
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversing selectivity of bambusuril macrocycles toward inorganic anions by installing spacious substituents on their portals†
Carola
Rando‡
ab,
Surbhi
Grewal‡
 ab,
Jan
Sokolov
ab,
Petr
Kulhánek
*c and
Vladimír
Šindelář
*ab
ab,
Jan
Sokolov
ab,
Petr
Kulhánek
*c and
Vladimír
Šindelář
*ab
aDepartment of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
bRECETOX, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic. E-mail: sindelar@chemi.muni.cz
cNational Centre for Biomolecular Research, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic. E-mail: kulhanek@chemi.muni.cz
First published on 6th December 2024
Abstract
Two chiral bambusurils, which are diastereomers to each other, show remarkable differences in their binding affinity and selectivity toward inorganic anions as determined by isothermal titration calorimetry. These differences are explained by quantum-chemical calculations.
Introduction
Artificial molecules with the ability to act as receptors of inorganic anions are investigated in many fields, including anion sensing,1,2 organocatalysis,3,4 anion remediation in industrial wastewater,5–7 and others. Supramolecular chemists dealing with the host–guest chemistry of inorganic anions are intrigued by the design of anion receptors with optimal binding affinity and selectivity toward the selected analyte.3,7–9 This design is particularly challenging in the case of receptors for inorganic anions featuring relatively small differences in their sizes. Among supramolecular host molecules, macrocycles are especially appreciated for their ability to discriminate particular anions because they are more rigid compared to their acyclic analogues.10 Macrocycles are valued for their high affinity, which is achieved thanks to multiple binding motifs. Moreover, the selectivity of macrocycles toward a particular anion can be tuned by the number of their repeating units, as can be illustrated on calix[n]pyrroles or hemicucurbit[n]urils. While calix[4]pyrroles show 100 times higher selectivity for fluoride compared to other halides and phosphates, calix[6]pyrroles preferentially bind iodide.11 Similarly, hemicucurbit[6]urils prefer to bind chloride over iodide, while hemicucurbit[8]urils form the most stable complexes with PF6− and SbF6−.12Bambus[n]urils are anion receptors consisting of n glycoluril units connected by n methylene bridges.13,14 Until now, only four (n = 4) and six (n = 6) membered bambusuril homologues have been reported.15,16 As the four-membered bambusurils do not bind to any anion, the six-membered homologues form stable complexes with many inorganic anions. The typical complex in the solution consists of a single anion included within the center of the macrocycle cavity. It has been reported that the binding affinity of bambus[6]urils (BU[6]s) to anions can be modulated by installing different substituents on the nitrogen atoms of the BU[6] portals.17,18 However, independently of their substituents, BU[6]s have shown essentially the same selectivity for all inorganic anions ranging from small F− to large ClO4− in the same solvent. This is mainly due to the flexibility of the BU[6] macrocyclic framework and its ability to adapt to the size of these anions. Thus, the selectivity of BU[6] is mostly dictated by anion solvation and correlates with the charge density of anions.13,19 Previously, we prepared enantiomerically pure bambusurils BU1 and BU2 (Fig. 1) and demonstrated their enantioselectivity in binding different chiral carboxylates.20 Here, we demonstrate that diastereomers BU1 and BU2 bind the same inorganic anions with very different affinities and selectivities. To the best of our knowledge, this is the first example of any receptor of inorganic anions, in which the type and position of substituents attached to the receptor binding site impact the receptor selectivity. The binding differences were rationalized with the help of theoretical calculations.
 | ||
| Fig. 1 Structures of bambusuril macrocycles investigated in this work. | ||
Results and discussion
The binding of BU1 and BU2 with a series of inorganic anions was investigated by isothermal titration calorimetry in chloroform. For all titrations, the experimental data fit well with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model, which is consistent with the 1:1 host–guest stoichiometry previously described for the number of bambusuril-anion complexes. The reported association constants are apparent, as we did not consider the competition of TBA cations and bambusuril macrocycles for anions.
