
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible addition of ethene to gallium(I) monomers and dimers†
Ryan J.
Schwamm‡
,
Malavika A.
Bhide‡
,
Gary S.
Nichol
 and
Michael J.
Cowley
*
and
Michael J.
Cowley
*
School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: michael.cowley@ed.ac.uk
First published on 15th April 2025
Abstract
Reversible interactions of organic substrates with transition metal compounds are a hallmark of their chemistry and its catalytic applications, but remain uncommon for low-valent p-block compounds. We report here the preparation of amidophosphine-supported gallium(I) compounds that exhibit equilibria between monomeric gallylene and dimeric digallene (Ga![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ga) states. The monomer–dimer equilibrium is controlled by the steric and electronic properties of the phosphine donor in the ligand employed. Regardless of their preference for monomeric or dimeric state, reactions of the Ga(I) systems with B(C6F5)3 affords monomeric gallylene adducts, whilst reactions with ethene produce 1,2-digallacyclobutanes via formal [2 + 2] cycloadditions. These ethene additions are reversible, with the digallacyclobutanes releasing ethene upon heating or treatment with Lewis acids to regenerate the gallium(I) species.
Ga) states. The monomer–dimer equilibrium is controlled by the steric and electronic properties of the phosphine donor in the ligand employed. Regardless of their preference for monomeric or dimeric state, reactions of the Ga(I) systems with B(C6F5)3 affords monomeric gallylene adducts, whilst reactions with ethene produce 1,2-digallacyclobutanes via formal [2 + 2] cycloadditions. These ethene additions are reversible, with the digallacyclobutanes releasing ethene upon heating or treatment with Lewis acids to regenerate the gallium(I) species.
Introduction
Reversible reactivity is a hallmark of transition-metal chemistry, in which the combination of reversible steps including ligand exchange, oxidative addition, and reductive elimination enables powerful catalytic processes. In main group chemistry, the widespread capability of low-oxidation state p-block compounds to engage in somewhat similar reactivity, particularly oxidative additions, has emerged relatively recently.1,2 It is still unclear how (and even if) this reactivity may serve as the basis of a general ‘redox-cycle catalysis’ at p-block element centres, even considering recent ground-breaking advances in redox-catalytic cycles at group 15 centres.3–6 Thus it remains a general challenge to engineer p-block systems in which oxidative addition reactions are readily reversible.7Reactions of main group species with unsaturated organic molecules such as alkenes and alkynes are relatively common, but normally irreversible.1,8–10 A few examples of reversible reactions have been reported. In group 14 chemistry, Kato reported reversible ethene binding at an amidophosphine supported Si(II) centre.11 Previous to that, Power reported reversible ethene binding by tin alkyne analogues,12 and more recently Tokitoh extended this reversibility to di germynes.13 In group 13 chemistry, the group of Crimmin has shown that the Al(I) monomer NacNacAl(I) can reversibly bind alkenes.14 Power reported terphenyl Ga(I) systems – which may adopt monomeric or dimeric structures in solution15,16 – that reversibly react with polyolefins.17 Dicationic digallenes also reversibly bind alkenes.18 Recently reported base-stabilised dialumenes react with alkenes and alkynes,19–21 in some cases dissociating to monomeric Al(I) species before doing so,19 but these reactions are not reversible. The related gallium analogues (base-stabilised digallenes) remain comparatively underexplored, and no reactivity with olefins is reported for them.22 The origins of the reversibility (or otherwise) in the reactions of Al(I) and Ga(I) with olefins remain unclear, as does its connection with the nuclearity of the reactive species.
Ga(I) chemistry is structurally rich, with compounds ranging from larger clusters to monomeric species.15–17,23–26 Dimeric systems can be divided into two classes: (i) weakly associated systems with low Ga–Ga bond order and (ii) ‘true’ dimers with Ga–Ga bond orders >1. Systems like Power's terphenyl Ga(I) series I or Stasch's [Ph2P(NDipp)2Ga]2 (ref. 27) exhibit weak interactions characterised by long Ga–Ga distances and significant trans-bending. The terphenyl Ga(I) systems I are almost wholly monomeric in solution.15,16,23 Nevertheless, indirect evidence implicates formation of transient GaGa dimers in reactions with olefins.17 In the related AriPr8Al(I) system, which is observed to exist as both monomers and/or AlAl bonded dimers, the mechanism of reactions with ethene are complex.28
Despite early reports of supported [R4Ga2]2− and [R4Ga2]˙ species featuring Ga–Ga bond orders ≥1 over 20 years ago,29–31 the first well-defined example of a neutral closed-shell digallene II was only recently presented.22 The NHC-coordinated digallene II is the gallium analogue of Inoue's landmark dialumene, III.20 The strong donor ligands of II imbue it with a strong GaGa bond and short Ga–Ga distance ((2.341(3) Å) vs. e.g. 2.627(1) Å for I). No reactivity of II with small molecules has been reported. Krossing and co-workers have investigated related dicationic digallene systems that react with a range of small molecules.31–33 In chemistry related to that of dialumene III, we recently reported the synthesis of the trans-bent base-coordinated dialumene IV, supported by an amidophosphine ligand system.19
The amidophosphine ligands of IV are conveniently prepared, provide a useful 31P NMR ‘handle’ for characterisation and mechanistic insight, and in principle are readily modified at the phosphorus centre to vary both electronic and steric properties. We were curious whether such amidophosphine ligands could shed light on Ga(I) chemistry and provide improved control or understanding of speciation within it.
Here, we show that the amidophosphine ligand used to support dialumene IV may be transferred successfully to Ga(I) chemistry, in the process developing a high-yielding preparative route to Ga(I) systems. By altering the size of the substituents on the phosphine, we demonstrate the ability to tune Ga(I) systems to favour monomeric (gallylene) or dimeric (digallene) states. Regardless of their preference for monomer/dimer, the amidophosphine Ga(I) species we report react with ethene to form digallacyclobutanes – products containing two Ga centres. Remarkably, these digallacyclobutanes reversibly release ethene upon heating or by treatment with Lewis acids (Fig. 1).
 | ||
| Fig. 1 Structures of low valent Al and Ga species. Ar = 2,6-Dipp2C6H3, Mes = C6H2Me3. | ||
Results and discussion
A digallene
For the preparation of digallene 1, the gallium analogue of dialumene IV, we envisaged a simple salt metathesis route with a gallium subhalide (e.g.“GaI”). However, with the lithiated amidophosphine ligand precursor LMes/tBuLi(OEt2) (see ESI†), these reactions led only to intractable mixtures. The substitution of Cp* ligands in Cp*Ga or (Cp*Al)4 with metalated ligands is emerging as a useful route to neutral or anionic Ga(I) or Al(I) compounds.34,35 We decided to trial this strategy for our amidophosphine ligands.The reaction of Cp*Ga with the lithiated amidophosphine ligand LMes/tBuLi(OEt2) in toluene at 80 °C (Fig. 2) generated a red solution. 31P{1H} NMR spectroscopy revealed the formation of a new phosphorus-containing species with a broad resonance at δ 26.0 and, after three hours, copious quantities of orange precipitate – the digallene LMes/tBuGa = GaLMes/tBu (1) – had formed. Following recrystallisation, digallene 1 was obtained as red crystals in 74% yield.
 | ||
| Fig. 2 Synthesis of digallene 1. X-ray structure of 1 (H atoms omitted for clarity). Thermal ellipsoids at 50% probability. Selected bond distances (Å) and angles (°): Ga1–Ga1′ 2.480(1), Ga1–N1 2.002(3), Ga1–P1 2.513(6), N1–Ga1–P1 82.55(15), θ = 55.6. | ||
Digallene 1 is a Ga(I) dimer in the solid-state. The structure of 1 (Fig. 2), determined by X-ray diffraction, very closely resembles that of dialumene IV, including in the 2-site disorder of the symmetry-related Ga centres (70/30% occupancy). The disorder arises from the heavily trans-bent GaGa bond (trans-bend angle θ = 55.6/57.6°), which displays Ga1–Ga1′ distances of 2.480(1) and 2.468(3) Å. The phosphorus substituents are trans to one another across the GaGa bond, meaning the digallene exists in E-configuration.
Dialumene IV, the aluminium analogue of digallene 1, features a notably longer AlAl bond than its NHC/silyl substituted counterpart dialumene III.20 The same pattern is observed in the gallium analogues: the Ga–Ga distance in 1 is longer than in Wang's NHC/silyl substituted digallene II (2.480(1)/2.468(3) Å vs. 2.341(3) Å).22
Density functional theory (DFT) calculations (BP86-D3/def2svp) on digallene 1 reveal the presence of a GaGa π-bonding interaction (Fig. S4†). As in dialumene IV, the HOMO of 1 has substantial Ga–Ga π-bonding character (Fig. S4†). Consistent with this, the calculated Wiberg bond index (WBI) for the GaGa bond is 1.24, which supports the conclusion of a degree of multiple bonding character. Compared to the same measure in Wang's NHC/silyl substituted digallene II, however, the WBI in 1 is much lower (1.71 vs. 1.24).22 This is consistent with the ∼0.13 Å shorter Ga–Ga bond distance in II. In fact, the Ga1–Ga1′ distance in digallene 1 falls approximately midway between that in II and that for Power's weakly associated terphenyl Ga(I) species I (2.627(1) Å),24 which dissociates in solution. With the stretched GaGa distance and low calculated bond-order in 1, we became curious about its solution behaviour.
Digallene 1 exists in solution as a mixture of monomers and dimers. Like its aluminium analogue IV,19 the E isomer of digallene 1 has three possible diastereomers, 1A, 1B and 1C. 1A and 1B are meso-compounds, and each have equivalent phosphorus centres; they both should thus have one phosphorus chemical shift. In contrast, the two phosphorus centres in 1C are chemically inequivalent, and so this compound should give rise to two 31P NMR signals. At 300 K, the 31P{1H} NMR spectrum of digallene 1 reveals a single broad resonance at δ 26.0, indicating that all three diastereomers 1A–C are interconverting rapidly on the NMR timescale.
Upon cooling, the exchange of diastereomers slows and the single broad 31P resonance becomes further resolved (Fig. 3a).
 | ||
| Fig. 3 (a) 1H and 31P{1H} VT NMR spectra of 1. (b) Ga inversion mechanism between 1A and 1B, and enantiomers of 1C. (c) Dissociation pathway for interconversion of 1A/1B and 1C. | ||
The dynamic processes involved in the exchange of diastereomers 1A–C are more clearly apparent in the 1H NMR spectrum (Fig. 3a). At room temperature, one signal at δ 3.03 for the NCCH bridgehead proton of all three diastereomers is observed. On cooling to 233 K, the resonance splits into two new singlets, at δ 3.00 and δ 3.05. We assign one of these signals to diastereomers 1A and 1B, which are interconverting by an intramolecular process that simultaneously inverts the stereochemistry at both Ga centres (Fig. 3b); a related “trans-flip” process is operative in the dialumene IV.19 The trans-flip exchanges the two inequivalent bridgehead CH environments of 1C, thus explaining the appearance of just one resonance for this diastereomer. On cooling still further, to 193 K, the signal observed at δ 3.00 at 233 K itself resolves into two new singlets at δ 2.99 and δ 2.95 due to a slowing of the trans-flip process between 1A/1B or for 1C. It is not possible to unequivocally assign whether it is exchange between 1A/1B or between CH environments in 1C which becomes slow at 193 K.
For the digallene 1 in solution at room temperature, the observation of just one signal for the NCCH bridgehead proton indicates all three diastereomers 1A–C are rapidly exchanging. Since the trans-flip process cannot interconvert 1A/1B with 1C, this exchange can only be through reversible dissociation of the digallene into monomers (Fig. 3c).
Although the NMR spectroscopic behaviour of digallene 1 can only be explained by invoking its reversible dissociation into monomers, the compound nevertheless exists predominantly as a dimer in solution. The UV/vis spectrum of 1 in Et2O reveals an intense absorption at λmax = 501 nm (ε = 9847 mol L−1 cm−1) (Fig. S54†). TD-DFT calculations (BP86-D3/Def2SVP) on 1 allowed us to assign the absorption to an excitation with substantial contributions from GaGa π to π*-type transition (HOMO → LUMO (Fig. S4†)), as well as a smaller contribution from a ligand-based orbital (HOMO−2 → LUMO). The spectroscopic measurements reveal that 1 has a marginally narrower HOMO–LUMO gap than the reported digallene II (λmax = 521 nm).22
A gallylene
In transition metal chemistry, ready steric and electronic tuning of phosphine ligands allows powerful control over the properties and reactivity of transition-metal complexes.36 We were curious whether the same effects might translate to low-valent p-block chemistry.We thus prepared HLMes/Mes, an analogue of the amidophosphine ligand of 1 in which the PtBu2 donor has been replaced with the diaryl phosphine fragment PMes2. We expected the PMes2 fragment should be both less electron-donating and more sterically hindered than PtBu2.
The lithium salt of the ligand, LMes/MesLi(OEt2), was readily obtained by treatment of HLMes/Mes with n-butyl lithium (see ESI†).
