
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural characterization of surface immobilized platinum hydrides by sensitivity-enhanced 195Pt solid state NMR spectroscopy and DFT calculations†
Benjamin A.
Atterberry‡
 ab,
Erik J.
Wimmer‡
c,
Sina
Klostermann
c,
Wolfgang
Frey
c,
Johannes
Kästner
c,
Deven P.
Estes
*c and
Aaron J.
Rossini
*ab
ab,
Erik J.
Wimmer‡
c,
Sina
Klostermann
c,
Wolfgang
Frey
c,
Johannes
Kästner
c,
Deven P.
Estes
*c and
Aaron J.
Rossini
*ab
aIowa State University, Department of Chemistry, Ames, IA 50011, USA. E-mail: arossini@iastate.edu; deven.estes@itc.uni-stuttgart.de
bUS DOE Ames National Laboratory, Ames, Iowa 50011, USA
cUniversity of Stuttgart, Department of Chemistry, Stuttgart, Baden-Württemberg 70569, Germany
First published on 20th November 2024
Abstract
Supported single-site platinum hydride compounds are promising heterogeneous catalysts for organic transformations. Few methods exist to describe the structures of single-site Pt catalysts with atomic resolution because of their disordered structures and low Pt loadings. Here, we study the compounds formed when bis(tri-tert-butylphosphino)platinum, Pt(PtBu3)2, is supported on dehydroxylated SiO2 or SiO2–Al2O3. First, we obtain magic angle spinning (MAS) 1H, 31P and 195Pt ssNMR spectra of four model Pt phosphine compounds with oxidation states of 0 or +2 and coordination numbers between 2 and 4. These compounds are analogs of potential structures present in the supported compounds. MAS 195Pt ssNMR spectra were obtained using 31P{195Pt} sideband selective J-resolved and J-HMQC experiments. The measured 1H and 31P chemical shifts, 31P–195Pt J-couplings and 195Pt chemical shift (CS) tensors are shown to be diagnostic of oxidation state and coordination number. Room temperature 1H ssNMR spectra of Pt(PtBu3)2 supported on SiO2 or SiO2–Al2O3 show diagnostic hydride NMR signals, suggesting that Pt(PtBu3)2 undergoes oxidative addition, resulting in surface hydrides and Pt–oxygen bonds to the support surface. MAS dynamic nuclear polarization (DNP) enables 31P{195Pt} correlation NMR experiments on the supported compounds. These experiments enable the measurement of the 31P–195Pt J-coupling constants and 195Pt CS tensors. Combined NMR and DFT analyses suggest that the primary surface platinum species are [HPt(PtBu3)2OSi] on SiO2 and [HPt(PtBu3)2]+[Si–O−–Al] on SiO2–Al2O3. The Pt–oxygen bond length is dependent on the support and estimated as 2.1–2.3 Å and 2.7–3.0 Å for SiO2 and SiO2–Al2O3, respectively.
Introduction
Single-site or single-atom platinum catalysts find widespread application in industrial processes such as fuel production, automotive catalytic converters for emissions control, hydrogenation reactions, hydrosilylation and other organic reactions.1–6 One such notable class are the platinum hydride catalysts, renowned for their exceptional catalytic performance in cycloisomerization, isomerization and hydroformylation reactions.7–9 However, Pt and Pd catalysts often suffer from a significant drawback in homogeneous catalysis: deactivation through dimerization.10–12 To overcome this limitation, heterogeneous catalysts can be used because they prevent dimerization and enable the reusability of the platinum hydride.13 Heterogeneous catalysts can be synthesized via surface organometallic chemistry (SOMC), which involves immobilizing well-defined metal complexes on metal oxide surfaces.14–18 In SOMC, metal oxide samples undergo controlled dehydroxylation at high temperature and high vacuum, resulting in a surface covered with a controlled density of chemically similar OH groups. These OH groups serve as ligands for organometallic complexes, typically achieved through protonolysis reactions where a basic ligand deprotonates the OH group. This process forms a new M–O bond directly to the surface while releasing HX.19Recently, some of the authors of this paper utilized surface OH groups on Brønsted acidic supports like SiO2–Al2O3 to immobilize Pt(PR3)2 complexes.13 Through solid-state nuclear magnetic resonance (ssNMR) spectroscopy, infrared spectroscopy (IR), and X-ray absorption spectroscopy (XAS) characterization, we discovered that the immobilization occurred via the apparent oxidative addition of OH groups to the Pt(0) center in the precursor, resulting in Pt(II)–H species with a new Pt–O bond to the surface. This finding is quite rare, with only two known examples reported in the literature so far.13,20 The Pt–O bond length (2.01 Å) measured by EXAFS in our study was indicative of a four-coordinate Pt–H with a slightly elongated Pt–O bond.21 However, the Pt–H 1H chemical shift (−36 ppm) and JPt–H (2400 Hz) values were more consistent with formation of cationic three-coordinate Pt–H, such as [(tBu3P)2Pt–H]+. Based on these observations, we proposed that the structure on the surface lies somewhere between three- and four-coordinate, where the nature of the Pt–O bond is something in between a true Pt–O bond and an ion pair. Thus, additional experimental methods are needed to characterize the structure of these surface species.
195Pt ssNMR spectroscopy is potentially an appealing method to investigate the structure of surface-supported platinum organometallics. Essential information about the chemical and electronic environments of Pt centers in catalysts are encoded in the 195Pt chemical shift (CS) tensor, which is sensitive to the bonding character and symmetry of the neighboring ligands.22–32 However, 195Pt NMR experiments in the solid-state are often challenging because Pt compounds often exhibit a large 195Pt chemical shift anisotropy (CSA) on the order of several thousand parts-per-million (ppm), especially for Pt(II) complexes, which typically adopt a square planar geometry.23,33,34 Several notable methods have been proposed to expedite the measurement of 195Pt ssNMR spectra and CS tensors. Schurko and coworkers have incorporated Wideband Uniform Rate Smooth Truncation (WURST) pulses35,36 and broadband adiabatic inversion cross-polarization (BRAIN-CP)37 into Carr–Purcell–Meiboom–Gill (CPMG)38 sequences that allow static 195Pt ssNMR spectra to be acquired with increased sensitivity on molecular complexes. Unfortunately, it is time-consuming to study single-site Pt catalysts using these methods because they have low Pt loadings (<5 wt%). To address this challenge, dynamic nuclear polarization surface enhanced NMR spectroscopy (DNP SENS) has been used to increase the sensitivity of static37,39 and magic angle spinning (MAS)27,40195Pt ssNMR experiments. We have also previously shown that a sensitive spy nucleus like 1H, 31P, or 13C can be used to enable acquisition of wideline 195Pt ssNMR spectra, including for detection of surface species.27,41,42 Further gains in sensitivity have been achieved by combining indirect detection MAS experiments with DNP SENS. Most indirect detection ssNMR experiments have used MAS frequencies >25 kHz, while slower MAS frequencies less than 12.5 kHz have not been employed yet.27
In this work, we use room temperature fast MAS 1H{195Pt} and 1H–31P{195Pt} J-resolved and J-HMQC experiments to investigate four different molecular bis(tri-tert-butylphosphino)platinum compounds (Fig. 1). These compounds feature Pt in oxidation states of 0 and +2 and some of the compounds have coordinated hydrides. We then use slow MAS (12.5 kHz) cryogenic DNP SENS 1H–31P{195Pt} J-resolved experiments to study two low Pt wt% (1.9 and 2 wt%) single-site catalysts. By comparing the 1H, 31P, and 195Pt ssNMR spectra of two-, three-, and four-coordinate Pt model complexes (1–4) with those of the surface complexes, we were able to gain insights into the nature of the Pt–O bond on the surface. These methods, combined with DFT calculations, offer a blueprint of the electronic and coordination spheres of the molecular and surface-supported complexes. Overall, these experiments allow us to put forward evidence-based structural models of surface-supported platinum hydride compounds.
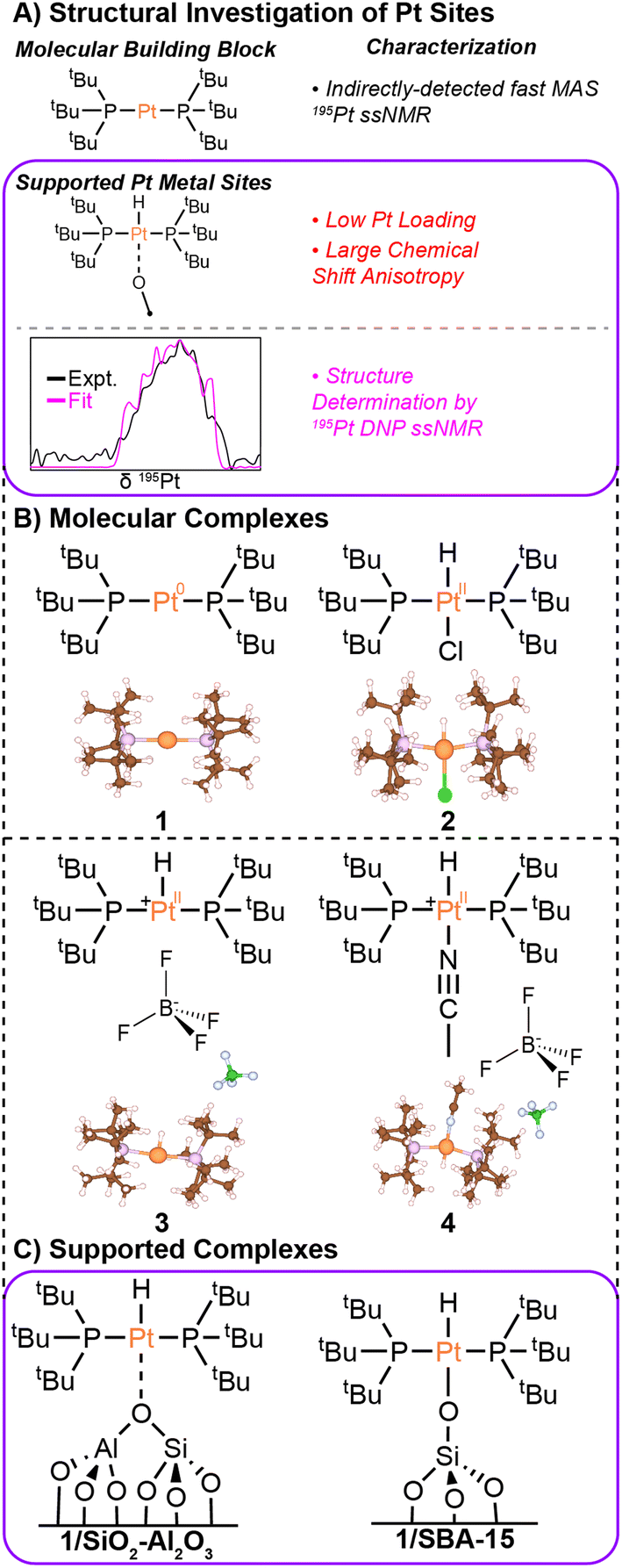 | ||
| Fig. 1 (A) Graphical summary of the characterization of molecular and surface-supported Pt complexes. (B) Molecular structures and X-ray crystal structures are shown for Pt(PtBu3)2 (1), HPt(PtBu3)2Cl (2),43 [HPt(PtBu3)2]BF4 (3), and [HPt(PtBu3)2NCCH3]BF4 (4). (C) Hypothesized structures are shown for the supported single-site Pt complexes discussed in this work. The Pt loadings in the supported single-site materials were 2 wt% (1/SiO2–Al2O3) and 1.9 wt% (1/SBA-15). | ||
Results & discussion
Overview
First, we applied ssNMR spectroscopy to molecular platinum hydride and phosphine compounds (Fig. 1). These compounds serve as analogs for different surface species that could be present when supporting bis(tri-tert-butylphosphino)platinum (1) on SBA-15 or SiO2–Al2O3. For each molecular compound, we measured the 1H, 31P, and 195Pt isotropic chemical shifts, 195Pt anisotropic chemical shift tensor parameters, and 31P–195Pt and 1H–195Pt J-couplings (when possible). These measured NMR parameters are diagnostic of the Pt oxidation state and coordination environment. These NMR parameters are also reproduced with DFT calculations. In the last part of the paper, we use DNP-enhanced 31P detected ssNMR experiments to study the immobilized platinum phosphine compounds supported on SBA-15 and on SiO2–Al2O3. Comparison of experimental and calculated 195Pt CS tensor parameters and hydride 1H chemical shifts are used to elucidate structural models for the surface species formed on the two different supports.1H, 31P and 195Pt ssNMR experiments on molecular complexes
We begin with a study of the molecular platinum phosphine compounds, 1–4. Fig. 2A shows the 31P CPMAS NMR spectrum and a rotor-synchronized 31P{195Pt} J-HMQC spectrum of 1. The 1D 31P CPMAS NMR spectrum shows two satellites that arise from a J-coupling of 4407 Hz between 195Pt and 31P. In the 2D J-HMQC spectrum, only the satellite peaks are visible. The 31P{195Pt} J-HMQC was recorded with the t1-increment set to a rotor period (40 μs), corresponding to the indirect dimension spectral width being equal to the MAS frequency (25 kHz). Rotor synchronization of the indirect dimension maximizes sensitivity of the 2D experiments because all the spinning sidebands will be aliased onto a single peak. From the 2D 31P{195Pt} J-HMQC spectrum, we can determine the offset of a single 195Pt spinning sideband, which was −605492 Hz from the reference 195Pt frequency in this case. Once we know the offset of a single sideband, we can then perform sideband selective 1D 31P{195Pt} J-HMQC or J-resolved NMR experiments that enable us to map out the intensity of each spinning sideband that makes up the entire 195Pt MAS ssNMR spectrum (Fig. 2B and C).41,42 The sideband selective NMR experiments use 195Pt selective long (SL) pulses that have durations of one rotor cycle or longer and RF fields less than 50 kHz.44 Each line represents the measured intensity or dephasing of the 31P NMR signal at the indicated 195Pt offset. Note that sideband selective experiments also require the RF-field strength of the pulses to be accurately calibrated because using an RF field 10 kHz higher-than-optimal will result in an NMR spectrum that does not accurately reconstruct the MAS 195Pt ssNMR spectrum.42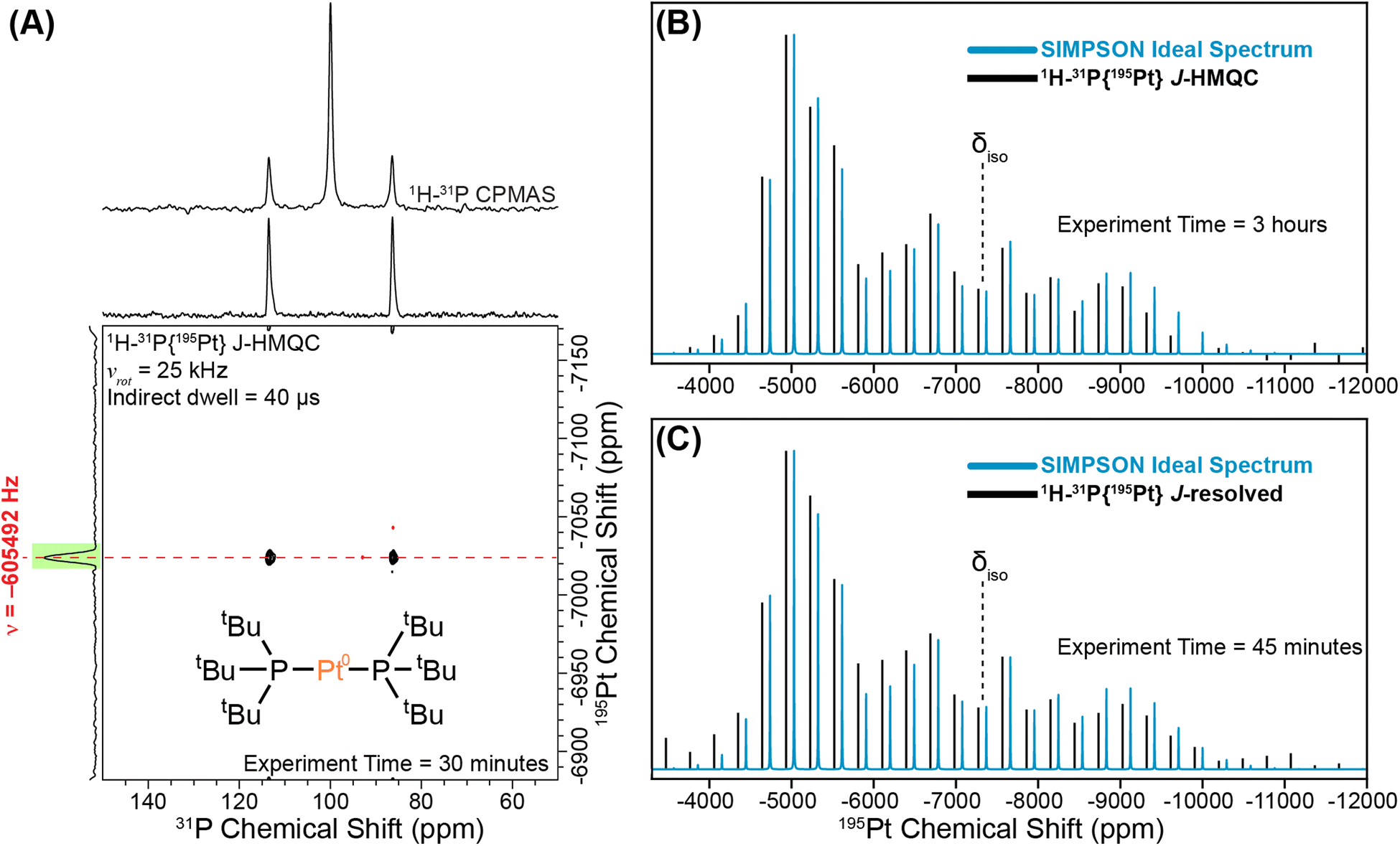 | ||
| Fig. 2 Graphical illustration of implementing a sideband selective experiment for compound 1. (A) 1D 1H–31P CPMAS spectrum overlaid on the rotor-synchronized 1H–31P{195Pt} J-HMQC spectrum. Reconstructed 195Pt ssNMR spectra obtained using (B) sideband selective 1H–31P{195Pt} J-HMQC and (C) 1H–31P{195Pt} J-resolved experiments (black lines). The intensities of the sidebands are compared to the SIMPSON-calculated ideal 195Pt MAS NMR spectrum (blue lines). All NMR experiments were performed with a 25 kHz MAS frequency and a magnetic field of 9.4 T. The 195Pt saturation pulses were 80 μs in duration with RF fields of 5 kHz and 9 kHz RF for J-HMQC and J-resolved, respectively. | ||
Fig. 2B and C show experimental 1H–31P{195Pt} J-HMQC and J-resolved sideband selective NMR spectra of 1 obtained with 80 μs 195Pt saturation pulses, with RF fields of 5 kHz and 9 kHz RF for J-HMQC and J-resolved, respectively.42 In both cases, the sideband selective spectra match closely with the overlaid ideal MAS 195Pt ssNMR spectrum. The NMR spectra shown in Fig. 2B and C were obtained in 3 hours and 45 minutes, respectively.
Fig. 3 shows 31P{195Pt} J-resolved and J-HMQC sideband selective experiments for 1, 2, and 4 obtained with a 25 kHz MAS frequency. For 3, 1H{195Pt} J-resolved and J-HMQC sideband selective experiments were performed at 50 kHz MAS. 1H spin echo and 1H–31P CPMAS NMR spectra for complexes 1–4 are shown in Fig. S1.† Overall, Fig. 3 illustrates that we can quickly and accurately measure the 195Pt ssNMR spectra of Pt–phosphine or hydride compounds. Below, we discuss trends in the measured 195Pt, 1H and 31P chemical shifts and J-coupling constants.
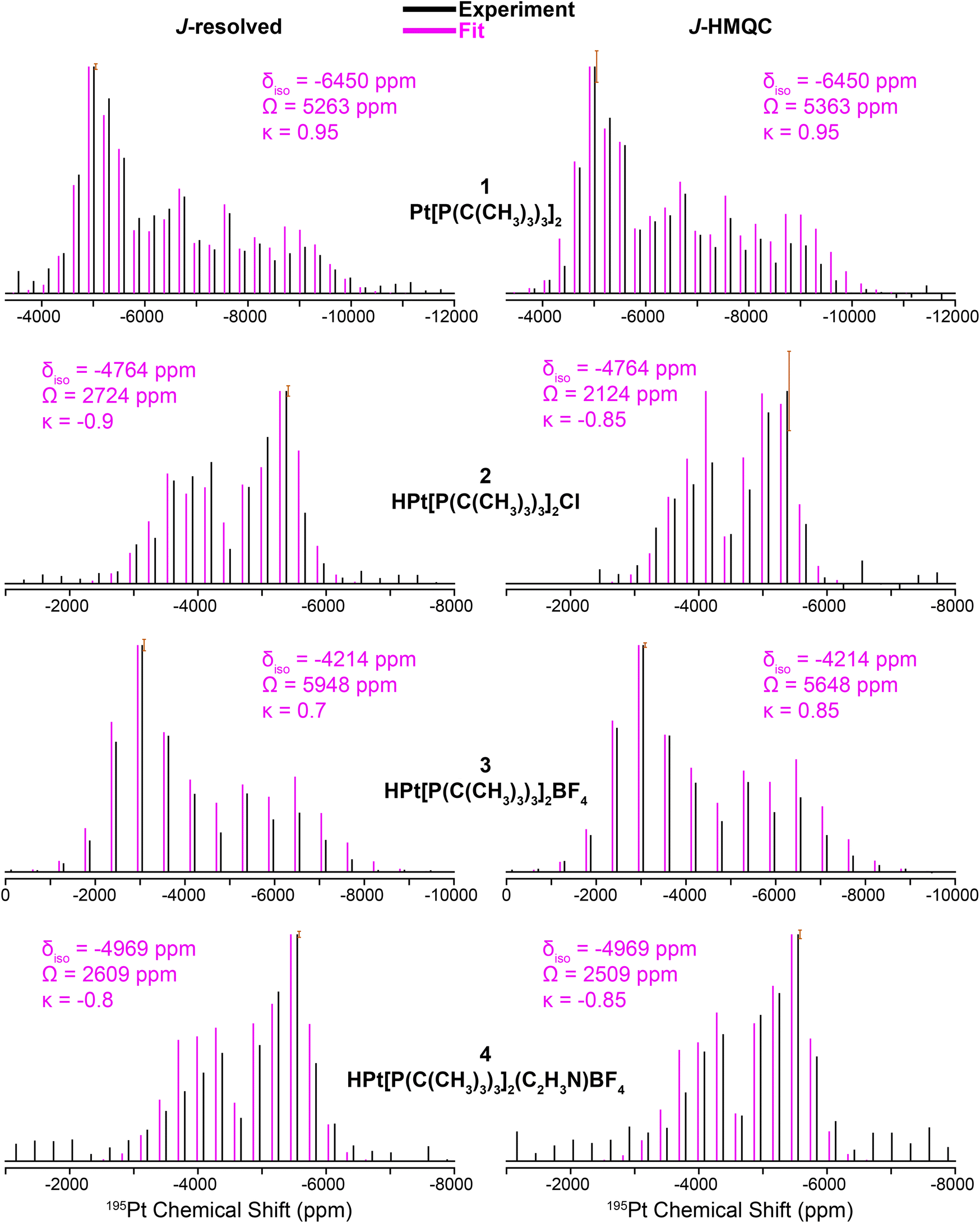 | ||
| Fig. 3 195Pt ssNMR spectra reconstructed using 31P{195Pt} J-resolved (left) and 31P{195Pt} J-HMQC (right) sideband selective experiments along with SIMPSON fits for structures 1, 2, and 4. 1H{195Pt} J-resolved and J-HMQC experiments are shown for 3. Sideband selective experiments and SIMPSON fits are colored black and pink, respectively. The chemical shift (CS) tensors from the SIMPSON fits are given with the Herzfeld–Berger convention.45 Error bars are overlaid on the most intense spinning sidebands. | ||
The clear identification of oxidation state and coordination sphere symmetry can be achieved by analyzing the trends in the 195Pt CS tensors. Comparing the 195Pt isotropic shifts reveals that complex 1 with an oxidation state of Pt(0) gives rise to the most negative shift (−6450 ppm) and complexes 2–4 with Pt(II) have more positive shifts (−4200 ppm to −5000 ppm). This observation is consistent with the results obtained from 195Pt solution NMR experiments.46–48 The second parameter that gives insight about geometry at the Pt atoms is the span (Ω). As shown in Fig. 3, Ω is much larger for the three-coordinate complex (3, ca. 5750 ppm) in comparison to the four-coordinate complexes (2, 4, ca. 2500 ppm). Although, we caution that the Ω is also sensitive to the electronic nature of the ligands, with stronger donors leading to smaller Ω.41 The last parameter to discuss would be the asymmetry parameter of the CS tensor (κ). For the four-coordinate compounds, κ is close to −1, indicating the presence of a mirror plane (or C2 symmetric or higher rotational axis) and the requirement for two of the three parameters in the CS tensor to be the same. Similarly, the three- and two-coordinate compounds exhibit a κ close to +1. The transition from κ = −1 to κ = +1 can be attributed to shifts in the electronic configuration due to differences in the coordination number (less electron donation perpendicular to the P–Pt–P axis), consequently altering the energies of the d orbitals and thus the positions of the corresponding tensor components. The 195Pt CS tensor parameters provide a reliable means of determining the oxidation state or coordination environment of an unknown complex.
In addition, the 31P and 1H ssNMR spectra are diagnostic of the Pt oxidation state. Specifically, a three-coordinate Pt–H complex exhibits a characteristic hydride chemical shift in the 1H NMR shift at ca. −38 ppm, whereas the four-coordinate complexes display shifts around −19 ppm (Fig. S1†). We also observed that the 31P shift for 1 is centered at 100 ppm (Pt0) and 2–4 have 31P shifts around 70–80 ppm (PtII). Finally, the 31P–195Pt J-couplings could be directly measured based on the difference in frequencies of the satellite peaks in the 31P NMR spectra, except for 4, for which we used a J-resolved evolution curve to measure the J-coupling (Fig. S2†). For complex 1, a sizeable 31P–195Pt J-coupling is observed (1J(31P–195Pt) = 4407 Hz), which is typical of Pt0 compounds.49 But for complexes 2–4, 1J(31P–195Pt) decreases to ∼2500 to ∼3000 Hz. The 1H–195Pt J-couplings for complexes 2–4 were also measured and 1J(1H–195Pt) is ∼1000 Hz for 4-coordinate complexes (2 & 4) and 2630 Hz for the 3-coordinate complex (3). These characteristic chemical shifts and coupling constants supply valuable information that can aid in the investigation of surface-supported samples. The root mean square error plots for the numerical fits of the 31P{195Pt} J-resolved and J-HMQC sideband selective experiments on 1–4 are shown in Fig. S3.†
DFT calculations of NMR observables
Modern computational chemistry methods can accurately predict CS tensors50,51 and J-couplings52,53 for heavy nuclei such as 195Pt. We geometrically optimized the H-atom positions in the X-ray crystal structures of complexes 1–4 using DFT calculations in CASTEP54 and then used molecular DFT calculations in the Amsterdam Modeling Suite55 along with a hybrid density functional56,57 to predict the NMR parameters. Overall, the calculated and experimental 1H, 31P and 195Pt NMR parameters are in good agreement for complexes 1, 2, and 4 (Table 1 and Fig. 4).| Compound | Pt oxidation state | Geometry | Method | Hydride δiso (1H) (ppm) | δiso (31P) (ppm) | 1 J (31P-195Pt) (Hz) | 1 J (1H-195Pt) (Hz) | 195Pt δiso (ppm) | 195Pt Ω (ppm) | 195Pt κ |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | +0 | Linear | Experiment | — | 100 | 4407 | — | −6450 | 5313 | 0.95 |
| X-ray structure | — | 100 | 5226 | — | −6392 | 5272 | 0.99 | |||
| 2 | +2 | Square planar | Experiment | −19 | 76 | 3025 | 1100 | −4764 | 2424 | −0.88 |
| X-ray structure | −17 | 81 | 3386 | 1071 | −4716 | 2078 | −0.81 | |||
| 3 | +2 | T-shaped | Experiment | −38 | 88 | 2624 | 2630 | −4214 | 5798 | 0.78 |
| X-ray structure | −35 | 92 | 3088 | 3117 | −3717 | 7325 | −0.10 | |||
| One trans | −27 | 62, 34 | 2114 | 2355 | −4154 | 5168 | −0.17 | |||
| Two orthogonal | −32 | 62, 47 | 3031 | 4278 | −3188 | 7109 | 0.33 | |||
| 4 | +2 | Square planar | Experiment | −19 | 95, 78 | 2793 | 1030 | −4969 | 2559 | −0.83 |
| X-ray structure | −15 | 90, 66 | 2822, 3049 | 1114 | −4885 | 2019 | −0.67 | |||
| 1/SiO2–Al2O3 | +2 | Square planar/T-shaped | Experiment | −35 | 82 | 2433 | 2400 | −4841 | 5680 | −0.6 |
| Optimized structure | −32 | 85 | 3434 | 1882 | −4385 | 6314 | −0.3 | |||
| 1/SBA-15 | +2 | Square planar/T-shaped | Experiment (hydride) | −26 | 72 | 2908 | 1034 | −4441 | 4080 | 0.0 |
| Experiment (C–H activated) | − | 52 | — | — | −3541 | 7980 | 0.0 | |||
| Optimized Structure | −26 | 99 | 3315 | 1213 | −4484 | 3775 | −0.8 |
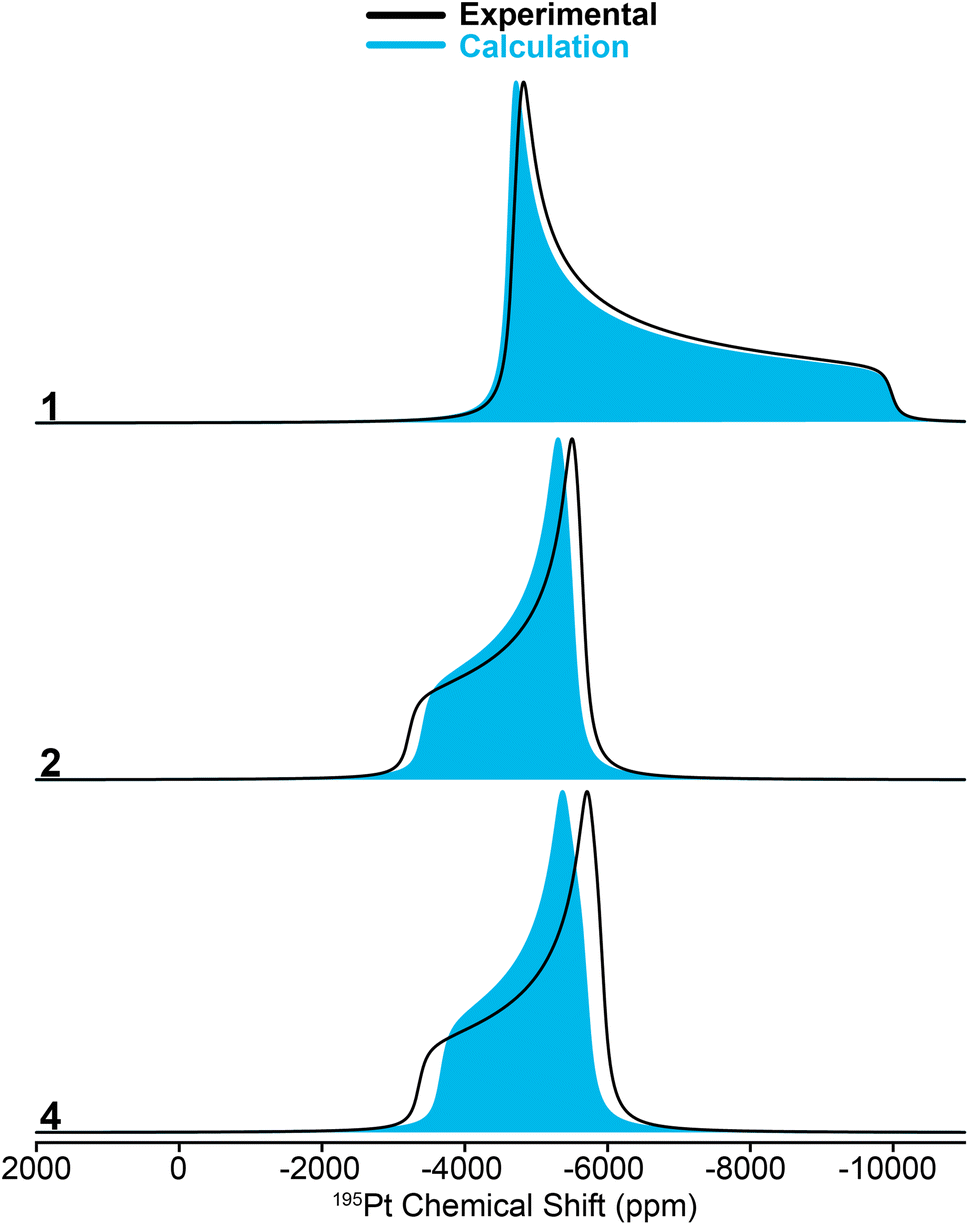 | ||
| Fig. 4 Simulated 195Pt static ssNMR spectra for compounds 1, 2, and 4. Black lines show the spectra simulated using 195Pt CS tensors measured with the MAS ssNMR experiments and solid blue patterns correspond to DFT calculated 195Pt CS tensors. | ||
While calculating the CS tensors with optimized H atom positions and crystallographic coordinates for heavy atoms (crystal structures for 2–4 are shown in Fig. S13–S15 and Tables S3–S5†) worked well for complexes 1, 2, and 4, the agreement for complex 3 was less than satisfactory. As for 1, 2, and 4 we calculated the 195Pt CS tensor using the single crystal X-ray diffraction structure (with DFT optimization of the H atom positions). This calculation gave a predicted 195Pt NMR spectrum with δiso = −3717 ppm, Ω = 7325 ppm, and κ = −0.10, which was significantly different from the experimental spectrum that we measured for 3 (δiso = −4214 ppm, Ω = 5798 ppm, and κ = 0.78) (Fig. 5, bottom). Examination of the experimental 1H and 31P NMR spectra shows that the complex has not decomposed before measurement (signals for Pt–H and of the PtBu3 groups are correct, Table 1 and Fig. S1†). However, we have observed that the crystal lattice of 3 is not stable over long time periods due to slow loss of the dichloromethane solvents of crystallization from the crystal lattice (Fig. S4†). Indeed, placing the crystals under vacuum even for short periods results in reduced crystallinity. We theorized that the local structure of 3 could change upon loss of the solvents of crystallization to give a different coordination environment – changing the CS tensor. One commonly observed interaction in similar T-shaped cationic Pt phosphine complexes is the formation of C–H agostic interactions, which is notably absent from the crystal structure of complex 3.58–62 Indeed, DFT calculations showed that optimization of the methyl positions resulted in a new structure, 3(trans), which features a C–H agostic interaction in the position trans to the hydride ligand is ca. 4.5 kcal mol−1 more stable than the structure with no agostic interactions (3(X-ray)). The optimized structure of 3(trans) showed a Pt–C distance of 2.77 Å and a ∠H–Pt–C = 157.30°, both of which are typical for such complexes.63 We therefore calculated the 195Pt NMR parameters of 3(trans) and found that the agreement with our experimental observations was now significantly better (δiso = −4154 ppm, Ω = 5168 ppm, and κ = −0.17). It is evident that if methyl groups participate in secondary bonding there is a change two of the principal components of the 195Pt CS tensor (δ11 and δ22) but not δ33. Still, the agreement between the calculated NMR parameters of 3(trans) and the experimental spectrum of 3 is not perfect. The δ22 tensor component shows the largest deviation, as demonstrated by the very different κ value for 3(trans). We also examined another model, 3(ortho), having agostic interactions in the apical position (orthogonal to the H–Pt–P plane). This led to a structure with two agostic interactions perpendicular to the T-shaped coordination structure and had an energy that is ca. 40 kcal mol−1 higher than 3(trans). The NMR parameters of this species were also quite different than the experimental spectrum. Calculations predict a large 1H–195Pt J-coupling for 3(ortho) (3031 Hz), which is much larger than what is predicted for 3(trans) (2114 Hz) and what is observed experimentally (2630 Hz). The 31P and 1H spectra predicted for 3(trans) also match the experimental spectrum better than what is seen for the other two models. However, we only see one chemically equivalent peak for the PtBu3 groups in 3, rather than the two inequivalent signals predicted for 3(trans). Therefore, we hypothesize that the experimental spectrum of 3 can be explained by formation of a trans-agostic interaction upon loss of solvated dichloromethane from the crystal lattice. The observed chemical equivalency of the P atoms of the two phosphines of 3 could be the result of rapid exchange between methyl groups in the agostic position.
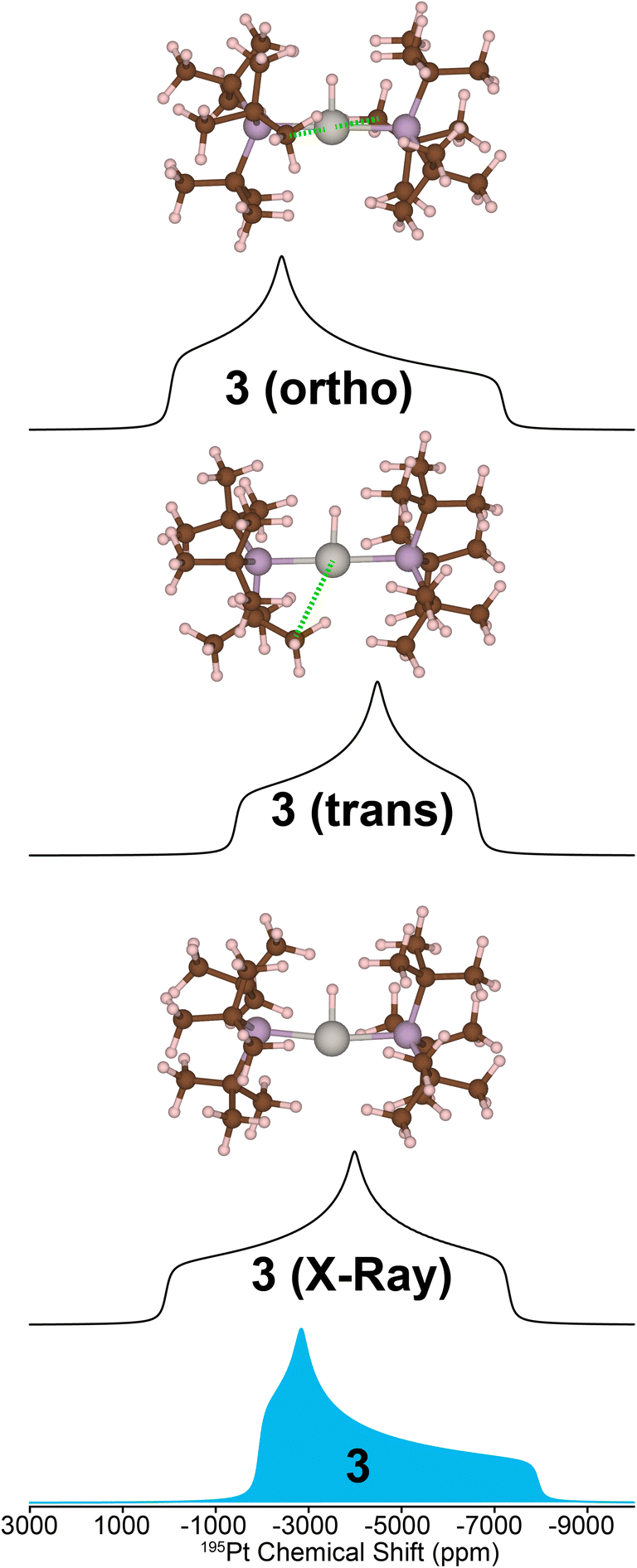 | ||
| Fig. 5 Comparison of the 195Pt ssNMR spectrum for complex 3 and the DFT-calculated 195Pt NMR spectra for different structural models. Calculations were performed with the single crystal X-ray diffraction structure (bottom), a modified structure featuring one agostic interaction between Pt and a methyl group C–H bond that was oriented trans to the platinum hydride bond (middle), and two Pt methyl C–H agostic interactions orthogonal to the platinum hydride bond (top). A green dashed line indicates an agostic interaction. | ||
In summary, NMR experiments on complexes 1–4 illustrate how 195Pt CS tensors can be measured and related back to the structural features and Pt oxidation states of the compounds. The 31P and 1H chemical shifts and 31P–195Pt J-coupling constants also provide valuable structural information. Using relativistic DFT calculations it is possible to accurately reproduce the NMR observables. With these capabilities in hand, we now turn to the structural characterization of surface-supported platinum phosphine compounds.
ssNMR experiments on surface-supported complexes
We sought to understand the chemical structure of the surface-supported complexes 1/SiO2–Al2O3 and 1/SBA-15. The surface immobilized complexes were synthesized by reacting a solution of 1 at room temperature with either SiO2–Al2O3 or SBA-15 for 18 hours, as we reported previously.1331P CPMAS NMR spectra show that the results of the immobilization are highly dependent on the level of dehydroxylation of the material (Fig. 6). | ||
| Fig. 6 Room temperature 31P CPMAS ssNMR spectra of 1/SBA-15 (green) and 1/SiO2–Al2O3 made with SiO2–Al2O3 had not been dehydroxylated (red), dehydroxylated at Td = 200 °C (purple), and dehydroxylated at Td = 500 °C (blue). | ||
We immobilized 1 on SiO2–Al2O3 that had not been dehydroxylated, as well as material dehydroxylated at 200 °C and at 500 °C. When the material had not been dehydroxylated, a major peak at 53.7 ppm corresponding to the protonated phosphine HPtBu3+ and a minor peak at 79.9 ppm corresponding to the four coordinate hydrated complex [HPt(PtBu3)2(OH2)]+ were observed.64,65 Additionally, two other minor peaks can be seen at 70 ppm and 24 ppm that have been more difficult to assign. Four coordinate platinum hydrides are known to activate the C–H bonds of ligated PtBu3 at relatively mild conditions to give the cyclometallated Pt complexes and loss of H2 (vide infra).11 We did not observe formation of tri-tert-butylphosphine oxide (66.5 ppm)66 in any of the grafted samples. However, the chemical shift of tri-tert-butylphosphine oxide can vary considerably due to hydrogen bonding to surface Brønsted and Lewis acid sites.67–71
The rate of C–H activation is highly dependent on the identity of the fourth ligand. For four-coordinated Pt complexes, the metalation rate decreases in the order I > Br > Cl > O2CCF3 ≈ NO3, with the trans–I complex undergoing cyclometallation in less than 24 hours.11 In contrast, for three-coordinated complexes with non-coordinating anions (e.g. BF4−), no cyclometallation is observed (Scheme 1).64 While the mechanism of the C–H activation in these cases is not completely clear, there seems to be a relationship between the rate of cyclometallation and the trans-influence of the fourth ligand, which matches the trend observed above. The signals of cyclometallated complexes of the type [Pt(–CH2CMe2PtBu2)(PtBu3)]+ in the 31P NMR spectrum also vary depending on the identity of the counterion/fourth ligand. The resonance corresponding to coordinated PtBu3 falls between 70 and 59 ppm while the cyclometallated phosphine can have values between 25 and −16 ppm. Thus, the observed NMR signals at 70 and 24 ppm in 1/SiO2–Al2O3 dehydroxylated at room temperature could be consistent with cyclometallation after the initial preparation (vide infra).11,72 However, upon dehydroxylation of the surface at temperatures up to 500 °C, all of these signals give way to a complex with spectral signals similar to that of three coordinate 3 (δ31P = 84.7 ppm, δPt–H = −35 ppm), which becomes the main species on the surface dehydroxylated at 500 °C. Degradation over time was observed towards forming more protonated phosphine, with only small amounts of C–H activation. Therefore, our subsequent analysis focused on 1/SiO2–Al2O3 prepared with SiO2–Al2O3 that was dehydroxylated at 500 °C. Since we mostly observe the four-coordinate [HPt(PtBu3)2(OH2)]+ complex on the surface of partly hydroxylated supports, this would explain its propensity for undergoing slow cyclometallation, which is not observed in the three-coordinate [HPt(PtBu3)2]+ species formed on the dehydroxylated surfaces.
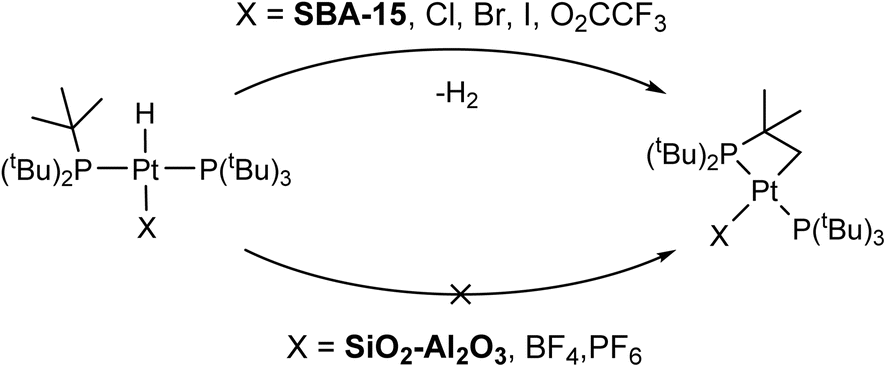 | ||
| Scheme 1 C–H activation and formation of cyclometallated Pt species on silica surfaces (top) and non-activated 3-coordinated Pt–H on SiO2–Al2O3 surfaces (bottom). | ||
Interestingly, regardless of the dehydroxylation temperature, only one species forms for 1/SBA-15 indicated by the presence of a 31P NMR signal at 80 ppm. However, over time, we also observed C–H activation, which might be accelerated under reduced pressure when the compounds are stored in sealed tubes under vacuum (see Fig. S6–S8†); two additional 31P peaks are visible at 56 and 24 ppm in the DNP-enhanced 31P ssNMR spectrum of 1/SBA-15. These chemical shifts are consistent with the literature-reported C–H activated Pt(PtBu3)2 complex.72
We used a combination of room temperature 25 kHz MAS frequency and 100 K DNP SENS NMR experiments (12.5 kHz MAS frequency) to assess the Pt coordination sphere of 1/SiO2–Al2O3 and 1/SBA-15. Due to the low concentration of the phosphine compounds on the surface, DNP was used to boost the sensitivity of the 31P{195Pt} J-resolved NMR experiments. Fig. 7A shows 1H spin echo NMR spectra of 1/SiO2–Al2O3 and 1/SBA-15 recorded with a 25 kHz MAS frequency. Both materials show negatively shifted 1H NMR signals attributed to hydride protons. The chemical shifts of the hydride peaks are −35 ppm for 1/SiO2–Al2O3 and –26 ppm for 1/SBA-15. The hydride 1H NMR signals also show splittings attributable to 1H–195Pt spin–spin couplings (2.6 and 1.7 kHz for 1/SiO2–Al2O3 and 1/SBA-15, respectively). For 1/SiO2–Al2O3 we confirmed that the satellite peaks dephase in a 1H{195Pt} J-resolved experiment (Fig. S5†), which confirms the presence of platinum hydrides in these supported compounds. A 1H{195Pt} J-resolved experiment was not conducted for 1/SBA-15 due to the poorer 1H signal-to-noise.