:1 binding model, which is consistent with the 1:1 host–guest stoichiometry previously described for the number of bambusuril-anion complexes. The reported association constants are apparent, as we did not consider the competition of TBA cations and bambusuril macrocycles for anions.
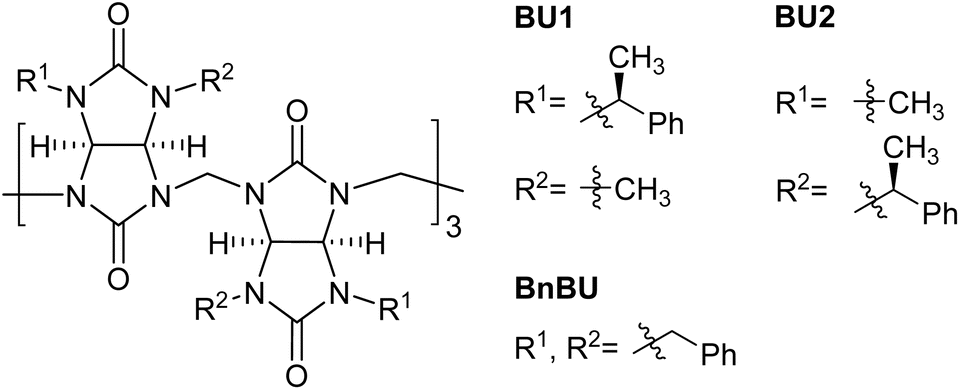
When BU1 and BU2 were compared, striking differences in their binding affinities of more than two orders of magnitude were observed (Table 1). For example, the binding selectivity of BU2 over BU1 for F−, Cl−, Br−, and I− is 592, 1273, 891, and 750. The highest selectivity of BU2 over BU1 of 5822 was observed for NO3−. We also compared the binding characteristics of BU1 and BU2 to those of the previously studied BnBU (Table 1 and Fig. 2).19 This macrocycle bears 12 benzyl substituents on its portals. We compared the binding affinities of all three bambusurils according to the volume of bound anions. The stability of complexes with BU1 and BU2 increases from F− to I−, while it decreases for anions with large diameters, such as ClO4−, ReO4−, and PF6−. In contrast, the binding affinity of BnBU increases from F− up to ClO4− and decreases for the larger anions. This clearly illustrates that the cavity of BnBU is more flexible and can even adapt to larger ClO4−, while the BU1 and BU2 cavities are best suited for smaller I− and bind ClO4− less efficiently.
| Anions | BU1 | BU2 | BnBU |
|---|---|---|---|
| a Published previously, see ref. 19. Standard deviations were calculated from two or three independent experiments and are lower than 17%, except for the BU2·ReO4− complex, for which it is 30%. | |||
| CH3CO2− | 1.9 × 104 | 2.0 × 106 | 5.6 × 105 |
| MeSO3− | 8.2 × 104 | 9.4 × 105 | 7.3 × 105 |
| NO3− | 5.1 × 105 | 3.0 × 109 | 1.8 × 109 |
| ReO4− | 1.7 × 107 | 3.0 × 107 | 1.1 × 108 |
| ClO4− | 6.8 × 107 | 1.7 × 109 | 2.1 × 1010 |
| PF6− | 1.2 × 107 | 4.1 × 108 | 8.7 × 108 |
| F− | 1.3 × 105 | 7.7 × 107 | 1.9 × 106 |
| Cl− | 3.3 × 105 | 4.2 × 108 | 1.3 × 107 |
| Br− | 1.1 × 107 | 9.8 × 109 | 6.7 × 108 |
| I− | 1.6 × 108 | 1.2 × 1011 | 1.6 × 1010 |
 | ||
| Fig. 2 Dependence of apparent association constants on anions size. | ||
The absolute values of association constants of BnBU complexes are comparable with those of BU2 but significantly higher than those of BU1 (Table 1). Further comparison of BnBU with BU2 revealed that all halides are bound by BU2 more strongly than by BnBU, but the reverse is observed for ClO4− and large anions. This clearly shows the preferential binding of BU2 to smaller spherical anions.
