Scaffolds from Flueggea virosa, Org. Lett., 2013, 15, 120–123 CrossRef CAS PubMed.
- H. Zhang, W. Wei and J.-M. Yue, From monomer to tetramer and beyond: the intriguing chemistry of Securinega alkaloids from Flueggea virosa, Tetrahedron, 2013, 69, 3942–3946 CrossRef CAS.
- H. Zhang, C.-R. Zhang, Y.-S. Han, M. A. Wainberg and J.-M. Yue, New Securinega alkaloids with anti-HIV activity from Flueggea virosa, RSC Adv., 2015, 5, 107045–107053 RSC.
- H. Zhang, Y.-S. Han, M. A. Wainberg and J.-M. Yue, Anti-HIV Securinega alkaloid oligomers from Flueggea virosa, Tetrahedron, 2015, 71, 3671–3679 CrossRef CAS.
- L.-S. Gan, C.-Q. Fan, S.-P. Yang, Y. Wu, L.-P. Lin, J. Ding and J.-M. Yue, Flueggenines A and B, Two Novel C,C-Linked Dimeric Indolizidine Alkaloids from Flueggea virosa, Org. Lett., 2006, 8, 2285–2288 CrossRef CAS.
- B. E. Hetzler, D. Trauner and A. L. Lawrence, Natural product anticipation through synthesis, Nat. Rev. Chem, 2022, 6, 170–181 CrossRef.
- M. Kim, S. Park, G. Kang, Y. S. Jang, K. H. Kim, S. Han and C. S. Kim, Natural product anticipation via chemical synthesis: Discovery of two new securinega alkaloids, iScience, 2024, 27, 110495 CrossRef CAS PubMed.
- B.-X. Zhao, Y. Wang, C. Li, G.-C. Wang, X.-J. Huang, C.-L. Fan, Q.-M. Li, H.-J. Zhu, W.-M. Chen and W.-C. Ye, Flueggedine, a novel axisymmetric indolizidine alkaloid dimer from Flueggea virosa, Tetrahedron Lett., 2013, 54, 4708–4711 CrossRef CAS.
- H. Wei, C. Qiao, G. Liu, Z. Yang and C. Li, Stereoselective Total Syntheses of (−)-Flueggine A and (+)-Virosaine B, Angew. Chem., Int. Ed., 2013, 52, 620–624 CrossRef CAS.
- N. Ma, Y. Yao, B.-X. Zhao, Y. Wang, W.-C. Ye and S. Jiang, Total synthesis of securinega alkaloids (−)-norsecurinine, (−)-niruroidine and (−)-flueggine A, Chem. Commun., 2014, 50, 9284–9287 RSC.
- G. Kang and S. Han, Synthesis of Dimeric Securinega Alkaloid Flueggeacosine B: From Pd-Catalyzed Cross-Coupling to Cu-Catalyzed Cross-Dehydrogenative Coupling, J. Am. Chem. Soc., 2022, 144, 8932–8937 CrossRef CAS PubMed.
- G. Kang and S. Han, Synthesis of Suffranidine B, J. Am. Chem. Soc., 2023, 145, 24493–24498 CAS.
- L. Liu, T. L. Olson and J. L. Wood, Total Synthesis of (−)-Flueggeacosine C, Org. Lett., 2024, 26, 7341–7346 CrossRef CAS PubMed.
- S. Jeon, J. Park and S. Han, Syntheses of Dimeric Securinega Alkaloids, Synlett, 2017, 28, 2353–2359 CrossRef CAS.
- G. Kang, S. Park and S. Han, Synthesis of High-Order and High-Oxidation State Securinega Alkaloids, Acc. Chem. Res., 2023, 56, 140–156 CrossRef CAS.
- S. Jeon and S. Han, An Accelerated Intermolecular Rauhut–Currier Reaction Enables the Total Synthesis of (−)-Flueggenine C, J. Am. Chem. Soc., 2017, 139, 6302–6305 CrossRef CAS.
- S. Jeon, J. Lee, S. Park and S. Han, Total synthesis of dimeric Securinega alkaloids (−)-flueggenines D and I, Chem. Sci., 2020, 11, 10934–10938 RSC.
- J. Park, S. Jeon, G. Kang, J. Lee, M.-H. Baik and S. Han, Dimerization Strategies for the Synthesis of High-Order Securinega Alkaloids, J. Org. Chem., 2019, 84, 1398–1406 CrossRef CAS PubMed.
- A. Fürst and P. A. Plattner, Über Steroide und Sexualhormone. 160. Mitteilung. 2α, 3α- und 2β, 3β-Oxido-chlolestane; Konfiguration der 2-Oxy-cholestane, Helv. Chim. Acta, 1949, 32, 275–283 CrossRef.
- S. Mannathan, S. Raoufmoghaddam, J. N. H. Reek, J. G. de Vries and A. J. Minnaard, Palladium(II) Acetate Catalyzed Reductive Heck Reaction of Enones; A Practical Approach, ChemCatChem, 2015, 7, 3923–3927 CrossRef CAS.
- R. B. Woodward, F. E. Bader, H. Bickel, A. J. Frey and R. W. Kierstead, THE TOTAL SYNTHESIS OF RESERPINE, J. Am. Chem. Soc., 1956, 78, 2023–2025 CrossRef CAS.
- H. Todoroki, M. Iwatsu, D. Urabe and M. Inoue, Total Synthesis of (−)-4-Hydroxyzinowol, J. Org. Chem., 2014, 79, 8835–8849 CrossRef CAS.
- J. Gong, H. Chen, X.-Y. Liu, Z.-X. Wang, W. Nie and Y. Qin, Total synthesis of atropurpuran, Nat. Commun., 2016, 7, 12183 CrossRef CAS PubMed.
- J. Zhang, M. Liu, C. Wu, G. Zhao, P. Chen, L. Zhou, X. Xie, R. Fang, H. Li and X. She, Total Synthesis of (−)-Pepluanol B: Conformational Control of the Eight-Membered-Ring System, Angew. Chem., Int. Ed., 2020, 59, 3966–3970 CrossRef CAS.
- T. Watanabe, K. Oga, H. Matoba, M. Nagatomo and M. Inoue, Total Synthesis of Taxol Enabled by Intermolecular Radical Coupling and Pd-Catalyzed Cyclization, J. Am. Chem. Soc., 2023, 145, 25894–25902 CrossRef CAS.
- S. Puri, N. Thirupathi and M. Sridhar Reddy, Iodo Meyer–Schuster Rearrangement of 3-Alkoxy-2-yn-1-ols for β-Mono (Exclusively Z-Selective)-/Disubstituted α-Iodo-α,β-Unsaturated Esters, Org. Lett., 2014, 16, 5246–5249 CrossRef CAS.
- M. S. Reddy, N. Thirupathi, M. Hari Babu and S. Puri, Synthesis of Substituted 3-Iodocoumarins and 3-Iodobutenolides via Electrophilic Iodocyclization of Ethoxyalkyne Diols, J. Org. Chem., 2013, 78, 5878–5888 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including characterization data and NMR spectra. CCDC 2387436, 2385997, 2385995, 2385999 and 2386004. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07525k |
| This journal is © The Royal Society of Chemistry 2025 |