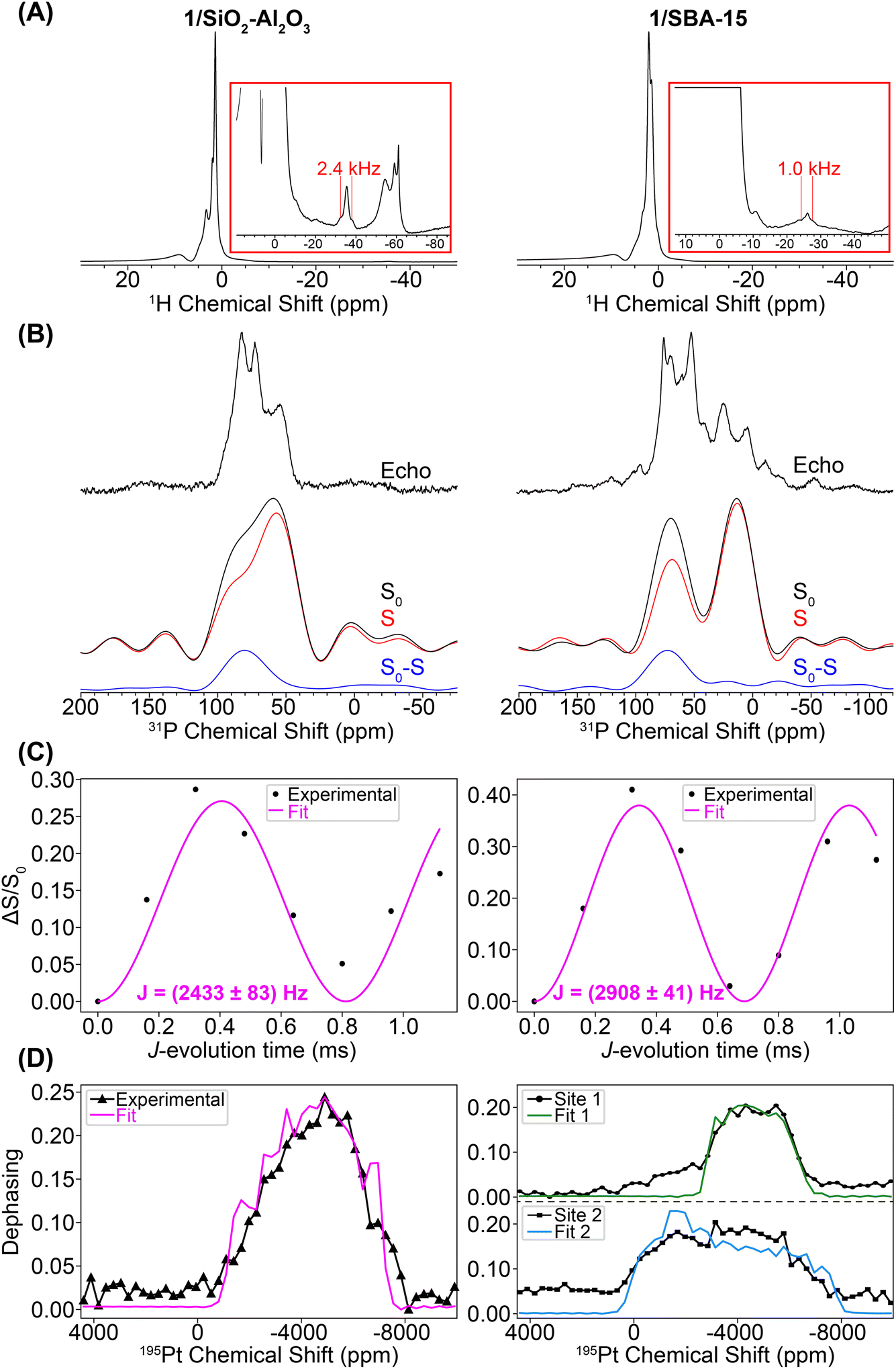 | ||
| Fig. 7 (A) Room temperature MAS 1H spin echo NMR spectra of 1/SiO2–Al2O3 (left) and 1/SBA-15 (right). (B) DNP-enhanced 31P{195Pt} J-resolved control (S0), dephasing (S) and difference (S0 − S) spectra. Spectra were obtained with CPMG detection and co-addition of the spin echoes in the time domain. Spectra were acquired with a 0.32 ms J-evolution time (spin echo duration). A higher resolution DNP-enhanced 31P CPMAS spin echo spectrum is overlaid. (C) 31P{195Pt} J-evolution plots. Black circles and pink lines correspond to experimental data points and least-squares fit, respectively. (D) Plot of 31P{195Pt} J-resolved signal dephasing as a function of 195Pt offset. The black lines with data markers correspond to experiments on complexes 1/SiO2–Al2O3 and 1/SBA-15, and the pink line is the SIMPSON fit of the experiment on complex 1/SiO2–Al2O3 (δiso = −4841 ppm, Ω = 5680 ppm, κ = −0.6). The green and blue lines are the SIMPSON fits for site 1 (δiso = −4441 ppm, Ω = 4080 ppm, κ = 0.0) and site 2 (δiso = −3541 ppm, Ω = 7980 ppm, κ = 0.0), respectively, of 1/SBA-15. | ||
We performed control room temperature 31P ssNMR experiments on samples of 1/SBA-15 and 1/SiO2–Al2O3 impregnated with TEMPO solutions (Fig. S6–S7†). We observed decreased 31P NMR sensitivity but no degradation towards C–H activated or other complexes. Therefore, the degradation to C–H activated species likely occurred while the samples were held under vacuum in sealed storage tubes for the weeks it took to ship the samples from Germany to the USA to perform DNP experiments. Exposure to the biradical does appear to cause some degradation to phosphine oxide and protonated phosphine decomposition products (vide infra). However, under the conditions needed for DNP enough of the Pt–H species are present for to measure the 195Pt ssNMR spectra.
Fig. 7B shows DNP-enhanced 31P{195Pt} J-resolved control (S0), dephasing (S), and difference (S0 − S) spectra. The 31P NMR spectrum of 1/SiO2–Al2O3 shows the same peak at 82 ppm due to the [HPt(PtBu3)2] fragment, along with new peaks at 71 ppm (OPtBu3) and 57 ppm (HP(tBu)3+).65,73 These new peaks were due to partial decomposition from the DNP biradical solution (Fig. S7–S8†). The 31P NMR of 1/SBA-15 shows the expected signal of the [HPt(PtBu3)2] fragment at 72 ppm along with new signals at 52, 25, and 4 ppm. The 31P–195Pt J-couplings of both of the Pt–H species were measured to be 2433 and 2908 Hz for 1/SiO2–Al2O3 and 1/SBA-15, respectively, which are close to the expected value of ca. 2500–3000 Hz for a 31P–195Pt(II) bond. The 31P NMR signals at 52 and 25 ppm (with close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) can be assigned as the C–H activated [κ2-tBu2P(CMe2CH2–)Pt(PtBu3)]+ complex based on the similarity to NMR spectra of known cyclometallated Pt complexes.72 While the exact nature of the decomposition peak at 4 ppm on 1/SBA-15 is at this point unclear, this is consistent with higher oxidation of the phosphine (e.g. to phosphites or phosphonates) upon reaction with TEKPol.74 Peak fits of the 1H–31P CPMAS spectra for 1/SiO2–Al2O3 and 1/SBA-15 are shown in Fig. S9 and Table S1.† These fits demonstrate that while the Pt–H is still the most prominent species in 1/SiO2–Al2O3 (ca. 60%), the decomposition of 1/SBA-15 is significantly greater, showing that the proportion of Pt–H to C–H activated species are both ca. 30% of the total phosphorus.72
:1 ratio) can be assigned as the C–H activated [κ2-tBu2P(CMe2CH2–)Pt(PtBu3)]+ complex based on the similarity to NMR spectra of known cyclometallated Pt complexes.72 While the exact nature of the decomposition peak at 4 ppm on 1/SBA-15 is at this point unclear, this is consistent with higher oxidation of the phosphine (e.g. to phosphites or phosphonates) upon reaction with TEKPol.74 Peak fits of the 1H–31P CPMAS spectra for 1/SiO2–Al2O3 and 1/SBA-15 are shown in Fig. S9 and Table S1.† These fits demonstrate that while the Pt–H is still the most prominent species in 1/SiO2–Al2O3 (ca. 60%), the decomposition of 1/SBA-15 is significantly greater, showing that the proportion of Pt–H to C–H activated species are both ca. 30% of the total phosphorus.72
Lastly, we performed variable 195Pt offset 31P{195Pt} J-resolved experiments to measure the 195Pt CS tensor. These experiments were challenging for a number of reasons. First, the 195Pt spinning sidebands are likely broadened by several kilohertz, meaning that at the MAS frequency of 12.5 kHz achievable in the DNP setup the sidebands will likely be overlapped with one another. Consequently, the dephasing profiles will roughly trace out the MAS spectrum sideband pattern, but it is not possible to measure sideband positions or isotropic 195Pt chemical shifts with high precision. Second, we also had to use 195Pt saturation pulses with durations of 120 μs and 35 kHz RF fields to induce enough dephasing in the 31P{195Pt} J-resolved experiments (Fig. S10†). The use of these pulse conditions causes distortions of the dephasing profile, making it challenging to accurately determine δ22 or κ. Finally, under DNP conditions, the 31P signals suffered from inhomogeneous broadening on the order of a few kHz, reducing NMR sensitivity. To overcome the inhomogeneous broadening, a 31P CPMG echo train detection was used in the 31P{195Pt} J-resolved experiment (Fig. S11†).
Fig. 7D shows the 195Pt NMR spectra reconstructed from the 31P{195Pt} J-resolved experiments for both 1/SiO2–Al2O3 and 1/SBA-15 along with SIMPSON-simulated fits. For the 1/SiO2–Al2O3, where the Pt–H is the primary surface species, the 195Pt NMR signature matches closely with complexes 2–4 (δiso = −4841 ppm, Ω = 5680 ppm, κ = −0.6). The spectrum of 1/SBA-15 is somewhat more complicated and had to be fit to two Pt sites, one with a small CSA (site 1: δiso = −4441 ppm, Ω = 4080 ppm, κ = 0.0) and the other with a large CSA (site 2: δiso = −3541 ppm, Ω = 7980 ppm, κ = 0.0). We note that for both site 1 and site 2, there is large uncertainty in the fit of κ, so it is set equal to 0.0 (Fig. S12†). The spectrum of site 1 was obtained by monitoring the dephasing of the spikelets in the 31P CPMG spectrum centered around 90 ppm (mainly from Pt–H surface complexes) while the spectrum of site 2 is from the lower frequency spikelets around 50 ppm (mainly from C–H activated complex). Hence, we believe that site 1 is the Pt hydride and site 2 is the C–H activated species (Fig. S9 and S11†). Dephasing of the 31P resonance from the cyclometallated phosphine at 25 ppm was not observed due to a lower J-coupling constant (JPt–P ≈ 2000 Hz) in comparison to Pt–H complexes (JPt–P ≈ 3000 Hz).