Another crucial difference between the investigated bambusurils is in their selectivity. Specifically, BnBU preferentially binds ClO4− over Cl− with selectivity exceeding 1600. In contrast, the selectivity for ClO4− over Cl− in the cases of BU1 and BU2 is reduced to 206 and 4. Moreover, BnBU and BU1 bind ReO4− more than 8 and 50 times more strongly than Cl−, while BU2 preferentially binds Cl− over ReO4− with a selectivity of 14. The reverse selectivity among the investigated bambusurils was observed also for the NO3−/Br− anion pair (Table 1). Inspired by the reviewers' comments, we decided to test if the reverse selectivity will be recorded in a solvent different from chloroform. We determined the association constants for the complexes of BU1 and TBA salts of Br− (2.6 × 107 M−1) and NO3− (2.9 × 106 M−1) in acetonitrile (Table S3†). The results showed that BU1 preferentially binds Br− over NO3− in both chloroform and acetonitrile with selectivities of 22 and 9, while BnBU preferentially binds NO3− over Br− with a selectivity of 3 in chloroform.
These results clearly show that replacing benzyl with (S)-1-phenylethyl substituents in the bambusuril structure leads to changes in binding affinity and, consequently, to reverse selectivity toward anions. This contrasts with previous studies,17,21 in which the selectivity toward anions remained the same for differently substituted bambusurils.
We also decided to test whether the observed differences in anion binding could be rationalized by the ability of bambusuril to act as a heterotopic ion-pair receptor interacting simultaneously with the anion and cation of the salt. We conducted an ITC titration of BU1 with tetraethylammonium (TEA) chloride in chloroform. We determined that Ka for the BU1·Cl− complex is about 4.6 times lower when TEA instead of TBA salt was used. This reflects the higher affinity of TEA towards Cl− compared to TBA and supports that the bambusuril does not act as a heterotopic ion-pair receptor.
We also performed 1H NMR titration of the BU2 solution using TBACl in chloroform (Fig. S24†). The results showed that TBA proton signals experience an upfield chemical shift in the presence of BU2. This is explained by shielding TBA+ from the chloride anion encapsulated inside the macrocycle, excluding the possibility of BU2 cooperatively interacting with the cation. The binding of chloride within the BU2 cavity is slow on the NMR time scale, in contrast to TBA+, which exhibits fast exchange upon interaction with the macrocycle. This indicates that the TBACl ion pair dissociates upon interacting with the macrocycle. The DOSY spectra of an equimolar solution of TBACl and BU2 in chloroform showed that the diffusion coefficient for TBA+ is higher compared to that of the macrocycle but lower than that of TBA+ measured in the absence of the macrocycle (Fig. S25 and S26†). This indicates that TBA+ and the BU2·Cl− complex interact to some extent. We performed dilution experiments of an equimolar mixture of BU2 and TBACl in chloroform to determine the ion-pair association constant (Kip) between the BU2·Cl− complex and TBA+ to be 1.2 × 105 M−1 (Fig. S27†). Although we do not know the value of Kip of TBACl in chloroform, we expect it to be about the same order of magnitude as the published Kip value of 9 × 109 M−1 for TEACl in the same solvent.22 The found Kip value of 1.2 × 105 M−1 for the BU2·Cl− complex and TBA+ is several orders of magnitude lower than that for TEACl, showing that the macrocycle dramatically weakened the ion pairing. Moreover, the ITC experiments performed in acetonitrile and chloroform showed similar preferences of BU1 for Br− over NO3−. As ion-pairing in acetonitrile is weak (Kip for TBACl is 5 M−1),22 we believe that this experiment further supports the insignificance of cations in the observed preferential anion binding.
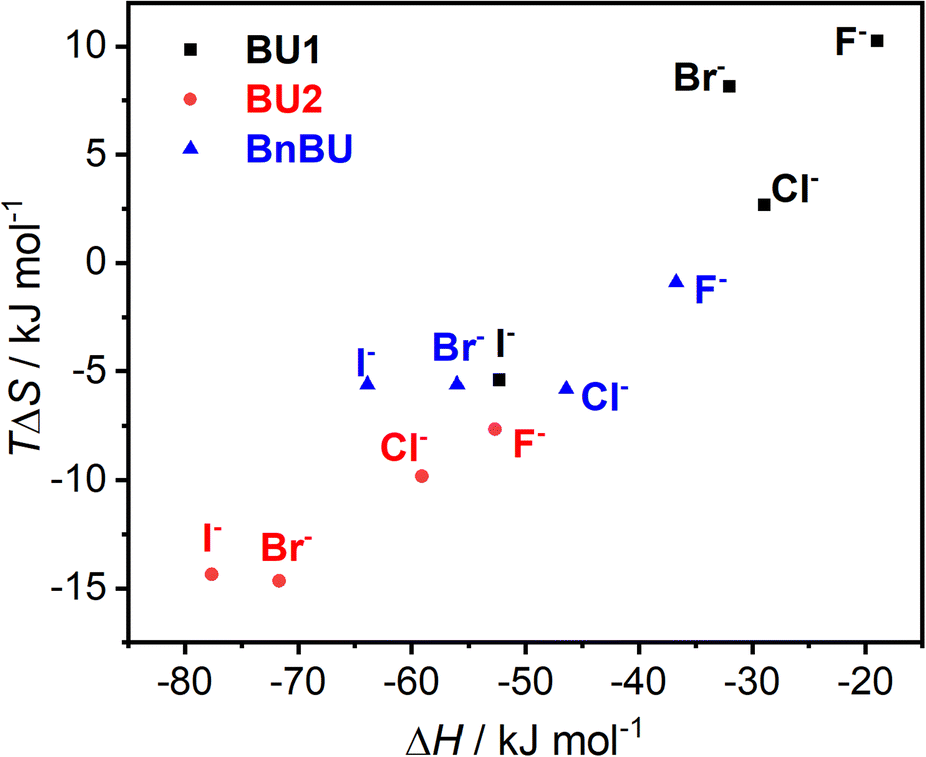
The observed differences in binding affinities between BnBU, BU1, and BU2 are reflected in their enthalpy–entropy compensation (Fig. 3). All complexes are strongly exothermically favorable, but most of them are counterbalanced by an entropic cost. The enthalpy–entropy compensation is most significant for BU2 halide complexes and the least for the complexes of BU1.
 | ||
| Fig. 3 Enthalpy–entropy compensation plot of the BU complexes with halides. | ||
We performed a computational study to better understand bambusuril binding affinity and selectivity differences. Quantum-chemical calculations were performed for the macrocycles with empty cavities and their complexes with halides, all in implicit chloroform solvent. Bambusurils BU1 and BU2 are diastereomers to each other. They differ only in the placement of the 1-phenylethyl and methyl groups, which are attached to the nitrogen atoms of the glycoluril building blocks in opposite positions (see R1 and R2 in Fig. 1). In addition to these distinct arrangements, the 1-phenylethyl groups can rotate along the N–C bond, resulting in different conformers of the macrocycles. Here, we tested two boundary conformations (Fig. 4), which differ by the orientation of the methyl and phenyl groups of the 1-phenylethyl substituent relative to glycoluril five-membered rings.
 | ||
| Fig. 4 Two boundary conformations for BU1 and BU2. | ||
In the closed form, the aromatic parts of 1-phenylethyl groups are aligned with the axial axis of the macrocycle, seemingly “closing” the cavity.
However, a short MD simulation reveals that the cavity is not effectively sealed but rather extended due to the tumbling of the phenyl groups. The cavity is closed upon halide binding. In contrast, the aromatic parts head out of the cavity in the open form, exposing the cavity to the solvent, similar to non-derivatized bambusurils.23 In all cases, the calculations showed that BU1 prefers the closed form, while BU2 prefers the open form (Fig. 4). The magnitude of the preference is comparable to the binding affinities, and it does not depend on the type of anion occupying their cavity (Table S5†). The calculation agrees with the previously determined crystal structure of the BU2 complex with Br−,20 in which the macrocycle adapts the open-form conformation. The stable conformations, the closed for BU1 and the open for BU2, while completely different, share the same structural feature. In both cases, the methine group of the 1-phenylethyl group interacts with carbonyl oxygens at the central belt through hydrogen bonding. This seems to be a key interaction already observed in the other BU derivatives, such as BnBU or dodecamethylbambusuril.19,20
Next, we calculated the binding affinities of halide anions to BU1 and BU2. In agreement with the experiment, we found that BU2 exhibits binding affinities higher than BU1 (Table 2). Compared to experimental values, the calculated values are overestimated because they do not include entropy contributions. Better agreement can be observed for the calculated (ΔΔEb) and experimental (ΔΔGb) binding energy differences between BU2 and BU1. In addition, to trace the origin of this preference, we decomposed the obtained binding affinities (ΔΔEb) into (i) the interaction of the halide anion with the bambusuril cavity (ΔΔEi), (ii) macrocycle deformation upon halide anion binding (ΔΔEd), and (iii) solvation contributions (ΔΔEs). Their values clearly reveal that the binding difference between BU1 and BU2 is predominantly the result of interaction factors, as solvation and deformation effects do not substantially differ for different anions.
| Anion | BU1 | BU2 | BU2–BU1 | BU1 | BU2 | BU2–BU1 | |||
|---|---|---|---|---|---|---|---|---|---|
| ΔGb | ΔGb | ΔΔGb | ΔEb | ΔEb | ΔΔEb | ΔΔEi | ΔΔEd | ΔΔEs | |
| F− | −29.7 | −43.5 | −13.7 | −45.4 | −65.1 | −19.7 | −31.4 | 8.0 | 3.7 |
| Cl− | −31.6 | −48.5 | −16.8 | −85.1 | −103.9 | −18.8 | −26.3 | 1.6 | 5.9 |
| Br− | −41.4 | −51.6 | −10.2 | −95.7 | −106.3 | −10.5 | −19.6 | 3.0 | 6.0 |
| I− | −47.5 | −61.9 | −14.4 | −108.0 | −113.7 | −5.7 | −10.5 | 3.4 | 1.5 |
We tried to decipher possible structural factors responsible for the observed binding preference. The differences in the preferred conformation of BU1 and BU2 are clearly caused by the placement of 1-phenylethyl groups and impact the cavity arrangement. The BU1 cavity is screw-like distorted, as indicated by the different lengths of C–H⋯X−, by about 0.3 Å for each of the two methine protons of glycoluril units (Table S6†). In contrast, all C–H⋯X− hydrogen bonds in BU2 are equalized with the same length. A similar situation can be found in the experimental structure of BnBU complexed with chloride,19 where all C–H⋯Cl− hydrogen bonds are equalized with the same length of 3.13 Å. This result confirms that the structure of the BU2 macrocycle is more flexible compared to BU1 and better adapts to the anion size. On the other hand, the spacious arrangement of the 1-phenylethyl units in BU1 induces distortion of the macrocycle cavity, resulting in a significantly lower anion binding affinity of this macrocycle compared to BU2 and BnBU.22–31
Conclusions
In conclusion, we showed that the position of methyl and 1-phenylethyl groups on the glycoluril building blocks of diastereomeric bambusurils BU1 and BU2 induces significant differences in their anion binding. BU1 binds inorganic anions up to three orders of magnitude less strongly than BU2. Quantum-chemical calculations revealed that these binding differences are due to distortion of the BU1 cavity, which is forced by the arrangement of spacious 1-phenylethyl groups, while the more relaxed arrangement is proposed for the BU2 diastereomer. Moreover, both BU1 and BU2 and previously reported BnBU feature very different or even reverse selectivity for pairs of inorganic anions. This is a unique achievement in bambusuril chemistry, as, until today, different substituents on the macrocycle portals resulted only in the modulation of their binding affinity, while selectivity among anions remained similar. Thus, our results show that bulky substituents could be used to rigidify the structures of any flexible macrocyclic receptor to tune its binding selectivity toward guest molecules.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
V. S. conceived the project. J. S. synthesized the macrocyclic compounds. C. R. and S. G. conducted ITC measurements. S. G. performed NMR experiments. P.K. carried out the computational studies. V. S. and P. K. drafted the manuscript. All authors contributed to the writing of the ESI† and to the revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (23-05271S). We thank the RECETOX Research Infrastructure (LM2023069) financed by the Ministry of Education, Youth and Sports. This project was supported by the European Union's Horizon 2020 Research and Innovation Programme under grant agreement no. 857560 (CETOCOEN Excellence). This publication reflects only the author's view and the European Commission is not responsible for any use that may be made of the information it contains. Computational resources were provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.Notes and references
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- D. A. McNaughton, W. G. Ryder, A. M. Gilchrist, P. Wang, M. Fares, X. Wu and P. A. Gale, Chem, 2023, 9, 3045–3112 CAS.
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- Anion-binding catalysis, ed. O. García Mancheño, Wiley-VCH, Weinheim, 2022 Search PubMed.
- L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chem, 2022, 8, 46–118 CAS.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- W. Zhou, A. Li, P. A. Gale and Q. He, Cell Rep. Phys. Sci., 2022, 3, 100875 CrossRef CAS.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, 2edn, 2009, repr. with corr Search PubMed.
- M. d'Ischia, A. Napolitano and A. Pezzella, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 353–388 Search PubMed.
- S. Kaabel, J. Adamson, F. Topić, A. Kiesilä, E. Kalenius, M. Öeren, M. Reimund, E. Prigorchenko, A. Lõokene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC.
- T. Lizal and V. Sindelar, Isr. J. Chem., 2018, 58, 326–333 CrossRef CAS.
- O. Reany, A. Mohite and E. Keinan, Isr. J. Chem., 2018, 58, 449–460 CrossRef CAS.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378–2381 CrossRef CAS.
- V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13, 4000–4003 CrossRef CAS PubMed.
- M. Chvojka, D. Madea, H. Valkenier and V. Šindelář, Angew. Chem., Int. Ed., 2024, 63, e202318261 CrossRef CAS PubMed.
- V. Havel, M. Babiak and V. Sindelar, Chem.–Eur. J., 2017, 23, 8963–8968 CrossRef CAS PubMed.
- V. Havel and V. Sindelar, ChemPlusChem, 2015, 80, 1601–1606 CrossRef CAS PubMed.
- J. Sokolov and V. Šindelář, Chem.–Eur. J., 2018, 24, 15482–15485 CrossRef CAS PubMed.
- H. Valkenier, O. Akrawi, P. Jurček, K. Sleziaková, T. Lízal, K. Bartik and V. Šindelář, Chem, 2019, 5, 429–444 Search PubMed.
- Y. Liu, A. Sengupta, K. Raghavachari and A. H. Flood, Chem, 2017, 3, 411–427 CAS.
- F. Neese, Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- J. G. Brandenburg, C. Bannwarth, A. Hansen and S. Grimme, J. Chem. Phys., 2018, 148, 064104 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Mov. Front. Quantum Chem., 2009, 356, 98–109 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07150f |
| ‡ Dr C. Rando and Dr S. Grewal have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |