
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study of the in situ formation of H2O2 by lytic polysaccharide monooxygenases: the reaction mechanism depends on the type of reductant†
Zhanfeng
Wang
 *a,
Xiaodi
Fu
a,
Wenwen
Diao
b,
Yao
Wu
*c,
Carme
Rovira
*de and
Binju
Wang
*c
*a,
Xiaodi
Fu
a,
Wenwen
Diao
b,
Yao
Wu
*c,
Carme
Rovira
*de and
Binju
Wang
*c
aCenter for Advanced Materials Research & Faculty of Arts and Sciences, Beijing Normal University, Zhuhai 519087, China. E-mail: zfwang@bnu.edu.cn
bOujiang Laboratory (Zhejiang Lab for Regenerative Medicine, Vision and Brain Health), Wenzhou, Zhejiang 325000, China
cState Key Laboratory of Physical Chemistry of Solid Surfaces and Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: wuyao@xmu.edu.cn; wangbinju2018@xmu.edu.cn
dDepartament de Química Inorgànica i Orgànica & IQTCUB, Universitat de Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain. E-mail: c.rovira@ub.edu
eInstitució Catalana de Recerca i Estudis Avançats (ICREA), Passeig Lluís Companys, 23, 08010 Barcelona, Spain
First published on 10th January 2025
Abstract
Lytic polysaccharide monooxygenases (LPMOs) are a unique group of monocopper enzymes that exhibit remarkable ability to catalyze the oxidative cleavage of recalcitrant carbohydrate substrates, such as cellulose and chitin, by utilizing O2 or H2O2 as the oxygen source. One of the key challenges in understanding the catalytic mechanism of LPMOs lies in deciphering how they activate dioxygen using diverse reductants. To shed light on this intricate process, we conducted in-depth investigations using quantum mechanical/molecular mechanical (QM/MM) metadynamics simulations, molecular dynamics (MD) simulations, and density functional theory (DFT) calculations. Specifically, our study focuses on elucidating the in situ formation mechanism of H2O2 by LPMOs in the presence of cellobiose dehydrogenase (CDH), a proposed natural reductant of LPMOs. Our findings reveal a proton-coupled electron transfer (PCET) process in generating the Cu(II)-hydroperoxide intermediate from the Cu(II)-superoxide intermediate. Subsequently, a direct proton transfer to the proximal oxygen of Cu(II)-hydroperoxide results in the formation of H2O2 and LPMO-Cu(II). Notably, this mechanism significantly differs from the LPMO/ascorbate system, where two hydrogen atom transfer reactions are responsible for generating H2O2 and LPMO-Cu(I). Based on our simulations, we propose a catalytic mechanism of LPMO in the presence of CDH and the polysaccharide substrate, which involves competitive binding of the substrate and CDH to the reduced LPMOs. While the CDH-bound LPMOs can activate dioxygen to generate H2O2, the substrate-bound LPMOs can employ the H2O2 generated from the LPMO/CDH system to perform the peroxygenase reactions of the polysaccharide substrate. Our work not only provides valuable insights into the reductant-dependent mechanisms of O2 activation in LPMOs but also holds implications for understanding the functions of these enzymes in their natural environment.
1 Introduction
Utilizing abundant and renewable polysaccharides, such as cellulose, starch, and chitin, to produce biofuels and commercial chemicals is highly appealing, serving as a promising solution to address future shortages and environmental threats associated with fossil-based energy and chemicals. However, there is a major obstacle to degrade polysaccharides into smaller units due to their inherent recalcitrance.1 Lytic polysaccharide monooxygenases (LPMOs), key elements of current commercial enzymatic mixtures for the processing of lignocellulosic biomass,1–4 have been well known for their roles in boosting degradation of polysaccharides since their identification in 2010.5LPMOs belong to a superfamily of mono-copper enzymes that oxidatively depolymerize polysaccharides through hydroxylation of the C1 or C4 position of the scissile glycosidic bond connecting the sugar units.6–9 The copper ion in LPMOs is coordinated by two conserved histidine residues, forming a structure known as the “histidine brace” (refer to Fig. 1).10–12 Mechanistically, LPMOs can activate both O2 and H2O2 (as a cosubstrate) to form an active species, Cu(II)-oxyl ([CuO]+), which is generally believed to be responsible for C4–H/C1–H hydroxylation.13–21 Initially, LPMOs were characterized as monooxygenases using molecular oxygen as the co-substrate.5,11,22–25 However, an increasing number of studies supported that LPMOs can also be H2O2-dependent peroxygenases.13,21,26–31 Interestingly, it has been established that substrate-free LPMOs can produce H2O2 (oxidase reactivity) through uncoupled turnover when exposed to O2 and a reducing agent,32–35 which further complicates the elucidation of their molecular mechanism. Recently, the H2O2 production capability of LPMOs has been harnessed for fueling enzyme cascade reactions.36,37 Therefore, gaining an in-depth understanding of their oxidase reactivity is essential for elucidating the atomic details of LPMOs' catalytic mechanism and for harnessing their potential applications.
 | ||
| Fig. 1 Interaction complex of LPMO and the CYT domain of CDH. The distance between CYT-Fe and LPMO-Cu is only ca. 15 Å, which can guarantee an efficient electron transfer between them. | ||
The molecular mechanism of in situ formation of H2O2 by LPMOs has been theoretically studied in several studies.20,38–40Scheme 1 provides an overview of the possible catalytic pathways for the formation of active species via the O2-dependent activity. In the resting state, the copper ion is in the Cu(II) state. It is well established that the Cu(II) center requires a priming reduction for subsequent activation of O2 or H2O2 (path a).6 This reduction can be facilitated by a reducing agent such as ascorbate or the enzyme cellobiose dehydrogenase (CDH).41,42 While ascorbate is suitable for homogeneous LPMO preparations, CDH ensures a specific and efficient electron transfer (ET) with LPMOs, making it more suitable for fermentation samples.32,41 Additionally, the use of CDH enables a kinetically controlled supply of electrons, providing advantages over small molecule reductants.43 Starting from the reduced Cu(I) ion, the binding of O2 to the active site leads to the formation of the Cu(II)-superoxide ([CuO2]+) intermediate (path b).24 The Cu(II)-superoxide species then accepts a proton and an electron (a hydrogen atom), resulting in the formation of the Cu(II)-hydroperoxide ([CuO2H]+) species (path c). The superoxide and hydroperoxide species have been captured in a neutron crystallography study, confirming the formation and stability of them.44 From the hydroperoxide species, several possible pathways have been suggested, depending on the oxygen atom being subsequently protonated. A QM/MM study by Hedegård and Ryde suggested that protonation of the distal oxygen atom of the Cu(II)-hydroperoxide species leads to the cleavage of the O–O bond, the dissociation of a water molecule, and the formation of the Cu(III)-oxyl species (path d).39 Subsequent one-electron reduction of Cu(III)-oxyl results in the formation of the active species Cu(II)-oxyl (path e).39 In contrast, a QM/MM study by Wang et al. suggested that protonation at the distal oxygen atom (path d) is not feasible and instead protonation occurs at the proximal position, coupled with an electron transfer from ascorbate to the copper ion (path f), yielding the Cu(I)–H2O2 complex.38 It is noteworthy that the energetic features depend on the functional used for the QM region and the choice of the QM region itself.39,40 A very recent QM/MM study by Hedegård and co-workers using a large QM region also excluded the feasibility of path d.40 Alternatively, the proton may transfer to the proximal oxygen atom of the Cu(II)-hydroperoxide species, forming the Cu(II)–H2O2 complex (path g),20 and subsequently one-electron reduction yields the Cu(I)–H2O2 complex (path h). From there, Cu(I)–H2O2 can readily evolve into the active species Cu(II)-oxyl with the dissociation of one water molecule (path i).19,20 Notably, the reaction of reduced LPMO with H2O2 in the absence of a substrate or excessive H2O2 can lead to enzyme damage and eventually inactivation.13,45–48
 | ||
| Scheme 1 Possible catalytic pathways for the formation of active species Cu(II)-oxyl via the O2-dependent activity in LPMOs. The proximal (p) and distal (d) oxygen atoms in Cu(II)-superoxide and Cu(II)-hydroperoxide species are labeled for clarity. | ||
It is worth noting that a consensus regarding the O2 activation mechanism by LPMOs has not been reached. The studies conducted by Hedegård and colleagues considered the involved ET process in an implicit way,20,39 while the work by Wang et al. explicitly accounted for it using the ascorbate monoanion (AscH−) as the reductant.38 Ascorbate is the most extensively studied small molecule reductant, whereas CDH is the enzymatic reductant considered as the natural electron donor for LPMOs and is more suitable for fermentation samples.32,41,49 It has been well established that the N-terminal heme b-containing cytochrome (CYT) domain of CDH serves as the electron donor and interacts directly with LPMOs.50,51 Additionally, it is well known that different reductants lead to different LPMO activities,41,52 which may be attributed to variations in the H2O2 generation rate.53–57 Mechanistically, AscH− possesses an active hydrogen atom capable of providing an electron and a proton simultaneously.38 In contrast, the reduced CYT can only supply electrons. Consequently, the use of CDH as a reducing agent is expected to result in a different O2 activation mechanism. In this study, we employ a combination of MD simulations and QM/MM MD simulations to (i) uncover the O2 activation mechanism by LPMOs when CDH is utilized as the reductant, (ii) compare this mechanism with the LPMO/ascorbate system and elucidate the reductant-dependent reactivity of LPMOs, and (iii) provide insights into the connection between O2-dependent and H2O2-dependent pathways.
2 Computational methods
2.1. System setup
The model system used in this study includes only the CYT domain of CDH and the LPMO enzyme. This choice, which lowers the computational cost, is supported by experimental evidence demonstrating that the reduced CYT domain is responsible for LPMO reduction.50 The LPMO structure was obtained from a high-resolution X-ray structure of Lentinus similis (LsAA9, PDB ID: 5ACF),58 while the CYT domain was taken from the structure of Neurospora crassa CDH (PDB ID: 4QI7).50 The interaction complex between LPMO and CYT (Fig. 1) was taken from our previous study.59 The model was initially obtained by protein–protein docking via the online program “Z-dock” and subsequently optimized by MD simulations. Based on the interaction complex, two systems, namely CYT-Fe(II) + LPMO-Cu(II)-superoxide (system I) and CYT-Fe(II) + LPMO-Cu(II)-hydroperoxide (system II), were constructed to investigate the two proton transfer (PT) processes involved in the O2 activation process. The metal-containing active sites of both LPMO and CYT, including LPMO-Cu(II)-superoxide, LPMO-Cu(II)-hydroperoxide, and CYT-Fe(II), were parameterized using the “MCPB.py” tool of AmberTools18.60,61 The Amber ff14SB force field was applied for the normal protein residues.62 The protonation states of titratable residues (His, Asp, and Glu) were consistent with our previous study,59 with the exception of histidine 147, which was doubly protonated to supply a proton during the activation process of O2 (more discussion of the protonation sate of His147 can be found in the Results and discussion section). Counter ions were added to neutralize the total charge of the system, which was solvated in a rectangular box of TIP3P waters extending up to a minimum cutoff of 16 Å from the protein surface.2.2. Classical molecular dynamics (MD) simulations
The built systems were first minimized keeping the enzymes fixed, whereas solvent molecules and ions are allowed to move, followed by a full minimization of the whole system with a combination of steepest descent and conjugate gradient methods. Afterwards, each system was gradually heated from 0 to 300 K over 300 ps with a weak restraint of 25 kcal mol−1 Å−2 on the protein atoms in the canonical ensemble. Subsequently, a 1 ns density equilibration (NPT) simulation was performed under an isothermal-isobaric Langevin thermostat63 and Berendsen barostat64 with a collision frequency of 2 ps−1 and a pressure relaxation time of 1 ps at a target temperature of 300 K and target pressure of 1.0 atm to achieve a uniform density. Following this, all restraints were removed, and each system was further equilibrated for 3 ns under the NPT ensemble. Finally, three replicas of 100 ns production simulations were performed for each system using different initial velocities. During all MD simulations, covalent bonds containing hydrogen atoms were constrained using the SHAKE algorithm,65 and an integration step of 2 fs was used. Nonbonded interactions were treated with the Particle Mesh Ewald method,66 with a cutoff set at 10 Å. All MD simulations were performed with the GPU version of the Amber 18 package.67–692.3. Constant pH MD simulations
Constant pH MD simulations using discrete protonation states in explicit solvent were performed in system II to determine the fraction of different protonation states of His147 at different pH,70 which is suggested to act as a proton supplier in this study and several others.34,39,71,72 The method involves standard MD simulations being propagated in explicit solvent and protonation state changes being attempted in implicit solvent at fixed intervals. Eight 100 ns constant pH MD simulations were carried out spanning the pH range from 4.0 to 7.5 at 0.5 pH-unit intervals. Protonation state changes were attempted every 200 fs for all constant pH MD simulations. The solvent relaxation time was set to 200 fs as suggested by Swails et al.702.4. QM/MM MD and metadynamics simulations
One representative snapshot extracted from the classical MD trajectories of each system was used for the subsequent QM/MM MD simulations, in which a hydrogen bond between doubly-protonated His147 and Gln162 is formed to ensure an efficient PT. All QM/MM MD simulations were performed with the CP2K package (2022.2 version),73 which combines the QM program QUICKSTEP74 and the MM driver FIST. A real space multigrid technique is used to compute the electrostatic coupling between the QM and MM regions.75,76 The QM region included the active site of LPMO, the reduced heme b cofactor, and several water molecules between them (refer to Fig. S1A and B† for the QM region of system I and system II, respectively). Specifically, the LPMO part consisted of the Cu cofactor, Hic1 residue, and the side chains of residues His78, His147, Gln162, and Tyr164, while the CYT part contained the heme b cofactor and the side chains of two residues that coordinate to the iron of heme, namely Met74 and His176. The remaining atoms were treated at the molecular mechanics (MM) level using the same force-field as in the classical MD simulations, whereas the dangling bonds between the QM and the MM regions were capped with hydrogen atoms. The QM region was described at the DFT(B3LYP-D3) level, using a dual basis set of Gaussian and plane-wave (GPW) formalism.74 The Gaussian double-ζ valence polarized (DZVP) basis set was used to expand the wave function,77 converging the electron density employing an auxiliary plane-wave basis set with a density cut-off of 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ry, along with Geodecker-Teter-Hutter (GTH) pseudopotentials.78,79 To accelerate the calculation of the Hartree–Fock exchange within B3LYP, the auxiliary density matrix method (ADMM) was used.80 All QM/MM MD simulations were performed in the NVT ensemble using an integration time step of 0.5 fs. The systems were first equilibrated without any restraint for 1.5 ps. Then, the metadynamics method81,82 was used to explore the PT and possibly involved ET processes. The collective variable (CV) was defined as the distance between the proton of His147 (H1) and the distal oxygen atom (O2) for the first PT process, while the difference distance between the H1 atom and the proximal oxygen (O1) atom and between the H1 atom and N1 atom of His147 was used as the CV for the second PT process (refer to Fig. S2†). The width of the Gaussian-shaped potential hills was taken between 0.1 and 0.2 Å. The Gaussian height was set to 0.6 kcal mol−1, while the time deposition interval between two consecutive Gaussians was set to 10 fs. The first re-crossing criterion, as recommended for chemical reactions, was used.83
Ry, along with Geodecker-Teter-Hutter (GTH) pseudopotentials.78,79 To accelerate the calculation of the Hartree–Fock exchange within B3LYP, the auxiliary density matrix method (ADMM) was used.80 All QM/MM MD simulations were performed in the NVT ensemble using an integration time step of 0.5 fs. The systems were first equilibrated without any restraint for 1.5 ps. Then, the metadynamics method81,82 was used to explore the PT and possibly involved ET processes. The collective variable (CV) was defined as the distance between the proton of His147 (H1) and the distal oxygen atom (O2) for the first PT process, while the difference distance between the H1 atom and the proximal oxygen (O1) atom and between the H1 atom and N1 atom of His147 was used as the CV for the second PT process (refer to Fig. S2†). The width of the Gaussian-shaped potential hills was taken between 0.1 and 0.2 Å. The Gaussian height was set to 0.6 kcal mol−1, while the time deposition interval between two consecutive Gaussians was set to 10 fs. The first re-crossing criterion, as recommended for chemical reactions, was used.83
Additional details regarding static QM/MM scanning calculations and small QM model calculations can be found in the ESI Methods section of the ESI.†
3 Results and discussion
3.1. The potential role of His147 as a proton supplier
As alluded to above, the activation of O2 requires two electrons and two protons. The electrons needed during dioxygen activation are supplied by the reduced CYT domain of CDH in this study, one at a time. The electron transfer (ET) efficiency between CYT and LPMO depends on the distance between the heme-Fe of CYT and the Cu ion of LPMO. In a prior study, we already obtained the interaction complex of CYT and LPMO using a combination of protein–protein docking calculations and classical MD simulations, showing that the distance between heme-Fe and LPMO-Cu is as short as ca. 15 Å (Fig. 1), enabling an efficient ET.59 For the PT reactions, it is clear that the proton acceptors are the Cu(II)-superoxide and the Cu(II)-hydroperoxide species for the first and the second PT, respectively. However, the identity of the proton source is still a matter of debate.34,38,39,44,71,72,84 A second-sphere histidine residue, His147, has been suggested as a potential proton donor by some experimental studies34,71,72 and theoretical studies.39,85 However, refinement of crystal structures (obtained at 100 K) indicated that this residue is in singly-protonated form even under acidic conditions due to the dimeric packing of enzymes.44,84 Therefore, it is important to investigate the protonation state of this residue under more biologically relevant conditions.Constant pH MD simulations70 are performed to determine the fraction of the doubly-protonated form of His147 at different pH values ranging from 4.0 to 7.5 under 300 K, which is shown in Fig. 2. It is seen that in solution the doubly-protonated form is predominant when pH ≤ 5.5. At pH 6, the doubly-protonated form still accounts for 36%. It is noteworthy that the optimal condition for most LPMOs (including the one used in this study) is slightly acidic85–87 and the best performance of the LPMO + CDH system was achieved at pH 6.0.41 Besides, the recommended pH range of one of the most popular commercial LPMO-containing cellulose-degrading enzyme cocktails is also slightly acidic, pH 5.0–5.5, for the best performance. Therefore, the condition of pH ≤ 6 is more relevant for LPMOs, under which the formation of the doubly-protonated form of His147 is a high-probability event. Consistent with a recent theoretical study,85 our simulations show a correlation between the protonation state and the conformation of His147. For the doubly protonated form, it adopts predominantly a solvent-exposed “outward” conformation, which is further away from the active site (Fig. S3†). The movement of this residue between bulk solvent and the active site may facilitate proton transfers to the active-site pocket, as suggested by Isaksen et al.85
 | ||
| Fig. 2 Probability of different protonation forms of His147 under different solvent pH estimated with constant pH MD simulations. The fraction of the doubly-protonated form (HIP) is shown with red dots, while the fraction of the singly-protonated form, either HIE (epsilon tautomer) or HID (delta tautomer), is shown with blue dots. | ||
Next, the potential role of the doubly protonated His147 residue as a proton supplier during the activation process of O2 is investigated with classical MD simulations. Regardless of the protonation state of the copper-oxide species, i.e., Cu(II)-superoxide and Cu(II)-hydroperoxide, the distance between the electron donor (CYT-Fe(II)) and acceptor (LPMO-Cu(II)) remains within 16 Å (see Fig. S4 and S5† for the time evolution of the Fe–Cu distance during classical MD simulations in the Cu(II)-superoxide and Cu(II)-hydroperoxide system, respectively), which is favourable for efficient ET.88 In the case of the Cu(II)-superoxide system (Fig. 3A), no direct H-bond is observed between His147 and the distal O2 atom during MD simulations. The distance between His147 and the distal O2 atom is approximately 8 Å for about four-fifths of the total simulation time (middle panel of Fig. 3A and S6†). Instead of forming a H-bond with Cu(II)-superoxide species, His147 is found to form a weak H-bond with the carbonyl group of the side chain of residue Gln162 (refer to Fig. 3A and S7†). This H-bond maintains a distance about 4 Å between H1 of His147 and the distal O2 atom. Similarly, in the case of Cu(II)-hydroperoxide (Fig. 3B), His147 forms a H-bond with the side chain of Gln162 (right panel of Fig. 3B and S8†), which is more stable compared to the Cu(II)-superoxide system. Due to the formation of this H-bond, the distance between His147 and the proximal oxygen atom (O1) of Cu(II)-hydroperoxide remains at around 3.0 Å for most of the simulation time (middle panel of Fig. 3B and S9†). Overall, our MD simulation results indicate that His147 could exist as a doubly-protonated form and serve as a proton supplier under LPMOs' working conditions, yet no direct H-bond forms between His147 and the proton destination copper-oxide species.
 | ||
| Fig. 3 No direct H-bond forms between His147 and copper-oxide species in (A) Cu(II)-superoxide and (B) Cu(II)-hydroperoxide systems. The structures illustrating the labels of key atoms are shown in the left panel. The statistics of the distance between the origin and destination of the proton transfer reaction in these two systems, the distance between the distal O2 atom and H1 atom in the Cu(II)-superoxide system (O2–H1) and the distance between the proximal O1 atom and H1 atom in the Cu(II)-hydroperoxide system (O1–H1) are shown in the middle panel. Statistics of the distance between the O3 atom of the Gln162 side chain and H1 atom are shown in the right panel, showing the H-bond formed between His147 and the carbonyl group of the Gln162 side chain. | ||
3.2. Formation of Cu(II)-hydroperoxide from Cu(II)-superoxide involves PCET
Most of the previous studies agree on the formation of Cu(II)-hydroperoxide species during the activation process of dioxygen. There is one exception worth mentioning, where the superoxide was suggested to react with a water molecule, leading to the formation of Cu(II)–OH and release of hydrosuperoxide, which may further evolve to H2O2 in solution.23 However, our QM scanning calculations show that dissociation of superoxide is highly thermodynamically unfavourable and no proton transfer from the water ligand to the dissociated superoxide is observed (Fig. S10†), which is consistent with a previous computational study.20 Moreover, the experimental result23 that a rapid regeneration (>0.15 s−1) from reduced Cu(I) to the resting Cu(II) state, in the absence of a substrate, was observed by EPR can be also well explained by the in situ formation of H2O2 by LPMOs. Additionally, the addition of superoxide dismutase was shown to have little effect on H2O2 production,72 which also refutes the dissociation of superoxide species. Therefore, the superoxide dissociation mechanism was not considered further. In principle, the formation of Cu(II)-hydroperoxide from Cu(II)-superoxide may involve either a hydrogen atom transfer or a stepwise PT and ET. In the presence of both the proton supplier (doubly protonated His147) and electron donor (reduced CYT protein), whether protonation of the Cu(II)-superoxide species is coupled with one ET is investigated using QM/MM MD and metadynamics simulations.Starting with a conformation in which the doubly-protonated His147 forms an H-bond with the side chain of Gln162, the QM/MM MD simulations confirm the stability of this H-bond, with the H1⋯O3 distance below 2 Å (Fig. S11†). Consistent with the results of classical MD simulations, no direct H-bond between Cu(II)-superoxide and His147 is observed. The distance between H1 and O2 increases from 3.5 Å to 4.5 Å during QM/MM MD simulations (Fig. S11†). The distance between the proximal O1 atom and the copper ion fluctuates around 2.0 Å (Fig. S12†), while the distance between the two oxygen atoms oscillates around 1.3 Å (Fig. S12†). The calculated spin density of the copper ion and the dioxygen moiety is ca. 0.65 and 1.15 (Fig. S13†), respectively, further confirming the starting species as Cu(II)-superoxide.59
A metadynamics simulation using the distance between the O2 and H1 atoms as the collective variable (CV) shows that the PT reaction is coupled with ET from CYT-Fe(II) to the dioxygen moiety of Cu(II)-superoxide, leading directly to the formation of the Cu(II)-hydroperoxide species. The free energy landscape reconstructed from the metadynamics simulation shows two free energy minima and one metastable intermediate (Fig. 4). In the initial state, RC1 species, the CV fluctuates around 3.5 Å. A metastable intermediate (IC1) forms when the CV decreases to approximately 2.4 Å due to the formation of a weak H-bond between the distal O2 atom and residue His147. As the CV further decreases to 1.0 Å at around 1000 fs during the metadynamics simulation (Fig. 4B), the PT occurs, triggering ET from CYT-Fe(II) to the dioxygen moiety and resulting in the formation of the Cu(II)-hydroperoxide intermediate (PC1 species in Fig. 4). The occurrence of electron transfer is supported by the increase in the spin density of iron from 0.00 to 1.05 and the decrease in the total spin densities of the two oxygen atoms from ca. 1.15 to 0.15 (Fig. 4C). After such PCET reaction, the distance between the O1 and O2 atoms increases from 1.3 Å to approximately 1.5 Å (Fig. 4B), consistent with the conversion from a superoxide to peroxide species. The free energy barrier of this PCET process is estimated to be 10.8 kcal mol−1 with metadynamics simulations, which is quite low. However, we should note that the estimated adiabatic barrier obtained from metadynamics simulations may not be entirely physical due to the involvement of long-range ET between CYT and LPMO.59 For this reason, we further analyse the corresponding ET rate using Marcus theory below.89
 | ||
| Fig. 4 The PT process from doubly-protonated His147 to Cu(II)-superoxide is coupled with an ET. (A) Free energy profile (kcal mol−1) of the PCET process involved in the formation of Cu(II)-hydroperoxide from Cu(II)-superoxide, reconstructed from QM(B3LYP)/MM-based metadynamics simulations. The collective variable (CV) is the distance between the distal O2 atom and H1 atom of His147. Legend: RC = reactant complex, IC = intermediate complex, and PC = product complex. (B) Time evolution of key distances along the simulation trajectory. (C) Time evolution of the spin density of key atoms or moiety along the simulation trajectory. (D) Representative snapshots of the QM region along the reaction pathway. Spin-up isodensity surfaces are plotted in yellow. | ||
3.3. Protonation of Cu(II)-hydroperoxide species occurs via the proximal oxygen
Starting from the conformation shown in Fig. 3B, where a strong H-bond forms between His147 and Gln162, proton transfer from doubly protonated His147 to the proximal oxygen O1 atom occurs spontaneously during QM/MM MD simulation (Fig. S14†), indicating the feasibility of this PT reaction. Prior to the reaction, the proton is located on the residue His147, and the iron remains in its reduced form (RC2 in Fig. 5). Unlike the case of Cu(II)-superoxide, this PT process is not found to be coupled with ET. The spin density on iron remains at 0.00 after the PT process (Fig. S15†), indicating the absence of ET. Metadynamics simulations further support that the facile PT from His147 to Cu(II)-hydroperoxide is not coupled with ET. Fig. 5A shows the free energy landscape of this pure PT reaction from Cu(II)-hydroperoxide (RC2) to Cu(II)–H2O2 (PC2) reconstructed from the metadynamics simulation. The small free energy barrier of 4.6 kcal mol−1 confirms the feasibility of this reaction. Scrutiny of the metadynamics trajectory clearly shows that the PT had occurred without ET. The proton transfers to the acceptor in the time window 150 fs to 350 fs (Fig. S16†), yet the iron atom remains in its reduced state (Fig. S17†). Fig. 5B shows representative structures along with spin density maps of the RC2 and PC2 species, confirming the formation of the Cu(II)–H2O2 complex (PC2 in Fig. 5). Subsequently, the newly formed hydrogen peroxide, located at the equatorial position, may be displaced by a water molecule and escape from the active site (Fig. S18†). This mechanism is consistent with the elongation of the Cu⋯O1 distance after the formation of H2O2 (Fig. S14†) and the experimental proposal that substrate-free LPMOs can provide H2O2 for substrate-bound LPMOs.13,25,90 The results obtained here, as well as previous studies,38,40,91 exclude path d in Scheme 1, previously suggested by Hedegård and Ryde,39 in which the proton transfers to the distal rather than the proximal oxygen atom. | ||
| Fig. 5 The PT process from doubly-protonated His147 to Cu(II)-hydroperoxide is not coupled with an ET when reduced CYT is used as the reductant. (A) Free energy profile (kcal mol−1) of the PT process involved in the formation of LPMO-Cu(II) and H2O2 from Cu(II)-hydroperoxide, reconstructed from QM(B3LYP)/MM-based metadynamics simulations. The CV is the difference distance between the proximal O1 atom and H1 atom of His147 and between the N1 atom and H1 atom. Legend: RC = reactant complex, PC = product complex. (B) Representative snapshots of the QM region along the reaction pathway. Spin-up isodensity surfaces are plotted in yellow. | ||
3.4. PT vs. PCET
Next, we seek to understand why the first PT of the reaction mechanism is coupled to an ET yet the second not. When no electron was supplied (i.e., the electron donor is excluded from the QM region), the QM/MM scanning calculations demonstrate that the energy steadily increases as the distance between H1 and O2 atoms (reactive coordinate) decreases (a pure PT process, red line in Fig. 6). The energy increases to 25.9 kcal mol−1 when the reactive coordinate reaches 1.02 Å. In contrast, when the electron donor of the reduced CYT was included in the QM region, the energy profile exhibits a sudden decrease when the reactive coordinate decreases to approximately 1.20 Å, indicating an ET from CYT to LPMO and thus a typical PCET process (blue line in Fig. 6). When the reactive coordinate equals 1.00 Å, the scanning energy has decreased to −9.3 kcal mol−1. Therefore, the QM/MM scanning calculations provided further confirmation that the first PT must be coupled with an ET. Additionally, it became evident that without ET, the product [Cu-OOH]2+ formed after the pure PT is an unstable species. This instability is the fundamental reason for the occurrence of PCET. These findings are generally consistent with a very recent QM/MM study by Hagemann et al. using a big QM region (up to 900 atoms)40 and shed light on the importance of ET in stabilizing the intermediate species during the PCET process. Our QM-scanning calculations (details can be found in the ESI Methods of ESI†) showed that the electron affinity increases during the two PT processes (Fig. 7). In the first PT (formation of Cu(II)-hydroperoxide from Cu(II)-superoxide), the electron affinity increases by 22.4 kcal mol−1, from 76.7 kcal mol−1 to 99.1 kcal mol−1 (blue line in Fig. 7) as the reaction coordinate (the distance between the distal oxygen O2 and the proton H1, labelled as d1 in Fig. 7) decreases from its initial optimized value of 1.75 Å to 1.00 Å. Similarly, in the second PT from Cu(II)–OOH− to Cu(II)–H2O2, the electron affinity increases by 12.6 kcal mol−1 from 75.0 kcal mol−1 to 87.6 kcal mol−1 as the reaction coordinate (d2, the distance between O1 and H1, inset of Fig. 7) decreases from 1.52 Å to 1.02 Å (red line in Fig. 7). Hence, the ability of the first PT to drive ET from the Cu-superoxide system is much stronger than that of the second PT from the Cu-hydroperoxide system. When the distance between the proton and its acceptor is approximately 1.0 Å, the electron affinity of the Cu-superoxide system is 11.5 kcal mol−1 higher than that of the Cu-hydroperoxide system. It is worth noting that the ionization energy of heme-Fe(II), calculated to be 91.3 kcal mol−1 at the same level of theory, is lower than the electron affinity of the Cu(II)-superoxide species yet higher than that of the Cu(II)-hydroperoxide species when the reaction coordinate is approximately 1.0 Å (refer to Fig. 7). Consequently, only the PT to Cu(II)-superoxide can induce long-range ET from heme-Fe(II), while the PT to Cu(II)-hydroperoxide does not. | ||
| Fig. 6 QM(UB3LYP)/MM scanning energies (kcal mol−1) of the first PT process from His147 to Cu(II)-superoxide with (blue line) or without (red line) the reductant (reduced CYT-Fe(II)) in the QM region. The reactive coordinate used here is the distance between the distal O2 atom and H1 atom of His147. When the reductant is included in the QM region, the PT can induce an ET, leading to a PCET process (blue line), which is much favored over the pure PT process (red line). | ||
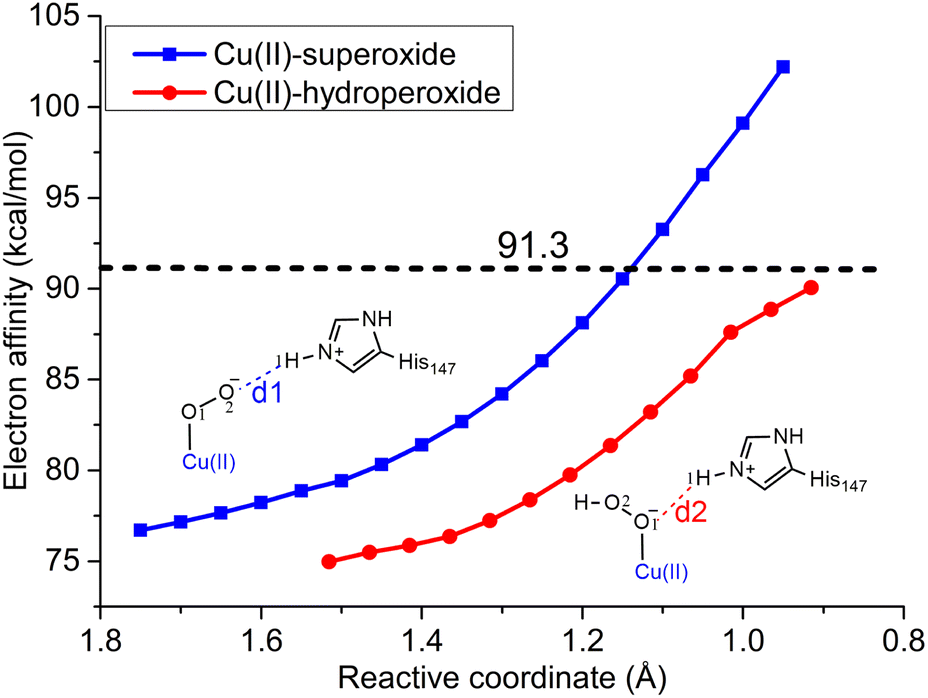 | ||
| Fig. 7 The change of electron affinity during the first (blue line) and second PT (red line) processes. For the first PT in the Cu(II)-superoxide system, the reactive coordinate is the distance between the distal O2 atom and the H1 atom (d1). For the second PT in the Cu(II)-hydroperoxide system, the reactive coordinate is the distance between the proximal O1 atom and the H1 atom (d2). The ionization energy of CYT-Fe(II) is calculated to be 91.3 kcal mol−1, which is shown by a black dashed line. | ||
The above discussion provides an explanation of why the first PT to Cu(II)-superoxide is coupled with an ET, while the second PT to Cu(II)-hydroperoxide species is a pure PT process when CYT is used as the reducing agent. We suggest that the coupling of the PT process with ET in the second PT depends on the properties of the reducing agent. For a strong reductant, with an ionization energy lower than 87.6 kcal mol−1, it may directly reduce Cu(II) to Cu(I),38 whereas a weak reductant, i.e., CDH investigated herein, cannot do it.
3.5. Estimation of the PCET rate
As noted in our previous study,59,92 the QM/MM MD simulations may overestimate the rate of a long-range electron transfer reaction. Thus, we estimate the rate of the long-range ET (kET) involved in the conversion process from Cu(II)-superoxide to Cu(II)-hydroperoxide with Marcus theory, eqn (1).89,93–95 | (1) |
In our previous study, the value of the electronic coupling between CYT and LPMO was obtained with a value of 0.47 meV.59 Using the experimentally determined ET rate of 20.6 s−1 and the typical experimental driving force of −150 meV for the ET from CYT-Fe(II) to LPMO-Cu(II) (step a in Scheme 1),1,41 we got the value of the total reorganization energy of 2.23 eV based on eqn (1).59 Furthermore, the total inner-sphere reorganization energy for the initial priming ET process was estimated to be 1.07 eV, and thus the outer-sphere energy for the CYT/LPMO system can be derived to be 2.23 eV − 1.07 eV = 1.16 eV.59 Herein, for the PCET process from Cu(II)-superoxide to Cu(II)-hydroperoxide (step c in Scheme 1), the value of ΔG can be obtained with the above metadynamics simulations, assumed to be −5.4 kcal mol−1 (−0.23 eV, refer to Fig. 4A). Consistent with our previous work,59 the electronic coupling HDA and the outer-sphere reorganization energy λo can be estimated as 0.47 meV and 1.16 eV, respectively. For the inner-sphere reorganization energy λi, we re-estimated it due to the change of the inner-sphere geometry, especially for the LPMO part (refer to Fig. S19†). For the CYT and LPMO parts, λi was estimated to be 0.05 eV and 1.16 eV, respectively (see the calculation details in the ESI†). With these values, the kET of the PCET process was estimated to be 20.4 s−1 with eqn (1) at 300 K, which is comparable to the ET rate of 20.6 s−1 in the initial reduction step (step a in Scheme 1). It is worth noting that the estimation of the λi for the LPMO part was based on a geometry in which a direct H-bond forms between the proton donor (H1) and acceptor (O2) (refer to Fig. S19†). However, our above MD simulations indicated that there is no direct H-bond formed between the proton donor and acceptor (refer to Fig. 3A). Thus, the inner-sphere reorganization energy was underestimated by the current calculations, leading to an overestimation of the rate of the PCET reaction from Cu(II)-superoxide to Cu(II)-hydroperoxide. Therefore, our above analysis suggested that the rate of the PCET reaction from Cu(II)-superoxide to Cu(II)-hydroperoxide is slower than the rate of the first priming ET reaction during the reduction of LPMO-Cu(II). This finding is consistent with a significant amount of experimental evidence indicating that the activity of LPMOs is limited by the rate of H2O2 formation when O2 is used as the oxygen source.13,37,53–57
3.6. Implication of the catalytic mechanism of LPMOs
As alluded to above, both ascorbate and CDH(CYT) can act as efficient reducing agents to supply electrons for LPMOs. However, there is a substantial difference between them. Ascorbic acid (AscH−) is a relatively poor electron donor yet a good hydrogen atom (an electron and a proton) supplier.96,97 In contrast, the CYT protein possesses a lower redox potential (ca. 0.099–0.163 V and ca. 0.35 V for CYT and ascorbate, respectively) yet no active hydrogen atom.98,99 Consequently, distinct O2 activation mechanisms have been revealed for these two reductants, summarized in Fig. 8. In the CDH-dependent pathway, the formation of Cu(II)-hydroperoxide from Cu(II)-superoxide proceeds through a PCET process. In this pathway, the electron is supplied by the reduced CYT, while a potential proton supplier, doubly protonated His147, provides the necessary proton. The electron and proton are transferred simultaneously, facilitating the conversion. On the other hand, in the ascorbate-dependent pathway, both the proton and the electron are supplied by an active hydrogen atom from ascorbic acid, resulting in a hydrogen atom transfer (HAT) process.38 For the subsequent formation of H2O2 from Cu(II)-hydroperoxide, the protonation of the proximal O1 atom cannot induce ET from CYT. Therefore, a pure PT process takes place in the CDH-dependent pathway, leading to the formation of LPMO-Cu(II) and the restoration of the resting state. In contrast, the ascorbate-dependent pathway involves a facile HAT process, resulting in the production of the reduced copper site.38 Overall, the specific O2 activation mechanism of LPMO is highly dependent on the properties of the reducing agent employed. | ||
| Fig. 8 Comparison between the CDH-dependent O2 activation mechanism (upper part) and the ascorbate-dependent one (lower part). | ||
Based on the O2 activation mechanism revealed herein, we further conjecture a catalytic mechanism of LPMO in the presence of CDH and the polysaccharide substrate (Scheme 2). It is generally accepted that the reduction of LPMO occurs before its binding to the polysaccharide substrate,9 due to the findings that the binding affinity to the polysaccharide substrate is enhanced by the reduction of LPMO51,100 and when it is bound to the substrate it is inaccessible to the reducing agent.50,51,59,101,102 Therefore, the binding affinity to the reducing agent (CDH) is expected to be slightly higher than that to the polysaccharide substrate. We note that it has been determined experimentally that the substrate binding affinity increases by only about 1 kcal mol−1 due to LPMO reduction.51 Therefore, it is reasonable to assume that CDH may competitively bind to LPMO even after LPMO reduction. As illustrated in Scheme 2, some reduced LPMO molecules may bind to the substrate and some others may bind to the reducing agent. Such competitive binding picture of the substrate and CDH to LPMO-Cu(I) can rationalize both the generation of H2O2 and the substrate oxidation. For LPMO-Cu(I) bound to CDH, it can act as an oxidase to generate H2O2. While for LPMO-Cu(I) bound to the substrate, it can employ the H2O2 generated from the LPMO/CDH system to perform substrate oxidation reactions via the H2O2-dependent pathway.13,19,25,90
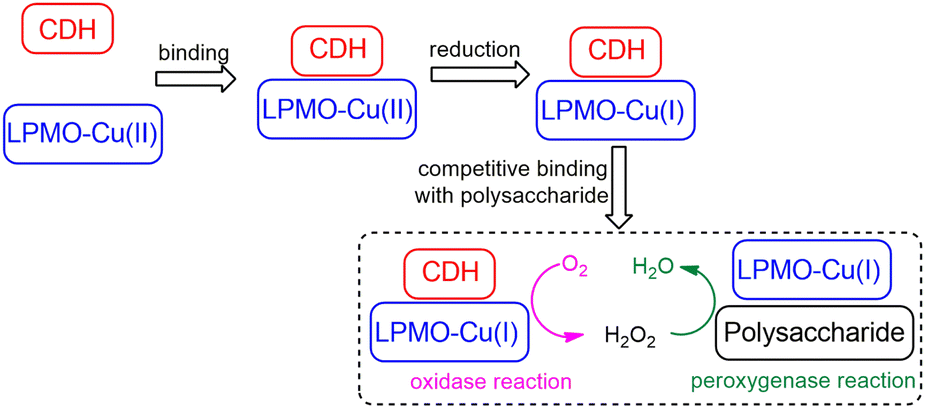 | ||
| Scheme 2 Proposed catalytic mechanism of LPMO in the presence of CDH and the polysaccharide substrate. Oxidized LPMOs prefer to bind with the reducing agent CDH, yet reduced LPMOs competitively bind with CDH and the polysaccharide substrate. LPMOs bound with CDH can use dioxygen to generate hydrogen peroxide, which can diffuse to the solvent and be used by substrate-bound LPMOs to perform substrate oxidation reactions. | ||
Of note, different LPMOs have been observed to possess divergent oxidase activities,35,55,103,104 likely due to their different structures and redox potentials.103 For instance, LPMOs from the AA9 and AA11 families can efficiently produce H2O2 through their intrinsic oxidase activities, while LPMOs from the AA10 family largely rely on H2O2 generated from the auto-oxidation of reductants.55,104 Additionally, the proton donors can vary across different LPMOs. In the AA9 family, a doubly-protonated histidine (His) residue has been suggested as the proton donor, while in the AA10 family, a neutral glutamic acid (Glu) residue has been proposed.20 Therefore, the exact O2 activation mechanism may vary in different LPMOs families, especially for the second PT reaction that can occur with or without an electron input.
4 Conclusions
In summary, we employ a combination of QM/MM metadynamics simulations, MD simulations, QM calculations, and static QM/MM calculations to investigate the mechanism of dioxygen activation by LPMOs in the presence of the enzymatic reductant CDH. Our study yields the following key findings: (i) dioxygen activation proceeds through a H2O2 formation pathway, involving first the formation of Cu(II)-superoxide (Cu(II)-OO˙−) and, subsequently, Cu(II)-hydroperoxide (Cu(II)-OOH−). (ii) The conversion from superoxide to hydroperoxide is a PCET process, where the electron is donated by CDH and the proton is donated by the doubly-protonated His147 residue. This step is identified as the rate-limiting step in the overall O2 activation process. (iii) The transformation from hydroperoxide to H2O2 occurs through a pure PT process. Mechanistically, we show that the protonation of the distal O atom of Cu(II)-superoxide, en route to the hydroperoxide species, is coupled with an electron input. This coupling can take place through either a PCET or hydrogen atom transfer (HAT) mechanism, depending on the identities of the electron and proton donors. Conversely, the protonation of the proximal oxygen atom of Cu(II)-hydroperoxide, en route to H2O2, may occur with or without an electron input, depending on the properties of the reductant. In the case of ascorbic acid, which is a poor electron donor yet an efficient H atom donor, two HAT processes are involved during the in situ formation of H2O2. Therefore, the O2 activation mechanism of LPMOs is highly dependent on the properties of the reductant. Based on our simulations and previous findings, we propose a catalytic mechanism of LPMO in the presence of CDH and the polysaccharide substrate, which involves the competitive binding of the substrate and CDH to the reduced LPMOs. While the CDH-bound LPMOs can activate dioxygen to generate H2O2, the substrate-bound LPMOs can employ the H2O2 generated from the LPMO/CDH system to perform the peroxygenase reactions of the polysaccharide substrate. These mechanistic insights contribute to our understanding of the chemistry of LPMOs in their natural environment.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. W.: conceptualization, investigation, formal analysis, methodology, resources, writing, supervision, funding acquisition, review & editing; X. F.: investigation, formal analysis, review & editing; W. D.: formal analysis, review & editing; Y. W.: investigation, formal analysis, methodology, review & editing; C. R.: funding acquisition, supervision, methodology, review & editing; B. W.: funding acquisition, resources, conceptualization, supervision, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22203007 to Z. W., no. 22073077 and 22122305 to B. W.), the Start-up Funding from Beijing Normal University (no. 310432104 to Z. W.), the Spanish Ministry of Science, Innovation and Universities (MICINN/AEI/FEDER, UE, PID2020-118893GB-100), the Spanish Structures of Excellence María de Maeztu (CEX2021-001202-M) and the Agency for Management of University and Research Grants of Catalonia (AGAUR, 2021-SGR-00680). Dr Z. Wang is supported by the Interdisciplinary Intelligence SuperComputer Center of Beijing Normal University Zhuhai.Notes and references
- K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars, Chem. Rev., 2018, 118, 2593–2635 CrossRef CAS PubMed.
- K. S. Johansen, Discovery and industrial applications of lytic polysaccharide mono-oxygenases, Biochem. Soc. Trans., 2016, 44, 143–149 CrossRef CAS PubMed.
- X. Cai, J. Hua, Z. M. Lin, C. Y. Sun, C. H. Hu, X. J. Zhang, J. D. Shen, H. Y. Zhou, H. Y. Wang, K. Q. Chen, D. S. Chen, X. P. Cheng, M. Li, Z. Q. Liu and Y. G. Zheng, Enhanced Enzymatic Hydrolysis of High-Solids Content Corncobs by a Lytic Polysaccharide Monooxygenase from Podospora anserina S Mat+ for Valuable Monosaccharides, ACS Sustainable Chem. Eng., 2023, 11, 9858–9867 CrossRef CAS.
- X. Qin, K. Yang, X. L. Wang, T. Tu, Y. Wang, J. Zhang, X. Y. Su, B. Yao, H. Q. Huang and H. Y. Luo, Insights into the H2O2-Driven Lytic Polysaccharide Monooxygenase Activity on Efficient Cellulose Degradation in the White Rot Fungus Irpex lacteus, J. Agric. Food Chem., 2023, 71, 8104–8111 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. L. Liu, H. Zhai, M. Sorlie and V. G. H. Eijsink, An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- P. Chylenski, B. Bissaro, M. Sorlie, Å. K. Rohr, A. Várnai, S. J. Horn and V. G. H. Eijsink, Lytic Polysaccharide Monooxygenases in Enzymatic Processing of Lignocellulosic Biomass, ACS Catal., 2019, 9, 4970–4991 CrossRef CAS.
- X. Guo, Y. J. An, F. F. Liu, F. P. Lu and B. Wang, Lytic polysaccharide monooxygenase-A new driving force for lignocellulosic biomass degradation, Bioresour. Technol., 2022, 362, 127803 CrossRef CAS PubMed.
- B. Bissaro, A. Várnai, A. K. Rohr and V. G. H. Eijsink, Oxidoreductases and Reactive Oxygen Species in Conversion of Lignocellulosic Biomass, Microbiol. Mol. Biol. Rev., 2018, 82, e00029 CrossRef CAS PubMed.
- A. Munzone, V. G. H. Eijsink, J. G. Berrin and B. Bissaro, Expanding the catalytic landscape of metalloenzymes with lytic polysaccharide monooxygenases, Nat. Rev. Chem, 2024, 8, 106–119 CrossRef CAS PubMed.
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Bracing copper for the catalytic oxidation of C-H bonds, Nat. Catal., 2018, 1, 571–577 CrossRef CAS.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J. C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jorgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components, Proc. Natl. Acad. Sci., India, 2011, 108, 15079–15084 CAS.
- G. Vaaje-Kolstad, Z. Forsberg, J. S. M. Loose, B. Bissaro and V. G. H. Eijsink, Structural diversity of lytic polysaccharide monooxygenases, Curr. Opin. Struct. Biol., 2017, 44, 67–76 CrossRef CAS PubMed.
- B. Bissaro, Å. K. Rohr, G. Müller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2, Nat. Chem. Biol., 2017, 13, 1123–1128 CrossRef CAS PubMed.
- M. M. Hagemann and E. D. Hedegard, Molecular Mechanism of Substrate Oxidation in Lytic Polysaccharide Monooxygenases: Insight from Theoretical Investigations, Chem.–Eur. J., 2023, 29, e202202379 CrossRef CAS PubMed.
- B. Bissaro, B. Streit, I. Isaksen, V. G. H. Eijsink, G. T. Beckham, J. L. DuBois and Å. K. Rohr, Molecular mechanism of the chitinolytic peroxygenase reaction, Proc. Natl. Acad. Sci., India, 2020, 117, 1504–1513 CAS.
- S. Kim, J. Ståhlberg, M. Sandgren, R. S. Paton and G. T. Beckham, Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism, Proc. Natl. Acad. Sci., India, 2014, 111, 149–154 CAS.
- E. D. Hedegård and U. Ryde, Targeting the reactive intermediate in polysaccharide monooxygenases, J. Biol. Inorg. Chem., 2017, 22, 1029–1037 CrossRef PubMed.
- L. Bertini, R. Breglia, M. Lambrughi, P. Fantucci, L. De Gioia, M. Borsari, M. Sola, C. A. Bortolotti and M. Bruschi, Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations, Inorg. Chem., 2018, 57, 86–97 CrossRef CAS PubMed.
- B. J. Wang, E. M. Johnston, P. F. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate, ACS Catal., 2018, 8, 1346–1351 CrossRef CAS.
- O. Caldararu, E. Oksanen, U. Ryde and E. D. Hedegård, Mechanism of hydrogen peroxide formation by lytic polysaccharide monooxygenase, Chem. Sci., 2019, 10, 576–586 RSC.
- H. Lim, M. T. Brueggemeyer, W. J. Transue, K. K. Meier, S. M. Jones, T. Kroll, D. Sokaras, B. Kelemen, B. Hedman, K. O. Hodgson and E. I. Solomon, Kβ X-ray Emission Spectroscopy of Cu(I)-Lytic Polysaccharide Monooxygenase: Direct Observation of the Frontier Molecular Orbital for H2O2 Activation, J. Am. Chem. Soc., 2023, 145, 16015–16025 CrossRef CAS PubMed.
- W. T. Beeson, C. M. Phillips, J. H. D. Cate and M. A. Marletta, Oxidative Cleavage of Cellulose by Fungal Copper-Dependent Polysaccharide Monooxygenases, J. Am. Chem. Soc., 2012, 134, 890–892 CrossRef CAS PubMed.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton, K. S. Johansen, K. O. Hodgson, B. Hedman and E. I. Solomon, Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases, Proc. Natl. Acad. Sci., India, 2014, 111, 8797–8802 CAS.
- G. Courtade, L. Ciano, A. Paradisi, P. J. Lindley, Z. Forsberg, M. Sorlie, R. Wimmer, G. J. Davies, V. G. H. Eijsink, P. H. Walton and F. L. Aachmann, Mechanistic basis of substrate-O2 coupling within a chitin-active lytic polysaccharide monooxygenase: An integrated NMR/EPR study, Proc. Natl. Acad. Sci., India, 2020, 117, 19178–19189 CAS.
- J. A. Hangasky, A. T. Iavarone and M. A. Marletta, Reactivity of O2versus H2O2 with polysaccharide monooxygenases, Proc. Natl. Acad. Sci., India, 2018, 115, 4915–4920 CAS.
- S. Kuusk, B. Bissaro, P. Kuusk, Z. Forsberg, V. G. H. Eijsink, M. Sorlie and P. Väljamäe, Kinetics of H2O2-driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase, J. Biol. Chem., 2018, 293, 523–531 CrossRef CAS PubMed.
- R. Kont, B. Bissaro, V. G. H. Eijsink and P. Väljamäe, Kinetic insights into the peroxygenase activity of cellulose-active lytic polysaccharide monooxygenases (LPMOs), Nat. Commun., 2020, 11, 5786 CrossRef CAS PubMed.
- T. M. Hedison, E. Breslmayr, M. Shanmugam, K. Karnpakdee, D. J. Heyes, A. P. Green, R. Ludwig, N. S. Scrutton and D. Kracher, Insights into the H2O2-driven catalytic mechanism of fungal lytic polysaccharide monooxygenases, FEBS J., 2021, 288, 4115–4128 CrossRef CAS PubMed.
- S. Brander, R. Tokin, J. O. Ipsen, P. E. Jensen, C. Hernández-Rollán, M. H. H. Norholm, L. Lo Leggio, P. Dupree and K. S. Johansen, Scission of Glucosidic Bonds by a Lentinus similis Lytic Polysaccharide Monooxygenases Is Strictly Dependent on H2O2 while the Oxidation of Saccharide Products Depends on O2, ACS Catal., 2021, 11, 13848–13859 CrossRef CAS.
- D. Kracher, Z. Forsberg, B. Bissaro, S. Gangl, M. Preims, C. Sygmund, V. G. H. Eijsink and R. Ludwig, Polysaccharide oxidation by lytic polysaccharide monooxygenase is enhanced by engineered cellobiose dehydrogenase, FEBS J., 2020, 287, 897–908 CrossRef CAS PubMed.
- W. Gao and H. Yin, Insight into the peroxygenase activity of lytic polysaccharide monooxygenases (LPMO): Recent progress and mechanistic understanding, Chem. Phys. Rev., 2023, 4, 031310 CrossRef CAS.
- R. Kittl, D. Kracher, D. Burgstaller, D. Haltrich and R. Ludwig, Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay, Biotechnol. Biofuels, 2012, 5, 79 CrossRef CAS PubMed.
- K. R. Hall, C. Joseph, I. Ayuso-Fernández, A. Tamhankar, L. Rieder, R. Skaali, O. Golten, F. Neese, Å. K. Rohr, S. A. V. Jannuzzi, S. DeBeer, V. G. H. Eijsink and M. Sorlie, A Conserved Second Sphere Residue Tunes Copper Site Reactivity in Lytic Polysaccharide Monooxygenases, J. Am. Chem. Soc., 2023, 145, 18888–18903 CrossRef CAS PubMed.
- K. Chorozian, A. Karnaouri, N. Georgaki-Kondyli, A. Karantonis and E. Topakas, Assessing the role of redox partners in TthLPMO9G and its mutants: focus on H2O2 production and interaction with cellulose, Biotechnol. Biofuels, 2024, 17, 19 CrossRef CAS PubMed.
- L. Rieder, A. A. Stepnov, M. Sorlie and V. G. H. Eijsink, Fast and Specific Peroxygenase Reactions Catalyzed by Fungal Mono-Copper Enzymes, Biochemistry, 2021, 60, 3633–3643 CrossRef CAS PubMed.
- F. Li, F. Y. Ma, H. L. Zhao, S. Zhang, L. Wang, X. Y. Zhang and H. B. Yu, A Lytic Polysaccharide Monooxygenase from a White-Rot Fungus Drives the Degradation of Lignin by a Versatile Peroxidase, Appl. Environ. Microbiol., 2019, 85, e02803–e02818 CAS.
- A. A. Stepnov, V. G. H. Eijsink and Z. Forsberg, Enhanced in situ H2O2 production explains synergy between an LPMO with a cellulose-binding domain and a single-domain LPMO, Sci. Rep., 2022, 12, 6129 CrossRef CAS PubMed.
- B. J. Wang, P. H. Walton and C. Rovira, Molecular Mechanisms of Oxygen Activation and Hydrogen Peroxide Formation in Lytic Polysaccharide Monooxygenases, ACS Catal., 2019, 9, 4958–4969 CrossRef CAS PubMed.
- E. D. Hedegård and U. Ryde, Molecular mechanism of lytic polysaccharide monooxygenases, Chem. Sci., 2018, 9, 3866–3880 RSC.
- M. M. Hagemann, E. K. Wieduwilt, U. Ryde and E. D. Hedegård, Investigating the Substrate Oxidation Mechanism in Lytic Polysaccharide Monooxygenase: H2O2- versus O2-Activation, Inorg. Chem., 2024, 63, 21929–21940 CrossRef CAS PubMed.
- D. Kracher, S. Scheiblbrandner, A. K. G. Felice, E. Breslmayr, M. Preims, K. Ludwicka, D. Haltrich, V. G. H. Eijsink and R. Ludwig, Extracellular electron transfer systems fuel cellulose oxidative degradation, Science, 2016, 352, 1098–1101 CrossRef CAS PubMed.
- L. Zhang, C. V. F. P. Laurent, L. Schwaiger, L. S. Wang, S. Ma and R. Ludwig, Interdomain Linker of the Bioelecrocatalyst Cellobiose Dehydrogenase Governs the Electron Transfer, ACS Catal., 2023, 13, 8195–8205 CrossRef CAS PubMed.
- J. S. M. Loose, Z. Forsberg, D. Kracher, S. Scheiblbrandner, R. Ludwig, V. G. H. Eijsink and G. Vaaje-Kolstad, Activation of bacterial lytic polysaccharide monooxygenases with cellobiose dehydrogenase, Protein Sci., 2016, 25, 2175–2186 CrossRef CAS PubMed.
- G. C. Schröder, W. B. O'Dell, S. P. Webb, P. K. Agarwal and F. Meilleur, Capture of activated dioxygen intermediates at the copper-active site of a lytic polysaccharide monooxygenase, Chem. Sci., 2022, 13, 13303–13320 RSC.
- G. Müller, P. Chylenski, B. Bissaro, V. G. H. Eijsink and S. J. Horn, The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail, Biotechnol. Biofuels, 2018, 11, 209 CrossRef PubMed.
- F. Filandr, D. Kavan, D. Kracher, C. V. F. P. Laurent, R. Ludwig, P. Man and P. Halada, Structural Dynamics of Lytic Polysaccharide Monooxygenase during Catalysis, Biomolecules, 2020, 10, 242 CrossRef CAS PubMed.
- A. Kadic, A. Várnai, V. G. H. Eijsink, S. J. Horn and G. Lidén, In situ measurements of oxidation-reduction potential and hydrogen peroxide concentration as tools for revealing LPMO inactivation during enzymatic saccharification of cellulose, Biotechnol. Biofuels, 2021, 14, 46 CrossRef CAS PubMed.
- I. A. Christensen, V. G. H. Eijsink, A. A. Stepnov, G. Courtade and F. L. Aachmann, Following the Fate of Lytic Polysaccharide Monooxygenases under Oxidative Conditions by NMR Spectroscopy, Biochemistry, 2023, 62, 1976–1993 CrossRef CAS PubMed.
- C. M. Phillips, W. T. Beeson, J. H. Cate and M. A. Marletta, Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora crassa, ACS Chem. Biol., 2011, 6, 1399–1406 CrossRef CAS PubMed.
- T. C. Tan, D. Kracher, R. Gandini, C. Sygmund, R. Kittl, D. Haltrich, B. M. Hällberg, R. Ludwig and C. Divne, Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation, Nat. Commun., 2015, 6, 7542 CrossRef PubMed.
- G. Courtade, R. Wimmer, Å. K. Rohr, M. Preims, A. K. G. Felice, M. Dimarogona, G. Vaaje-Kolstad, M. Sorlie, M. Sandgren, R. Ludwig, V. G. H. Eijsink and F. L. Aachmann, Interactions of a fungal lytic polysaccharide monooxygenase with β-glucan substrates and cellobiose dehydrogenase, Proc. Natl. Acad. Sci., India, 2016, 113, 5922–5927 CAS.
- M. Frommhagen, M. J. Koetsier, A. H. Westphal, J. Visser, S. W. A. Hinz, J. P. Vincken, W. J. H. van Berkel, M. A. Kabel and H. Gruppen, Lytic polysaccharide monooxygenases from Myceliophthora thermophila C1 differ in substrate preference and reducing agent specificity, Biotechnol. Biofuels, 2016, 9, 186 CrossRef PubMed.
- O. Golten, I. Ayuso-Fernández, K. R. Hall, A. A. Stepnov, M. Sorlie, Å. K. Rohr and V. G. H. Eijsink, Reductants fuel lytic polysaccharide monooxygenase activity in a pH-dependent manner, FEBS Lett., 2023, 597, 1363–1374 CrossRef CAS PubMed.
- F. Filandr, P. Man, P. Halada, H. H. Chang, R. Ludwig and D. Kracher, The H2O2-dependent activity of a fungal lytic polysaccharide monooxygenase investigated with a turbidimetric assay, Biotechnol. Biofuels, 2020, 13, 37 CrossRef CAS PubMed.
- A. A. Stepnov, Z. Forsberg, M. Sorlie, G. S. Nguyen, A. Wentzel, A. K. Rohr and V. G. H. Eijsink, Unraveling the roles of the reductant and free copper ions in LPMO kinetics, Biotechnol. Biofuels, 2021, 14, 28 CrossRef CAS PubMed.
- O. A. Hegnar, D. M. Petrovic, B. Bissaro, G. Alfredsen, A. Várnal and V. G. H. Eijsink, pH-Dependent Relationship between Catalytic Activity and Hydrogen Peroxide Production Shown via Characterization of a Lytic Polysaccharide Monooxygenase from Gloeophyllum trabeum, Appl. Environ. Microbiol., 2019, 85, e02612–e02618 CrossRef CAS PubMed.
- A. A. Stepnov, I. A. Christensen, Z. Forsberg, F. L. Aachmann, G. Courtade and V. G. H. Eijsink, The impact of reductants on the catalytic efficiency of a lytic polysaccharide monooxygenase and the special role of dehydroascorbic acid, FEBS Lett., 2022, 596, 53–70 CrossRef CAS PubMed.
- K. E. H. Frandsen, T. J. Simmons, P. Dupree, J. C. N. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases, Nat. Chem. Biol., 2016, 12, 298–303 CrossRef CAS PubMed.
- Z. F. Wang, S. S. Feng, C. Rovira and B. J. Wang, How Oxygen Binding Enhances Long-Range Electron Transfer: Lessons From Reduction of Lytic Polysaccharide Monooxygenases by Cellobiose Dehydrogenase, Angew. Chem. Int. Ed., 2021, 60, 2385–2392 CrossRef CAS PubMed.
- P. F. Li and K. M. Merz, MCPB.py: A Python Based Metal Center Parameter Builder, J. Chem. Inf. Model., 2016, 56, 599–604 CrossRef CAS PubMed.
- P. F. Li and K. M. Merz, Metal Ion Modeling Using Classical Mechanics, Chem. Rev., 2017, 117, 1564–1686 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. A. Izaguirre, D. P. Catarello, J. M. Wozniak and R. D. Skeel, Langevin stabilization of molecular dynamics, J. Chem. Phys., 2001, 114, 2090–2098 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, Molecular-Dynamics with Coupling to an External Bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- V. Krautler, W. F. Van Gunsteren and P. H. Hunenberger, A fast SHAKE: Algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations, J. Comput. Chem., 2001, 22, 501–508 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- A. W. Götz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef PubMed.
- S. Le Grand, A. W. Götz and R. C. Walker, SPFP: Speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- J. M. Swails, D. M. York and A. E. Roitberg, Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation, J. Chem. Theory Comput., 2014, 10, 1341–1352 CrossRef CAS PubMed.
- W. B. O'Dell, P. K. Agarwal and F. Meilleur, Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase, Angew. Chem. Int. Ed., 2017, 56, 767–770 CrossRef PubMed.
- E. A. Span, D. L. M. Suess, M. C. Deller, R. D. Britt and M. A. Marletta, The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase, ACS Chem. Biol., 2017, 12, 1095–1103 CrossRef CAS PubMed.
- T. D. Kuhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schutt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borstnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Muller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Gloss, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. Laino, F. Mohamed, A. Laio and M. Parrinello, An efficient real space multigrid OM/MM electrostatic coupling, J. Chem. Theory Comput., 2005, 1, 1176–1184 CrossRef CAS PubMed.
- A. Laio, J. VandeVondele and U. Rothlisberger, A Hamiltonian electrostatic coupling scheme for hybrid Car-Parrinello molecular dynamics simulations, J. Chem. Phys., 2002, 116, 6941–6947 CrossRef CAS.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Relativistic separable dual-space Gaussian pseudopotentials from H to Rn, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- M. Guidon, J. Hutter and J. VandeVondele, Auxiliary Density Matrix Methods for Hartree-Fock Exchange Calculations, J. Chem. Theory Comput., 2010, 6, 2348–2364 CrossRef CAS PubMed.
- A. Barducci, G. Bussi and M. Parrinello, Well-tempered metadynamics: A smoothly converging and tunable free-energy method, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- A. Laio and M. Parrinello, Escaping free-energy minima, Proc. Natl. Acad. Sci., India, 2002, 99, 12562–12566 CAS.
- B. Ensing, A. Laio, M. Parrinello and M. L. Klein, A recipe for the computation of the free energy barrier and the lowest free energy path of concerted reactions, J. Phys. Chem. B, 2005, 109, 6676–6687 CrossRef CAS PubMed.
- S. Banerjee, S. J. Muderspach, T. Tandrup, K. E. H. Frandsen, R. K. Singh, J. O. Ipsen, C. Hernández-Rollán, M. H. H. Norholm, M. J. Bjerrum, K. S. Johansen and L. Lo Leggio, Protonation State of an Important Histidine from High Resolution Structures of Lytic Polysaccharide Monooxygenases, Biomolecules, 2022, 12, 194 CrossRef CAS PubMed.
- I. Isaksen, S. Jana, C. M. Payne, B. Bissaro and Å. K. Rohr, The rotamer of the second-sphere histidine in AA9 lytic polysaccharide monooxygenase is pH dependent, Biophys. J., 2024, 123, 1139–1151 CrossRef CAS PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Fungal Cellulases, Chem. Rev., 2015, 115, 1308–1448 CrossRef CAS PubMed.
- W. Purahong, T. Wubet, G. Lentendu, B. Hoppe, K. Jariyavidyanont, T. Arnstadt, K. Baber, P. Otto, H. Kellner, M. Hofrichter, J. Bauhus, W. W. Weisser, D. Krüger, E. D. Schulze, T. Kahl and F. Buscot, Determinants of Deadwood-Inhabiting Fungal Communities in Temperate Forests: Molecular Evidence From a Large Scale Deadwood Decomposition Experiment, Front. Microbiol., 2018, 9, 2120 CrossRef PubMed.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature of Biological Electron-Transfer, Nature, 1992, 355, 796–802 CrossRef CAS PubMed.
- R. A. Marcus and N. Sutin, Electron Transfers in Chemistry and Biology, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- H. C. Zhou, Y. B. Zhang, T. Li, H. D. Tan, G. H. Li and H. Yin, Distinct Interaction of Lytic Polysaccharide Monooxygenase with Cellulose Revealed by Computational and Biochemical Studies, J. Phys. Chem. Lett., 2020, 11, 3987–3992 CrossRef CAS PubMed.
- B. J. Wang, Z. F. Wang, G. J. Davies, P. H. Walton and C. Rovira, Activation of O2 and H2O2 by Lytic Polysaccharide Monooxygenases, ACS Catal., 2020, 10, 12760–12769 CrossRef CAS.
- W. H. Fang, S. S. Feng, Z. H. Jiang, W. Z. Liang, P. F. Li and B. J. Wang, Understanding the Key Roles of pH Buffer in Accelerating Lignin Degradation by Lignin Peroxidase, JACS Au, 2023, 3, 536–549 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. V. Soudackov, Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry, J. Phys. Chem. B, 2008, 112, 14108–14123 CrossRef CAS PubMed.
- M. C. Kessinger, J. Xu, K. Cui, Q. Loague, A. V. Soudackov, S. Hammes-Schiffer and G. J. Meyer, Direct Evidence for a Sequential Electron Transfer-Proton Transfer Mechanism in the PCET Reduction of a Metal Hydroxide Catalyst, J. Am. Chem. Soc., 2024, 146, 1742–1747 CrossRef CAS PubMed.
- A. Nilsen-Moe, C. R. Reinhardt, P. Huang, H. Agarwala, R. Lopes, M. Lasagna, S. Glover, S. Hammes-Schiffer, C. Tommos and L. Hammarstroem, Switching the proton-coupled electron transfer mechanism for non-canonical tyrosine residues in a de novo protein, Chem. Sci., 2024, 15, 3957–3970 RSC.
- D. Njus and P. M. Kelley, Vitamin-C and Vitamin-E Donate Single Hydrogen-Atoms Invivo, FEBS Lett., 1991, 284, 147–151 CrossRef CAS PubMed.
- D. Njus, P. M. Kelley, Y. J. Tu and H. B. Schlegel, Ascorbic acid: The chemistry underlying its antioxidant properties, Free Radical Biol. Med., 2020, 159, 37–43 CrossRef CAS PubMed.
- C. Sygmund, D. Kracher, S. Scheiblbrandner, K. Zahma, A. K. G. Felice, W. Harreither, R. Kittl and R. Ludwig, Characterization of the Two Neurospora crassa Cellobiose Dehydrogenases and Their Connection to Oxidative Cellulose Degradation, Appl. Environ. Microbiol., 2012, 78, 6161–6171 CrossRef CAS PubMed.
- T. Matsui, Y. Kitagawa, M. Okumura and Y. Shigeta, Accurate Standard Hydrogen Electrode Potential and Applications to the Redox Potentials of Vitamin C and NAD/NADH, J. Phys. Chem. A, 2015, 119, 369–376 CrossRef CAS PubMed.
- D. Kracher, M. Andlar, P. G. Furtmüller and R. Ludwig, Active-site copper reduction promotes substrate binding of fungal lytic polysaccharide monooxygenase and reduces stability, J. Biol. Chem., 2018, 293, 1676–1687 CrossRef CAS PubMed.
- C. V. F. P. Laurent, E. Breslmayr, D. Tunega, R. Ludwig and C. Oostenbrink, Interaction between Cellobiose Dehydrogenase and Lytic Polysaccharide Monooxygenase, Biochemistry, 2019, 58, 1226–1235 CrossRef CAS PubMed.
- B. Bissaro, I. Isaksen, G. Vaaje-Kolstad, V. G. H. Eijsink and Å. K. Rohr, How a Lytic Polysaccharide Monooxygenase Binds Crystalline Chitin, Biochemistry, 2018, 57, 1893–1906 CrossRef CAS PubMed.
- L. Rieder, D. Petrovic, P. Väljamäe, V. G. H. Eijsink and M. Sorlie, Kinetic Characterization of a Putatively Chitin-Active LPMO Reveals a Preference for Soluble Substrates and Absence of Monooxygenase Activity, ACS Catal., 2021, 11, 11685–11695 CrossRef CAS PubMed.
- L. Rieder and M. Sorlie, Recent Advances in Understanding LPMO Catalysis, Biochemistry, 2023, 62, 3170–3172 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06906d |
| This journal is © The Royal Society of Chemistry 2025 |