Dropwise addition of a solution of LMes/MesLi(OEt2) to Cp*Ga in toluene initially formed a yellow solution, which became dark orange and deposited a colourless precipitate (Cp*Li) after it was heated to 80 °C for 3 hours. 31P{1H} NMR spectroscopy of the crude reaction mixture revealed the formation of a new compound exhibiting a singlet at δ −38.7. The new compound was isolated by crystallisation from pentane as a yellow solid in 45% yield.
X-ray diffraction reveals the yellow crystalline material obtained from pentane to be the monomeric gallylene species LMes/MesGa (2), which features a two-coordinate gallium centre (Fig. 4). The Ga–P distance, at 2.7093(5) Å, is substantially (∼0.2 Å) elongated compared to that in digallene 1 (2.47–2.51 Å). Concurrently, the bite angle P1–Ga1–N1 in gallylene 2 is narrowed compared to that in 1 (78.92(5)° vs. 82.66(15)°). The increased Ga–P distance in 2 is consistent with the greater steric bulk and lower donating ability of the PMes2 (vs. PtBu2 in digallene 1).
 | ||
| Fig. 4 Synthesis of gallylene 2. X-ray structure of 2 (H atoms omitted for clarity). Thermal ellipsoids at 50% probability. Selected bond distances (Å) and angles (°): Ga1–N1 1.962(2), Ga1–P1 2.7093(5), N1–Ga1–P1 78.92(5). | ||
Consistent with the monomeric structure observed for gallylene 2 in the solid state, NMR spectroscopy reveals no evidence for formation a dimeric digallene ([2]2) in toluene solutions. Thus, the room temperature 31P{1H} spectrum of gallylene 2 showed a single broad resonance at δ −37.8. The formation of the putative digallene [2]2 would be expected to be revealed by the presence of 31P resonances for multiple diastereomers (in the absence of any reversible dissociation). Even on cooling a solution of gallylene 2 in d8-toluene to 193 K, the signal at δ −37.8 remained a singlet. If the dimer [2]2 forms under such conditions it must therefore be in low concentrations, or remain in equilibrium with the monomeric gallylene 2.
The room temperature UV/vis spectrum of the monomeric gallylene 2 in Et2O revealed an absorption at λmax at 440 nm (ε = 2081 mol L−1 cm−1) (Fig. S55†). In comparison to digallene 1, the absorption is blue shifted and less intense (for 1: λmax = 501 nm (ε = 9847 mol L−1 cm−1)). The shorter wavelength absorption and the weaker extinction coefficient observed for 2 are both consistent with the n → p transition expected for a gallylene monomer rather than the π to π* transition observed for digallene 1.
Visual inspection revealed that upon cooling an Et2O solution of monomeric gallylene 2 to 223 K, a colour change from yellow to red was observed. A UV/vis spectrum recorded at 223 K reveals a new intense absorption, at λmax = 510 nm, substantially red-shifted from the one observed at room temperature (Fig. S55†). The low-temperature absorption falls in the region expected for a GaGa π to π* transition in a trans-bent digallene (cf.1, λmax = 501 nm).
DFT and TD-DFT calculations support the assignment of the low-temperature 510 nm absorption to a dimer of 2, the trans-bent digallene [2]2. Using DFT calculations (BP86-D3/Def2SVP) we identified a dimeric structure [2]2 (Fig. S1†) as a minimum on the potential energy surface located 1.2 kcal mol−1 higher than the monomer(s) at 298 K (see later). The geometry calculated for [2]2 reveals a trans-bent GaGa core. A long Ga–Ga distance compared to that determined experimentally for 1 (2.612 Å vs. 2.480(1) Å) and much-reduced WBI (0.89 vs. 1.24) indicate a significantly weaker bonding interaction for [2]2 as compared to 1.
TD-DFT calculations on the optimised structure of [2]2 predict an absorbance at 569 nm arising from a transition akin to that found for digallene 1, with substantial GaGa π to π* character (HOMO → LUMO (Fig. S4†)). At 569 nm the absorption is red shifted by ∼60 nm compared to that observed experimentally, but is consistent with the trend between experiment and TD-DFT for digallene 1.
In benzene or toluene solutions, gallylene 2 appears essentially monomeric; even at lower temperatures, no evidence of dimerisation could be observed using NMR spectroscopy. In Et2O solution, however, UV/vis spectroscopy revealed the presence of the dimer [2]2. Gallylene 2 thus occupies the opposite end of a continuum it shares with digallene 1.
The finely-balanced energetics of the monomer/dimer equilibria of 1 and 2/[2]2 (see below) are further emphasised by the observation that gallylene 2 may also be crystallised as an elongated dimer in the solid state. Crystallisation of 2 from toluene (rather than pentane as described above) solution affords dark red-purple crystals of the weakly associated elongated dimer LMes/MesGa⋯GaLMes/Mes (Fig. 5) as a toluene solvate.
 | ||
| Fig. 5 X-ray structure of LMes/MesGa⋯GaLMes/Mes (H atoms omitted for clarity). Thermal ellipsoids at 50% probability. Selected bond distances (Å) and angles (°): Ga1–Ga2 2.8566(4), Ga1–N1 2.040(2), Ga1–P1 2.7282(8), N1–Ga1–P1 78.42(6), Ga2–N2 1.994(2), Ga2–P2 2.6492(7), N2–Ga2–P2 80.23(6). | ||
X-ray crystallography revealed long Ga⋯Ga interactions between two crystallographically inequivalent gallium(I) centres. The Ga⋯Ga distance, at 2.8566(4) Å, is almost 0.4 Å longer than that of the amidophosphine digallene 1 and NHC/silyl digallene II.22 It is however comparable to Power's weakly associated gallium(I) dimer I in which the Ga⋯Ga distance is 2.627(1) Å.24
In the elongated LMes/MesGa⋯GaLMes/Mes dimer, the relative arrangement of amidophosphine ligands about the Ga⋯Ga interaction reveal considerable trans-bending (θ: 78.37°) and twisting (NGaP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NGaP interplanar angle, τ = 89.10°). Ga1 is trigonal pyramidal, whilst Ga2 has an unusual T-shaped geometry, with P2 and Ga1 disposed trans to each other (Ga1–Ga2–P2 = 162.23(2)°). The Ga–P distance at T-shaped Ga2 is shorter than at Ga1 (2.6492(7) Å vs. 2.7282(8)). Both Ga–P distances are longer than in the dimeric digallene 1 (2.472(2)/2.513(6) Å), but remain comparable to those in monomeric gallylene 2 (2.7093(5) Å).
:NGaP interplanar angle, τ = 89.10°). Ga1 is trigonal pyramidal, whilst Ga2 has an unusual T-shaped geometry, with P2 and Ga1 disposed trans to each other (Ga1–Ga2–P2 = 162.23(2)°). The Ga–P distance at T-shaped Ga2 is shorter than at Ga1 (2.6492(7) Å vs. 2.7282(8)). Both Ga–P distances are longer than in the dimeric digallene 1 (2.472(2)/2.513(6) Å), but remain comparable to those in monomeric gallylene 2 (2.7093(5) Å).
To evaluate the role of ligand effects on the Ga–Ga bond dissociation energies of digallenes 1 and [2]2, we calculated dissociation energies for the two dimers using DFT (BP86-D3/def2svp). Dissociation for digallene 1 (bearing LtBu/Mes ligands) was calculated to be thermodynamically unfavourable; ΔH = 30.1, ΔG° = 13.9 kcal mol−1. In contrast [2]2, with bulkier LMes/Mes ligands has a thermodynamic preference for the monomeric state; ΔH = 17.9, ΔG° = −1.2 kcal mol−1. These values are consistent with the experimentally observed behaviour of 1 and 2/[2]2.
There is a considerable difference in the calculated ΔH for dissociation of 1 and [2]2 to monomers (30.1 and 17.9 kcal mol−1 respectively). An account by Mears and Power highlighted the importance of attractive dispersion interactions in multiply-bonded heavier main-group systems.37 To estimate their contribution to the stability (or otherwise) of the digallenes 1 and [2]2, we examined the simple model digallene [Me3P(Me2N)Ga]2. By comparing the bond dissociation enthalpies of the full digallene systems with those of the minimal models, we gain insight into stabilising contributions of the larger ligands, which in part will include dispersion. The Ga–Ga bond length in the minimal model was fixed to 2.482 Å or 2.612 Å to represent 1 or [2]2 respectively. ΔH for dissociation of the minimal models of 1 and [2]2 proved very similar, at 13.7 and 14.7 kcal mol−1 respectively, despite the longer Ga–Ga distance in the model for [2]2. In fact, a relaxed potential energy surface scan of the minimal model revealed that the system is rather insensitive to extension of the Ga–Ga distance, with the energy varying by <1 kcal mol−1 over the range 2.48–3 Å (Fig. S3†). The contribution of the ligand (LMes/tBuvs. LMes/Mes) to the stability of the Ga dimers is much more substantial than the Ga–Ga distance. For [2]2 and its model, there was minimal difference in ΔH for dissociation (14.7 vs. 17.9 kcal mol−1), in contrast to the comparison between 1 and its model (13.7 vs. 30.1 kcal mol−1). We conclude that digallene 1 is, to a large degree, stabilised by attractive dispersion interactions between the LtBu/Mes ligands that are absent in the digallene [2]2 bearing the much bulkier LMes/Mes ligand.
Reactions of 1 and 2 with B(C6F5)3
Having established that accessible gallylene–digallene equilibria existed (from opposite sides) in solution for 1 and 2, we sought to investigate the implications of these equilibria on reactivity.Treatment of 1 or 2 with tris(pentafluorophenyl)borane, B(C6F5)3, proceeded with immediate colour changes to pale yellow. The respective Lewis adducts (LMes/R)Ga–B(C6F5)3 (3, R = tBu, 4, R = Mes) were isolated in high yields as pale-yellow crystalline solids after work-up (Fig. 6).
 | ||
| Fig. 6 Synthesis of compounds 3 and 4 (top). X-ray crystal structures of 3 and 4 (H atoms are omitted). Thermal ellipsoids at 50% probability. Selected bond distances (Å) and angles (°) 3: Ga1–B1 2.121(2), Ga1–N1 1.904(2), Ga1–P1 2.4401(6), N1–Ga1–P1 87.51(5). 4: Ga1–B1: 2.128(3), Ga1–N1 1.915(2), Ga1–P1 2.4489(7), N1–Ga1–P1 87.38(6). | ||
Formation of compounds 3 and 4 was confirmed by NMR spectroscopy. 31P{1H} NMR spectra of 3 and 4 revealed resonances at δP 15.3 and −47.2, respectively. The 11B NMR spectra of 3 and 4 showed singlets at δB −19.0 and −18.2, respectively, shifted upfield from free B(C6F5)3 (δ 58.0).
The single crystal X-ray structures of 3 and 4 confirmed coordination of Ga to B, with Ga–B distances (3, 2.121(2) Å; 4, 2.128(3) Å) consistent with previously reported Ga–B(C6F5)3 dative interactions (range 2.108(1)–2.185(2) Å).15,38 Notably, the Ga(1)–P(1) distance in 4 is substantially contracted – by 0.26 Å – compared to in the free monomer 2. This is consistent with the observed upfield shift of 9.4 ppm in the 31P NMR upon coordination of B(C6F5)3.
Whilst the formation of the gallylene adduct 4 is expected, the analogous adduct 3 arising from the digallene 1 and B(C6F5)3 is additional evidence that the corresponding monomer is energetically accessible in solution.
Reactions of 1 and 2 with an oxidant
Tan, Wang and coworkers reported that the NHC-coordinated digallene II can be singly or doubly oxidised by treatment with an appropriate amount of [Ph3C][B(3,5-(CF3)2C6H3)4].22 We were curious to know whether compounds 1 and 2 could be oxidised in similar fashion.The reaction of digallene 1 with two equivalents of [Ph3C][B(C6F5)4] in fluorobenzene at room temperature initially formed a brown solution, which quickly turned green. Within minutes, crystalline dark green needles precipitated from the solution. X-ray crystallography revealed the product of the reaction to be [Mes/tBuLGa-GaLMes/tBu]2+ (5), the result of a 2-electron oxidation of 1. The dication 5 crystallised as the fluorobenzene solvate [Mes/tBuLGa-GaLMes/tBu][B(C6F5)4]2·2(C6H5F) (Fig. 7).
 | ||
| Fig. 7 Synthesis of [Mes/tBuLGa-GaLMes/tBu][B(C6F5)4]2 (top). X-ray structure of 5 from crystals of [Mes/tBuLGa-GaLMes/tBu][B(C6F5)4]2·2(C6H5F) (H atoms, [B(C6F5)4]− counter anions and PhF molecules omitted for clarity). Thermal ellipsoids at 50%. Bond distances: 5: Ga–Ga 2.390(2) Å. | ||
The double oxidation of digallene 1 to form the dication 5 induces substantial structural changes around the Ga2 unit, which in 5 adopts a planar conformation in contrast to the trans-bent GaGa unit of 1. Ga1 and Ga1′ deviate from the N1, P1, N1′,P1′ plane by just 0.05 Å. This is in contrast to the dication of digallene II, [II]2+ reported by Wang and Tan, which is twisted with a dihedral angle of 73.77°.22 The Ga–Ga bond length in dication 5 is 2.390(2) Å, which is shorter than that in the corresponding digallene 1 (2.480(1) Å). DFT calculations on dication 5 (BP86/Def2SVP) return an optimised structure with a Ga–Ga distance (2.398 Å) that closely replicates that determined experimentally. NBO calculations reveal the Wiberg bond index of the Ga–Ga bond in 5 is 0.90, (vs. 1.02 for [II]2+), consistent with a single bond. The Ga–Ga σ bonding orbital is found at −11.771 eV as HOMO−6. The contraction of the Ga–Ga distance in 1 upon oxidation (vs. the lengthening observed for II) is largely consequence of the highly trans-bent structure adopted by 1.
Subsequent preparations of 5 resulted in crystallisation of the dication as a different solvate, [Mes/tBuLGa-GaLMes/tBu][B(C6F5)4]2·4(C6H5F). Notably, these crystals were blue rather than green. Close inspection of the structures (see Table S8†) revealed no significant differences in geometry around the central Ga2 unit and N,P ligand. Neither structure displays close Ga–F interactions with fluorobenzene or [B(C6F5)4]− anion, with the closest “contact” in either being 4.673 Å (sum of Ga + F VdW radii = 3.34 Å). However, in both structures the shortest Ga–F distances are located directly above/below the plane of the Ga2 unit. The two structures differ in the number of F atoms distributed around that plane, with four present in the ·2(C6H5F) solvate and two in the ·4(C6H5F) solvate (Fig. S57†). These differing patterns of Ga–F may serve to explain the difference in colour.
The intense green/blue colour of the dication 5 in PhF solution or as crystalline solvates is surprising, given that [II]2+ is colourless. The extremely low solubility of 5 precluded UV/vis spectroscopic studies. We turned to TD-DFT calculations to understand the electronic structure of 5.
The LUMO of 5 has Ga–Ga π-bonding character and, at −8.824 eV, is low-lying; its HOMO is essentially ligand based and has CC π-bonding and N-lone pair character. HOMO−1 has delocalised N–Ga–Ga–N π-bonding character (Fig. S58†). In the UV-vis spectrum predicted by TD-DFT (Fig. S56†) the most intense absorption, at 784 nm, arises from a HOMO → LUMO transition. Such a low-energy transition is not observed in the colourless NHC-coordinated dication [II]2+because its twisted structure raises the energy of Ga–Ga π-bonding LUMO.
When we treated the monomeric gallylene 2 with [Ph3C][B(C6F5)4] (0.5 or 1 equiv.) we observed colour changes to red, brown or green, but to date we have been unable to isolate or characterise any gallium-containing products from this reaction. Similarly, when attempting the 1-electron oxidations of 1 or 2 to prepare the radical cation, or attempting comproportion-based routes from 5 and 1, we have also been unable to obtain isolable products.
Reversible reactions with ethene
Having shown that the digallene 1 can react as a monomer (as well as the already-predominantly-monomeric gallylene 2), we wanted to investigate the reactivity of 1 and 2 with ethene, considering previously reported Ga(I)/alkene chemistry17,25 and the reaction of dialumenes with ethene to form dialuminacyclobutanes.19Treatment of toluene solutions of 1 or 2 with ethene (1 atm) at ambient temperature resulted in immediate formation of the 1,2-digallacyclobutanes, 5 and 6 respectively, as apparent [2 + 2] cycloaddition products. The isolation of the 1,2-digallacyclobutane products is in contrast with previous experiments that revealed bis(terphenyl)digallenes react with 2 equivalents of ethene to form 1,4-digallacyclohexanes.15 Even after heating 1 or 2 to 80 °C under 1 atm ethene for 3 days, we did not observe further incorporation of ethene. Slow cooling of concentrated toluene solutions of 6 or 7 allowed for formation of crystals suitable for analysis via single crystal X-ray diffraction (Fig. 8). In both solid-state structures, the phosphorus donors are orientated anti across the Ga–Ga bonds, suggesting that the products may arise from reactions of ethene with E isomers of the digallene 1 or [2]2. Ga–Ga and Ga–C bond distances in the metallacycles are consistent with single bonds (6: Ga–Ga 2.4799(7) Å, Ga–C 2.027(4)–2.043(4) Å; 7: Ga–Ga 2.5011(8) Å, Ga–C 2.023(5)–2.031(5) Å). C–C bond distances are also consistent with single bonds (1.533(6) Å in 6, 1.555(6) Å in 7. Narrow Ga–Ga–C and C–C–Ga angles give rise to distorted Ga2C2 rings with significant out-of-plane twisting (sum of internal angles: 6, 348.1°, 7, 354.3°).
 | ||
| Fig. 8 Synthesis of compounds 6 and 7 (top). X-ray structures of compounds 6 and 7 (H atoms omitted for clarity). Thermal ellipsoids at 50%. 6: Ga–Ga 2.4799(7) Å, Ga–C 2.027(4)–2.043(4) Å; 7: Ga–Ga 2.5011(8) Å, Ga–C 2.023(5)–2.031(5) Å. | ||
As in the digallenes 1 and [2]2, the gallium atoms in digallacyclobutanes 6 and 7 are stereogenic. In the structure of 6, the disordered positions of carbon atoms of the norbornene bridge thus indicate that 6 crystallises as a mixture of 3 diastereomers (corresponding to digallene diastereomers 1A, 1B and 1C (Fig. S56†)). That mixture would be expected to give rise to four signals in the 31P NMR spectrum, which we indeed observed in C6D6 solutions prepared from crystalline 6 (6A/6Bδ 17.9, 17.6 (both d, 3JPP = 17 Hz; 6Cδ 17.6(s), 17.7(s)).
When we exposed digallene 1 in C6D6 to 1 atm of ethene and immediately acquired 31P{1H} NMR spectra, besides the four resonances assigned to 6A–C, we observed four additional signals at δ 16.1 (d, 3JPP = 5 Hz), 14.8 (s), 14.3 (s), 13.3 (d, 3JPP = 5 Hz). We assign these signals to the diastereomers 6D–F, in which the phosphorus centres are syn across the Ga–Ga bonds. Consistent with the much different P–Ga–Ga–P torsion angles in 6C and 6F, we note the reduced size of 3JPP in the latter (5 Hz vs. 17 Hz).
In the initial (kinetically determined) product mixture formed from 1 and ethene the anti (6A–C) and syn (6D–F) isomers are present in a ratio of ∼4:1. This ratio is determined by a combination of the equilibrium concentrations of the digallene diastereomers (which likely includes, although not observed in our variable temperature NMR studies on 1, Z digallene isomers 1D–F), the barriers to interconversion of 1A–F, and those to ethene addition.
On heating the mixture to 80 °C for 12 hours, the syn isomers 6D–F are converted to the thermodynamically preferred anti isomers 6A–C, resulting in an anti:syn ratio of ∼12:1 (Fig. 9).
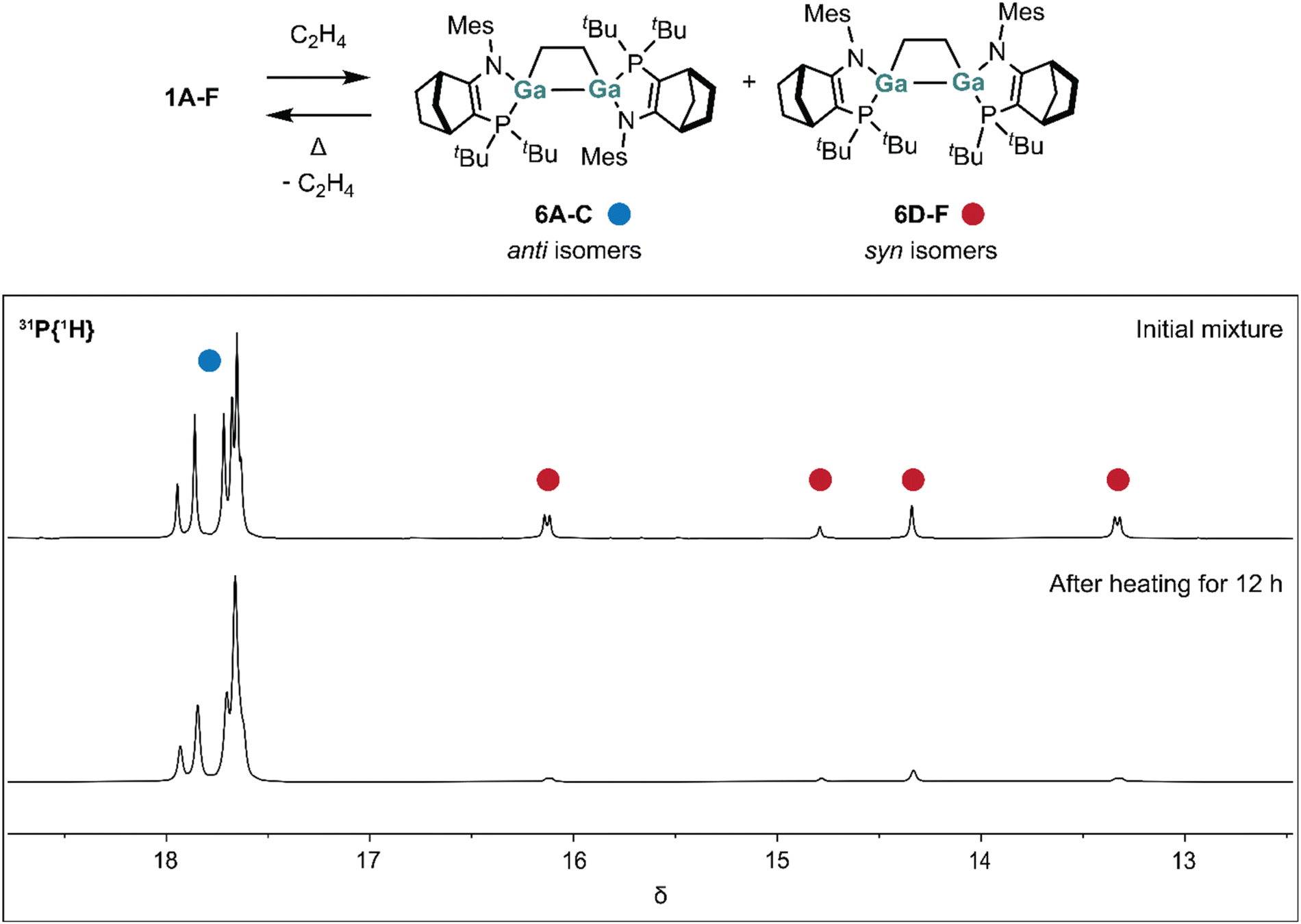 | ||
| Fig. 9 Reversible ethene binding to 1 (top). 31P{1H} NMR spectra showing thermally induced conversion of syn isomers 6D–F to anti isomers 6A–C. | ||
In contrast to 1, when digallene 2/[2]2 is treated with 1 atm ethene, 31P{1H} NMR spectroscopy revealed just one major diastereomer of digallacyclobutane 7 in the initial product mixture (δ −40.0, s). This is consistent with the solid-state structure of 6 which contains a single diastereomer.
For digallacyclobutane 6, the thermally induced conversion of syn-isomers 6D–5F to the more stable anti-isomers 6A–5C suggests the possibility of reversibility in the formation of 6 (and potentially the LMes/Mes system 7). Indeed, when toluene solutions of analytically pure samples of 6 or 7 were heated to 80 °C under an argon atmosphere, the dissociation of ethene was observed (Fig. S49 and S50†). On cooling to room temperature, rapid recombination of 1 and ethene resulted in formation of a mixture of anti- and syn-isomers (∼12:1 ratio). The same regeneration of the digallacyclobutane product was observed when previously heated samples of 7 were returned to room temperature.
Further demonstrating the ready reversibility of ethene addition to digallenes 1 and [2]2, the addition of B(C6F5)3·Et2O to isolated samples of 6 and 7 resulted in the complete dissociation of ethene and formation of the Lewis adducts 3 and 4, respectively.
Likely mechanism of reactions with ethene
We investigated the reactions of 1 and [2]2 with ethene using DFT methods (BP86/Def2SVP) (Fig. 10). Two obvious mechanisms exist for the formation of digallcyclobutanes: (i) a direct “[2 + 2]” cycloaddition of ethene to digallene, or (ii) [1 + 2] cycloaddition of ethene with gallylene monomer to form a gallacyclopropane intermediate, which then undergoes a C–Ga insertion reaction with a second gallylene monomer.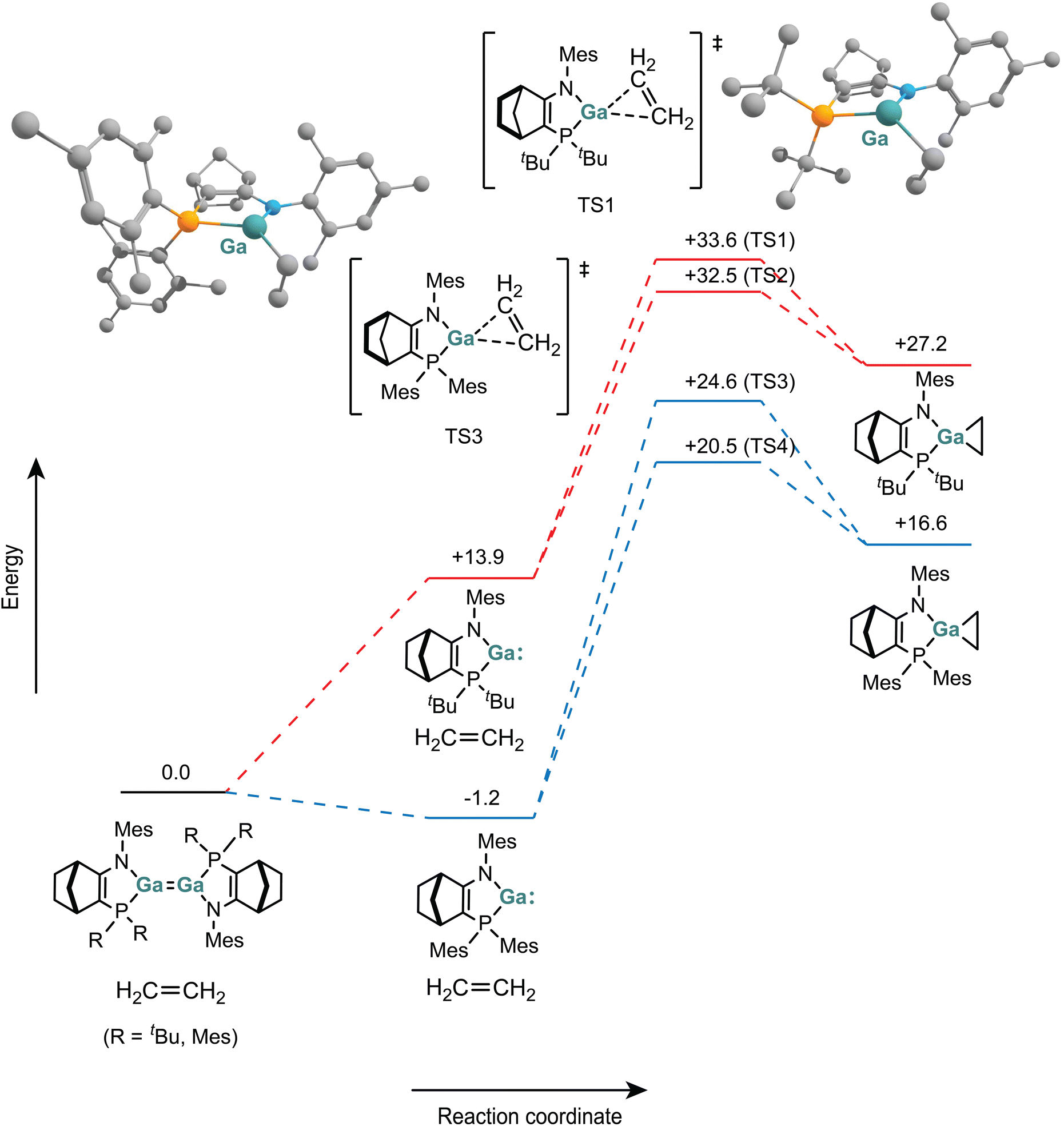 | ||
| Fig. 10 Calculated reaction pathways for the addition of ethene to monomers (energies in kcal mol−1). Transition states TS2 and TS4 are included in the ESI.† | ||
For reaction of ethene with monomeric gallylene, 1 must first dissociate to the monomer 1M, which lies 13.9 kcal above the digallene. In the LMes/Mes system, the monomer 2 is lower in energy than digallene [2]2 anyway (and thus the major species in solution). For both digallene 1 and gallylene 2, the formation of the gallacyclopropane products was calculated to be substantially endergonic (+27.2 kcal mol−1 and +17.8 kcal mol−1 respectively).
We were able to locate transition states for the addition of ethene to gallylene 1M and 2. The CH2 and CH2CH2 bridges of the norbornenyl ligand backbone mean that the approach of ethene from either “face” of the ligand are energetically differentiated, which leads to two transition states for each gallylene monomer. For the LMes/Mes gallylene 2, barriers to ethene addition are +21.7 and 25.8 kcal mol−1. Approach from the CH2CH2 “face” has the higher barrier. The same pattern is found for addition to gallylene 1M, with barriers of +18.6 and +19.7 kcal mol−1.
Combined with the energetic cost of monomerisation, the complete barriers for the reaction of digallene 1 with ethene to form the gallacyclopropane are +32.5/33.6 kcal mol−1. Such barriers would be incompatible with the observed reaction rates (within seconds at room temperature), making formation of digallacyclobutanes 6 and 7via initial [1 + 2] addition unlikely.
By DFT, ΔG0 for formation of the experimentally observed digallacyclobutane products 6 and 7 (ΔG0 = −9.1 kcal mol−1 and −27.0 kcal mol−1 respectively) is substantially more favourable than that calculated for formation of the gallacyclopropanes. Unfortunately, despite substantial effort, we were unable to locate transition states for direction addition of ethene to the digallenes 1 or [2]2 using DFT methods. Our failure to locate such transition states may be due to radical reaction pathways. For both dialumenes and digallenes, DFT methods have been found to imperfectly represent electronic and geometric structures because of substantial diradical character.39,40 In the closely-related disilenes (R2Si = SiR2), which themselves have some diradical character, addition of alkynes or other unsaturated compounds to form disilacyclobutenes (or similar) is known to proceed stepwise through radical intermediates.41,42
We suggest that the mechanism of the reaction of ethene with digallene 1 and gallylene 2 may proceed via a radical pathway involving addition to the digallenes 1 or [2]2 (akin to disilenes) (particularly considering the mismatch between experimentally observed rates of reaction and the calculated barriers for the reactions of ethene with the monomeric gallylenes). We are currently undertaking a more detailed experimental and computational study of the mechanism of these reactions.
Conclusions
We have demonstrated straightforward synthetic access to amidophosphine-supported gallium(I) systems. Digallene 1 is a trans-bent dimer in the solid-state, reversibility dissociating to monomer in solution. Gallylene 2 crystallises as a monomer or weakly associated dimer, but in solution can dimerise to form the digallene [2]2.Compounds 1 and 2 demonstrate dual functionality by displaying reactivity consistent with both monomeric gallylenes or dimeric digallenes in solution. The accessibility of monomeric species 1M and 2 is demonstrated by their reactivity with tris(pentafluorophenyl)borane, forming the gallylene adducts 3 and 4. By contrast, the reactions of 1 and 2 with ethene proceed through apparent [2 + 2] cycloaddition reactions, demonstrating that these species can also react (at least formally) as digallenes. Furthermore, the latter reactions are reversible, reforming free ethene and regenerating the Ga(I) species 1 and 2 at elevated temperatures.
The reversible activation of ethene by metal–metal multiply-bonded compounds has been observed in digermynes and distannynes,13,43 but the reversible reactions of 1 and 2 with ethene are the first unequivocal example of such reactivity of homometallic group 13 multiple bonds. Reversible cycloadditions with alkenes and alkynes have also been observed for monomeric Al(I) species,14 whilst polyolefins have been observed to react reversibly with terphenyl Ga(I) systems.17 Reversibility of the type shown by these compounds, and gallium systems 1 and 2, is a probable prerequisite for the development of transition-metal-like catalysis at main-group centres. Another component towards realising such a goal will be the ability to control reactivity at the main-group centre. Here, we have also shown how variation in the electronic and steric properties of the donor ligand can shift the equilibrium between predominantly dimeric (1) or monomeric (2) systems.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data has been deposited at the CCDC under CCDC records 2384332–2384338, 2433601, and 2433602, and can be obtained from https://www.ccdc.cam.ac.uk/structures/Author contributions
RJS and MJC proposed and initiated the study. Synthetic work and experimental characterisation was conducted by RJS and MAB. DFT calculations were carried out by RJS and MAB. GSN performed crystallography. RJS, MAB, and MJC co-wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received funding from the Leverhulme Trust in the form of a Research Project Grant, and from the European Research Council (ERC) under the EU Horizon 2020 research and innovation programme (ERC-2016-STG-716315).Notes and references
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- S. Bonfante, C. Lorber, J. M. Lynam, A. Simonneau and J. M. Slattery, J. Am. Chem. Soc., 2024, 146, 2005–2014 CrossRef CAS PubMed.
- H. Fujimoto, T. Kodama, M. Yamanaka and M. Tobisu, J. Am. Chem. Soc., 2020, 142, 17323–17328 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- D. J. D. Young and R. West, Chem. Lett., 1986, 15, 883–884 CrossRef.
- C. Cui, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 5062–5063 CrossRef CAS PubMed.
- R. Kinjo, M. Ichinohe, A. Sekiguchi, N. Takagi, M. Sumimoto and S. Nagase, J. Am. Chem. Soc., 2007, 129, 7766–7767 CrossRef CAS PubMed.
- R. Rodriguez, D. Gau, T. Kato, N. Saffon-Merceron, A. De Cózar, F. P. Cossío and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 10414–10416 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef CAS PubMed.
- T. Sugahara, J.-D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Chem. Commun., 2018, 54, 519–522 RSC.
- C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452–2458 RSC.
- N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 2667–2679 CrossRef CAS PubMed.
- Z. Zhu, R. C. Fischer, B. D. Ellis, E. Rivard, W. A. Merrill, M. M. Olmstead, P. P. Power, J. D. Guo, S. Nagase and L. Pu, Chem.–Eur. J., 2009, 15, 5263–5272 CrossRef CAS PubMed.
- C. A. Caputo, J.-D. Guo, S. Nagase, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2012, 134, 7155–7164 CrossRef CAS PubMed.
- P. Dabringhaus, H. Scherer and I. Krossing, Nat. Synth., 2024, 3, 732–743 CrossRef CAS.
- R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 CrossRef CAS PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed.
- C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC.
- Z. Feng, Y. Fang, H. Ruan, Y. Zhao, G. Tan and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 6769–6774 CrossRef CAS PubMed.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC.
- N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 2842–2844 CrossRef CAS.
- C. A. Caputo, Z. Zhu, Z. D. Brown, J. C. Fettinger and P. P. Power, Chem. Commun., 2011, 47, 7506 RSC.
- C. Dohmeier, D. Loos and H. Schnöckel, Angew Chem. Int. Ed. Engl., 1996, 35, 129–149 CrossRef CAS.
- A. L. Hawley, C. A. Ohlin, L. Fohlmeister and A. Stasch, Chem.–Eur. J., 2017, 23, 447–455 CrossRef CAS PubMed.
- A. Lehmann, J. D. Queen, C. J. Roberts, K. Rissanen, H. M. Tuononen and P. P. Power, Angew. Chem., 2024, e202412599 CAS.
- W. Uhl, U. Schütz, W. Kaim and E. Waldhör, J. Organomet. Chem., 1995, 501, 79–85 CrossRef CAS.
- X. He, R. A. Bartlett, M. M. Olmstead, K. Ruhlandt-Senge, B. E. Sturgeon and P. P. Power, Angew Chem. Int. Ed. Engl., 1993, 32, 717–719 CrossRef.
- J. Su, X.-W. Li, R. C. Crittendon and G. H. Robinson, J. Am. Chem. Soc., 1997, 119, 5471–5472 CrossRef CAS.
- A. Barthélemy, H. Scherer, M. Daub, A. Bugnet and I. Krossing, Angew. Chem., Int. Ed., 2023, 62(47), e202311648 CrossRef PubMed.
- A. Barthélemy and I. Krossing, Inorg. Chem., 2024, 64(46), 21763–21787 CrossRef PubMed.
- O. Kysliak, H. Görls and R. Kretschmer, Dalton Trans., 2020, 49, 6377–6383 RSC.
- D. Sarkar, P. Vasko, A. F. Roper, A. E. Crumpton, M. M. D. Roy, L. P. Griffin, C. Bogle and S. Aldridge, J. Am. Chem. Soc., 2024, 146, 11792–11800 CrossRef CAS PubMed.
- R. H. Crabtree, The organometallic chemistry of the transition metals, Wiley, Hoboken, N.J, 5th edn, 2009 Search PubMed.
- K. L. Mears and P. P. Power, Acc. Chem. Res., 2022, 55, 1337–1348 CrossRef CAS PubMed.
- N. J. Hardman, P. P. Power, J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, Chem. Commun., 2001, 1866–1867 RSC.
- K. M. Byrne, R. Bjornsson and T. Krämer, Phys. Chem. Chem. Phys., 2024, 26, 30018–30034 RSC.
- J. Moilanen, P. P. Power and H. M. Tuononen, Inorg. Chem., 2010, 49, 10992–11000 CrossRef CAS PubMed.
- A. T. Henry, J. L. Bourque, I. Vacirca, D. Scheschkewitz and K. M. Baines, Organometallics, 2019, 38, 1622–1626 CrossRef CAS.
- K. K. Milnes, L. C. Pavelka and K. M. Baines, Chem. Soc. Rev., 2016, 45, 1019–1035 RSC.
- S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590–593 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2384332–2384338, 2433601, and 2433602. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06894g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |