
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputing chemical potentials with machine-learning-accelerated simulations to accurately predict thermodynamic properties of molten salts†‡
Luke D.
Gibson
 *a,
Rajni
Chahal
b and
Vyacheslav S.
Bryantsev
*b
*a,
Rajni
Chahal
b and
Vyacheslav S.
Bryantsev
*b
aComputational Sciences and Engineering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: gibsonld@ornl.gov
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: bryantsevv@ornl.gov
First published on 24th January 2025
Abstract
The successful design and deployment of next-generation nuclear technologies heavily rely on thermodynamic data for relevant molten salt systems. However, the lack of accurate force fields and efficient methods has limited the quality of thermodynamic predictions from atomistic simulations. Here we propose an efficient free energy framework for computing chemical potentials, which is the central free energy quantity behind many thermodynamic properties. We accelerate our simulations without sacrificing accuracy by using machine learning interatomic potentials trained on density functional theory (DFT) data. Using lithium chloride as our model system, we compute chemical potentials with DFT-accuracy for solid and liquid phases by transmuting ions into noninteracting particles. Notably, in the liquid phase, we demonstrate consistency whether we transmute one ion pair or the entire system into ideal gas particles. By locating the temperature where the chemical potential of solid and liquid phases cross, we predict a melting point of 880 ± 18 K for lithium chloride, which is remarkably close to the experimental value of 883 K. With this successful demonstration, we lay the foundation for high-throughput thermodynamic predictions of many properties that can be derived from the chemical potentials of the minority and majority components in molten salts.
Introduction
Molten salts play a critical role in next-generation nuclear technologies. Specifically, in many next-generation reactor designs, molten salts are used for one or more core functionalities, where it can operate as a coolant, medium for dissolved fuel, and/or blanketing around solid fuel. At the tail end of the fuel cycle, strategies for recycling spent fuel, such as pyrochemical reprocessing, utilize molten salts as the solvent during electrochemical refinement of unspent fissile materials. The successful design and deployment of a spectrum of nuclear energy technologies rely on the existence of high-quality, validated property data for relevant molten salts and their mixtures. Although experimental measurements exist for many of the important molten salt systems, some properties still exhibit a high level of uncertainty, which is currently being addressed by the scientific community. Furthermore, the properties of important minor components in molten salts are still largely unknown. However, the acquisition of experimental data is time-consuming, requires specialized equipment, and is constrained by high costs and challenging operating conditions.Atomistic simulations offer a potential solution to this data scarcity problem with molecular dynamics (MD) simulations. Due to its atomic-scale resolution, MD simulations are often used to clarify the local structure of molten salts and their mixtures by comparing theoretical spectroscopic measurements (e.g., X-ray and neutron scattering, Raman spectroscopy, extended X-ray absorption fine structure (EXAFS) spectroscopy) against experimental spectra.1–15 These structural details validated against the experimental observables help explain the macroscopic behavior of molten salts. This information can also be incorporated into thermodynamic models to improve CALPHAD predictions. In addition to local structure, many theoretical studies also report thermophysical properties (e.g., density, diffusivity, viscosity, specific heat capacity) since they can be easily extracted from a single MD simulation. However, the number of studies on thermochemical properties (e.g., solubilities, redox potentials, and phase boundaries) of molten salts is far fewer.16–22 These thermochemical properties are challenging to predict because they require knowledge of how the system's chemical potentials change for a given thermodynamic process, which often necessitates free energy calculations of both solid and liquid phases using advanced sampling methods.
For many of the thermodynamic studies of molten salts, more challenging free energy simulations have been conducted primarily using inexpensive, classical force fields.18,20–23 Notably, Anwar, et al. used the rigid ion model (RIM) and the thermodynamic integration (TI) method to predict the melting point of sodium chloride by computing the chemical potentials in both solid and liquid phases.20 In their study, they used the Einstein crystal method24,25 to compute solid phase chemical potentials. The liquid phase methods involved computing the free energy of transforming either two ideal gas (IG) particles into a NaCl pair (i.e., inserting a NaCl pair into liquid NaCl) or an entire system of Lennard-Jones (LJ) particles into interacting NaCl ions. The authors found that the most reliable method was to transform the entire system into NaCl instead of inserting a single NaCl pair into molten NaCl. After addressing issues associated with differing Na+ and Cl− spring constants in the Einstein crystal calculations, the RIM-predicted NaCl melting point was reported to be ∼70 K higher than the experimental value of 1073 K.23 The poor performance of the single ion pair insertion method was attributed to difficulties with sampling the interion distance during TI calculations, resulting in large error bars and inaccurate chemical potentials. This underscores the necessity of developing reliable methodologies to compute the thermodynamic properties of impurities in molten salts which rely on the single ion pair insertion method.
Thermodynamic properties of more complex, multicomponent molten salt systems have also been successfully studied using RIM and the more advanced polarizable ion model (PIM).18,21,22 Zhou and Zhang22 leveraged RIM simulations to compute concentration dependent activity coefficients of UCl3 in LiCl–KCl mixtures through TI calculations. Using PIM, Salanne and coworkers18 demonstrated great agreement between experimental and predicted standard redox potentials of MCl3 salts (M3+: U3+, La3+, Y3+, Tb3+, and Sc3+) in LiCl–KCl mixtures with alchemical free energy methods. In another PIM study, DeFever and Maginn21 predicted the liquidus and eutectic point of KCl–LiCl and KCl–MgCl2 mixtures using both alchemical free energy methods and direct, two-phase simulations. Despite their success in constructing solid–liquid phase diagrams, their predictions differed from experiment because PIM failed to accurately predict the pure component melting points of both LiCl and MgCl2. Although classical force fields like RIM and PIM have enabled various thermodynamic studies, their ability to accurately predict thermodynamic properties is limited, emphasizing the importance of using accurate methods for thermodynamic predictions.
More expensive methods, such as ab initio molecular dynamics (AIMD), have been used predominantly to study structural motifs and, to a lesser extent, select thermophysical properties of molten salts that are difficult to converge given small system sizes and short simulation times.3–6,9,26–29 AIMD has also been reported to predict the Na solubility and redox potential in molten NaCl, but the simulations were again constrained by high statistical uncertainty.16 To circumvent the prohibitive computational cost of AIMD, recent studies have leveraged machine learning interatomic potentials (MLIPs) trained on density functional theory (DFT) data to probe thermodynamic properties of molten salts, such as solubilities,30 redox potentials,30 and phase boundaries.19 The prediction of chemical potentials of a molten salt system with a MLIP was first demonstrated by Shi, et al., where they trained a deep neural network interatomic potential (NNIP) for a molten NaCl system and used the quasi-chemical theory (QCT) method to predict the excess chemical potentials of Na+ and Cl− ions.17 However, to achieve close agreement with the experimental value, the QCT method necessitated several, separately-trained NNIPs and the NNIP-predicted excess chemical potentials ultimately required quantum chemical corrections to account for the deficiencies of the reference DFT method used to train their NNIP.17 In another study on Na/NaCl solubilities and redox potentials, Sun, et al.30 trained a moment tensor potential (MTP) and employed the Widom particle insertion method31 to compute excess chemical potentials of Na and Cl in molten NaCl and Na in liquid Na. However, the MTP-based free energy predictions differed from their DFT-based predictions by ∼10 kcal mol−1 with unacceptably large error bars (±10 kcal mol−1).30 Interestingly, their MTP and DFT predictions are also inconsistent with the findings of an earlier AIMD study16 (by 7 and 17 kcal mol−1, respectively), which employed the same DFT and free energy methods. Overall, while MLIPs offer significant improvements in efficiency over AIMD, thermodynamic predictions with DFT-trained MLIPs remain unreliable for molten salt systems.
In this work, we present a predictive framework for accurately computing chemical potentials in both solid- and liquid-phases by leveraging the efficiency of DFT-trained MLIPs. Through this ML-accelerated, predictive framework, we demonstrate the ability to accurately predict the melting point of lithium chloride at ambient pressure using the computed chemical potentials. We validate our chemical potential predictions in both liquid and solid phases by benchmarking against DFT calculations for smaller systems. We also highlight the unique challenges that arise when using MLIPs with these predictive methods and demonstrate effective and practical solutions that overcome the challenges faced by the existing approaches.17,20,30 Importantly, this ML-accelerated, predictive framework lays the foundation for large-scale predictions of molten salt thermodynamic properties with low statistical uncertainty, which can be used to guide experimental measurements and thus support the next generation of nuclear technologies.
Results & discussion
Machine learning force field benchmarks
To demonstrate our predictive framework for free energies and chemical potentials, we first trained separate machine learning force fields (MLFFs) for the solid and liquid phases of lithium chloride. Based on the test set errors (shown in the Fig. S1 of the ESI‡), both potentials displayed good performance in reproducing DFT energies, forces, and stresses. The liquid structure produced by the molten LiCl MLFF was also confirmed to remain stable (Fig. S3‡). The radial distribution function, g(r), for the Li–Cl pair in Fig. 1 shows nearly perfect agreement between the MLFF and DFT (AIMD with PBE-D3). Additionally, the MLFF exhibits similar liquid structure to experiment32 based on the good agreement with the experimental g(r) in Fig. 1. All free energy calculations were performed in the NVT ensemble at the equilibrium volume determined from NpT simulations at 1 bar. The predicted densities from both solid and liquid potentials exhibit good agreement with experiment: the MLFFs yield a liquid phase density of 1.53 g cm−3 at 943 K (ρExp.(l) [943 K] = 1.47 g cm−3, ref. 33) and solid phase density of 1.85 g cm−3 at 850 K (ρExp.(s) [850 K] = 1.92 g cm−3, ref. 34).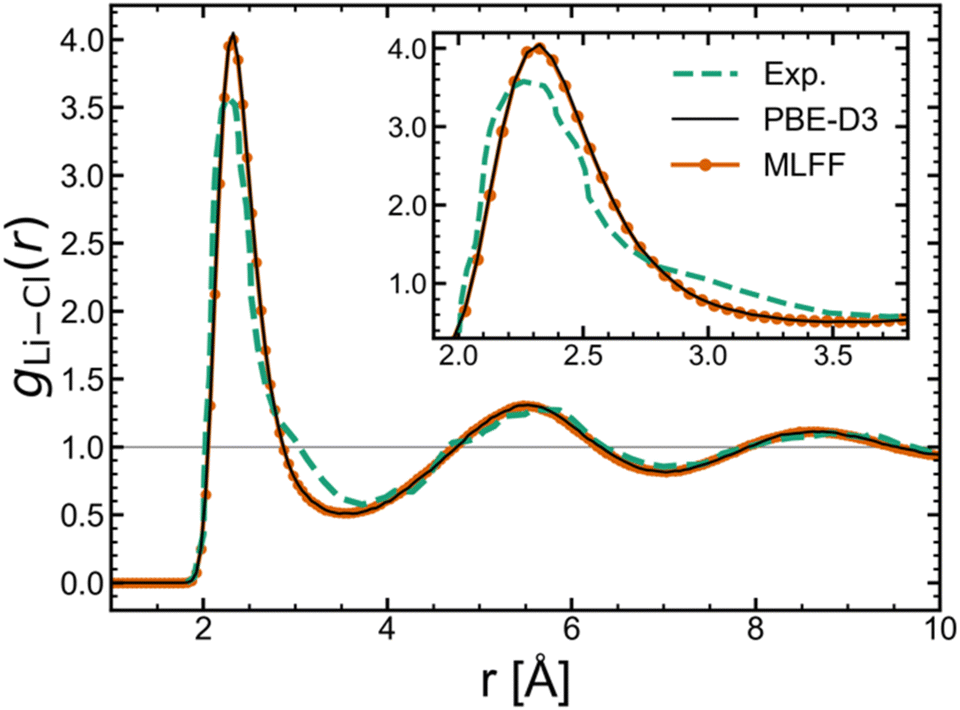 | ||
| Fig. 1 Comparison of the Li–Cl radial distribution function between experiment,32 DFT, and MLFF. | ||
Computing chemical potentials of molten lithium chloride
To compute the chemical potential of LiCl in the liquid phase, we used alchemical transformation to convert either one or all LiCl pairs from ideal gas particles into fully interacting Li+ and Cl− ions (selected pairs highlighted in Fig. 2a and b, respectively) via thermodynamic integration (TI). Herein, these two thermodynamic pathways are respectively referred to as ΔN = 1 and ΔN = N. As shown in the energy diagram in Fig. 2d, for a system with 150 LiCl pairs, both pathways yield the same value at 943 K for the chemical potential of molten LiCl. Since the two thermodynamic pathways are inherently different, their integrand plots (〈dH/dλ〉 vs. λ, Fig. 2c) have slightly different trends. Notably, the excess chemical potential computed with ΔN = 1 has a larger magnitude than ΔN = N by approximately 3.5 kcal mol−1 (or 1.9kBT); however, this difference is fully compensated by the difference in ideal chemical potentials. Although both pathways yield the same chemical potential value, the ΔN = N pathway has better statistics with uncertainty that is lower than the ΔN = 1 pathway by an order of magnitude. This reduced uncertainty stems from the fact that all ions contribute to 〈dH/dλ〉; whereas, for ΔN = 1, 〈dH/dλ〉 is only based on the local environment around the single Li+ and Cl− ions. Naturally, this also manifests in the time required to converge calculations using both pathways, where only ∼200 ps of simulation were required for the ΔN = N pathway and ∼2 ns for the ΔN = 1 pathway.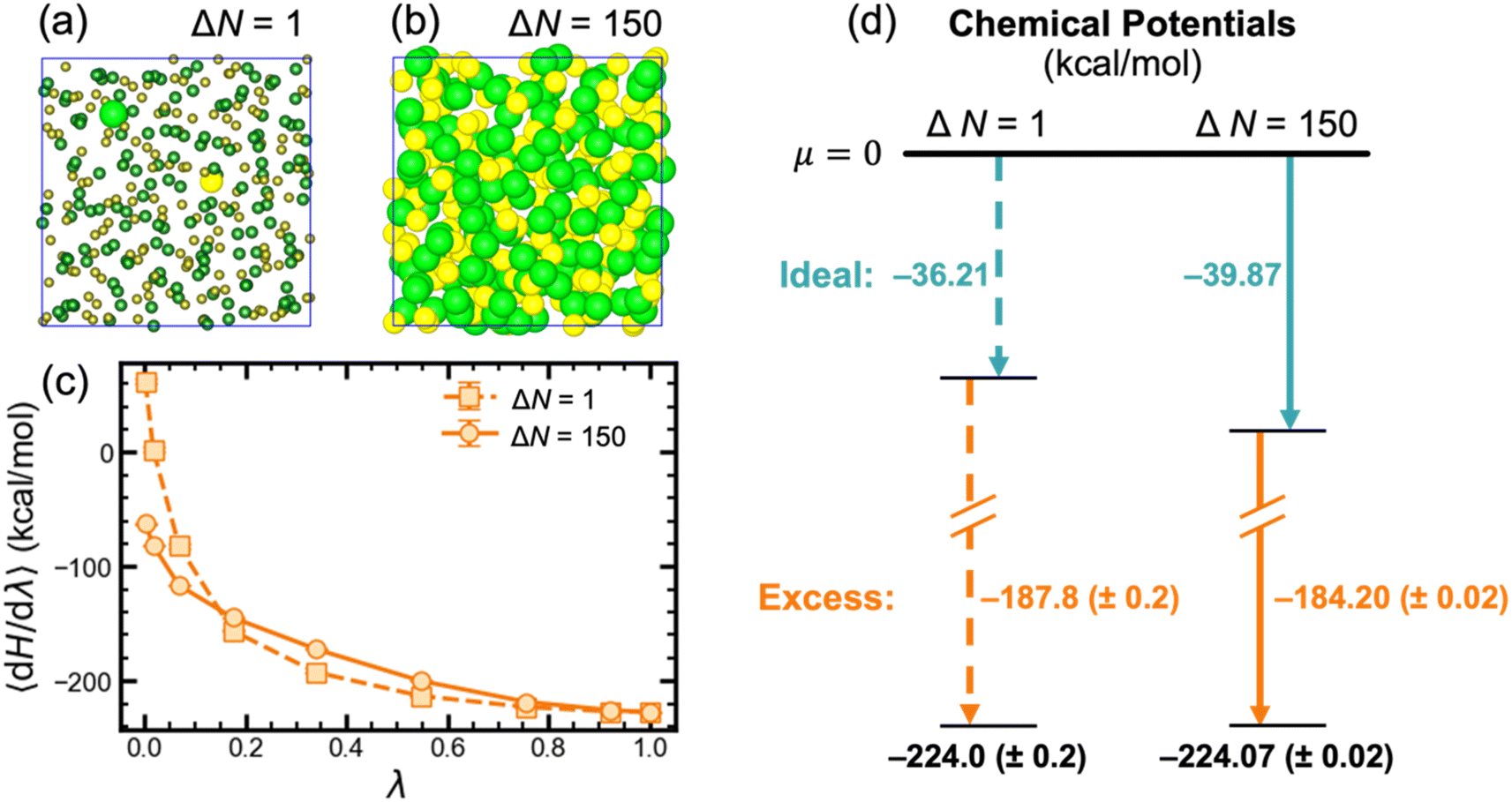 | ||
| Fig. 2 Comparison of chemical potentials computed via TI for ΔN = 1 and ΔN = 150 pathways on a system with 150 LiCl pairs at 943 K. Highlighted ions denote that their interactions were modified via TI in the cases of (a) a single pair and (b) the full system. (c) Comparison of the integrand at each λ point for both approaches. Error bars represent 95% confidence intervals, which are smaller than the plotted markers. (d) Breakdown of the contributions to the chemical potential, which showcases the agreement across both approaches. Li+ and Cl− ions are respectively colored yellow and green in (a) and (b). | ||
Although the chemical potentials computed with ΔN = 1 and ΔN = N pathways closely matched, previous studies20,35 have reported challenges with the ΔN = 1 pathway. In a study of KF solubility in water, Ferrario, et al. noted difficulties with sufficiently sampling the interionic distance distribution for the simultaneously inserted K+ and F− ions, which caused them to instead insert K+ and F− separately.35 In a study on predicting the NaCl melting point, Anwar, et al. also experienced similar challenges when computing chemical potentials of liquid NaCl via single NaCl ion pair insertion.20 The authors considered two different pathways for inserting a single NaCl pair: (i) two ideal gas particles were directly transformed into a NaCl ion pair by simultaneously scaling both the van der Waals (VDW) and charge parameters; and (ii) two ideal gas particles were first transformed into VDW particles and then into a NaCl ion pair. Both approaches ultimately yielded similar values that differed from their calculations using a ΔN = N pathway by approximately −2kBT. Although this error closely matches the difference in ideal chemical potentials for the two pathways, the correct ideal chemical potentials were already employed. Additionally, larger uncertainties were reported for the two pathways for inserting single NaCl ion pair. Ultimately, this deviation was again attributed to the poor sampling of the Na–Cl distances during TI calculations.
To better understand the origins of these challenges with single ion pair insertion, we computed the excess chemical potential of LiCl with the Li–Cl distance fixed at four separate distances (r = 2.3, 3.5, 5.5, and 9.5 Å). In Fig. 3, the Li–Cl potential of mean force (PMF) and the fixed-distance excess chemical potentials are overlaid with matching relative scales, revealing the direct relationship between the Li–Cl distance and excess chemical potentials. Given this trend, without sufficient sampling, one or more TI windows could oversample short interionic distances and consequently overestimate the excess chemical potential. Notably, at a fixed distance of 9.5 Å, beyond the second solvation shell, the computed excess chemical potential closely matched the value in Fig. 2d when the ion distances were not constrained, which was also confirmed to have sampled the equilibrium distance distribution (Fig. S4‡). We interpret this phenomenon to be caused by the vanishingly small probability of the two ions directly interacting in the thermodynamic limit, where it naturally follows that the excess chemical potential will converge to the same value with ions fixed at large distances. For molten salt melts where short-range ordering is relatively short-lived (e.g., LiCl), excess chemical potential calculations can be converged more rapidly by fixing the ion pair at a distance where g(r) approaches unity. However, for more acidic melts that exhibit significant short- and intermediate-range ordering,3,6,11,14,29 sufficiently sampling the important configurations around the chosen cation and anion(s) will become more challenging as ion-exchange kinetics become slower.4
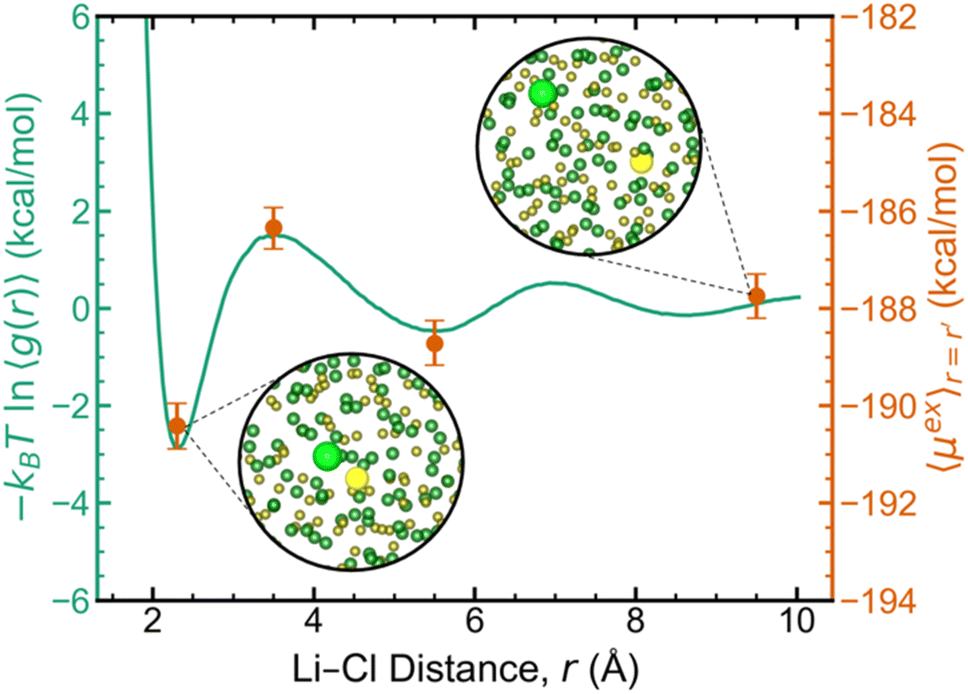 | ||
| Fig. 3 Dual-axis plot of the potential of mean force between Li+ and Cl− based on g(r) (left axis, solid green line) and the excess chemical potential computed at multiple fixed Li–Cl distances (right axis, orange markers). Insets show representative configurations at the corresponding Li–Cl distance with the targeted ions highlighted. System conditions for each calculation match those in Fig. 2. | ||
Given that the ΔN = N pathway provides lower uncertainty, we computed molten LiCl chemical potentials at 943 K for multiple system sizes (N = 50, 100, 150, and 300 LiCl pairs; shown in Table 1) to assess the extent of finite size effects. The ideal, excess, and total chemical potentials were considered converged by 150 LiCl pairs since there was no appreciable change when the system size was doubled to 300 LiCl pairs. Thus, we used the ΔN = N pathway with a system of 150 LiCl pairs to compute the chemical potential of liquid phase LiCl at T = 850, 880, 910, and 943 K, which are also listed in Table 1. In fact, the results are essentially converged even for 50 LiCl ion pairs, where the absolute difference with respect to N = 300 is only 0.11 kcal mol−1.
| Source | T (K) | N LiCl | Ideal free energy, Fid (kcal mol−1) | Excess free energy, Fex (kcal mol−1) | Chemical potential, μ (kcal mol−1) |
|---|---|---|---|---|---|
| a Computed using soft core potentials as shown in Fig. 4a. b Computed using the ΔN = 1 thermodynamic pathway. | |||||
| MLFF | 850 | 150 | −35.33 | −185.58 ± 0.02 | −220.91 ± 0.02 |
| 880 | 150 | −36.79 | −185.17 ± 0.02 | −221.96 ± 0.02 | |
| 910 | 150 | −38.25 | −184.67 ± 0.02 | −222.92 ± 0.02 | |
| 943 | 50 | −39.74 | −184.25 ± 0.04, −184.37 ± 0.01a | −223.99 ± 0.04, −224.11 ± 0.01a | |
| 100 | −39.84 | −184.27 ± 0.03 | −224.10 ± 0.03 | ||
| 150 | −39.87, −36.21b | −184.20 ± 0.02, −187.8 ± 0.2b | −224.07 ± 0.02, −224.0 ± 0.2b | ||
| 300 | −39.91 | −184.19 ± 0.01 | −224.10 ± 0.01 | ||
| DFT | 943 | 50 | −39.74 | −184.2 ± 0.2a | −224.0 ± 0.2a |
Importantly, with the use of MLIPs instead of physical models, electrostatic interactions are combined with van der Waals interactions and all interactions are truncated to short-range based on the cutoffs used in the MLIP. Additionally, the short Debye length in pure molten LiCl limits the impact of long-range electrostatic interactions in bulk phase calculations. Fortunately, since the total system charge remains neutral in all training configurations and thermodynamic pathways considered, we do not need to account for energetic corrections arising from interactions with the surface potential or Ewald electrostatics.36,37
Given that the MLFF architecture has the coupling parameter λ integrated directly into the Hamiltonian (eqn (18)), MLFFs are much more well-suited for these free energy calculations than other MLIP architectures. To demonstrate the ability to perform these free energy calculations using a different MLIP architecture, we have trained a NNIP for the liquid LiCl system using the DeePMD-kit package.38 Since the coupling parameter is not integrated into the NNIP architecture, thermodynamic pathways other than ΔN = N are not possible because the pairwise interactions for a subset of atoms cannot be independently scaled by λ. As a result, for the NNIP, the hybrid Hamiltonian that couples the ideal gas state to a fully interacting LiCl liquid becomes,
| HIG→NNIP(λ) = (1 − λ)HIG + λHNNIP, | (1) |
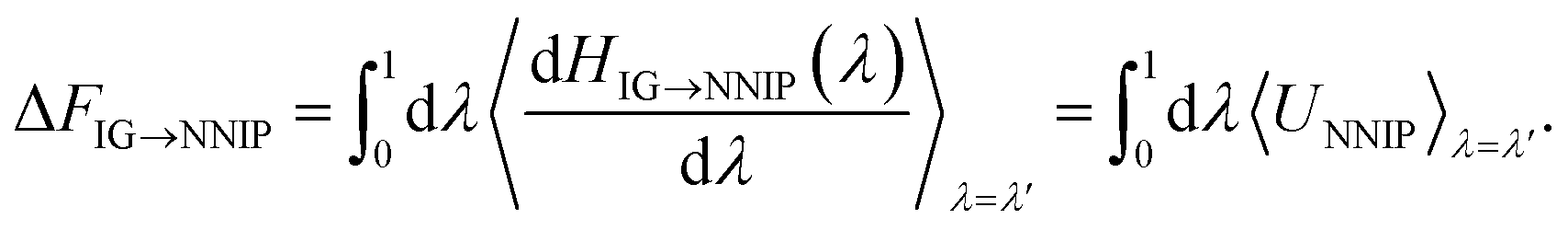 | (2) |
The integrand of the second integral simplifies to the potential energy of the NNIP, UNNIP, since the kinetic energy terms in HIG and HNNIP are identical. However, unique problems can arise in simulations with λ < 1 since the forces in HIG→NNIP are scaled by λ during MD, thereby increasing the likelihood of sampling nonphysical or highly unlikely configurations the NNIP has never seen. Such structures are much less problematic with physics-based models (e.g., classical force fields) and can still be evaluated; however, MLIPs cannot reliably extrapolate to novel configurations, which can lead to unrealistic energies and forces that cause catastrophic failures during MD. In these untrained regions of configuration space, MLIPs will often predict massive energies and forces that either crash the simulation or stabilize highly non-physical configurations (shown in inset of Fig. 4b). For simulations that do not crash, these artificially stabilized structures cause discontinuities in 〈dH/dλ〉, corrupting the free energy calculation. Indeed, we observed that the NNIP exhibited such behavior during TI calculations (Fig. 4b), leading to an incorrect excess chemical potential prediction (by >5 kcal mol−1). Interestingly, the MLFF does not experience the same problem when performing TI in the same manner (i.e., λHMLFF instead of HMLFF(λ)).
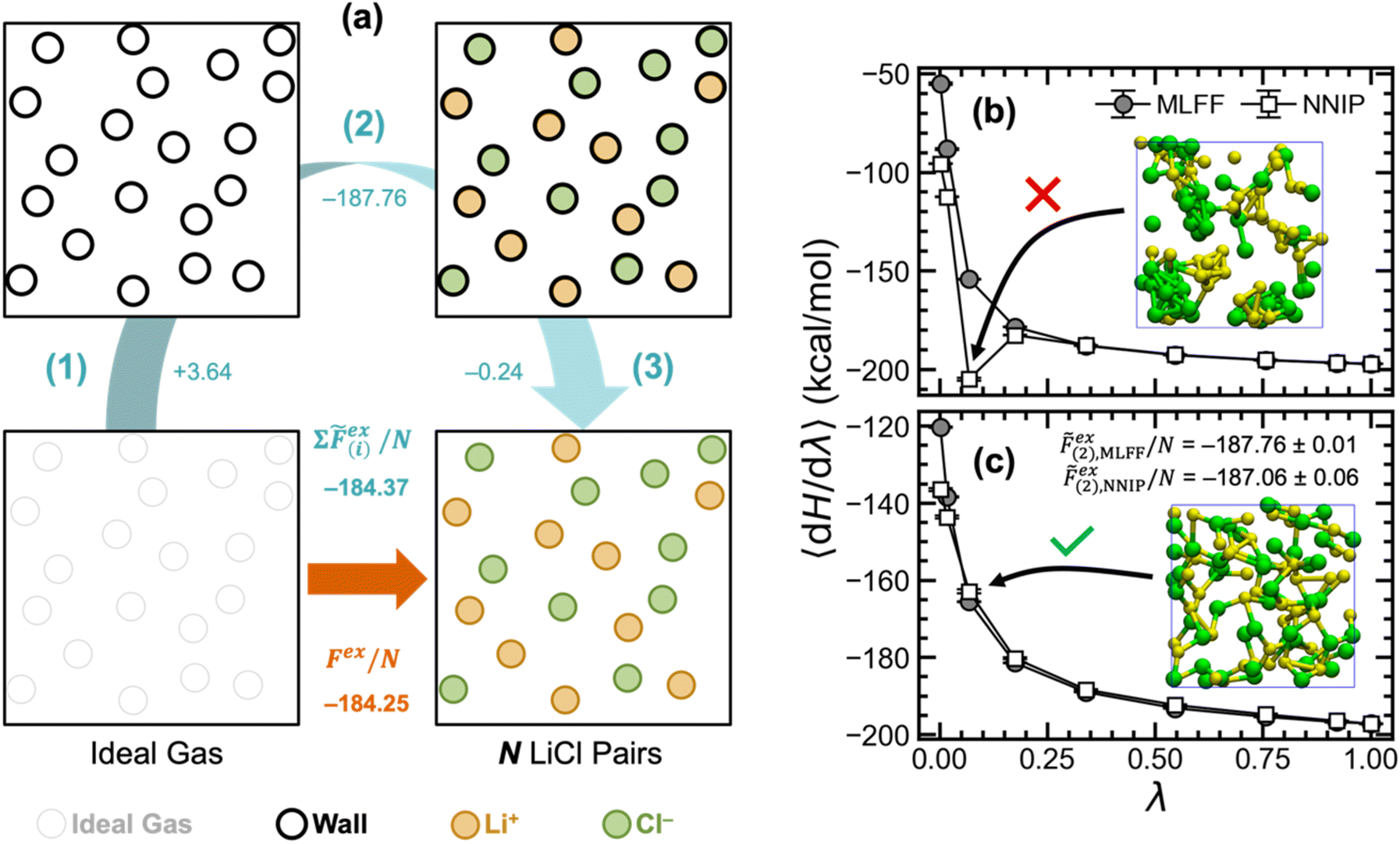 | ||
| Fig. 4 (a) Workflow for transforming ideal gas particles into Li+ and Cl− ions using soft core potentials. Free energy changes for each step in the workflow are shown in blue, whereas the excess free energy for direct transformation is shown in orange. The overall free energy change for the workflow is shown below the excess free energy of direct transformation. Excess free energies computed with both approaches used 50 LiCl pairs at 943 K. Uncertainties are omitted for clarity but can be found in Table 2. Comparison of integration curves using both MLFF and NNIP models (b) without and (c) with soft core potentials. All energy units are kcal mol−1. | ||
To address this instability, we utilize the stepwise approach shown in Fig. 4a, where a soft-core potential is used as a prophylactic tool to prevent non-physical configurations from occurring (e.g., overlapping atoms). In this way, the excess chemical potential can be computed in three steps: (1) ideal gas particles are transformed into soft-core particles, ![[F with combining tilde]](https://www.rsc.org/images/entities/i_char_0046_0303.gif) ex(1); (2) without adjusting the soft-core potential, the particles are transformed into fully interacting Li+ and Cl− ions, ex(2); and lastly (3) the soft-core potential is removed, ex(3). In this work, we employed the Fermi potential implemented in VASP as the soft-core potential on all pairwise interactions, which has the functional form,
ex(1); (2) without adjusting the soft-core potential, the particles are transformed into fully interacting Li+ and Cl− ions, ex(2); and lastly (3) the soft-core potential is removed, ex(3). In this work, we employed the Fermi potential implemented in VASP as the soft-core potential on all pairwise interactions, which has the functional form,
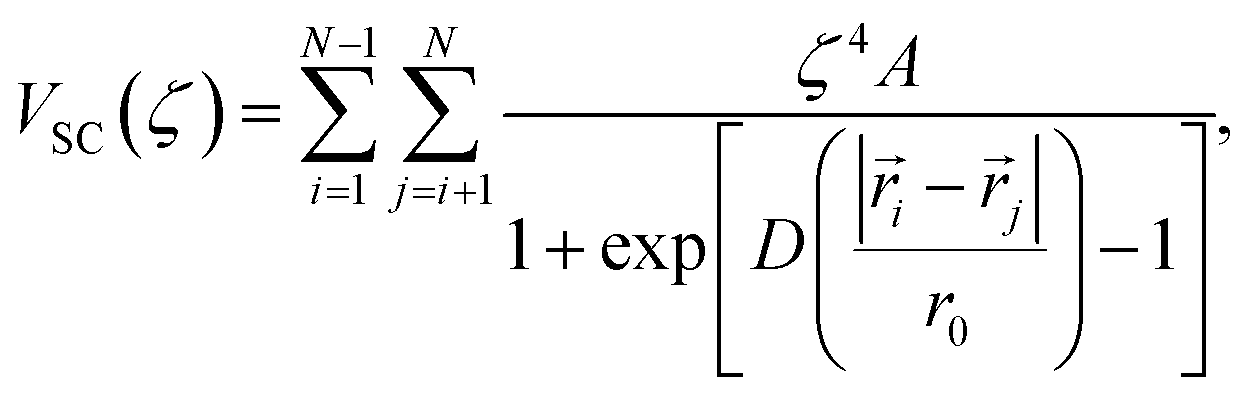 | (3) |
By first introducing this soft-core potential on top of the MLIP, we demonstrate in Fig. 4c that the previous instabilities can be avoided during TI calculations. We computed the free energy changes for steps (1) and (3) in Fig. 4avia the Bennett acceptance ratio method42 from 16 separate simulations per step 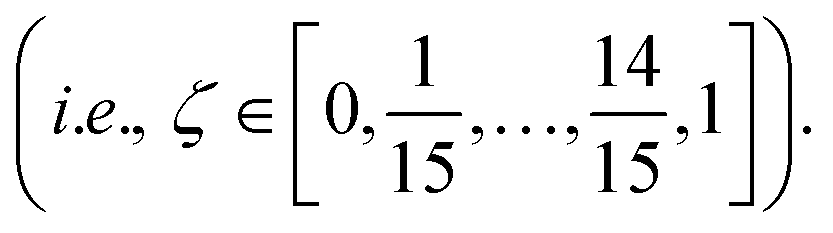 Using the MLFF, we showcase in Fig. 4a that the excess chemical potential computed using soft-core potentials matches the value from direct TI, confirming that this stepwise approach is a viable alternative. Moreover, by demonstrating good agreement between the NNIP- and MLFF-computed values for ex(2) in Table 2, we show that excess chemical potentials can be computed using multiple MLIP architectures.
Using the MLFF, we showcase in Fig. 4a that the excess chemical potential computed using soft-core potentials matches the value from direct TI, confirming that this stepwise approach is a viable alternative. Moreover, by demonstrating good agreement between the NNIP- and MLFF-computed values for ex(2) in Table 2, we show that excess chemical potentials can be computed using multiple MLIP architectures.
| Potential |
ex(1)/N |
ex(2)/N |
ex(3)/N |
F ex/N | |
|---|---|---|---|---|---|
| Stepwise | Direct TI | ||||
|
a Stepwise Fex approximated using MLFF-computed values for ex(1) and ex(3).
b Calculations at low λ using the direct TI approach encounter discontinuities when employing NNIP and fail to converge when using PBE-D3.
|
|||||
| MLFF | 3.64 ± 0.01 | −187.76 ± 0.01 | −0.2410 ± 0.0002 | −184.37 ± 0.01 | −184.25 ± 0.04 |
| NNIP | — | −187.06 ± 0.06 | — | −183.66 ± 0.06a | —b |
| PBE-D3 | — | −187.6 ± 0.2 | — | −184.2 ± 0.2a | —b |
By leveraging soft-core potentials, we were also able to compute ex(2) using PBE-D3 with AIMD simulations, shown in Table 2. However, despite the system only containing 50 LiCl pairs, the number of integration points for Gauss–Lobatto quadrature was reduced from 10 to 5 to account for the large computational expense. Given that n-point Gauss–Lobatto quadrature is accurate for polynomials up to degree 2n − 3, this reduction in integration points was not expected to negatively impact our results.43 Indeed, we confirmed with MLFF calculations that this change had a negligible effect on computed values. Without soft-core potentials, DFT-based TI calculations were not feasible due to challenges at low λ with converging self-consistent field calculations during wavefunction optimization. In fact, even with soft-core potentials, the TI window with the smallest λ (0.029816) occasionally sampled configurations for which the wavefunction could not be optimized. By computing ex(2) with AIMD on a reduced system of 50 LiCl, we demonstrate consistency with the MLFF- and NNIP-based calculations, further highlighting the accuracy and viability of the stepwise approach for computing excess free energies with MLIPs.
Computing chemical potentials of solid phase lithium chloride
For solid phase free energy calculations, we used the Einstein crystal method first proposed by Frenkel and Ladd,24,25 which uses the Einstein crystal as a reference state with a known free energy. By transforming the Einstein crystal into the real crystal with TI, the absolute free energy of the real crystal can be computed. The Einstein crystal reference state consists of non-interacting atoms whose positions are harmonically restrained to their equilibrium lattice positions of the real crystal. In the first iteration of this method, the harmonic restraints of all atoms were identical, but later the method was extended to allow different spring constants for different atom types.23 As long as the centers of mass of the Einstein and real crystals were constrained, it was shown that the computed free energies of a NaCl crystal were consistent regardless of the choice of spring constants. However, the lowest uncertainties were achieved when Na+ and Cl− spring constants were chosen to respectively match their mean squared displacements (MSD).23In this work, our Einstein crystal calculations employed spring constants based on the Li+ and Cl− MSD in a 3 × 3 × 3 supercell at 800 K (kLi+ = 47.168 J m−2, kCl− = 98.550 J m−2). We also confirmed that free energy calculations were independent of our choice of spring constants for Li+ and Cl−. To compute the free energy between an Einstein crystal and the real LiCl crystal, we used 16-point Gauss–Legendre quadrature for our TI calculations. Additional technical details of our Einstein crystal calculations can be found in the Methods section.
Due to the finite size effects that arise from simulations with constrained centers of mass,23 we computed the absolute free energies of LiCl crystals constructed from 3 × 3 × 3, 4 × 4 × 4, and 5 × 5 × 5 supercells comprising 108, 256, and 500 LiCl pairs, respectively. The free energy components of each calculation are shown in Table S2.‡ After extrapolating to infinite system sizes 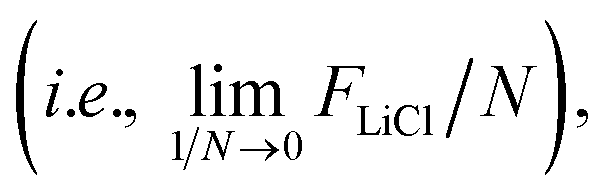 we report the absolute free energies of LiCl at T = 800, 850, 880, and 900 K in Table 3. The free energy plot of the finite systems at each temperature shown in Fig. S5‡ reveals a weak dependence on system size for our Einstein crystal calculations.
we report the absolute free energies of LiCl at T = 800, 850, 880, and 900 K in Table 3. The free energy plot of the finite systems at each temperature shown in Fig. S5‡ reveals a weak dependence on system size for our Einstein crystal calculations.
| Source | T (K) | Method | Chemical potential, μ (kcal mol−1) |
|---|---|---|---|
| MLFF | 800 | Einstein crystal | −219.704 ± 0.007 |
| 850 | Einstein crystal | −221.093 ± 0.007 | |
| 880 | Einstein crystal | −221.949 ± 0.008 | |
| 900 | Einstein crystal | −222.513 ± 0.008 | |
| DFT | 700 | Harmonic + anharmonic | −217.00 ± 0.02 |
| 750 | Harmonic + anharmonic | −218.34 ± 0.02 | |
| 800 | Harmonic + anharmonic | −219.75 ± 0.01 |
To validate the free energies computed with the MLFF and Einstein crystal method, we applied the method described in ref. 44 for computing absolute free energies of solid LiCl entirely with DFT calculations. Briefly, to compute the free energy of a solid, this approach first approximates the solid as a set of coupled harmonic oscillators, and then transforms the harmonic crystal into the real crystal with TI to capture anharmonic contributions. Using this approach, we computed the free energies of 3 × 3 × 3 and 4 × 4 × 4 LiCl supercells at T = 700, 750, and 800 K, which are reported in Table S3.‡ Calculations above 800 K were not considered with this approach because they exhibited instabilities (i.e., imaginary normal modes) in the harmonic approximation, breaking down for larger equilibrium cell volumes at higher temperatures. Additionally, due to the significant computational cost of these DFT and AIMD calculations, larger supercells were not considered, but absolute free energies extrapolated to infinite system sizes are reported in Table 3. Yet again, we demonstrate excellent agreement between DFT- and MLFF-based free energy calculations, reporting only a 0.05 kcal mol−1 difference at 800 K, with the DFT-computed free energies closely following the same trend as the MLFF-computed free energies displayed in Fig. 5.
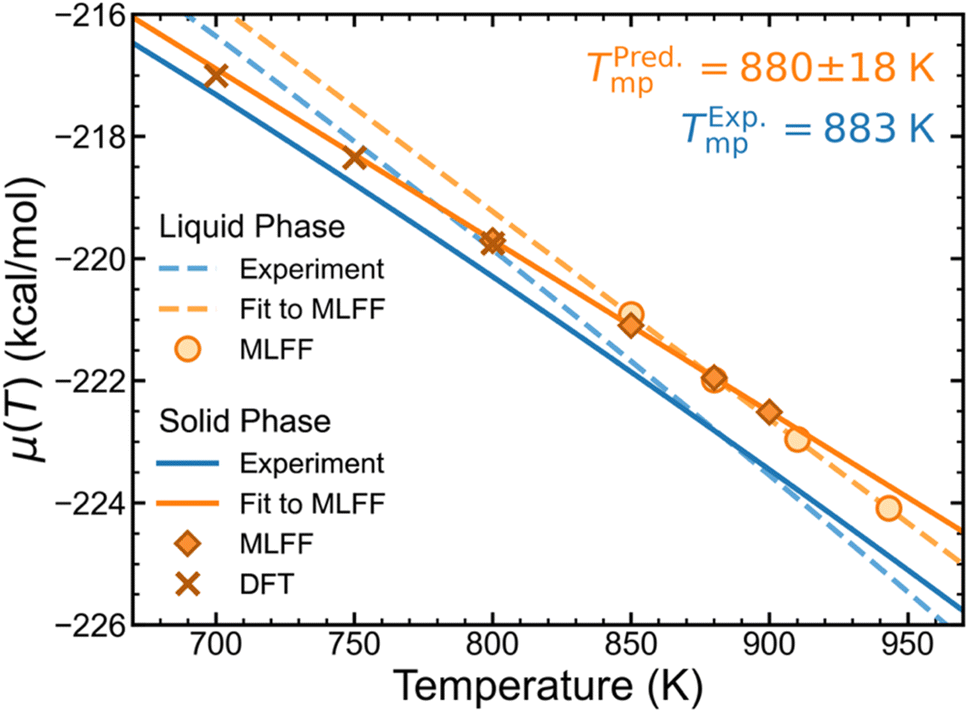 | ||
| Fig. 5 Solid and liquid phase chemical potentials from MLFF and DFT predictions compared to experiment.45 Uncertainties of the fitted lines are omitted for clarity, but error bars of MLFF and DFT data points are smaller than the size of the markers. | ||
Lithium chloride melting point prediction
We have computed chemical potentials of LiCl in both solid and liquid phases across a range of temperatures near the experimental melting point (883 K),45 which were also validated by DFT- and AIMD-based calculations. These chemical potentials are plotted against temperature in Fig. 5, which also shows nearly linear trends for both phases based on MLFF calculations. Using thermodynamic tables,45 we have also plotted experimental chemical potentials for both solid- and liquid-phase LiCl. By locating the temperature at which the solid- and liquid-phase trends intersect, the ambient pressure melting point of LiCl is predicted to be 880 ± 18 K, which is remarkably close to the experimental value of 883 K.Conclusions
The next generation of nuclear technologies heavily relies on the utility of molten salts for a variety of applications, ranging from coolants in nuclear reactors to solvents for recycling spent nuclear fuel. Despite great strides being made in experimental measurements of important molten salt mixtures, atomistic modeling can inform—and even accelerate—experiments by leveraging MD simulations for thermodynamic property predictions. However, poorly described interatomic interactions will lead to inaccurate predictions, and unreliable free energy methods frequently cause large uncertainties. Therefore, the overarching goal of this work is to demonstrate how our MLIP-accelerated, predictive framework can be leveraged to interrogate challenging thermodynamic properties of molten salts. To this end, we predicted the melting point of lithium chloride because previous studies have shown that previously employed21 empirical methods fail to accurately predict this important thermodynamic property.Given that the chemical potential is the basis for predicting the melting point of LiCl, as well as many other thermochemical properties, we performed several chemical potential calculations of both molten and solid LiCl through TI-based approaches with DFT-trained MLIPs. For select cases, we demonstrated excellent agreement between our MLIP-based predictions and DFT free energy benchmarks, with absolute errors of 0.16 and 0.05 kcal mol−1 for liquid- and solid-phase calculations, respectively.
Liquid phase chemical potentials are notoriously difficult to compute, and perhaps even more challenging to achieve consistent results across the many free energy methods.16,17,20,30 We explored two commonly used thermodynamic pathways for liquid phase chemical potential calculations where either one or all LiCl ion pairs are transformed from noninteracting particles into fully interacting ions. Significantly, we achieved consistent results across both pathways for a system of 150 LiCl pairs at 943 K, with an absolute deviation of only 0.11 kcal mol−1, when accounting for the difference in the ideal terms of eqn (7) and (10). To achieve this consistency, we found that significantly more sampling (2.0 vs. 0.2 ns per TI window) was required for the single ion pair insertion pathway to avoid oversampling of short interionic distances, which can appreciably alter excess chemical potential calculations (±2 kcal mol−1). This can possibly explain the difficulties seen in previous theoretical studies20,35 that used single ion pair insertion. We also found that less sampling was required to converge calculations when the ions were fixed at large interionic distances, but we note that other sampling challenges may arise for molten salts that exhibit significant short- and intermediate-range ordering.3,6,11,14,29 These insights will become particularly important when single ion pair insertion is the most appropriate pathway for studying a thermodynamic process for minor components in molten salts. We also demonstrated how soft-core potentials can address the unique challenges that arise for TI calculations with MLIPs when two or more atoms become too close at small coupling parameter, λ. Using a stepwise TI approach, we were able to achieve good agreement for excess chemical potentials computed with both MLFF and NNIP architectures (−184.37 and −183.66 kcal mol−1, respectively) and AIMD (−184.21 kcal mol−1), which were also consistent with the value from direct TI calculations (−184.25 kcal mol−1). This agreement showcases how our predictive framework is both accurate and extensible to a variety of MLIP architectures.
Solid-phase free energy calculations were markedly less challenging than in the liquid phase. We used the Einstein crystal method for all solid LiCl free energy calculations. By applying this method for varying system sizes, we reported absolute free energies of solid LiCl extrapolated to infinite system sizes at a range of temperatures. Free energies from Einstein crystal calculations were also validated against DFT calculations, which involve computing the harmonic vibrational contributions and accounting for anharmonic effects through TI for a hybrid Hamiltonian that couples a harmonic crystal to a real crystal via a series of AIMD simulations. Excellent agreement was observed at 800 K between MLFF and DFT, with values of the total chemical potentials of −219.704 and −219.75 kcal mol−1, respectively.
Using the array of solid- and liquid-phase LiCl chemical potentials computed at varying temperatures, we predicted the melting point of LiCl to be 880 ± 18 K, which matches exceptionally well with the experimental value of 883 K. Interestingly, our computed chemical potentials only differ from experimental values by 0.5–1.0 kcal mol−1, suggesting that PBE-D3 excels at describing LiCl systems. Indeed, this is consistent with a previous benchmarking study on various electronic structure methods.46 Thus, for a system where existing models have been shown to fail,21 using the predictive framework presented herein, we have demonstrated a dramatic improvement in the accuracy of thermodynamic predictions by leveraging MLIPs to achieve DFT-level accuracy. This exciting result demonstrates the utility of our MLIP-accelerated, predictive framework for studying the challenging thermodynamic properties of molten salts. By validating against DFT at several points and demonstrating predictions with multiple MLIP architectures, we demonstrate both the broad utility and accuracy of this computational approach. More importantly, in doing so, we also lay the foundation for high-throughput predictions of thermodynamic properties to guide experimentation and ultimately support the next generation of nuclear technologies.
In a broad context, this work facilitates the computation of free energy quantities from condensed phase simulations with ab initio accuracy, without making any assumptions about the underlying interatomic potentials. Examples of properties that can, in principle, be computed for arbitrary systems—such as molten salts, aqueous and nonaqueous electrolytes, brines, and radioactive liquid waste—are the solvation free energies, solubilities, phase diagrams, activity coefficients, redox potentials, vapor pressure, to name a few. The first practical challenge with these simulations is to develop robust and accurate MLIPs that adequately cover the relevant configurational space—a task that becomes increasingly difficult as the complexity of the system of interest grows. The second challenge lies in attaining the accuracy necessary for these thermodynamic calculations to be truly predictive, allowing them to effectively guide measurements, improve their efficiency, and maximize the value of experiments. While achieving DFT accuracy for thermodynamic properties of liquids and complex fluids is now attainable, the field of science is likely to move towards pushing these simulations to reach a level of accuracy comparable to that of small molecules in the gas phase using advanced electronic structure methods.
Methods
Methodology for computing chemical potentials of liquid systems
The chemical potential of a pure system is thermodynamically defined as,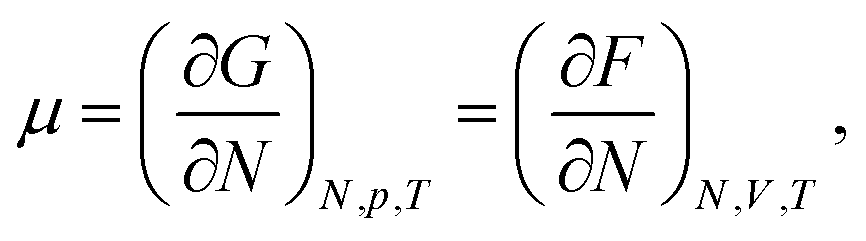 | (4) |
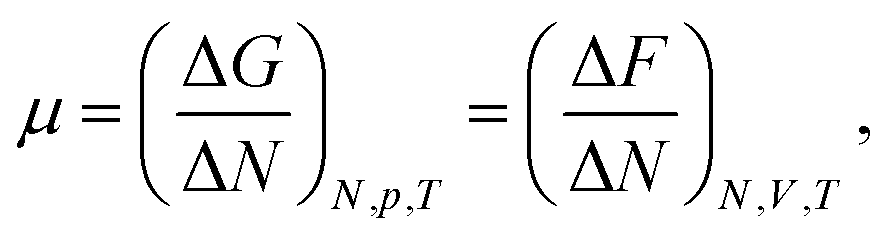 | (5) |
In the liquid phase, the chemical potential of a species can be represented as a sum of an ideal chemical potential (μid) and an excess chemical potential (μex),
| μ = μid + μex, | (6) |
Alchemical transformation uses thermodynamic integration (TI) to gradually transform one or more ideal gas particles into fully interacting species in one or more steps. When transforming a single LiCl pair (ΔN = 1) from ideal gas particles to LiCl ions (N − 1 → N) in the canonical ensemble (NVT), the chemical potential can be computed by,
 | (7) |
| Nμ = G = F + pV. | (8) |
Therefore, we can also compute the chemical potential in the NVT ensemble by computing F at the equilibrium volume from NpT simulations at 1 bar. Importantly, by simulating at these conditions, the pV term in eqn (8) becomes vanishingly small (<0.01 kcal mol−1 for the smallest system with N = 50 LiCl pairs) and is not included further. Like the ΔN = 1 pathway, the alchemical transformation method can also be used to transform the entire system from an ideal gas into N LiCl pairs (ΔN = N) to directly compute the excess free energy (Fex) of the entire system. However, since the thermodynamic pathway differs from the single LiCl pair transformation, the expression for computing the chemical potential in the canonic ensemble instead becomes,
| NLiClμLiCl = Fid + Fex, | (9) |
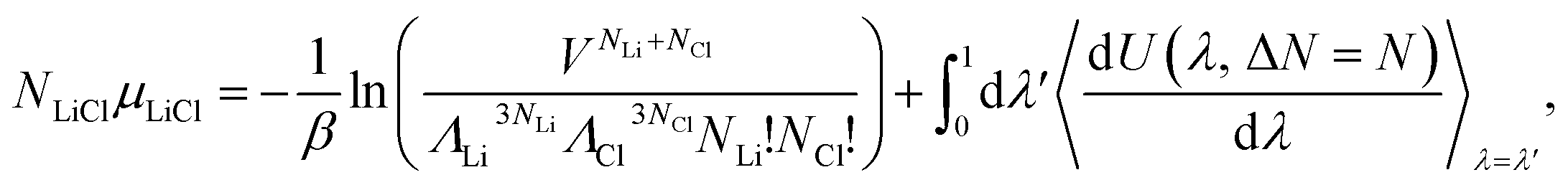 | (10) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnNLiNCl)/(NLiNCl), which is reflected in the computed excess terms. As a separate issue, Anwar, et al. noted errors in chemical potentials computed using a single pair, which were attributed to difficulties in sampling the full inter-ion distance distribution.20 Similar challenges were noted by Ferrario, et al. in a solubility study of KF in water.35 Mester and Panagiotopoulos49,50 did not report these problems for ion pair insertion in water, but their calculations were based on simulations with tens of nanoseconds and likely sampled the necessary degrees of freedom sufficiently. The all-atom alchemical transformation approach has successfully been used to compute the chemical potentials of molten NaCl using RIM20 and liquid Si using AIMD.44 Zhang, et al.51 developed a robust NNIP for water and employed TI to predict the full phase diagram of water. Non-equilibrium thermodynamic methods have also been used to predict changes in free energies.52,53
lnNLiNCl)/(NLiNCl), which is reflected in the computed excess terms. As a separate issue, Anwar, et al. noted errors in chemical potentials computed using a single pair, which were attributed to difficulties in sampling the full inter-ion distance distribution.20 Similar challenges were noted by Ferrario, et al. in a solubility study of KF in water.35 Mester and Panagiotopoulos49,50 did not report these problems for ion pair insertion in water, but their calculations were based on simulations with tens of nanoseconds and likely sampled the necessary degrees of freedom sufficiently. The all-atom alchemical transformation approach has successfully been used to compute the chemical potentials of molten NaCl using RIM20 and liquid Si using AIMD.44 Zhang, et al.51 developed a robust NNIP for water and employed TI to predict the full phase diagram of water. Non-equilibrium thermodynamic methods have also been used to predict changes in free energies.52,53
Methodology for computing chemical potentials of solid systems with the Einstein crystal method
For solid systems, the absolute free energy can be computed using the Einstein crystal method.24,25 An Einstein crystal (EC) consists of non-interacting atoms that are tethered to their ideal lattice positions with a harmonic restraint. The analytical expression for the absolute free energy of an Einstein crystal is given by,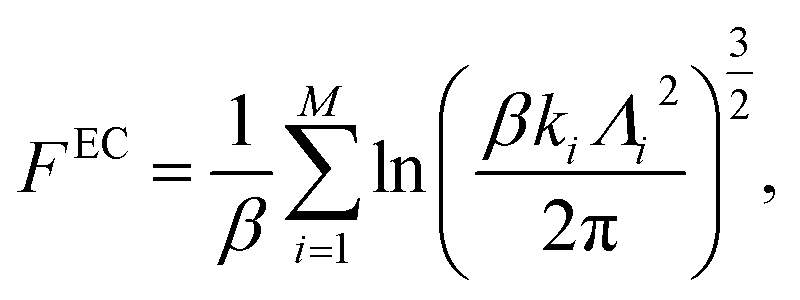 | (11) |
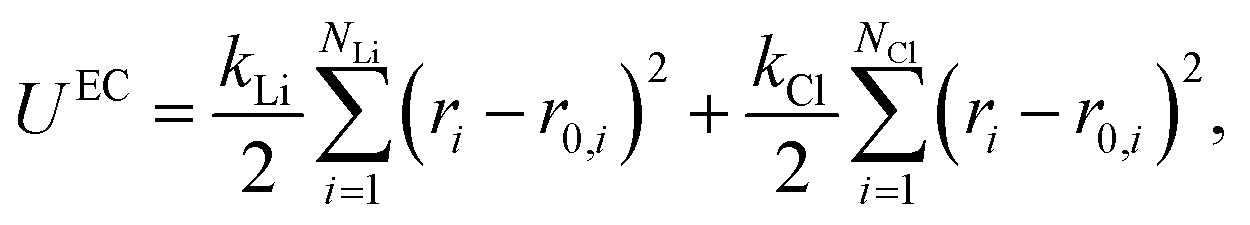 | (12) |
| HTI(λ) = λHRC + (1 − λ)HEC, | (13) |
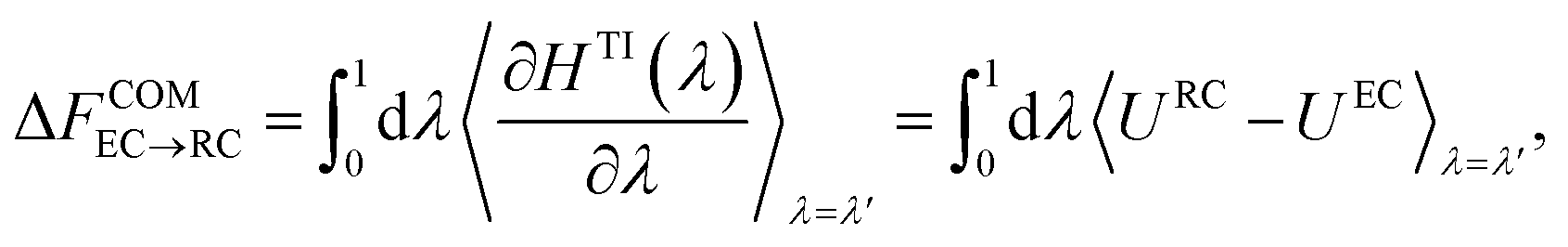 | (14) |
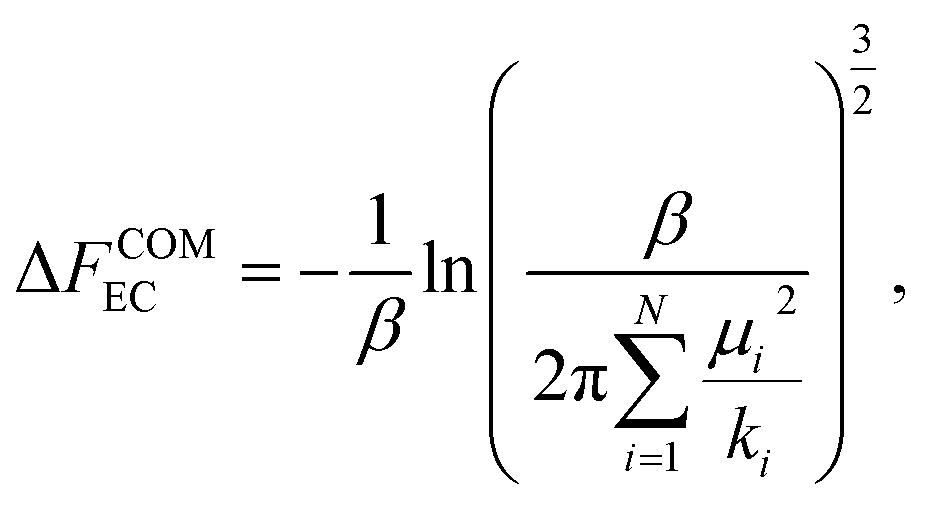 | (15) |
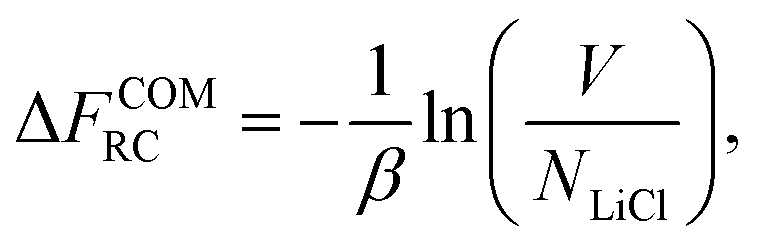 | (16) |
Finally, the absolute free energy of the real crystal can then be expressed as,
| NLiClμLiCl(s) = FEC + ΔFCOMEC + ΔFCOMEC→RC + ΔFCOMRC. | (17) |
Density functional theory (DFT) simulation details
All DFT calculations in this work were performed with the Vienna Ab initio Simulation Package (VASP)54–57 using the gradient-corrected exchange–correlation functional developed by Perdew–Burke–Ernzerhof (PBE).58–61 Dispersion interactions were included via Grimme's DFT-D3 method.62 The PBE-D3 method has been shown to perform well at describing various properties of lithium chloride in solid46 and liquid63 phases. The projector augmented-wave (PAW) method64,65 was used to describe core-valence electron interactions and valence electrons were expanded with plane waves. Self-consistent field (SCF) calculations were considered converged once the energy difference between consecutive iterations was less than 10−5 eV. Charge mixing was controlled during SCF calculations with the Pulay mixing scheme.66 A kinetic energy plane wave cutoff of 650 eV was employed in all calculations, except the generated neural network interatomic potential training data which used a cutoff of 500 eV. However, negligible differences in energies and forces were observed between 500 and 650 eV cutoffs. All calculations were performed at the Γ-point with a 1 × 1 × 1 k-point mesh grid.Machine learning interatomic potentials
The MLIP architecture implemented in VASP, known as a machine learning force field (MLFF), is a highly data efficient kernel-based method and can be trained using an on-the-fly algorithm that uses Bayesian linear regression to both optimize model parameters and provide uncertainty estimates.67,68 As a result, only structures with high error estimates are used for training, thereby greatly reducing the number of DFT calculations required to train the potential when compared to neural network-based architectures. Another benefit of the MLFF architecture is the compatibility with the thermodynamic integration (TI) method due to the coupling parameter, λ, that is incorporated directly into the Hamiltonian of the potential,67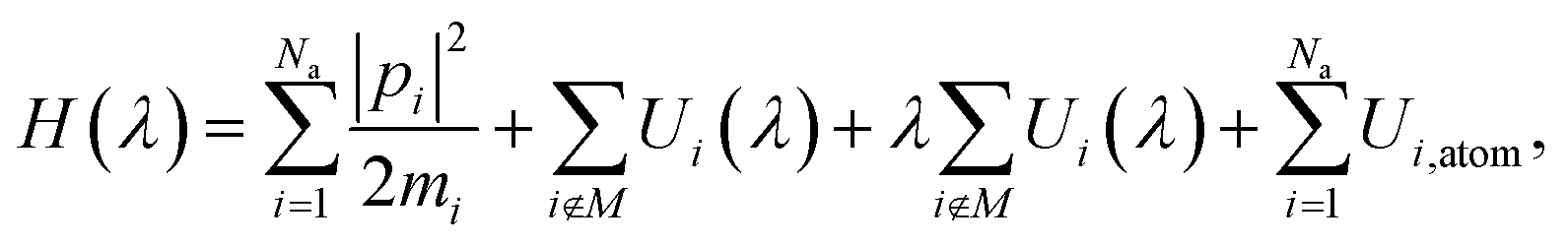 | (18) |
Since eqn (18) only scales interatomic interactions during TI, it consequently becomes important when training the MLFF to provide values for Ui,atom of each atom type. Otherwise, the resulting free energies from TI calculations will incorrectly include contributions from Ui,atom when evaluating 〈∂H(λ)/∂λ〉. Using the same DFT settings from training, Ui,atom was computed for both isolated Li+ and Cl− ions rather than neutral atoms to properly quantify interaction energies during training. The energies of isolated ions were computed by extrapolating to an infinitely large cell size by repeating calculations for cubic cells with box lengths of 25, 35, and 45 Å while correcting for monopole interactions between periodic images (using VASP setting IDIPOL = 4), yielding values of 5.28877 and −3.98253 eV for Li+ and Cl−, respectively.
A full description of the MLFF training process and model parameters can be found in the ESI.‡ For comparison with the kernel-based MLFF, we also developed a neural network-based interatomic potential (NNIP). The details of the NNIP training and model parameters are also discussed in the ESI.‡
While the models developed herein are for pure LiCl systems, careful considerations will be necessary for multicomponent systems to ensure the transferability of developed models across a compositional range.69
Molecular dynamics simulations and free energy calculations
Molecular dynamics (MD) simulations with MLFFs and DFT (AIMD) were performed using VASP, whereas NNIP-based MD was performed using LAMMPS.70 All MD simulations employed a 1 fs time step. Equilibrium densities were determined for all solid and liquid phase systems using isobaric–isothermal (NpT) simulations with at least 500 ps. In VASP, the Parrinello–Rahman barostat71,72 was used to maintain system pressure at 1 bar using a friction coefficient of 5 ps−1 and a fictitious mass of 1000 amu for the lattice degrees-of-freedom. Unless otherwise stated, all isothermal MD simulations (NpT or NVT) in VASP employed the Langevin thermostat with a fiction coefficient of 10 ps−1. After equilibrating the density for each system size and temperature, another 500 ps of equilibration was performed with fixed volume, isothermal (NVT) simulations. NVT simulations in LAMMPS with the NNIP employed the Nose–Hoover thermostat73 to maintain system temperature.All liquid-phase free energy calculations were performed in the NVT ensemble. For MLFF- and NNIP-based TI calculations using the ΔN = N thermodynamic pathway, additional equilibration was performed over 200 ps and the following 300 ps were used for statistical averaging at 880, 910, and 943 K. Using the ΔN = 1 thermodynamic pathway, equilibration was instead performed over 1 ns and the following 2 ns was used for statistical averaging at 943 K. For MLFF-based liquid-phase free energy calculations, the values of λ for each TI window were determined from 10-point Gauss–Lobatto quadrature after applying a variable transformation to convert the integration variable from x ∈ [−1, 1] to λ ∈ [0, 1] with λ(x) = [(x + 1)/2]2, following the same approach for liquid calculations in ref. 44. The resulting λ values used in liquid-phase TI calculations were: [0.000000, 0.001619, 0.017060, 0.068141, 0.174190, 0.339469, 0.546066, 0.755834, 0.921153, 1.000000]. The free energies associated with introducing or removing the soft-core potentials were computed using the Bennett acceptance ratio method42 from 16 separate simulations, where ζ in eqn (3) ranged from 0 to 1 with 16 equally spaced windows 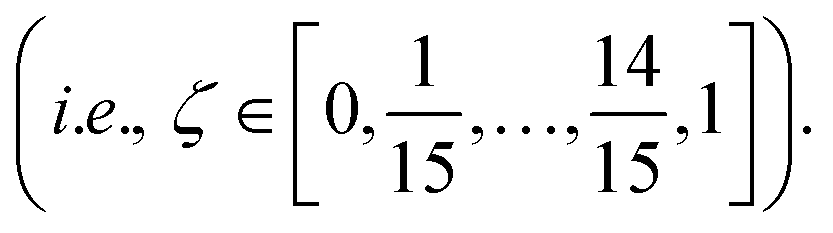 The soft-core potential in eqn (3) was implemented using the PLUMED library39,40 for NNIP-based simulations in LAMMPS. Due to the large computational expense, AIMD-based chemical potential calculations at 943 K were only performed for 10 ps and instead used the 5-point Gauss–Lobatto quadrature rule, where (after variable transformation from x to λ) λ = [0.0, 0.029816, 0.25, 0.68447, 1.0].
The soft-core potential in eqn (3) was implemented using the PLUMED library39,40 for NNIP-based simulations in LAMMPS. Due to the large computational expense, AIMD-based chemical potential calculations at 943 K were only performed for 10 ps and instead used the 5-point Gauss–Lobatto quadrature rule, where (after variable transformation from x to λ) λ = [0.0, 0.029816, 0.25, 0.68447, 1.0].
MLFF-based Einstein crystal calculations were performed in the NVT ensemble at 800, 850, 880, and 900 K with the Andersen thermostat74 to ensure the center of mass was constrained during MD. As opposed to free energy calculations in the liquid phase, Einstein crystal calculations converged significantly faster and only required 100 ps of sampling. Free energies were confirmed to match calculations based on 1 ns of MD. For solid-phase calculations, the values of λ for each TI window were determined from 16-point Gauss–Legendre quadrature after applying a variable transformation to convert the integration variable from x ∈ [−1, 1] to λ ∈ [0, 1] with λ(x) = [(x + 1)/2]2, following the same approach for solid calculations in ref. 44. The resulting λ values used in solid-phase TI calculations were: [0.00529953, 0.02771249, 0.0671844, 0.1222978, 0.19106188, 0.27099161, 0.35919822, 0.45249375, 0.54750625, 0.64080178, 0.72900839, 0.80893812, 0.8777022, 0.9328156, 0.97228751, 0.99470047].
Data availability
Computational data are available from the corresponding authors upon reasonable request.Author contributions
Luke D. Gibson: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft, writing – review & editing. Rajni Chahal: investigation, writing – original draft, writing – review & editing. Vyacheslav S. Bryantsev: funding acquisition, resources, conceptualization, methodology, investigation, writing – review & editing.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors would like to thank Thomas L. Beck (ORNL), Dilip Asthagiri (ORNL), and Juliano Schorne-Pinto (U. of South Carolina) for helpful discussions. The authors would also like to personally thank the NERSC staff, especially Neil Mehta, for his software support in assisting with parallel installation of DeePMD-kit with required modules on Perlmutter. This work was supported by the Office of Materials and Chemical Technologies within the Office of Nuclear Energy, U.S. Department of Energy. This research used resources of the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC05-00OR22725. This research also used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under contract no. DE-AC02-05CH11231.References
- L. C. Dewan, C. Simon, P. A. Madden, L. W. Hobbs and M. Salanne, Molecular dynamics simulation of the thermodynamic and transport properties of the molten salt fast reactor fuel LiF-ThF4, J. Nucl. Mater., 2013, 434, 322–327 CrossRef CAS.
- K. Machado, D. Zanghi, M. Salanne and C. Bessada, Structural, Dynamic, and Thermodynamic Study of KF–AlF3 Melts by Combining High-Temperature NMR and Molecular Dynamics Simulations, J. Phys. Chem. C, 2019, 123, 2147–2156 CrossRef CAS.
- M. S. Emerson, S. Sharma, S. Roy, V. S. Bryantsev, A. S. Ivanov, R. Gakhar, M. E. Woods, L. C. Gallington, S. Dai, D. S. Maltsev and C. J. Margulis, Complete Description of the LaCl3–NaCl Melt Structure and the Concept of a Spacer Salt That Causes Structural Heterogeneity, J. Am. Chem. Soc., 2022, 144, 21751–21762 CrossRef CAS PubMed.
- L. D. Gibson, S. Roy, R. Khanal, R. Chahal, A. Sedova and V. S. Bryantsev, Tracing mechanistic pathways and reaction kinetics toward equilibrium in reactive molten salts, Chem. Sci., 2024, 15, 3116–3129 RSC.
- S. Roy, M. Brehm, S. Sharma, F. Wu, D. S. Maltsev, P. Halstenberg, L. C. Gallington, S. M. Mahurin, S. Dai, A. S. Ivanov, C. J. Margulis and V. S. Bryantsev, Unraveling Local Structure of Molten Salts via X-ray Scattering, Raman Spectroscopy, and Ab Initio Molecular Dynamics, J. Phys. Chem. B, 2021, 125, 5971–5982 CrossRef CAS PubMed.
- S. Roy, Y. Liu, M. Topsakal, E. Dias, R. Gakhar, W. C. Phillips, J. F. Wishart, D. Leshchev, P. Halstenberg, S. Dai, S. K. Gill, A. I. Frenkel and V. S. Bryantsev, A Holistic Approach for Elucidating Local Structure, Dynamics, and Speciation in Molten Salts with High Structural Disorder, J. Am. Chem. Soc., 2021, 143, 15298–15308 CrossRef CAS PubMed.
- R. J. Heaton, R. Brookes, P. A. Madden, M. Salanne, C. Simon and P. Turq, A first-principles description of liquid BeF2 and its mixtures with LiF: 1. Potential development and pure BeF2, J. Phys. Chem. B, 2006, 110, 11454–11460 CrossRef CAS PubMed.
- M. Salanne, C. Simon, P. Turq, R. J. Heaton and P. A. Madden, A first-principles description of liquid BeF2 and its mixtures with LiF: 2. Network formation in LiF-BeF2, J. Phys. Chem. B, 2006, 110, 11461–11467 CrossRef CAS.
- M.-T. Nguyen, V.-A. Glezakou, J. Lonergan, B. McNamara, P. D. Paviet and R. Rousseau, Ab initio molecular dynamics assessment of thermodynamic and transport properties in (K,Li)Cl and (K, Na)Cl molten salt mixtures, J. Mol. Liq., 2021, 326, 115262 CrossRef CAS.
- Y. Okamoto, P. A. Madden and K. Minato, X-ray diffraction and molecular dynamics simulation studies of molten uranium chloride, J. Nucl. Mater., 2005, 344, 109–114 CrossRef CAS.
- Y. Okamoto, S. Suzuki, H. Shiwaku, A. Ikeda-Ohno, T. Yaita and P. A. Madden, Local Coordination about La3+ in Molten LaCl3 and Its Mixtures with Alkali Chlorides, J. Phys. Chem. A, 2010, 114, 4664–4671 CrossRef CAS.
- A. L. Smith, E. Capelli, R. J. M. Konings and A. E. Gheribi, A new approach for coupled modelling of the structural and thermo-physical properties of molten salts. Case of a polymeric liquid LiF-BeF2, J. Mol. Liq., 2020, 299, 112165 CrossRef CAS.
- G. I. L. van Oudenaren, J. A. Ocadiz-Flores and A. L. Smith, Coupled structural-thermodynamic modelling of the molten salt system NaCl-UCl3, J. Mol. Liq., 2021, 342, 117470 CrossRef CAS.
- R. Chahal, S. Roy, M. Brehm, S. Banerjee, V. Bryantsev and S. T. Lam, Transferable Deep Learning Potential Reveals Intermediate-Range Ordering Effects in LiF-NaF-ZrF4 Molten Salt, JACS Au, 2022, 2, 2693–2702 CrossRef CAS PubMed.
- S. Fayfar, R. Chahal, H. Williams, D. N. Gardner, G. Zheng, D. Sprouster, J. C. Neuefeind, D. Olds, A. Hwang, J. Mcfarlane, R. C. Gallagher, M. Asta, S. Lam, R. O. Scarlat and B. Khaykovich, Complex Structure of Molten FLiBe (2LiF-BeF2) Examined by Experimental Neutron Scattering, X-Ray Scattering, and Deep-Neural-Network Based Molecular Dynamics, PRX Energy, 2024, 3, 013001 CrossRef.
- M. Y. Liu, P. Masset and A. Gray-Weale, Solubility of Sodium in Sodium Chloride: A Density Functional Theory Molecular Dynamics Study, J. Electrochem. Soc., 2014, 161, E3042–E3048 CrossRef CAS.
- Y. Shi, S. T. Lam and T. L. Beck, Deep neural network based quantum simulations and quasichemical theory for accurate modeling of molten salt thermodynamics, Chem. Sci., 2022, 13, 8265–8273 RSC.
- M. Salanne, C. Simon, P. Turq and P. A. Madden, Calculation of activities of ions in molten salts with potential application to the pyroprocessing of nuclear waste, J. Phys. Chem. B, 2008, 112, 1177–1183 CrossRef CAS PubMed.
- T. Shah, K. Fazel, J. Lian, L. Huang, Y. Shi and R. Sundararaman, First-principles molten salt phase diagrams through thermodynamic integration, J. Chem. Phys., 2023, 159, 124502 CrossRef CAS PubMed.
- J. Anwar, D. Frenkel and M. G. Noro, Calculation of the melting point of NaCl by molecular simulation, J. Chem. Phys., 2003, 118, 728–735 CrossRef CAS.
- R. S. DeFever and E. J. Maginn, Computing the Liquidus of Binary Monatomic Salt Mixtures with Direct Simulation and Alchemical Free Energy Methods, J. Phys. Chem. A, 2021, 125, 8498–8513 CrossRef CAS.
- W. Zhou and J. Zhang, Direct Calculation of Concentration-Dependent Activity Coefficient of UCl3 in Molten LiCl-KCl, J. Electrochem. Soc., 2015, 162, E199–E204 CrossRef CAS.
- V. Khanna, J. Anwar, D. Frenkel, M. F. Doherty and B. Peters, Free energies of crystals computed using Einstein crystal with fixed center of mass and differing spring constants, J. Chem. Phys., 2021, 154, 164509 CrossRef CAS PubMed.
- D. Frenkel and A. J. C. Ladd, New Monte Carlo method to compute the free energy of arbitrary solids. Application to the fcc and hcp phases of hard spheres, J. Chem. Phys., 1984, 81, 3188–3193 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding Molecular Simulation: From Algorithms to Applications, Elsevier Science, 2001 Search PubMed.
- D. A. Andersson and B. W. Beeler, Ab initio molecular dynamics (AIMD) simulations of NaCl, UCl3 and NaCl-UCl3 molten salts, J. Nucl. Mater., 2022, 568, 153836 CrossRef CAS.
- K. Duemmler, Y. Lin, M. Woods, T. Karlsson, R. Gakhar and B. Beeler, Evaluation of thermophysical properties of the LiCl-KCl system via ab initio and experimental methods, J. Nucl. Mater., 2022, 559, 153414 CrossRef CAS.
- M. Schreuder, J. A. Ocadiz Flores, A. E. Gheribi, O. Benes, J. C. Griveau, E. Colineau, R. J. M. Konings and A. L. Smith, Experimental and Computational Exploration of the NaF-ThF(4) Fuel System: Structure and Thermochemistry, J. Phys. Chem. B, 2021, 125, 8558–8571 CrossRef CAS PubMed.
- M. S. Emerson, A. S. Ivanov, L. C. Gallington, P. Halstenberg, S. Dai, D. S. Maltsev, S. Roy, V. S. Bryantsev and C. J. Margulis, Heterogeneous Structure, Mechanisms of Counterion Exchange, and the Spacer Salt Effect in Complex Molten Salt Mixtures Including LaCl3, J. Phys. Chem. B, 2024, 128, 3972–3980 CrossRef CAS PubMed.
- H. Sun, C. Maxwell, E. Torres and L. K. Béland, Interatomic potential for sodium and chlorine in both neutral and ionic states, Phys. Rev. B, 2024, 109, 174113 CrossRef CAS.
- B. Widom, Some Topics in the Theory of Fluids, J. Chem. Phys., 1963, 39, 2808–2812 CrossRef CAS.
- M. A. Howe and R. L. Mcgreevy, A Neutron-Scattering Study of the Structure of Molten Lithium-Chloride, Philos. Mag. B, 1988, 58, 485–495 CAS.
- G. J. Janz, F. W. Dampier, G. R. Lakshminarayanan, P. K. Lorenz and R. P. T. Tomkins, Molten Salts: Volume 1, Electrical Conductance, Density, and Viscosity Data, Nat. Stand. Ref. Data Ser., 1967, vol. 15 Search PubMed.
- D. B. Sirdeshmukh, L. Sirdeshmukh and K. G. Subhadra, Alkali Halides: A Handbook of Physical Properties, Springer Berlin, Heidelberg, 2001 Search PubMed.
- M. Ferrario, G. Ciccotti, E. Spohr, T. Cartailler and P. Turq, Solubility of KF in water by molecular dynamics using the Kirkwood integration method, J. Chem. Phys., 2002, 117, 4947–4953 CrossRef CAS.
- Y. Shi and T. L. Beck, Absolute ion hydration free energy scale and the surface potential of water via quantum simulation, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30151–30158 CrossRef CAS.
- T. T. Duignan, M. D. Baer, G. K. Schenter and C. J. Mundy, Electrostatic solvation free energies of charged hard spheres using molecular dynamics with density functional theory interactions, J. Chem. Phys., 2017, 147, 161716 CrossRef.
- H. Wang, L. F. Zhang, J. Q. Han and W. N. E, DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics, Comput. Phys. Commun., 2018, 228, 178–184 CrossRef CAS.
- M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Banás, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bottaro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. C. Chen, W. Chen, F. Colizzi, S. De, M. De La Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Ferguson, M. Filizola, J. S. Fraser, H. H. Fu, P. Gasparotto, F. L. Gervasio, F. Giberti, A. Gil-Ley, T. Giorgino, G. T. Heller, G. M. Hocky, M. Iannuzzi, M. Invernizzi, K. E. Jelfs, A. Jussupow, E. Kirilin, A. Laio, V. Limongelli, K. Lindorff-Larsen, T. Löhr, F. Marinelli, L. Martin-Samos, M. Masetti, R. Meyer, A. Michaelides, C. Molteni, T. Morishita, M. Nava, C. Paissoni, E. Papaleo, M. Parrinello, J. Pfaendtner, P. Piaggi, G. Piccini, A. Pietropaolo, F. Pietrucci, S. Pipolo, D. Provasi, D. Quigley, P. Raiteri, S. Raniolo, J. Rydzewski, M. Salvalaglio, G. C. Sosso, V. Spiwok, J. Sponer, D. W. H. Swenson, P. Tiwary, O. Valsson, M. Vendruscolo, G. A. Voth and A. White, Promoting transparency and reproducibility in enhanced molecular simulations, Nat. Methods, 2019, 16, 670–673 CrossRef.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, PLUMED 2: new feathers for an old bird, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- T. C. Beutler, A. E. Mark, R. C. Vanschaik, P. R. Gerber and W. F. Vangunsteren, Avoiding Singularities and Numerical Instabilities in Free-Energy Calculations Based on Molecular Simulations, Chem. Phys. Lett., 1994, 222, 529–539 CrossRef CAS.
- C. H. Bennett, Efficient Estimation of Free Energy differences from Monte Carlo Data, J. Comput. Phys., 1976, 22, 245–268 CrossRef.
- G. Hummer and A. Szabo, Calculation of free-energy differences from computer simulations of initial and final states, J. Chem. Phys., 1996, 105, 2004–2010 CrossRef CAS.
- F. Dorner, Z. Sukurma, C. Dellago and G. Kresse, Melting Si: Beyond Density Functional Theory, Phys. Rev. Lett., 2018, 121, 195701 CrossRef CAS PubMed.
- M. W. Chase, NIST-JANAF Thermochemical Tables, American Chemical Society and American Institute of Physics for National Institute of Standards and Technology, Washington, D.C., 4th edn, 1998 Search PubMed.
- F. Tran, J. Stelzl and P. Blaha, Rungs 1 to 4 of DFT Jacob's ladder: extensive test on the lattice constant, bulk modulus, and cohesive energy of solids, J. Chem. Phys., 2016, 144, 204120 CrossRef PubMed.
- T. L. Beck, M. E. Paulaitis and L. R. Pratt, The Potential Distribution Theorem and Models of Molecular Solutions, Cambridge University Press, Cambridge, 2006 Search PubMed.
- J. G. Kirkwood, Statistical Mechanics of Fluid Mixtures, J. Chem. Phys., 1935, 3, 300–313 CrossRef CAS.
- Z. Mester and A. Z. Panagiotopoulos, Temperature-dependent solubilities and mean ionic activity coefficients of alkali halides in water from molecular dynamics simulations, J. Chem. Phys., 2015, 143, 044505 CrossRef PubMed.
- Z. Mester and A. Z. Panagiotopoulos, Mean ionic activity coefficients in aqueous NaCl solutions from molecular dynamics simulations, J. Chem. Phys., 2015, 142, 044507 CrossRef PubMed.
- L. Zhang, H. Wang, R. Car and W. E, Phase Diagram of a Deep Potential Water Model, Phys. Rev. Lett., 2021, 126, 236001 CrossRef CAS PubMed.
- S. Menon, Y. Lysogorskiy, J. Rogal and R. Drautz, Automated free-energy calculation from atomistic simulations, Phys. Rev. Mater., 2021, 5, 103801 CrossRef CAS.
- K. Shinohara, T. Shibayama, H. Imamura, K. Nishimra, C. Shinagawa, S. Takamoto and J. Li, Presented in Part at the 2024 MRS Fall Meeting, Boston, MA, 2024 Search PubMed.
- G. Kresse and J. Furthmuller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics for Open-Shell Transition-Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Comment on “Generalized gradient approximation made simple” – Reply, Phys. Rev. Lett., 1998, 80, 891 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)], Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Local and gradient-corrected density functionals, Chemical Applications of Density-Functional Theory, 1996, vol. 629, pp. 453–462 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Sivaraman, J. Guo, L. Ward, N. Hoyt, M. Williamson, I. Foster, C. Benmore and N. Jackson, Automated Development of Molten Salt Machine Learning Potentials: Application to LiCl, J. Phys. Chem. Lett., 2021, 12, 4278–4285 CrossRef CAS PubMed.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. Pulay, Convergence Acceleration of Iterative Sequences – the Case of Scf Iteration, Chem. Phys. Lett., 1980, 73, 393–398 CrossRef CAS.
- R. Jinnouchi, F. Karsai and G. Kresse, Making free-energy calculations routine: combining first principles with machine learning, Phys. Rev. B, 2020, 101, 060201 CrossRef CAS.
- R. Jinnouchi, F. Karsai and G. Kresse, On-the-fly machine learning force field generation: application to melting points, Phys. Rev. B, 2019, 100, 014105 CrossRef CAS.
- Z. A. H. Goodwin, M. B. Wenny, J. H. Yang, A. Cepellotti, J. Ding, K. Bystrom, B. R. Duschatko, A. Johansson, L. Sun, S. Batzner, A. Musaelian, J. A. Mason, B. Kozinsky and N. Molinari, Transferability and Accuracy of Ionic Liquid Simulations with Equivariant Machine Learning Interatomic Potentials, J. Phys. Chem. Lett., 2024, 15, 7539–7547 Search PubMed.
- S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular-Dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: a new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- M. Parrinello and A. Rahman, Crystal Structure and Pair Potentials: A Molecular-Dynamics Study, Phys. Rev. Lett., 1980, 45, 1196–1199 CrossRef CAS.
- D. J. Evans and B. L. Holian, The Nose-Hoover Thermostat, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- H. C. Andersen, Molecular dynamics simulations at constant pressure and/or temperature, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
Footnotes |
| † This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://www.energy.gov/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07253g |
| This journal is © The Royal Society of Chemistry 2025 |