
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast synthesis of DNA origami single crystals at room temperature†
Yifan
Yu
a,
Min
Ji
a,
Yong
Wang
a,
Xuehui
Yan
a,
Lizhi
Dai
 a,
Ningning
Ma
a,
Zhaoyu
Zhou
a,
Hang
Xing
b and
Ye
Tian
*a
a,
Ningning
Ma
a,
Zhaoyu
Zhou
a,
Hang
Xing
b and
Ye
Tian
*a
aCollege of Engineering and Applied Sciences, State Key Laboratory of Analytical Chemistry for Life Science, National Laboratory of Solid State Microstructures, Jiangsu Key Laboratory of Artificial Functional Materials, Chemistry and Biomedicine Innovation Center, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210023, China. E-mail: ytian@nju.edu.cn
bInstitute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan Provincial Key Laboratory of Biomacromolecular Chemical Biology, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China
First published on 4th December 2024
Abstract
Structural DNA nanotechnology makes the programmable design and assembly of DNA building blocks into user-defined microstructures feasible. However, the formation and further growth of these microstructures requires slow heat treatment in precise instruments, as otherwise amorphous aggregates result. Here, we used an organic solute, urea, as the catalyst for the crystallization of DNA origami building blocks to achieve the fast synthesis of DNA origami single crystals with a cubic Wulff shape at room temperature. The ordered assemblies can be formed within 4 hours at room temperature, which further grew into cubic microcrystals with an average size of about 5 micrometers within 2 days. Furthermore, the phase diagram provides an inverse logic that allows users to proactively customize the melting temperature (Tm) of crystallization according to the target temperature conditions, rather than requiring de novo design of DNA sequences or painstakingly difficult trial-and-error attempts. On this basis, even under random fluctuating outdoor temperature conditions, DNA origami crystals can still grow and maintain high quality and high yield comparable to those of crystals synthesized in precise instruments, creating a basis for the development of adaptive self-assemblies and the industrialization of functional DNA microstructures.
Introduction
Assembling atoms, molecules, and nanoparticles into two-dimensional (2D) or three-dimensional (3D) arrays in a periodic arrangement can give rise to a series of distinctive collective properties that are not normally achieved in naturally occurring materials.1–8 These artificial structures, which often consist of plentiful components and controllable nanoscale architectures, are of great importance in applications such as optical applications,9–11 mechanical metamaterials,12–14 and catalysis.15,16 Since the last century, the rise of structural DNA nanotechnology has enabled a programmable route for bottom-up self-assembly, allowing the customization of structural and functional properties in artificial crystals at the nanoscale level.17–19 Based on this structural design route, numerous 3D colloidal superlattices with dozens of symmetries have been successfully fabricated,20 some of which exhibit crystal habits resembling those of atomic crystals.21–24 However, in terms of fabrication methods, the synthesis of these 3D DNA crystals is mostly achieved through slow heat treatment, especially for micrometer-scale DNA single crystals with specific habits, which necessitate more rigorous annealing processes, for example, slowing the annealing rate or conducting multiple annealing cycles. These refined heat treatments are typically conducted in advanced temperature-control instruments, and the duration of crystallization process is generally around a week with the objective of promoting crystal growth. To date, a fast technique for producing 3D DNA single crystals directly at room temperature with high yield remains to be developed; such a technique may be helpful to simplify the synthesis process and improve the production capacity of DNA single crystals.In comparison to the use of heat treatment to facilitate the assembly towards thermodynamic stability, the crystallization of DNA building blocks at room temperature presents several challenges that may result in reduced yield and quality of the desired microstructures: (i) the inability of room temperature conditions to drive the ordered assembly of building blocks; (ii) the potential formation of metastable products due to kinetic traps; and (iii) the slower thermal movement rate of building blocks at room temperature, which may hinder assembly efficiency. For DNA-based assemblies, one theoretically feasible approach to tackle these challenges is adjusting the Tm of crystallization to near room temperature through sequence design of sticky ends, which enables the formation of the desired assemblies rather than kinetic by-products at room temperature. However, the Tm of single-stranded DNA is normally not equal to the Tm of crystallization, as the crystallization process often involves the synergistic effects of multiple sticky ends and kinetic behavior such as orientation rearrangement between the DNA building blocks.25,26 Consequently, customizing the Tm of crystallization is challenging, and often requires the repeated de novo design of sticky ends between DNA building blocks and painstakingly difficult trial-and-error attempts during experiments. In addition, in many cases, altering the recognition sequence of sticky ends also introduces negative consequences such as reduction in crystal quality and yield.27–29 Therefore, establishing a more general approach with inverse design logic that allows customization of Tm depending on the target temperature conditions without adjusting the original DNA sequences is necessary, as it would promote efficient crystallization at room temperature or in more common environments, such as fluctuating outdoor conditions. In previous works, researchers have explored the assembly of DNA nanostructures without heat treatments,30 but these DNA nanostructures were often DNA tiles or origami with relatively small molecular weights, and the synthesis conditions were usually isothermal.31 The universal synthesis of larger and more complex DNA crystals has rarely been reported. From an assembly process perspective, the synthesis of micron-sized 3D DNA crystals is different from that of nano-sized DNA origami or tiles. The assembly of the latter is simply driven by the hybridization of single-stranded DNA, while the former also involves the change of orientation between thousands of colloid building blocks and the synergistic effect between multiple sticky ends, which has traditionally been achieved by annealing.
In this study, taking the crystallization of DNA origami building blocks as an example, we propose a urea-mediated method to achieve fast synthesis of high-quality DNA origami single crystals with considerable yield at room temperature. The key to this method is utilizing the competition of hydrogen bonds between urea molecules and DNA base pairs to weaken the binding strength of adjacent DNA origami building blocks, thereby promoting the orientation rearrangement between adjacent building blocks at room temperature rather than kinetic trapping. In addition, due to their stability, the structure of DNA origami was confirmed not to be damaged even under high urea concentrations in a previous work,32 which ensures that the DNA origami building blocks can be efficiently assembled in urea solution at room temperature. By introducing an appropriate concentration of urea into the assembly systems, we successfully achieved high-quality crystallization of DNA origami blocks at room temperature. Under the catalysis of urea, ordered assemblies can be rapidly formed within 4 hours at room temperature, and further grow into cubic microcrystals with an average size of about 5 micrometers within 2 days. The whole crystallization process is completely spontaneous at room temperature, and requires no extra human intervention during the incubation. To explore the effect of urea on the crystallization of the DNA origami building blocks, we plotted a phase diagram that exhibits the evolution of the crystal structure in response to variations in urea concentration. The phase transition temperature between ordered assemblies and the liquid phase was defined as the Tm of crystallization, which exhibited a linear relationship with the urea concentration derived from the phase diagram, providing a robust foundation for inverse customization of Tm based on the target temperature conditions. By employing this inverse design strategy, we further successfully achieved high-quality and high-yield synthesis of DNA origami single crystals not only at room temperature, but also at outdoor temperature (random fluctuating temperature conditions). Our approach for crystallizing DNA origami building blocks at room temperature provides a universal method for the 3D assembly of DNA nanostructures into microcrystals, potentially advancing the functional capabilities and large-scale production of DNA crystals.
Results and discussion
The DNA origami building blocks used in this work are regular-octahedral DNA origami frameworks embedded with a 10 nm gold nanoparticle at the body center (Fig. 1a, left panel and S1†). To enable 3D assembly, four identical DNA sticky ends comprising a block spacer and a complementary recognition sequence region are respectively encoded on the six vertices of two separate building blocks, thereby establishing a binary assembly system (Fig. 1a, right panel). Upon mixing the two types of DNA origami building blocks, they can theoretically connect with each other in a vertex-to-vertex binding mode as shown in Fig. 1b. Notably, although the building blocks possess the ability to interconnect, this does not guarantee that they will form an orderly arrangement in 3D space. A strong binding strength between sticky ends results in the formation of amorphous aggregates, while weak interactions lead to the building blocks remaining in a monodisperse liquid state. Only a moderate binding strength enables the building blocks to adjust their orientations during crystallization, thereby promoting the formation of ordered assemblies (Fig. 1c). In the past, regulating the binding strength between DNA building blocks to a moderate level has typically been achieved through heat treatment,2,22 which weakens the hydrogen bonding between base pairs due to the heat (Fig. 1d, left panel). Conversely, when the DNA origami building blocks are directly subjected for incubation at room temperature, the strong binding strength causes the assemblies to develop completely into amorphous aggregates (Fig. 1d, right panel). Therefore, to weaken the originally strong binding strength at room temperature to a moderate level, we propose the utilization of an organic solute, urea, to compete with hydrogen bonding between base pairs, thereby achieving the dehybridization of sticky ends to avoid their undesired connection. In this way, DNA origami building blocks can nucleate within 4 hours at room temperature, and these ordered assemblies can further grow into cubic microcrystals with an average size of about 5 micrometers within 2 days (Fig. 1e).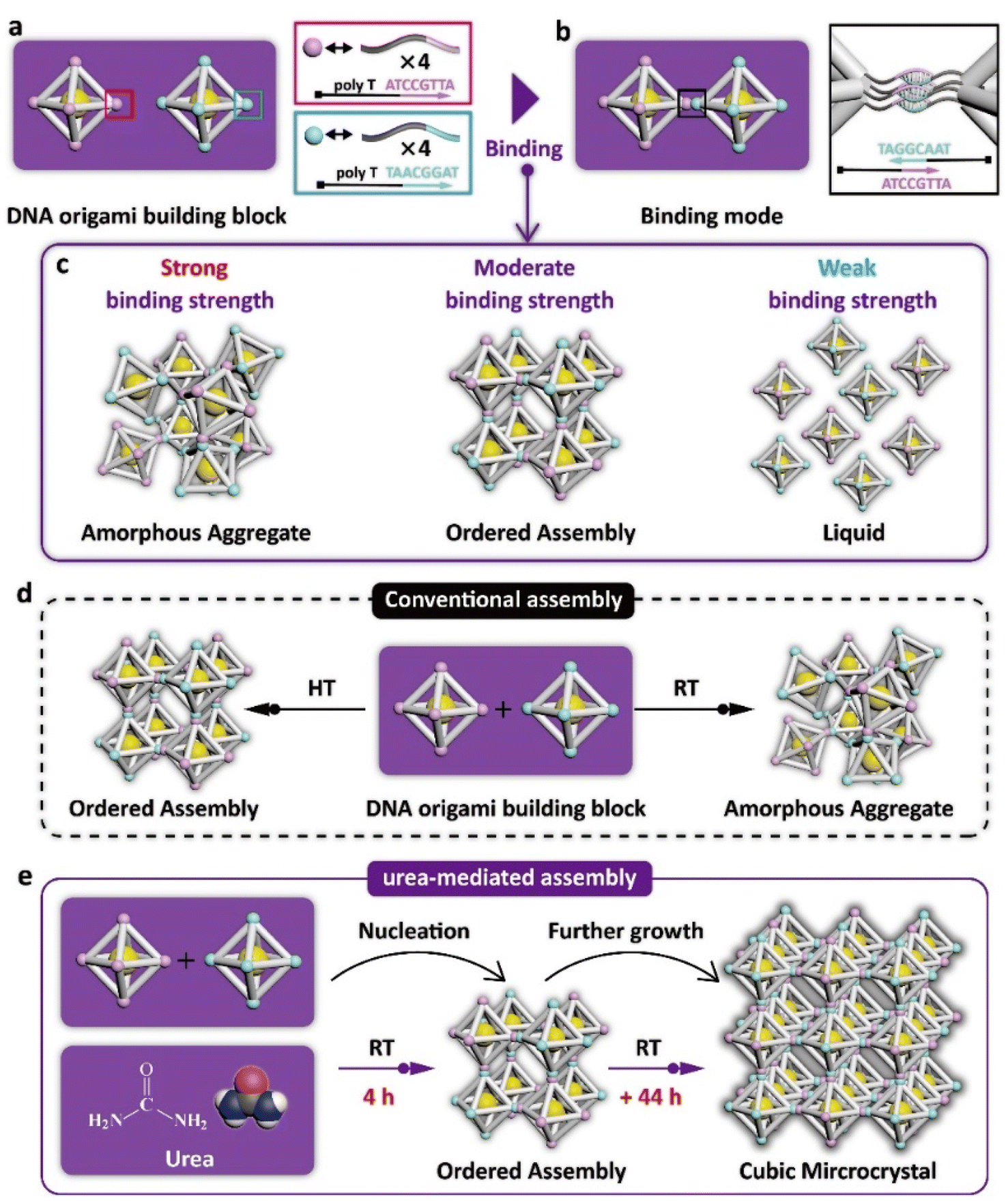 | ||
| Fig. 1 Scheme of the crystallization of DNA origami building blocks at room temperature. (a) Model of the binary regular-octahedral DNA origami building blocks. (b) The vertex-to-vertex binding mode between the adjacent building blocks. (c) Structure properties of the assemblies with strong, moderate, and weak binding strengths, respectively. (d) In conventional assembly, heat treatment (HT) drives the formation of ordered assemblies from DNA origami building blocks, while crystallization at room temperature (RT) leads to amorphous aggregates. (e) In this study, urea-mediated crystallization can form ordered assemblies within 4 hours, which further grow into cubic microcrystals with considerable size within 2 days. | ||
To demonstrate the effectiveness of urea in crystallizing DNA origami components without heat treatment, a comparative analysis of the assembly products of systems with and without urea was first conducted. We first mixed the synthesized regular-octahedral DNA origami building blocks together in equal proportions and adjusted the urea concentration to 0 M (no urea) or 3.8 M. The two samples were then directly placed on the workbench in our laboratory (the room temperature was approximately 24 °C during this experiment) for 48 hours, without performing any other operations during this period (Fig. 2a, top panel and Fig. 2b, top panel). During incubation, an increasing number of red precipitates were generated at the bottom of the tube in both samples. We characterized the micro-morphology and structure properties of these precipitates through confocal laser scanning microscopy (CLSM) and small angle X-ray scattering (SAXS), respectively. For the sample incubated without urea, all the precipitates were observed to be amorphous aggregates under CLSM, preliminarily confirming the disordered assembly (Fig. 2a, bottom left and S2†). Additionally, the 1D SAXS curve derived from the 2D scattering pattern exhibited typical amorphous structure characteristics, that is, a small number of scattering peaks with wide peak width, which suggested that the DNA origami building blocks did not have the ability to crystallize at room temperature (Fig. 2a, bottom right). In comparison, for the urea-mediated assembly, all the precipitates exhibited a cubic micro-morphology of around 5 micrometers rather than amorphous shapes under CLSM, illustrating the high yield and high quality of the crystals (Fig. 2b, bottom left and S3†). Moreover, the sharp and numerous SAXS peaks indicated the ordered arrangement of the DNA origami building blocks inside these crystals (Fig. 2b, bottom right). The experimental scattering peaks matched well with the standard scattering peaks of a simple cubic structure, and the lattice parameter was 58.5 nm, which was consistent with our proposed model (Fig. S4†), confirming that urea-mediated crystallization at room temperature could retain programmable properties.
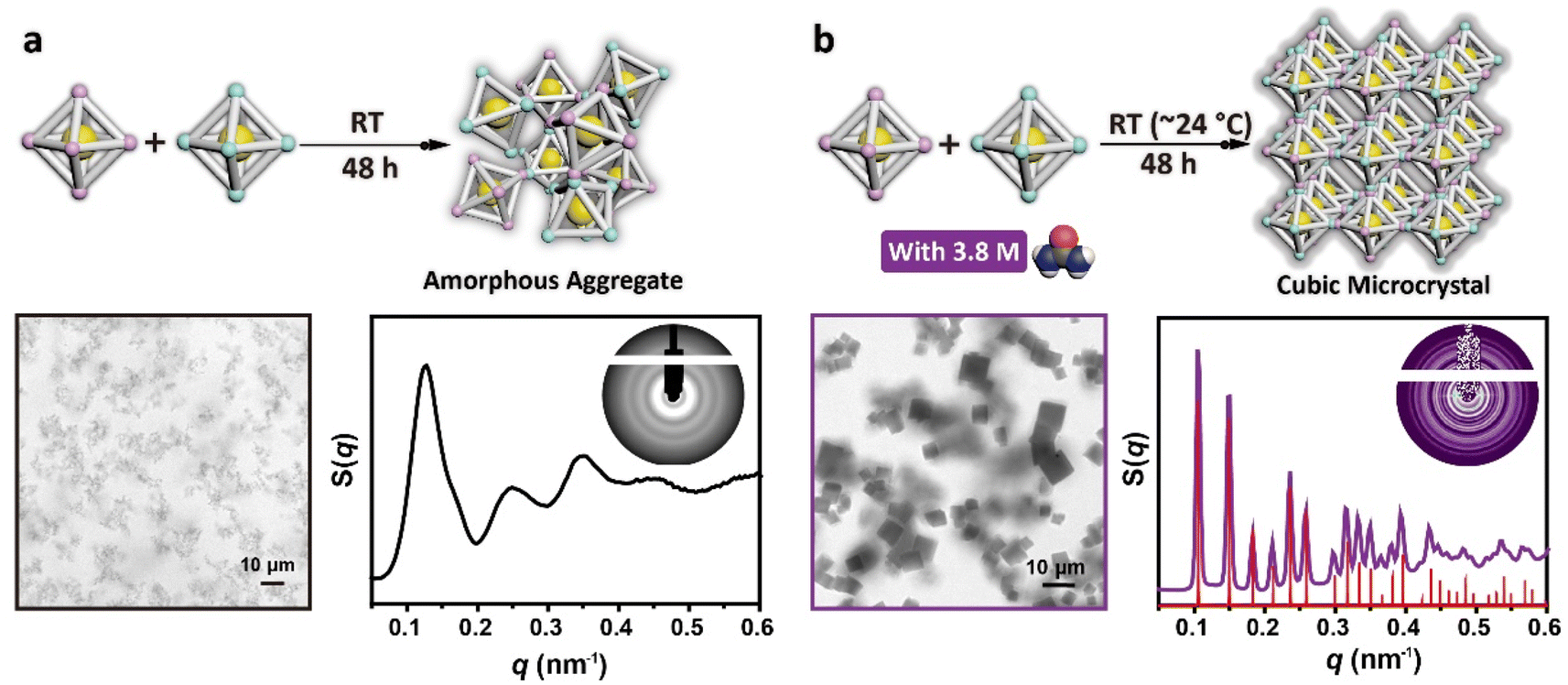 | ||
| Fig. 2 Comparison of the micro-morphologies and inner structures of the products assembled without and with urea at room temperature (∼24 °C). (a) Assembly products incubated at room temperature for 48 hours without urea. A representative CLSM image is shown at the bottom left, and the 1D SAXS curve and 2D SAXS pattern (inset) are shown at the bottom right. (b) Assembly products incubated at room temperature for 48 hours with 3.8 M urea. A representative CLSM image is shown at the bottom left, and the 1D SAXS curve and 2D SAXS pattern (inset) are shown at the bottom right. | ||
Having achieved the synthesis of DNA origami single crystals of comparable size within 48 hours at room temperature, we further investigated their formation process. To determine the start time of the formation of ordered assemblies at room temperature, we utilized SAXS to continuously collect the structure information of assemblies at different time points. We first adjusted the urea concentration in the binary system to 3.5 M, and then placed the capillary in the X-ray path, exposing the sample every 2 hours (the room temperature was measured to be about 25.7 °C in this experimental environment). The change in the 1D SAXS curves of the sample structures with time is shown in Fig. 3a. At 2 hours, the DNA origami building blocks were still discrete, as no scattering peak was observed in the SAXS curves. At 4 hours, obvious characteristic scattering peaks of a simple cubic structure appeared, indicating the onset of the crystallization of the DNA origami building blocks. As time progressed, the number of scattering peaks gradually increased and the original peaks became sharper, confirming the further growth of the DNA origami single crystals. To intuitively reflect the process of crystal growth, we further analysed the development of the crystal size with time by measuring the crystal size under CLSM (Fig. S5†). At 2 hours, no crystals were observed under CLSM; the crystals appeared at 4 hours. We were unable to measure the crystal size at 4 hours or 6 hours because the crystals were too small. At 8 hours, the crystals had grown and exhibited a cubic morphology that could be intuitively observed under CLSM. We counted the sizes of over 70 crystals at subsequent time points, and the increase of crystal size over time confirmed the gradual growth of DNA origami single crystals. During the whole crystallization process, DNA origami building blocks first formed ordered assemblies with simple cubic structures, and these assemblies then further grew into cubic microcrystals in the subsequent incubation time.
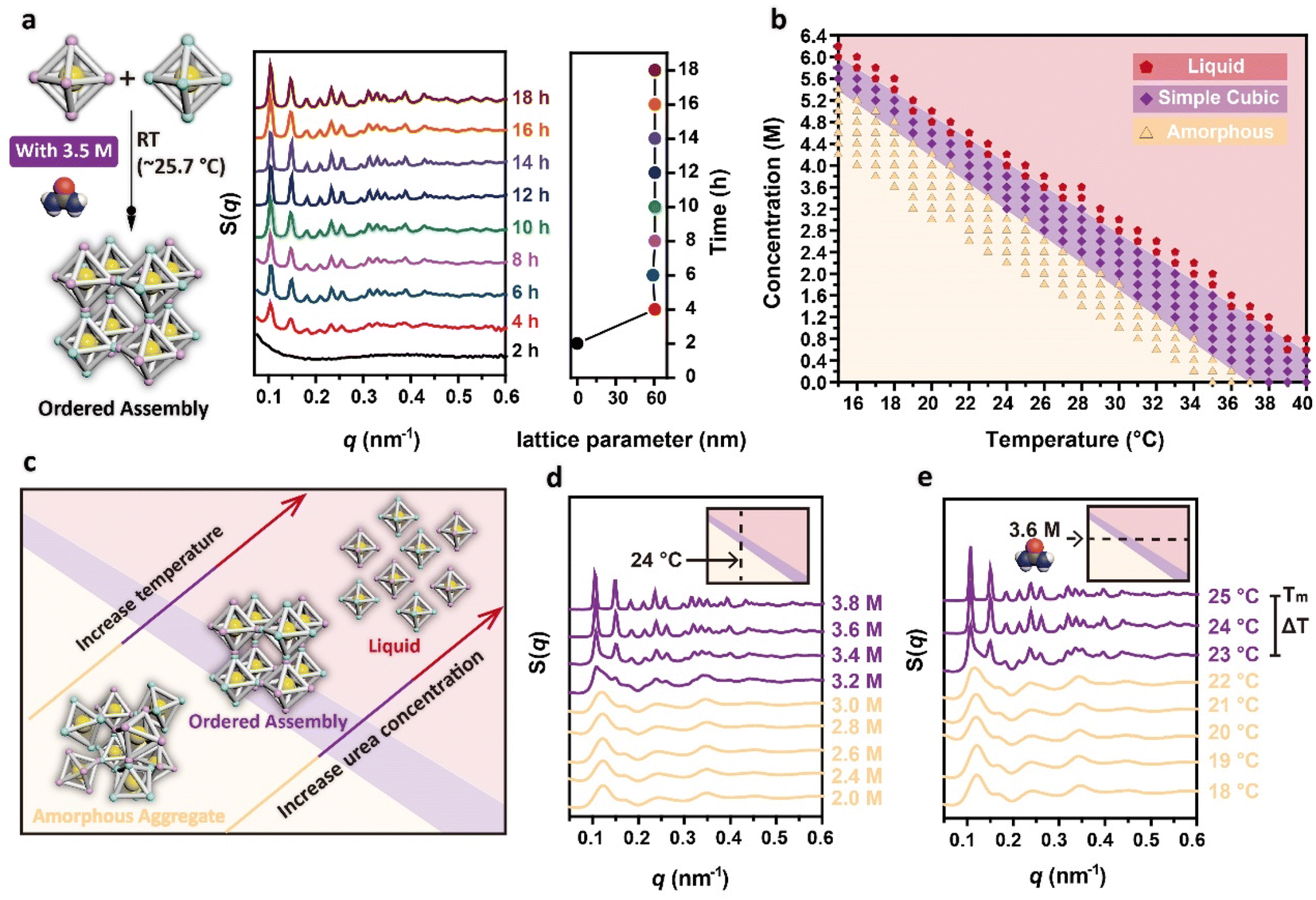 | ||
| Fig. 3 Time-structure transformation trends and phase diagram. (a) 1D SAXS curves of the assemblies at different times under the catalysis of 3.5 M urea at room temperature (∼25.7 °C). Right panel: change in the lattice parameters derived from the SAXS curves over time. (b) Phase diagram of the assemblies after isothermal incubation for 48 hours at a series of fixed temperature and urea concentrations (red pentagons: liquid, purple quadrangles: simple cubic, yellow triangles: amorphous aggregates). (c) Change in the structure of the assemblies with urea concentration and temperature. (d) Change in the structures of the assemblies with urea concentration at 24 °C. (e) Change in the structures of the assemblies with temperature at a urea concentration of 3.6 M. The temperature range for orderly assembly is defined as ΔT, and the transition point from the simple cubic structure to a liquid phase is defined as Tm. | ||
Despite the ability of the DNA origami building blocks to rapidly crystallize within 4 hours at room temperature, the formation of ordered assemblies was not observed when the urea concentrations were changed, as shown in Fig. S6,† indicating that the urea concentration was critical in determining whether the DNA origami building blocks could crystallize at room temperature. Thus, to explore the effect of urea on crystallization and facilitate determination of the required urea concentrations for crystallization at different temperatures, we carried out isothermal incubation for 48 hours using various fixed temperatures and urea concentrations. The structures of all the samples were characterized through SAXS (Fig. S7†), and the corresponding structure types were represented in a phase diagram (Fig. 3b), in which red pentagons represent the liquid phase, purple quadrangles represent the simple cubic phase, and yellow triangles represent disordered aggregates. As either the temperature or the urea concentration was increased, the type of structure obtained evolved from amorphous aggregates to simple cubic structures and finally to a liquid (Fig. 3c). Taking the crystallization of the DNA origami building blocks at 24 °C as an example, as the urea concentration was increased from 2.0 M to 3.8 M, the structure of the assembly products gradually transitioned from amorphous aggregates to simple cubic structures (Fig. 3d). Since the samples with urea concentrations greater than 3.8 M did not precipitate after 48 hours of incubation, we did not characterize them using SAXS, and directly defined them as a liquid phase. The crystallization of the DNA origami building blocks can be achieved in the urea concentration range of 3.2 M to 3.8 M, with the higher urea concentration resulting in more ordered assemblies. The same trend in the structures can also be found in the samples incubated with fixed urea concentrations but varying incubation temperatures, as shown in Fig. 3e. At a urea concentration of 3.6 M, 25 °C was the phase transition point from simple cubic structures to a liquid phase. We defined this temperature point as the Tm of crystallization for the binary system in 3.6 M urea. It should be emphasized that this Tm of crystallization is different from the Tm of double-stranded DNA, due to fact that the crystallization process involves the synergistic effects of multiple sticky ends and the reorientation between building blocks. Additionally, there also existed a temperature interval (23 °C to 25 °C) for crystallization, which was defined as ΔT. Within this ΔT, the structures of the assemblies also exhibited more ordered properties when the incubation temperature was closer to the Tm.
To visually observe the arrangement of the DNA origami building blocks inside the assemblies, we selected a sample in the simple cubic phase region and coated the crystal with a thin layer of silica to observe the details of the binding mode between adjacent DNA origami building blocks using scanning electron microscopy (SEM). As shown in the middle panel of Fig. 4a, the regular-octahedral origami frameworks were arranged regularly with the same orientations in 3D space, and the number of the vertices were maximized for binding (Fig. S8†), illustrating the moderate binding strength that makes the reorientation between adjacent building blocks possible. For the assemblies in the amorphous region, the representative SEM images showed that the amorphous aggregates exhibited irregular orientation of the DNA origami building blocks with many underutilized vertices, confirming the strong binding strength that results in the inability of building blocks to rearrange (Fig. 4a, left panel and S9†). Since the samples in the liquid phase region did not precipitate, we used TEM to observe the state of the DNA origami building blocks. The representative TEM images showed that there was almost no connection between the DNA origami building blocks in the liquid phase, which was also demonstrated by particle size analysis using dynamic light scattering (DLS) (Fig. 4a, right panel and S10†).
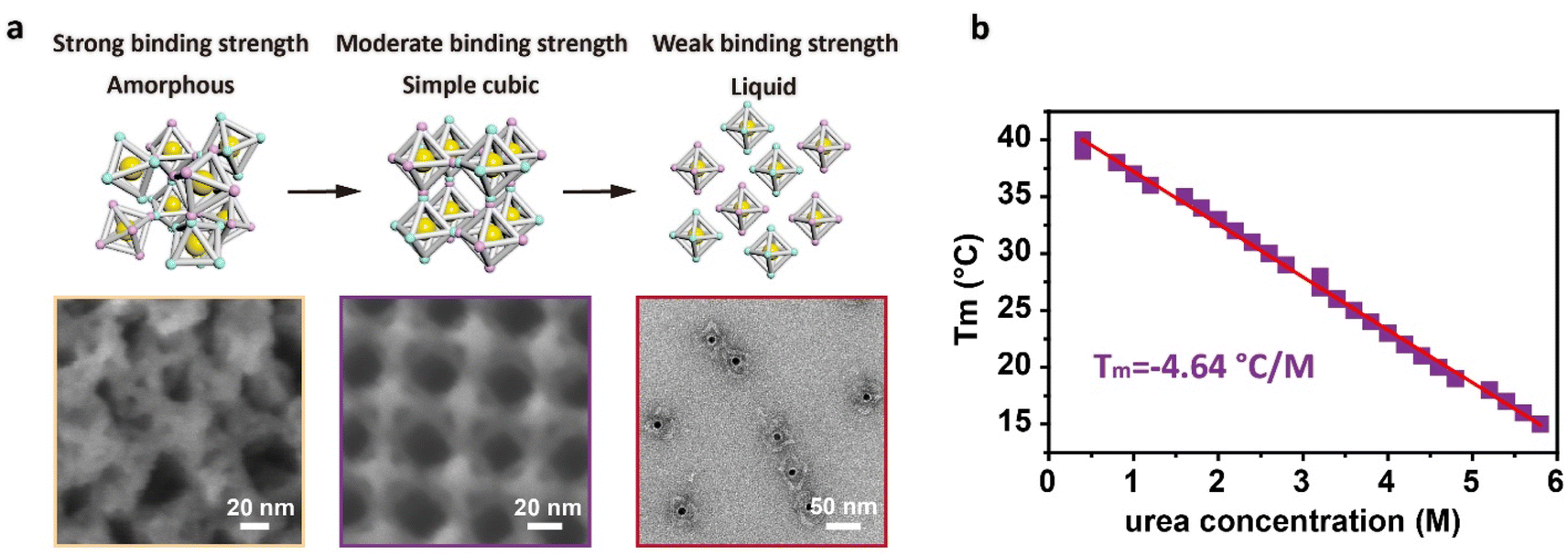 | ||
| Fig. 4 Morphology of the three phases and the variation of Tm with urea concentration. (a) Proposed models of the three phases (top panel) and the corresponding representative SEM and TEM images. (b) Tm of the system at different urea concentration derived from the phase diagram. Red solid line: linear fitting curve with a slope of −4.64 °C M−1. | ||
From the phase diagram, we can also clearly extract the relationship between Tm and urea concentration. The rate of variation of Tm with the urea concentration was −4.64 °C M−1 (Fig. 4b), which was established based on the 3D crystallization of DNA building blocks, rather than simple hybridization between sticky ends. Although the Tm of crystallization involves kinetic factors such as reorientation among the building blocks, it still maintains a linear relationship with the urea concentration. This linear relationship between urea concentration and Tm provides a good foundation for inverse customization of Tm based on the target temperature conditions. Using this linear conversion, the Tm of crystallization can be quickly customized by adding the appropriate amount of urea without requiring de novo design of the sticky ends, conveniently enabling the DNA origami building blocks to crystallize under various temperature conditions. To confirm the universality of this approach, we attempted to crystallize other systems at room temperature. We changed the sequence of the sticky ends for the regular-octahedral DNA origami framework, and crystals with cubic shapes were again formed at room temperature (24 °C) in the presence of 4.0 M urea (Fig. S11†). In addition to the sequence of the sticky ends, we also changed the shape of the DNA origami frameworks to an elongated-octahedral shape. These alterations were also compatible with this room-temperature synthesis approach. Crystals with cuboid habits formed at room temperature (24 °C) in the presence of 3.6 M urea within 48 hours (Fig. S12†). In addition, we further changed the flexible region of the sticky ends to a hairpin DNA conformation for the regular-octahedral DNA origami framework and increased the length of the sequence of the sticky ends to 10 bases along the Z axis. The introduction of a hairpin DNA conformation makes the assembly behavior of the system more complex, and in previous work, the resulting assemblies were confirmed to lack regular morphologies.28 However, even when these complex designs were integrated into the crystallization systems, we were still able to use this approach to crystallize the building blocks into ordered assemblies at room temperature (24 °C) in a solution with a 4.0 M urea concentration (Fig. S13†). It is worth emphasizing that the urea concentration of each system could be directly determined via a linear relationship between the Tm and urea concentration like that shown in Fig. 4b, confirming the universality and stability of our method.
The feasibility of this strategy was further demonstrated by the ability of DNA origami building blocks to crystallize at more ordinary temperatures, such as outdoor temperature. Compared to room temperatures, outdoor temperatures fluctuate unevenly with irregular temperature variation rates. To facilitate the growth of crystals under these temperature conditions, we predicted the suitable urea concentration using the average temperature during the incubation time as Tm, which could make ΔT fully fall into the temperature variation range. Specifically, we first consulted the temperature fluctuations over the next 2 days and determined that the average temperature would be about 29 °C. Then, we calculated a required urea concentration of about 2.8 M based on the fitting curve of Tm. After mixing the binary system in equal proportion and adjusting the urea concentration to 2.8 M, we placed the samples outdoors, without performing any other operations during the incubation period. The recorded temperature variation near the sample is shown in Fig. 5a; as previously hypothesized, the temperature exhibited an uneven fluctuation with irregular temperature variation rates. The control samples incubated without urea demonstrated that the binary system alone lacked the capacity to assemble in an orderly manner under such temperature conditions. Representative CLSM images showed that the precipitates exhibited an amorphous aggregate micro-morphology (Fig. 5b and S14†); additionally, the 1D SAXS curves and inset 2D SAXS pattern shown in Fig. 5d further confirmed the disordered arrangement of the DNA origami building blocks within these aggregates (Fig. 5d, black curves and black pattern). In contrast, the precipitates formed in the 2.8 M urea solution presented a single crystal cubic morphology under CLSM (Fig. 5c and S15†), and the sharp scattering peaks of the 1D SAXS curve confirmed the simple cubic crystal structures, indicating that the DNA origami single crystals could be synthesized outdoors with the help of urea under non-temperature-controlled conditions. Furthermore, high-quality cubic DNA origami single crystals were stably produced under a variety of outdoor temperature conditions, further confirming the effectiveness and generalizability of this inverse design strategy in crystallization (Fig. S16†).
 | ||
| Fig. 5 Quality comparation of crystals incubated at outdoor temperature and simulated outdoor temperature conditions in an instrument. (a) Temperature variation curves of the outdoor temperature over 48 hours. (b) Representative CLSM image of crystals incubated without urea at outdoor temperature. (c) Representative CLSM image of crystals incubated with 2.8 M urea at outdoor temperature. (d) 1D SAXS curves and 2D patterns of the assemblies incubated without urea (black) and with 2.8 M urea (green) at outdoor temperature. (e) Temperature variation curves of the simulated outdoor temperature conditions over 48 hours. (f) Representative CLSM image of crystals incubated without urea under the simulated outdoor temperature conditions. (g) Representative CLSM image of crystals incubated with 2.8 M urea under the simulated outdoor temperature conditions. (h) Histogram of the size distribution of the crystals incubated at outdoor temperature (green) and simulated outdoor temperature (blue) for 48 hours. | ||
The fluctuations of the outdoor temperatures were inhomogeneous; such conditions frequently result in assemblies being trapped by kinetics, thereby leading to a degradation of the crystal quality or yield. Therefore, to identify whether this situation occurred, we repeated this incubation process in a temperature-controlled instrument. In comparison to the outdoor temperature conditions, in the simulated outdoor temperature conditions, the same temperature variation ranges were used, but the temperature change rates were uniform (Fig. 5e). The binary system without urea incubated under this temperature condition still could not achieve orderly assembly. The assemblies observed under CLSM again exhibited the form of amorphous aggregates (Fig. 5f and S17†), demonstrating that the formation of amorphous aggregates in the absence of urea at outdoor temperature was not attributable to the inhomogeneous temperature fluctuations. Rather, the underlying cause was the inability of the DNA origami building blocks to crystallize within this temperature range. Conversely, under the catalysis of 2.8 M urea, the binary systems showed the ability to crystallize and produced micron-sized cubic single crystals in high yield (Fig. 5g and S18†). The size distribution of the crystals synthesized at outdoor temperature and under the simulated outdoor temperature conditions were statistically analyzed, as shown in Fig. 5h. The size distribution and the average size of the crystals were found to be approximately equivalent, suggesting that the urea-mediated crystallization of the DNA origami building blocks could circumvent the kinetic impact of uneven temperature fluctuations without compromising the quality or yield of the crystals.
Conclusions
In this work, we employed urea as a catalyst to facilitate the crystallization process of DNA origami building blocks and achieved the fast synthesis of DNA origami single crystals at room temperature in high yield, overcoming the previous requirements for long-duration and slow annealing in an instrument for the preparation of DNA origami single crystals. Using an appropriate urea concentration, ordered assemblies could be formed within 4 hours at room temperature, and further grew into cubic microcrystals with an average size of about 5 micrometers within 2 days. During the crystallization process, urea effectively regulated the assembly kinetics between adjacent DNA origami building blocks, thus promoting the rearrangement of their orientation at room temperature. As the urea concentration was increased, the assemblies gradually transformed from amorphous aggregates to ordered assemblies and finally to the liquid phase. The temperature of phase transition from ordered assemblies to the liquid phase was defined as Tm and exhibited a linear relationship with the urea concentration, providing a robust foundation for inverse customization of the Tm of crystallization. The change in Tm with increasing urea concentration was −4.64 °C M−1, which was established due to the crystallization behavior of the DNA origami building blocks rather than simple hybridization of single-stranded DNA, and involved the hybridization of multiple DNA sticky ends and orientation rearrangement; thus, it may be applicable for the synthesis of various DNA crystals at room temperature. Based on this urea-mediated strategy, we successfully achieved the crystallization of DNA origami building blocks at outdoor temperatures, with comparable crystal quality to those synthesized under precise temperature-controlled conditions in instruments. Our proposed method for the fabrication of DNA origami crystals at room temperature frees high-quality DNA crystal synthesis from its dependence on slow heat treatments and simplifies the crystallization process, facilitating the large-scale production of DNA origami microstructures. In addition, the advantages of the low-energy and room-temperature assembly mode will optimize the assembly conditions of micron-sized DNA origami devices, thereby promoting the functionalization of DNA origami single crystals.Data availability
All detailed experimental and characterization data associated with this work are available in the ESI†.Author contributions
Y. Y. and Y. T. conceived the project; Y. T. supervised the project; Y. Y., X. Y., and Z. Z. performed research; Y. Y., M. J., Y. W., L. D., N. M., and Y. T. analyzed data; Y. Y. and Y. T. wrote the paper; Y. Y., M. J., Y. W., X. Y., L. D., N. M., Z. Z., H. X., and Y. T. discussed the project and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Natural Science Foundation of Jiangsu Province (Grant No. BK20220124), the Fundamental Research Funds for the Central Universities (2024300319), the National Natural Science Foundation of China (Grant No. 22372077, Grant No. 92356304, and Grant No. 22374043), Hunan Provincial Science & Technology Department (2022SK2003, 2022JJ10007), the Science & Technology Innovation Program of Hunan Province (2022RC3046), and the funding from State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM2301). We thank the staff members at the BL19U2 beamline of the National Facility for Protein Science in Shanghai (NFPS), Shanghai Advanced Research Institute, Chinese Academy of Sciences, for providing technical support and assistance in data collection and analysis. We thank the staff from BL16B1 beamline at Shanghai Synchrotron Radiation Facility for assistance during data collection.References
- M. B. Ross, J. C. Ku, V. M. Vaccarezza, G. C. Schatz and C. A. Mirkin, Nanoscale form dictates mesoscale function in plasmonic DNA-nanoparticle superlattices, Nat. Nanotechnol., 2015, 10, 453–458 CrossRef CAS
.
- Y. Tian, J. R. Lhermitte, L. Bai, T. Vo, H. L. Xin, H. Li, R. Li, M. Fukuto, K. G. Yager, J. S. Kahn, Y. Xiong, B. Minevich, S. K. Kumar and O. Gang, Ordered three-dimensional nanomaterials using DNA-prescribed and valence-controlled material voxels, Nat. Mater., 2020, 19, 789–796 CrossRef CAS
- Q. Liu, K. W. Wan, Y. X. Shang, Z. G. Wang, Y. Y. Zhang, L. R. Dai, C. Wang, H. Wang, X. H. Shi, D. S. Liu and B. Q. Ding, Cofactor-free oxidase-mimetic nanomaterials from self-assembled histidine-rich peptides, Nat. Mater., 2021, 20, 395–402 CrossRef CAS PubMed
- J. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. Sha, P. E. Constantinou, S. L. Ginell, C. Mao and N. C. Seeman, From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal, Nature, 2009, 461, 74–77 CrossRef CAS PubMed
- J. Song, Z. Li, P. F. Wang, T. Meyer, C. D. Mao and Y. G. Ke, Reconfiguration of DNA molecular arrays driven by information relay, Science, 2017, 357, eaan3377 CrossRef PubMed
- Y. H. Zhou, J. Y. Dong, C. Zhou and Q. B. Wang, Finite Assembly of Three-Dimensional DNA Hierarchical Nanoarchitectures through Orthogonal and Directional Bonding, Angew. Chem. Int. Ed., 2022, 61, e202116416 CrossRef CAS
- L. F. Liu, Z. Li, Y. L. Li and C. D. Mao, Rational Design and Self-Assembly of Two-Dimensional, Dodecagonal DNA Quasicrystals, J. Am. Chem. Soc., 2019, 141, 4248–4251 CrossRef CAS
- X. Sun, S. H. Ko, C. Zhang, A. E. Ribbe and C. Mao, Surface-Mediated DNA Self-Assembly, J. Am. Chem. Soc., 2009, 131, 13248–13249 CrossRef CAS PubMed
- A. Kuzyk, R. Schreiber, Z. Y. Fan, G. Pardatscher, E. M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response, Nature, 2012, 483, 311–314 CrossRef CAS
- X. Jin, Y. T. Sang, Y. H. Shi, Y. G. Li, X. F. Zhu, P. F. Duan and M. H. Liu, Optically Active Upconverting Nanoparticles with Induced Circularly Polarized Luminescence and Enantioselectively Triggered Photopolymerization, ACS Nano, 2019, 13, 2804–2811 CrossRef CAS PubMed
- J. T. Zhang, K. Zhou, Y. J. Zhang, M. M. Du and Q. B. Wang, Precise Self-Assembly of Nanoparticles into Ordered Nanoarchitectures Directed by Tobacco Mosaic Virus Coat Protein, Adv. Mater., 2019, 31, 1901485 CrossRef
- Y. W. Li, H. X. Jin, W. J. Zhou, Z. Wang, Z. W. Lin, C. A. Mirkin and H. D. Espinosa, Ultrastrong colloidal crystal metamaterials engineered with DNA, Sci. Adv., 2023, 9, eadj8103 CrossRef CAS
- X. Liu, F. Zhang, X. Jing, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang, J. Lin, Y. Liu, D. Zhao, H. Yan and C. Fan, Complex silica composite nanomaterials templated with DNA origami, Nature, 2018, 559, 593–598 CrossRef CAS PubMed
- M. X. Zheng, Z. Li, C. Z. Zhang, N. C. Seeman and C. D. Mao, Powering ≈ 50 μm Motion by a Molecular Event in DNA Crystals, Adv. Mater., 2022, 34, 2200441 CrossRef CAS
- C. L. Bai and M. H. Liu, From Chemistry to Nanoscience: Not Just a Matter of Size, Angew. Chem. Int. Ed., 2013, 52, 2678–2683 CrossRef CAS
- S.-T. Wang, B. Minevich, J. Liu, H. Zhang, D. Nykypanchuk, J. Byrnes, W. Liu, L. Bershadsky, Q. Liu, T. Wang, G. Ren and O. Gang, Designed and biologically active protein lattices, Nat. Commun., 2021, 12, 3702 CrossRef CAS
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, DNA-programmable nanoparticle crystallization, Nature, 2008, 451, 553–556 CrossRef CAS PubMed
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, DNA-guided crystallization of colloidal nanoparticles, Nature, 2008, 451, 549–552 CrossRef CAS PubMed
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Challenges and opportunities for structural DNA nanotechnology, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Nanoparticle Superlattice Engineering with DNA, Science, 2011, 334, 204–208 CrossRef CAS PubMed
- E. Auyeung, T. I. N. G. Li, A. J. Senesi, A. L. Schmucker, B. C. Pals, M. O. de la Cruz and C. A. Mirkin, DNA-mediated nanoparticle crystallization into Wulff polyhedra, Nature, 2014, 505, 73–77 CrossRef
- Y. Wang, L. Dai, Z. Ding, M. Ji, J. Liu, H. Xing, X. Liu, Y. Ke, C. Fan, P. Wang and Y. Tian, DNA origami single crystals with Wulff shapes, Nat. Commun., 2021, 12, 3011 CrossRef PubMed
- L. Dai, X. Hu, M. Ji, N. Ma, H. Xing, J.-J. Zhu, Q. Min and Y. Tian, Programming the morphology of DNA origami crystals by magnesium ion strength, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, 2302142120 CrossRef PubMed
- S. E. Seo, M. Girard, M. O. de la Cruz and C. A. Mirkin, Non-equilibrium anisotropic colloidal single crystal growth with DNA, Nat. Commun., 2018, 9, 4558 CrossRef PubMed
- R. J. Macfarlane, B. Lee, H. D. Hill, A. J. Senesi, S. Seifert and C. A. Mirkin, Assembly and organization processes in DNA-directed colloidal crystallization, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10493–10498 CrossRef CAS
- S. Huang, M. Ji, Y. Wang and Y. Tian, Geometry guided crystallization of anisotropic DNA origami shapes, Chem. Sci., 2023, 14, 11507–11514 RSC
- R. J. Macfarlane, R. V. Thaner, K. A. Brown, J. Zhang, B. Lee, S. T. Nguyen and C. A. Mirkin, Importance of the DNA “bond” in programmable nanoparticle crystallization, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14995–15000 CrossRef CAS
- X. H. Yan, Y. Wang, N. N. Ma, Y. F. Yu, L. Z. Dai and Y. Tian, Dynamically Reconfigurable DNA Origami Crystals Driven by a Designated Path Diagram, J. Am. Chem. Soc., 2023, 145, 3978–3986 CrossRef CAS
- N. Ma, L. Dai, Z. Chen, M. Ji, Y. Wang and Y. Tian, Environment-Resistant DNA Origami Crystals Bridged by Rigid DNA Rods with Adjustable Unit Cells, Nano Lett., 2021, 21, 3581–3587 CrossRef CAS PubMed
- R. Jungmann, T. Liedl, T. L. Sobey, W. Shih and F. C. Simmel, Isothermal assembly of DNA origami structures using denaturing agents, J. Am. Chem. Soc., 2008, 130, 10062–10063 CrossRef CAS
- J.-P. J. Sobczak, T. G. Martin, T. Gerling and H. Dietz, Rapid Folding of DNA into Nanoscale Shapes at Constant Temperature, Science, 2012, 338, 1458–1461 CrossRef CAS PubMed
- S. Ramakrishnan, G. Krainer, G. Grundmeier, M. Schlierf and A. Keller, Structural stability of DNA origami nanostructures in the presence of chaotropic agents, Nanoscale, 2016, 8, 10398–10405 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07267g |
| This journal is © The Royal Society of Chemistry 2025 |