
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective nickel-catalyzed electrochemical reductive conjugate alkenylation of α,β-unsaturated ketones†
Siriphong
Somprasong
 *,
Bin
Wan
and
Syuzanna R.
Harutyunyan
*
*,
Bin
Wan
and
Syuzanna R.
Harutyunyan
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 3, 9747 AG, Groningen, The Netherlands. E-mail: s.harutyunyan@rug.nl; s.somprasong@rug.nl
First published on 20th November 2024
Abstract
Catalytic electrochemical asymmetric catalysis is emerging as a promising strategy for the synthesis of chiral compounds. Herein, we report an asymmetric electrochemical nickel-catalysed reductive conjugate addition of alkenyl bromides/aryl iodides to α,β-unsaturated ketones in an undivided cell, leading to addition products with high yields and excellent enantioselectivities (up to 96% yield and 96% ee).
Introduction
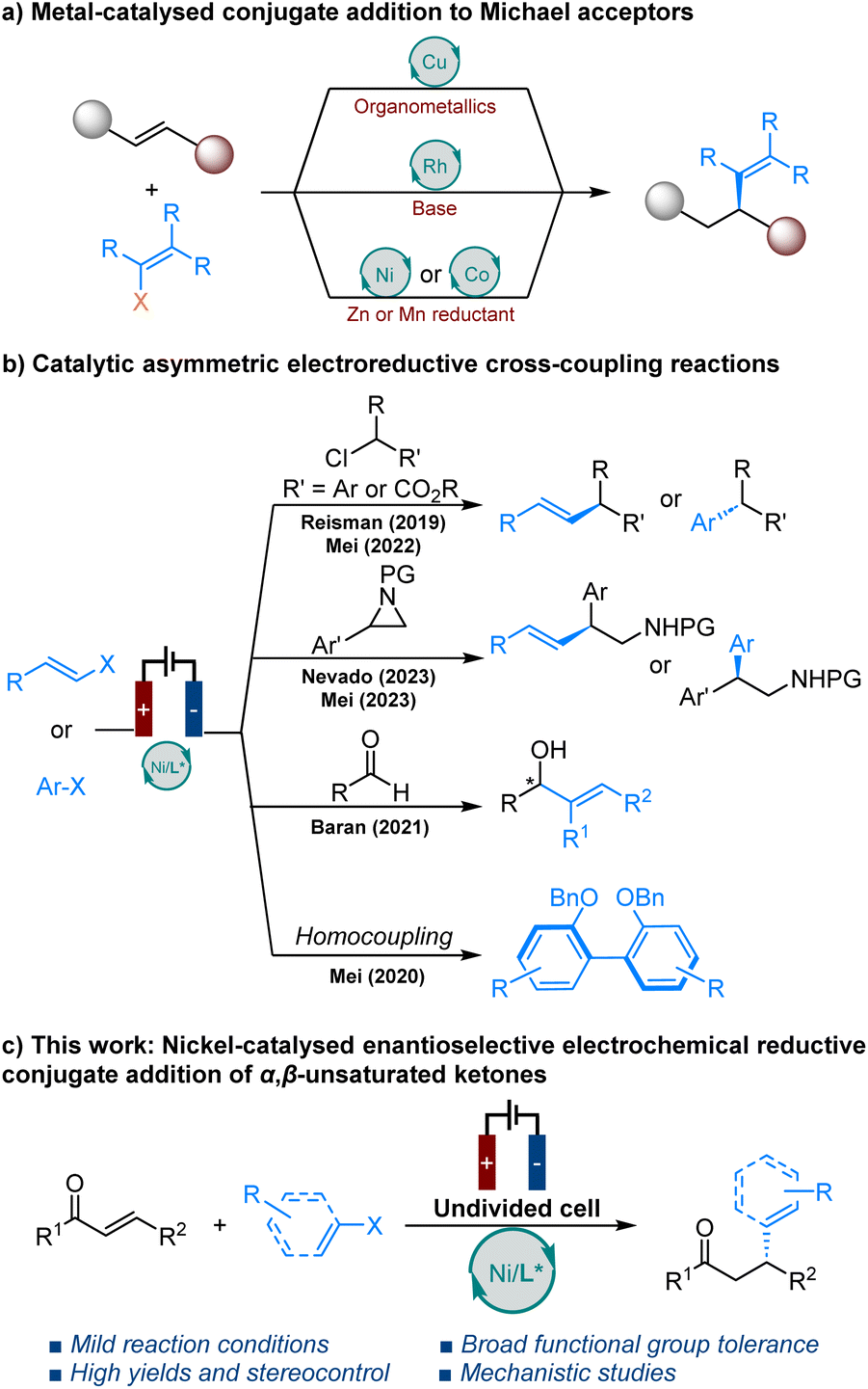
Catalytic enantioselective conjugate addition to α,β-unsaturated carbonyl compounds, forming carbon–carbon bonds, constitutes one of the central pillars of modern-day organic synthesis.1 This important transformation has been extensively developed in the last few decades and widely used in the synthesis of chiral products.2 Among various catalytic systems, chiral copper-based complex catalysts have successfully promoted asymmetric conjugate additions of organometallic reagents to various Michael acceptors.3 However, this approach requires prior synthesis of reactive organometallic reagents from the corresponding organohalides, is sensitive to both air and humidity and presents a narrow scope for both alkenylation and arylation reactions. In contrast, rhodium catalysis has been successfully applied in the asymmetric conjugate alkenylation of alkenylboronic,4 silicon,5 zirconium,6 tin7 and potassium organotrifluoroborate nucleophiles.8 Nevertheless, most of these reactions require harsh conditions and additional steps for the synthesis of alkenyl reagents. Recently, Zhou et al. were the first to report on the nickel- and cobalt-catalysed enantioselective reductive conjugate addition of aryl/alkenyl halides to enones.9–11 This transformation requires the use of super-stoichiometric amounts of zinc or manganese powder and must be carried out under an inert atmosphere in a glove box.With the resurgence of organic electrolysis,12 asymmetric electrocatalysis involving anodic oxidation has attracted considerable attention in recent years.13 However, catalytic asymmetric electrochemical reduction reactions have received comparatively less attention (Scheme 1b). Chiral nickel based catalytic systems for the enantioselective coupling of alkenyl and benzyl halides,14 α-chloroesters,15 aryl aziridines,16,17 and Nozaki–Hiyama–Kishi coupling18 without the need for super-stoichiometric metal powder reductants have been developed by the groups of Reisman, Baran, Mei and Nevado.
 | ||
| Scheme 1 Design of enantioselective electrochemical reductive conjugate addition. | ||
Additionally, Mei et al. reported enantioselective nickel-catalysed electrochemical reductive homocoupling of aryl bromides.19 The recent report by Zhang et al. represents a significant advance in this area,20 but limited examples of alkenylations emphasise the current challenges faced by this chemistry. Electroreductive alkenylations remain challenging due to the potential for side reactions, such as electrochemical difunctionalisation of alkenes, due to the presence of double bonds in both the starting materials and the resulting alkene products.21 Herein, we present enantioselective nickel-catalysed electrochemical conjugate alkenylation of α,β-unsaturated ketones with alkenyl bromides/aryl iodides (Scheme 1c).
Results and discussion
We started our investigation (Table 1) by exploring the asymmetric electrochemical reductive conjugated addition of trans-chalcone 1a with 2-bromopropene 2a in the presence of a catalyst derived from the combination of a nickel salt and Quinox-L1 in an undivided electrochemical cell (IKA Electrasyn 2.0). After systematic evaluation of the reaction parameters (see the ESI†), we were delighted to find that the desired product 3a could be obtained in 72% yield and 96% ee (entry 1) when employing zinc as the anode and reticulated vitreous carbon (RVC) as the cathode in the presence of 5 mol% NiBr2·DME, 6 mol% chiral quinoline–oxazoline (Quinox) ligand L1, 1.5 equiv. of tBuOH and 1.0 equiv. of nBu4NBF4 in DMSO/DMF as solvent under 10 mA constant current electrolysis. Further investigation of different chiral quinoline–oxazoline ligands revealed that Quinox-L2 with a bulky tert-butyl substituent allows for a similar outcome, while with isopropyl (L3) and phenyl (L4) substituents the corresponding product was obtained with a decreased yield and enantiopurity. Other chiral ligands such as pyridine-oxazoline (L5), phosphine-oxazoline (L6) and bis-oxazolines (L7 and L8) all afforded the addition product 3a with lower yields.|
|
|||
|---|---|---|---|
| Entry | Deviation from reaction conditions | Yieldb (%) | eec (%) |
a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.4 mmol, 2.0 equiv.), NiBr2 DME (5 mol%), chiral ligand L* (6 mol%), additive (0.3 mmol, 1.5 equiv.), electrolyte (0.2 mmol, 1.0 equiv.) in DMSO/DMF (0.1 M, 2 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under a N2 atmosphere and 10 mA constant current in an undivided cell at room temperature.
b The yield of 3a was determined from the H NMR spectra of the reaction crude. Isolated yield is reported in parentheses.
c Enantiomeric excess (ee) was determined by SFC with a chiral stationary phase.
d In this case 1a (0.4 mmol, 1.0 equiv.) and 2a (0.8 mmol, 2.0 equiv.) were used. :1) under a N2 atmosphere and 10 mA constant current in an undivided cell at room temperature.
b The yield of 3a was determined from the H NMR spectra of the reaction crude. Isolated yield is reported in parentheses.
c Enantiomeric excess (ee) was determined by SFC with a chiral stationary phase.
d In this case 1a (0.4 mmol, 1.0 equiv.) and 2a (0.8 mmol, 2.0 equiv.) were used.
|
|||
| 1 | None | 72 | 96 |
| 2 | 8 h instead of 6 h | 41 | 95 |
| 3 | 4 h instead of 6 h | 61 | 94 |
| 4 | NaBr instead of nBu4NBF4, 4 h | 12 | 94 |
| 5 | MgBr2 instead of nBu4NBF4, 4 h | 17 | 92 |
| 6 | n Bu4NBr instead of nBu4NBF4, 4 h | 12 | 94 |
| 7 | n Bu4NPF6 instead of nBu4NBF4, 4 h | 55 | 95 |
| 8 | H2O instead of tBuOH | 71 | 94 |
| 9 | EtOH instead of tBuOH | 40 | 95 |
| 10 | iPrOH instead of tBuOH | 74 | 96 |
| 11 | iPrOH instead of tBuOH, 0.06 M | 49 | 95 |
| 12 | i PrOH instead of t BuOH, 0.2 M | 75 (74) | 96 |

To improve the results further, we examined other reaction parameters. When the reaction was extended beyond 6 hours or stopped before 6 hours, lower yields were obtained (entries 2 and 3). The use of different supporting electrolytes such as NaBr, MgBr2, nBu4NBr, or nBu4NPF6 also proved detrimental to the reaction outcome (entries 4–7). At this point, we investigated the effect of additives. While reactions with H2O and EtOH resulted in a reduced yield of 3a (entries 8 and 9), iPrOH was found to be the most suitable additive for this reaction, yielding 74% of 3a with 96% ee (entry 10). Reducing the concentration to 0.06 M did not improve the reaction results (entry 11). A slight increase in yield was achieved when the reaction was performed on a 0.4 mmol scale with a concentration of 0.2 M (entry 12).
Based on these results, we adopted the following optimised reaction conditions for further substrate scope studies: NiBr2·DME (5 mol%) and Quinox-L1 (6 mol%) as chiral nickel catalysts, with iPrOH (1.5 equiv.) as the additive and nBu4NBF4 as the electrolyte in a DMSO/DMF (0.2 M) solvent mixture. The reaction was carried out under a N2 atmosphere using a zinc anode and an RVC cathode in an undivided cell with 10 mA constant current electrolysis for 6 hours at room temperature.
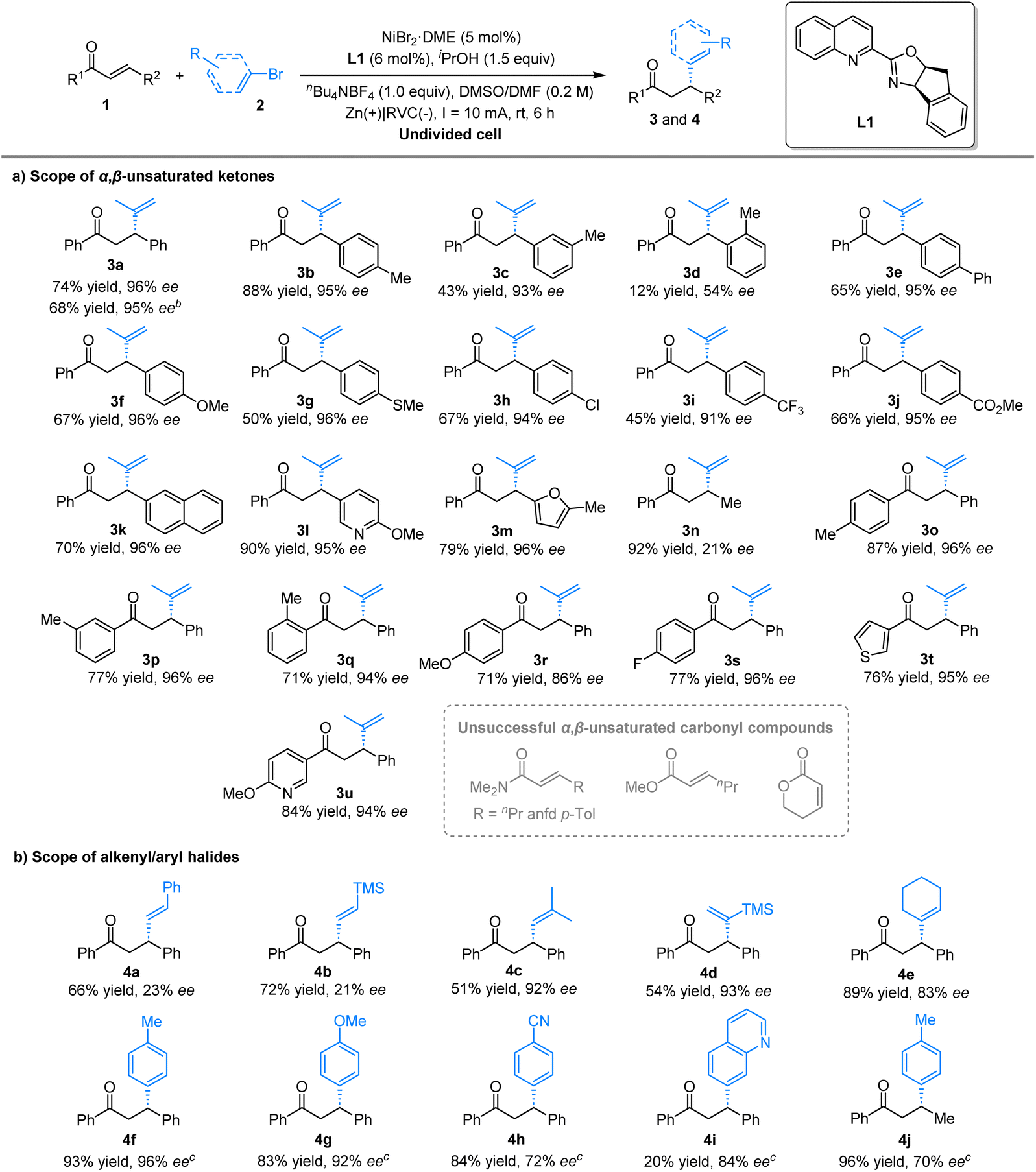
With the optimised reaction conditions in hand, we explored the scope of this transformation with respect to α,β-unsaturated ketone substrates (Table 2(a)). First, the reaction was successfully scaled up to 1.2 mmol, yielding product 3a in 68% yield with 95% ee. A wide range of aryl, heteroaryl, and alkyl substituents at the β-position of chalcones were well tolerated. Methyl substituents at the meta or para positions were compatible, yielding products 3b and 3c in 88% and 43% yields, respectively, hardly affecting the enantioselectivity. However, using the aryl moiety with an ortho-methyl substituent resulted in the corresponding product 3d with a lower yield and enantiopurity (12% yield and 54% ee). In contrast, this transformation exhibited minimal sensitivity to the electronic nature of substituents on the aromatic ring.
|
a Reaction conditions: 1a (0.4 mmol, 1.0 equiv.), alkenyl bromides 2 (0.8 mmol, 2.0 equiv.), NiBr2·DME (5 mol%), chiral ligand L1 (6 mol%), iPrOH (0.6 mmol, 1.5 equiv.), nBu4NBF4 (0.4 mmol, 1.0 equiv.) in DMSO/DMF (0.2 M, 2 mL, 1:1) under a N2 atmosphere and 10 mA constant current in an undivided cell at room temperature for 6 h.
b 1.2 mmol scale.
c Aryl iodides were used. Reported yields correspond to the isolated yields. For each compound, ee values were determined by SFC on a chiral stationary phase.
|
|---|
|
A variety of functional groups, including phenyl (3e), electron-donating (3f and 3g), halogen (3h), and electron-withdrawing (3i and 3j) groups, were well tolerated, delivering the desired products in moderate to good yields (45–67%) with excellent enantiomeric excess (91–96% ee). Moreover, the method accommodated more sterically hindered substrates, such as 2-naphthyl-substituted ketone, which afforded product 3k in 70% yield with 96% ee. The method also proved effective for enones containing heteroaromatic groups, such as pyridinyl and furfuryl, yielding products 3l and 3m in high yields (79–90%) with excellent enantioselectivities (96% ee). In particular, the use of trans-crotonophenone gave 3n in an excellent yield of 92%, although with low enantiopurity (21% ee). The position of substituents on the phenyl ring α relative to the carbonyl moiety—whether ortho, meta, or para—had little impact on the outcome, resulting in products 3o–q with high yields and excellent enantioselectivities. A variety of aryl groups containing electron-donating and -withdrawing substituents, such as methoxy (3r) and fluoro (3s), as well as heteroaryl substituents like thiophen (3t) and 2-methoxypyridine (3u), were well accommodated. However, under the standard conditions, α,β-unsaturated carboxamides and esters did not yield the corresponding alkenylated products.
We then examined the scope of alkenyl bromides using this electrochemical protocol (Table 2(b)). Both β-bromostyrene and β-(trimethylsilyl)vinyl bromide afforded products 4a and 4b in good yields (66–72%), albeit with low enantioselectivities (21–23% ee). However, an alkenyl bromide bearing a β-substituent provided product 4c in moderate yield with excellent enantiopurity. Additionally, an α-substituted alkene reacted smoothly, yielding product 4d in 54% yield with 93% ee. Cyclohexenyl bromide gave product 4e in 89% yield with 83% ee, while 4-iodotoluene successfully yielded the arylated product 4f with excellent yield and enantioselectivity (93% yield and 96% ee). The reaction also accommodated both electron-donating and electron-withdrawing groups, yielding products 4g and 4h in good yields (83–84%) with moderate to excellent enantioselectivity (72–92% ee). Furthermore, N-heteroaryl iodides yielded product 4i in low yields but with good enantioselectivity. In contrast, the less sterically hindered product 4j was obtained with lower enantiopurity.
To gain further insight into the asymmetric nickel-catalysed electrochemical reductive conjugate addition, a series of control experiments were performed to verify the necessity of each reaction component (Fig. 1a). The process was completely suppressed in the absence of the nickel-ligand complex, ligand, or the electric current source, demonstrating that both the nickel-ligand catalyst and the electrical current were necessary for this transformation with no contribution from a competitive racemic background pathway. In addition, a low yield was obtained in the absence of alcohol and the deuterated product 3a was observed when using deuterated iPrOD as the additive (Fig. 1b), suggesting that alcohol is crucial for hydrolysing nickel enolates and facilitating product formation after reductive elimination. Next, we conducted cyclic voltammetry experiments (Fig. 1c). The mixture of NiBr2·DME and L1 in a ratio of 1:1 exhibits three reduction peaks (−1.13 V, −1.50 V and −1.72 V vs. Ag/Ag+), corresponding to the successive formation of Ni(I), Ni(0), and [Ni(0)-L1]˙− species.22 Ni(0) can undergo oxidative addition with alkenyl bromide. The cyclic voltammetry of NiBr2-L1 was carried out in the presence of 1.0 equiv. of 2-bromopropene 2a. New reductive peaks and the reduction of the oxidation peak (−0.68 V vs. Ag/Ag+) suggest that oxidative addition of alkenyl bromide to Ni(0) occurred readily.
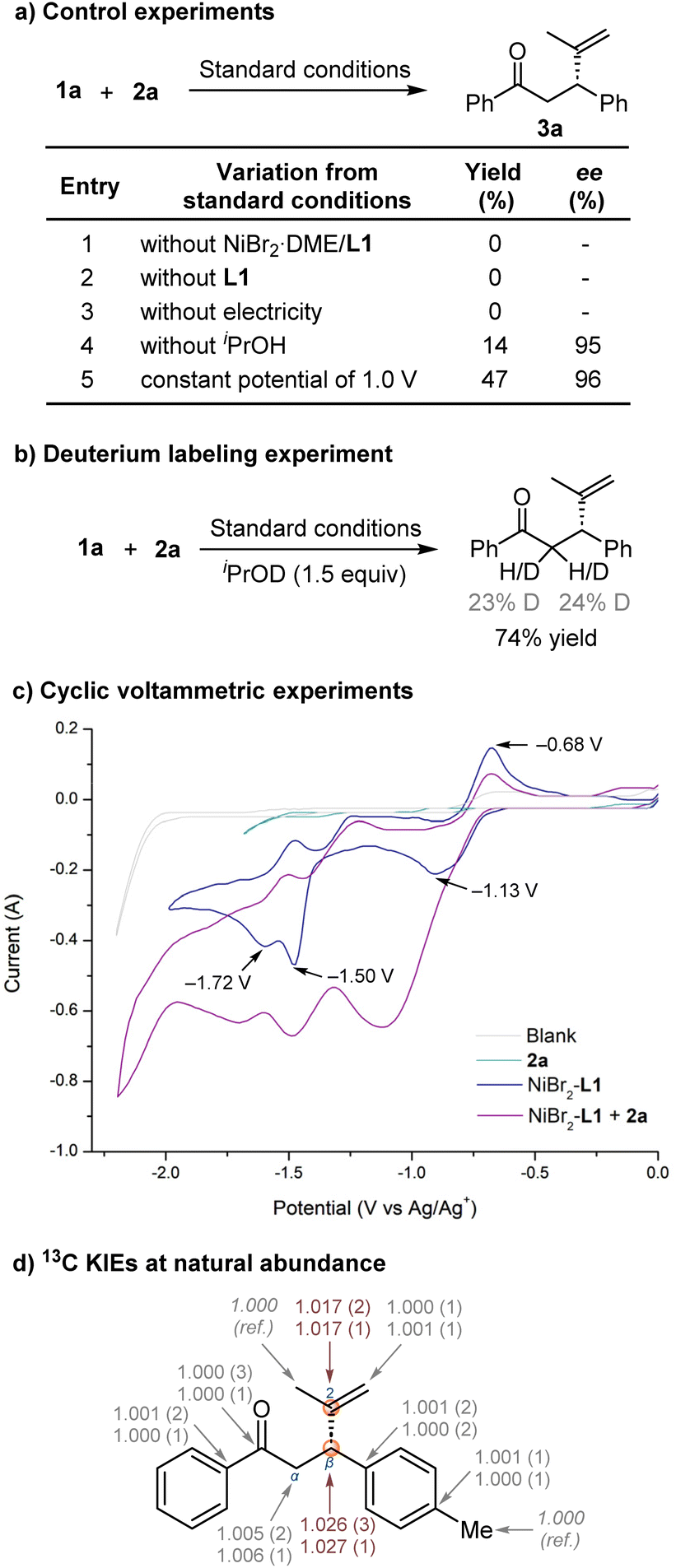 | ||
| Fig. 1 Mechanistic proposal for asymmetric nickel-catalysed electrochemical reductive conjugate alkenylation. (a) Control experiments. (b) Deuterium labelling experiment. (c) Cyclic voltammetry studies of 2a, NiBr2-L1, and the mixture of 2a and NiBr2-L1. (d) Experimental 13C KIEs at natural abundance for the conjugate addition of trans-4-methylchalcone 1b with 2a from product analysis. Two experiments were carried out yielding up to 19 and 30% yields of the product 2b to determine the KIEs at dihydrochalcone and propenyl moieties. The numbers in parentheses represent the standard deviation of the last digit as determined from seven independent measurements. | ||
Next, we investigated intramolecular 13C isotope effects by product analysis (Fig. 1d).23 Determining KIEs can reveal major bonding changes in the rate-limiting step of a reaction.24 We used high-precision NMR measurements of 13C KIEs at natural isotopic abundance, developed by Singleton.25 We conducted two independent reactions of trans-4-methylchalcone 1b and 2a under optimised reaction conditions on a 5 mmol scale and stopped the reaction at low conversions to the product 3b (19 and 30%). The product 3b was isolated, and its 13C isotopic composition was compared to those of unreacted samples of 1b and 2a.
From changes in relative isotopic composition and fractional conversion, we determined 13C KIEs (see details in the ESI†). We observed a primary KIE of ∼2.7% at Cβ of the dihydrochalcone moiety and ∼1.7% at C2 of the propenyl moiety, suggesting that Cβ and C2 are involved in the rate-determining step in the catalytic process. In contrast, all other carbons in the molecule exhibited near-unity KIEs, indicating their lack of involvement in the rate-determining step. The observed KIEs on both carbons strongly support that the migration of the alkenyl group from organonickel species to the electrophilic site Cβ of α,β-unsaturated ketones is the rate-determining step of the catalytic cycle.
Based on literature reports9,10 and our studies, we suggest the plausible catalytic cycle (Scheme 2). Asymmetric electrochemical catalysis is initiated by the cathodic reduction of the LnNi(II)X2 precatalyst to form the active LnNi(0) catalyst that subsequently undergoes oxidative addition with an alkenyl halide leading to LnNi(II)RBr. The subsequent cathodic reduction produces LnNi(I)R, which binds the α,β-unsaturated ketone. Migratory insertion leads to the formation of a nickel O-enolate, likely in the rate-determining step. Subsequent hydrolysis by alcohol releases the desired product and regenerates LnNi(I) species. Finally, the cathodic reduction of LnNi(I) (OR) or LnNi(I)Br completes the catalytic cycle.
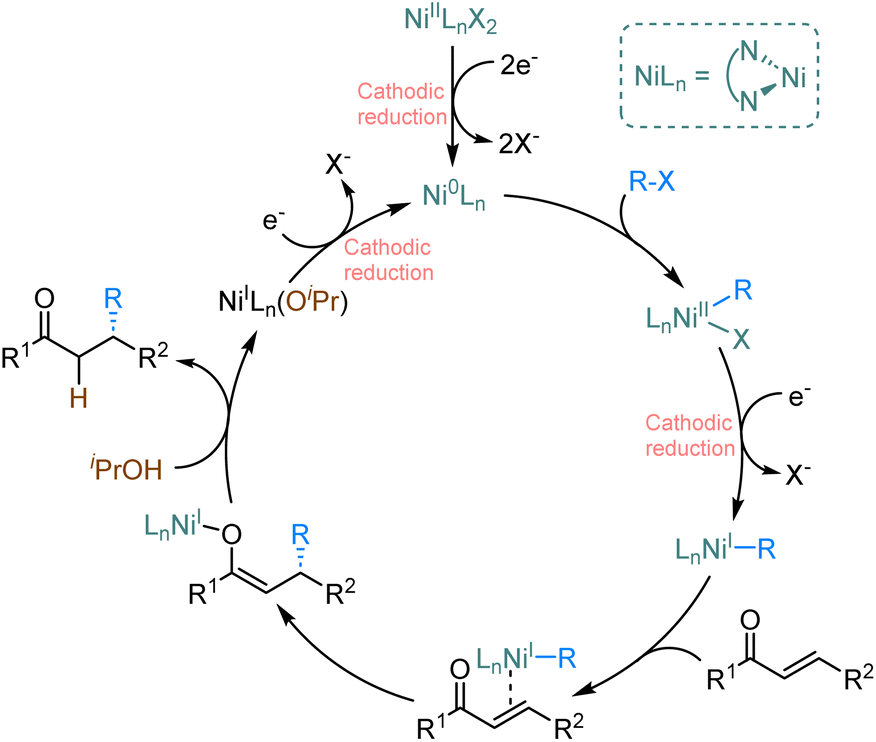 | ||
| Scheme 2 Proposed reaction mechanism for the enantioselective nickel-catalysed electrochemical reductive conjugate addition. | ||
Conclusions
In conclusion, we have developed asymmetric nickel-catalysed conjugate addition of alkenyl bromides and aryl iodides to α,β-unsaturated ketones in an undivided cell by merging electrochemistry and chiral nickel catalysis, affording a wide range of α,β-unsaturated ketones under mild reaction conditions. Additional investigations indicated that alkenyl halides get activated through oxidative addition to Ni(0) species and the rate-determining step of the transformation is likely reductive elimination that leads to the enolate product.Data availability
Experimental and analytical data supporting this article are available in the ESI.†Author contributions
Siriphong Somprasong designed the project, optimised the reaction conditions, investigated the substrate scope, analysed the data, performed the mechanistic experiments and wrote the draft of the manuscript. Bin Wan expanded the substrate scope and analysed the data. Syuzanna R. Harutyunyan supervised the project and edited the manuscript. All the authors contributed to and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (S. R. H. grant no. 773264, LACOPAROM) and to the China Scholarship Council (CSC, to B. W.) are acknowledged. We thank P. van der Meulen and F. de Vries for help with structural elucidations using NMR spectroscopy and R. Sneep for help with the HRMS measurements.Notes and references
- (a) P. Perlmutter, Conjugate Addition Reactions in Organic Synthesis, Pergamon, Oxford, 1st edn, 1992 Search PubMed; (b) A. Alexakis, N. Krause and S. Woodward, Copper-Catalyzed Asymmetric Synthesis, Wiley-VCH, Weinheim, Germany, 1st edn, 2014 CrossRef; (c) G. P. Howell, Org. Process Res. Dev., 2012, 16, 1258–1272 CrossRef CAS; (d) L. Ge and S. R. Harutyunyan in Catalytic Asymmetric Synthesis, ed. T. Akiyama and I. Ojima, John Wiley & Sons, Inc., 4th edn, 2022, ch. 16, p. 617 Search PubMed.
- (a) R. W. Bates and S. Sridhar, J. Org. Chem., 2008, 73, 8104–8105 CrossRef CAS PubMed; (b) M. K. Brown and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 12904–12906 CrossRef CAS; (c) Y.-J. Chin, S.-Y. Wang and T.-P. Loh, Org. Lett., 2009, 11, 3674–3676 CrossRef CAS PubMed; (d) C. L. Pereira, Y.-H. Chen and F. E. McDonald, J. Am. Chem. Soc., 2009, 131, 6066–6067 CrossRef CAS.
- For selected examples of copper-catalysed asymmetric conjugate additions, see: (a) K.-S. Lee, M. K. Brown, A. W. Hird and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 7182–7184 CrossRef CAS; (b) F. López, A. J. Minnaard and B. L. Feringa, Acc. Chem. Res., 2007, 40, 179–188 CrossRef; (c) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed; (d) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed; (e) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039–1075 RSC; (f) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC; (g) R. M. Maksymowicz, P. M. C. Roth and S. P. Fletcher, Nat. Chem., 2012, 4, 649–654 CrossRef CAS; (h) B. Maciá, Formation of Quaternary Stereocentres by Copper-Catalysed Enantioselective Conjugate Addition Reaction, in Progress in Enantioselective Cu(I)-catalyzed Formation of Stereogenic Centers, ed. S. R. Harutyunyan, Springer International Publishing, Cham, 2016, pp. 41–98 Search PubMed; (i) P. Ortiz, F. Lanza and S. R. Harutyunyan, 1,2-Versus 1,4-Asymmetric Addition of Grignard Reagents to Carbonyl Compounds, in Progress in Enantioselective Cu(I)-catalyzed Formation of Stereogenic Centers, ed. S. R. Harutyunyan, Springer International Publishing, Cham, 2016, pp. 99–134 Search PubMed; (j) A. V. Brethomé, R. S. Paton and S. P. Fletcher, ACS Catal., 2019, 9, 7179–7187 CrossRef; (k) Y. Guo and S. R. Harutyunyan, Beilstein J. Org. Chem., 2020, 16, 1006 CrossRef CAS PubMed.
- (a) Y. Takaya, M. Ogasawara and T. Hayashi, J. Am. Chem. Soc., 1998, 120, 5579–5580 CrossRef CAS; (b) T. Hayashi, T. Senda and M. Ogasawara, J. Am. Chem. Soc., 2000, 122, 10716–10717 CrossRef CAS; (c) P. Mauleón and J. C. Carretero, Chem. Commun., 2005, 4961–4963 RSC; (d) J. L. Zigterman, J. C. S. Woo, S. D. Walker, J. S. Tedrow, C. J. Borths, E. E. Bunel and M. M. Faul, J. Org. Chem., 2007, 72, 8870–8876 CrossRef CAS; (e) N. Tokunaga and T. Hayashi, Adv. Synth. Catal., 2007, 349, 513–516 CrossRef CAS; (f) C. Defieber, H. Grutzmacher and E. M. Carreira, Angew Chem. Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed.
- (a) S. Oi, A. Taira, Y. Honma and Y. Inoue, Org. Lett., 2003, 5, 97–99 CrossRef CAS; (b) Y. Nakao, J. Chen, H. Imanaka, T. Hiyama, Y. Ichikawa, W. Duan, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2007, 29, 9137–9143 CrossRef PubMed; (c) R. Shintani, Y. Ichikawa, T. Hayashi, J. Chen, Y. Nakao and T. Hiyama, Org. Lett., 2007, 9, 4643–4645 CrossRef CAS PubMed.
- (a) S. Oi, T. Sato and Y. Inoue, Tetrahedron Lett., 2004, 45, 5051–5055 CrossRef CAS; (b) K. C. Nicolaou, W. Tang, P. Dagneau and R. Faraoni, Angew Chem. Int. Ed., 2005, 44, 3874–3879 CrossRef CAS PubMed.
- S. J. Mahoney, A. M. Dumas and E. Fillion, Org. Lett., 2009, 11, 5346–5349 CrossRef CAS PubMed.
- (a) M. Pucheault, S. Darses and J.-P. Genêt, Eur. J. Org Chem., 2002, 3552–3557 CrossRef CAS; (b) A. Duursma, J.-G. Boiteau, L. Lefort, J. Boogers, A. H. de Vries, J. G. de Vries, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2004, 69, 8045–8052 CrossRef CAS; (c) T. Cendrineau, J.-P. Genêt and S. Darses, Org. Lett., 2009, 11, 3486–3489 CrossRef; (d) X. Hu, M. Zhuang, Z. Cao and D. Du, Org. Lett., 2009, 11, 4744–4747 CrossRef CAS PubMed.
- L. Zhang, M. Zhao, M. Pu, Z. Ma, J. Zhou, C. Chen, Y.-D. Wu, Y. R. Chi and J. S. Zhou, J. Am. Chem. Soc., 2022, 144, 20249–20257 CrossRef CAS PubMed.
- M. Zhao, L. Zhang and J. S. Zhou, ACS Catal., 2024, 14, 6228–6235 CrossRef CAS.
- M. Zhao, W. Xu, Y.-D. Wu, X. Yang, J. Wang and J. S. Zhou, J. Am. Chem. Soc., 2024, 146, 20477–20493 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS; (b) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS; (c) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (d) J. L. Roeckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef CAS PubMed; (e) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed; (f) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (g) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (h) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC.
- For selected examples of asymmetric electrocatalysis involving anodic oxidation, see: (a) D. Minato, Y. Nagasue, Y. Demizu and O. Onomura, Angew. Chem., Int. Ed., 2008, 47, 9458–9461 CrossRef CAS; (b) K. L. Jensen, P. T. Franke, L. T. Nielsen, K. Daasbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 129–133 CrossRef CAS; (c) N. Fu, L. Song, J. Liu, Y. Shen, J. C. Siu and S. Lin, J. Am. Chem. Soc., 2019, 141, 14480–14485 CrossRef CAS PubMed; (d) Q. Zhang, X. Chang and C. Guo, Angew. Chem., Int. Ed., 2019, 58, 6999–7003 CrossRef CAS; (e) X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Nat. Catal., 2019, 2, 34–40 CrossRef CAS; (f) X. Chang, Q. Zhang and C. Guo, Angew. Chem., Int. Ed., 2020, 132, 12712–12722 CrossRef; (g) U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 13451–13457 CrossRef CAS; (h) X. Chang, J. Zhang, Q. Zhang and C. Guo, Angew. Chem., Int. Ed., 2020, 59, 18500–18504 CrossRef CAS; (i) L. Li, Y. Li, N. Fu, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2020, 59, 14347–14351 CrossRef CAS; (j) Z.-H. Wang, P.-S. Gao, X. Wang, J.-Q. Gao, X.-T. Xu, Z. He, C. Ma and T.-S. Mei, J. Am. Chem. Soc., 2021, 143, 15599–15605 CrossRef CAS PubMed; (k) K. Liang, Q. Zhang and C. Guo, Sci. Adv., 2022, 8, eadd7134 CrossRef CAS PubMed; (l) Q. Zhang, K. Liang and C. Guo, Angew. Chem., Int. Ed., 2022, 61, e202210632 CrossRef CAS; (m) P. Xiong, M. Hemming, S. I. Ivlev and E. Meggers, J. Am. Chem. Soc., 2022, 144, 6964–6971 CrossRef CAS; (n) K. Liang, Q. Zhang and C. Guo, Nat. Synth., 2023, 2, 1184–1193 CrossRef CAS; (o) D. Mazzarella, C. Qi, M. Vanzella, A. Saetorel, G. Pelosi and L. Dell'Amico, Angew. Chem., Int. Ed., 2024, 63, e202401361 CrossRef CAS PubMed; (p) J. Zhang, W. Zhu, Z. Chen, Q. Zhang and C. Guo, J. Am. Chem. Soc., 2024, 146, 1522–1531 CrossRef CAS.
- T. J. DeLano and S. E. Reisman, ACS Catal., 2019, 9, 6751–6754 CrossRef CAS PubMed.
- D. Liu, Z.-R. Liu, Z.-H. Wang, C. Ma, S. Herbert, H. Schirok and T.-S. Mei, Nat. Commun., 2022, 13, 7318 CrossRef CAS PubMed.
- X. Hu, I. Cheng-Sánchez, S. Cuesta-Galisteo and C. Nevado, J. Am. Chem. Soc., 2023, 145, 6270–6279 CrossRef CAS PubMed.
- Y.-Z. Wang, Z.-H. Wang, I. L. Eshel, B. Sun, D. Liu, Y.-C. Gu, A. Milo and T.-S. Mei, Nat. Commun., 2023, 14, 2322 CrossRef CAS PubMed.
- Y. Gao, D. E. Hill, W. Hao, B. J. McNicholas, J. C. Vantourout, R. G. Hadt, S. E. Reisman, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 9478–9488 CrossRef CAS.
- H. Qiu, B. Shuai, Y.-Z. Wang, D. Liu, Y.-G. Chen, P.-S. Gao, H.-X. Ma, S. Chen and T.-S. Mei, J. Am. Chem. Soc., 2020, 142, 9872–9878 CrossRef CAS.
- Z. Ye, W. Ma, X. Zhang, H. Liu and F. Zhang, Adv. Sci., 2024, 2405926 CrossRef CAS PubMed.
- For selected examples of electrochemical difunctionalisation of alkenes, see: (a) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS; (b) C.-Y. Cai and H.-C. Xu, Nat. Commun., 2018, 9, 3551 CrossRef PubMed; (c) P. Xiong, H. Long, J. Song, Y. Wang, J.-F. Li and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS PubMed; (d) P. Xiong, H. Long and H.-C. Xu, Asian J. Org. Chem., 2019, 8, 658–660 CrossRef CAS; (e) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS; (f) S. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef; (g) L. Zhang, G. Zhang, P. Wang, Y. Li and A. Lei, Org. Lett., 2018, 20, 7396–7399 CrossRef CAS; (h) K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef CAS; (i) Z.-J. Wu, S.-R. Li and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 14070–14074 CrossRef CAS; (j) J. C. Siu, J. B. Parry and S. Lin, J. Am. Chem. Soc., 2019, 141, 2825–2831 CrossRef CAS PubMed; (k) N. Fu, Y. Shen, A. R. Allen, L. Song, A. Ozaki and S. Lin, ACS Catal., 2019, 9, 746–754 CrossRef CAS PubMed; (l) M.-J. Luo, B. Liu, Y. Li, M. Hu and J.-H. Li, Adv. Synth. Catal., 2019, 361, 1538–1542 CrossRef CAS; (m) K. Liang, Q. Zhang and C. Guo, Sci. Adv., 2022, 8, eadd7134 CrossRef CAS.
- For selected examples on cyclic voltammetric analysis for NiII(bpy)X2, see: (a) Y. H. Budnikova, J. Perichon, D. G. Yakhvarov, Y. M. Kargin and O. G. Sinyashin, J. Organomet. Chem., 2001, 630, 185–192 CrossRef CAS; (b) D. G. Yakhvarov, E. G. Samieva, D. I. Tazeev and Y. G. Budnikova, Russ. Chem. Bull., 2002, 51, 796–804 CrossRef CAS; (c) A. Klein, Y. H. Budnikova and O. G. Sinyashin, J. Organomet. Chem., 2007, 692, 3156–3166 CrossRef CAS; (d) K. Urgin, R. Barhdadi, S. Condon, E. Leonel, M. Pipelier and V. Blot, Electrochim. Acta, 2010, 55, 4495–4500 CrossRef CAS.
- For selected examples, see: (a) D. E. Frantz, D. A. Singleton and J. P. Snyder, J. Am. Chem. Soc., 1997, 119, 3383–3384 CrossRef CAS; (b) D. E. Frantz and D. A. Singleton, J. Am. Chem. Soc., 2000, 122, 3288–3295 CrossRef CAS; (c) M. J. Vetticatt and D. A. Singleton, Org. Lett., 2012, 14, 2370–2373 CrossRef CAS PubMed; (d) S. Xiang and M. P. Mayer, J. Am. Chem. Soc., 2014, 136, 5832–5835 CrossRef CAS; (e) S. Somprasong, M. Castiñeira Reis and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2023, 62, e2022173 CrossRef; (f) S. Somprasong, M. Castiñeira Reis and S. R. Harutyunyan, ACS Catal., 2024, 14, 13030–13039 CrossRef CAS PubMed.
- H. J. A. Dale, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2021, 143, 21079–21099 CrossRef CAS.
- D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06891b |
| This journal is © The Royal Society of Chemistry 2025 |