DFT modeling of surface-supported complexes
To understand the nature of the Pt–O bonds of the surface supported platinum hydrides, we created nine DFT structural models for 1/SBA-15 (Fig. 8) and six for 1/SiO2–Al2O3 (Fig. 9) with varying Pt–O bond lengths. For these calculations, [HPt(PtBu3)2]+ fragments were ligated by [(MeO)3Al–O–Si(OMe)3]− or [OSi(OMe)3]− (structural analogs of the SiO2–Al2O3 and SBA-15 surface oxygen atoms, respectively) and were geometrically optimized by DFT, then the NMR parameters were calculated. However, these model ligands do not have the same steric and electronic behavior as the true surface sites. Therefore, we subsequently geometrically optimized the same structure with constrained Pt–O bond lengths and calculated their NMR parameters. The Pt–O bond length was successively increased from the DFT calculated equilibrium distances for each model ligand in order to see the effect of lengthening the Pt–O bond on the NMR spectra and more accurately predict the true Pt–O bond length. We also carried out the same calculations for Pt(0) complexes coordinating to either surface OH or anionic surface sites using the same surface model ligands. Table S2† contains a detailed list of the DFT-calculated hydride 1H and 31P chemical shifts, and the 1H–195Pt and 31P–195Pt J-couplings for all structural models. The 31P chemical shifts and 31P–195Pt J-couplings will not be discussed further because they change little between the structural models. The 1H–195Pt J-couplings differ by several hundred Hertz for each model, but due to the poor signal-to-noise in the room temperature 1H spin echo spectra (Fig. 7A), it was difficult to accurately measure the 1H–195Pt J-couplings, so they will not be discussed either. Based on our calculations, the 195Pt CS tensors and 1H chemical shift of the hydride provide crucial structural information about the surface supported complexes. | ||
| Fig. 8 (Top left) Possible structural models of 1/SBA-15 used for DFT calculations. The Pt–O bond lengths are indicated. (Top right) Comparison of DFT calculated 195Pt static lineshapes for each model and experimental 195Pt static lineshape for 1/SBA-15. Green bars that are 600 ppm wide are shown at δ11 and δ33 to show the approximate combined error based upon fits of experimental dephasing profiles and standard deviation of DFT calculations. Green bars are not shown for δ22 due to the large uncertainty in the skew (κ). (Lower panel) Comparison of experimental (black) and calculated hydride 1H chemical shifts (pink). | ||
 | ||
| Fig. 9 (Top left) Possible structural models of 1/SiO2–Al2O3 used for DFT calculations. The Pt–O bond lengths are indicated. (Top right) Comparison of DFT calculated 195Pt static lineshapes for each model and experimental 195Pt static lineshape for 1/SiO2–Al2O3. Green bars that are 600 ppm wide are shown at δ11, δ22, and δ33 to show the approximate combined error based upon fits of experimental dephasing profiles and standard deviation of DFT calculations. (Lower panel) Comparison of experimental (black) and calculated hydride 1H chemical shifts (pink). | ||
Fig. 8 shows the comparison of the experimental 195Pt static lineshape to the calculated lineshapes of the model structures as well as the calculated hydride chemical shifts for 1/SBA-15. We note that there is large uncertainty in the least-squares fit of κ/δ22 for 1/SBA-15, so it is not considered here. The calculations predict that the 195Pt CSA increases as the Pt–O distance increases past the calculated equilibrium distance of 2.131 Å. This prediction is consistent with our experiments that showed the three-coordinate complex 3 has a much larger CSA than either of the four-coordinate complexes 2 or 4. The hydride shift also becomes more negative as the Pt–O distance increases (−23 ppm for model III and −37 ppm for model IX). The least squares fit of the 195Pt CSA indicates that the calculated 195Pt lineshape of model III matches most closely with the experimental spectrum, while the hydride 1H chemical shift matches best with model IV. Therefore, we conclude the surface supported Pt–H complex present in 1/SBA-15 most likely has Pt–O distances between 2.1 Å and 2.3 Å.
The DFT structural models for 1/SiO2–Al2O3 are shown in Fig. 9. DFT calculations predicted an equilibrium Pt–O bond length of 2.383 Å. The comparison of the experimental 195Pt static lineshape to the calculated lineshapes of the model structures as well as the calculated hydride chemical shifts are shown. DFT calculations predict similar trends between the Pt–O bond length and the 195Pt CSA and hydride 1H chemical shift as was seen above for 1/SBA-15. The least squares fit of the 195Pt CSA indicates that the 195Pt lineshape of model XIV matches best with the experimental spectrum, and the hydride chemical shift of model XV matches with the experimental chemical shift. We therefore conclude that the Pt–O distance is likely between 2.7 and 3.0 Å for 1/SiO2–Al2O3.
Our experimental observations and DFT calculations demonstrate a few key differences between the Pt–H complexes on the different supports. Previous NMR and FTIR studies suggest that ca. 90% of surface Si–OH groups on SiO2–Al2O3 neighbor nearby Al lewis acid sites, explaining the enhanced acidity of SiO2–Al2O3 as compared to SiO2.75–79 This higher acidity of leads to a higher Pt loading per unit surface area on SiO2–Al2O3 by making the thermodynamics of the O–H oxidative addition to Pt more favorable. Consequently, the Pt loadings on SBA-15 and SiO2–Al2O3 are similar, despite SBA-15 having a much larger surface area than SiO2–Al2O3. Consistent with this finding, the IR spectra of SiO2–Al2O3 before and after immobilization of 1 shows a decrease in the intensity of νOH by about half (Fig. S16†). In contrast, the intensity of νOH on SBA-15 shows a smaller intensity change after immobilization of 1, suggesting that only a small percentage of isolated Si–OH groups react with 1 (Fig. S17†). In addition, we expect that the increased acidity of the OH groups on SiO2–Al2O3 leads to longer Pt–O bond distances than for 1/SBA-15. This is because the higher acidity of the [Al–OH–Si] groups makes [Al–O–Si]− more weakly coordinating than [Si–O]−, leading to longer Pt–O bond lengths on SiO2–Al2O3. Longer Pt–O distances on SiO2–Al2O3 also correlate with the propensity for the complexes to undergo C–H activation of the tBu groups, which is dependent on the trans-influence of the X-type ligand (vide supra).11 This fact explains why 1/SiO2–Al2O3 shows no C–H activation while 1/SBA-15 does undergo cyclometallation (Scheme 1).
We previously measured a Pt–O distance of 2.01(3) Å via Pt L3 edge EXAFS of 1/SiO2–Al2O3, which is seemingly at odds with our results here.13 However, given that the complex undergoes C–H activation, it may be that the 2.01(3) Å bond distance actually corresponded to the Pt–C bond of the cyclometallated complex, which formed either during shipment of the sample under high vacuum or (more likely) in the X-ray beam of the XAS measurements. This explanation is supported by the experimental Pt–C bond length of ca. 2.06 Å of a similar cyclometallated Pt(PtBu3) complex.72 This result highlights the need for complementary characterization strategies such as NMR spectroscopy and EXAFS in order to arrive at a clear understanding of surface structure.
Conclusions
In conclusion, this study demonstrates that sideband selective experiments with selective long pulses can be used for the rapid reconstruction of 195Pt ssNMR spectra of supported platinum hydrides on different metal oxides (SiO2 and SiO2/Al2O, ∼2 Pt wt% loading). With 1H{195Pt} and 1H–31P{195Pt} J-resolved and J-HMQC experiments it was possible to rapidly measure 195Pt ssNMR spectra of the molecular complexes 1–4. The 1H, 31P and 195Pt NMR fingerprints provide direct insight into the oxidation state and coordination environment of the molecular complexes. With this information it was possible to probe the structure of surface species formed by supporting 1 on SBA-15 or SiO2–Al2O3.Monitoring the 31P signal dephasing as a function of 195Pt offset in J-resolved experiments allowed for the determination of the 195Pt CS tensor of the surface Pt species in 1/SiO2–Al2O3 and 1/SBA-15. Numerical fits of 1H–31P{195Pt} J-resolved curves also allowed the accurate determination of 31P–195Pt J-coupling constants in the supported complexes (2433 Hz and 2908 Hz for 1/SiO2–Al2O3 and 1/SBA-15, respectively). These values align closely with the expected value of approximately 2500 Hz for a 31P–195Pt(II) bond. Comparing experimental and DFT calculated NMR parameters suggested that the bond distance between Pt and O for 1/SiO2–Al2O3 was about 2.7 Å. This distance does not correspond to that of a typical covalent bond, which is around 2 Å, indicating the presence of a 3-coordinated complex on the surface of 1/SiO2–Al2O3 (Fig. 10). Several additional data, including the hydride 1H NMR signal at −36 ppm, the 195Pt CSA, and the 31P–195Pt J-coupling constants, show the characteristic features of a 3-coordinate complex. Room temperature 31P ssNMR spectra indicate the presence of two surface species on 1/SBA-15, one of which is a 4-coordinated bisphosphine hydride complex with a Pt–O bond length between 2.1 Å to 2.3 Å and a proton shift of −27 ppm (Fig. 10). The other species is a decomposition product, which was assigned to a cyclometallated compound, formed by C–H activation. The use of sideband selective 195Pt ssNMR experiments allowed the differentiation of two platinum species.
 | ||
| Fig. 10 Summary of structural information for supported compounds. Major surface sites after dehydroxylation at 500 °C on the respective metal oxide before reaction with 1. When 1 is supported on SBA-15 two different Pt surface species are formed: a bisphosphine hydride and a CH-activated phosphine complex. Calculations of the 195Pt CSA suggest that the Pt–O bond length is between 2.1 to 2.3 Å for the bisphosphine hydride. When 1 is supported on SiO2–Al2O3 a bisphosphine hydride is the major surface species. The Pt–O bond length is between 2.7 and 3.0 Å. | ||
Overall, the combination of experimental and computational techniques presented in this paper offers a comprehensive characterization of these surface-supported Pt complexes. The knowledge gained from this study contributes to the understanding of the coordination geometry, Pt oxidation states, and electronic environments of these complexes. This knowledge can guide the design and optimization of heterogeneous catalysts based on supported Pt species, facilitating the development of efficient catalytic systems for various chemical transformations.
Experimental section
General
All air- and moisture-sensitive materials were manipulated under an atmosphere of oxygen-free dry argon using a MBraun glovebox and standard schlenkline techniques.80 Spectrograde acetone was stored over molecular sieve (3 A) 24 h prior to use. Dichloromethane and pentane were used directly form the SPS and dry benzene was used as bought from Sigma Aldrich (anhydrous benzene, 99.99%). Materials: bis(tri-tert-butyl-phosphine)platinum(0) 1 and the platinum hydrides 2–4 were synthesized according to an adapted literature procedures.64Preparation of complex 2: trans-PtHCl[P(tBu)3]2
To a stirred solution of Pt(0)(PtBu3)21 (650 mg, 1.1 mmol) in toluene, HCl dissolved in toluene (2 eq., 0.2 mmL, 12 M) was added slowly. It was stirred for 20 minutes and dried under vacuum and crystallized from n-pentane to give a white powder (570 mg, 0.9 mmol, 83%). 1H NMR (400 MHz, benzene-d6): δ = 1.58–1.51 (m, 54H), −19.20 (tt, JH–Pt = 1072 Hz, JH–P 12.9 Hz, 1H) ppm. 31P NMR (162 MHz, benzene-d6): δ = 75.69 (t, JP–Pt = 2951 Hz).Preparation of complex 3: trans-PtH[P(tBu)3]2 BF4
Complex 2 (450 mg, 0.7 mmol) was dissolved in dry acetone (3 mL) and AgBF4 (137.70 mg, 0.7 mmol) dissolved in acetone (2 mL) was added slowly. The precipitate AgCl was filtered off and the filtrate was dried under vacuum. After precipitation from CH2Cl2/n-pentane a bright yellow powder was obtained (230 mg, 0.33 mmol, 50%). It was crystalized from CH2Cl2/n-pentane. 1H NMR (400 MHz, chloroform-d) δ 1.29–1.15 (m, 54H), −35.63 (t, JH–Pt = 2638 Hz, 1H) ppm. 31P NMR (162 MHz, chloroform-d): δ = 85.58 ppm (t, JP–Pt = 2625) ppm.Preparation of complex 4: trans-PtH[CH3CN][P(tBu)3]2 BF4
3 was dissolved in CH2Cl2 and an excess of CH3CN (>1 eq.) was added. The solution was dried under vacuum and a white powder was yield quantitatively. It was crystalized from CH2Cl2/n-pentane. 1H NMR (400 MHz, CDCl3): δ = 1.48–1.37 (m, 54H), −19.20 (t, JH–Pt = 1049 Hz, JH–P = 12.4 Hz, 1H) ppm. 31P NMR (162 MHz, chloroform-d): δ = 80.07 (t, JP–Pt = 2810) ppm.Preparation of 1/SBA-15
The reaction of 1 with SBA-15 was done using the double-Schlenk technique. A solution of 1 (30 mg, 0.058 mmol) in toluene (3 mL) was frozen in one finger of the double Schlenk. The other finger was filled with SBA-15 (dehydroxylated at 500 °C, 500 mg). The Schlenk was evacuated and under static vacuum the solution was unfrozen and added to the powder. The reaction was stirred for 20 h at room temperature. The mixture was filtered and washed with n-pentane (3 × 5 mL) before dried under high vacuum (10−5 mbar). An off-white powder was obtained (loading: 0.1 mmol g−1, determined by ICP-OES). Following synthesis, room temperature MAS 1H ssNMR spectra were acquired with an MAS frequency of 7 kHz. The 1H ssNMR spectrum showed a diagnostic hydride 1H NMR signal at −26.6 ppm and a 1H-195Pt J-coupling of 1034 Hz.Preparation of 1/SiO2–Al2O3
A solution of 1 (30 mg, 0.058 mmol) in pentane (1 mL) was added to suspension of SiO2–Al2O3 (dehydroxylated at 500 °C) in pentane (2 mL). After 20 h stirring at room temperature the mixture was filtered and washed with n-pentane (3 × 5 mL) before drying under high vacuum (10−5 mbar). An off-white powder was obtained (loading: 0.1 mmol g−1, determined by ICP-OES). Following synthesis, a room temperature 1H MAS ssNMR spectrum was acquired with a 10 kHz MAS frequency. The 1H ssNMR spectrum showed a diagnostic hydride 1H NMR signal at −36.3 ppm and a 1H-195Pt J-coupling of 2510 Hz.Room temperature solid-state NMR experiments
All experiments were performed with a 9.4 T wide bore magnet equipped with a Bruker Avance III HD console. Chemical shifts were indirectly referenced to adamantane using the 1H shift at 1.82 with respect to TMS in CDCl3 ppm using the IUPAC-recommended chemical shift convention.81 The probe was not retuned for any sideband selective experiment. The 195Pt RF field was calibrated using the Bloch–Siegert shift method.82 The RF field strength of the 195Pt saturation pulses for the sideband selective experiments varied depending on the MAS frequency and the saturation pulse length that was used. Numerical simulations suggest that the optimal RF fields are within a few kilohertz of those that were used experimentally (Fig. S18 and Table S6†).DNP-SENS solid-state NMR experiments
All experiments were performed with a wide bore 9.4 T/263 GHz DNP spectrometer83 equipped with a Bruker Avance III HD console. Chemical shifts were indirectly referenced to the 13C chemical shift of tetrakis(trimethylsilyl)silane at 2.75 ppm (with respect to TMS (δ(1H) = 0 ppm) in 98% CDCl3). The 195Pt RF field was calibrated by scaling the 13C π/2 pulse length, which was calibrated directly on tetrakis(trimethylsilyl)silane by nutating the pulse length.Room temperature 1H{195Pt} experiments
Complex 3 was packed into a 1.3 mm zirconia rotor in a glovebox then spun with nitrogen gas. A 1.3 mm Bruker HX probe and a MAS frequency of 50 kHz was used. Before experiments began, the magic angle was precisely calibrated by minimizing the splitting in the 2H NMR spectrum of deuterated oxalic acid.841H 90 and 180° pulse used durations of 2.5 and 5 μs, respectively. A 1H{195Pt}2D J-HMQC with a rotor-synchronized t1-evolution period and hyper-complex states-TPPI acquisition85 was done to determine the exact frequency of a 195Pt spinning sideband so that sideband selective experiments could be performed. The sideband selective experiments were performed with 32 scans, 1.55 s recycle delay (1.3 × T1), 0.4 ms J-evolution period (1J(1H–195Pt) = ∼2500 Hz), and 31 sub-spectra. 60 μs SL pulses with an 8 kHz and 6 kHz RF field were employed for J-resolved and J-HMQC, respectively.Room temperature 1H–31P{195Pt} experiments on 1
Complex 1 was packed into a 2.5 mm zirconia rotor in a glovebox then spun with nitrogen gas. A Bruker 2.5 mm HXY probe configured in 1H–31P–195Pt mode and a MAS frequency of 25 kHz were used. Before experiments began, the magic angle was precisely calibrated by maximizing the intensity of the second spinning sideband of the 79Br spectrum of KBr.861H 90 and 180° pulse used durations of 2.5 and 5 μs, respectively. The 1H → 31P CP contact time was set to 2.5 ms and optimal 1H and 31P spinlock RF powers of 105 and 70 kHz, respectively, were used; the 1H RF power was ramped from 90 to 100%. A 1H–31P{195Pt} 2D J-HMQC experiment with a rotor-synchronized t1-evolution period and hyper-complex states-TPPI acquisition85 was used to determine the exact frequency of a 195Pt spinning sideband so that sideband selective experiments could be performed. Sideband selective experiments used 80 μs SL 195Pt pulses with a 6 kHz RF field or 5 kHz RF field for J-resolved and J-HMQC, respectively. 128 scans and 512 scans were acquired for J-resolved and J-HMQC, respectively The recycle delay was 0.66 s (1.3 × T1), the J-evolution period was 0.22 ms (1J(31P–195Pt) = 4400 Hz), and 31 sub-spectra were acquired. 100 kHz 1H SPINAL-64 decoupling was applied during the J-evolution periods and signal acquisition.87 The same 31P CP conditions and 1H decoupling conditions were used for room temperature ssNMR experiments on compound 2–4.Room temperature 1H–31P{195Pt} experiments on 2
Sideband selective experiments used 120 μs SL pulses with 7 kHz and 4 kHz RF fields for J-resolved and J-HMQC, respectively. The recycle delay was 1.17 s (1.3 × T1), the J-evolution period was 0.22 ms (1J(31P–195Pt) = ∼3000 Hz), and 56 sub-spectra were acquired. 32 scans were acquired at each 195Pt offset.Room temperature 1H{195Pt} experiments on 3
Sideband selective experiments used 60 μs SL pulses with 8 kHz and 6 kHz RF fields for J-resolved and J-HMQC, respectively. The recycle delay was 1.55 s (1.3 × T1), the J-evolution period was 0.4 ms (1J(31P–195Pt) = ∼2500 Hz), and 31 sub-spectra were acquired. 32 scans were acquired at each 195Pt offset.Room temperature 1H–31P{195Pt} experiments on 4
Sideband selective experiments used 80 μs SL pulses with 7 kHz and 5 kHz RF for J-resolved and J-HMQC, respectively. The recycle delay was 1.43 s (1.3 × T1), the J-evolution period was 0.16 ms (1J(31P–195Pt) = ∼6250 Hz), which was not optimal, and 66 sub-spectra were acquired. 64 scans were acquired at each 195Pt offset. A CPMG echo train was added at the end of the sideband selective pulse sequences for detection to increase sensitivity.DNP-SENS 1H–31P{195Pt} experiments on 1/SiO2–Al2O3 and 1/SBA-15
The DNP samples of 1/SiO2–Al2O3 and 1/SBA-15 were prepared in a glovebox and impregnated with a solution of 10 mM TEKPol88 in 1,1,2,2 tetrachloroethane (TCE) via the incipient wetness method and packed into a 3.2 mm sapphire rotor. The rotor was then inserted into a low-temperature Bruker 3.2 mm HXY MAS DNP probe configured in 1H–31P–195Pt mode which was precooled to 100 K, then spun with an MAS frequency of 12.5 kHz. The 1H → 31P CP contact time was set to 2 ms and optimal 1H and 31P spinlock RF powers of 40 and 72 kHz, respectively, were used; the 1H RF power was ramped from 90 to 100%. 1H–31P{195Pt} J-resolved experiments to measure 31P–195Pt J-couplings were performed with a WURST-80 (ref. 35) saturation pulse on 195Pt. The WURST-80 pulse was 25 μs duration, used a RF field of ca. 120 kHz, and swept over a total frequency range of 1.25 MHz. The WURST pulse was found to give superior dephasing as compared to a rectangular pulse. 100 kHz 1H SPINAL-64 decoupling was applied during the J-evolution periods and signal acquisition.87 For 195Pt variable offset 1H–31P{195Pt} J-resolved experiments rectangular 195Pt saturation pulses that were 120 μs (1.5 × τr) in duration were used with an RF field of 35 kHz. The 195Pt saturation pulse offset was incremented in steps of 25 kHz while retuning the 195Pt channel on the probe every 250 kHz; 69 sub-spectra were acquired. The total echo duration was set to 0.32 ms which is approximately the inverse of the 31P–195Pt J-coupling. For all experiments, the recycle delay was 2.21 s for 1/SiO2–Al2O3 and 4.43 s for 1/SBA-15. 128 scans were acquired at each 195Pt offset.Room temperature ssNMR experiments on 1/SiO2–Al2O3 and 1/SBA-15
1/SiO2–Al2O3 and 1/SBA-15 were packed into 2.5 mm zirconia rotors inside of a glovebox and then spun with nitrogen gas. A Bruker 2.5 mm HX probe configured in double mode was used. The 1H spin echo (π/2–τ–π–τ) spectra were acquired with a (1.3 × T1) recycle delay (2 s for 1/SiO2–Al2O3 and 1.42 s for 1/SBA-15). The 1H spectra were acquired with 4096 and 16384 scans for 1/SiO2–Al2O3 and 1/SBA-15, respectively.
Numerical SIMPSON simulations
Numerical simulations were performed using SIMPSON v4.2.1.89–91 The SIMPSON input codes are provided with the archived data. All pulses in the files were of finite duration, except the 1H and 31P π/2 pulses, which were ideal. The rep320 crystal file was used for all simulations. The number of gamma angles was set to 13. The heat maps in the ESI† were constructed using MATLAB_R2021B. The 1D spectra were processed with Python using Microsoft Visual Studio Code as the interpreter.DFT calculations
For complexes 1–4, hydrogen atom coordinates were geometrically optimized in CASTEP54 using the PBE-GGA functional,92 TS dispersion correction scheme,93 and ultra-soft pseudopotentials.94,95 The coordinates for the models of 1/SiO2–Al2O3 and 1/SBA-15 were geometrically optimized with AMS 2021. All NMR calculations were performed using AMS 2021 with the hybrid PBE0 functional56,57 and a Slater-type basis set of triple-ζ with two polarization functions.55 Relativistic effects were treated by zero order regular approximation (ZORA)96–99 with spin–orbit relativity level.Data availability
The data that support the findings of this study are openly available at https://doi.org/10.5281/zenodo.12773732 or https://zenodo.org/records/12773732. Data includes experimental NMR datasets and pulse programs (TopSpin format), SIMPSON simulation files and AMS 2021 files. Solution NMR spectra of molecular compounds, 1D 1H and 31P MAS NMR spectra, crystallographic data and infrared spectra are available at https://doi.org/10.18419/darus-4580.Author contributions
B. A. A. performed solid-state NMR experiments, performed DFT calculations and analyzed data. E. J. W. synthesized compounds, characterized them by standard techniques and assisted with solid-state NMR experiments. S. K. and J. K. performed DFT calculations. W. F. obtained single-crystal X-ray structures of compounds. A. J. R. and D. P. E. directed and oversaw all research. The manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
B. A. A. and A. J. R. were supported by the National Science Foundation under grant no. CBET-1916809. This work was also funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation). Project number 358283783-SFB 1333/2.References
- X. Cui, K. Junge, X. Dai, C. Kreyenschulte, M.-M. Pohl, S. Wohlrab, F. Shi, A. Brückner and M. Beller, ACS Cent. Sci., 2017, 3, 580–585 CrossRef CAS.
- Z. Xiang, W. Li, F. Liu, F. Tan, F. Han, X. Wang, C. Shao, M. Xu, W. Liu and X. Yang, Electrochem. Commun., 2021, 127, 107039 CrossRef CAS.
- P. Bourges, S. Lunati and G. Mabilon, in Studies in Surface Science and Catalysis, Elsevier, 1998, vol. 116, pp. 213–222 Search PubMed.
- J. Hou, M. Yang, C. Ke, G. Wei, C. Priest, Z. Qiao, G. Wu and J. Zhang, EnergyChem, 2020, 2, 100023 CrossRef.
- A. V. Da Rosa, in Fundamentals of Renewable Energy Processes, Elsevier/AP, Amsterdam, 3rd edn, 2013 Search PubMed.
- J. Kijenski and P. Winiarek, in Studies in Surface Science and Catalysis, Elsevier, 2000, vol. 143, pp. 787–794 Search PubMed.
- M. Gomez, G. Muller, D. Sainz, J. Sales and X. Solans, Organometallics, 1991, 10, 4036–4045 CrossRef CAS.
- L. Charruault, V. Michelet, R. Taras, S. Gladiali and J.-P. Genêt, Chem. Commun., 2004, 850–851 RSC.
- A. B. Permin and V. S. Petrosyan, Appl. Organomet. Chem., 1990, 4, 329–333 CrossRef CAS.
- D. Gregson, J. A. K. Howard, M. Murray and J. L. Spencer, J. Chem. Soc., Chem. Commun., 1981, 716–717 RSC.
- R. G. Goel, W. O. Ogini and R. C. Srivastava, Organometallics, 1982, 1, 819–824 CrossRef CAS.
- T. Yoshida, T. Yamagata, T. H. Tulip, J. A. Ibers and S. Otsuka, J. Am. Chem. Soc., 1978, 100, 2063–2073 CrossRef CAS.
- S. Maier, S. P. Cronin, M.-A. Vu Dinh, Z. Li, M. Dyballa, M. Nowakowski, M. Bauer and D. P. Estes, Organometallics, 2021, 40, 1751–1757 CrossRef CAS.
- P. J. Ayare, S. A. Gregory, R. J. Key, A. E. Short, J. G. Tillou, J. D. Sitter, T. Yom, D. W. Goodlett, D.-C. Lee, F. M. Alamgir, M. D. Losego and A. K. Vannucci, Green Chem., 2021, 23, 9523–9533 RSC.
- S. L. Wegener, T. J. Marks and P. C. Stair, Acc. Chem. Res., 2012, 45, 206–214 CrossRef CAS PubMed.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed.
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D'Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J.-M. Basset, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef.
- A. W. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072–5085 CrossRef CAS.
- P. Laurent, L. Veyre, C. Thieuleux, S. Donet and C. Copéret, Dalton Trans., 2013, 42, 238–248 RSC.
- E. J. W. Austin, P. J. Barrie and R. J. H. Clark, J. Chem. Soc., Chem. Commun., 1993, 1404–1405 Search PubMed.
- B. E. G. Lucier, A. R. Reidel and R. W. Schurko, Can. J. Chem., 2011, 89, 919–937 Search PubMed.
- B. E. G. Lucier, K. E. Johnston, W. Xu, J. C. Hanson, S. D. Senanayake, S. Yao, M. W. Bourassa, M. Srebro, J. Autschbach and R. W. Schurko, J. Am. Chem. Soc., 2014, 136, 1333–1351 Search PubMed.
- T. Kobayashi, F. A. Perras, T. W. Goh, T. L. Metz, W. Huang and M. Pruski, J. Phys. Chem. Lett., 2016, 7, 2322–2327 CrossRef CAS PubMed.
- M. Soorholtz, L. C. Jones, D. Samuelis, C. Weidenthaler, R. J. White, M.-M. Titirici, D. A. Cullen, T. Zimmermann, M. Antonietti, J. Maier, R. Palkovits, B. F. Chmelka and F. Schüth, ACS Catal., 2016, 6, 2332–2340 Search PubMed.
- Z. Wang, L. A. Völker, T. C. Robinson, N. Kaeffer, G. Menzildjian, R. Jabbour, A. Venkatesh, D. Gajan, A. J. Rossini, C. Copéret and A. Lesage, J. Am. Chem. Soc., 2022, 144, 21530–21543 CrossRef CAS.
- S. Todisco, G. Saielli, V. Gallo, M. Latronico, A. Rizzuti and P. Mastrorilli, Dalton Trans., 2018, 47, 8884–8891 Search PubMed.
- G. J. Rees, S. T. Orr, L. O. Barrett, J. M. Fisher, J. Houghton, G. H. Spikes, B. R. C. Theobald, D. Thompsett, M. E. Smith and J. V. Hanna, Phys. Chem. Chem. Phys., 2013, 15, 17195 Search PubMed.
- C. D. Makowka, C. P. Slichter and J. H. Sinfelt, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 5663–5679 CrossRef CAS PubMed.
- C. P. Slichter, Surf. Sci., 1981, 106, 382–396 CrossRef CAS.
- S. W. Sparks and P. D. Ellis, J. Am. Chem. Soc., 1986, 108, 3215–3218 CrossRef CAS.
- M. J. Jaroszewicz, A. R. Altenhof, R. W. Schurko and L. Frydman, J. Am. Chem. Soc., 2021, 143, 19778–19784 CrossRef CAS PubMed.
- T. Pawlak, M. L. Munzarová, L. Pazderski and R. Marek, J. Chem. Theory Comput., 2011, 7, 3909–3923 CrossRef CAS PubMed.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97–102 CrossRef.
- A. W. MacGregor, L. A. O'Dell and R. W. Schurko, J. Magn. Reson., 2011, 208, 103–113 CrossRef CAS.
- K. J. Harris, A. Lupulescu, B. E. G. Lucier, L. Frydman and R. W. Schurko, J. Magn. Reson., 2012, 224, 38–47 CrossRef CAS PubMed.
- I. Hung, A. J. Rossini and R. W. Schurko, J. Phys. Chem. A, 2004, 108, 7112–7120 CrossRef CAS.
- J. Camacho-Bunquin, M. Ferrandon, H. Sohn, D. Yang, C. Liu, P. A. Ignacio-de Leon, F. A. Perras, M. Pruski, P. C. Stair and M. Delferro, J. Am. Chem. Soc., 2018, 140, 3940–3951 CrossRef CAS.
- Y. Ishizaka, N. Arai, K. Matsumoto, H. Nagashima, K. Takeuchi, N. Fukaya, H. Yasuda, K. Sato and J. Choi, Chem.–Eur. J., 2021, 27, 12069–12077 CrossRef CAS.
- A. Venkatesh, D. Gioffrè, B. A. Atterberry, L. Rochlitz, S. L. Carnahan, Z. Wang, G. Menzildjian, A. Lesage, C. Copéret and A. J. Rossini, J. Am. Chem. Soc., 2022, 144, 13511–13525 CrossRef CAS PubMed.
- B. A. Atterberry, E. Wimmer, D. P. Estes and A. J. Rossini, J. Magn. Reson., 2023, 352, 107457 CrossRef CAS PubMed.
- E. Prack, C. A. O'Keefe, J. K. Moore, A. Lai, A. J. Lough, P. M. Macdonald, M. S. Conradi, R. W. Schurko and U. Fekl, J. Am. Chem. Soc., 2015, 137, 13464–13467 CrossRef CAS.
- P. Paluch, A. G. M. Rankin, J. Trébosc, O. Lafon and J.-P. Amoureux, Solid State Nucl. Magn. Reson., 2019, 100, 11–25 CrossRef CAS PubMed.
- J. Herzfeld and A. E. Berger, J. Chem. Phys., 1980, 73, 6021–6030 CrossRef CAS.
- F. D. Rochon and A. Morneau, Magn. Reson. Chem., 1991, 29, 120–126 CrossRef CAS.
- S. J. S. Kerrison and P. J. Sadler, Inorg. Chim. Acta, 1985, 104, 197–201 CrossRef CAS.
- L. Schwartsburd, R. Cohen, L. Konstantinovski and D. Milstein, Angew. Chem., 2008, 120, 3659–3662 CrossRef.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- M. Sterzel and J. Autschbach, Inorg. Chem., 2006, 45, 3316–3324 CrossRef CAS PubMed.
- J. Autschbach and S. Zheng, Magn. Reson. Chem., 2008, 46, S45–S55 CrossRef.
- A. V. Buevich, J. Saurí, T. Parella, N. De Tommasi, G. Bifulco, R. T. Williamson and G. E. Martin, Chem. Commun., 2019, 55, 5781–5784 RSC.
- J. S. Fabián, J. M. García De La Vega and E. San Fabián, J. Chem. Theory Comput., 2014, 10, 4938–4949 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- J. Campos, L. Ortega-Moreno, S. Conejero, R. Peloso, J. López-Serrano, C. Maya and E. Carmona, Chem.–Eur. J., 2015, 21, 8883–8896 CrossRef CAS PubMed.
- H. Braunschweig, K. Radacki and K. Uttinger, Chem.–Eur. J., 2008, 14, 7858–7866 CrossRef CAS PubMed.
- W. Baratta, S. Stoccoro, A. Doppiu, E. Herdtweck, A. Zucca and P. Rigo, Angew. Chem., Int. Ed., 2003, 42, 105–109 CrossRef CAS PubMed.
- N. Carr, L. Mole, A. G. Orpen and J. L. Spencer, J. Chem. Soc., Dalton Trans., 1992, 2653–2662 Search PubMed.
- M. J. Ingleson, M. F. Mahon and A. S. Weller, Chem. Commun., 2004, 2398–2399 RSC.
- M. A. Ortuño, P. Vidossich, G. Ujaque, S. Conejero and A. Lledós, Dalton Trans., 2013, 42, 12165 RSC.
- R. G. Goel and R. C. Srivastava, Can. J. Chem., 1983, 61, 1352–1359 Search PubMed.
- J. Rodriguez, D. B. Culver and M. P. Conley, J. Am. Chem. Soc., 2019, 141, 1484–1488 CrossRef CAS PubMed.
- A. Bowden, S. J. Coles, M. B. Pitak and A. W. G. Platt, Inorg. Chem., 2012, 51, 4379–4389 CrossRef CAS.
- M. Dyballa, Energy Fuels, 2023, 37, 18517–18559 CrossRef CAS.
- P. E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1996, 118, 4442–4450 CrossRef CAS.
- T. M. Gilbert, I. Hristov and T. Ziegler, Organometallics, 2001, 20, 1183–1189 CrossRef CAS.
- S. Niu and M. B. Hall, J. Am. Chem. Soc., 1998, 120, 6169–6170 CrossRef CAS.
- B. L. Simms and J. A. Ibers, J. Organomet. Chem., 1987, 330, 279–289 CrossRef CAS.
- T. Troadec, S. Tan, C. J. Wedge, J. P. Rourke, P. R. Unwin and A. B. Chaplin, Angew. Chem., Int. Ed., 2016, 55, 3754–3757 CrossRef CAS.
- S. J. Coles, S. J. Fieldhouse, W. T. Klooster and A. W. G. Platt, Polyhedron, 2019, 161, 346–351 CrossRef CAS.
- S. E. Maier, T. Nagel, M. Turan, E. Kaya, W. Frey, M. Dyballa and D. P. Estes, Organometallics, 2024, 43, 233–241 CrossRef CAS.
- G. Crépeau, V. Montouillout, A. Vimont, L. Mariey, T. Cseri and F. Maugé, J. Phys. Chem. B, 2006, 110, 15172–15185 CrossRef.
- W. Daniell, U. Schubert, R. Glöckler, A. Meyer, K. Noweck and H. Knözinger, Appl. Catal., A, 2000, 196, 247–260 CrossRef CAS.
- F. Rascón, R. Wischert and C. Copéret, Chem. Sci., 2011, 2, 1449 RSC.
- W. S. Salvia, T. Y. Zhao, P. Chatterjee, W. Huang and F. A. Perras, Chem. Commun., 2023, 59, 13962–13965 RSC.
- F. A. Perras, Z. Wang, T. Kobayashi, A. Baiker, J. Huang and M. Pruski, Phys. Chem. Chem. Phys., 2019, 21, 19529–19537 RSC.
- D. F. Shriver and M. A. Drezdzon, The Manipulation of Air-Sensitive Compounds, Wiley, New York, 2nd edn, 1986 Search PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- I. Hung, P. Gor’kov and Z. Gan, J. Magn. Reson., 2020, 310, 106636 CrossRef CAS.
- M. Rosay, L. Tometich, S. Pawsey, R. Bader, R. Schauwecker, M. Blank, P. M. Borchard, S. R. Cauffman, K. L. Felch, R. T. Weber, R. J. Temkin, R. G. Griffin and W. E. Maas, Phys. Chem. Chem. Phys., 2010, 12, 5850 RSC.
- S. Antonijevic and G. Bodenhausen, Angew. Chem., Int. Ed., 2005, 44, 2935–2938 CrossRef CAS.
- D. Marion, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1989, 85, 393–399 CAS.
- J. S. Frye and G. E. Maciel, J. Magn. Reson., 1982, 48, 125–131 CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS.
- Z. Tošner, R. Andersen, B. Stevensson, M. Edén, N. C. Nielsen and T. Vosegaard, J. Magn. Reson., 2014, 246, 79–93 CrossRef.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS PubMed.
- Z. Tošner, T. Vosegaard, C. Kehlet, N. Khaneja, S. J. Glaser and N. C. Nielsen, J. Magn. Reson., 2009, 197, 120–134 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- E. Van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. V. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- E. Van Lenthe, R. Van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- E. Van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional SIMPSON simulation results and solid-state NMR spectra. CCDC 2339745–2339747. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06450j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |