
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning-driven discovery of highly selective antifungal peptides containing non-canonical β-amino acids†
Douglas H.
Chang‡
 a,
Joshua D.
Richardson‡
a,
Myung-Ryul
Lee
a,
David M.
Lynn
*ab,
Sean P.
Palecek
*a and
Reid C.
Van Lehn
*ab
a,
Joshua D.
Richardson‡
a,
Myung-Ryul
Lee
a,
David M.
Lynn
*ab,
Sean P.
Palecek
*a and
Reid C.
Van Lehn
*ab
aDepartment of Chemical and Biological Engineering, University of Wisconsin–Madison, Madison, WI, USA. E-mail: david.lynn@wisc.edu; sppalecek@wisc.edu; vanlehn@wisc.edu
bDepartment of Chemistry, University of Wisconsin–Madison, Madison, WI, USA
First published on 20th February 2025
Abstract
Antimicrobial peptides (AMPs) are promising compounds for the treatment and prevention of multidrug-resistant infections because of their ability to directly disrupt microbial membranes, a mechanism that is less likely to lead to resistance compared to antibiotics. Unfortunately, natural AMPs are prone to proteolytic cleavage in vivo and have relatively low selectivity for microbial versus human cells, motivating the development of synthetic peptidomimetics of AMPs with improved peptide stability, activity, and selectivity. However, a lack of understanding of structure–activity relationships for peptidomimetics constrains development to rational design or experimental predictors, both of which are cost and time prohibitive, especially when the design space of possible sequences scales exponentially with the number of amino acids. To address these challenges, we developed an iterative Gaussian process regression (GPR) approach to explore a large design space of 336![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 synthetic α/β-peptide analogues of a natural AMP, aurein 1.2, based on an initial training set of 147 sequences and their biological activities against microbial pathogens and selectivity for microbes vs. mammalian cells. We show that the quantification of prediction uncertainty provided by GPR can guide the exploration of this design space via iterative experimental measurements to efficiently discover novel sequences with up to a 52-fold increase in antifungal selectivity compared to aurein 1.2. The highest selectivity peptide discovered using this approach features an unconventional substitution of cationic amino acids in the hydrophobic face and would be unlikely to be explored by conventional rational design. Overall, this work demonstrates a generalizable approach that integrates computation and experiment to accurately predict the selectivity of AMPs containing synthetic amino acids, which we employed to discover new α/β-peptides that hold promise as selective antifungal agents to combat the antimicrobial resistance crisis.
000 synthetic α/β-peptide analogues of a natural AMP, aurein 1.2, based on an initial training set of 147 sequences and their biological activities against microbial pathogens and selectivity for microbes vs. mammalian cells. We show that the quantification of prediction uncertainty provided by GPR can guide the exploration of this design space via iterative experimental measurements to efficiently discover novel sequences with up to a 52-fold increase in antifungal selectivity compared to aurein 1.2. The highest selectivity peptide discovered using this approach features an unconventional substitution of cationic amino acids in the hydrophobic face and would be unlikely to be explored by conventional rational design. Overall, this work demonstrates a generalizable approach that integrates computation and experiment to accurately predict the selectivity of AMPs containing synthetic amino acids, which we employed to discover new α/β-peptides that hold promise as selective antifungal agents to combat the antimicrobial resistance crisis.
Introduction
Since the introduction of insulin to treat type 1 diabetes 100 years ago, large strides have been made in the field of peptide therapeutics to treat a range of other diseases such as cancer, multiple sclerosis, and HIV.1,2 Antimicrobial peptides (AMPs), a component of the innate immune system, have garnered particular attention as potential agents to combat the alarming increase in multi-drug-resistant bacterial and fungal infections. Many natural AMPs are cationic, α-helical peptides that work via disruption of microbial cell membranes, making them less likely to lead to resistance than antibiotics with a specific molecular target.3–6 AMPs are typically amphiphilic in nature, with opposing hydrophilic and hydrophobic faces that are important for membrane disruption.5 However, natural α-helical AMPs typically have low stability in vivo because of their susceptibility to proteolytic cleavage7,8 and also exhibit low selectivities for microbial versus human cells; these two limitations have served as obstacles to their translation into clinical therapies.9 Novel approaches to design peptides and their analogues are necessary to tackle these challenges.One strategy to address these issues is to substitute traditional α-amino acids with noncanonical β-amino acids, which have an additional carbon in the backbone relative to α-amino acids, in specific patterns and positions of the original sequence, as shown in Fig. 1.9–13 This class of synthetic peptides, known as α/β-peptides, can exhibit enhanced proteolytic resistance while maintaining side chain presentations similar to those of α-peptides, enabling the templating of sequences on naturally occurring AMPs.14–18 In addition, using β-amino acids enables the exploration of a much larger array of amino-acid combinations compared to naturally occurring peptides (such as repeating ααβ or ααβαααβ backbone patterning or the presence of the helix-stabilizing component ‘ACPC’ shown in Fig. 1), which could lead to enhanced stability and selectivity profiles. For example, our previous studies introduced α/β-peptides developed as analogues of aurein 1.2, an AMP found in Australian bell frogs, to enhance their antifungal selectivity against Candida albicans (Fig. 1).9,19C. albicans is the most prevalent hospital-acquired fungal pathogen and is known to cause bloodstream infections,20 leading to a mortality rate of up to 70% in the case of sepsis.21 Despite an alarming increase in antifungal resistance, only a handful of antifungal drug classes are approved for use in the clinic, motivating a drive for the development of new treatments, including AMP-based therapeutics.22–24 Recent rational design efforts by our group have resulted in α/β-peptides that exhibit significantly enhanced antifungal selectivity, with up to a 22-fold improvement over aurein 1.2.19
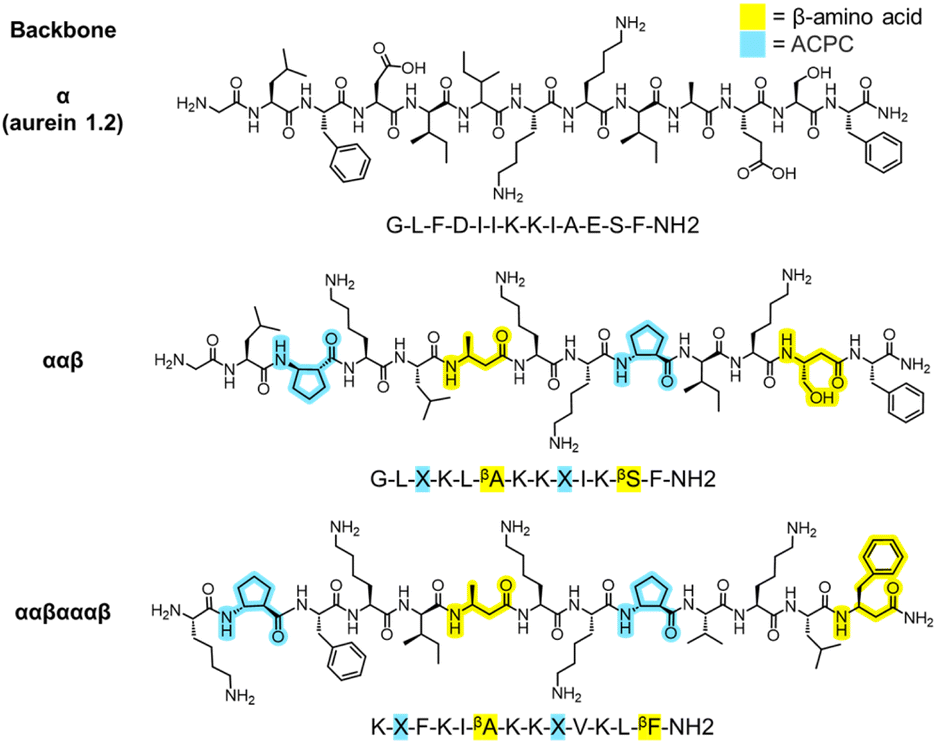 | ||
| Fig. 1 Example chemical structures of aurein 1.2 and template α/β-peptides used in this study. “Backbone” denotes β-amino acid patterning, yellow highlights correspond to β-amino acids, and light blue highlights correspond to the β-amino acid ‘ACPC’ (trans-2-aminocyclopentane-carboxylic acid, simplified as ‘X’). α-Amino acids are unhighlighted. α/β-Peptides were previously rationally designed as analogues of aurein 1.2 to improve its antifungal selectivity. | ||
The antifungal selectivities of α/β-peptides in our prior work were quantified using a selectivity index (SI). This parameter is the ratio between mammalian cell toxicity (often defined as ‘HC10’, or the concentration of peptide required to cause 10% hemolysis, or the lysis of human red blood cells) and antifungal activity (defined as the minimum inhibitory concentration, or ‘MIC’, needed to inhibit at least 90% growth of fungal cells).9 The SI is used as a metric to guide the design of highly selective (high-SI) antimicrobial AMPs and mimetics.9,25,26 However, obtaining AMPs and peptidomimetics with high antifungal SI remains challenging compared to the design of highly selective antibacterial AMPs because antifungal toxicity correlates more strongly with hemolysis than does antibacterial toxicity.19,27 This challenge is largely attributed to the more similar membrane compositions of eukaryotic fungal and mammalian cells compared to prokaryotic bacterial cells, which affect the membrane-active mechanism of most AMPs and mimetics.28–30 While many peptides can disrupt membranes, the most valuable therapeutic options would be those that are most selective.
Our past studies have shown that experimentally determined peptide physicochemical properties such as helical rigidity and hydrophobicity can be used to accurately predict fungal activity and hemolysis metrics, in addition to providing specific ranges over which high selectivity could be expected.9,19 However, experimentally determined physicochemical properties require low-throughput biophysical characterization. In addition, most physicochemical properties of short peptides are highly correlated, rendering rational design a difficult strategy because altering a single amino acid can result in changes in multiple physicochemical properties.31 Consequently, rational design approaches require educated guesses about the results of a single amino acid substitution followed by α/β-peptide synthesis, an overall approach that is often prohibitive at large scale in terms of time and financial cost. Therefore, a predictive method to navigate the sequence space and discover novel α/β-peptide sequences with improved antifungal selectivity would be useful and could significantly expedite development. Unfortunately, the prediction of novel α/β-peptide sequences is hindered by the relative lack of experimental data available in databases compared to natural α-peptide AMPs (such as APD3,32 CAMPR3,33 and DRAMP34). This data scarcity limits the application of large database-driven machine learning (ML) methods, such as deep learning,35–37 natural language models,38 and variational autoencoding,39 which have proven useful in past studies for the prediction of novel canonical AMP sequences. Additionally, models trained on quickly generated sequence-based data representations for α-peptides involving only the 20 canonical amino acids (e.g., one-hot encoding40,41) are not directly transferable to AMPs containing synthetic amino acids due to these data scarcity limits. Consequently, predicting new sequences of synthetic peptidomimetics remains challenging.
In this study, we demonstrate that highly selective α/β-peptidomimetics can be discovered by iteratively predicting their biological activities in silico using an ML model with computationally derived descriptors and then measuring selectivity using in vitro experimental measurements to test model predictions and update model parameters. Using an initial dataset of 147 α/β-peptide sequences as a starting point,9,19 we developed an iterative Gaussian process regression (GPR) workflow to predict HC10 and MIC values of novel α/β-peptide sequences templated on the most selective sequences in this initial training set. As input to the GPR model, we used 2D molecular descriptors that can be quickly computed from peptide chemical structures without relying on large databases or computationally expensive predictive techniques. Peptides were selected for synthesis based upon GPR predictions, with experimentally measured HC10 and MIC values then used to update GPR model parameters to guide the next prediction round. Because GPR estimates the uncertainty of regression predictions,42–46 this approach permits evaluation of promising new sequences with low uncertainty and high predicted SI while also testing sequences with high uncertainty to expand the design space (e.g., sequences with new amino acids) for future prediction rounds. After six rounds, we identified new selective α/β-peptides with up to a 52-fold improvement in SI compared to aurein 1.2. Although these new peptides possess desirable physicochemical properties typically indicative of high antifungal selectivity, their sequences, which largely contain unconventional amino acid substitutions, would be less likely to be identified through rational design. Our results demonstrate the effectiveness of an iterative GPR-guided strategy to screen through a large sequence space and identify multiple new α/β-peptides with significantly enhanced antifungal selectivity. These peptides could, with further development, prove valuable as therapeutic agents to combat the rising incidence of drug-resistant fungal infections.
Results and discussion
Iterative GPR workflow implementation
Fig. 2 summarizes the iterative workflow developed and used in this study. The goal of this approach was to guide the selection of peptide sequences for experimental testing, and thus model training and AMP design, by quantifying the uncertainty of model predictions obtained from GPR, building on studies that have successfully implemented iterative Gaussian-based techniques.47,48 The initial training set for the iterative GPR predictive workflow included 147 13-amino-acid α/β-peptides designed as analogues of aurein 1.2 (Fig. 2a)9,19 in our past studies to maximize antifungal selectivity, measured through a metric called the selectivity index (SI).9 The SI is defined as the ratio of experimentally determined peptide HC10 to MIC measurements as shown in eqn (1):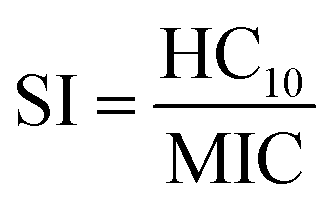 | (1) |
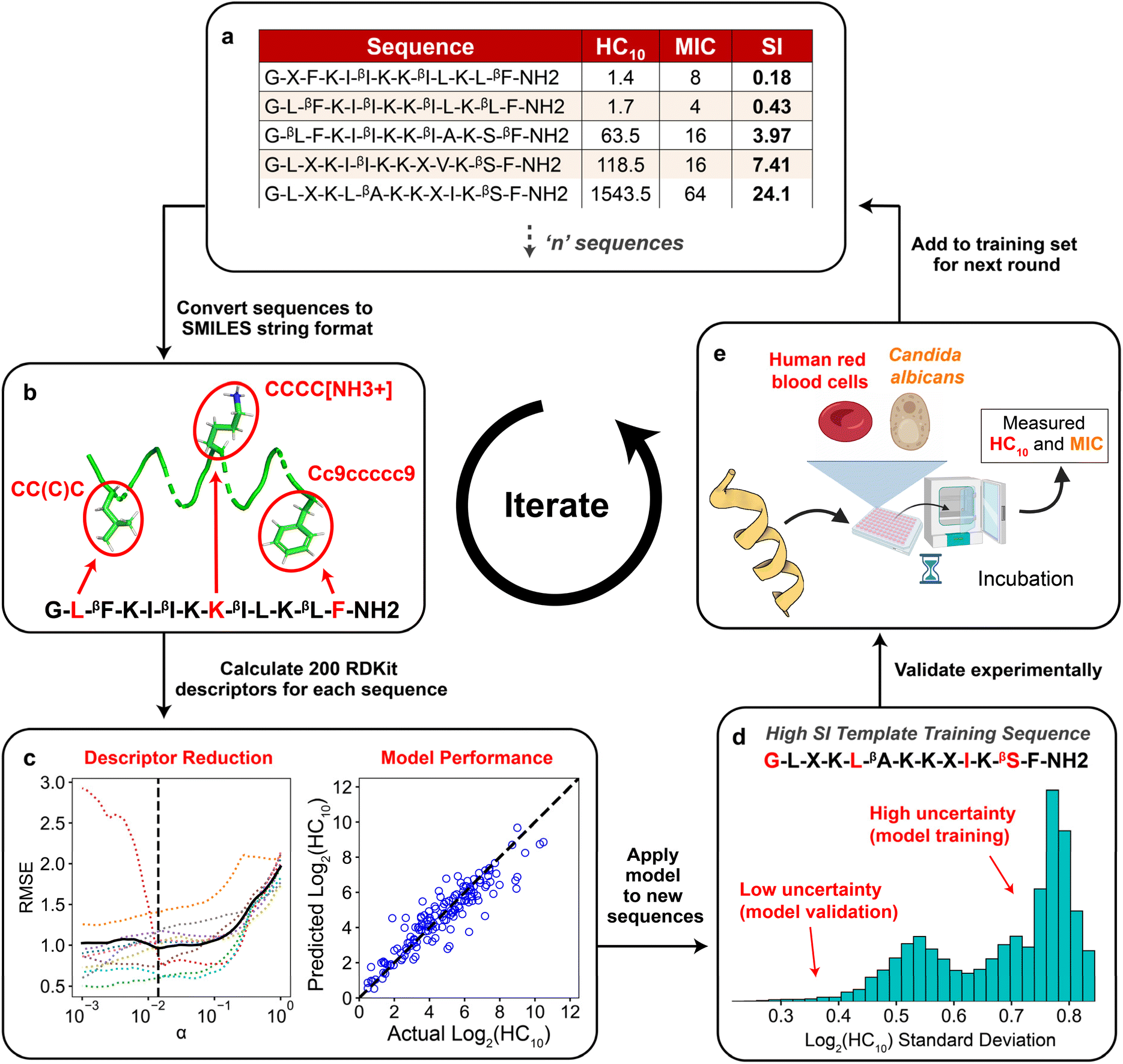 | ||
| Fig. 2 Schematic demonstrating the integrated computational and experimental iterative approach used in this study. (a) Training set of ‘n’ sequences with corresponding HC10 and MIC labels for Gaussian process regression (GPR). (b) All sequences were converted to SMILES string representations (Section S1†), in which selected amino acids are highlighted in red for visualization. (c) After calculating 200 descriptors for each sequence and removing all constant descriptors, LASSO cross validation was used to reduce the set of descriptors. The reduced set of descriptors was then used to train separate GPR models for HC10 and MIC using data in the training set (right plot, see Table S4† for model parameters). (d) The trained GPR model was applied to predict HC10 and MIC for all peptides in the design space generated from the template sequence. New sequences were selected for experimental synthesis based on a balance between sequences with low uncertainty predictions for model validation and high uncertainty predictions for further model training. (e) HC10 and MIC values for new sequences were quantified experimentally, compared to model predictions, and added to the training set (a) for the next round. This figure includes graphics imported from https://www.biorender.com. | ||
From two-fold serial dilutions, HC10 was interpolated continuously, whereas MIC was discretely determined to stay consistent with conventional methodologies.9,19 All HC10 and MIC data were log2 scaled to accommodate the serially diluted nature of the experimental measurements. Of the 147 sequences, fifteen are newly reported in this study (Appendix I) and were evaluated with the same methodology as that of prior studies to ensure reliable SI quantification. As depicted in Fig. 2a, the training set consisted of peptides with β-amino acids prefixed with a “β” in the amino-acid sequence, in addition to the β-amino acid ‘ACPC’ (trans-2-aminocyclopentane-carboxylic acid), simplified as ‘X’ (Fig. 1 and 2a) which is a cyclic 5-membered ring that improves helical folding and stability.14,49–51 For our approach, each sequence in the training set (Fig. 2a; i.e., the sequences with corresponding experimentally measured HC10 and MIC) was first transformed into its simplified molecular-input line-entry system (SMILES) representation (Fig. 2b), which uniquely represents the bonding, branching, ring groups, and aromaticity of each peptide52 through the definition of both backbone and sidechain fragments (see Section S1 and Fig. S1† for more details). Unlike popular sequence encodings that can only categorically distinguish between amino acids in peptide sequences (e.g., binary vectors in one-hot encoding40,41), the SMILES data representation captures important molecular properties of these amino acids in the context of their local bonding environment. We utilized these SMILES strings as input to the RDKit Cheminformatics toolkit to calculate 200 molecular descriptors for each sequence.53 Descriptors are numerical values associated with each sequence that quantify properties such as atomic van der Waals surface areas (VSA), partial charges, bonding and branching complexity, and chemical group counts (Section S2†).
The total number of descriptors was large compared to the number of training sequences, indicating that model training could potentially lead to overfitting, which would not generate robust predictions. Therefore, the total number of descriptors used during model training in each prediction round was reduced in two steps. First, all descriptors that had constant numerical values for all sequences in the training set were excluded because they cannot distinguish between properties of different sequences (Fig. S2†). Second, we performed 10-fold LASSO cross validation (CV) (left plot in Fig. 2c) to minimize the average root-mean-square error of model predictions compared to the measured HC10 and MIC values (further details in Section S2 and Fig. S3†). This approach quantifies the trade-off between the number of descriptors and prediction accuracy to identify a reduced set of descriptors that yields acceptable prediction accuracy. This procedure resulted in between 13 and 40 descriptors used for model training for each round. Utilizing this reduced descriptor set per round (Tables S1 and S2†), the highest accuracy (R2) GPR model (right plot in Fig. 2c) from a hyperparameter search was then selected (see Table S4†) for HC10 and MIC predictions of novel α/β-peptide sequences. We note that separate models, using separate sets of descriptors, were trained to independently predict HC10 and MIC values. GPR model selection criteria are described in Section S3.† For all regression steps, HC10 and MIC values were log2 scaled (Fig. S6†) to simplify comparisons to experimental values of hemolysis and antifungal activity, which are typically assessed, by convention, using 2-fold serial dilutions of peptide concentrations.
The two trained GPR models (for HC10 and MIC) were then used to assess peptides from the design space of 168000 possible sequences (for a single ‘template sequence’), which we refer to as ‘test sequences.’ The ‘template sequences’ were the three highest SI sequences in the initial training set (Fig. 2a) for each backbone (specifically ααβ and ααβαααβ, depicted in orange in Fig. 3c and d) that were selected to provide a well-defined sequence space to explore in the step shown in Fig. 2d using the iterative GPR approach (details on design space generation are explained in depth in the next section, ‘properties of training sequences and design space generation’). Test sequences were converted to SMILES representations and corresponding descriptors were calculated. Due to the large range of descriptor values for the test sequences (up to six times larger than the upper bound of descriptor values for training sequences, see Fig. S7 and S8†), we only used the GPR models to predict HC10 and MIC values for test sequences for which all descriptor values were within the bounds of the values computed for training sequences to prevent large prediction inaccuracies due to extrapolation (Table S5†). We then utilized the ability of GPR to quantify prediction uncertainty through standard deviations (σ) to inform the selection of test sequences for experimental synthesis. To compare prediction uncertainties across prediction rounds by accounting for the changes in standard deviation distributions that resulted from updating the descriptors (Section S2†) and the GPR models (Section S3†) every round, we define the normalized standard deviation (NSD), which varies from 0 to 1, in eqn (2):
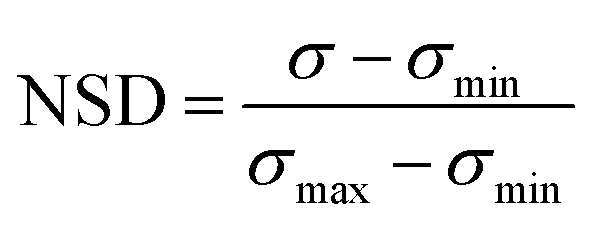 | (2) |
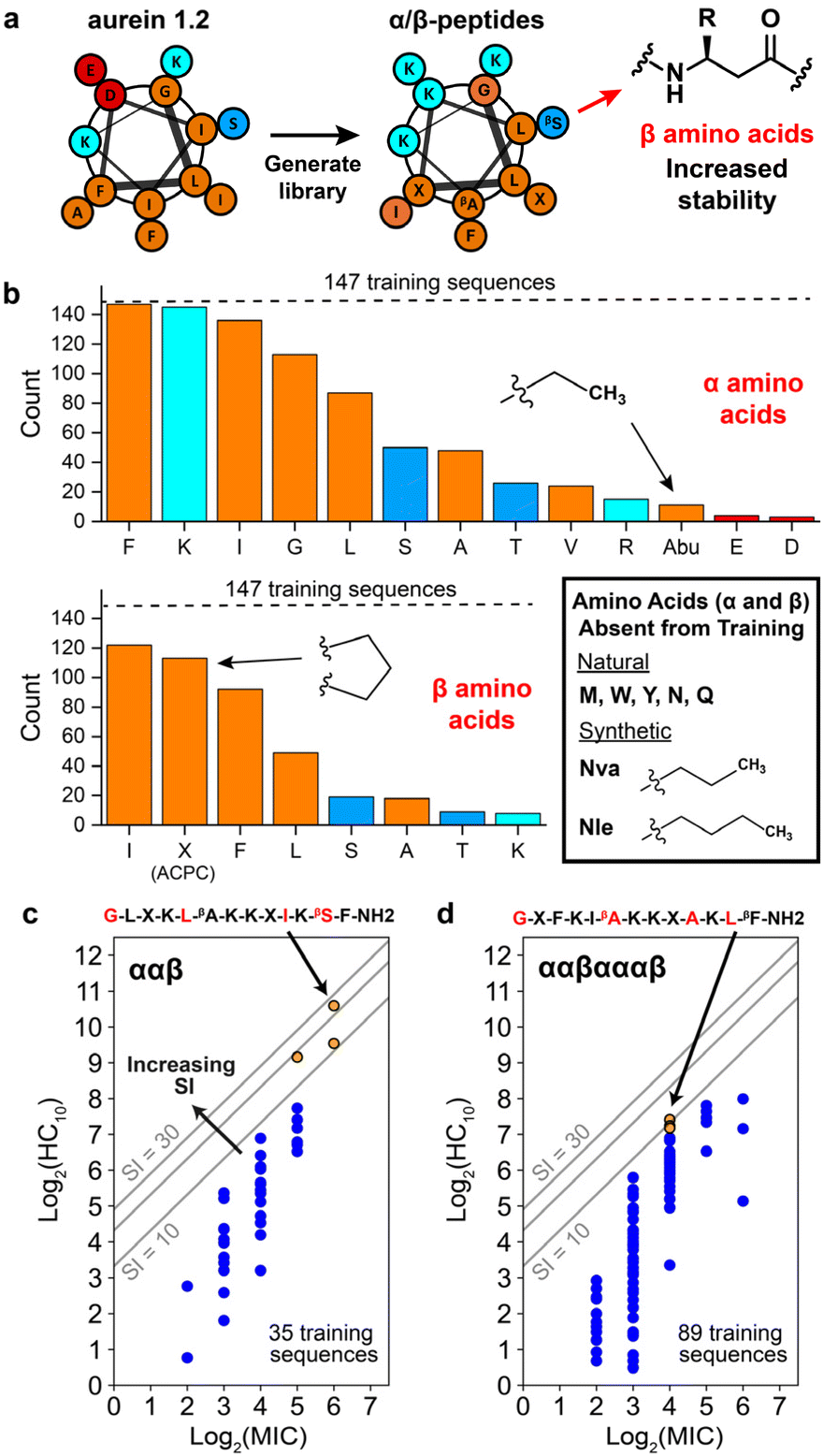 | ||
| Fig. 3 Overview of α/β-peptides and training design space for the Gaussian process regression workflow. (a) Helical wheel representation of aurein 1.2, which was used as the starting sequence to generate a library of 147 α/β-peptide sequences (used as the initial training set in our study) in previous rational design studies. Amino acid types are color-coded as follows: nonpolar (orange), polar (dark blue), basic (cyan), and acidic (red). “β” refers to amino acids with an additional backbone carbon to increase stability compared to traditional α-amino acids and R denotes side chain. (b) Number of training sequences (out of 147, denoted with dashed line) that contain at least one of each α- (top) and β- (bottom) amino acid. Amino acids (α and β) absent from the training peptides but utilized in this study are boxed at lower right. Amino acids are color coded in the same manner as in the previous panel. (c and d) Distribution of training sequences for the (c) ααβ and (d) ααβαααβ backbones according to their hemolysis (log2(HC10)) and antifungal activity (log2(MIC)). Grey lines indicate constant SI values equal to 10, 20, and 30. The top three SI template peptides for each backbone are shown as orange points. The corresponding amino-acid template sequence is shown with positions corresponding to varying amino acids in red. “—NH2” refers to an amidated C-terminus, whereas the N-terminus is +1-charged (these properties are shared for all peptide sequences). All other training sequences are shown as blue points. log2(HC10) vs. log2(MIC) distributions for the other two backbones used for model training (αααβ and αβαβααβ) are shown in Fig. S9,† and the combined activity distribution for all 4 backbone types is plotted in Fig. S10.† | ||
Properties of training sequences and design space generation
As described in the previous section, the initial training set comprised 147 rationally designed α/β-peptides derived from the natural sequence of aurein 1.2,9,19 incorporating both natural amino acids and synthetic β-amino acids to enhance proteolytic stability and antifungal selectivity (Fig. 3a). These peptides consisted of four distinct α/β-peptide backbone motifs—either ααβ, αααβ, ααβαααβ, or αβαβααβ—all predicted to adopt helical conformations resembling α-helices (i → i + 4 folding), as supported by prior high-resolution and circular dichroism (CD) structural studies.10,13,54 The rational design-based library generation, illustrated in Fig. 3a, was guided by helical wheel diagrams that qualitatively depict the expected folding behaviour of the α/β-peptides, assuming they would mimic the structure of aurein 1.2. This assumption was corroborated by CD spectral alignment between the i → i + 4 folding GCN4-pLI-based α/β-peptides and the training set peptides,9,17,19 the inclusion of helix-stabilizing ACPC β-amino acids,14 and the extended 13-residue sequence length,55 although we acknowledge that this does not preclude the possibilities of other bonding states such as i → i + 3 folding. This prior rational design took into account the expected distribution of amino acids in the helical wheels to maintain or enhance amphiphilicity as a strategy to increase antifungal selectivity through extensive trial and error.9,19Fig. 3b visualizes the occurrence of each α- and β-amino acid in the initial 147 sequence training set (dashed horizontal line) in terms of their polarity: nonpolar (orange), polar (dark blue), charged basic (cyan), and charged acidic (red). Only 13 of the 20 possible canonical α-amino acids were present in the 147-peptide training dataset. Additionally, only five α-amino acids (phenylalanine ‘F’, lysine ‘K’, isoleucine ‘I’, glycine ‘G’, leucine ‘L’) and three β-amino acids (β-homoisoleucine ‘βI’, ACPC ‘X’, β-homophenylalanine ‘βF’) were represented in at least half of the training sequences. Therefore, a large sequence design space remains unexplored, even for short 13-amino-acid peptides. To reduce this design space, we focused on templating and improving on high selectivity (high-SI) peptides in the training set based on amino-acid positions that vary amongst them (red amino acids in Fig. 3c and d), where the higher-SI peptides trend towards the top left of the plots in Fig. 3c and d.
The three highest-SI peptides of the 147 training sequences consisted of the ααβ backbone (indices #241, #239, #231) with SI values ranging from 11.6 to 24.1; we selected these three peptides as template peptides for the ααβ backbone. The sequences of these peptides varied in amino-acid positions 5, 10, and 12, and we additionally chose to vary amino-acid position 1 during the iterative GPR workflow to investigate the effects of N-terminal substitutions on peptide hemolysis and antifungal activity. Previous experimental structure–activity studies have found that substituting the N-terminal ‘G’ in aurein 1.2 with ‘A’ significantly decreased activity against C. albicans,56 whereas a ‘G’ to ‘A’ substitution increased broad-spectrum activity against bacteria.57 The three template ααβ-peptide sequences are shown as orange points in Fig. 3c along with the other 32 training sequences on the ααβ backbone (dark blue points) for reference. The 4–6th highest SI peptides in the training set (#133, #029, #131) consisted of the ααβαααβ backbone with SI values ranging from 9.1 to 10.6 (orange points in Fig. 3d); we selected these three peptides as template peptides for the ααβαααβ backbone. We chose to consider amino acid variations in these three peptides (at positions 1, 6, 10, 12) given their high antifungal activity, albeit with lower selectivity compared to the ααβ-peptides. Fig. 3c and d shows the two resulting template sequences (for the ααβ and ααβαααβ backbones, respectively) with the four amino-acid positions in each sequence that were varied during the GPR approach indicated in red.
For α positions in the template sequences, we considered the 13 amino acids included in the training data (top histogram in Fig. 3b) plus five natural amino acids (methionine ‘M’, tryptophan ‘W’, tyrosine ‘Y’, asparagine ‘N’, glutamine ‘Q’) and two hydrophobic synthetic amino acids (norvaline ‘Nva’, norleucine ‘Nle’) for a total of 20 possible substitutions. Nva and Nle are extensions of the α-aminobutyric acid (Abu) amino acid present in training peptides and were included based on previous studies suggesting that these amino acids increase membrane binding by promoting positive membrane curvature58 and improve antimicrobial activity by increasing peptide hydrophobicity.59 Three natural amino acids were not considered: proline 'P' due to destabilization of AMP folding by inducing kinks in helices,60,61 cysteine ‘C’ due to its capability to form disulfide bridges,62 and histidine ‘H' due to its sensitivity to physiological pH, leading to changes in charge.63 For β positions in the template sequences, the β version of the 20 α-amino acids described above plus ACPC ‘X’ were considered for a total of 21 possible substitutions. Given the three α- and one β-amino acid positions varied for each backbone type (red amino acids in Fig. 3c and d), two different design spaces of 168000 (203 × 21) possible sequences were considered for this study for a total of 336000 sequences. As described in the previous section, ‘iterative GPR workflow implementation’, these design spaces were reduced each round (Fig. 2d, S18 and Table S5†), only considering test sequences for which all descriptor values were within the bounds of training descriptor values (Fig. S7 and S8†).
Iterative GPR model training
Upon implementation of the iterative GPR workflow demonstrated in Fig. 2 based on the initial training set and design space outlined in Fig. 3, we observed that the approach demonstrates stable accuracy per prediction round for both HC10 and MIC (black lines in Fig. 4a), and R2 values for the comparison of GPR-predicted and experimentally-determined HC10 and MIC were comparable to previous efforts to predict MIC for a variety of antimicrobial compounds using simple sequence- or SMILES-based input data representations.64–66 As expected, MIC accuracy was consistently lower than HC10 given the nature of antifungal activity assays, where, by convention, the maximum of three replicates from serially diluted experiments, rather than the average, is taken as the MIC9,67,68 (Section S10†). Therefore, continuous log2(MIC) GPR predictions were rounded up to the nearest whole number to match this experimental methodology. For rounds 1 to 3, the selected set of descriptors remained the same (see ‘Initial’ column in Tables S1 and S2†). However, given a large increase in the maximum error between experimentally measured HC10 and MIC versus GPR predictions for both quantities in round 3 (red lines in Fig. 4a) after the introduction of a large HC10 and MIC sequence to the training data (sequence 2-4, see Fig. 5a, S11 and S12†), we revised the workflow to update the selected set of descriptors through LASSO CV during each subsequent round from round 4 onwards as described above. Although this approach led to an initial increase in prediction RMSE between GPR predictions and experimental measurements (Fig. S13†), AMPs with high HC10 and MIC values were more accurately predicted for newly introduced sequences, particularly compared to the consistent underprediction of high-value MIC labels in rounds 1 to 3 (Fig. S12†).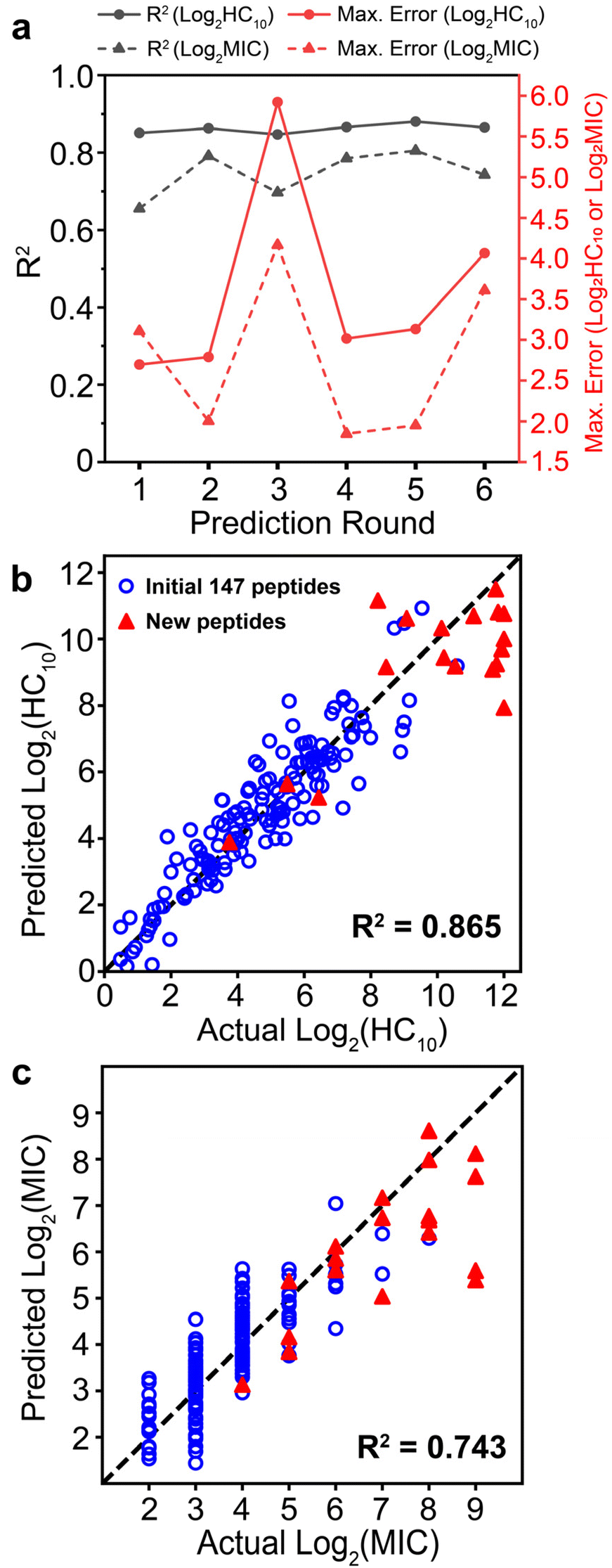 | ||
| Fig. 4 (a) R2 (black lines) and maximum error (red lines) metrics calculated per prediction round for separate HC10 (solid) and MIC (dashed) prediction workflows. (b and c) Comparison of GPR-predicted vs. actual experimentally measured values of (b) log2(HC10) and (c) log2(MIC). GPR predictions used the final trained GPR model for the 6th prediction round. Points are validation set predictions from 10-fold cross-validation; that is, each point is the predicted value from when the corresponding sequence is not used for model training. The original 147 training peptides are shown as open blue circles and new peptides discovered with GPR are shown as red triangles. Coefficients of determination (R2) are included in bold. | ||
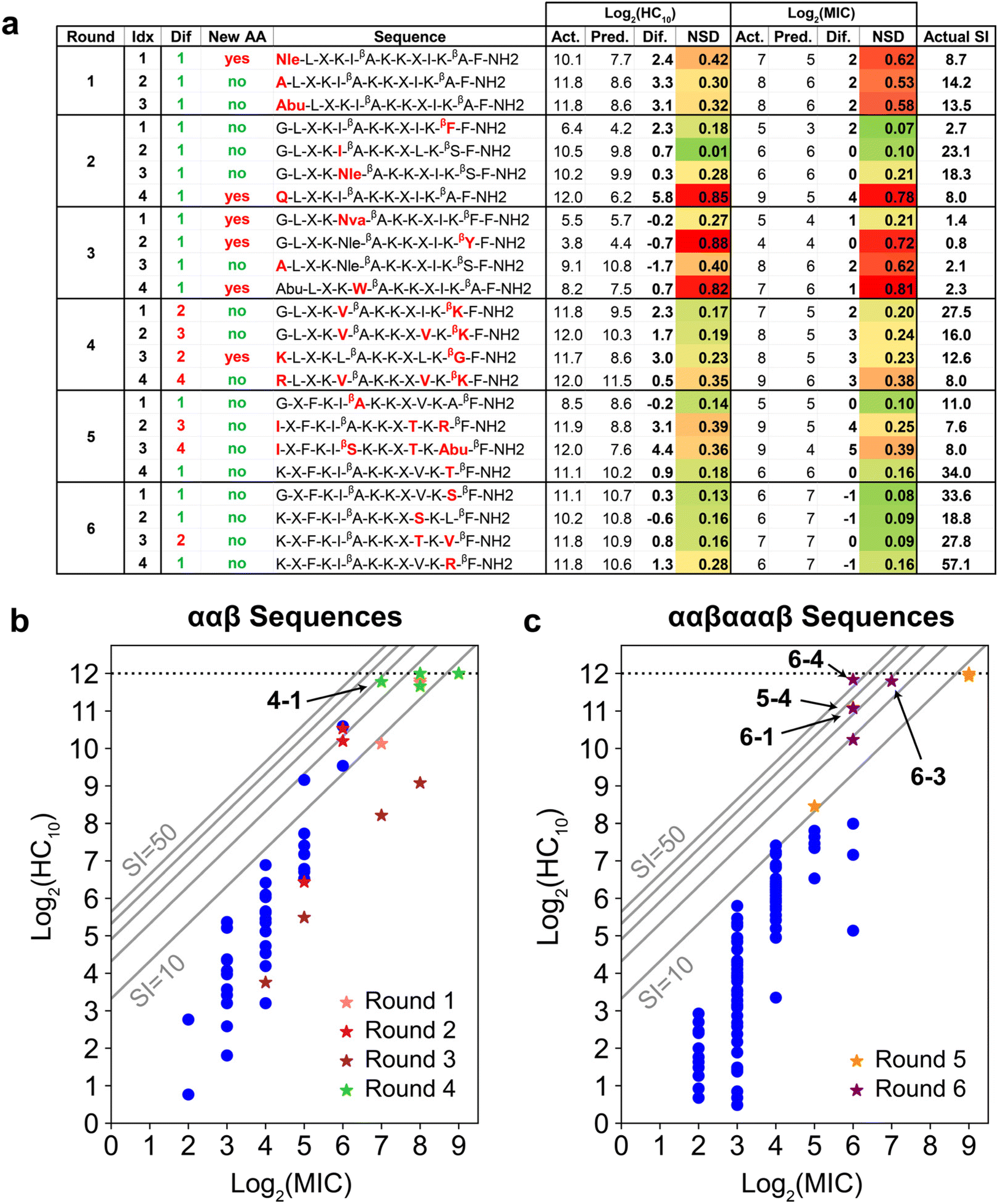 | ||
| Fig. 5 (a) Summary of GPR prediction results. Amino-acid substitutions relative to any training sequence are in red. The ‘Dif’ column quantifies the number of these substitutions, and the ‘New AA’ column denotes if any substitution is an amino acid not present in the training data for that round. Experimentally measured (Act.), GPR-predicted (Pred.), and their difference are shown for both log2(HC10) and log2(MIC). NSD refers to the normalized GPR standard deviation and ranges from 0 (dark green; low uncertainty) to 1 (dark red; high uncertainty). SI computed from the experimentally measured HC10 and MIC is also shown. (b and c) log2(HC10) vs. log2(MIC) distributions comparing original training (dark blue dots) vs. new peptides added during the iterative workflow (stars) for prediction rounds (b) 1 to 4 and (c) 5 to 6. Grey diagonal lines denote SI bands with values of 10, 20, 30, 40, and 50. All peptides discovered with larger SI values than the original highest-SI peptide from the initial training set (SI = 24.1) are indicated in bold. The horizontal dotted line at log2(HC10) = 12 denotes the upper limit of the hemolysis assay, corresponding to a peptide concentration of 4096 μg mL−1. The test design space and corresponding standard deviation ranges for all GPR prediction results are visualized in Fig. S18.† | ||
Fig. 4b and c shows parity plots for log2(HC10) and log2 (MIC) predictions for the final GPR model used for round 6, which not only visualize the final prediction accuracy, but also highlight the novel sequences introduced (red triangles) and their distributions of log2(HC10) and log2(MIC) values compared to the original 147 training sequences (blue open circles). As part of our evaluation of model robustness across prediction rounds (Fig. S14–S16†), we also probed the predictive power of the GPR models through y-randomization (Fig. S16†),69,70 which compares randomly shuffled labels with the ones used for model development. Overall, these results verified that the predictions made by the model were not from random chance, thereby validating the rigor of our descriptor and GPR model selection approach.
GPR guides the discovery of α/β-peptide sequences with novel amino acids and motifs
Fig. 5a shows the results of the GPR workflow for the 23 newly discovered peptides experimentally tested during the six prediction rounds (Fig. S21 and S22†), which are plotted as stars in Fig. 5b and c and compared to the initial training sequences in blue (Fig. 3c and d). In general, NSD values aligned well with the difference in actual (Act.) vs. predicted (Pred.) labels, and for newly introduced amino acids or motifs, there was an initial prediction inaccuracy that was resolved when reintroduced in later prediction rounds. For instance, sequences 1-1 (new Nle) and 2-1 (new βF-F C-terminal motif) were underpredicted by a factor of approximately two for both log2(HC10) and log2(MIC). Sequences 2-3 (containing Nle) and 3-1 (containing βF-F), however, were predicted with excellent accuracy relative to experimental measurements. Additionally, NSD values are associated with how chemically disparate newly introduced amino acids were from amino acids present in training sequences. For instance, sequence 3-1 was predicted with relatively low uncertainty (NSD < 0.30 for both log2(HC10) and log2(MIC) predictions) despite the new Nva amino acid. This is consistent with Nva having linear alkyl side chain that is intermediate in length compared to Abu and Nle (reference chemical structures in Fig. 3b), which were present in the training set by round 3. In contrast, peptides 2-4, 3-2, and 3-4, each of which each introduced amino acids ‘Q,’ ‘βY,’ and ‘W,’ containing amine, phenol, and indole chemical groups, respectively, had higher prediction uncertainty (NSD > 0.70), even with a single amino-acid substitution relative to training sequences.To further support the ability of the GPR model to capture relationships between input physicochemical properties of new amino acids and target activity measurements, we calculated the NSD distribution of each newly introduced amino acid in rounds 1 to 3 since the set of descriptors for HC10 and MIC predictions remained the same for these prediction rounds (Tables S1 and S2†). Bar and whisker plots of these NSD distributions are plotted in Fig. S17† for the Nle, ‘Q’, Nva, ‘βY,’ and ‘W’ amino acids, which show that, in general, there was a large decrease in the average NSD (therefore, a decrease in prediction uncertainty) for these amino acids in prediction rounds following their introduction. Interestingly, Nva (Fig. S17c†) largely followed the same trends as Nle (Fig. S17a†), despite not being introduced prior to the changes observed in NSD. This observation supports the model's ability to transfer learned information from amino acids present in the training sequences to new amino acids that have similar chemical structures, and highlights the advantage of 2D molecular descriptors over more traditional data representations for peptide sequences such as one-hot or orthogonal encoding,37,71 which would distinguish Nva and Nle as separate amino acids with no intuition for their chemical similarity.
GPR approach yields multiple highly selective α/β-peptides and identifies an unconventional cationic amino acid substitution
Fig. 6 visualizes amino acid substitutions of aurein 1.2 that resulted in the highest-SI peptides discovered through the iterative GPR approach. We first note that the highest-SI template peptides (#241, SI = 24.1; #131, SI = 9.08) previously developed through rational design exhibited improved selectivity compared to aurein 1.2.9,19 In the initial prediction rounds (1 to 4), peptides were synthesized with the focus on model training by introducing new amino acids and previously utilized amino acids in novel positions. In this process, two high-SI peptides were discovered, one of which featured an unconventional cationic amino acid (4-1, ‘βK’ substitution) on the hydrophobic face, a substitution that was not explored in our prior rational design attempts because of concerns that reductions in amphiphilicity and hydrophobicity would result in excessive losses in antifungal activity.19,72 As expected, these newly discovered high-SI peptides did not exhibit increased antifungal activity. Instead, the decrease in hemolytic activity was significant, which drove an increase in selectivity. Therefore, the initial rounds demonstrate that peptides discovered by the model improved selectivity by reducing hemolysis rather than by increasing antifungal activity.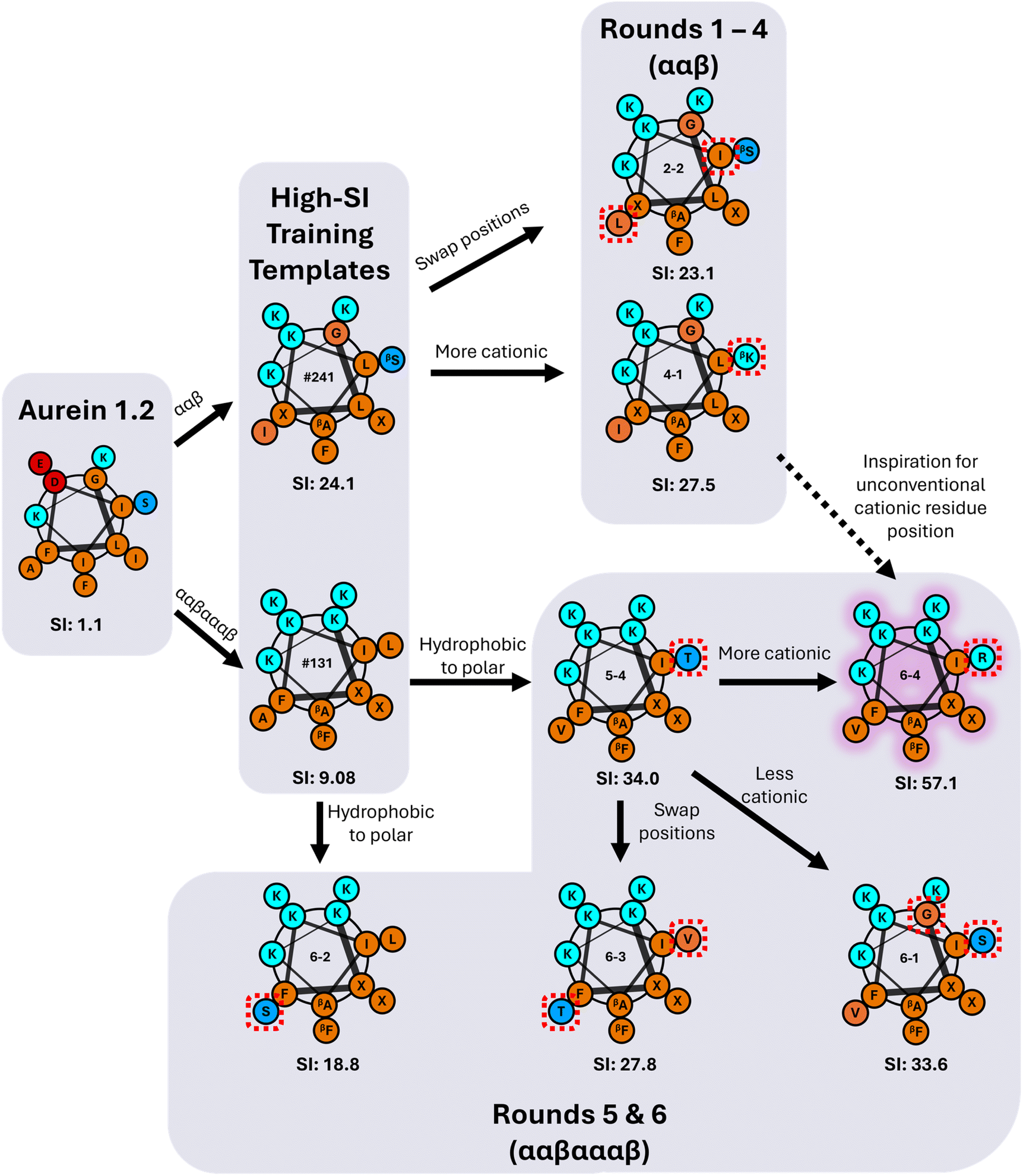 | ||
| Fig. 6 Flowchart of the iterative GPR-guided sequence-substitutions of α/β-peptides templated on aurein 1.2 that resulted in high selectivity against C. albicans. Hydrophobic amino acids are shown in orange, cationic amino acids in cyan, anionic amino acids in red, and neutral polar amino acids in blue. β-Amino acids are visualized with a subscript and ACPC is represented as X. Red dashed boxes represent single amino-acid substitutions between each peptide. Each peptide is labelled with its experimentally measured SI. The highest-SI peptide discovered by the iterative GPR approach is highlighted in purple. | ||
In rounds 5 and 6, the more antifungal ααβαααβ backbone was explored as the template (Fig. 3d) using the same workflow as in rounds 1 to 4, but by switching the design space to both evaluate the transferability of our approach and to obtain higher-SI peptides. This change enabled us to use the large amount of low hemolysis (high log2(HC10)) training data from rounds 1 to 4 (Fig. 5a) to probe whether the model could be applied to a different peptide backbone with inherently higher antifungal activities (lower log2(MIC)) to find new high-SI peptides. Although there was an initial increase in the overall prediction RMSE in round 5 with the switch in backbone template from ααβ to ααβαααβ and selection of 2 test sequences with moderate uncertainty (5-2 and 5-3, Fig. 5a), the selection of 4 low uncertainty sequences to probe new high SI peptides in round 6 led to the lowest prediction RMSE across all prediction rounds (Fig. S13†). Interestingly, this approach was successful in discovering four additional high-SI peptides (5-4, 6-1, 6-3, 6-4; 27.8 < SI < 57.1) with high prediction accuracy and low uncertainty (low NSD) while drawing on learned trends from previous prediction rounds on the ααβ backbone (Fig. 5c). For instance, as seen in peptides 4-3 and 4-4, 1st position cationic (K, R) amino acids tend to produce peptides with low hemolysis (high log2(HC10)) and good selectivity (high-SI), which was reflected by many high-SI ααβαααβ candidate sequences in rounds 5 and 6 (5-4, 6-2, 6-3, 6-4) with a 1st position ‘K.’ Additionally, drawing on the ability of peptide 4-1 to increase selectivity with a cationic amino-acid substitution on the hydrophobic face of the ααβ backbone, an arginine (R) substitution on the hydrophobic face of a high-SI ααβαααβ template (#131) was predicted and experimentally validated, resulting in an over 6-fold improvement in selectivity (6-4 in Fig. 6). With an SI of 57.1, peptide 6-4 was the highest-SI peptide identified in this study and showcases a substantial increase in antifungal selectivity compared to the training data. Overall, this approach was able to discover 13 novel high-SI peptides (11.0 < SI < 57.1) with comparable or higher SIs than the template peptides (9.08 < SI < 24.1) (Fig. 5a). We further note that our SIs are significantly higher or compare favorably to the SIs of other antifungal AMP mimics reported in the literature (often around 16 for hRBC HC10 over C. albicans MIC),26,73–75 as well as certain clinically used small molecules such as amphotericin B (which has an SI of 1 (ref. 76 and 77)). Additional studies in animal models of fungal infection will be necessary to determine the extent to which these substantial increases in SI may also translate to therapeutic significance in different pre-clinical contexts.
The number of test sequences considered per round (between 8764 and 17238 sequences depending on prediction round, see Table S5†) is sufficient to explore large variations in amino acids and motifs as demonstrated in Fig. 5a, given that this design space is approximately 2 orders of magnitude larger than the initial peptide training set (147 sequences). We also note that the number of α/β-peptides that has been characterized experimentally is significantly smaller than α peptides, such that this design space represents a substantial number of novel sequences. To further increase the potential design space considered in our approach, we note that the decrease in the number of considered test peptide sequences (Table S5,† ‘total’ column) is largely a result of the increasing number of descriptors kept with LASSO CV from rounds 1-6 (Fig. S3†) because we require each potential test sequence to have all descriptor values fall within the lower and upper bounds of the training set per round. Therefore, although it is out of the scope of this study, a potential avenue for increasing the possible number of test sequences would be to truncate the number of descriptors kept with LASSO CV (e.g., keeping only the top 20 descriptors by LASSO coefficient weights) to increase the design space, particularly since the average RMSE for 10-fold LASSO CV only slightly increases when fewer descriptors are used (Fig. S3†). For instance, truncating the descriptor set to the top 20 descriptors increases the design space for round 6 for HC10 predictions from 10111 to 34419 sequences and for MIC predictions from 19057 to 43650 sequences while maintaining similar cross-validation prediction accuracies compared to the full descriptor set. Because our present approach successfully identified new peptides with improved selectivity indices that are relevant in potential therapeutic contexts (as discussed above), we do not foresee this approach as necessary, but one could utilize this method (or similar methods to relax the constraints on the number of descriptors) to expand the design space in future work or other related applications.
Overall, these results demonstrate that an iterative ML approach can successfully discover high-SI peptides through substitutions that would not have been rationally anticipated. The advantage of the SMILES representation for peptide sequences is that it is agnostic to the backbone type (combination of α and β amino acids), thereby promoting the transferability of the model across backbones in this study and, potentially, for other synthetic motifs (e.g., γ-amino acids with two additional backbone hydrocarbons compared to α-amino acids78) in the future.
Improved antifungal selectivity in GPR-screened α/β-peptides is dependent on charge, hydrophobicity, and helical rigidity
To better understand the relationship between sequence, structure, and activity for the newly discovered high-SI α/β-peptides in comparison to aurein 1.2 and the high-SI template peptides, we experimentally measured their physicochemical properties (Table 1) and visualized representative comparisons in Fig. 7. Notably, the highest-SI peptides discovered by the iterative GPR approach (4-1, 5-4, 6-1, 6-4; 27.5 < SI < 57.1) exhibited lower hydrophobicity (i.e., smaller HPLC-retention times; RT) and reduced helical rigidity compared to the highest-SI template peptides (#029-#241; 9.08 < SI < 24.1) (Table 1 and Fig. 7). Peptide net charge either remained the same or increased by one compared to the template peptides, demonstrating the usefulness of such substitutions in certain positions. In general, all high-SI peptides in this study possessed lower hydrophobicity and helical rigidity, while possessing a higher cationic charge compared to aurein 1.2. These are well-documented characteristics of highly selective antifungal peptides.26,79| Peptide type | Peptide Idx | Sequence | αβ motif | Retention time (min ± SD) | Charge | [θ] 100% TFE ± SD | [θ] 15% TFE ± SD | Helical rigidity % ± SD | Measured molecular weight (Da) |
|---|---|---|---|---|---|---|---|---|---|
| Wild type | Aurein 1.2 | G-L-F-D-I-I-K-K-I-A-E-S-F-NH2 | α | 23.93 ± 0.01 | 1 | −16.8 ± 0.4 | −17.1 ± 0.1 | 102 ± 2 | 1479.79 |
| Template peptides | #241 | G-L-X-K-L-βA-K-K-X-I-K-βS-F-NH2 | ααβ | 15.45 ± 0.01 | 5 | −27.2 ± 1.7 | −9.1 ± 1.1 | 34 ± 5 | 1481.94 |
| #239 | G-L-X-K-L-βA-K-K-X-L-K-βS-F-NH2 | ααβ | 16.11 ± 0.01 | 5 | −27.4 ± 2.9 | −10.5 ± 0.2 | 38 ± 4 | 1481.94 | |
| #231 | G-L-X-K-I-βA-K-K-X-I-K-βA-F-NH2 | ααβ | 14.95 ± 0.01 | 5 | −20.5 ± 1.3 | −12.3 ± 1.6 | 60 ± 9 | 1465.94 | |
| #133 | G-X-F-K-I-βA-K-K-X-A-K-L-βF-NH2 | ααβαααβ | 16.61 ± 0.01 | 5 | −30.9 ± 1.1 | −14.2 ± 2.0 | 46 ± 7 | 1499.96 | |
| #131 | K-X-F-K-I-βA-K-K-X-V-K-L-βF-NH2 | ααβαααβ | 15.55 ± 0.01 | 6 | −26.2 ± 0.8 | −11.6 ± 1.4 | 44 ± 5 | 1599.13 | |
| #29 | G-X-F-K-I-βI-K-K-X-A-K-S-βF-NH2 | ααβαααβ | 18.17 ± 0.01 | 5 | −35.2 ± 1.6 | −13.1 ± 0.9 | 37 ± 3 | 1515.96 | |
| Newly discovered peptides | 4-1 | G-L-X-K-V-βA-K-K-X-I-K-βK-F-NH2 | ααβ | 13.50 ± 0.02 | 6 | −23.1 ± 0.8 | −7.2 ± 0.6 | 31 ± 3 | 1509.01 |
| 5-4 | K-X-F-K-I-βA-K-K-X-V-K-T-βF-NH2 | ααβαααβ | 14.02 ± 0.04 | 6 | −36.0 ± 0.6 | −8.5 ± 1.6 | 24 ± 4 | 1587.06 | |
| 6-1 | G-X-F-K-I-βA-K-K-X-V-K-S-βF-NH2 | ααβαααβ | 15.38 ± 0.08 | 5 | −33.7 ± 1.0 | −8.5 ± 0.9 | 25 ± 3 | 1501.96 | |
| 6-4 | K-X-F-K-I-βA-K-K-X-V-K-R-βF-NH2 | ααβαααβ | 13.33 ± 0.04 | 7 | −27.6 ± 2.2 | −7.3 ± 1.7 | 27 ± 6 | 1642.10 |
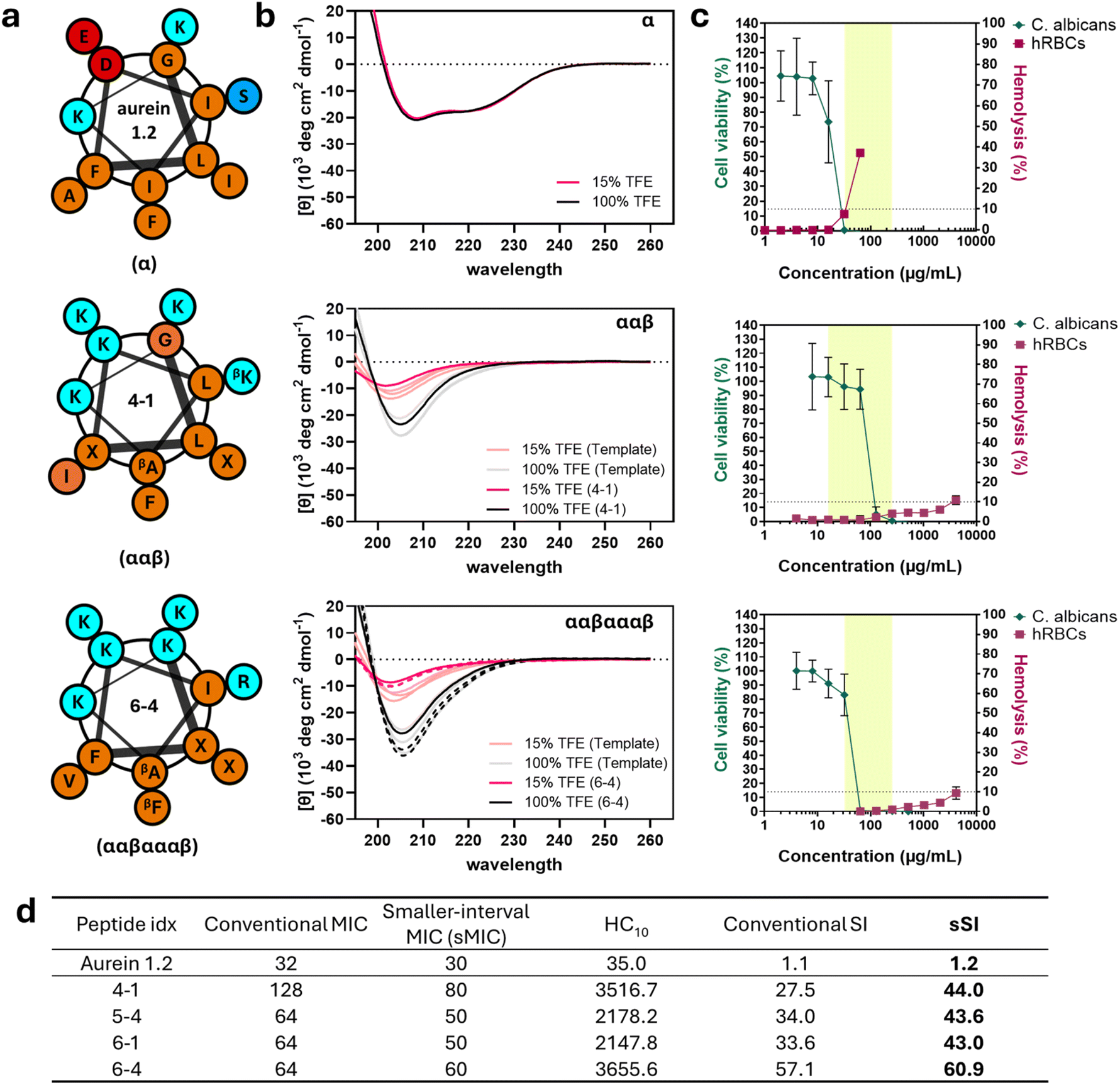 | ||
| Fig. 7 Characterization of the highest-SI newly discovered peptides for each backbone. Aurein 1.2 (top), ααβ-backbone peptide 4-1 (middle), and ααβαααβ-backbone peptide 6-4 (bottom) are shown. (a) Helical wheel representations of sequences and expected amphiphilic amino acid arrangement. Hydrophobic amino acids are shown in orange, cationic amino acids in cyan, anionic amino acids in red, and non-charged polar amino acids in blue. β-Amino acids are visualized with a subscript and ACPC is represented as X. (b) Circular dichroism spectra of the three peptides in 15% (red) and 100% trifluoroethanol (black). The CD curves of other high-SI peptides with a ααβαααβ backbone are represented as dashed lines. (c) Comparisons between antifungal (C. albicans, green) and hemolytic (hRBC, red) activities of peptides along with their broad-spectrum MIC range (highlighted). Highlighted regions represent the effective range of MICs of each peptide against all fungal and bacterial cells tested. Data points are the averages of three independent experiments with at least two technical duplicates each and error bars represent the standard deviation. (d) Table of C. albicans MIC, smaller-interval MIC (sMIC; see text), hRBC HC10, SI and smaller-interval SI (sSI) for the four highest SI peptides in comparison to aurein 1.2. sMIC was measured at 5 μg mL−1 intervals and sSI was calculated by HC10/sMIC. | ||
Generally, decreases in hydrophobicity are correlated with decreased mammalian cell toxicity,80–82 which, in turn, leads to higher selectivity. Supporting these trends, newly discovered high-SI peptides exhibited significantly decreased hydrophobicity in all cases compared to their respective high-SI templates (i.e., RT of 13-15 min compared to 15-18 min for ααβαααβ backbones). When compared to aurein 1.2, the differences were even greater, with an at least 10.5 minute reduction in RT, illustrating the importance of low hydrophobicity for antifungal selectivity. Similarly, the helical rigidities of newly discovered high-SI peptides were lower than those of the template peptides and aurein 1.2 (Table 1 and Fig. 7b). Quantified as the ratio between the molar ellipticity of peptide (minimum amplitude [θ]) in 15% and in 100% trifluoroethanol (TFE) and measured through circular dichroism (CD), helical rigidity describes the ability of a peptide to remain helical in aqueous conditions in comparison to its maximum helical conformation, which TFE is known to induce.83 The amplitude at the minimum wavelength was utilized as a measurement of helicity to remain consistent with prior structural studies coupling crystallography and NMR with CD.17,84 TFE is often used to mimic the microbial cell membrane environment,26,79,85 while 15% TFE in water has been used to obtain quantifiable differences in low-helicity peptides in aqueous conditions.9,19 Similar to hydrophobicity, high helical rigidity results in toxicity against human cells.19,26,86–93 For example, aurein 1.2 presented classic α-helical CD spectra in both aqueous and organic solvent conditions (15% and 100% TFE, Fig. 7b), with a high helical rigidity of 102 ± 2% (Table 1).
Both template and newly discovered high-SI peptides exhibited CD curves characteristic of helical α/β-peptides,9,13,17 with lower helicity in aqueous conditions (red line, [θ]15) compared to those in organic conditions (black line, [θ]100) (Table 1, Fig. 7b and S24†). While the presence of an additional carbon in β-amino acids in α/β-peptides can lead to an altered arrangement of the amide chromophores and lead to deviations from the classic α-helical CD spectra, high-resolution structural data coupled with CD analysis of previously studied helical α/β-peptides17,84 suggest that the newly discovered peptides in this study also likely fold in a manner similar to aurein 1.2, but with significantly reduced helical rigidities. These results support the use of α/β-peptides as a strategy to enhance antimicrobial selectivity of naturally sourced AMPs without compromising their side-chain presentation pattern.
Newly discovered peptides from our approach were also less helical than the template peptides regardless of backbone type. For example, peptide 4-1 (ααβ-motif) exhibited around 31% helical rigidity while ααβ template peptides had 34–60%; similarly, peptides 5-4, 6-1, and 6-4 (ααβαααβ-motif) exhibited helical rigidities between 24 and 27%, while ααβαααβ template peptides had 37–46%. This decrease in helical rigidity can be attributed largely to the significant reduction in the helicity of peptides in aqueous conditions ([θ]15) rather than changes in helicity in organic conditions ([θ]100) (Fig. 7b and S25†). Similar trends have been observed for homogeneous α-amino acid-containing antifungal26 and antibacterial AMPs,86–89 where the ability of the peptides to adopt a non-helical-to-helical transition upon exposure to helix-inducing conditions (such as at the cell membrane interface, or in TFE) was a critical contributor to selectivity, predominantly driven by the high correlation between helicity and hemolysis.90–93 Overall, these results suggest that the iterative GPR approach discovers higher selectivity peptides largely by decreasing their hydrophobicity and increasing their helical flexibility.
α/β-Peptides with high antifungal selectivity induce significantly reduced hemolysis and kill other fungal and bacterial microbes
Fig. 7c summarizes cell viability differences, highlighting how variations in sequence, secondary structure, and other physicochemical properties characterized in Table 1 influence selectivity. In addition, the peptides were evaluated against other clinical fungal (Candida glabrata, Candida tropicalis, Candida parapsilosis) and model bacterial strains (S. aureus, E. coli) to assess their potential applications against other microbial species in comparison to aurein 1.2 (Tables S8 and S9†). Aurein 1.2 demonstrated activity against C. albicans at an MIC of 32 μg mL−1 (green line), with other microbial MICs ranging from 16 to 256 μg mL−1 (highlighted region, Fig. 7c) (Table S8 and Fig. S26†). However, aurein 1.2 exhibited 37.2 ± 1.8% hemolysis at 64 μg mL−1 (red line), resulting in a narrow SI of just 1.1 against C. albicans, and was unable to kill other microbial cells at the concentrations tested. In comparison, peptides 4-1 (ααβ backbone) and 6-4 (ααβαααβ backbone) exhibited comparable MICs (128 μg mL−1 and 64 μg mL−1 against C. albicans, respectively, and 16–256 μg mL−1 and 32–256 μg mL−1 broad-spectrum, Table S8 and Fig. S26†) to aurein 1.2, while only exhibiting hemolytic activities of 10.9 ± 2.2% and 9.4 ± 3.1% at a concentration of 4096 μg mL−1, respectively. This represents an almost 128-fold reduction in hemolysis while maintaining similar antifungal and antibacterial activity (highlighted region, Fig. 7c). Other newly discovered high-SI peptides (5-4 and 6-1) demonstrated similar trends (Fig. S22, S26, Tables S8 and S9†). Notably, some peptides also exhibited a significant improvement in antibacterial selectivity, such as peptide 4-1, with an E. coli-specific SI of 219.8 compared to an SI of 1.1 using aurein 1.2, representing a 200-fold increase. These results not only indicate that our model effectively discovers peptides with increased C. albicans selectivity compared to human red blood cells (hRBCs), but also demonstrate that these peptides are active against other microbial cells.Finally, we sought to quantify more precisely the antifungal activities of lead peptides against C. albicans, the original target of our model, to better understand the extent of improvement in selectivity achieved using our approach. Here, we note that, in all the determinations of MIC in the studies above, we used two-fold serial dilutions to determine the lowest concentration inhibiting over 90% of microbial growth. While conventional, this method yields less accurate representations of true antimicrobial activity that may lie in between the discrete concentrations that are actually tested and is exacerbated at higher ranges (e.g., two compounds with MICs of 64 versus 128 μg mL−1 misses more precise activity comparisons in contrast to MICs between 1 μg mL−1 and 2 μg mL−1). Because MIC is used to calculate SI, reported SI values may underestimate the improvement in selectivity resulting from our approach. Therefore, we measured the MICs of aurein 1.2 and high-SI test peptides at smaller intervals of 5 μg mL−1 (sMIC), as opposed to two-fold serial dilutions (Fig. 7d and S27†) and calculated corresponding ‘smaller interval SIs’ (sSI = HC10/sMIC). As expected, all peptides tested including aurein 1.2 exhibited higher sSI than SI, with 4-1 showing the largest increase. Compared to aurein 1.2, which had an sSI of 1.2, all four newly discovered high-SI peptides demonstrated significantly improved selectivities, ranging from 43.0 to 60.9, indicating up to a 51-fold enhancement in selectivity.
Peptides 4-1 and 6-4, which contain cationic amino acids on their hydrophobic faces, demonstrated the highest sSI despite different backbones (ααβ for 4-1, ααβαααβ for 6-4). These results further support the idea that the unconventional cationic amino acid substitution in the hydrophobic face of α/β-peptides could be a productive strategy to enhance their antifungal selectivity. While unintuitive from a rational design standpoint, similar substitutions, denoted as “specificity determinants” by Hodges and coworkers,94–96 have been reported previously to increase the selectivity of AMPs29 and D-enantiomer analogues94–96 for antibacterial applications, where (1) increases in charge, (2) decreases in hydrophobicity, and (3) decreases in helical rigidity led to a drastic reduction of hemolysis and, in turn, enhanced antibacterial selectivity.94–96 These studies mainly attributed the enhanced selectivity of these AMPs to their altered membrane-active action, citing differences between prokaryotic and eukaryotic membranes, such as net charge, in the context of reduced helical rigidity of peptides. We observed in this study that C. albicans selectivity was also enhanced through a comparable amino-acid substitution, even though C. albicans is eukaryotic. A potential explanation for this is that the higher content of anionic phospholipids in C. albicans cell membranes compared to those of red blood cells (around 20% difference)97,98 may have similarly contributed to the observed increase in selectivity. However, considering that C. albicans cells contain many other components different from human red blood cells, such as the presence of a cell wall97 and different sterols in cell membranes (ergosterol in fungi vs. cholesterol in hRBCs),97 further mechanistic studies on interactions between α/β-peptides and cell membranes are needed. Nevertheless, these results suggest that substitutions of cationic amino acids into the more hydrophobic face of α/β-peptides can be a strategy to improve their antifungal selectivity. A potential next step to enhance the antifungal selectivity of highly antifungal but hemolytic α/β-peptides could involve exploring cationic-amino acid substitutions at other positions in their hydrophobic faces, which can be greatly accelerated by iterative GPR.
Conclusions
In this work, we demonstrate the advantages of an iterative approach that combines model prediction with experimental evaluation to discover highly selective antifungal peptides containing noncanonical amino acids. By utilizing a Gaussian process regression model capable of quantifying prediction uncertainty, we explored a vast sequence space of peptidomimetic α/β-peptides (336000 sequences) based on a relatively small initial training set (147 sequences) to enhance their antifungal selectivity. The uncertainties provided by the model enabled us to make informed decisions on the selection of novel α/β-peptide sequences to synthesize and evaluate experimentally each round. After six prediction rounds, in which 23 new peptide sequences were characterized, we discovered peptides with high selectivity (sSI between 43 to 61), which had up to a 52-fold improvement compared to the wild-type aurein 1.2 (sSI = 1.2). In addition, these peptides also showed antimicrobial activity against other microbial species including bacteria (SI of 220 against E. coli), further broadening their potential applications.
Experimental characterization of newly discovered high-SI peptides demonstrated that they exhibit physicochemical properties typical of other highly selective natural AMPs, such as high cationic charge, low hydrophobicity, and low helical rigidity. Furthermore, the most selective α/β-peptides for both α/β-backbones contain unconventional cationic amino acid substitutions near the hydrophobic face, suggesting that incorporating such substitutions could be a potential strategy to enhance antifungal selectivity. This confirms that our approach, based on only sequence and activity data, successfully discovers highly selective peptides that would be far less likely to be identified through conventional rational design. Further understanding the impact of these substitutions on interactions with cell membranes to provide mechanistic insight into peptide activity (e.g., by modelling AMP-induced membrane disruption with computational modelling techniques99) will be a subject of future work. Given the challenges in physically interpreting the descriptors used in this work (Section S9†), physiochemical descriptors extracted from membrane binding and pore formation molecular dynamics simulations100 of model α/β-AMPs (including descriptors related to changes in hydrophobicity and helicity), for instance, could directly support experimental HC10 and MIC measurements in this study.
Overall, our approach circumvents the need for large sets of low-throughput and costly experimental physicochemical data while maintaining good prediction accuracy. Additionally, since the SMILES data representation captures the structural complexities of both backbone and side chain elements in a compound-agnostic manner, such a workflow is transferrable to other classes of antimicrobial peptides and mimetics, such as, but not limited to, those of peptides with γ-amino acids78 or with hydrocarbon-staples.101 We anticipate that our approach, which enabled the discovery of highly selective α/β-peptides with potential for antifungal therapy, can similarly stimulate the discovery of other highly selective peptidomimetics.
Data availability
A PDF of all computational and experimental methodology for the iterative GPR workflow is included in the ESI.† Python scripts to run GPR model training and analysis as well as all peptide sequences, SMILES strings, calculated RDKit descriptors, and antimicrobial and hemolytic activity curve datasheets have been uploaded to a GitHub repository associated with this manuscript (github.com/jdrichardson97/Peptide-GPR) to facilitate reproducibility of the results in this work.Author contributions
Conceptualization: DHC, JDR, RCV, SPP, DML; computational methodology: JDR; experimental methodology: DHC; sequence selection: JDR, DHC; peptide synthesis: DHC; investigation: JDR, DHC; funding acquisition: DML, SPP, RCV, supervision: DML, SPP, RCV; writing: DHC, JDR, RCV, DML, SPP.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 27th February 2025, which contained an error in the caption for Table 1.Acknowledgements
This study was supported by National Institutes of Health grant R33AI127442 to SPP and DML and by the National Science Foundation under award DMR-2044997 to RCV. DHC was supported in part by the UW–Madison NIH Biotechnology Training Program (5T32GM135066-02). The authors thank Prof. Samuel Gellman (UW−Madison) for providing resources for α/β-peptide synthesis, purification, and characterization. The authors gratefully acknowledge the use of facilities and instrumentation supported by the National Science Foundation through the University of Wisconsin Materials Research Science and Engineering Center (DMR-2309000). The purchase of the Bruker Impact II and Thermo Q Exactive Plus, which were used to quantify peptide molecular weight, was funded by the Bender gift to the Department of Chemistry and NIH Award 1S10 OD020022-1 to the Department of Chemistry, respectively.References
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- H. Jenssen, P. Hamill and R. E. W. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS PubMed.
- A. H. Benfield and S. T. Henriques, Front. Med. Technol., 2020, 2, 610997 CrossRef PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- L. Yu, K. Li, J. Zhang, H. Jin, A. Saleem, Q. Song, Q. Jia and P. Li, ACS Appl. Bio Mater., 2022, 5, 366–393 CrossRef CAS PubMed.
- T. Rozek, K. L. Wegener, J. H. Bowie, I. N. Olver, J. A. Carver, J. C. Wallace and M. J. Tyler, Eur. J. Biochem., 2000, 267, 5330–5341 CrossRef CAS PubMed.
- Y. Zhu, C. Shao, G. Li, Z. Lai, P. Tan, Q. Jian, B. Cheng and A. Shan, J. Med. Chem., 2020, 63, 9421–9435 CrossRef CAS PubMed.
- C. G. Starr and W. C. Wimley, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 2319–2326 CrossRef CAS PubMed.
- M. R. Lee, N. Raman, S. H. Gellman, D. M. Lynn and S. P. Palecek, ACS Chem. Biol., 2017, 12, 2975–2980 CrossRef CAS PubMed.
- L. M. Johnson and S. H. Gellman, Methods Enzymol., 2013, 523, 407–429 CAS.
- D. F. Hook, P. Bindschädler, Y. R. Mahajan, R. Šebesta, P. Kast and D. Seebach, Chem. Biodiversity, 2005, 2, 591–632 CrossRef CAS PubMed.
- D. L. Steer, R. A. Lew, P. Perlmutter, A. I. Smith and M.-I. Aguilar, Lett. Pept. Sci., 2001, 8, 241–246 CrossRef CAS.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS PubMed.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751–14756 CrossRef CAS PubMed.
- J. W. Checco, E. F. Lee, M. Evangelista, N. J. Sleebs, K. Rogers, A. Pettikiriarachchi, N. J. Kershaw, G. A. Eddinger, D. G. Belair, J. L. Wilson, C. H. Eller, R. T. Raines, W. L. Murphy, B. J. Smith, S. H. Gellman and W. D. Fairlie, J. Am. Chem. Soc., 2015, 137, 11365–11375 CrossRef CAS PubMed.
- J. L. Price, W. S. Horne and S. H. Gellman, J. Am. Chem. Soc., 2010, 132, 12378–12387 Search PubMed.
- W. S. Horne, J. L. Price and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9151–9156 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071–13072 CrossRef CAS.
- D. H. Chang, M.-R. Lee, N. Wang, D. M. Lynn and S. P. Palecek, ACS Infect. Dis., 2023, 9, 2632–2651 Search PubMed.
- S. Haider, C. Rotstein, D. Horn, M. Laverdiere and N. Azie, Can. J. Infect. Dis. Med. Microbiol., 2014, 25, 308169 Search PubMed.
- P. G. Pappas, M. S. Lionakis, M. C. Arendrup, L. Ostrosky-Zeichner and B. J. Kullberg, Nat. Rev. Dis. Primers, 2018, 4, 1–20 Search PubMed.
- R. E. Lewis, Mayo Clin. Proc., 2011, 86, 805–817 CrossRef CAS PubMed.
- L. E. Cowen, J. B. Anderson and L. M. Kohn, Annu. Rev. Microbiol., 2002, 56, 139–165 CrossRef CAS PubMed.
- L. E. Cowen, FEMS Microbiol. Lett., 2001, 204, 1–7 CrossRef CAS PubMed.
- R. N. Murugan, B. Jacob, M. Ahn, E. Hwang, H. Sohn, H.-N. Park, E. Lee, J.-H. Seo, C. Cheong, K.-Y. Nam, J.-K. Hyun, K.-W. Jeong, Y. Kim, S. Y. Shin and J. K. Bang, PLOS ONE, 2013, 8, e80025 CrossRef PubMed.
- Y. Lyu, Y. Yang, X. Lyu, N. Dong and A. Shan, Sci. Rep., 2016, 6, 27258 CrossRef CAS PubMed.
- Z. Jiang, B. J. Kullberg, H. Van Der Lee, A. I. Vasil, J. D. Hale, C. T. Mant, R. E. W. Hancock, M. L. Vasil, M. G. Netea and R. S. Hodges, Chem. Biol. Drug Des., 2008, 72, 483–495 CrossRef CAS PubMed.
- R. F. Epand, M. A. Schmitt, S. H. Gellman and R. M. Epand, Biochim. Biophys. Acta, 2006, 1758, 1343–1350 CrossRef CAS PubMed.
- A. Hawrani, R. A. Howe, T. R. Walsh and C. E. Dempsey, J. Biol. Chem., 2008, 283, 18636–18645 Search PubMed.
- M. Kodedová, M. Valachovič, Z. Csáky and H. Sychrová, Cell. Microbiol., 2019, 21, e13093 CrossRef PubMed.
- S. Bobone and L. Stella, Adv. Exp. Med. Biol., 2019, 1117, 175–214 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- F. H. Waghu, R. S. Barai, P. Gurung and S. Idicula-Thomas, Nucleic Acids Res., 2016, 44, D1094–D1097 CrossRef CAS PubMed.
- G. Shi, X. Kang, F. Dong, Y. Liu, N. Zhu, Y. Hu, H. Xu, X. Lao and H. Zheng, Nucleic Acids Res., 2022, 50, D488–D496 CrossRef CAS PubMed.
- G. Cordoves-Delgado and C. R. García-Jacas, J. Chem. Inf. Model., 2024, 64, 4310–4321 CrossRef CAS PubMed.
- D. Veltri, U. Kamath and A. Shehu, Bioinformatics, 2018, 34, 2740–2747 CrossRef CAS PubMed.
- H. ElAbd, Y. Bromberg, A. Hoarfrost, T. Lenz, A. Franke and M. Wendorff, BMC Bioinf., 2020, 21, 235 CrossRef CAS PubMed.
- W. Dee, Bioinf. Adv., 2022, 2, vbac021 Search PubMed.
- S. N. Dean and S. A. Walper, ACS Omega, 2020, 5, 20746–20754 CrossRef CAS PubMed.
- M. L. Bileschi, D. Belanger, D. H. Bryant, T. Sanderson, B. Carter, D. Sculley, A. Bateman, M. A. DePristo and L. J. Colwell, Nat. Biotechnol., 2022, 40, 932–937 CrossRef CAS PubMed.
- N. S. Detlefsen, S. Hauberg and W. Boomsma, Nat. Commun., 2022, 13, 1914 CrossRef CAS PubMed.
- M. Krynski and M. Rossi, NPJ Comput. Mater., 2021, 7, 169 Search PubMed.
- P. A. Romero, A. Krause and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E193–E201 Search PubMed.
- J. Parkinson and W. Wang, J. Chem. Inf. Model., 2023, 63, 4589–4601 Search PubMed.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- T. Vornholt, M. Mutný, G. W. Schmidt, C. Schellhaas, R. Tachibana, S. Panke, T. R. Ward, A. Krause and M. Jeschek, ACS Cent. Sci., 2024, 10, 1357–1370 CrossRef CAS PubMed.
- A. S. Kelkar, B. C. Dallin and R. C. Van Lehn, J. Chem. Phys., 2022, 156, 024701 CrossRef CAS PubMed.
- Y. Khalak, G. Tresadern, D. F. Hahn, B. L. de Groot and V. Gapsys, J. Chem. Theory Comput., 2022, 18, 6259–6270 CrossRef CAS PubMed.
- J. L. Price, W. S. Horne and S. H. Gellman, J. Am. Chem. Soc., 2010, 132, 12378–12387 CrossRef CAS PubMed.
- E. A. Porter, X. Wang, H.-S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS PubMed.
- M. Szefczyk, K. Ożga, M. Drewniak-Świtalska, E. Rudzińska-Szostak, R. Hołubowicz, A. Ożyhar and Ł. Berlicki, RSC Adv., 2022, 12, 4640–4647 RSC.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- RDKit: Open-Source Cheminformatics, Available from https://www.rdkit.org Search PubMed.
- W. S. Horne, M. D. Boersma, M. A. Windsor and S. H. Gellman, Angew. Chem., Int. Ed. Engl., 2008, 47, 2853–2856 CrossRef CAS PubMed.
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2004, 126, 6848–6849 CrossRef CAS PubMed.
- D. Migoń, M. Jaśkiewicz, D. Neubauer, M. Bauer, E. Sikorska, E. Kamysz and W. Kamysz, Probiotics Antimicrob. Proteins, 2019, 11, 1042–1054 CrossRef PubMed.
- S. Soufian and L. Hassani, Pak. J. Biol. Sci., 2011, 14, 729–735 CrossRef CAS PubMed.
- D. S. Radchenko, S. Kattge, S. Kara, A. S. Ulrich and S. Afonin, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2019–2027 CrossRef CAS PubMed.
- M. Rajabi, B. Ericksen, X. Wu, E. de Leeuw, L. Zhao, M. Pazgier and W. Lu, J. Biol. Chem., 2012, 287, 21615–21627 CrossRef CAS PubMed.
- G. von Heijne, J. Mol. Biol., 1991, 218, 499–503 CrossRef CAS PubMed.
- J. Y. Suh, Y. T. Lee, C. B. Park, K. H. Lee, S. C. Kim and B. S. Choi, Eur. J. Biochem., 1999, 266, 665–674 CrossRef CAS PubMed.
- R. d. O. Dias and O. L. Franco, Peptides, 2015, 72, 64–72 CrossRef CAS PubMed.
- M. A. Hitchner, L. E. Santiago-Ortiz, M. R. Necelis, D. J. Shirley, T. J. Palmer, K. E. Tarnawsky, T. D. Vaden and G. A. Caputo, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 182984 CrossRef CAS PubMed.
- K. Nesmerak, A. Toropov and I. Yildiz, Front. Biosci., 2022, 27(4), 112 CrossRef PubMed.
- M. Yasir, A. M. Karim, S. K. Malik, A. A. Bajaffer and E. I. Azhar, Saudi J. Biol. Sci., 2022, 29, 3687–3693 CrossRef CAS PubMed.
- A. Szabóová, O. Kuželka and F. Železný, IEEE International Conference on Bioinformatics and Biomedicine Workshops, 2012 Search PubMed.
- N. Raman, M. R. Lee, D. M. Lynn and S. P. Palecek, Pharmaceuticals, 2015, 8, 483–503 CrossRef CAS PubMed.
- M.-R. Lee, N. Raman, S. H. Gellman, D. M. Lynn and S. P. Palecek, ACS Chem. Biol., 2014, 9, 1613–1621 CrossRef CAS PubMed.
- P. Király, R. Kiss, D. Kovács, A. Ballaj and G. Tóth, Mol. Inf., 2022, 41, 2200072 CrossRef PubMed.
- C. Rücker, G. Rücker and M. Meringer, J. Chem. Inf. Model., 2007, 47, 2345–2357 CrossRef PubMed.
- K. Lin, A. C. W. May and W. R. Taylor, J. Theor. Biol., 2002, 216, 361–365 CrossRef CAS PubMed.
- Y. Akkam, Jordan J. Pharm. Sci., 2016, 9, 51–75 CrossRef.
- Y. Yang, C. Wang, N. Gao, Y. Lyu, L. Zhang, S. Zhang, J. Wang and A. Shan, Front. Microbiol., 2020, 11, 548620 CrossRef PubMed.
- M. Kumar, A. K. Chaturvedi, A. Kavishwar, P. K. Shukla, A. P. Kesarwani and B. Kundu, Int. J. Antimicrob. Agents, 2005, 25, 313–320 CrossRef CAS PubMed.
- S. S. Snyder, J. W. Gleaton, D. Kirui, W. Chen and N. J. Millenbaugh, Int. J. Pept. Res. Ther., 2021, 27, 281–291 CrossRef CAS.
- J. P. Adler-Moore and R. T. Proffitt, in Long Circulating Liposomes: Old Drugs, New Therapeutics, ed. M. C. Woodle and G. Storm, Springer Berlin Heidelberg, Berlin, Heidelberg, 1998, pp. 185–206 Search PubMed.
- R. Liu, X. Chen, S. P. Falk, B. P. Mowery, A. J. Karlsson, B. Weisblum, S. P. Palecek, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 4333–4342 CrossRef CAS PubMed.
- Y.-H. Shin and S. H. Gellman, J. Am. Chem. Soc., 2018, 140, 1394–1400 CrossRef CAS PubMed.
- J. Wang, S. Chou, Z. Yang, Y. Yang, Z. Wang, J. Song, X. Dou and A. Shan, J. Med. Chem., 2018, 61, 3889–3907 CrossRef CAS PubMed.
- S. E. Blondelle and R. A. Houghten, Biochemistry, 1991, 30, 4671–4678 CrossRef CAS PubMed.
- L. H. Kondejewski, M. Jelokhani-Niaraki, S. W. Farmer, B. Lix, C. M. Kay, B. D. Sykes, R. E. Hancock and R. S. Hodges, J. Biol. Chem., 1999, 274, 13181–13192 CrossRef CAS PubMed.
- T. Tachi, R. F. Epand, R. M. Epand and K. Matsuzaki, Biochemistry, 2002, 41, 10723–10731 CrossRef CAS PubMed.
- M. K. Luidens, J. Figge, K. Breese and S. Vajda, Biopolymers, 1996, 39, 367–376 CrossRef CAS PubMed.
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 417–428 CrossRef CAS PubMed.
- Z. Lai, X. Yuan, W. Chen, H. Chen, B. Li, Z. Bi, Y. Lyu and A. Shan, J. Med. Chem., 2024, 67, 10891–10905 CrossRef CAS PubMed.
- A. J. Beevers and A. M. Dixon, Chem. Soc. Rev., 2010, 39, 2146–2157 RSC.
- L. Chen, X. Li, L. Gao and W. Fang, J. Phys. Chem. B, 2015, 119, 850–860 CrossRef CAS PubMed.
- B. Deslouches, S. M. Phadke, V. Lazarevic, M. Cascio, K. Islam, R. C. Montelaro and T. A. Mietzner, Antimicrob. Agents Chemother., 2005, 49, 316–322 CrossRef CAS PubMed.
- A. A. Strömstedt, M. Pasupuleti, A. Schmidtchen and M. Malmsten, Antimicrob. Agents Chemother., 2008, 593–602 Search PubMed.
- Y. Shai and Z. Oren, J. Biol. Chem., 1996, 271, 7305–7308 CrossRef CAS PubMed.
- Y. Shai and Z. Oren, Peptides, 2001, 22, 1629–1641 CrossRef CAS PubMed.
- Y. Huang, L. He, G. Li, N. Zhai, H. Jiang and Y. Chen, Protein Cell, 2014, 5, 631–642 CrossRef CAS PubMed.
- M. A. Cherry, S. K. Higgins, H. Melroy, H. S. Lee and A. Pokorny, J. Phys. Chem. B, 2014, 118, 12462–12470 CrossRef CAS PubMed.
- Z. Jiang, C. T. Mant, M. Vasil and R. S. Hodges, Chem. Biol. Drug Des., 2018, 91, 75–92 CrossRef CAS PubMed.
- Z. Jiang, A. I. Vasil, M. L. Vasil and R. S. Hodges, Pharmaceuticals, 2014, 7, 366–391 CrossRef CAS PubMed.
- Z. Jiang, A. I. Vasil, L. Gera, M. L. Vasil and R. S. Hodges, Chem. Biol. Drug Des., 2011, 77, 225–240 CrossRef CAS PubMed.
- M. Rautenbach, A. M. Troskie and J. A. Vosloo, Biochimie, 2016, 130, 132–145 CrossRef CAS PubMed.
- A. Singh, T. Prasad, K. Kapoor, A. Mandal, M. Roth, R. Welti and R. Prasad, OMICS: J. Integr. Biol., 2010, 14, 665–677 CrossRef CAS PubMed.
- J. D. Richardson and R. C. Van Lehn, J. Phys. Chem. B, 2024, 128, 8737–8752 CrossRef CAS PubMed.
- S. Amrhein, S. A. Oelmeier, F. Dismer and J. Hubbuch, J. Phys. Chem. B, 2014, 118, 1707–1714 CrossRef CAS PubMed.
- Y. You, H. Liu, Y. Zhu and H. Zheng, Amino Acids, 2023, 55, 421–442 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06689h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |