
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReconstructing the phase of vanadium oxides enables redox-catalysis manipulated reversible sulfur conversion for stable Zn–S batteries†
Hao
Luo‡
 *ae,
Fan
Li‡
ab,
Mingli
Wang
*d,
Shang
Sun
b,
Min
Zhou
b,
Wenjing
Zhang
b,
Hengrui
Guo
ae,
Xueyin
Su
ae,
Xiaolong
Li
*c and
Lina
Ma
*b
*ae,
Fan
Li‡
ab,
Mingli
Wang
*d,
Shang
Sun
b,
Min
Zhou
b,
Wenjing
Zhang
b,
Hengrui
Guo
ae,
Xueyin
Su
ae,
Xiaolong
Li
*c and
Lina
Ma
*b
aSchool of Materials Science and Engineering, Xiamen University of Technology, Xiamen 361024, China. E-mail: luohao_hit@163.com
bCollege of Chemistry and Chemical Engineering, Qingdao University, Qingdao 266071, China. E-mail: malina@qdu.edu.cn
cState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute, Sichuan University, Chengdu 610065, P.R. China. E-mail: xiaolongli@scu.edu.cn
dSchool of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: mingliw2000@163.com
eSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, China
First published on 18th November 2024
Abstract
The naturally sluggish redox kinetics and limited utilization associated with the sulfur conversion in Zn/S electrochemistry hinder its real application. Herein, we report an in situ phase reconstruction strategy that activates the catalytic activity of vanadium oxides for invoking redox-catalysis to manipulate reversible sulfur conversion. It was identified that the V2O3@C/S precursor derived from metal organic frameworks could be transformed into V2O5−m·nH2O@C/S by a facile electrochemical induction process. Vanadium oxides can realize a faster zinc ion storage process than sulfur components during the discharging process, thereby the pre-zincified ZnxV2O5·nH2O behaves as a redox medium to catalyze the sulfur reduction via a spontaneous reaction (Znx+1V2O5 + S = ZnxV2O5 + ZnS, △G = −6.4 kJ mol−1). For the reverse battery recharging, the electrodeposited ZnS around the active sites can be easily activated and the facile Zn2+ transport between ZnxV2O5·nH2O and ZnS enables the reversible conversion of ZnS back to S (ZnxV2O5 + ZnS = Znx+1V2O5 + S, ΔG = −7.02 kJ mol−1). Accordingly, the composite cathode delivers a high capacity of 1630.7 mA h g−1 and maintains stable capacity retention after 150 cycles at 4 A g−1. The proposed redox catalytic effect sheds light on the tunable Zn–S chemistry.
1 Introduction
Aqueous zinc–sulfur (Zn–S) batteries feature the merits of being cost-efficient, high theoretical energy density, and non-toxicity, and have been considered as one of the candidates for the next generation of energy storage equipment. Although achievements have been made in Zn–S batteries, their notorious problems have hampered their further development.1–4 Specifically, the inherent low conductivity of sulfur and its discharge product ZnS greatly jeopardize the redox kinetics, thereby offering an inferior electrochemical response and limited utilization of active materials.5–7 Furthermore, during the zincation process, the significant expansion of the sulfur species could inflict mechanical damage on the electrode, further deteriorating the cycling performance.To solve the above-mentioned problems, some strategies have been developed. For example, recent studies indicate that carbon-based materials supported by sulfur are promising cathode materials for Zn–S batteries, but the nonpolar carbon matrix fails to promote the redox kinetics of sulfur. Thus, one can further incorporate the electrocatalytic sites and/or components (e.g., metal oxides) into the composites to facilitate the redox kinetics of sulfur species, thereby endowing the battery with a higher reversible capacity.8–15 However, for the metal-based catalytic components, it is usually difficult to achieve uniform dispersion in the carbon matrix. In this context, metal–organic frameworks (MOFs) have attracted much attention due to their high porosity, large surface area, and uniform distribution of metal centers and organic ligands at the molecular level.16,17 After heat treatment, a composite of the carbon matrix with a uniform distribution of metal oxides can be obtained (i.e., catalytic sites), which has the potential to act as the functional matrix for hosting sulfur. Moreover, most reported studies usually focus on accelerating the one-direction conversion (reduction or oxidation) of sulfur, which cannot realize the high utilization of active materials.18,19 Therefore, it is significant to explore comprehensive cathode materials with bidirectional catalysis for sulfur conversion.
In this contribution, a composite sulfur cathode composed of V2O5 and porous carbon (VOC/S) has been prepared by an in situ electrochemical-induction strategy, in which the inactive V2O3 component is converted to sulphophilic V2O5−m·nH2O. Benefiting from a unique two-electron redox reaction during the (de) zincification process, the ZnxV23+/5+O5 hotspots can serve as a redox medium to invoke the redox catalytic effect for tuning the bidirectional sulfur redox (Scheme 1), thereby greatly enhancing the electrochemical performance of the Zn–S batteries. Accordingly, a highly reversible discharge capacity of 1630.7 mA h g−1 and excellent cycling stability can be achieved. This work sheds light on the feasibility of redox catalysis in tuning the sulfur speciation pathway toward high-performance Zn–S batteries.
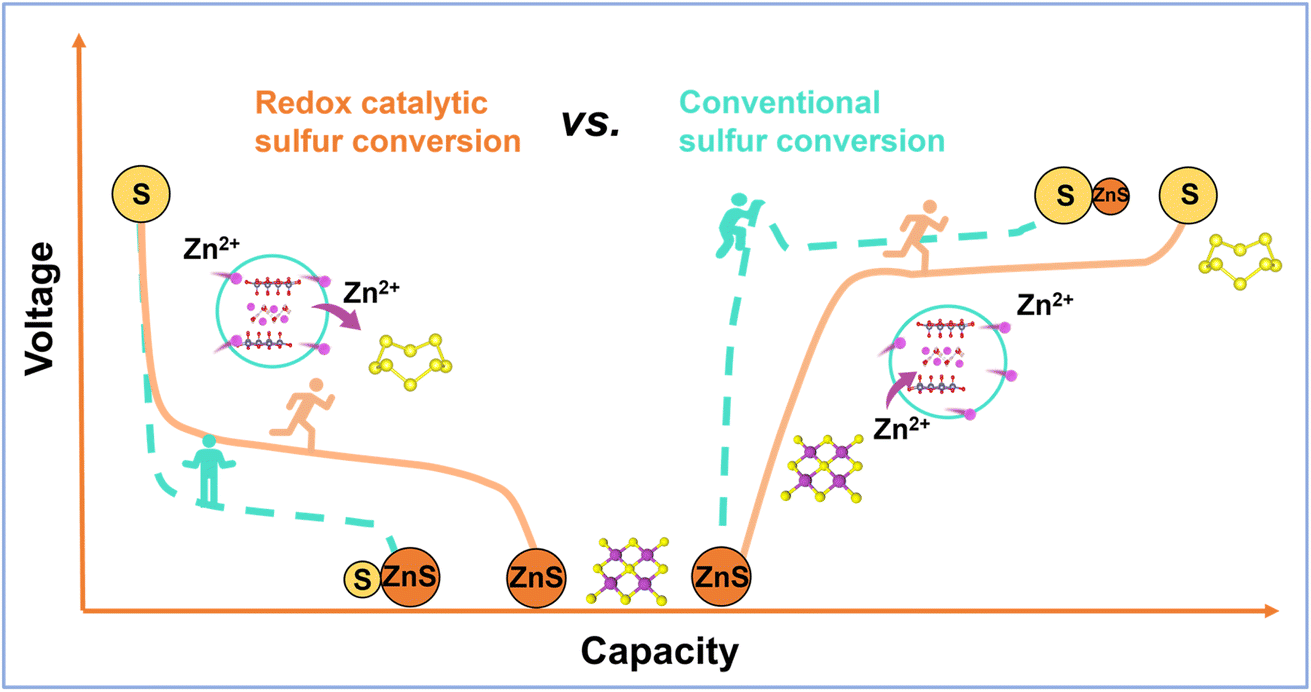 | ||
| Scheme 1 Schematic illustration of different sulfur speciation pathways enabled by the different matrices. | ||
2 Results and discussion
2.1 Design and characterization of the functional host
Fig. 1a demonstrates the schematic formation process of the V2O3@C/S precursor. In a typical procedure, the vanadium-based metal–organic framework (V-MOF) was prepared by the solvothermal method,20 which showed a cube structure with a variable length (Fig. S1†). After carbonization, the organic component was decomposed into the carbon matrix with a porous structure while the V-centers were oxidized to V2O3, which overall led to the transformation of the V-MOF into the vanadium-based oxide and carbon composite materials (V2O3@C). Of note, to ensure that the V2O3@C inherited the morphological characteristics of the V-MOF without the structure collapsing, we chose to heat the vanadium-based MOFs at a slow heating rate (2 °C min−1) up to 700 °C and keep them there for 4 h to obtain the V2O3-containing porous carbon materials (Fig. S2†). Then, the active sulfur was encapsulated into the V2O3@C matrix (V2O3@C/S) through a conventional melting-diffusion method and the cube structure was retained well, which also realized a sulfur loading of 35 wt% (Fig. 1b and S3†). As shown in Fig. 1c, the high-resolution transmission electron microscopy (HR-TEM) images displayed the lattice fringes of 0.29 nm and 0.32 nm, corresponding to the (104) plane of V2O3 and the (206) plane of sulfur (Fig. 1d).21 The crystalline structure of V2O3@C/S was also analyzed by the X-ray diffraction (XRD) technique, and the corresponding results agreed well with the above TEM analysis (Fig. S4†). Furthermore, energy dispersive X-ray spectroscopy (EDS) mapping of V2O3@C/S unveiled the homogeneous dispersion of the sulfur element within the carbon scaffold (Fig. 1e). And the overlapping of the C and V elements also confirmed the uniform distribution of vanadium oxides, which is beneficial for facilitating the sulfur redox conversion. The surface characteristics and pore structure of V2O3@C before and after sulfur impregnation were evaluated by nitrogen adsorption–desorption measurements (Fig. 1f). V2O3@C possesses a high specific surface area of about 201.3 m2 g−1 and a mesopore dominant structure (Table S1†), which can achieve a relatively high sulfur loading while maintaining a good balance of electrochemical performance.22 After sulfur loading, the sharp decrease in the specific surface area and pore size confirmed the encapsulation of sulfur into the nanopores of V2O3@C (Fig. S5†). The above results reveal the successful synthesis of the V2O3@C matrix and the good encapsulation of sulfur.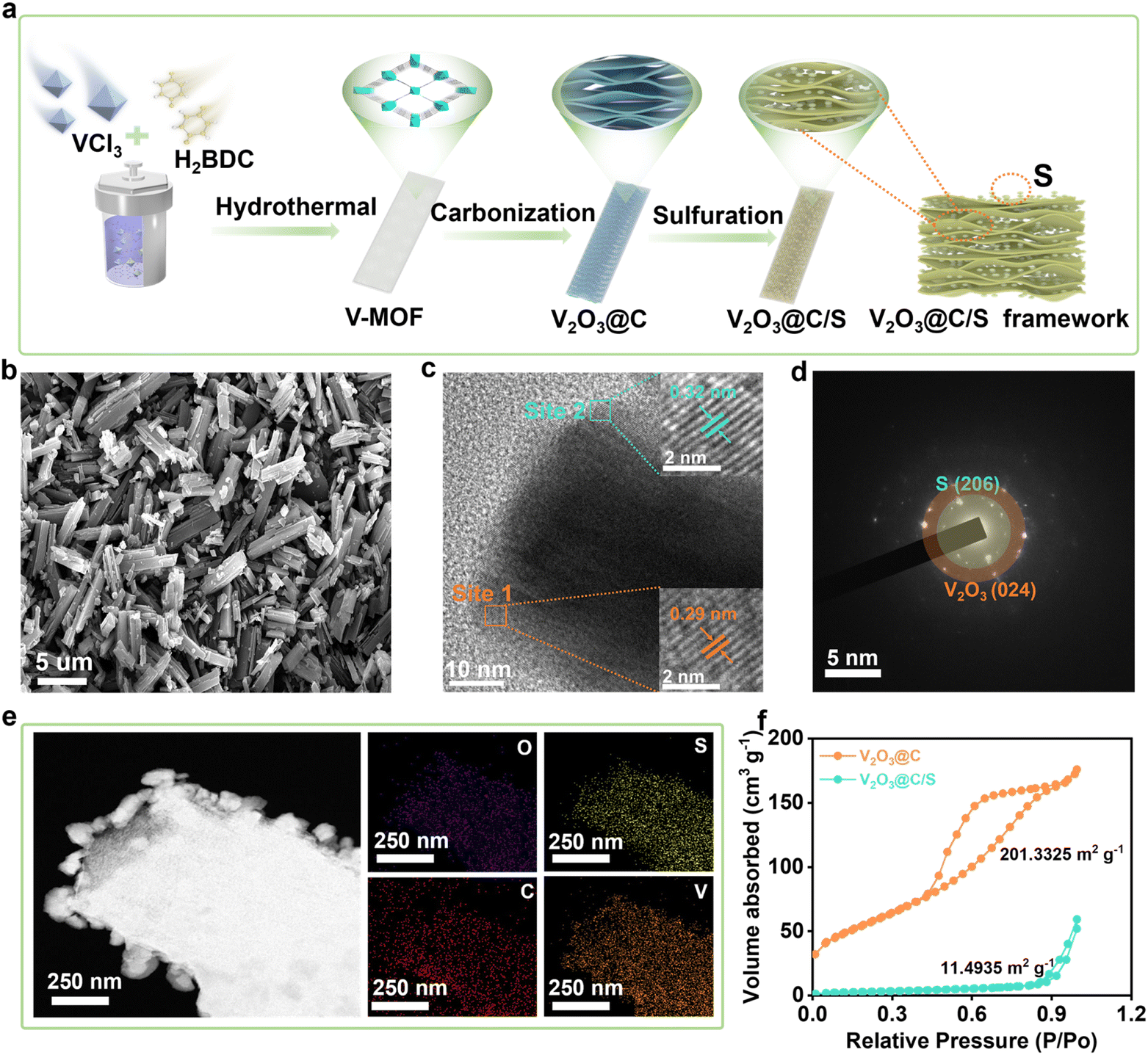 | ||
| Fig. 1 Structural and compositional characterization of the as-prepared V2O3@C/S precursor. (a) Schematic illustration of the synthesis procedure for the V2O3@C/S composite. (b) SEM images of V2O3@C/S. (c) TEM images of V2O3@C/S. (d) SAED pattern of V2O3@C/S. (e) SEM image of V2O3@C/S and corresponding elemental mappings. (f) Nitrogen adsorption–desorption isotherms of different samples. | ||
The low valence state of V and the insufficient active sites in V2O3 motivated us to further activate its activity by increasing the valence state of vanadium species. Thus, the in situ electrochemical induction strategy was employed to reconstruct the phase of V2O3 in V2O3@C. As shown in Fig. 2a, the V2O3@C electrodes underwent electrochemical oxidation during the first charge process as revealed by the high initial charge capacity, suggesting the transformation of V2O3@C into V2O5@C (VOC). It is worth noting that the phase conversion of vanadium oxides is irreversible because the highly efficient anodic oxidation of V2O3@C was achieved only during the first charging process. Correspondingly, as shown in the CV curves of VOC (Fig. 2b), an obvious oxidation peak at ∼1.27 V appeared during the first positive scan and then disappeared in the subsequent cycles, which indicates the irreversible electrochemical oxidation of V2O3 and also consistent with the charge–discharge curves in Fig. 2a. Furthermore, during the subsequent cycles, two redox peaks at ∼1.09/1.1 V and ∼0.55/0.74 V were found to be in good agreement with the redox peaks of V5+/V4+ and V4+/V3+, which further indicated the formation of V2O5.20 To further explore the structural evolution of VOC during the initial discharge/charge cycle, XRD patterns at different voltages were collected (Fig. 2c). When the VOC cathode is recharged to 1.5 V, a peak of V2O5−m·nH2O appears (JCPDS no. 00-025-1006) while the characteristic peak moves slightly to the left, indicating the expanded lattice spacing. As summarized in Fig. 2d, the phase transformation from V2O3 to V2O5−m·nH2O occurs during the first charging process, and the VOC then functions as the cathode for hosting the ion (de)intercalation in subsequent cycles. Moreover, it is worth mentioning that the in situ formed V2O5−m·nH2O has faster ion (de)intercalation kinetics and a higher ion storage capacity than commercial V2O5. And, the lattice water within the V2O5−m·nH2O can act as a pillar to maintain the stability of the layered V2O5, thus potentially enhancing the durability of the catalytic matrix.23,24 Theoretical calculations were also employed to verify the activity of two types of vanadium oxides toward zinc ion storage. As shown in Fig. 2e, adsorption energies of V2O3 and V2O5−m·2.4H2O towards Zn2+ are calculated, and it is found that the adsorption energy of V2O5−m·nH2O (−3.33 eV) is much higher than that of V2O3 (−0.51 eV). This difference may be ascribed to the more exposed negatively charged O of the layered V2O5−m·nH2O that shows stronger adsorption ability toward zinc ions. This result indicates that V2O5−m·nH2O could play a key role in facilitating reversible sulfur conversion by accelerating zinc transport.
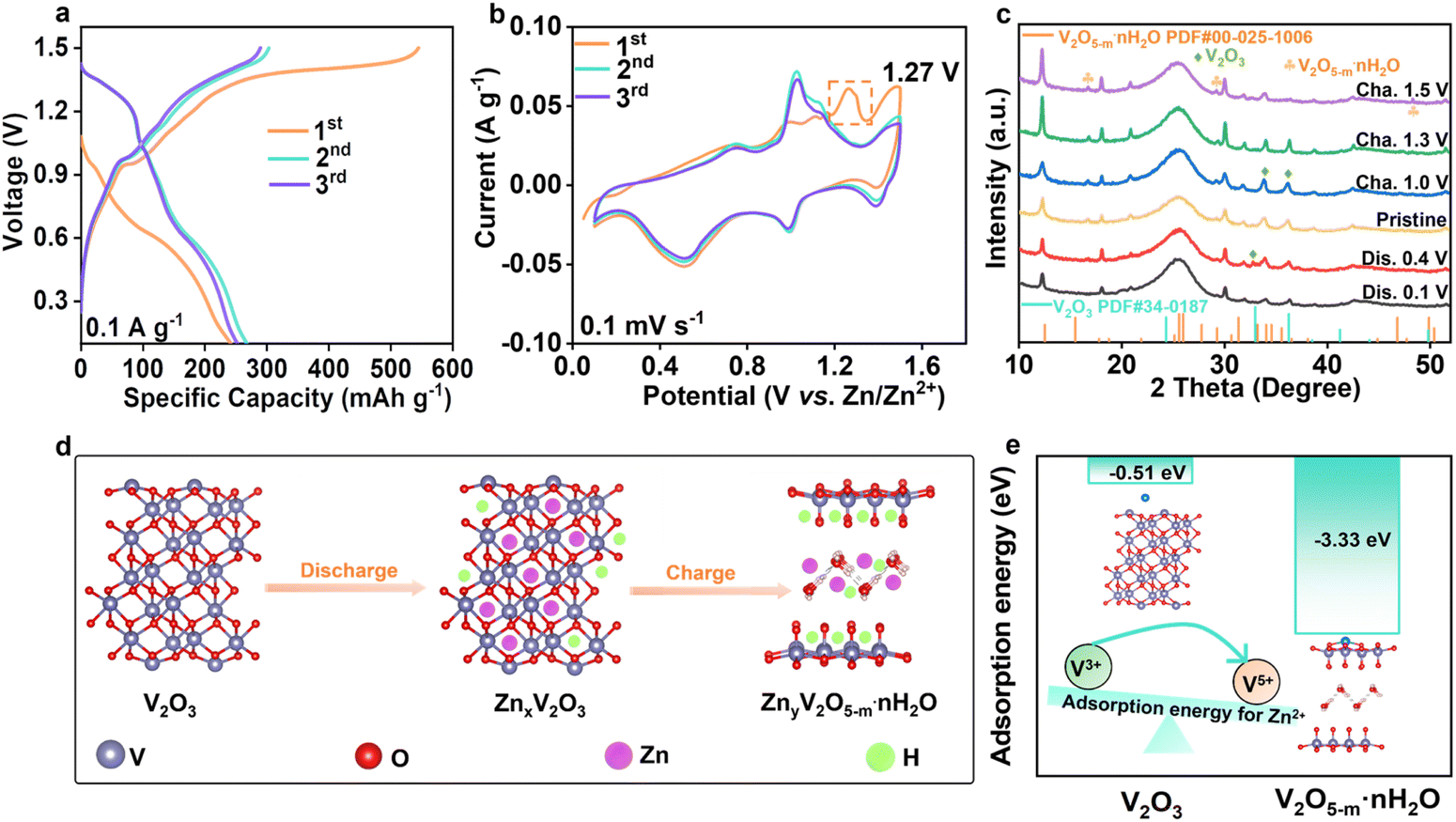 | ||
| Fig. 2 In situ electrochemical reconstruction of the phase of vanadium oxides. (a) Galvanostatic charge/discharge curves of VOC at a current density of 0.1 A g−1. (b) CV curves of VOC at 0.1 mV s−1. (c) XRD patterns of the VOC cathode at different states. (d) Schematic illustration of the oxidation of V2O3 during the first cycle. (e) Adsorption energies of V2O3 and V2O5−m·nH2O towards zinc ions. | ||
2.2 Cation reservoir-mediated redox catalytic sulfur speciation pathway
As discussed above, the VOC functional matrix has shown merits for hosting active sulfur. To directly reflect the matrix-dependent sulfur redox behavior, cyclic voltammetry (CV) curves of Zn–S cells assembled with different cathodes were compared in Fig. 3a. The bare VOC matrix exhibits two pairs of redox peaks, which correspond to the conversion between V5+ and V3+. It was identified that the potential of reduction peaks for VOC was higher than that of S/C and the oxidation potential was lower, indicating that the produced ZnxV23+/5+O5 possessed the spontaneous redox catalytic activity toward sulfur species during discharge/charge cycling. Specifically, the pre-encapsulated Zn2+ ions within the interlayer of VOC transfer from Znx+1V23+O5 to the sulfur component via a spontaneous chemical reaction (Znx+1V2O5 + S = ZnxV2O5 + ZnS, ΔG = −6.4 kJ mol−1).25 For the charging process, the reoxidation of ZnS is realized by ZnxV25+O5 (ZnxV2O5 + ZnS = Znx+1V2O5 + S, ΔG = −7.02 kJ mol−1), which overall facilitates the reversible bidirectional sulfur speciation pathway. The VOC/S cathode indeed exhibited small polarization and higher response current compared with the S/C cathode, suggesting accelerated redox kinetics and efficient sulfur conversion. We then conducted galvanostatic charge/discharge (GCD) tests to verify the improved sulfur utilization and redox kinetics. As expected, the VOC/S cathode delivered better redox kinetics and efficient sulfur conversion, as indicated by its lower voltage polarization and high capacity compared with the S/C cathode (Fig. 3b). The specific capacity of VOC/S reached 1630.73 mA h g−1 at 0.2 A g−1 and a low polarization of 0.55 V. Of note, for the reverse charging process, the VOC/S cathode displayed a lower charging voltage plateau and activation potential, suggesting the facile ZnS reoxidation process. Such a result confirms that the redox catalytic effect invoked by the vanadium active centers could significantly accelerate the bidirectional sulfur conversion.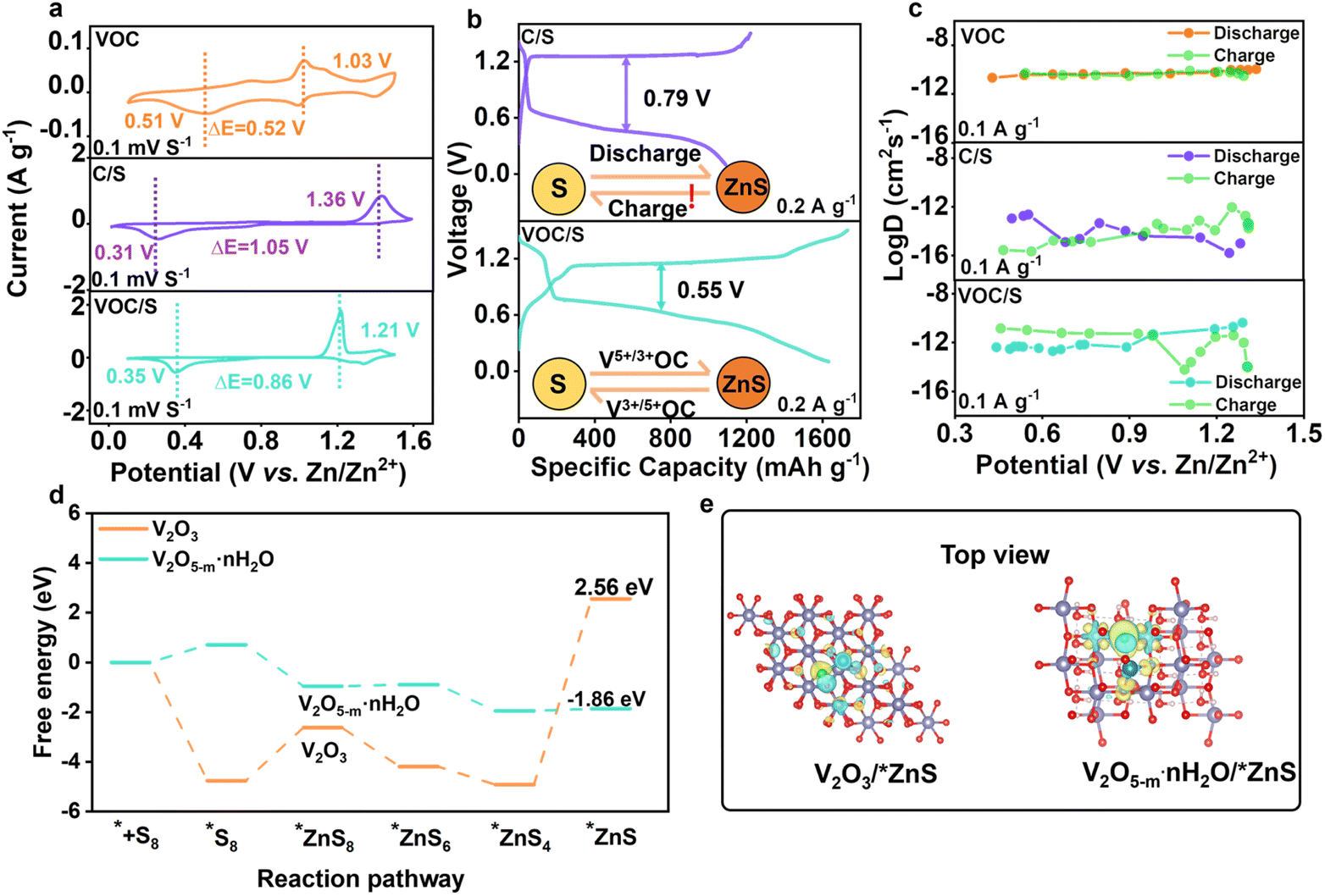 | ||
| Fig. 3 Mechanistic investigation on the redox catalysis facilitated sulfur conversion and DFT results. (a) CV curves of different cathodes as noted. (b) GCD profiles of C/S and VOC/S cathodes. (c) GITT curves of different cathodes. (d) Gibbs free energy for the sulfur reduction path. (e) Top views of the charge density distribution of V2O3 and V2O5−m·nH2O with decomposition in the *ZnS adsorption system. | ||
Then, the galvanostatic intermittent titration technique (GITT) was employed to determine the diffusion coefficient of Zn2+(DZn) thereby revealing the origin of the improved redox kinetics (Fig. S6†).26–31 As shown in Fig. 3c, the calculated DZn of VOC was as high as 10−12 to 10−9 cm2 s−1, which is higher than that of the S/C cathode, suggesting the substantially faster Zn2+ (de)insertion kinetics of V2O5−m·nH2O than sulfur species. Accordingly, the VOC/S cathode indeed exhibited higher DZn than the S/C cathode, which confirms the accelerated cation transport enabled by the VOC matrix. This also suggests that V2O5 served as a cation reservoir for rapid Zn2+ transfer, endowing the VOC/S cathode with significantly enhanced redox kinetics. To further validate the catalytic activity of V2O5 towards sulfur conversion at the molecular or atomic scale, the Gibbs free energies of each zincification step were calculated and are described in Fig. 3d. As shown, the rate-determining step for the whole reduction process was identified as the conversion of *ZnS4 to *ZnS.32,33 Compared with V2O3, V2O5 possesses a lower free energy of 1.86 eV, indicating more efficient sulfur conversion. Furthermore, the charge density distribution of ZnS adsorbed on different matrices was calculated to reveal the electron transport properties in this work. As shown in Fig. 3e, it can be noted that the charge density between V2O5−m·nH2O and ZnS was richer, indicating the formation of an electron conduction pathway between V2O5−m·nH2O and polysulfide. Such electron transport could enable the V2O5 sites to better utilize their intrinsic catalytic properties.
2.3 Electrochemical performance of aqueous Zn–S batteries
To validate the accessibility of the as-prepared VOC/S cathode, the rate performance of the as-prepared Zn–S cells was evaluated. The VOC/S cathode indeed exhibits an excellent rate performance of 1040–422 mA h g−1 at current densities ranging from 1–10 A g−1, substantially better than that of the S/C cathodes. Note that the reversible capacity of the VOC/S cathode retains over 40% of its initial value when the current density increases 10 times from 1 to 10 A g−1, suggesting that the V2O5 components could substantially accelerate the ion transport. The corresponding GCD profiles of VOC/S at various current densities are presented in Fig. S7,† in which these voltage plateaus are visible and the polarization increases slowly, further indicating superior redox kinetics.Of note, the cycling stability of aqueous Zn–S batteries is a critical issue because the sluggish redox kinetics and irreversible sulfur conversion would cause the fast capacity fade. Interestingly, the VOC/S//Zn cell delivers a good capacity retention of 82% after 50 cycles at a current density of 2 A g−1 (Fig. 4b), while the C/S//Zn cell nearly loses its capacity. Such strong differences highlight that the redox catalytic effect enabled by the VOC matrix could significantly improve the reversibility of bidirectional sulfur conversion. Note that the irreversible side reaction (e.g. 2ZnS + 4H2O + 10e− → 2Zn2+ + SO42− + S + 8H+) during battery recharging is the main reason for the relatively low coulombic efficiency (CE) of the VOC/S//Zn cell.34
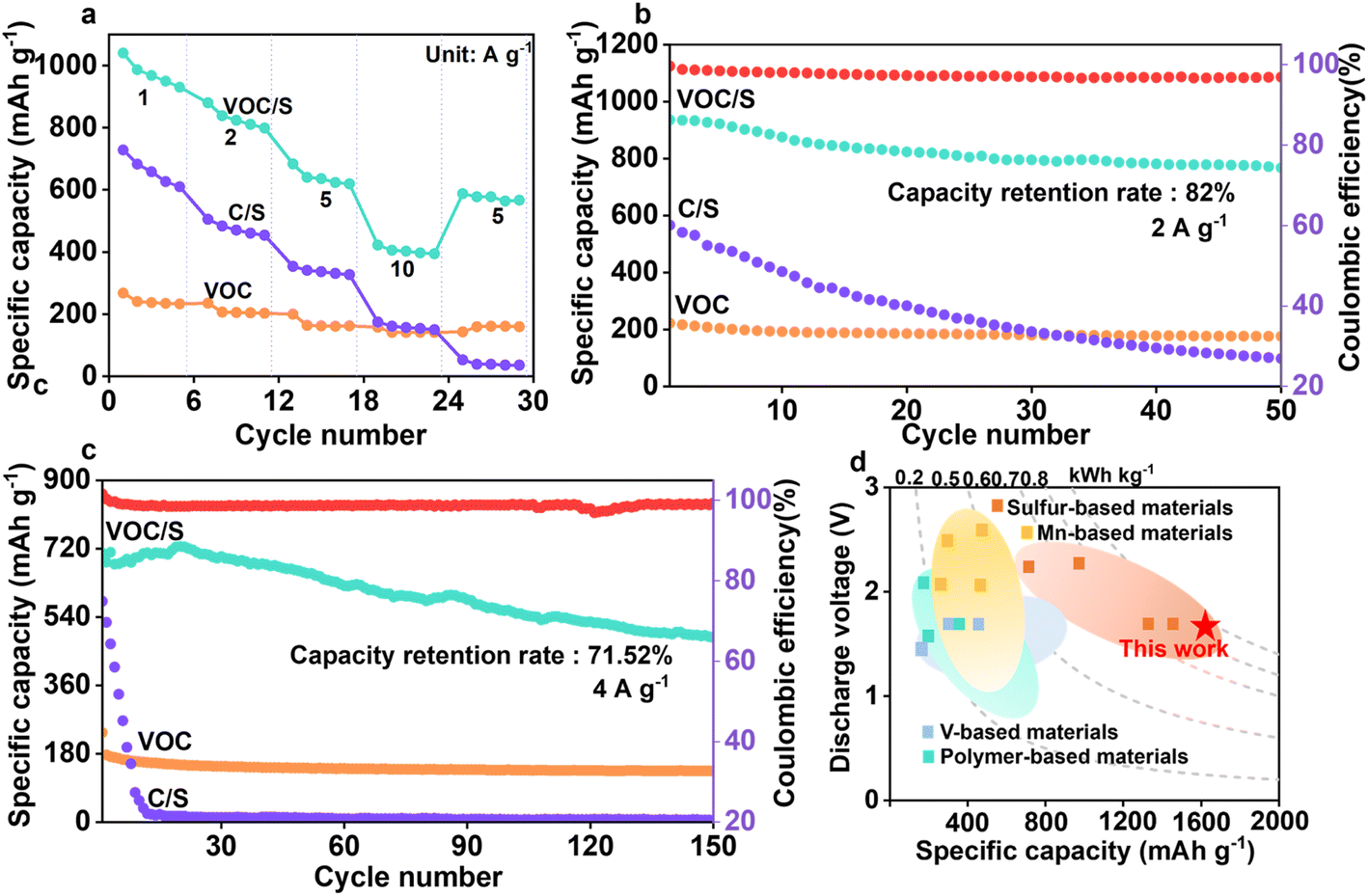 | ||
| Fig. 4 Electrochemical performance of the VOC/S cathode. (a) Rate performance. Cycle performance at a current density of (b) 2 A g−1 and (c) 4 A g−1. (d) Electrochemical performances of our work compared with those of previously reported aqueous Zn-ion batteries. | ||
After further cycling at a high current density of 4 A g−1, the VOC/S cathode can still obtain a capacity retention rate of 71.52% after 150 cycles (Fig. 4c), and the coulombic efficiency is nearly 100%. Moreover, it can be observed that there was almost no capacity attenuation in the VOC/S cathode during the first 30 cycles (Fig. S8†). Electrochemical impedance spectra of the cathodes before and after cycling were collected, in which the VOC/S cathode still maintained a low internal resistance compared with the C/S cathode, indicating that the VOC matrix greatly enhances redox kinetics (Fig. S9†). Moreover, the cycled C/S cathode displayed more agglomeration after cycling compared with the fresh C/S cathode, while the VOC/S cathode retained a relatively complete structure, confirming its structural stability (Fig. S10†). Such structural robustness enables the as-prepared Zn–S cells with a long cycling life. These results suggest that the V2O5 cation reservoir plays a key role in facilitating the ion transfer and also demonstrates the robustness of the VOC matrix.
A proof-of-concept Zn–S pouch cell was assembled to further validate the application potential of the VOC/S cathode (Fig. S11†). The as-prepared pouch cell could brighten a small light bulb and run a fan, demonstrating its promise in real applications. Fig. 4d also compares the overall electrochemical performance of this work with the reported results, which is comparable to or even better than that of previously reported literature data.35–48
2.4 Investigation of the reversible sulfur redox conversion
To reveal the Zn2+ ion storage mechanism, the phase changes of the VOC/S cathode were recorded. Fig. 5a displays the XRD patterns of VOC/S electrodes at different charge/discharge states. The characteristic peak of ZnS gradually became obvious during discharge, while the peak gradually decreased during the charging process, indicating the reversible conversion between ZnS and S.49–52 Of note, during one charge/discharge cycle (Fig. 5b), the vibrational Raman peaks of Zn–S also followed the same trend with the XRD results, further confirming the reversible sulfur speciation paths. Such complete sulfur conversion originates from the V-redox couple accelerated redox kinetics. Thus, the valence-state changes of the VOC/S cathode were further inspected to demonstrate the reason for the origin of the reversible sulfur conversion reactions. As shown in Fig. 5c, the original state (163.0 and 167.48 eV) can be assigned to S8. When discharged to 0.1 V, a new peak (161.2 eV) appears, representing ZnS. After recharging to 1.5 V, the peak corresponding to S8 is the same as the original state, further confirming the complete reversible sulfur conversion. The vanadium oxides function as the cation reservoir that can accommodate fast zinc ion transport.53–56 During the battery discharging process, the reduction of ZnxV25+O5 and sulfur with the formation of Znx+1V23+O5 and ZnS takes place simultaneously (Fig. 5d and e). The pre-zincified Znx+1V23+O5 could integrate zinc ions with sulfur, thereby facilitating the sulfur reduction kinetics. For reverse charging, the vanadium-active centers enable facile ZnS reoxidation, overall enabling the one-step reversible conversion between sulfur and ZnS.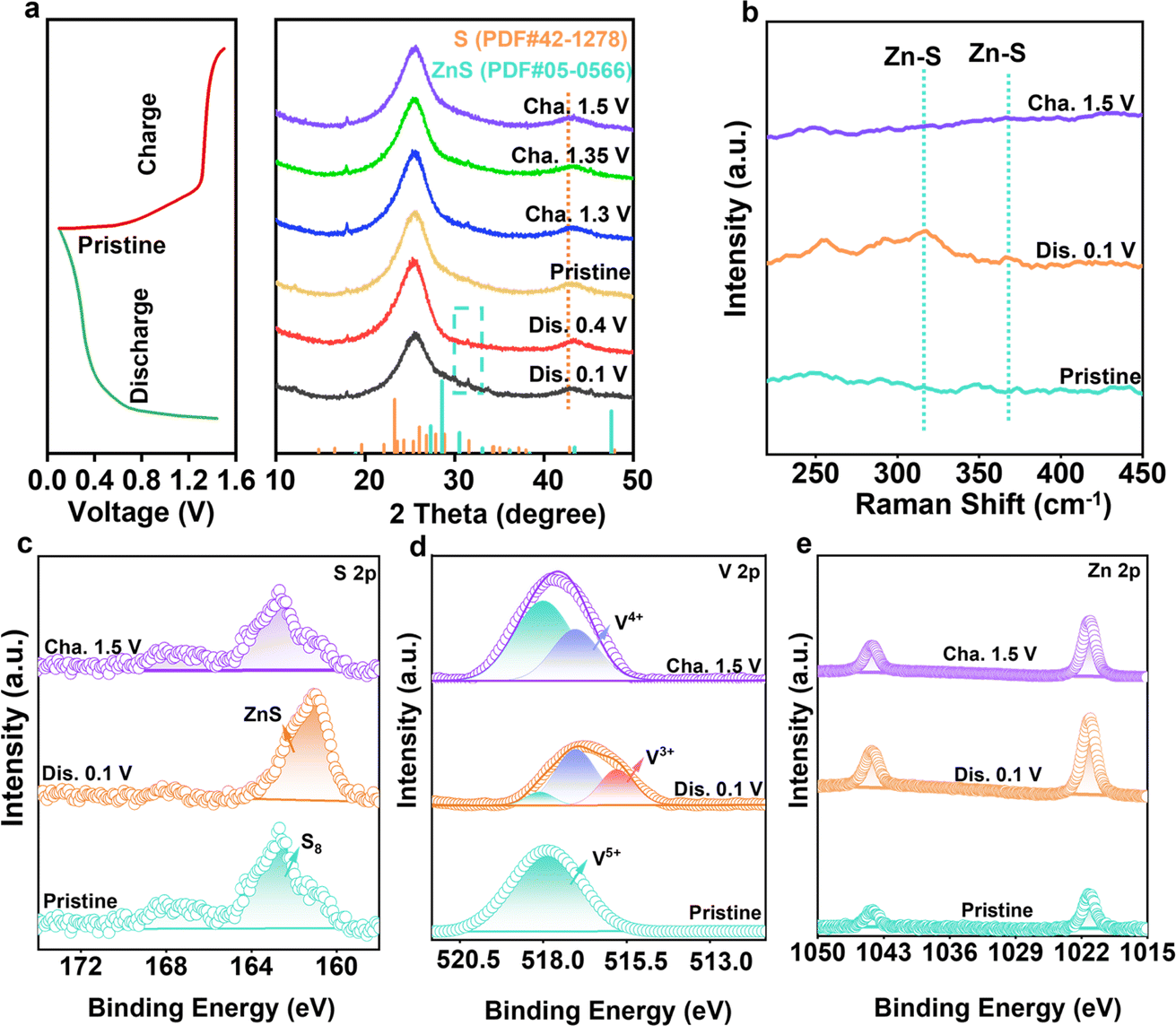 | ||
| Fig. 5 Analysis of reversible sulfur redox conversion. (a) Ex situ XRD patterns collected at different states. (b) Raman spectra at different charge and discharge states. XPS spectra of (c) S 2p, (d) V 2p, and (e) Zn 2p at different states. | ||
3 Conclusion
In summary, an in situ reconstruction strategy was developed to fabricate a catalytic matrix for hosting sulfur species and tune its conversion pathway. The phase transition process of the VOC was analyzed by systematic spectroscopic techniques and electrochemical tests, while the theoretical results revealed that the V2O5 features more merits for facilitating sulfur conversion. Therefore, the as-prepared V2O5 functions as a cation-transport medium, which can direct the reversible sulfur conversion. Specifically, the zincified ZnxV23+/5+O5 redox couple served as the cation reservoir to enable redox catalytic reversible sulfur conversion during dynamic battery discharge–charge. Consequently, the as-obtained sulfur cathode exhibits a lower voltage hysteresis of 0.55 V, a high discharge capacity of 1630.7 mA h g−1, and an excellent rate performance of 422 mAh g−1 at 10 A g−1. This work demonstrates a new avenue for manipulating sulfur redox, which could unlock the full potential of aqueous zinc–sulfur batteries.Data availability
Data will be made available on request.Author contributions
Hao Luo and Fan Li contributed equally to this work. Hao Luo: conceptualization, funding acquisition, writing-review & editing. Fan Li: investigation, writing-original draft. Mingli Wang: conceptualization, investigation, writing-original draft. Shang Sun: investigation, formal analysis. Min Zhou: writing-review & editing. Wenjing Zhang: formal analysis, investigation. Hengrui Guo: investigation, methodology. Xueyin Su: formal analysis, methodology. Xiaolong Li: methodology, writing-review & editing. Lina Ma: conceptualization, funding acquisition, resources.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was financially supported by the China Postdoctoral Science Foundation (Grant No. 2022TQ0282), the Natural Science Foundation of Fujian Province, China (No. 2024J011210) the High-level talent start-up Foundation of Xiamen Institute of Technology (grant No. YKJ23017R), and the Natural Science Foundation of Shandong Province of China (No. ZR2023QE071).References
- M. L. Wang, H. Zhang, W. L. Zhang, Q. W. Chen and K. Lu, Small Methods, 2022, 6, 2200335 CrossRef CAS.
- L. K. Hencz, H. Chen, Z. Z. Wu, S. S. Qian, S. Chen, X. X. Gu, X. H. Liu, C. Yan and S. Q. Zhang, Exploration, 2022, 2, 20210131 CrossRef CAS PubMed.
- H. Zhang, B. Song, W. W. Zhang, B. An, L. Fu, S. T. Lu, Y. W. Cheng, Q. W. Chen and K. Lu, Angew. Chem., Int. Ed., 2023, 62, e202217009 CrossRef CAS PubMed.
- X. L. Li, N. Li, Z. D. Huang, Z. Chen, G. J. Liang, Q. Yang, M. Li, Y. W. Zhao, L. T. Ma, B. B. Dong, Q. Huang, J. Fan and C. Y. Zhi, Adv. Mater., 2021, 33, 2006897 CrossRef CAS.
- M. Salama, Rosy, R. Attias, R. Yemini, Y. Gofer, D. Aurbach and M. Noked, ACS Energy Lett., 2019, 4, 436–446 CrossRef CAS.
- H. Zhang, Z. T. Shang, G. Luo, S. H. Jiao, R. G. Cao, Q. W. Chen and K. Lu, ACS Nano, 2022, 16, 7344–7351 CrossRef CAS PubMed.
- D. D. Liu, B. He, Y. Zhong, J. Chen, L. X. Yuan, Z. Li and Y. H. Huang, Nano Energy, 2022, 101, e107474 CrossRef.
- R. H. Gao, M. T. Zhang, Z. Y. Han, X. Xiao, X. R. Wu, Z. H. Piao, Z. J. Lao, L. Nie, S. G. Wang and G. M. Zhou, Adv. Mater., 2024, 36, 2303610 CrossRef CAS.
- H. Li, M. Y. Chuai, X. Xiao, Y. Y. Jia, B. Chen, C. Li, Z. H. Piao, Z. G. Lao, M. T. Zhang, G. H. Gao, B. K. Zhang, Z. Y. Han, J. L. Yang and G. M. Zhou, J. Am. Chem. Soc., 2023, 145, 22516–22526 CrossRef CAS PubMed.
- K. Lu, H. Zhang, S. Y. Gao, H. Y. Ma, J. Z. Chen and Y. W. Cheng, Adv. Funct. Mater., 2019, 29, 1807309 CrossRef.
- B. Q. Li, H. J. Peng, X. Chen, S. Y. Zhang, J. Xie, C. X. Zhao and Q. Zhang, CCS Chem., 2019, 1, 128–137 CrossRef CAS.
- V. Marangon, C. Hernández-Rentero, M. Olivares-Marín, V. Gómez-Serrano, Á. Caballero, J. Morales and J. Hassoun, ChemSusChem, 2021, 14, 3333 CrossRef CAS.
- A. Manthiram, Y. Z. Fu, S.-H. Chung, C. X. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS.
- R. Wang, S. Xin, D. L. Chao, Z. X. Liu, J. D. Wan, P. Xiong, Q. Q. Luo, K. Hua, J. N. Hao and C. F. Zhang, Adv. Funct. Mater., 2022, 32, 2207751 CrossRef CAS.
- Y. Yang, H. J. Yang, R. J. Zhu and H. S. Zhou, Energy Environ. Sci., 2023, 16, 2723–2731 RSC.
- X. Wang, L. Y. Liu, Z. W. Hu, C. C. Peng, C. Han and W. J. Li, Adv. Energy Mater., 2023, 13, 2302927 CrossRef CAS.
- K. N. Wang, J. W. Wang, Z. Q. Zhang, W. L. Zhang, F. Feng and Y. P. Du, Sci. China: Chem., 2023, 66, 2711–2718 CrossRef CAS.
- L. L. Feng and S. P. Wang, Sci. China: Chem., 2023, 66, 2267–2273 CrossRef CAS.
- P. Zeng, H. Zou, C. Cheng, L. Wang, C. Yuan, G. L. Liu, J. Mao, T.-S. Chan, Q. Y. Wang and L. Zhang, Adv. Funct. Mater., 2023, 33, 2214770 CrossRef CAS.
- H. Luo, B. Wang, F. Wang, J. Yang, F. D. Wu, Y. Ning, Y. Zhou, D. L. Wang, H. K. Liu and S. X. Dou, ACS Nano, 2020, 14, 7328–7337 CrossRef CAS.
- S. S. Tan, Y. L. Jiang, Q. L. Wei, Q. M. Huang, Y. H. Dai, F. Y. Xiong, Q. D. Li, Q. Y. An, X. Xu, Z. Z. Zhu, X. D. Bai and L. Q. Mai, Adv. Mater., 2018, 30, 1707122 CrossRef PubMed.
- Y. R. Qi, Q.-J. Li, Y. K. Wu, S.-J. Bao, C. M. Li, Y. M. Chen, G. X. Wang and M. W. Xu, Nat. Commun., 2021, 12, 6347 CrossRef CAS.
- D. Kundu, B. Adams, V. Duffort, S. H. Vajargah and L. Nazar, Nat. Energy, 2016, 1, 1611 Search PubMed.
- M. Y. Yan, P. He, Y. C, S. Y. W, Q. L. W, K. N. Zhao, X. Xu, Q. Y. An, Y. Shuang, Y. Y. Shao, K. T. Mueller, L. Q. Mai, J. Liu and J. H. Yang, Adv. Mater., 2017, 1703725 Search PubMed.
- X. Y. Wu, A. Markir, L. Ma, Y. K. Xu, H. Jiang, D. P. Leonard, W. Shin, T. P. Wu, J. Lu and X. L. Ji, Angew. Chem., Int. Ed., 2019, 58, 12640 CrossRef CAS PubMed.
- Y. C. Chien, H. D. Liu, A. S. Menon, W. R. Brant, D. Brandell and M. J. Lacey, Nat. Commun., 2023, 14, 2289 CrossRef CAS.
- W. Wang, Y. Tang, J. Liu, H. B. Li, R. Wang, L. H. Zhang, F. Liang, W. Bai, L. Zhang and C. F. Zhang, Chem. Sci., 2023, 14, 9033–9040 RSC.
- R. Wang, Q. W. Ma, L. H. Zhang, Z. X. Liu, J. D. Wan, J. F. Mao, H. B. Li, S. L. Zhang, J. N. Hao, L. Zhang and C. F. Zhang, Adv. Energy Mater., 2023, 13, 2302543 CrossRef CAS.
- H. J. Yang, R. J. Zhu, Y. Yang, Z. Y. Lu, Z. Chang, P. He, C. Y. Zhu, S. Kitano, Y. Aoki, H. Habazaki and H. S. Zhou, Energy Environ. Sci., 2023, 16, 2133–2141 RSC.
- Y. Yang, W. H. Yang, R. J. Zhu, G. Wu, Y.-K. Choe, K. Sho, H. J. Yang and E. Yoo, Energy Storage Mater., 2024, 72, 103691 CrossRef.
- Y. Yang, R. J. Zhu, G. Wu, W. H. Yang, H. J. Yang and E. Yoo, ACS Nano, 2024, 18, 19003–19013 CrossRef CAS.
- W. J. Xue, Z. Shi, L. M. Suo, C. Wang, Z. Q. Wang, H. Z. Wang, K. P. So, A. Maurano, D. W. Yu and Y. M. Chen, Nat. Energy, 2019, 4, 374–382 CrossRef CAS.
- W. L. Wu, S. B. Wang, L. Lin, H.-Y. Shia and X. Q. Sun, Energy Environ. Sci., 2023, 16, 4326 RSC.
- M. L. Wang, H. Zhang, T. H. Ding, F. J. WuL. Fu, B. Song, P. F. Cao and K. Lu, Sci. China Chem., 2024, 67, 1531–1538 CrossRef CAS.
- Y. Q. Guo, R. Chua, Y. Q. Chen, Y. Cai, E. J. J. Tang, J. J. N. Lim, T. H. Tran, V. Verma, M. W. Wong and M. Srinivasan, Small, 2023, 19, 2207133 CrossRef CAS PubMed.
- Y. C. Ding, Y. Q. Peng, S. H. Chen, X. X. Zhang, Z. Q. Li, L. Zhu, L.-E. Mo and L. H. Hu, ACS Appl. Mater. Interfaces, 2019, 11, 44109–44117 CrossRef CAS.
- C. Sun, J. H. Zhu, B. Liu, M. W. Xu, J. Jiang and T. Yu, ACS Energy Lett., 2023, 8, 772–779 CrossRef CAS.
- Z. D. Yang, B. Y. Wang, Y. J. Chen, W. H. Zhou, H. P. Li, R. Z. Zhao, X. R. Li, T. S. Zhang, F. X. Bu, Z. W. Zhao, W. Li, D. L. Chao and D. Y. Zhao, Natl. Sci. Rev., 2023, 10, nwac268 CrossRef CAS.
- K. K. Sonigara, J. V. Vaghasiya, C. C. Mayorga-Martinez C and P. Martin, npj 2D Mater. Appl., 2023, 7, 45 CrossRef CAS.
- Y. Fei and G. Li, Adv. Funct. Mater., 2024, 34, 2312550 CrossRef CAS.
- Y. X. Li, Y. Lu, Y. X. Ni, S. B. Zheng, Z. H. Yan, K. Zhang, Q. Zhao and J. Chen, J. Am. Chem. Soc., 2022, 144, 8066–8072 CrossRef CAS PubMed.
- X. X. Jia, C. F. Liu, Z. Wang, D. Huang and G. Z. Cao, Nano-Micro Lett., 2024, 16, 129 Search PubMed.
- Y. F. Huang, Y. Z. Peng, Q. Ouyang, Q. Feng, H. Wang, D. Z. Zheng, F. X. Wang, X. H. Lu and Q. Liu, Energy Storage Mater, 2024, 70, 103476 CrossRef.
- J. L. Yang, W. B. Gong and F. X. Geng, Adv. Funct. Mater., 2023, 33, 2301202 Search PubMed.
- D. X. Geng, H. Zhang, Z. J. Fu, Z. M. Liu, Y. F. An, J. Yang, D. W. Sha, L. Pan, C. Yan and Z. M. Sun, Adv. Sci., 2024, 2407073 Search PubMed.
- S. B. Zheng, D. J. Shi, D. Yan, Q. R. Wang, T. J. Sun, T. Ma, L. Li, D. He, Z. L. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117511 CrossRef CAS PubMed.
- H. Zhang, D. X. Xu, L. W. Wang, Z. L. Ye, B. Chen, L. Y. Pei, Z. Y. Wang, Z. Y. Cao, J. F. Shen and M. X. Ye, Small, 2021, 17, 2100902 Search PubMed.
- Z. L. Han, S. P. Li, M. J. Sun, R. J. He, W. Zhong, C. Yu, S. J. Cheng and J. Xie, J. Energy Chem., 2022, 68, 752–761 Search PubMed.
- W. W. Zhang, M. L. Wang, J. K. Ma, H. Zhang, L. Fu, B. Song, S. T. Lu and K. Lu, Adv. Funct. Mater., 2023, 33, 2210899 Search PubMed.
- M. Yang, Z. C. Yan, J. Xiao, W. L. Xin, L. Zhang, H. L. Peng, Y. H. Geng, J. W. Li, Y. X. Wang, L. Liu and Z. Q. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202212666 Search PubMed.
- X. Z. Yang, C. Li, Z. T. Sun, S. Yang, Z. X. Shi, R. Huang, B. Z. Liu, S. Li, Y. H. Wu, M. L. Wang, Y. W. Su, S. X. Dou and J. Y. Sun, Adv. Mater., 2021, 33, 210595 Search PubMed.
- M. J. Chen, Q. Y. Zhao, H. R. Wang, T. Long, X. Ma, Q. Wu, W. X. Zhou, X. W. W and X. X. Zeng, Sci. China Chem., 2023, 66, 289–296 Search PubMed.
- J. D. Guo, J. X. Liu, W. B. Ma, Z. Y. Sang, L. X. Yin, X. Q. Zhang, H. Chen, J. Liang and D. A. Yang, Adv. Funct. Mater., 2023, 33, 2302659 CrossRef CAS.
- T. Zhou, D. Xuan and G. Guo, J. Energy Chem., 2024, 95, 9–19 CrossRef CAS.
- Z. Y. Feng, Y. L. Zhang, Z. M. Gao, D. D. Hu, H. M. Jiang and T. Hu, Adv. Powder Mater., 2024, 3, 100167 CrossRef.
- Z. H. Song, Y. Zhao, H. R. Wang, A. B. Zhou, X. Y. Jin, Y. X. Huang, L. Li, F. Wu and R. J. Chen, Energy Environ. Sci., 2024, 17, 6666–6675 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06593j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |